 Susanna C. Fagerholm
Susanna C. Fagerholm Carla Guenther
Carla Guenther Marc Llort Asens1
Marc Llort Asens1 Liisa M. Uotila
Liisa M. Uotila- 1Molecular and Integrative Biosciences Research Program, Faculty of Bio- and Environmental Sciences, University of Helsinki, Helsinki, Finland
- 2Iho- ja Allergiasairaala, HUS, Helsinki, Finland
- 3Research Services, University of Helsinki, Helsinki, Finland
Beta2-integrins are complex leukocyte-specific adhesion molecules that are essential for leukocyte (e.g., neutrophil, lymphocyte) trafficking, as well as for other immunological processes such as neutrophil phagocytosis and ROS production, and T cell activation. Intriguingly, however, they have also been found to negatively regulate cytokine responses, maturation, and migratory responses in myeloid cells such as macrophages and dendritic cells, revealing new, and unexpected roles of these molecules in immunity. Because of their essential role in leukocyte function, a lack of expression or function of beta2-integrins causes rare immunodeficiency syndromes, Leukocyte adhesion deficiency type I, and type III (LAD-I and LAD-III). LAD-I is caused by reduced or lost expression of beta2-integrins, whilst in LAD-III, beta2-integrins are expressed but dysfunctional because a major integrin cytoplasmic regulator, kindlin-3, is mutated. Interestingly, some LAD-related phenotypes such as periodontitis have recently been shown to be due to an uncontrolled inflammatory response rather than to an uncontrolled infection, as was previously thought. This review will focus on the recent advances concerning the regulation and functions of beta2-integrins in leukocyte trafficking, immune suppression, and immune deficiency disease.
Integrins and integrin regulation
Integrins are heterodimeric type I transmembrane proteins consisting of alpha and beta subunits (1). Integrins are expressed in all nucleated cells and play a key role in adhesion, cell communication, and migration. They mediate adhesion to the extracellular matrix, by binding to the RGD motif of fibronectin, collagen, and laminin, among others (2). Integrins in leukocytes also bind to soluble ligands such as the complement component iC3b, and to other cells, by binding to ICAMs (Intercellular adhesion molecules) and VCAM-1 (Vascular cell adhesion molecule) (3, 4). Additionally, integrins link to the actin cytoskeleton inside the cell and thereby connect the inside of the cell with the outside.
Integrins have large extracellular domains which contain the ligand-binding sites, and short cytoplasmic domains which are important for integrin regulation. The ability of the integrin to bind to ligands is regulated through conformational changes as well as by integrin clustering. Integrins can be found in three main conformational states: inactive (bent-closed), intermediate (extended-closed), and active state (extended-open) (5). The predominant state seems to be the inactive (bent-closed) state based on affinity and thermodynamics studies with K562 cells (alpha5beta1-integrins, bent-closed: 99.75%; extended-closed: 0.10%; extended-open: 0.15%) (6). The active conformation (extended-open) has a 4,000-fold increase in ligand affinity compared to the other two states (7). Also on resting peripheral T cells the vast majority of LFA-1 (Lymphocyte function-associated antigen-1, alphaLbeta2-integrin) appear to be in the inactive conformation, as stabilizing the active conformation leads to a 1,000-fold increase in affinity of the integrin (8). The LFA-1 conformational change (integrin extension) on the surface of migrating T cells has recently been directly measured by super-resolution microscopy [interferometric photoactivation, and localization microscopy (iPALM)] (9).
Integrin activation takes place upon cell stimulation through various cell surface receptors such as chemokine receptors or the T cell receptor. Cell stimulation triggers an inside-out signaling pathway that ultimately recruits cytoplasmic factors such as talin and kindlin to the NPxY motifs of the cytoplasmic tail of the integrin's beta-chain, which causes the cytoplasmic tails of the integrin subunits to separate (10) and switches the integrin to the active (extended-open) conformation (11, 12). Kindlin and talin connect the integrin to the actin cytoskeleton and stabilize the extended-open conformation of the integrin through actin cytoskeleton exerted tensile force (6, 13). In addition, many other proteins, such as 14-3-3 proteins, alpha-actinin, coronin 1A, cytohesin 1, filamin A, and Dok1 can interact directly with the integrin beta-chain and modulate integrin function (14–16). These interactions are often regulated by phosphorylation of the integrin beta-chain cytoplasmic domain (15–18).
In addition to their ability to respond to the environment through inside-out signaling, integrins can take part in a variety of signaling cascades following ligand binding (outside-in signaling). Integrins take part in the formation of adhesion complexes and focal adhesions in cells such as fibroblasts, modulation of actin cytoskeleton dynamics, cell migration, differentiation, proliferation, angiogenesis, and apoptosis (19).
Beta2-Integrins in Leukocyte Trafficking
Beta2-integrins (CD11a/CD18, alphaLbeta2, LFA-1; CD11b/CD18, alphaMbeta2, Mac-1, CR3; CD11c/CD18, alphaXbeta2, p150.95, CR4; and CD11d/CD18, alphaDbeta2) are a subgroup of integrins which share a common beta2- or CD18-chain but have different alpha-chains and ligands. Beta2-integrins are expressed exclusively in leukocytes, but the different members have their own distinct expression pattern. CD11a/CD18 is expressed on all leukocytes, while CD11b/CD18, CD11c/CD18, and CD11d/CD18 are mainly expressed on myeloid cells, but at varying levels (19, 20). CD11a/CD18 has a more restricted ligand binding capacity than the other beta2-integrins, and binds ligands such as ICAM-1-5 found on the surface of other cells. In contrast, CD11b/CD18 is a very promiscuous integrin with more than 40 reported ligands, including ICAMs, iC3b, fibrinogen, RAGE (receptor for advanced glycation end products), and CD40L (20). Interestingly, ligand-specific blockade of CD11b/CD18 has recently been shown to protect against bacterial sepsis, while blocking all CD11b/CD18 functions potentiates it, showing that CD11b/CD18 indeed has very complicated roles in immunity due to its many ligands (21). In addition to leukocytes, beta2-integrins are also found in extracellular vesicles (EVs), and integrins in EVs may play novel roles in development of pathogenic conditions such as sepsis (22).
It is undisputed that beta2-integrins are of fundamental importance for leukocyte trafficking. This is because they are required for the firm adhesion to the endothelial layer surrounding the blood vessels under conditions of shear flow (blood flow) and for leukocyte extravasation into tissues (23). The leukocyte adhesion cascade (Figure 1) is a multistep process involving rolling, firm adhesion or arrest, spreading/crawling, and finally extravasation (24). This complex process is accomplished by several proteins acting in parallel and succession, as the leukocyte proceeds to its destination. Initially contacts between the leukocyte and the endothelial cells allows selectins and ICAM-1 on endothelial cells to mediate leukocyte rolling on the endothelium. The close contact between the cells during rolling allows the leukocyte to sense chemokines present on the endothelium. In neutrophils, both selectins and chemokine receptors activate beta2-integrins via a signaling pathway involving the small GTPase Rap1a and phosphatidylinositol-4-phosphate 5-kinase (PIP5Kγ90). The activation of beta2-integrins involves conversion into the intermediate affinity state that mediates slow rolling, followed by conversion into the high affinity state, which mediates leukocyte arrest (25). Both selectins and integrins can form slip bonds, whose lifetime is shortened by applied shear force, as well as catch bonds, which strengthen under shear force (26–28), inducing further changes downstream of the integrins. During these leukocyte-endothelial contacts numerous integrin-ligand bonds are continuously broken and formed and further reinforced by the recruitment of more integrins and downstream cytoskeletal proteins such as talin, kindlin-3, focal adhesion kinase, and paxillin to form adhesion complexes which strengthen cell adhesion and induce actin reorganization and cell spreading (26). Following adhesion, cells crawl along the endothelium looking for a suitable extravasation site, a process critically dependent on the beta2-integrin CD11b/CD18 (29). As integrins act as mechanosensors in cells (30), it is likely that integrins are also central for the subsequent steps of probing the endothelium for suitable points of exit, either through a paracellular or transcellular route.
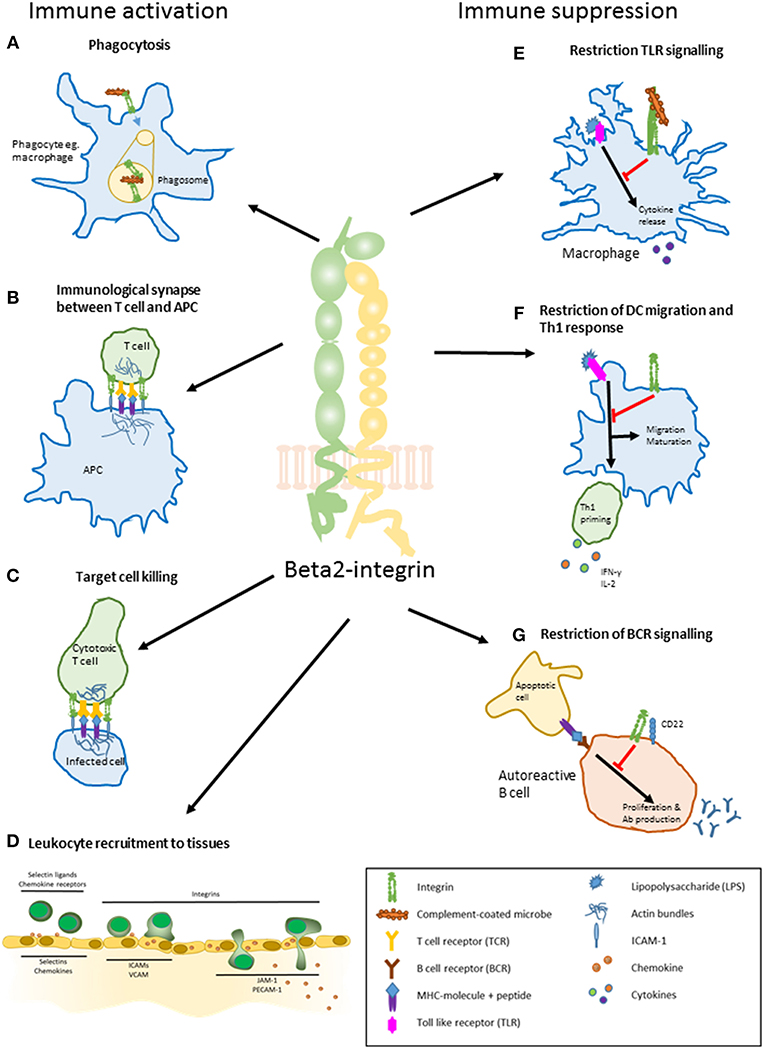
Figure 1. The main roles of beta2-integrins in immune activation and suppression. (A) phagocytosis. Beta2-integrins mediate phagocytosis by binding to iC3b on the surface of complement-coated bacteria. (B) regulation of T cell activation by a dendritic cell. Beta2-integrins participate in the formation of an immunological synapse between a T cell and an antigen presenting cell such as DC. The synapse stabilizes the interaction between and regulates signaling in the two participating cells. (C) target cell killing. Beta2-integrins participate in forming and maintaining an immunological synapse between a cytotoxic T cell and an infected cell. (D) leukocyte recruitment to tissues. Leukocytes are activated by selectins and chemokines on the surface of activated endothelial cells close to a site of inflammation. This slows down the leukocyte speed and induces integrins to change their conformation through inside-out signaling, allowing them to bind ICAMs on the endothelium. Beta2-integrins are essential in the slow rolling and firm adhesion of a leukocyte, after which the cells transmigrate to the inflamed tissue. Leukocytes use the chemokine gradient to navigate toward the site of inflammation. (E) regulation of TLR-signaling. Beta2-integrin CD11b/CD18 restrains macrophage activation and cytokine production upon TLR (Toll-like receptor) activation by LPS (lipopolysaccharide). (F) restriction of dendritic cell migration, maturation, and Th1 priming. Proper beta2-integrin—cytoskeleton linkage controls DC maturation toward a migratory phenotype and restricts priming of Th1 cells. (G) restriction of B cell receptor signaling. Interaction of CD11b/CD18 and CD22 on the surface of an autoreactive B cell leads to constraint in BCR signaling. This decreases auto-reactive B cell proliferation and antibody production.
Talin has long been known to be indispensable for leukocyte trafficking (31–34). More recently, also kindlin-3 and its interaction with the beta2-integrin tail has been shown to be vital for neutrophil and effector T cell firm adhesion under shear flow and for neutrophil and T cell trafficking in vivo (35–38), and for homing of progenitor T cells to the vascularized thymus (39). However, talin and kindlin-3 regulate different aspects of leukocyte trafficking. Talin is required for the conformational change of the integrin to the extended, intermediate affinity conformation which mediates slow rolling. In contrast, both talin and kindlin-3 are required for the induction of the high-affinity conformation, full integrin activation and neutrophil arrest (33, 38, 40). Recently, Src kinase-associated phosphoprotein 2 (Skap2) has been shown to be essential for the recruitment of talin and kindlin-3 to the beta2-integrin tail, and for neutrophil trafficking in vivo (41). Interestingly, a bent-open conformation of beta2-integrins has been reported on neutrophils, which limits neutrophil recruitment by binding to ICAM-1 in cis, but the molecular mechanisms regulating this process are currently unknown (42).
In contrast to talin and kindlin-3, filamin A has been suggested to negatively regulate integrin functions in vitro (15, 43, 44). However, it has also been reported to be required for platelet shear flow adhesion because it stabilizes the links between the plasma membrane and the underlying actin cytoskeleton (45). Recent studies utilizing T cell-specific filamin A-deficient mice have shown that filamin A is required for the optimal firm adhesion of T cells under shear flow conditions, trafficking of T cells into lymph nodes, and to the inflamed skin (46). These results demonstrate that in T cells, filamin A does not function as an integrin inhibitor but rather is required for cell trafficking in vivo. However, filamin A is not required for neutrophil adhesion under shear flow conditions, but instead filamin A-deficient neutrophils display enhanced adhesion, spreading, and defects in uropod retraction, thereby revealing cell-type specific functions of this integrin interacting protein (47, 48).
In contrast to leukocyte trafficking from blood stream to lymph nodes and tissues, leukocyte trafficking within tissues (e.g., in a confined 3D environment in the absence of shear flow) is a mechanistically different process that can occur even in the absence of integrins (49). In lymph nodes, integrins, and chemokine receptors contribute partly to naïve T cell migration speed (50). In this environment the integrin CD11a/CD18 (LFA-1) is required as a frictional interface with the substrate (the so called “integrin clutch”) by generating traction forces, but does not mediate substantial adhesion to the substrate (50). In some cases, integrins can even restrict leukocyte migration in tissues. Indeed, the beta2-integrin-kindlin-3 interaction negatively regulates DC migration to lymph nodes both under steady state and inflammatory conditions (36, 51). beta2-integrins restrict DC migration through a downstream mechanism which involves regulation of the transcriptional program and migratory phenotype of these cells (Figure 1).
Beta2-Integrins in Other Immune-Related Functions
In addition to their fundamentally important role in leukocyte trafficking, beta2-integrins also mediate other cell-cell contacts that are essential for immunological processes (Figure 1). Beta2-integrins (e.g., CD11a/CD18-integrin; LFA-1) are central components of the immunological synapse which forms between an antigen presenting cell (APC) and a T cell [reviewed in Dustin (52)], between a B cell and a T cell (53) and between an NK cell and its target cell (54). In brief, the cell-cell interactions mediated by CD11a/CD18 on the T cell enables T cell activation, by binding to ICAM-1 on the APC. T cells sample antigens on dendritic cells in lymph nodes via short term contacts, termed kinapses (52). When antigen is found, T cells stop migrating and form an immunological synapse with the dendritic cell (52). LFA-1 on the T cell binding to ICAM-1 on the DC play a crucial role in this structure. LFA-1, together with talin, kindlin-3, and Rap1, is positioned in the p-SMAC (peripheral supramolecular activation cluster), thereby stabilizing the interaction between the T cell receptor and peptide-MHC II at the center of the contact (c-SMAC) (52, 55). Optimal T cell activation in vivo requires talin and kindlin-3 to bind to LFA-1 (32, 56). Upon activation, LFA-1 can the signal into the T cell and thereby contribute to T cell activation and polarization of the T cell response (57). For example, LFA-1 ligation in T cells has been shown to promote Th1 polarization through a pathway involving Erk and Akt-mediated GSK3beta-inhibition, in turn leading to activation of the Notch pathway (58), and LFA-1 can also be regulated by, and engage in crosstalk with TGF-beta signaling in T cells (59, 60). In addition, a role for an intracellular pool of beta2-integrins in T cell activation and differentiation has recently been reported (61).
In addition to T cell activation, CD11a/CD18 is involved in the killing of infected target cells by cytotoxic T cells, by stabilizing the contact between the T cell and the target cell, and by sealing the contact zone so that cytolytic granules cannot escape (57). LFA-1 furthermore plays a role in the generation of T cell memory (57), survival of T follicular helper cells (62) and regulatory T cells (63) and B cell-mediated antibody production, by mediating cell-cell contacts, but also by initiating intracellular signaling cascades (57, 64). LFA-1 is important for CD8+ T cell trafficking (65) and for Th2 (but not Th1) homing, as well as Th2-induced allergic lung disease (66). Interestingly, certain CD11a polymorphisms critically influence Th2 homing (67).
In myeloid cells such as macrophages, beta2-integrins can initiate intracellular signaling pathways leading to cytokine secretion, either by themselves or together with Toll like receptors (TLRs) (21, 68, 69). In addition, many neutrophil functions such as cytokine release and oxidative burst are dependent on beta2-integrins (70–73). CD11b/CD18 and CD11c/CD18 are receptors for complement component iC3b and are essential for phagocytosis of opsonized pathogens in neutrophils and other phagocytic cells, where they induce a RhoA-dependent phagocytic pathway (74–76). The differential roles of these two highly similar integrins have been studied in vivo in CD11b−/− and CD11c−/− mice. The results indicate that CD11b/CD18 is involved in neutrophil functions and in the anti-inflammatory functions of macrophages, whereas CD11c/CD18 is more relevant in the regulation of macrophage inflammatory functions (77). Recently, beta2-integrins has been shown to be required for recruitment of monocytes, as well as hematopoiesis of these cells during Schistosome infection, and a low expression of beta2-integrins correlates with increased parasite burden in a murine model of the disease (78).
Beta2-Integrins in Immune Suppression
In addition to their well-characterized role in mediating cellular interactions and promoting pro-inflammatory signaling, beta2-integrins have also been associated with many immunosuppressive functions (20) (Figure 1). Beta2-integrins can inhibit TLR signaling in macrophages through negative feedback loops, either directly or indirectly, through the anti-inflammatory cytokine IL-10 (79, 80). TLR stimulation leads to PI(3)K- and RapL-mediated inside-out activation of CD11b/CD18. Integrin outside-in signaling activates Src/Syk, leading ultimately to degradation of the important TLR signaling transducers MyD88 and TRIF and downregulation of TLR signaling (80). The mechanism of CD11b/CD18-dependent modulation of TLR responses has been shown to involve inhibition of the NF-κB pathway and activation of the p38 MAPK pathway (81). Beta2-integrins have been found to repress DC-mediated T cell activation (82–84), and the presence of CD11b/CD18 on APCs has been demonstrated to suppress Th17 differentiation and lead to immune tolerance (85, 86). Recently, CD11b/CD18-expressing neutrophils have been shown to suppress T cell-dependent influenza pathology in vivo by limiting T cell proliferation (87). The immunoregulatory role of leukocyte integrins may be taken advantage of by the macrophage-infecting bacterium Francisella tularensis, which is phagocytosed in a CD11b-dependent manner and uses the CD11b-driven inhibition of inflammasome activation to evade the innate immune system (88). In addition to opsonized bacteria, CD11b/CD18, and CD11c/CD18 also recognize iC3b-opsonized apoptotic cells, which leads to inhibition of proinflammatory cytokine production through NF-κB inhibition (89).
A series of important findings of the immunoregulatory roles of beta2-integrins has been produced using mice where the kindlin-3 binding site in the CD18-chain has been mutated, leading to expressed but inactive integrins on the surface of immune cells (TTT/AAA-beta2-integrin KI mice) (35). DCs from these mice mature toward a migratory phenotype, accumulate in lymphoid organs, and induce increased Th1 immune responses in vivo (51). In addition, functional integrins are essential for restricting the accumulation of mast cells in inflamed skin and mast cell responses in vitro, and inflammatory cytokine production in the inflamed skin in vivo (36). In the context of obesity-associated inflammation, mice on a high fat diet display increased numbers of neutrophils in white adipose tissue, increased insulin resistance and elevated inflammatory profile (90). However, the total deletion of an individual beta2-integrin, e.g., CD11b in mice led to increased weight gain on a high fat diet and lowered insulin sensitivity but to decreased inflammatory gene expression compared to WT mice in vivo, suggesting that the CD11b-integrin specifically is pro-inflammatory under diet-induced obesity conditions (91).
Interestingly, variations at the ITGAM gene, which encodes for CD11b, is one of the strongest genetic risk factors for systemic lupus erythematosus (SLE). These nucleotide polymorphisms confer amino acid changes in the CD11b protein, leading to deficient ligand binding, and a reduced ability to restrict cellular cytokine expression (92–94). Interestingly, activation of CD11b/CD18 with a CD11b agonist LA1 is able to overcome the effects of CD11b/CD18 malfunction in the carriers of the SLE-associated polymorphisms (95).
While most of the findings concerning the immunoregulatory role of beta2 integrins have been made in myeloid cells, also some lymphocyte subgroups express CD11b/CD18. Indeed, in B cells, CD11b/CD18 has been shown to negatively regulate B cell receptor signaling to maintain autoreactive B cell tolerance (96). Together, these results show that, while it is clear that beta2-integrins are important for immune cell activation and function, beta2 integrins (especially CD11b) have an equally significant role in repressing the body's reactions against self. Therefore, manipulating integrin activation pharmacologically could be an efficient therapeutic approach in treating certain inflammatory or autoimmune diseases.
Leukocyte Adhesion Deficiencies
The importance of beta2-integrins in immunity is highlighted by the rare genetic diseases known as Leukocyte adhesion deficiencies type I and type III (LAD-I and LAD-III) (Table 1). LAD syndromes are a group of congenital autosomal-recessive diseases with immune deficiency condition resulting in impaired leukocyte adhesion and migration. In LAD-I, the expression of CD18 (the beta2-integrin-chain) is either diminished or abolished. In LAD-III, mutations in kindlin-3 prevents it from activating beta2-integrins. Both conditions present partly with similar symptoms, which include leukocytosis and a lack of neutrophil extravasation from the blood stream into tissues. Consequently, the patients end up suffering from recurrent life-threatening infections, unless they receive a hematopoietic stem cell transplant (HSCT) (97). LAD-II is a selectin- (rather than integrin) related disease which is caused by a failure in selectin ligand expression (98) and will not be discussed further here.
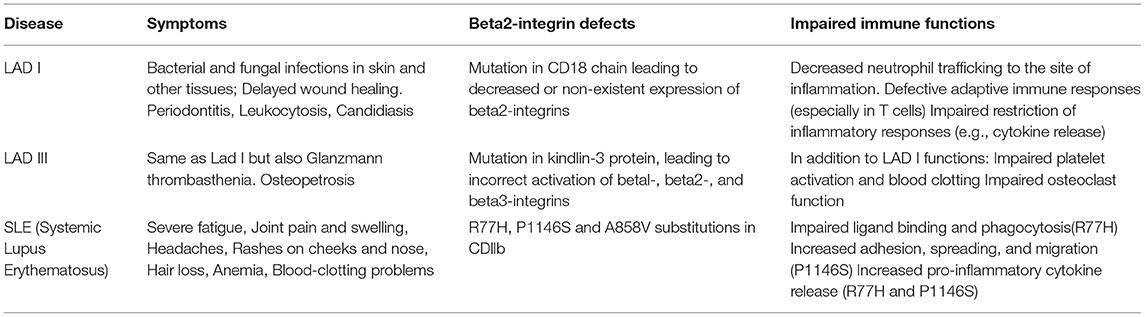
Table 1. Beta2-integrins in immunodeficiency and inflammatory disease.
LAD I—Over 200 mutations have been identified in LAD-I patients which cause decreased expression of CD18. The severity of the disease varies according to the functionality of the beta2-integrin (99). LAD-I patients suffer from life threatening bacterial and fungal infections early in life, and especially neutrophil trafficking is reduced into the inflamed tissue. In a recent (2018) review of all published LAD cases before 2017 (323 cases) (100) it was reported that the most common infections in severe LAD-I (<2% CD18 expression) were respiratory tract infections (including pneumonia), sepsis, and otitis media whilst in LAD-I with moderate CD18 expression the most common infections were periodontal infection, otitis media and sepsis. Perianal skin infections and necrotic skin ulcers were reported in both groups. Delayed umbilical cord detachment is common. In addition, patients suffer from symptoms such as delayed wound healing.
For severe LAD-I, survival beyond 2 years of age was only 39%, showing that severe LAD-I remains a life-threatening condition (100). The prognosis for LAD-I with moderate CD18 expression is much better, with survival over 2 years and beyond (to adulthood) for over 90% of cases with >4% CD18 expression (100, 101).
HSCT remains the only cure for patients expressing very low (<1–2 %) levels of CD18 protein in leukocytes, but unfortunately transplant-related mortality remains high (19% for all groups in LAD-I) (100).
Beta2-integrin deficient mice have similar immune defects as LAD-I patients (102). These mice have been useful to investigate the role of beta2-integrins and their function in different leukocytes (102, 103).
LAD-III—LAD-III is a much rarer disease than LAD-I, with <40 patients reported worldwide (104). Patients suffer from similar symptoms as LAD-I patients, e.g., recurrent bacterial infections including bacteremias, pulmonary infections, omphalitis, and other soft tissue infections. Also fungal infections have been reported. However, unlike LAD-I patients, LAD-III patients additionally have Glanzmann type thrombasthenia, a bleeding disorder. Transfusions have been performed in >90% of cases as bleeding is a hallmark of the disease and remains a serious complication (105). In addition, recombinant factor VIIa has been used successfully in LAD-III to treat bleeding events (105). Furthermore, patients can suffer from osteopetrosis, due to deficient integrin-mediated osteoclast bone resorption.
LAD-III patients have normal integrin expression but carry mutations in the FERMT3 gene encoding kindlin-3 protein (106). Since kindlin-3 binds to beta1, beta2, and beta3-integrins and regulates their function, patients display more complex symptoms compared to LAD-I patients. In platelets kindlin-3 is required for αIIbβ3-integrin-mediated formation of blood clots. Kindlin-3 further regulates normal bone regeneration by several integrins. As for LAD-I, the only curative treatment for LAD-III is HSCT, and HSCT-related mortality remains high [22%, (105)].
Kindlin-3 has a central role in immunity which is shown by the phenotype of the kindlin-3 deficient mice (12, 38). These mice die early after birth because of excessive bleeding. These mice, as well as mice carrying a mutation in the kindlin-3 binding site in beta2-integrin cytoplasmic tail (TTT/AAA-beta2-integrin KI mice) have shown a crucial role of kindlin-3 and beta2-integrins in the regulation of immune cells (35, 36, 51, 56).
LAD and Inflammation
Many of the symptoms in LAD patients are thought to be caused by defective leukocyte (especially neutrophil) trafficking into inflamed tissue. However, not all symptoms in LAD-I are due to defective leukocyte-mediated immune surveillance. Instead, periodontitis and associated bone loss in LAD-I has recently been shown to be associated with an increased inflammatory response, with excessive production of IL-17 and related cytokines (107), and blocking the IL-17 cytokine response reduces symptoms in a LAD-I patient (108). In addition, particular inflammatory disorders (e.g., colitis) have been reported in LAD-patients (109–111). This indicates that at least some pathological symptoms in LAD-I patients are caused by dysregulated inflammatory responses. The increased IL-17 production in LAD-I patients may be—at least in part—due to defective neutrophil recruitment into tissues e.g., dysregulation of the so called “neutrostat,” which senses and regulates neutrophil numbers in vivo (107). However, beta2-integrins have been shown to directly restrict cytokine responses in many types of immune cells, such as macrophages (80), DCs (51), and mast cells (36), and to restrict Th1 (51) and Th17 (85) polarization in vivo. In addition, functional beta2-integrins restrict expression of cytokines in a skin inflammation model, although neutrophil trafficking is relatively normal in this model (36). Dysregulated cytokine responses may therefore contribute to the paradoxical increase in inflammation (periodontitis, colitis) in LAD-I patients (107, 109, 110, 112).
Therapeutic Targeting of Beta2-Integrins
Because of the crucial role of beta2-integrins in leukocyte functions such as leukocyte recruitment, beta2-integrins have been considered attractive targets in inflammatory disease such as psoriasis, arthritis, and multiple sclerosis [reviewed in Mitroulis et al. (113)]. Indeed, an antibody against alphaL-integrin chain, efalizumab, has previously been in clinical use in psoriasis (113). However, the drug was withdrawn from the market in 2009 because it was associated with serious side effects, e.g., reactivation of latent John Cunningham (JC) virus infection and resulting progressive multifocal leukoenphalopathy (PML). Therefore, therapeutic blocking of beta2-integrins in disease may be difficult because these molecules play such multifaceted roles in central immune reactions.
Conclusions and Future Perspectives
Beta2-integrins are of crucial importance for leukocyte trafficking and immune cell activation, but interestingly play a role in immune suppression as well. Consequently, dysfunctional or absent integrins are linked not only to immune deficiency disease but also to inflammatory disease, thereby contributing to both ends of the spectrum of immune-related diseases. A better understanding of the disease processes where dysfunctional beta2-integrins are involved may provide novel drug targets for immunodeficiency and inflammatory disease symptoms (95, 108).
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author's laboratory has funding from Academy of Finland, Sigrid Juselius foundation, HiLIFE/University of Helsinki, Liv och Hälsa, Svenska Kulturfonden, and Magnus Ehrnrooth foundation. We thank Heidi Harjunpää for useful comments on the manuscript.
References
1. Hynes RO. Integrins: a family of cell surface receptors. Cell (1987) 48:549–54. doi: 10.1016/0092-8674(87)90233-9
2. Pierschbacher MD, Ruoslahti E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature (1984) 309:30–3. doi: 10.1038/309030a0
3. Rothlein R, Dustin ML, Marlin SD, Springer TA. A human intercellular adhesion molecule (ICAM-1) distinct from LFA-1. J Immunol. (1986) 137:1270–4.
4. Patarroyo M, Clark EA, Prieto J, Kantor C, Gahmberg CG. Identification of a novel adhesion molecule in human leukocytes by monoclonal antibody LB-2. FEBS Lett. (1987) 210:127–31. doi: 10.1016/0014-5793(87)81321-2
5. Nishida N, Xie C, Shimaoka M, Cheng Y, Walz T, Springer TA. Activation of leukocyte beta2 integrins by conversion from bent to extended conformations. Immunity (2006) 25:583–94. doi: 10.1016/j.immuni.2006.07.016
6. Li J, Springer TA. Integrin extension enables ultrasensitive regulation by cytoskeletal force. Proc Natl Acad Sci USA. (2017) 114:4685–4690. doi: 10.1073/pnas.1704171114
7. Li J, Su Y, Xia W, Qin Y, Humphries MJ, Vestweber D, et al. Conformational equilibria and intrinsic affinities define integrin activation. EMBO J. (2017) 36:629–45. doi: 10.15252/embj.201695803
8. Schurpf T, Springer TA. Regulation of integrin affinity on cell surfaces. EMBO J. (2011) 30:4712–27. doi: 10.1038/emboj.2011.333
9. Moore TI, Aaron J, Chew TL, Springer TA. Measuring integrin conformational change on the cell surface with super-resolution microscopy. Cell Rep. (2018) 22:1903–12. doi: 10.1016/j.celrep.2018.01.062
10. Kim M, Carman CV, Springer TA. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science (2003) 301:1720–5. doi: 10.1126/science.1084174
11. Campbell ID, Ginsberg MH. The talin-tail interaction places integrin activation on FERM ground. Trends Biochem Sci. (2004) 29:429–35. doi: 10.1016/j.tibs.2004.06.005
12. Moser M, Nieswandt B, Ussar S, Pozgajova M, Fassler R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med. (2008) 14:325–30. doi: 10.1038/nm1722
13. Nordenfelt P, Elliott HL, Springer TA. Coordinated integrin activation by actin-dependent force during T-cell migration. Nat Commun. (2016) 7:13119. doi: 10.1038/ncomms13119
14. Morse EM, Brahme NN, Calderwood DA. Integrin cytoplasmic tail interactions. Biochemistry (2014) 53:810–20. doi: 10.1021/bi401596q
15. Takala H, Nurminen E, Nurmi SM, Aatonen M, Strandin T, Takatalo M, et al. Beta2 integrin phosphorylation on Thr758 acts as a molecular switch to regulate 14–3-3 and filamin binding. Blood (2008) 112:1853–62. doi: 10.1182/blood-2007-12-127795
16. Thome S, Begandt D, Pick R, Salvermoser M, Walzog B. Intracellular beta2 integrin (CD11/CD18) interacting partners in neutrophil trafficking. Eur J Clin Invest. (2018) 48:e12966. doi: 10.1111/eci.12966
17. Chatterjee D, Zhiping LL, Tan SM, Bhattacharjya S. Interaction analyses of the integrin beta2 cytoplasmic tail with the F3 FERM domain of talin and 14–3-3zeta reveal a ternary complex with phosphorylated tail. J Mol Biol. (2016) 428:4129–42. doi: 10.1016/j.jmb.2016.08.014
18. Fagerholm SC, Hilden TJ, Nurmi SM, Gahmberg CG. Specific integrin alpha and beta chain phosphorylations regulate LFA-1 activation through affinity-dependent and -independent mechanisms. J Cell Biol. (2005) 171:705–15. doi: 10.1083/jcb.200504016
19. Tan SM. The leucocyte beta2 (CD18) integrins: the structure, functional regulation and signalling properties. Biosci Rep. (2012) 32:241–69. doi: 10.1042/BSR20110101
20. Schittenhelm L, Hilkens CM, Morrison VL. beta2 integrins as regulators of dendritic cell, monocyte, and macrophage function. Front Immunol. (2017) 8:1866. doi: 10.3389/fimmu.2017.01866
21. Wolf D, Anto-Michel N, Blankenbach H, Wiedemann A, Buscher K, Hohmann JD, et al. A ligand-specific blockade of the integrin Mac-1 selectively targets pathologic inflammation while maintaining protective host-defense. Nat Commun. (2018) 9:525. doi: 10.1038/s41467-018-02896-8
22. Kawamoto E, Masui-Ito A, Eguchi A, Soe ZY, Prajuabjinda O, Darkwah S, et al. Integrin and PD-1 ligand expression on circulating extracellular vesicles in systemic inflammatory response syndrome and sepsis. Shock (2018). doi: 10.1097/SHK.0000000000001228. [Epub ahead of print].
23. Gahmberg CG, Fagerholm SC, Nurmi SM, Chavakis T, Marchesan S, Gronholm M. Regulation of integrin activity and signalling. Biochim Biophys Acta (2009) 1790:431–44. doi: 10.1016/j.bbagen.2009.03.007
24. Klaus Ley CL, Myron I. Cybulsky, sussan nourshargh getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. (2007) 7:678–89. doi: 10.1038/nri2156
25. Yago T, Zhang N, Zhao L, Abrams CS, McEver RP. Selectins and chemokines use shared and distinct signals to activate β2 integrins in neutrophils. Blood Adv. (2018) 2:731–44. doi: 10.1182/bloodadvances.2017015602
26. McEver RP, Zu C. Rolling cell adhesion. Annu Rev Cell Dev Biol. (2010) 26:363–96. doi: 10.1146/annurev.cellbio.042308.113238
27. Jiang G, Giannone G, Critchley DR, Fukumoto E, Sheetz MP. Two-piconewton slip bond between fibronectin and the cytoskeleton depends on talin. Nature (2003) 424:334–7. doi: 10.1038/nature01805
28. Kong F, Garcia AJ, Mould AP, Humphries MJ, Zhu C. Demonstration of catch bonds between an integrin and its ligand. J Cell Biol. (2009) 185:1275–84. doi: 10.1083/jcb.200810002
29. Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med. (2006) 203:2569–75. doi: 10.1084/jem.20060925
30. Sun Z, Guo SS, Fässler R. Integrin-mediated mechanotransduction. J Cell Biol. (2016) 215:445–56. doi: 10.1083/jcb.201609037
31. Manevich-Mendelson E, Grabovsky V, Feigelson SW, Cinamon G, Gore Y, Goverse G, et al. Talin1 is required for integrin-dependent B lymphocyte homing to lymph nodes and the bone marrow but not for follicular B-cell maturation in the spleen. Blood (2010) 116:5907–18. doi: 10.1182/blood-2010-06-293506
32. Wernimont SA, Wiemer AJ, Bennin DA, Monkley SJ, Ludwig T, Critchley DR, et al. Contact-dependent T cell activation and T cell stopping require talin1. J Immunol. (2011) 187:6256–67. doi: 10.4049/jimmunol.1102028
33. Lefort CT, Rossaint J, Moser M, Petrich BG, Zarbock A, Monkley SJ, et al. Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood (2012) 119:4275–82. doi: 10.1182/blood-2011-08-373118
34. Klapholz B, Brown NH. Talin - the master of integrin adhesions. J Cell Sci. (2017) 130:2435–46. doi: 10.1242/jcs.190991
35. Morrison VL, Macpherson M, Savinko T, San Lek H, Prescott A, Fagerholm SC. The beta2 integrin-kindlin-3 interaction is essential for T-cell homing but dispensable for T-cell activation in vivo. Blood (2013) 122:1428–36. doi: 10.1182/blood-2013-02-484998
36. Savinko TS, Morrison VL, Uotila LM, Wolff CH, Alenius HT, Fagerholm SC. Functional Beta2-integrins restrict skin inflammation in vivo. J Invest Dermatol. (2015) 135:2249–57. doi: 10.1038/jid.2015.164
37. Moretti FA, Moser M, Lyck R, Abadier M, Ruppert R, Engelhardt B, et al. Kindlin-3 regulates integrin activation and adhesion reinforcement of effector T cells. Proc Natl Acad Sci USA. (2013) 110:17005–10. doi: 10.1073/pnas.1316032110
38. Moser M, Bauer M, Schmid S, Ruppert R, Schmidt S, Sixt M, et al. Kindlin-3 is required for beta2 integrin-mediated leukocyte adhesion to endothelial cells. Nat Med. (2009) 15:300–5. doi: 10.1038/nm.1921
39. Moretti FA, Klapproth S, Ruppert R, Margraf A, Weber J, Pick R, et al. Differential requirement of kindlin-3 for T cell progenitor homing to the non-vascularized and vascularized thymus. eLife (2018) 7:e35816. doi: 10.7554/eLife.35816
40. Calderwood DA, Zent R, Grant R, Rees DJ, Hynes RO, et al. The Talin head domain binds to integrin beta subunit cytoplasmic tails and regulates integrin activation. J Biol Chem. (1999) 274:28071–4. doi: 10.1074/jbc.274.40.28071
41. Boras M, Volmering S, Bokemeyer A, Rossaint J, Block H, Bardel B, et al. Skap2 is required for beta2 integrin-mediated neutrophil recruitment and functions. J Exp Med. (2017) 214:851–74. doi: 10.1084/jem.20160647
42. Fan Z, McArdle S, Marki A, Mikulski Z, Gutierrez E, Engelhardt B, et al. Neutrophil recruitment limited by high-affinity bent beta2 integrin binding ligand in cis. Nat Commun. (2016) 7:12658. doi: 10.1038/ncomms12658
43. Kiema T, Lad Y, Jiang P, Oxley CL, Baldassarre M, Wegener KL, et al. The molecular basis of filamin binding to integrins and competition with talin. Mol Cell. (2006) 21:337–47. doi: 10.1016/j.molcel.2006.01.011
44. Liu J, Das M, Yang J, Ithychanda SS, Yakubenko VP, Plow EF, et al. Structural mechanism of integrin inactivation by filamin. Nat Struct Mol Biol. (2015) 22:383–9. doi: 10.1038/nsmb.2999
45. Falet H, Pollitt AY, Begonja AJ, Weber SE, Duerschmied D, Wagner DD, et al. A novel interaction between FlnA and Syk regulates platelet ITAM-mediated receptor signaling and function. J Exp Med. (2010) 207:1967–79. doi: 10.1084/jem.20100222
46. Savinko T, Guenther C, Uotila LM, Llort Asens M, Yao S, Tojkander S, et al. Filamin a is required for optimal T cell integrin-mediated force transmission, flow adhesion, and T cell trafficking. J Immunol. (2018) 200:3109–16. doi: 10.4049/jimmunol.1700913
47. Uotila LM, Guenther C, Savinko T, Lehti TA, Fagerholm SC. Filamin a regulates neutrophil adhesion, production of reactive oxygen species, and neutrophil extracellular trap release. J Immunol. (2017) 199:3644–53. doi: 10.4049/jimmunol.1700087
48. Sun C, Forster C, Nakamura F, Glogauer M. Filamin-A regulates neutrophil uropod retraction through RhoA during chemotaxis. PLoS ONE (2013) 8:e79009. doi: 10.1371/journal.pone.0079009
49. Lammermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Soldner R, Hirsch K, et al. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature (2008) 453:51–5. doi: 10.1038/nature06887
50. Hons M, Kopf A, Hauschild R, Leithner A, Gaertner F, Abe J, et al. Chemokines and integrins independently tune actin flow and substrate friction during intranodal migration of T cells. Nat Immunol. (2018) 19:606–16. doi: 10.1038/s41590-018-0109-z
51. Morrison VL, James MJ, Grzes K, Cook P, Glass DG, Savinko T, et al. Loss of beta2-integrin-mediated cytoskeletal linkage reprogrammes dendritic cells to a mature migratory phenotype. Nat Commun. (2014) 5:5359. doi: 10.1038/ncomms6359
52. Dustin ML. Cell adhesion molecules and actin cytoskeleton at immune synapses and kinapses. Curr Opin Cell Biol. (2007) 19:529–33. doi: 10.1016/j.ceb.2007.08.003
53. Carrasco YR, Fleire SJ, Cameron T, Dustin ML, Batista FD. LFA-1/ICAM-1 interaction lowers the threshold of B cell activation by facilitating B cell adhesion and synapse formation. Immunity (2004) 20:589–99. doi: 10.1016/S1074-7613(04)00105-0
54. Osman MS, Burshtyn DN, Kane KP. Activating Ly-49 receptors regulate LFA-1-mediated adhesion by NK cells. J Immunol. (2007) 178:1261–7. doi: 10.4049/jimmunol.178.3.1261
55. Kondo N, Ueda Y, Kita T, Ozawa M, Tomiyama T, Yasuda K, et al. NDR1-dependent regulation of kindlin-3 controls high-affinity LFA-1 binding and immune synapse organization. Mol Cell Biol. (2017) 37:e00424-16. doi: 10.1128/MCB.00424-16
56. Morrison VL, Uotila LM, Llort Asens M, Savinko T, Fagerholm SC. Optimal T cell activation and B cell antibody responses in vivo require the interaction between leukocyte function-associated antigen-1 and kindlin-3. J Immunol. (2015) 195:105–15. doi: 10.4049/jimmunol.1402741
57. Walling BL, Kim M. LFA-1 in T cell migration and differentiation. Front Immunol. (2018) 9:952. doi: 10.3389/fimmu.2018.00952
58. Verma NK, Fazil MH, Ong ST, Chalasani ML, Low JH, Kottaiswamy A, et al. LFA-1/ICAM-1 ligation in human T cells promotes Th1 polarization through a GSK3beta signaling-dependent notch pathway. J Immunol. (2016) 197:108–18. doi: 10.4049/jimmunol.1501264
59. Verma NK, Dempsey E, Long A, Davies A, Barry SP, Fallon PG, et al. Leukocyte function-associated antigen-1/intercellular adhesion molecule-1 interaction induces a novel genetic signature resulting in T-cells refractory to transforming growth factor-beta signaling. J Biol Chem. (2012) 287:27204–16. doi: 10.1074/jbc.M112.376616
60. Boutet M, Gauthier L, Leclerc M, Gros G, de Montpreville V, Theret N, et al. TGFbeta signaling intersects with CD103 integrin signaling to promote T-lymphocyte accumulation and antitumor activity in the lung tumor microenvironment. Cancer Res. (2016) 76:1757–69. doi: 10.1158/0008-5472.CAN-15-1545
61. Capece T, Walling BL, Lim K, Kim KD, Bae S, Chung HL, et al. A novel intracellular pool of LFA-1 is critical for asymmetric CD8(+) T cell activation and differentiation. J Cell Biol. (2017) 216:3817–29. doi: 10.1083/jcb.201609072
62. Meli AP, Fontes G, Avery DT, Leddon SA, Tam M, Elliot M, et al. The Integrin LFA-1 controls T follicular helper cell generation and maintenance. Immunity (2016) 45:831–46. doi: 10.1016/j.immuni.2016.09.018
63. Klann JE, Remedios KA, Kim SH, Metz PJ, Lopez J, Mack LA, et al. Talin plays a critical role in the maintenance of the regulatory T cell pool. J Immunol. (2017) 198:4639–51. doi: 10.4049/jimmunol.1601165
64. Verma NK, Kelleher D. Not just an adhesion molecule: LFA-1 contact tunes the T lymphocyte program. J Immunol. (2017) 199:1213–21. doi: 10.4049/jimmunol.1700495
65. Schmits R, Kundig TM, Baker DM, Shumaker G, Simard JJ, Duncan G, et al. LFA-1-deficient mice show normal CTL responses to virus but fail to reject immunogenic tumor. J Exp Med. (1996) 183:1415–26. doi: 10.1084/jem.183.4.1415
66. Lee SH, Prince JE, Rais M, Kheradmand F, Ballantyne CM, Weitz-Schmidt G, et al. Developmental control of integrin expression regulates Th2 effector homing, J Immunol. (2008) 180:4656–67. doi: 10.4049/jimmunol.180.7.4656
67. Knight JM, Lee SH, Roberts L, Smith CW, Weiss ST, Kheradmand F, et al. CD11a polymorphisms regulate TH2 cell homing and TH2-related disease. J Allergy Clin Immunol. (2014) 133:189–97 e1–8. doi: 10.1016/j.jaci.2013.03.049
68. Rezzonico R, Imbert V, Chicheportiche R, Dayer JM. Ligation of CD11b and CD11c beta(2) integrins by antibodies or soluble CD23 induces macrophage inflammatory protein 1alpha (MIP-1alpha) and MIP-1beta production in primary human monocytes through a pathway dependent on nuclear factor-kappaB. Blood (2001) 97:2932–40. doi: 10.1182/blood.V97.10.2932
69. Ling GS, Bennett J, Woollard KJ, Szajna M, Fossati-Jimack L, Taylor PR, et al. Integrin CD11b positively regulates TLR4-induced signalling pathways in dendritic cells but not in macrophages. Nat Commun. (2014) 5:3039. doi: 10.1038/ncomms4039
70. Anderson KE, Boyle KB, Davidson K, Chessa TA, Kulkarni S, Jarvis GE, et al. CD18-dependent activation of the neutrophil NADPH oxidase during phagocytosis of Escherichia coli or Staphylococcus aureus is regulated by class III but not class I or II PI3Ks. Blood (2008) 112:5202–11. doi: 10.1182/blood-2008-04-149450
71. Van Ziffle JA, Lowell CA. Neutrophil-specific deletion of Syk kinase results in reduced host defense to bacterial infection. Blood (2009) 114:4871–82. doi: 10.1182/blood-2009-05-220806
72. Zhou MJ, Brown EJ. CR3 (Mac-1, alpha M beta 2, CD11b/CD18) and Fc gamma RIII cooperate in generation of a neutrophil respiratory burst: requirement for Fc gamma RIII and tyrosine phosphorylation. J Cell Biol. (1994) 125:1407–16. doi: 10.1083/jcb.125.6.1407
73. Wilson ZS, Ahn LB, Serratelli WS, Belley MD, Lomas-Neira J, Sen M, et al. Activated beta2 integrins restrict neutrophil recruitment during murine acute Pseudomonal Pneumonia. Am J Respir Cell Mol Biol. (2017) 56:620–7. doi: 10.1165/rcmb.2016-0215OC
74. Dupuy AG, Caron E. Integrin-dependent phagocytosis: spreading from microadhesion to new concepts. J Cell Sci. (2008) 121:1773–83. doi: 10.1242/jcs.018036
75. Underhill DM, Goodridge HS. Information processing during phagocytosis. Nat Rev Immunol. (2012) 12:492–502. doi: 10.1038/nri3244
76. Rosales C, Uribe-Querol E. Phagocytosis: a fundamental process in immunity. Biomed Res Int. (2017) 2017:9042851. doi: 10.1155/2017/9042851
77. Jawhara S, Pluskota E, Cao W, Plow EF, Soloviev DA. Distinct effects of integrins alphaXbeta2 and alphaMbeta2 on leukocyte subpopulations during inflammation and antimicrobial responses. Infect Immun. (2017) 85:e00644-16. doi: 10.1128/IAI.00644-16
78. Souza COS, Espindola MS, Fontanari C, Prado MKB, Frantz FG, Rodrigues V, et al. CD18 regulates monocyte hematopoiesis and promotes resistance to experimental Schistosomiasis. Front Immunol. (2018) 9:1970. doi: 10.3389/fimmu.2018.01970
79. Wang L, Gordon RA, Huynh L, Su X, Park Min KH, Han J, et al. Indirect inhibition of Toll-like receptor and type I interferon responses by ITAM-coupled receptors and integrins. Immunity (2010) 32:518–30. doi: 10.1016/j.immuni.2010.03.014
80. Han C, Jin J, Xu S, Liu H, Li N, Cao X. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat Immunol. (2010) 11:734–42. doi: 10.1038/ni.1908
81. Yee NK, Hamerman J. A. beta(2) integrins inhibit TLR responses by regulating NF-kappaB pathway and p38 MAPK activation. Eur J Immunol. (2013) 43:779–92. doi: 10.1002/eji.201242550
82. Varga G, Balkow S, Wild MK, Stadtbaeumer A, Krummen M, Rothoeft T, et al. Active MAC-1 (CD11b/CD18) on DCs inhibits full T-cell activation. Blood (2007) 109:661–9. doi: 10.1182/blood-2005-12-023044
83. Balkow S, Heinz S, Schmidbauer P, Kolanus W, Holzmann B, Grabbe S, et al. LFA-1 activity state on dendritic cells regulates contact duration with T cells and promotes T-cell priming. Blood (2010) 116:1885–94. doi: 10.1182/blood-2009-05-224428
84. Podgrabinska S, Kamalu O, Mayer L, Shimaoka M, Snoeck H, Randolph GJ, et al. Inflamed lymphatic endothelium suppresses dendritic cell maturation and function via Mac-1/ICAM-1-dependent mechanism. J Immunol. (2009) 183:1767–79. doi: 10.4049/jimmunol.0802167
85. Ehirchiou D, Xiong Y, Xu G, Chen W, Shi Y, Zhang L. CD11b facilitates the development of peripheral tolerance by suppressing Th17 differentiation. J Exp Med. (2007) 204:1519–24. doi: 10.1084/jem.20062292
86. Nowatzky J, Manches O, Khan SA, Godefroy E, Bhardwaj N. Modulation of human Th17 cell responses through complement receptor 3 (CD11b/CD18) ligation on monocyte-derived dendritic cells. J Autoimmun. (2018) 92:57–66. doi: 10.1016/j.jaut.2018.05.005
87. Tak T, Rygiel TP, Karnam G, Bastian OW, Boon L, Viveen M, et al. Neutrophil-mediated suppression of influenza-induced pathology requires CD11b/CD18 (MAC-1). Am J Respir Cell Mol Biol. (2018) 58:492–9. doi: 10.1165/rcmb.2017-0021OC
88. Hoang KV, Rajaram MVS, Curry HM, Gavrilin MA, Wewers MD, Schlesinger LS. Complement receptor 3-mediated inhibition of inflammasome priming by Ras GTPase-activating protein during francisella tularensis phagocytosis by human mononuclear phagocytes. Front Immunol. (2018) 9:561. doi: 10.3389/fimmu.2018.00561
89. Amarilyo G, Verbovetski I, Atallah M, Grau A, Wiser G, Gil O, et al. iC3b-opsonized apoptotic cells mediate a distinct anti-inflammatory response and transcriptional NF-kappaB-dependent blockade. Eur J Immunol. (2010) 40:699–709. doi: 10.1002/eji.200838951
90. Meakin PJ, Morrison VL, Sneddon CC, Savinko T, Uotila L, Jalicy SM, et al. Mice lacking beta2-integrin function remain glucose tolerant in spite of insulin resistance, neutrophil infiltration and inflammation. PLoS ONE. (2015) 10:e0138872. doi: 10.1371/journal.pone.0138872
91. Wolf D, Bukosza N, Engel D, Poggi M, Jehle F, Anto Michel N, et al. Inflammation, but not recruitment, of adipose tissue macrophages requires signalling through Mac-1 (CD11b/CD18) in diet-induced obesity (DIO). Thromb Haemost. (2017) 117:325–38. doi: 10.1160/TH16-07-0553
92. Fagerholm SC, MacPherson M, James MJ, Sevier-Guy C, Lau CS. The CD11b-integrin (ITGAM) and systemic lupus erythematosus. Lupus (2013) 22:657–63. doi: 10.1177/0961203313491851
93. Rosetti F, Mayadas TN. The many faces of Mac-1 in autoimmune disease. Immunol Rev. (2016) 269:175–93. doi: 10.1111/imr.12373
94. MacPherson M, Lek HS, Prescott A, Fagerholm SC. A systemic lupus erythematosus-associated R77H substitution in the CD11b chain of the Mac-1 integrin compromises leukocyte adhesion and phagocytosis. J Biol Chem. (2011) 286:17303–10. doi: 10.1074/jbc.M110.182998
95. Faridi MH, Khan SQ, Zhao W, Lee HW, Altintas MM, Zhang K, et al. CD11b activation suppresses TLR-dependent inflammation and autoimmunity in systemic lupus erythematosus. J Clin Invest. (2017) 127:1271–83. doi: 10.1172/JCI88442
96. Ding C, Ma Y, Chen X, Liu M, Cai Y, Hu X, et al. Integrin CD11b negatively regulates BCR signalling to maintain autoreactive B cell tolerance. Nat Commun. (2013) 4:2813. doi: 10.1038/ncomms3813
97. von Andrian UH, Berger EM, Ramezani L, Chambers JD, Ochs HD, Harlan JM, et al. In vivo behavior of neutrophils from two patients with distinct inherited leukocyte adhesion deficiency syndromes. J Clin Invest. (1993) 91:2893–7. doi: 10.1172/JCI116535
98. Etzioni A, Frydman M, Pollack S, Avidor I, Phillips ML, Paulson JC, et al. Brief report: recurrent severe infections caused by a novel leukocyte adhesion deficiency. N Engl J Med. (1992) 327:1789–92. doi: 10.1056/NEJM199212173272505
99. van de Vijver E, Maddalena A, Sanal O, Holland SM, Uzel G, Madkaikar M, et al. Hematologically important mutations: leukocyte adhesion deficiency (first update). Blood Cells Mol Dis. (2012) 48:53–61. doi: 10.1016/j.bcmd.2011.10.004
100. Almarza Novoa E, Kasbekar S, Thrasher AJ, Kohn DB, Sevilla J, Nguyen T, et al. Leukocyte adhesion deficiency-I: a comprehensive review of all published cases. J Allergy Clin Immunol Pract. (2018) 6:1418–20 e10. doi: 10.1016/j.jaip.2017.12.008
101. Cox DP, Weathers DR. Leukocyte adhesion deficiency type 1: an important consideration in the clinical differential diagnosis of prepubertal periodontitis. A case report and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. (2008) 105:86–90. doi: 10.1016/j.tripleo.2007.02.026
102. Scharffetter-Kochanek K, Lu H, Norman K, van Nood N, Munoz F, Grabbe S, et al. Spontaneous skin ulceration and defective T cell function in CD18 null mice. J Exp Med. (1998) 188:119–31. doi: 10.1084/jem.188.1.119
103. Grabbe S, Varga G, Beissert S, Steinert M, Pendl G, Seeliger S, et al. Beta2 integrins are required for skin homing of primed T cells but not for priming naive T cells. J Clin Invest. (2002) 109:183–92. doi: 10.1172/JCI0211703
104. Etzioni A. Leukocyte adhesion deficiency III - when integrins activation fails. J Clin Immunol. (2014) 34:900–3. doi: 10.1007/s10875-014-0094-4
105. Saultier P, Szepetowski S, Canault M, Falaise C, Poggi M, Suchon P, et al. Long-term management of leukocyte adhesion deficiency type III without hematopoietic stem cell transplantation. Haematologica (2018) 103:e264–e267. doi: 10.3324/haematol.2017.186304
106. Rognoni E, Ruppert R, Fassler R. The kindlin family: functions, signaling properties and implications for human disease. J Cell Sci. (2016) 129:17–27. doi: 10.1242/jcs.161190
107. Moutsopoulos NM, Konkel J, Sarmadi M, Eskan MA, Wild T, Dutzan N, et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci Transl Med. (2014) 6:229ra40. doi: 10.1126/scitranslmed.3007696
108. Moutsopoulos NM, Zerbe CS, Wild T, Dutzan N, Brenchley L, DiPasquale G, et al. Interleukin-12 and interleukin-23 blockade in leukocyte adhesion deficiency type 1. N Engl J Med. (2017) 376:1141–6. doi: 10.1056/NEJMoa1612197
109. Uzel G, Kleiner DE, Kuhns DB, Holland SM. Dysfunctional LAD-1 neutrophils and colitis. Gastroenterology (2001) 121:958–64. doi: 10.1053/gast.2001.28022
110. Uzel G, Tng E, Rosenzweig SD, Hsu AP, Shaw JM, Horwitz ME, et al. Reversion mutations in patients with leukocyte adhesion deficiency type-1 (LAD-1). Blood (2008) 111:209–18. doi: 10.1182/blood-2007-04-082552
111. Wolach B, Gavrieli R, Wolach O, Stauber T, Abuzaitoun O, Kuperman A, et al. Leucocyte adhesion deficiency-A multicentre national experience. Eur J Clin Invest. 49:e13047. doi: 10.1111/eci.13047
112. D'Agata ID, Paradis K, Chad Z, Bonny Y, Seidman E. (1996). Leucocyte adhesion deficiency presenting as a chronic ileocolitis. Gut (2018) 39:605–8. doi: 10.1136/gut.39.4.605
Keywords: integrin, trafficking, kindlin-3, leukocyte adhesion deficiency, leukocyte adhesion cascade
Citation: Fagerholm SC, Guenther C, Llort Asens M, Savinko T and Uotila LM (2019) Beta2-Integrins and Interacting Proteins in Leukocyte Trafficking, Immune Suppression, and Immunodeficiency Disease. Front. Immunol. 10:254. doi: 10.3389/fimmu.2019.00254
Received: 01 October 2018; Accepted: 29 January 2019;
Published: 19 February 2019.
Edited by:
Deirdre R. Coombe, Curtin University, AustraliaReviewed by:
Elena Monica Borroni, Humanitas Research Hospital, ItalyNavin Kumar Verma, Nanyang Technological University, Singapore
Copyright © 2019 Fagerholm, Guenther, Llort Asens, Savinko and Uotila. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susanna C. Fagerholm, c3VzYW5uYS5mYWdlcmhvbG1AaGVsc2lua2kuZmk=