 Sabine Groeger
Sabine Groeger Joerg Meyle
Joerg Meyle
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 14 February 2019
Sec. Mucosal Immunity
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00208
This article is part of the Research TopicRecent Advances in Oral ImmunityView all 12 articles
Cellular Phenotype and Apoptosis: The function of epithelial tissues is the protection of the organism from chemical, microbial, and physical challenges which is indispensable for viability. To fulfill this task, oral epithelial cells follow a strongly regulated scheme of differentiation that results in the formation of structural proteins that manage the integrity of epithelial tissues and operate as a barrier. Oral epithelial cells are connected by various transmembrane proteins with specialized structures and functions. Keratin filaments adhere to the plasma membrane by desmosomes building a three-dimensional matrix.
Cell-Cell Contacts and Bacterial Influence: It is known that pathogenic oral bacteria are able to affect the expression and configuration of cell-cell junctions. Human keratinocytes up-regulate immune-modulatory receptors upon stimulation with bacterial components. Periodontal pathogens including P. gingivalis are able to inhibit oral epithelial innate immune responses through various mechanisms and to escape from host immune reaction, which supports the persistence of periodontitis and furthermore is able to affect the epithelial barrier function by altering expression and distribution of cell-cell interactions including tight junctions (TJs) and adherens junctions (AJs).
In the pathogenesis of periodontitis a highly organized biofilm community shifts from symbiosis to dysbiosis which results in destructive local inflammatory reactions.
Cellular Receptors: Cell-surface located toll like receptors (TLRs) and cytoplasmatic nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) belong to the pattern recognition receptors (PRRs). PRRs recognize microbial parts that represent pathogen-associated molecular patterns (PAMPs).
A multimeric complex of proteins known as inflammasome, which is a subset of NLRs, assembles after activation and proceeds to pro-inflammatory cytokine release.
Cytokine Production and Release: Cytokines and bacterial products may lead to host cell mediated tissue destruction. Keratinocytes are able to produce diverse pro-inflammatory cytokines and chemokines, including interleukin (IL)-1, IL-6, IL-8 and tumor necrosis factor (TNF)-α. Infection by pathogenic bacteria such as Porphyromonas gingivalis (P. gingivalis) and Aggregatibacter actinomycetemcomitans (A. actinomycetemcomitans) can induce a differentiated production of these cytokines.
Immuno-modulation, Bacterial Infection, and Cancer Cells: There is a known association between bacterial infection and cancer. Bacterial components are able to up-regulate immune-modulatory receptors on cancer cells. Interactions of bacteria with tumor cells could support malignant transformation an environment with deficient immune regulation.
The aim of this review is to present a set of molecular mechanisms of oral epithelial cells and their reactions to a number of toxic influences.
The oral mucosal epithelium is a barrier that separates the underlying tissues from their environment. It consists of two layers, the surface stratified squamous epithelium and the deeper lamina propria. In keratinized oral mucosa, the epithelium is composed of the four layers stratum basale, stratum spinosum, stratum granulosum, and stratum corneum. In nonkeratinised epithelium, the stratum basale is followed by the stratum filamentosum and the stratum distendum. In the oral mucosa distinct phenotypes are differentiated, lining mucosa, masticatory mucosa, and specialized mucosa (1). Lining mucosa is localized over mobile structures such as soft palate, cheeks, lips, alveolar mucosa, vestibular fornix and floor of the mouth and is extensible and loosely bound to adjacent structures by an elastin rich connective tissue and has a non-keratinizing squamous epithelium. Masticatory mucosa is the rigid and tough protecting cover of the gingiva and the hard palate, tightly bound by dense connective tissue to the underlying bone. This epithelium is keratinized. Specialized mucosa is located on the dorsum of the tongue, shows a keratinized epithelium and includes lingual papillae and taste buds as specialized structures (2).
Junctional epithelium (JE) maintains the direct attachment to the tooth surface. The basal cells of the JE are attached to the connective tissue by the external basal lamina while the suprabasal cells are anchored to the tooth surface by an internal basal lamina that is produced by the JE. JE contains fewer cell-junctions as the oral gingival epithelium, but well developed gap junctions and some small adherens junctions can be detected (2). The JE has wide intercellular spaces, is highly permeable for water-soluble substances and serves as the primary pathway for the transmigration of polymorph nuclear leukocytes (3, 4). JE does not exhibit phenotypic stratification, but the outermost cells appear elongated and align with their long axis parallel to tooth surface (3).
The oral epithelial barrier is the outcome of numerous structural and functional protein interactions resulting in the ability to respond to a various exogenous, possibly toxic, influences. Squamous epithelia possess structural properties like stratification and cornification of the keratinocytes and specific cell-to-cell interactions to maintain its barrier function. It is now recognized that epithelial cells are not passive bystanders, but rather are metabolically active and capable of reacting to external stimuli by synthesizing a number of cytokines, adhesion molecules, growth factors, chemokines, and matrix-metalloproteases (5). Gingival tissues provide defense to resist frictional forces of mastication as well as to defend the soft tissues against chemical or microbial challenge (3).
The stratified epithelium of the oral mucosa belongs, together with the epithelium of the skin, to the most protective and resistant epithelia. It is composed of two layers, first epithelial cells with a basement membrane and second an underlying connective tissue, the lamina propria (4). The gingiva is combined of epithelial and connective tissues forming a collar of masticatory mucosa attached to the teeth and the alveolar bone. Gingival epithelium constitutes of a stratified squamous keratinized epithelium while the oral sulcular epithelium appears to be stratified and non-keratinized (Figure 1).
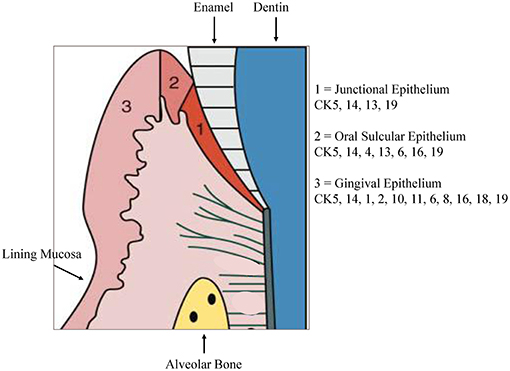
Figure 1. Cytokeratin distribution patterns. Cytokeratin (CK) distribution patterns in oral epithelia. Modified according to Pöllänen et al. (6).
The non-keratinized JE shows no true phenotypic stratification (3). In contrast to the ortho-keratinized epidermis of the skin, oral epithelia normally express all three major differentiation patterns of keratinocytes. As an anatomical and functional unit, the gingival keratinization pattern shows variations that origin partly from adaptive processes of the tissue to the special site around fully erupted teeth. A keratinized epithelium similar to the epidermis is exhibited in regions that encounter masticatory and other mechanical forces. The muco-gingival junction designates the boundary of the gingiva from the movable alveolar mucosa and the mucosa of the floor of the mouth. The floor of the mouth and the buccal part need to be flexible for speech, swallowing or chewing and are covered with a lining mucosa that doesn't keratinize. The specialized mucosa on the dorsum of the tongue includes a number of papillae and is covered by an epithelium, which may be either keratinized or non-keratinized. Under physiological conditions, the barrier of polarized epithelia allows regulated paracellular fluxes of solutes and nutrients as well as the collection of antigens and surveillance by mucosal immune cells. During inflammation, this protective mechanism may be compromised by different stimuli originating from both sides of the epithelial barrier.
Keratins are one major component of the epithelial cytoskeleton. They belong to the intermediate filament group of cytoskeletal proteins. A gene family of approximately 30 members encode keratins. They have a common structure composed of about 310-amino-acid central o-helical rod domain flanked by non-helical end-domains which are highly variable in sequence and structure (7). Based on the amino acid sequence and charge the keratin proteins are divided into two groups, acidic type I keratins including keratins K9-K20 and the basic or neutral type II keratins including K1–K8. Two keratin proteins, one type I and one type II, are always co-expressed and build heteropolymers to form the 10-nm keratin intermediate filaments (Ifs) that are part of the cytoskeleton. In the basal proliferative layer the keratin pair K5/K14 is expressed in stratified epithelia. Keratin 19 is detectable in simple epithelia and basal cells of non-keratinizing epithelia (8, 9). The keratin pair that is expressed in the post-mitotic layers of differentiating suprabasal cells differs depending on the localization. Cytokeratin distribution is highly specific and varies with type of epithelium, site, differentiation grade, so keratin expression is a sensitive and specific marker of differentiation in epithelial cells (10). Gingival and epidermal tissues as examples for cornifying epithelia, the keratins K1 and K10 are present while epithelia of buccal mucosa or esophagus K4 and K13 are the mainly expressed keratins (11). Suprabasal epithelial cells of the hard palate and gingiva furthermore express K2, designated as K2p in contrast to the epidermal K2e. The genes of K2p and K2e are related but separate (12). Other than the keratin pattern expressed from the attached gingiva some specialized epithelial cells within the gingiva show a distinct keratin pattern. The sulcular epithelium and cells of the gingival margin express K4 and K13. In contrast, the junctional epithelium adjacent to the tooth surface synthesizes K8, K13, K16, K18, and K19 (11, 13). Figure 1 shows the regional cytokeratin distribution pattern of the gingiva. Oral epithelia can exhibit one of 2 patterns of epithelial maturation, (1) they keratinize thus the mucosa matures by formation of a surface layer of keratin. This includes orthokeratinization which refers to the absence of nuclei in the superficial layer of scales on maturation and parakeratinization which designates the retention of pyknotic nuclei in the surface layer of squames during maturation (14). Nonkeratinization is the second possibility which means maturation with absence of a keratin layer which denotes that the nuclei remain, with scarce keratin filaments, in the cytoplasm of the most superficial cells (15). Depending on the functional demands, different types of keratinization are present in the gingival tissue. Oral gingival epithelium is keratinized while the sulcular and junctional epithelia (JE) as well as the lining mucosa are not keratinized (14).
Keratin filament assembly starts by parallel association. One type I chain form a paired dimer with its type II counterpart, two dimers associate in an antiparallel fashion to a tetramer. Two tetramers connect laterally resulting in a protofilament, from which eight are twisted into a rope building the keratin filament. Each keratin filament therefore possesses a cross section of 32 individual α helical coils. The polypeptide chains are further stabilized by strong lateral hydrophobic interactions. Bundled keratin filaments are associated to macromolecular networks that are oriented radial in the cytoplasm (2, 16). The regional specificity of keratin expression may be attributed to intrinsic specialization of regional keratinocyte stem cells. Disorders in keratin may be genetic or acquired. Numerous keratin mutations were identified as cause of several skin and mucosal disorders. Abnormal keratinization is part of several oral diseases. This topic is reviewed by Rao et al. (17).
Biofilms differentially modulate the epithelial cellular immune response based on their properties and composition. The bacterial biofilm located on the tooth surface and in the gingival crevice is considered as the primary causative agent involved in the pathogenesis of gingivitis and periodontitis and involves polymicrobial synergy and dysbiosis (18). Dysbiosis is based on the relative abundance of different bacterial species compared to their low presence in health, causing a modification of host–microbe interactions that can mediate destructive inflammation and bone loss (19, 20).
Keystone pathogens, such as Porphyromonas gingivalis (P. gingivalis), are able to subvert host response and promote breakdown of the homeostatic state, while further bacterial species show properties of pathobionts that can trigger destructive inflammation including both innate and adaptive immune response (21, 22). In addition the onset of gingivitis and periodontitis requires a susceptible host governing the complex inflammatory interactions.
Keratinocytes of the gingival epithelium form a barrier against bacterial infection and invasion (23). They are interconnected by a number of specialized transmembrane molecular complexes, among them cell-cell junctions comprising tight junctions adherens junctions, and gap junctions (Figure 2).
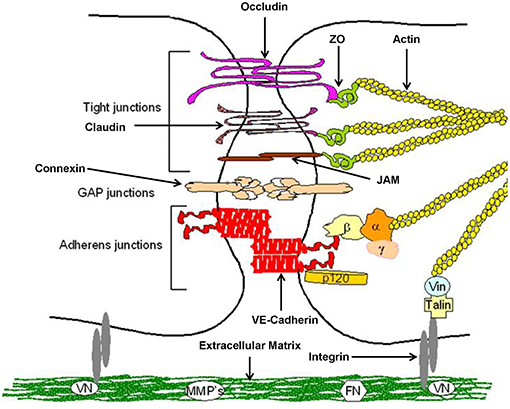
Figure 2. Structure of epithelial cell-cell contacts. Model of cellular junctions, modified according to Metha and Malik (24). Arrangement of junctions comprising tight junctions, adherens junctions, gap junctions, and integrin. Occludin, claudins, and junctional adhesion molecules (JAMs) required for tight junctions, whereas vascular endothelial (VE)-cadherin forms adherens junctions. Connexins are part of gap junctions. The extracellular domains of occludin, claudins, and VE-cadherin maintain cell-cell contacts. Intracellular domains provide junctional stability linking to the actin cytoskeleton via catenins (β,β-catenin, α,α-catenin; γ,γ-catenin; p120, p120-catenin) or zonula occludens-1 protein (ZO-1). Gap junctions are responsible for fast exchange of information by low-molecular-mass second messengers such as Ca2+ and IP3 between contiguous cells. Integrin receptors link endothelial cells with the extracellular matrix (ECM) through matrix proteins like fibronectin (FN) or vitronectin (VN). The cytosolic domains of integrins are linked to the actin cytoskeleton through the proteins talin and vinculin (Vin), involved in integrin-mediated signaling.
Normal expression of these molecular complexes in the gingival tissues is essential for maintaining epithelial integrity. Once integrity is disturbed by biofilm-derived noxious influences, the associated bacteria may invade into the deeper periodontal tissues, triggering an inflammatory response. Thus these cell-cell connections are a crucial part of the innate immune response to resist microbial and toxic challenge.
Tight junctions are complex protein structures, forming a belt like pattern among neighboring cells, encircling cells at the apical side of the lateral membrane (25). One of the major functions of the TJ complex is to form a barrier to regulate the passage through the paracellular pathway of water, ions, solutes and other small molecules (26–28). The structure of TJs appears as a string of continuous particles inlaid into the membrane, forming TJ strands. The strand is a fibril-like structure built by the assembly of claudin and tight junction–associated MARVEL proteins (TAMPs). TAMPS are composed of the tight junction-associated MAL = myelin and lymphocyte domain and the MARVEL = related proteins for vesicle trafficking and membrane link domain. The assembled structure represents the functional unit of TJs, formed by adjacent plasma membranes (29, 30).
A number of different signaling and trafficking molecules, that regulate cell differentiation, proliferation and polarity, are coordinated by TJs (31, 32). TJ topology consists of three protein domains, a helical transmembrane domain, a cytosolic scaffolding domain and cytosolic tail featuring cellular signaling. TJ strands are formed by the transmembrane proteins, a class that consists of multiple integral membrane proteins, including the groups of tissue- and cell-specific claudins, (29, 33) the TAMP family and the junction adhesion molecules (JAMs). The structure of claudin includes four transmembrane domains, two extracellular domains forming two loops, in which the N-terminus and C-terminus are located intracellularly. Claudins fulfill barrier properties (30, 34, 35) and are able to regulate the gate function as paracellular tight junction channels (PTJC). Their biological and physical properties are comparable to traditional ion channels (36). They also include occludin into the junctions (29). In adjacent mouse liver cells it was shown that various members of the claudin family form homophilic or heterophilic polymers. Furthermore, claudins may form paired strands to the membrane of adjacent cells (37). Differences in barrier properties between cell types are probably caused by different combinations of claudins (38).
The TAMP family includes MARVEL D1, also called occludin, MARVEL D2 (tricellulin) and MARVEL D3 protein. These molecules possess four transmembrane domains and two extracellular loops, similar to claudin (39, 40). It is not clear whether occludin composed strands have the same functions as strands formed by claudins but in vitro and in vivo studies demonstrated that occludin is of importance in TJ barrier function and intercellular adhesive interactions (39, 41–43). Claudin 1 and occludin were detected in the gingival but not in the sulcular and junctional epithelium. Furthermore, it was found that the adherens junction proteins P-cadherin and α-catenin are detectable in all three epithelia while E-cadherin was not present in junctional epithelium (44). The expression of claudin-4 was detected in the human oral squamous cell carcinoma epithelial cell line H413 (45) and in immortalized human gingival keratinocytes (46). Genetic investigations of adhesion proteins in stratified multi-layered gingival epithelial cell cultures showed strong expression of claudin-4, claudin-1, JAM-1, claudin-25, claudin-17, occludin and claudin-12 (47). Occludin is able to associate with different signaling molecules such as the non-receptor tyrosine kinase c-Yes, atypical protein kinase C (aPKC) and phosphoinositide 3-kinase (PI3K), as well as protein phosphatases 2A and 1 and appears to have signal transmitter functions (48, 49).
MARVEL D2 (also called tricellulin) is detectable at the tricellular contact sites. It is assembled to strands that form a tubular structure vertical to the bicellular TJ belt (50). It probably controls the flow of macromolecules but it is also needed for TJ organization. In the mouse mammalian epithelial cell line Eph4 tricellulin knock-out led to impairment of the structure of bicellular and tricellular contacts (51). The third TAMP, MARVEL D3, is expressed in many epithelial cells and its function was found not to be essential for TJ formation (52).
The members of the junctional adhesion molecule family (JAM), JAM-A, JAM-B, JAM-4, JAM-L and coxsackie and adenovirus receptor (CAR) belong to the immunoglobulin superfamily. JAMs seem to be less important for the regulation of the junctional structure, but rather contribute to adhesion and signaling. Studies mostly focused on the junctional role of JAM-A, that was shown to be localized to claudin-based tight junction fibrils in epithelial cells (53). JAM-A protein contains two extracellular immunoglobulin-like loops, a single transmembrane and a cytoplasmic domain ending in a PDZ binding motif that has been reported to interact with AF-6/afadin and zonula occludens protein (ZO)-1 (54) and ZO-2 (55). CAR appears to be a cell adhesion molecule that contributes to the formation of cell–cell contacts. In cultured epithelial cells, CAR molecules on adjacent cells form homotypic interactions (56). CAR is locally concentrated in TJs at the most apical regions of the lateral surfaces of polarized epithelial cells and its overexpression in cultured polarized cells increased TER (57), while soluble CAR and anti-CAR antibodies were shown to disrupt TJs, (56) suggesting that CAR is involved in the barrier function of TJs.
All members of the cytosolic scaffolding proteins have one or multiple post synaptic density proteins (PSD95), drosophila disc large tumor suppressor (Dlg1) domains, and zonula occludens-1 protein (PDZ) domains. They are able to bind to various integral membrane proteins like claudins, occludin or JAMs and also can bind to actin filaments. In this manner they connect TJ to the actin filaments and stabilize the protein complexes. An increasing number of PDZ containing proteins is known including membrane associated guanylate kinase (MAGUK)-like proteins, protein associated with Lin7 (Pals1), AF-6/afadin, atypical protein kinase C (aPKC), isotype-specific interacting protein (ASIP), partitioning-defective protein 3 (PAR-3), multi-PDZ protein 1 (MUPP1) and protein associated with tight junctions (PATJ) (58). These scaffolding proteins seem to be important for the organization and localization of TJs because blockade of PDZ domains results in poorly organized TJs that in consequence are distributed to other areas (59). Zonula occludens (ZO) proteins belong to the MAGUK family and include the three members ZO-1 (60), ZO-2 (61), and ZO-3 (62). Through their three PDZ domains ZO proteins interact with several proteins such as claudins, MARVEL D1 or JAMs and F-actin. This association with multiple proteins makes the formation of large complexes possible which are linking the cytoskeleton to the TJ strands (58).
The barrier function and structure of TJs is regulated by intracellular signaling proteins which include protein kinase A, protein kinase C, Rho kinase, myosin light chain kinase, GTPase Rab13, tyrosin kinase and mitogen activated protein kinase. All these proteins are not specific for TJs but essential for their establishment and function. The signal transduction of TJs is reviewed in Takano et al. (63). Further reviews are available addressing TJ physiology and function (64), regulation (65) and the specific components such as tricellular tight junctions (66). In gingival tissue, TJs were observed only in the granular and cornified layer, were they did not form complex strands, in contrast to cultured gingival keratinocytes in vitro that showed a largely extended framework of TJ strands (67).
Measurement of the transepithelial electrical resistance (TER) is a method for investigations of the permeability of mucosal barriers in vitro and alterations of TER values are directly related to the integrity and function of the paracellular occluding barrier (68, 69). TER measurements are a useful tool to assess integrity of tight junctions. The strength of the transmucosal resistance is closely related to the number of junctional strands and junctional tightness (70). This correlation was demonstrated in primary human gingival keratinocytes (Figure 3) (67). The development of TER depends on the intracellular Ca2+ concentration (72).
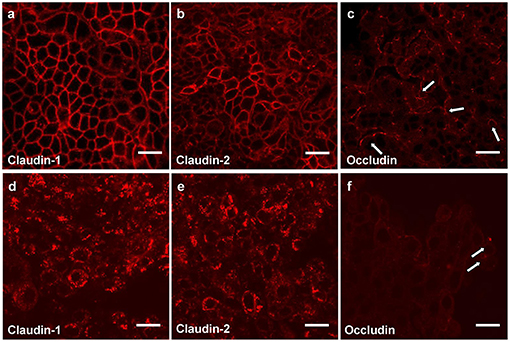
Figure 3. Tight junction proteins of primary keratinocytes after infection with P. gingivalis. Immunostaining of the tight junction proteins in primary human gingival keratinocytes, claudin 1 (a,d) claudin 2 (b,e) and occludin (c,f); (a–c) cells in culture medium; (d–f) cells infected apically plus basolaterally with Pophyromonas gingivalis (P. gingivalis) W83 (MOI 104) for 4 h. Arrows (e,f) show curved occludin strains in the walls of non-infected cells, in infected cells the arrows point to occludin aggregations, scale bar = 20 μm (71).
Immortalized human gingival keratinocytes (IHGK) (73, 74) used in a 3D culture model, were infected with gingipain-producing P. gingivalis strains and a RGP/KGP defect mutant (75). This caused a significant decrease of TER after 24 h by the gingipain producing strains, but not by the defect mutant. Investigations of tight junction proteins in the same experimental setting using immunostaining revealed infection-induced alterations in claudin-1, claudin-2 and in occludin expressions. After infection the typical chicken wire pattern of claudin-1 and claudin-2 (Figures 3a,b) disappeared and the proteins formed conglomerates (Figures 3d,e). The curved strands of occludin present in the control assays (Figure 3c) were degraded as well (Figure 3f). Soluble virulence factors such as gingipains disrupt the epithelial barrier in vitro, which is correlated with the disintegration of junctional cell-cell complexes. Invasion and damage of the epithelial layer by infective agents is an important step and may result in bacterial invasion and destruction of the underlying connective tissue.
The results of this study give some insights into the initial stages of oral bacterial infections leading to gingivitis and periodontitis.
A further mechanism besides damage may be active internalization of epithelial adhesion complexes. In intestinal epithelial cells (T84 cells), IFN-γ induces a process of TJ protein internalization (claudin-1, occludin, JAM-A) by micropinocytosis, which results in leakage of the epithelial layer (76). Guo et al. (77) determined the impact of P. gingivalis, P. gingivalis LPS and eATP on TJ proteins in an oral epithelial cell culture model. Quantified real time polymerase chain reaction (RT-PCR), immunostaining and immunoblots of gene and protein expression in TJs revealed that P. gingivalis infection led to temporary upregulation of the genes encoding occludin, claudin- 1, and claudin-4 but not JAM-A, claudin-15, or ZO-1, while P. gingivalis LPS increased claudin-1, claudin-15, and ZO-1 and decreased occludin, JAM-A, and claudin-4. Significant upregulation of tight junction proteins was demonstrated when cells were pretreated with eATP. These results indicate that P. gingivalis induced early defense mechanisms of the host. P. gingivalis LPS stimulates the destruction of the epithelial barrier more potently than P. gingivalis. ATP stimulation further increased the effect on TJ proteins after P. gingivalis infection and LPS-induced disruption of epithelial integrity (77).
The adherens junctions (AJs) or zonula adherens, intermediate junction, or “belt desmosome,” are a defining feature of all epithelia, forming apical localized structures of adhesion closely aligned to the membranes of neighboring epithelial cells, that play an essential role in the regulation of the junctional complex. AJs are protein complexes that appear at cell–cell junctions in epithelial and endothelial tissues. Their localization is more basal than tight junctions. AJs are like bands that encircle the cell (zonula adherens) or appear as attachment spots to the extracellular matrix (adhesion plaques). The cell-to-cell adhesion sites are composed of cadherins which are connected to the actin cytoskeleton by catenins and other constituents like actinin and vinculin (78). AJs are formed by homophilic binding of the extracellular cadherin domains in a calcium-dependent manner. The cell-to cell apposition is maintained and reinforced by the homophilic interactions of single-pass transmembrane E-Cadherin (E-Cad) molecules. This process is stabilized by accumulating a tight network of actin filaments and by linking molecules that fix E-Cad clusters on the inner cytoskeleton (79). The E-Cad cytoplasmic domain consists of the β-catenin (β-Cat) interacting with p120-catenin (p120-Ctn). β-Cat associates with α-Catenin (α-Cad), maintaining the link to the actin cytoskeleton. Cadherin directly binds to β-catenin or plakoglobin, followed by binding to α-catenin that afterwards binds to vinculin, α-actinin, ZO-1 and actin (80). Also α-Cat is able to interact with further actin-binding proteins such as formin, AF6/afadin, or EPLIN (81). Nectin and its associated AF6/afadin protein L-afadin occurs concentrated at AJs (82). The transmembrane protein vezatin is localized at sites of cadherin-based cell-to-cell adhesion in cultured cells and anchors myosin VIIa to the cadherin-catenin complex (83). Due to their dynamic structure, adherens junctions physically connect adjacent epithelial cells, bridge intercellular adhesive contacts to the cytoskeleton, and are involved in the definition of each cell's apical–basal axis. E-Cads as well as catenins are substrates of phosphatases and kinases that are key regulators of AJs and modify the interactions between the proteins enabling them to regulate the interaction grade between E-Cad and the catenin complex and their concentrations in the membrane, which is essential for the modulation of adhesive strength and AJ remodeling [reviewed in (84)].
As a result of proteolytic disruption by putative periodontal pathogens such as P. gingivalis E-Cads are affected in periodontitis (85, 86). P. gingivalis is able to produce a variety of proteolytic enzymes, including eight endopeptidases and numerous exopeptidases (87). Gingipains are extracellular cysteine proteinases, which can impair endothelial cell adhesion (88, 89). Gingipains are also able to enhance collagenolysis by inducing matrix metalloproteinases of the host. Sheets et al. (90) also demonstrated cleavage of cellular receptors. The cleavage of adherence junction proteins (shown in HOK-16 cells) affects N-cadherin, VE-cadherin, β-integrin and reduces the adhesion of the cells to the extracellular matrix proteins (91). This may result in the detachment of the endothelial cells.
It was shown that P. gingivalis fimbriae bind to cellular α5β1-integrin, which mediates bacterial adherence to host cells (92–94). Cellular integrins are heterodimeric receptors for extracellular matrix proteins and are essentially involved in cellular physiological processes that are related to metabolism, activation, differentiation, motility, and proliferation (95). These functions depend on the α5β1-integrin binding to its ligand fibronectin (96).
P. gingivalis can degrade cellular signaling molecules and inactivate a variety of cellular functions, which are important for healing and regeneration as well as homeostatic properties of periodontal tissues (91, 97–99).
Invasion of epithelial cells disrupts the epithelial barrier and the intracellular pathogens affect cellular functions by the usage of dynamin, actin fibers, microtubules, PI3K, and lipid rafts of the host cells.
Intracellular localization enables pathogens to penetrate deep into tissues by spreading from cell to cell, a process that seems to be mediated by membrane protrusions based on actin polymerization. This avoids the need of bacterial release into the extracellular space, i.e., periodontal pathogens like P. gingivalis spread in between cells without entering the extracellular space which may allow colonization of oral tissues avoiding revelation to the humoral immune response (86).
The importance of junctional proteins in the immune response to bacterial biofilms has been demonstrated by Belibasakis et al. (47). The group investigated the effects of a 10-species subgingival biofilm model on gene expression of all known cellular contacts (tight junctions, desmosomes, gap junctions and adherens junctions), and evaluated the involvement of the 3 “red-complex” species [P. gingivalis, Treponema denticola (T. denticola), and Tannerella forsythia (T. forsythia)] in a multi-layered gingival epithelial cell culture. The results of this study showed different effects on the junctional expression of the 2 biofilm (BF) models (one with and one without the “red complex”). It was found that BF including the “red complex” did not affect the expression of any of the studied tight junction genes. Absence of the “red complex” (RC) from the biofilm resulted in significantly higher Claudin-4 expression compared to the control after 3 and 24 h. Assessment of gene expression of desmosomes, adherens junctions, and gap junction proteins in response to biofilms without the “red complex” resulted in up-regulated desmocollin-2 expression after 3 h while the “red complex” including biofilm did not induce this effect. After 24 h, this expression was significantly downregulated by both biofilm variants. After 3 h of biofilm challenge, the gene expression of none of the investigated junctional adapter proteins was regulated while after 24 h, the expression of desmoplakin and plakoglobin were down-regulated in response to both biofilms (47).
Toll-like receptors (TLRs) (Figures 4, 5) belong to the best characterized family of cellular effectors for the detection of pathogens (101). TLRs are widely expressed in eukaryotic cells. They are trans-membrane proteins that recognize molecular structures classified as “pathogen associated molecular patterns” (PAMPs) and thus belong to the pattern recognition receptors (PRRs). These patterns are present in nearly all types of microorganisms (102). Toll-like receptors contain a horseshoe-shaped extracellular leucine-rich repeat (LRR) and an intra-cytoplasmic toll/IL-1R (TIR) domain that are connected by a single trans-membrane domain. The LRR domain is responsible for ligand recognition and intracellular signal transfer is maintained by the TIR domain. TLRs represent not only the most important but also one of the first mechanisms in immune-defense against fungal, bacterial and viral pathogens. After binding the TLR downstream signaling pathway is activated playing an important role in innate and adaptive immune responses. In the oral cavity a great number of microorganisms is constantly present, therefore expression and function of TLRs is essential for the maintenance of oral tissue homeostasis. In humans, currently 10 TLRs have been identified, including extracellular as well as intracellular receptors. All exhibit a number of specific ligands, except for the orphan receptor TLR10, where the specific ligand has yet not been discovered (101, 103). TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10 are expressed on the cell surface for recognition of extracellular microorganisms and ligands. TLR3, TLR7, TLR8, and TLR9 are intracellularly localized in the cytosolic endosomal compartment, binding microorganisms and ligands which passed the membrane of the host cell (104). Figure 4 shows the location of TLRs and the identity of their ligands/agonists. TLR11 has been identified in the human genome but does not translate into a protein, because its open reading frame contains a stop codon (105). TLR2 forms heterodimers with TLR1 or TLR6 and recognizes peptidoglycan, lipopeptide and lipoproteins while lipopolysaccharide of Gram-negative bacteria is the specific ligand of TLR4 (106, 107). TLR3 recognizes double-stranded RNA (dsRNA), TLR 5 can detect bacterial flagellin, TLR7 and TLR8 were shown to recognize imidazoquinilins and single-stranded RNA and TLR9 detects bacterial and viral DNA over their cytosine and guanine basepairing (108–113). LRR binding by ligands induces conformational changes of TIR resulting in interactions between TIR domains of adjacent TLRs and binding of additional adaptor proteins that are needed for the initiation of the intracellular signaling cascade. The most important adaptor molecules are the myeloid differentiation factor 88 (MyD88), the MyD88 adaptor-like (Mal) (TIR domain-containing adaptor protein, TIRAP), the TIR domain-containing adaptor protein inducing interferon-β (TRIF) (TIR-containing adaptor molecule, TICAM as synonym) and the TRIF-related adaptor molecule (TRAM) (114–119). TLR signaling can be negatively regulated by a variety of inhibitory molecules, including the toll-interacting protein (Tollip), interleukin-1 receptor (IL-1R) associated protein kinase (IRAK)-M, the sterile a- and HEAT-Armadillo- motif-containing protein (SARM), and the B cell adaptor or PI3K (BCAP), which inhibit downstream phases in the TLR- dependent signaling cascades. IL-1R associated protein kinases (IRAKs) IRAK4, IRAK1, and IRAK2 are activated by MyD88 followed by activation of tumor necrosis factor receptor-associated factor 6 (TRAF6) and RIP, that proceed by activation of transforming growth factor (TGF)-β-activated kinase 1 (TAK1) and TAK1-binding protein (TAB1, TAB2, and TAB3) complex (120–123). Subsequently, gene expression regulatory factors of the mitogen activated protein kinases (MAPK) family (ERK, JNK, p38) and NF-kB are activated, regulating cell survival and proliferation, and induce immune cell activation, production of pro-/anti-inflammatory mediators (cytokines and chemokines), interferons, and anti-microbial products. Activation of the intracellularly located TLR7, TLR8, and TLR9 is forwarded through MyD88 as well, but can also initiate TRAF6, IRAK4, and TRAF3-dependent activation of IRF7, which translocates to the nucleus and induces the production of type-I interferon (114, 124).
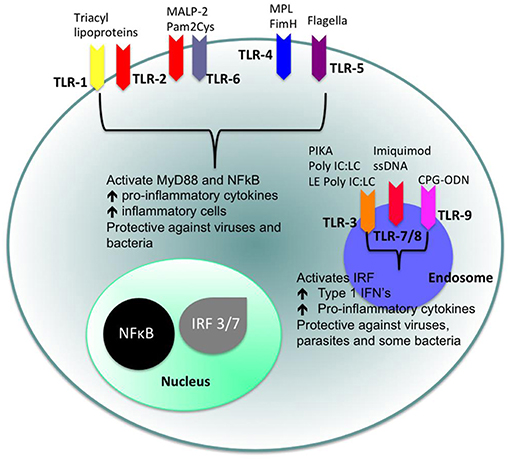
Figure 4. Cellular location of toll like receptors (TLRs) and the identity of their ligands/agonists. The stimulation of surface TLRs (TLR-2, TLR-4, and TLR-5) with appropriate ligands results in the activation of nuclear factor (NF)-κB. The ensuing increase in levels of pro-inflammatory cytokines and the influx of inflammatory cells then provides an environment, which protects against both virus and bacterial challenge. Activation of intracellular TLRs (TLR-3, TLR-7, TLR-8, and TLR-9) leads to interferon regulating factor (IRF) activation and the production of Type 1 interferons (IFNs) and pro-inflammatory cytokines, again providing an environment not conducive for pathogens (100).
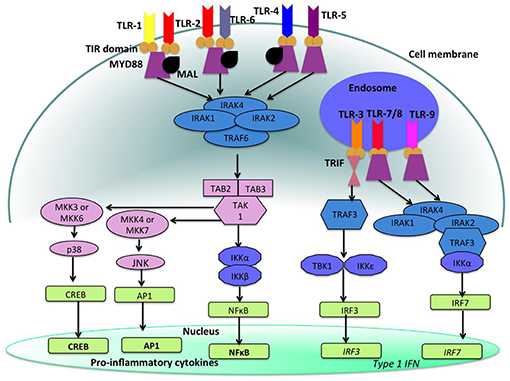
Figure 5. Toll like receptor (TLR)-signaling pathways. TLR-4, TLR-5, and the heterodimers TLR-1/TLR-2 and TLR-2/TLR-6 are located on the cell surface where they are activated by the appropriate ligand. Conversely, TLR-3, TLR-7, TLR-8, and TLR-9 are located within endosomal compartments of the cell and recognize microbial and viral nucleic acids. Stimulation of TLR-1/TLR-2, TLR-2/TLR-6, TLR-4, and TLR-5 leads to the engagement of myeloid differentiation primary response protein (MyD88) and MYD88-adapter-like protein (MAL) with the toll/interleukin-1 receptor (TIR) domain-containing adapter proteins. This stimulates downstream signaling pathways that involve the interactions between IL-1R-associated kinases (IRAKs) and the adapter molecules tumor necrosis factor (TNF) receptor-associated factors (TRAFs) and activates mitogen-activated protein kinases (MAPKs) JUN N-terminal kinases (JNK) and p38. Activation of these kinases leads to the activation of transcriptional factors such as nuclear factor-κB (NF-κB), cyclic adenosine mono phosphate (AMP)-responsive element binding protein (CREB), and activator protein-1 (AP-1). A major consequence of activation of surface TLRs is the induction of pro-inflammatory cytokines. Activation of TLR-7, TLR-8, and TLR-9 also leads to the engagement of MyD88, MAL, IRAKs, and NF-κB inhibitor kinase (IKK)α, however, interferon-regulatory factors (IRFs) are activated, which leads to the production of type 1 interferons (IFN). Stimulation of TLR-3 results in the association of TIR domain-containing adapter protein inducing IFNβ (TRIF). This leads to the downstream signaling of TNF receptor-associated factors (TRAFs) and IKK leading to the activation of IRF3 and the production of type 1 IFNs (100).
Figure 5 shows the signaling pathways of TLRs. mRNA of all 10 TLRs was detected in oral epithelial cells, but the actual expression and cellular localization of TLR proteins varies and is inducible. TLR2 is highly expressed in the basal layer of the gingival epithelium, levels are lower in the superficial layers that are more exposed to microorganisms and environmental influences. Apart from the detection of colonizing microorganisms in the superficial part of the epithelium, this may be regarded as a mechanism that facilitates TLR-depending inflammatory response only when pathogens are recognized in the basal layer. For TLR1, TLR3, TLR4, TLR5, and TLR9 a similar expression pattern was demonstrated (125, 126). The expression of TLR7 and TLR8 shows the same pattern in healthy and inflamed tissue. TLR2 and TLR4 expression is increased in acute and persistent gingival inflammation, though stimulation with TLR agonists did not induce production of pro-inflammatory cytokines, but β-defensin-2 generation in epithelial cells, and thus favored local downstream immune response (127).
Under chronic inflammatory conditions such as periodontitis in contrast to the TLR2 upregulation expression of TLR4 decreased, which may prevent from inflammatory exacerbation, i.e., tissue and bone destruction through containment of the inflammatory response (128). It was demonstrated that human healthy and inflamed oral tissues express TLR2, TLR4, NOD1 and NOD2 molecules, where cell-surface localizations of TLR2 and TLR4 could be more clearly detected in the inflamed than in healthy gingiva. It was furthermore demonstrated that human oral epithelial cell lines HSC-2, HO-1-u-1, and KB cells as well as primary cultured oral epithelial cells constitutively express TLR2, TLR4, NOD1, and NOD2. Stimulation of these cells with TLR and NOD agonists caused up-regulation of the antimicrobial peptide β-defensin (129). Oral epithelial cells, in contrast to colonic epithelial cells, did not secrete cytokines such as IL-8, monocyte chemoattractant protein-1 (MCP-1), granulocyte colony stimulating factor (G-CSF), granulocyte macrophage colony-stimulating factor (GM-CSF), and vascular endothelial growth factor (VEGF) after stimulation with bacterial components but upregulated expression of peptidoglycan recognition proteins (PGRPs), a further family of pattern recognition molecules (130, 131). These results suggest that part of the cells are desensitized to prevent tissue destruction over excessive innate immune responses to bacterial stimuli, because cells and bacteria interact constitutively (130, 131).
In periodontitis an abnormal immune response known as a “hyper-responsive” phenotype was demonstrated by investigations of peripheral blood leukocytes that were stimulated with TLR2 and TLR4 agonists. The stimulation resulted in elevated levels of pro-inflammatory cytokines produced by leukocytes that were derived from patients with localized aggressive periodontitis. This altered immune response may result in rapid loss of connective tissue and periodontal attachment as well as alveolar bone, which could result in early tooth loss already in young individuals (132). A cross-sectional study examined the role of epigenetic regulation, specifically DNA methylation status, of genes in the TLR pathway in patients with localized aggressive periodontitis (LAP). Peripheral blood stimulated with Escherichia coli (E. coli) LPS was analyzed for DNA methylation of seven TLR signaling genes. At specific CpG positions in LAP patients compared to healthy controls, differences in the methylation status were observed, as well as between severe and moderate LAP. Individuals with moderate LAP presented hypermethylation of both the upregulating and downregulating genes, while severe LAP presented hypomethylation of these genes. The methylation status correlated with an increased pro-inflammatory cytokine profile in LAP patients suggesting that epigenetic modifications in TLR signaling may modulate disease progression and tissue destruction (133).
A meta-analysis assessing the association between TLR4 polymorphisms and chronic periodontitis (CP) detected an association between TLR4C > G (rs7873784) allele and CP in Asians (134). The association between TLR4 polymorphisms and gastric cancer was investigated in the meta-analysis by Jin et al. (134). This group detected an increased gastric cancer risk in TLR4 + 896A/G and TLR4 + 1196C/T polymorphism in a Caucasian population (135).
Nucleotide-binding oligomerization domain receptors (NODs) (Figure 6) are cytosolic pattern recognition molecules that bind to peptidoglycan (PGN), a component of bacterial cell walls. They belong to the NOD-like receptor (NLRs) family including also NACHT-LRR (leucine-rich repeat) and pyrin-domain-containing proteins (NALPs), neuronal apoptosis inhibitor factors (NAIPs), and ICE-protease activating factor (IPAF) (137–139). The NOD1 ligand is PGN-derived γ-D-glutamylmesodiaminopimelicacid (iE-DAP) while muramyl dipeptide (MDP) is a NOD2 ligand (140, 141). MDP is detectable in Gram-negative and also in Gram-positive bacterial PGN, while iE-DAP is present in Gram-negative bacterial PGN and in PGN of particular Gram-positive bacteria such as Bacillus subtilis and Listeria monocytogenes (142). Hence, NOD1 is particularly involved in recognizing components from Gram-negative bacterial cell walls, while NOD2 can sense both (143, 144). A number of different cell types including oral epithelial cells express NOD1 that plays an essential role in innate immune responses (127, 129, 145). NOD1 binding and downstream signaling elicits an inflammatory reaction, inducing the production of cytokines, chemokines and antimicrobial peptides. Among these products, some are pro-inflammatory, such as interleukin (IL)-6, IL-8, tumor necrosis factor (TNF)-α and human beta defensin (hBD)-2, while others have immuno-regulatory or antimicrobial properties, such as interferon (IFN)-γ and human β-defensin-1 (hBD-1).The effects of iE-DAP on cytokine production have also been investigated with conflicting results: while it was reported that iE-DAP stimulated various human epithelial cells to produce anti-microbial peptides, but not pro-inflammatory cytokines like IL-6 and IL-8 (127, 129, 131, 145), it was also shown in human intestinal epithelial cells and dental pulp fibroblasts that NOD1 activation induces the production of pro-inflammatory cytokines (127, 142, 146–148).
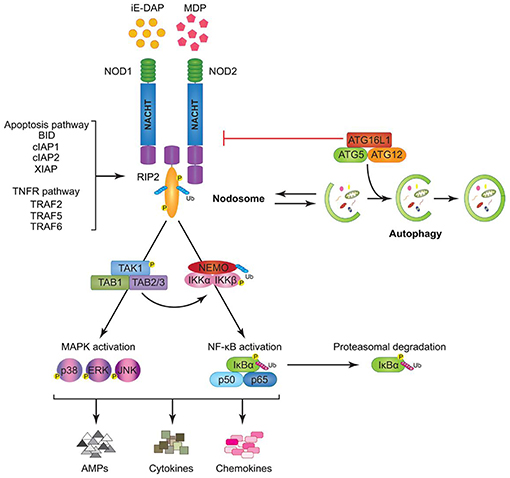
Figure 6. Model of Nucleotide-binding oligomerization domain-containing protein (NOD)1 and NOD2 signaling cascades. NOD1 and NOD2 recognize bacterial peptidoglycans (PGNs), (iE-DAP), and muramyl dipeptide (MDP), respectively. Following ligand sensing the NODs recruit their common adaptor receptor-interacting serine/threonine-protein kinase (RIP)2 by caspase activation and recruitment domains (CARD)–CARD interactions and induce RIP2 to undergo phosphorylation. The members of the tumor necrosis factor receptor-associated factor (TRAF) family (TRAF2, TRAF5, and TRAF6), the inhibitor of apoptosis (IAP) family (XIAP, cIAP1, and cIAP2), and the B-cell lymphoma (BCL)2 family (BID) bind to RIP2 and facilitate its ubiquitination allowing the recruitment of transforming growth factor-β-activated kinase (TAK)1 and ubiquitinated nuclear factor (NF-κB) essential modulator (NEMO) to the nodosome. On one hand, NEMO instigates activation of the canonical NF-κB pathway by phosphorylating NF-κB inhibitor kinase (IKK)α and IKKβ, by inducing nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha (IκBα) phosphorylation and proteasomal degradation, and by freeing p50 and p65 NF-κB subunits. On the other hand, transforming growth factor-β-activated kinase 1 (TAK1) recruits, transforming growth factor-β-activated kinase binding protein (TAB)1 and TAB2/3 inducing both (p38, extracellular-signal Regulated Kinases = ERK, and JUN N-terminal kinases = JNK) mitogen activated protein kinases (MAPK) and NF-κB activation. Stimulation of both arms culminates in the induction of anti-microbial peptides (AMPs), cytokines, and chemokines. The formation of the nodosome promotes autophagy and conversely, a fully functional autophagy machinery helps in signal transduction through the nodosome. Autophagy-related protein (ATG)16L1 along with ATG5 and ATG12 is required for autophagosome formation, however, independently of its autophagy functions, ATG16L1 negatively regulates NOD/RIP2 signaling (136).
In a human oral mucosal epithelial cell line (Leuk-1) upon stimulation activation of NOD1, receptor-interacting serine/threonine-protein kinase 2 (RIP2) and P-NF-κB was demonstrated, which was significantly inhibited by pretreatment of the cells with cigarette smoke extract (CSE). The suppressive effect of CSE on NOD1 expression was reversed following iE-DAP treatment. Combination of CSE stimulation with iE-DAP treatment prevented the further enhancement of RIP2 and P-NF-κB levels, i.e., iE-DAP reversed the inhibitory effect of CSE on NOD1 expression and prevented the over-activation of RIP2 and P-NF-κB due to CSE exposure. CSE furthermore upregulated levels of IL-6, IL-8, and TNF-α and downregulated IFN-γ level while iE-DAP enhanced the levels of IL-6, TNF-α, and IFN-γ, indicating that iE-DAP augmented gene expression and release of IL-6, TNF-α, and IFN-γ in Leuk-1 cells but diminished the mRNA level of IL-8 without affecting the production of IL-8 at protein level. These results indicate that iE-DAP is able to antagonize CSE-mediated effects on NOD1 expression and downstream signaling to a certain extent (149). In Figure 6 a model of NOD1 and NOD2 signaling cascades is depicted.
Protease-activated receptors (PARs) are a family of G-protein-coupled receptors (GPCRs) that include four members, PAR-1, PAR-2, PAR-3, and PAR-4, that play an important role in wound healing, inflammation, hemostasis, thrombosis, cancer progression, and embryonic development (150). PARs are activated by proteolytic cleavage of the N-terminal extracellular sequence of the receptors by a proteinase. This cleavage exposes a new N-terminal sequence, operating as a tethered ligand which, after binding to the receptor, initiates multiple signaling cascades (151–153). Although all PARs show the same mechanism of function, it has been demonstrated that different PARs can be activated by different proteinases and show diverse distributions and biological activities (154). One main activator of PAR-1, PAR-2, and PAR-3 is thrombin, further essential activators of PAR-1 comprise activated protein C (APC) and matrix metalloproteinase-1 (MMP-1). Trypsin and human mast cell tryptase activate PAR-2 while trypsin and cathepsin G activate PAR-4. Analysis of the downstream signaling responses after activation of PARs revealed that PAR-1, PAR-2, and PAR-4 may signal autonomously, while PAR-3 rather seem to be a co-receptor for PAR-1 and PAR-4 (155–158). PARs are expressed in a number of different cell types and it has been suggested that they influence physiological processes, such as growth, development, inflammation, tissue repair, and pain. In gingival epithelial cells (GEC) the presence of PAR-1,-2 and 3 mRNA and protein expression could be demonstrated while PAR-4 was not detected. Pre-incubation of the cells with P. gingivalis supernatant containing proteolytic activity, induced PAR-2 mRNA up-regulation. In contrast, PAR-1 and -3 were down-regulated. The authors concluded from these results that GECs recognize P. gingivalis by PARs and mediate innate immunity cell responses (159). PARs, NODs, and TLR were found to exhibit a complex interplay. In silencing experiments it was shown that knock-out of one receptor type may affect the others. GEC with silenced PAR-1 and -2 reacted with up-regulated NOD1 and NOD2 expression upon stimulation with P. gingivalis or Fusobacterium nucleatum (F. nucleatum). Expression of TLR2 decreased after infection with P. gingivalis when PAR2 was knocked down but was not affected after stimulation with F. nucleatum, while TLR4 expression was increased after PAR2 silencing and subsequent stimulation with F. nucleatum. F. nucleatum activates TLR4, while P. gingivalis, due to its unique LPS structure, is able to utilize TLR2 and shut down TLR4. These data suggest that if PAR receptors are absent, expression of TLRs is modified in response to bacteria following their activation level. PRRs can function as substitute in epithelial immune-response to bacterial challenge. These responses show variations depending on the properties of the bacterial stimuli (160).
One further mechanism of the immune system to initialize a pro-inflammatory response is the so called inflammasome (Figures 7, 8), a protein complex composed as multimer which develops in the cytoplasm participating in the immune response to pathogenic microbes or danger signals. The inflammasome induces the production and secretion of mature pro-inflammatory cytokines, IL-1β and IL-18 eventually leading to pyroptosis, a special kind of cell death (162–165). Inflammasomes can be grouped into canonical and noncanonical pathways (162, 164–167). Typically, a functional canonical inflammasome complex consists of a nucleotide-binding domain leucine-rich repeat (NLR) protein, an adaptor molecule apoptosis-associated speck-like protein containing a CARD (ASC) domain, and caspase-1 (167). The composition of the upstream regulators and specific molecules of the inflammasome depends on the type of the danger signals and the microbial inducers (164). The best characterized NLR, the pyrin domain containing 3 (NLRP3) inflammasome (Figure 8) for example is activated by extracellular adenosine triphosphate (eATP) danger signaling by the purinic receptor 2X7 (P2X7) and reactive oxygen species (ROS). Infection with bacterial pathogens can induce the NLRP3 inflammasome. Specific activation of the absent in melanoma (AIM)2 inflammasome is triggered by sensing double-stranded DNA in the cytosol while the IL-1β-converting enzyme (ICE) protease-activating factor (IPAF) inflammasome is activated by Gram-negative bacteria (e.g., Salmonella typhimurium, Shigella flexneri, Legionella pneumophila, and Pseudomonas aeruginosa) which have type III or IV secretion systems (164). In gingival epithelial cells F. nucleatum also activates the NLRP3 inflammasome, which in turn activates caspase-1 and stimulates secretion of mature IL-1β (168).
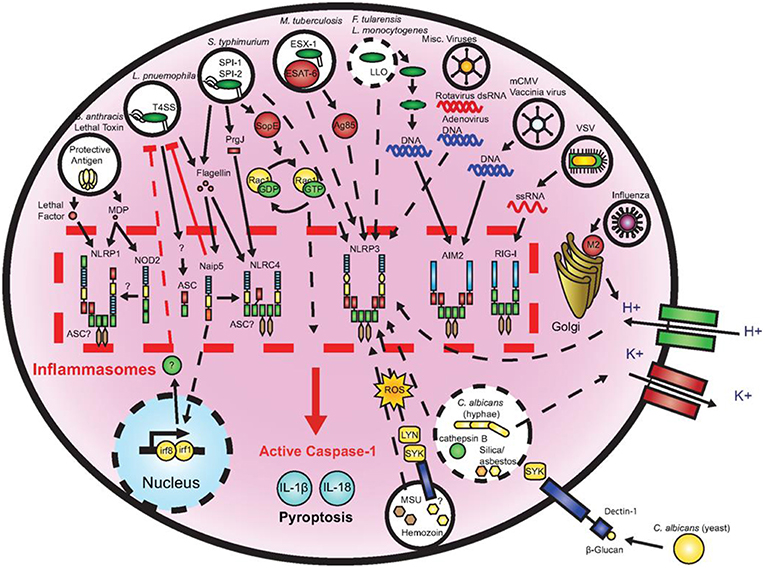
Figure 7. Microbial activation of the inflammasomes. Pathogenic microorganisms activate the inflammasomes through multiple agonists and pathways. Salmonells typhimurium (S. typhimurium), Legionells pneumophila (L. pneumophila), and Mycobacterium tuberculosis (M. tuberculosis) reside within the host cell phagosome and are capable of activating inflammasomes through secreted flagellin, effectors, or undefined NACHT, LRR, and PYD domains-containing protein (NLRP)3 agonists. NACHT = NAIP, neuronal apoptosis inhibitor protein; C2TA, class 2 transcription activator, of the MHC; HET-E, heterokaryon incompatibility; TP1, telomerase-associated protein 1; LRR, leucine-rich repeat; PYD, PYRIN domain. Francisella tularensis (F. tularensis) and Listeria monocytogenes (L. monocytogenes), which escape the phagosome activate absent in melanoma (AIM)2 that senses cytosolic deoxyribonucleic acid (DNA). Bacillus anthracis (B. anthracis) lethal toxin activates the NLRP1 inflammasome. Candida. albicans (C. albicans) and hemozoin activate NLRP3 through Spleen tyrosine kinase (SYK) signaling. Viral-mediated inflammasome activation is heavily dependent on the detection of nucleic acids by NLRP3, AIM2, and retinoic acid-inducible gene (RIG)-I. Dotted lines indicate signaling through an unknown mechanism (161).
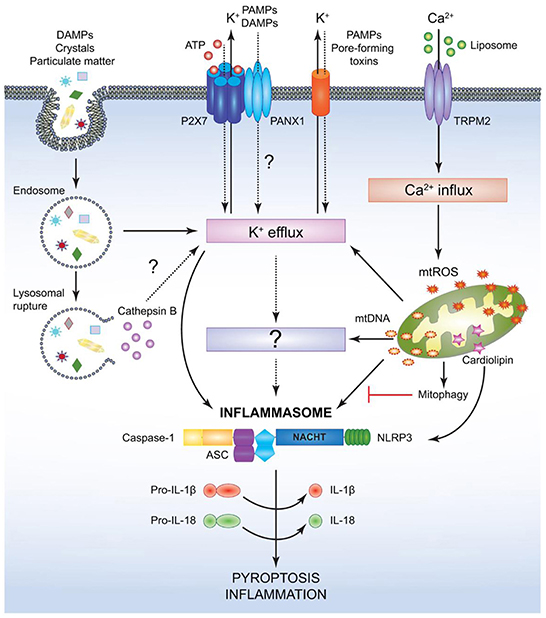
Figure 8. Mechanism for canonical NACHT, LRR and PYD domains-containing protein (NLRP)3- inflammasome activation. NACHT = NAIP, neuronal apoptosis inhibitor protein; C2TA, class 2 transcription activator, of the MHC; HET-E, heterokaryon incompatibility; TP1, telomerase-associated protein 1; LRR, leucine-rich repeat; PYD, PYRIN domain. Various pathogen associated molecular patterns (PAMPs) and damage associated molecular patterns (DAMPs) provide the signal 2 required to assemble and activate the NLRP3 inflammasome comprised of NLRP3, apoptosis-associated speck-like protein (ASC), and caspase-1. Although the precise mechanism leading to NLRP3 activation is still controversial, it is speculated that potassium ion K+ efflux may be the common cellular response that triggers inflammasome activation. However, this notion has not been fully verified and it is possible that an unidentified or intermediate adaptor may be required for transmitting signals between K+ efflux and the NLRP3 inflammasome. Crystals and particulate DAMPs enter the cell via endocytosis directly inducing K+ efflux and NLRP3-inflammasome formation. In addition, the endo- lysosomes carrying these DAMPs undergo lysosomal rupture and release cathepsin B, which acts as an intracellular DAMP and can induce K+ efflux. However, contradicting studies indicate that lysosomal rupture may cause K+ efflux and inflammasome activation even in the absence of cathepsin B. Adenosin triphosphate (ATP) binds to the P2X purinoceptor 7 (P2X7) receptor on the cell membrane and causes opening of the annexin 1 (PANX1) channels allowing K+ efflux and influx of any PAMPs and DAMPs present in the extracellular space. PAMPs such as pore-forming toxins activate the NLRP3 inflammasome and facilitate K+ efflux. Liposomes instigate Ca2+ influx through opening of (TRPM2) channels. Accumulation of excessive Ca2+ in the cytosol causes mitochondrial dysfunction and release of mitochondrial reactive oxygen species (mtROS) and oxidized mitochondrial deoxyribonucleic acid (mtDNA), which may activate the NLRP3 inflammasome either directly or by inducing K+ efflux. Clearance of distressed mitochondria by mitophagy serves to evade such inflammasome activation. Mitochondrial cardiolipin binds to NLRP3 and is required for the NLRP3-inflammasome activation. Following NLRP3-inflammasome assembly, caspase-1 undergoes proximity driven proteolytic cleavage and further processes pro-interleukin (IL)-18 and pro-IL-1β into their mature active forms. Activation of the NLRP3-caspase-1 axis results in inflammation and pyroptotic cell death (136).
P. gingivalis is able to inhibit the innate immune response using a nucleoside-diphosphate kinase (NDK) after stimulation with extracellular (e)ATP. The danger signal eATP binds to P2X7 receptors leading to activation of the inflammasome and caspase-1. Thus, exposure of gingival epithelial cells (GECs) to wild-type P. gingivalis was demonstrated to result in blockade of ATP-induced caspase-1 activation while NDK-deficient P. gingivalis showed fewer effects.
P. gingivalis NDK was shown to modify release of high-mobility group protein B1 (HMGB1), another pro-inflammatory danger signal, which, in non-infected cells, remains linked to chromatin. Infection with wild-type or NDK-deficient P. gingivalis induced release of HMGB1 from the nucleus to the cytosol. HMGB1 was delivered to the extracellular space when non-infected GECs were stimulated with ATP. HMGB1 was released in higher extend, when ATP-treated cells were infected with NDK-deficient mutants instead of wild-type P. gingivalis. These results suggest that NDKs are significantly involved in inhibiting P2X7-dependent inflammasome activation and HMGB1 release from bacterially infected GECs (169).
A multitude of inflammasomes exists, which can be activated by varying mechanisms resulting in the maturation and secretion of pro-inflammatory cytokines (170, 171). Extracellular ATP, one of the first activators that was discovered to induce NLRP3 inflammasome formation, is considered to belong to the group of endogenous damage-associated molecular patterns (DAMPs) released by dying or injured cells (172, 173). It exhibits minor presence in healthy tissues, but may increase to high micromolar concentrations at inflamed sites following tissue damage (174). Studies demonstrated that eATP caused caspase-1 activation that was followed by IL-1β release (175, 176). Yilmaz et al. (177) revealed that LPS-treated or infected gingival epithelial cells (GECs) did not secrete IL-1β unless they were stimulated with eATP and that eATP did not alter NLRP3 or apoptosis-associated speck like protein (ASC) expression in P. gingivalis infected gingival epithelial cells. NLRP10 is the smallest human NLR protein. It is different from the other NLR proteins because of its lack of the leucine-rich repeat domain, which is involved in ligand sensing or binding. Upon infection with two periodontal pathogens, T. forsythia and F. nucleatum the human oral epithelial cell line HOK-16B reacted with up-regulated mRNA and protein expression of NLRP10 while infection with Streptococcus oralis (S. oralis) did not induce this effect. These results demonstrate that NLRP10 up-regulation in HOK-16B cells is pathogen-specific (178). Figure 7 demonstrates microbial activation of the inflammasomes.
Interleukin (IL)-8 response of gingival epithelial cells after exposure to different multispecies biofilms was differentially regulated (179). Characterization of the whole secretome after biofilm challenge with species of the red complex demonstrated that more proteins were downregulated than up-regulated (180).
Keratinocytes are able to produce a variety of cytokines such as IL-1, IL-6, IL-8, and tumor necrosis factor (TNF)-α. They maintain normal homeostatic mechanisms and can induce proliferative effects upon injury. Mucosal cytokines may have pro-inflammatory as well as anti-inflammatory functions. An imbalance in the cytokine levels can support inflammatory diseases. Cytokines provide a paracrine (between adjacent cells), an endocrine (cells at distant sites) and an autocrine (intercellular) cell-to-cell communication system. Cytokines, based on different functions, origins, and chemical structures, are classified into the following groups: ILs, TNFs, chemokines, colony-stimulating factors (CSF), interferons (IFNs), and growth factors (GF). Cytokines share a multitude of activities and functions (pleiotropic and redundant), thus they could be classified in more than one group. IL-3 for example may also be classified in the CSF group (181). The IL-1 cytokines (IL-1α, IL-1β, and IL-1Ra) are important in regulation of immune response and inflammation because they induce the expression of many effector proteins, e.g., cytokines/chemokines, nitric oxide synthetase, and MMPs (182).
The immortalized human oral epithelial cell line OKF6-TERT2 responds to co-incubation with in vitro cultured biofilms of single- and mixed-bacterial species consisting of P. gingivalis, F. nucleatum, Aggregatibacter actinomycetemcomitans (A. actinomycetemcomitans) and Streptococcus mitis (S. mitis) with increased expression of mRNA for IL-8, C-X-C motif chemokine ligand 3 (CXCL3), CXCL1, IL-1, IL-6, CSF-2, and TNF-α. The response was biggest after stimulation with mixed-species biofilms (183).
The chemokine IL-8 shows crucial importance in oral health because it supports the transition of activated immune cells into and through gingival tissues, and promotes immune cell adhesion, tissue remodeling, and angiogenesis (184). IL-8 is increased in the saliva of patients with oral carcinomas. It could probably be a biomarker for the detection of oral and oropharyngeal squamous cell carcinomas (185). In patients with severe periodontitis, IL-8 was also detected in high levels in crevicular fluid at healthy sites (186). Schueller et al. investigated the basal release levels of IL-8, and linked it to the bacterial community, personal oral hygiene and nutrition in persons with a healthy gingival situation (187). It was shown that the basal IL-8 release was between 9.9 and 98.2 pg/ml, and bacterial biofilms were distinctive for healthy oral microbiota. An association between basal IL-8 levels and the oral microbiota was detected, suggesting a link between oral bacteria and the inflammatory state. A link between nutrition, personal oral hygiene, oral microbiota and IL-8 levels was also reported. The identification of indicator bacteria in healthy subjects with high levels of IL-8 release was regarded as important as they possibly are promising indicators for the onset of oral diseases (187). Fujimura et al. (188), reported that the hemophoric hemoglobin receptor (HbR), that binds hemoglobin and captures porphyrin and heme (P. gingivalis needs iron to grow), in interaction with host cells, affected cellular signal transduction of these cells, followed by inhibited differentiation of osteoclasts from bone marrow macrophages (188). The IL-8 inducing function of HbR from host epithelial cells was demonstrated to be maintained by activation of cellular signal transduction. Increased expression of IL-8 by gingival epithelial cells was induced by HbR in a dose dependent manner. This process is associated with activation of p38 MAPK and Erk1/2 using silencing (si)RNAs and inhibitors (189).
IL-33 belongs to the IL-1 cytokine family and is constitutively expressed in the nuclei of epithelial and endothelial cells (190). Epithelial cell-derived IL-33 augments T helper cell (Th)2 cytokine-mediated inflammatory immune response upon bacterial challenge (191). IL-33 was detected in inflamed gingival epithelium from chronic periodontitis patients. Enhanced IL-33 expression induced by P. gingivalis was detected in the cytoplasm of human gingival epithelial cells in vitro. In contrast, P. gingivalis fimbriae, lipopolysaccharide or lipopeptide did not induce this effect. Inhibition of P. gingivalis proteases (gingipains) blocked IL-33 mRNA induction. Also the P. gingivalis gingipain-null mutant KDP136 did not up-regulate IL-33 expression. Silencing of PAR-2 and inhibition of phospholipase C, p38 and NF-κB restrained the P. gingivalis induced IL-33 expression. These results indicate activation of the PAR-2/IL-33 axis in human gingival epithelial cells by P. gingivalis via a gingipain-dependent mechanism (192).
The angiopoietin-like protein (ANGPTL), belongs to a family of eight secreted glycoproteins, but doesn't bind to the tyrosine kinase with immunoglobulin-like and EGF-like domains (Tie)2 angiopoietin receptor or to the related protein Tie1, and, classified as orphan ligand, appears to exhibit biological functions different from angiopoietins (193–195). ANGPTL2 manages tissue homeostasis by induction of inflammation and angiogenesis (194, 196). Elevated ANGPTL2 concentrations are present in gingival crevicular fluid (GCF) from chronic periodontitis patients and stimulation with P. gingivalis LPS up-regulated ANGPTL2 mRNA and protein levels in gingival squamous cell carcinoma Ca9-22 cells. Recombinant human ANGPTL2 caused augmented IL-1β, IL-8, TNF-α mRNA and protein levels in Ca9-22 cells. Silencing of ANGPTL2 and blocking antibodies against the ANGPTL receptor integrin α5β1 inhibited the IL-1β, IL-8, and TNF-α mRNA and protein up-regulation, which suggests that ANGPTL secretion induces inflammatory cytokines in gingival epithelial cells through an autocrine loop. Thus, a new inflammatory cytokine induction cascade featuring sequential P. gingivalis LPS- ANGPTL2-integrin α5β1 activation was detected which might be responsible for periodontal destructive processes induced by gingival epithelial cells. Thus ANGPTL2 participates in the pathogenesis of periodontitis and may promote continuous chronic inflammation (197).
An immortalized human gingival cell line reacted with enhanced IL-8 and IL-6 mRNA concentrations and supported phosphorylation of ERK and p38 MAP kinase upon infection with A. actinomycetemcomitans (198).
A. actinomycetemcomitans, a member of the taxonomic family Pasteurellaceae, has been related to the development of aggressive periodontitis and may also promote chronic periodontitis (199–201). Amongst other pathogenic members of the periodontal biofilm, A. actinomycetemcomitans produces various substances that are able to damage cells and tissues in a direct or indirect manner. As a member of the oral biofilm this bacterium is known to express complex operons for two cytotoxins, leukotoxin (Lkt) and cytolethal distending toxin (Cdt) (202, 203). These toxins are able to impair the host's immune response and thus may promote the pathogenesis of periodontitis (204). Human gingival epithelial cells (HGECs) were stimulated with 50 clinical strains and 7 reference strains of A. actinomycetemcomitans, including various serotypes and non-serotypeable strains, strains from deep or shallow pockets, and reference serotype strains, and investigated for the expression of IL-1β, IL-6, IL-8, and TNF-α mRNAs. Results showed that IL-8 mRNA was strongly up-regulated after stimulation with clinically obtained A. actinomycetemcomitans and also with reference strains. Serotype f induced the highest expression in comparison to the other serotypes. The JP2-like leukotoxin promoter gene and non-serotypeable (NS) 1 and NS2 caused lesser IL-8 induction compared to serotypeable strains, and IL-8 up-regulation after stimulation with clinical strains from deep pockets showed also significantly lower levels than those isolated from shallow pockets. These results indicate that JP2-like leukotoxin NS1 and NS2 from clinical isolates of A. actinomycetemcomitans, obtained from deep pockets, are able to affect neutrophil function by lowering the IL-8 responses, which results in immunosuppression that may support virulence and survival of these bacteria (205).
T. denticola is a Gram-negative anaerobic oral spirochete that is known as a member of periodontal pathogens and is associated with chronic periodontitis (206, 207). It possesses a variety of virulence factors including dentilisin, an active cell-surface-located protease that cleaves at phenylalanyl/alanyl and prolyl/alanyl bonds, trypsin-like protease activity and the capability for motility and chemotaxis via periplasmic flagella [for Review see Dashper et al. (208)].
On primary gingival epithelial cells it has been demonstrated that T. denticola fails to induce IL-8 production that can't be explained by IL-8 degradation, as a protease mutant that does not degrade IL-8 also didn't induce IL-8 production. T. denticola furthermore failed to promote transcription of IL-8 and hβD-2 mRNA. This impaired epithelial cell response to T. denticola suggests contribution to the pathogenesis of periodontitis by deficient chemotaxis initiation of neutrophils into the periodontal pocket (209). The mechanism of IL-8 suppression by T. denticola was investigated using immortalized human gingival epithelial (HOK-16B) cells. Dentisilin degraded TNF-α, an IL-8-inducing cytokine, suggesting modulation of IL-8 (210). In monocytes derived from human peripheral blood mononuclear cells the role of T. denticola periplasmic flagella (PF) was investigated. Stimulation of the innate immune response via PAMPs revealed, that flagella-exhibiting wild type T. denticola induced the production of the cytokines TNF-α, IL-1, IL-6, IL-10, and IL-12 over activation of nuclear factor (NF)-κB through toll like receptor (TLR)2. These results suggest that T. denticola activates the innate immune response in a TLR2-dependent way and that flagella are involved as key bacterial components (211).
The IL-17 family, consisting of IL-17A–IL-17F, plays an important role in host defense against microbial challenge and has also been demonstrated to be crucial in pathogenesis of periodontitis (212). Initially IL-17A was regarded as a cytokine exclusively expressed by Th17 cells (213) but subsequent studies revealed that other cellular sources are capable to express IL-17A, including γδ T cells, natural killer cells, neutrophils, eosinophils, mast cells and macrophages (212). In gingival tissues of periodontitis patients (214–217) presence of IL-17 producing cells correlates with severity of inflammation in periodontitis lesions (218). Furthermore, elevated IL-17A levels were detected in GCF of patients with periodontitis. The IL-17A levels are reduced after non-surgical therapy (219, 220). Awang et al. (221) analyzed clinical linkage between cytokines of the IL-17 family and periodontitis and the biological effect of IL-17A and IL-17E using in vitro model systems. According to their studies serum, saliva and GCF IL-17A levels are increased in periodontitis patients and correlate with the clinical parameters attachment loss, pocket depth and bleeding on probing. Periodontitis patients exhibit lower IL-17E serum levels and the IL-17A-IL-17E ratio in serum also correlates positively with clinical parameters. In vitro, IL-17E suppressed IL-17A and P. gingivalis induced chemokine-expression by inhibiting phosphorylation of the NF-kB p65 subunit, which indicated that in the pathogenesis of periodontitis the serum IL-17A-IL-17E ratio might be a marker of disease severity while IL-17E is opposing IL-17A. IL-17E produced by oral keratinocytes may down-regulate IL-17A in the periodontium (221).
The growth arrest-specific 6 (GAS6) and Protein S (PROS1) are ligands of the tyrosin-protein kinase receptor (TYRO)3, AXL, and proto-oncogene (MERTK or TAM) receptor tyrosine kinases (222), which are involved in a number of biological processes including immune regulation (223). GAS6 is constitutively expressed in oral epithelial cells and was shown to downregulate epithelial activation at equilibrium state in order to sustain homeostasis (224). In the oral mucosa, the superficial layers of the epithelium express GAS6 together with its predominant receptor AXL. After birth GAS6 expression is induced in a MYD88-dependent way by the developing microbiota. GAS6 expressed by dendritic cells (DCs) was shown to inhibit IL-6 production by supporting development of T regulatory (Treg) cells and diminishing Th17 cell generation. This provides a more tolerogenic immunological environment for the oral microbiota (224). GAS6/AXL signaling seems to play a crucial role in the regulation of homeostasis in the oral mucosa. Thus, pathogens affecting the GAS6/AXL axis might generate a dysbiotic state and subsequent oral pathology. The induction of oral adaptive immune responses by specific pathogens is abolished in Gas6–/– mice and GAS6 is able to induce simultaneously pro- and anti-inflammatory regulatory pathways after mice were infected with P. gingivalis. GAS6 not only upregulates the expression of adhesion molecules in blood vessels which supports extravasation of immune cells belonging to the innate immune response, it also increases the expression of CCL19 and CCL21 chemokines and thus supports oral DCs to migrate to the lymph nodes. In addition the expression of the pro-inflammatory molecules P-selectin, intercellular adhesion molecule 1 (ICAM-1), and vascular cell adhesion molecule 1 (VCAM-1) in the oral mucosa is downregulated by GAS6. Furthermore, GAS6 blocks DC maturation and decreases antigen presentation by DCs to T cells. The authors concluded that GAS6 facilitates migration of inflammatory cells and DCs through the endothelium in both directions, while T-cell stimulation and activation of the mucosa is inhibited. This highly regulated activity of GAS6 supports a rapid but still moderate mucosal immunity to oral pathogens (225).
Evidence suggests an increased risk for cancer in chronic infections and inflammation. Bacterial infections and carcinogenesis seem to be connected (226). Periodontitis, one of the most common chronic human inflammatory diseases, is caused by microorganisms in the oral biofilm that trigger local inflammation. Periodontitis induces epithelial proliferation and apical migration along the root surface of the tooth and leads to a constant release of inflammatory cytokines, growth factors, prostaglandins, and enzymes, which all of them are closely associated with the development of cancer (226). In previous studies by Tezal et al. (226, 227) it was reported that the assumed association between periodontitis and oral neoplasms is significant. High PD-L1 expression has been linked to different types of human malignancies like lung cancer, pancreatic cancer, oral cancer, kidney cancer, breast and gastric cancer (228, 229). Head and neck cancer belongs to the 10 most frequent cancers worldwide (230). About 95% of the cases are of the squamous cell carcinoma (SCC) type. Tumors can only grow if their tissue environment provides them with a milieu that sustains their growth and spread. Alterations of tissue homeostasis by infection or inflammation can compromise stromal structural integrity and support tumorigenesis (231).
Programmed death ligand 1 (PD-L1, also called B7-H1) belongs to the B7 family and plays an important role in the regulation of cell-mediated immune response (232, 233). PD-L1 mediated signals are essential in co-signaling of T cell activation and tolerance (234). PD-L1 signals are also able to downregulate T cell functions and survival (228, 235). Modification of immune responses in cancer sites is a crucial mechanism that is linked to immune evasion of tumors. In the tumor microenvironment PD-L1 and programmed death receptor 1 (PD-1) may interact and induce tumor-protective mechanisms by activation of multiple specific pathways including ligation of PD-1 by PD-L1 on antigen specific T cells. This in turn may lead to functional anergy and/or apoptosis of these effector T cells. Ligation of PD-1 by PD-L1 possibly promotes tolerance and directly protects the tumor from apoptosis by reverse signaling through PD-L1 (228, 236, 237). It was demonstrated that P. gingivalis W83 up-regulates PD-L1 in oral cancer cells and in primary as well as in immortalized human gingival keratinocytes (238). High levels of PD-L1 were demonstrated in invasive oral squamous carcinoma cells (239). Also, positive PD-L1 expression was detected in tissue samples of oral squamous cell carcinomas ex vivo (240). Membrane proteins of P. gingivalis are responsible for the up-regulation while cytosolic proteins failed to induce PD-L1 (241). Primary human gingival keratinocytes (PHGK) and oral squamous cell carcinoma (SCC-25) cells up-regulated a number of inflammation-related genes upon infection with P. gingivalis membranes, amongst them members of the downstream NF-κB signaling pathway, TLR signaling and MAPK pathways. These data not only suggest that P. gingivalis membrane induces a pro-inflammatory response in malignant and non-malignant oral epithelial cells, but also indicates a possible link between infection and oral carcinomas, since p38 MAPK and MEK4-JNK1 signaling pathways were shown to be involved in context of tumor microenvironment and the control of cancer growth (242).
In a Chinese population an increased risk for head and neck carcinomas in individuals with oral submucous fibrosis, oral leukoplakia and repetitive dental ulcers was demonstrated suggesting a strong association between these diseases and cancer (243). Also patients with colorectal cancer carry strains of F. nucleatum in the cancerous lesions. F. nucleatum is one of the most densely colonized bacterial species in the oral cavity and known to be associated with periodontitis (244). The effect of oral pathogens on the development of oral tumors was investigated by Hoppe et al. (245). Stimulation of oral tumor cells with P. gingivalis led to increased cell proliferation while in contrast, A. actinomycetemcomitans promoted cell death. Bacteria as well as anti-microbial peptides induced diverse effects on the transcription levels of the oncogenic defensin genes and epidermal growth factor receptor (EGFR) signaling. The primary impact of the two oral pathogens were opposite on the proliferation behavior of oral tumor cells. In contrast, both induced similar secondary effects on the proliferation rate by modulating the extend of oncogenic important α-defensin gene expression. Human defensins differently modify epidermal growth factor receptor signaling, supporting the assumption that these anti-microbial peptides are possible ligands of EGFR. Interaction of these two molecules may cause the modification of the proliferative behavior of oral tumor cells (245). A newly established model of long term infection by stimulating human immortalized oral epithelial cells (HIOECs) with P. gingivalis at a low multiplicity of infection (MOI) for 5–23 weeks was used to investigate possible alterations in tumors. Persistent infection with P. gingivalis induced changes in cell morphology, enhanced proliferative capability and promoted cell migratory and invasive properties. Furthermore, tumor-related genes including GAS6 and PD-L1 that possibly act as key regulators in transformation from non-cancerous to tumor cells were upregulated as a reaction to long-term exposure of P. gingivalis. The authors concluded that P. gingivalis is able to support tumorigenic characteristics of HIOECs, suggesting that chronic P. gingivalis infection possibly represents a risk factor for oral cancers (246). Using a newly-established murine oral tumorigenesis model that is associated with periodontitis, it was reported that chronic bacterial infection supports development of OSCC, inducing enhanced signaling of the IL-6- signal transducer and activator of transcription 3 (STAT3) pathway. The results indicate that periodontal pathogens like P. gingivalis and F. nucleatum promote tumorigenesis by interacting directly with oral epithelial cells through TLR2. Furthermore, these pathogens stimulate the proliferation of human OSCC and induce expression of molecular key factors that take part in tumorigenesis, such as STAT3, that becomes activated in response to interferons, EGF, IL-5, IL-6, and cyclin D1, which is required for progression through the G1 phase of the cell cycle and induces cell migration. These findings represent a function of oral bacteria in the mechanism of chemically induced OSCC tumorigenesis (247).
The epithelial–mesenchymal transition (EMT) is critical in the conversion of normal epithelial cells into carcinoma cells during carcinogenesis. Downregulation of E-cadherin and up-regulation of N-cadherin and the transcription factors zinc finger protein SNAI1 (= Snail), SNAI2 (= Slug) and zinc finger e-box-binding homeobox (Zeb)1 are typical markers for this conversion (248). In a newly established long term infection in vitro model Lee et al. (249) demonstrated that primary oral epithelial cells after 120 h of infection with P. gingivalis develop an EMT phenotype including decrease of E-cadherin and increase of Slug, Snail, and Zeb1 expression. Infection of primary rat epithelial cell cultures with periodontal pathogens for 8 days caused an enhanced percentage of vimentin-positive cells, 20% after stimulation with P. gingivalis and 30% after infection with F. nucleatum. Furthermore, the periodontal pathogens induced augmented activation of Snail and the electrical impedance in comparison with unexposed controls of the cultures was reduced. The capability of the cells to migrate was extended as reaction to bacterial stimulation, demonstrated by the number of migrating cells and scratch-wound closure rates. In conclusion, persistent stimulation of primary rat oral keratinocyte cultures to periodontal pathogens elicited EMT-like properties, which indicates that this process may promote loss of epithelial integrity (250).
The composition of the gingival epithelial barrier is quite complex since its structure composes of a huge number of different molecules. Keratins are the major component of the keratinizing stratified epithelial cytoskeleton. Depending on localization and function different epithelia express a distinct cytokeratin pattern. In order to sustain their function, stratified epithelia, including the oral mucosa, have to sustain tight cell-cell adhesion in the viable cells that involves intercellular tight and adherens junctions which connect to the actin cytoskeleton. Oral tissue immune response is able to recognize microbial infection and colonization and is able to manage it. The epithelial cells express a number of pattern recognition receptors, including TLRs, NOD1, NOD2 and PARs and are able to assemble different kinds of inflammasomes and express a variety of pro-inflammatory cytokines and chemokines. Resident oral bacteria permanently influence epithelial host cells. Depending on their composition biofilms differentially modify cellular immune responses of the epithelium. Major periodontal pathogens like P. gingivalis possess a number of different strategies to escape from innate immunity and survive in the tissues, which affects the epithelial barrier by modifying the expression and integrity of the different cell-cell junctions.
The balanced immune-inflammatory state of the host with its biofilm in health may be disturbed by distinct species, such as P. gingivalis and F. nucleatum that are able to destroy this equilibrium, causing a dysbiotic microbiota (251). Chronic infections and persistent inflammation are associated with an increased risk of cancer. Persisting bacterial agents may induce up-regulation of immune-inhibitory receptors which in turn facilitate the ability of cancer cells to evade from anti-tumor immune responses of the host. Furthermore, long term infection possibly supports carcinogenesis by regulating gene expressions of the infected epithelial cells in a way that leads to development into a phenotype indicating cellular transformation from normal to cancerous.
SG wrote the manuscript. JM corrected the manuscript and assisted in writing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
3. Schroeder HE, Listgarten MA. The gingival tissues: the architecture of periodontal protection. Periodontol (1997)13:91–120.
4. Squier CA, Kremer MJ. Biology of oral mucosa and esophagus. J Natl Cancer Inst Monogr. (2001) 29:7–15.
5. Groeger SE, Meyle J. Epithelial barrier and oral bacterial infection. Periodontol (2015) 69:46–67. doi: 10.1111/prd.12094
6. Pollanen MT, Salonen JI, Uitto VJ. Structure and function of the tooth-epithelial interface in health and disease. Periodontol (2003) 31:12–31. doi: 10.1034/j.1600-0757.2003.03102.x
7. Steinert PM, Roop DR. Molecular and cellular biology of intermediate filaments. Annu Rev Biochem. (1988) 57:593–625.
8. Moll R, Franke WW, Schiller DL, Geiger B, Krepler R. The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell (1982) 31:11–24. doi: 10.1016/0092-8674(82)90400-7
9. Su L, Morgan PR, Lane EB. Protein and mRNA expression of simple epithelial keratins in normal, dysplastic, and malignant oral epithelia. Am J Pathol. (1994) 145:1349–57.
10. Moll R, Divo M, Langbein L. The human keratins: biology and pathology. Histochem Cell Biol. (2008) 129:705–33. doi: 10.1007/s00418-008-0435-6
11. Dale BA, Salonen J, Jones AH. New approaches and concepts in the study of differentiation of oral epithelia. Crit Rev Oral Biol Med. (1990) 1:167–90. doi: 10.1177/10454411900010030201
12. Collin C, Ouhayoun JP, Grund C, Franke WW. Suprabasal marker proteins distinguishing keratinizing squamous epithelia: cytokeratin 2 polypeptides of oral masticatory epithelium and epidermis are different. Differentiation (1992) 51:137–48. doi: 10.1111/j.1432-0436.1992.tb00690.x
13. Mackenzie IC, Rittman G, Gao Z, Leigh I, Lane EB. Patterns of cytokeratin expression in human gingival epithelia. J Periodontal Res. (1991) 26:468–78. doi: 10.1111/j.1600-0765.1991.tb01797.x
14. Berkovitz BKB HG, Moxham BJ. Oral Anatomy, Histology and Embryology. Edinbergh: Elsevier limited. (2009).
15. Nanci A. Ten Cate's Oral Histology, Development, Structure and Function. Mosby, MO: Elsevier Mosby (2008).
16. Alberts B JA, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. New York, NY: Garland Science (2002).
17. Rao RS, Patil S, Ganavi BS. Oral cytokeratins in health and disease. J Contemp Dent Pract. (2014) 15:127–36. doi: 10.5005/jp-journals-10024-1502
18. Hajishengallis G, Lamont RJ. Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol Oral Microbiol. (2012) 27:409–19. doi: 10.1111/j.2041-1014.2012.00663.x
19. Abusleme L, Dupuy AK, Dutzan N, Silva N, Burleson JA, Strausbaugh LD, et al. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J. (2013) 7:1016–25. doi: 10.1038/ismej.2012.174
20. Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol. (2012) 10:717–25. doi: 10.1038/nrmicro2873
21. Hajishengallis G. Immunomicrobial pathogenesis of periodontitis: keystones, pathobionts, and host response. Trends Immunol. (2014) 35:3–11. doi: 10.1016/j.it.2013.09.001
22. Hajishengallis G. The inflammophilic character of the periodontitis-associated microbiota. Mol Oral Microbiol. (2014) 29:248–57. doi: 10.1111/omi.12065
23. Presland RB, Jurevic RJ. Making sense of the epithelial barrier: what molecular biology and genetics tell us about the functions of oral mucosal and epidermal tissues. J Dent Educ. (2002) 66:564–74.
24. Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. (2006) 86:279–367. doi: 10.1152/physrev.00012.2005
25. Niessen CM. Tight junctions/adherens junctions: basic structure and function. J Invest Dermatol. (2007) 127:2525–32. doi: 10.1038/sj.jid.5700865
26. Anderson JM. Molecular structure of tight junctions and their role in epithelial transport. News Physiol Sci. (2001) 16:126–30.
27. Tsukita S, Furuse M, Itoh M. Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol. (2001) 2:285–93. doi: 10.1038/35067088
28. Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol. (2004) 5:261–70. doi: 10.1038/nrm1357
29. Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol. (1998) 141:1539–50. doi: 10.1083/jcb.141.7.1539
30. Furuse M, Sasaki H, Fujimoto K, Tsukita S. A single gene product, claudin-1 or−2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J Cell Biol. (1998) 143:391–401. doi: 10.1083/jcb.143.2.391
31. Matter K, Aijaz S, Tsapara A, Balda MS. Mammalian tight junctions in the regulation of epithelial differentiation and proliferation. Curr Opin Cell Biol. (2005) 17:453–8. doi: 10.1016/j.ceb.2005.08.003
32. Kohler K, Zahraoui A. Tight junction: a co-ordinator of cell signalling and membrane trafficking. Biol Cell (2005) 97:659–65. doi: 10.1042/BC20040147
33. Tsukita S, Furuse M. Overcoming barriers in the study of tight junction functions: from occludin to claudin. Genes Cells (1998) 3:569–73. doi: 10.1046/j.1365-2443.1998.00212.x
34. Furuse M, Hata M, Furuse K, Yoshida Y, Haratake A, Sugitani Y, et al. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J Cell Biol. (2002) 156:1099–111. doi: 10.1083/jcb.200110122
35. Tsukita S, Furuse M. Claudin-based barrier in simple and stratified cellular sheets. Curr Opin Cell Biol. (2002) 14:531–6. doi: 10.1016/S0955-0674(02)00362-9
36. Tang VW, Goodenough DA. Paracellular ion channel at the tight junction. Biophys J. (2003) 84:1660–73. doi: 10.1016/S0006-3495(03)74975-3
37. Furuse M, Sasaki H, Tsukita S. Manner of interaction of heterogeneous claudin species within and between tight junction strands. J Cell Biol. (1999) 147:891–903. doi: 10.1083/jcb.147.4.891
38. Furuse M. Knockout animals and natural mutations as experimental and diagnostic tool for studying tight junction functions in vivo. Biochim Biophys Acta (2009) 1788:813–9. doi: 10.1016/j.bbamem.2008.07.017
39. Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, et al. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. (1993) 123(6 Pt 2):1777–88. doi: 10.1083/jcb.123.6.1777
40. Muresan Z, Paul DL, Goodenough DA. Occludin 1B, a variant of the tight junction protein occludin. Mol Biol Cell (2000) 11:627–34. doi: 10.1091/mbc.11.2.627
41. Shen L, Turner JR. Actin depolymerization disrupts tight junctions via caveolae-mediated endocytosis. Mol Biol Cell (2005) 16:3919–36. doi: 10.1091/mbc.e04-12-1089
42. Utech M, Mennigen R, Bruewer M. Endocytosis and recycling of tight junction proteins in inflammation. J Biomed Biotechnol. (2010) 2010:484987. doi: 10.1155/2010/484987
43. Yu AS, McCarthy KM, Francis SA, McCormack JM, Lai J, Rogers RA, et al. Knockdown of occludin expression leads to diverse phenotypic alterations in epithelial cells. Am J Physiol Cell Physiol. (2005) 288:C1231–41. doi: 10.1152/ajpcell.00581.2004
44. Hatakeyama S, Yaegashi T, Oikawa Y, Fujiwara H, Mikami T, Takeda Y, et al. Expression pattern of adhesion molecules in junctional epithelium differs from that in other gingival epithelia. J Periodontal Res. (2006) 41:322–8. doi: 10.1111/j.1600-0765.2006.00875.x
45. Ye P, Yu H, Simonian M, Hunter N. Expression patterns of tight junction components induced by CD24 in an oral epithelial cell-culture model correlated to affected periodontal tissues. J Periodontal Res. (2014) 49:253–9. doi: 10.1111/jre.12102
46. Groeger S, Jarzina F, Windhorst A, Meyle J. Influence of retinoic acid on human gingival epithelial barriers. J Periodontal Res. (2016) 51:748–57. doi: 10.1111/jre.12351
47. Belibasakis GN, Kast JI, Thurnheer T, Akdis CA, Bostanci N. The expression of gingival epithelial junctions in response to subgingival biofilms. Virulence (2015) 6:704–9. doi: 10.1080/21505594.2015.1081731
48. Chen YH, Lu Q, Goodenough DA, Jeansonne B. Nonreceptor tyrosine kinase c-Yes interacts with occludin during tight junction formation in canine kidney epithelial cells. Mol Biol Cell (2002) 13:1227–37. doi: 10.1091/mbc.01-08-0423
49. Seth A, Sheth P, Elias BC, Rao R. Protein phosphatases 2A and 1 interact with occludin and negatively regulate the assembly of tight junctions in the CACO-2 cell monolayer. J Biol Chem. (2007) 282:11487–98. doi: 10.1074/jbc.M610597200
50. Ikenouchi J, Furuse M, Furuse K, Sasaki H, Tsukita S. Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J Cell Biol. (2005) 171:939–45. doi: 10.1083/jcb.200510043
51. Krug SM, Amasheh S, Richter JF, Milatz S, Gunzel D, Westphal JK, et al. Tricellulin forms a barrier to macromolecules in tricellular tight junctions without affecting ion permeability. Mol Biol Cell (2009) 20:3713–24. doi: 10.1091/mbc.e09-01-0080
52. Raleigh DR, Marchiando AM, Zhang Y, Shen L, Sasaki H, Wang Y, et al. Tight junction-associated MARVEL proteins marveld3, tricellulin, and occludin have distinct but overlapping functions. Mol Biol Cell (2010) 21:1200–13. doi: 10.1091/mbc.e09-08-0734
53. Itoh M, Sasaki H, Furuse M, Ozaki H, Kita T, Tsukita S. Junctional adhesion molecule (JAM) binds to PAR-3: a possible mechanism for the recruitment of PAR-3 to tight junctions. J Cell Biol. (2001) 154:491–7. doi: 10.1083/jcb.200103047
54. Ebnet K, Schulz CU, Meyer Zu, Brickwedde MK, Pendl GG, Vestweber D. Junctional adhesion molecule interacts with the PDZ domain-containing proteins AF-6 and ZO-1. J Biol Chem. (2000) 275:27979–88. doi: 10.1074/jbc.M002363200
55. Monteiro AC, Sumagin R, Rankin CR, Leoni G, Mina MJ, Reiter DM, et al. JAM-A associates with ZO-2, afadin, and PDZ-GEF1 to activate Rap2c and regulate epithelial barrier function. Mol Biol Cell (2013) 24:2849–60. doi: 10.1091/mbc.e13-06-0298
56. Cohen CJ, Shieh JT, Pickles RJ, Okegawa T, Hsieh JT, Bergelson JM. The coxsackievirus and adenovirus receptor is a transmembrane component of the tight junction. Proc Natl Acad Sci USA. (2001) 98:15191–6. doi: 10.1073/pnas.261452898
57. Walters RW, Freimuth P, Moninger TO, Ganske I, Zabner J, Welsh MJ. Adenovirus fiber disrupts CAR-mediated intercellular adhesion allowing virus escape. Cell (2002) 110:789–99. doi: 10.1016/S0092-8674(02)00912-1
58. Zhang GH, Wu LL, Yu GY. Tight junctions and paracellular fluid and ion transport in salivary glands. Chin J Dent Res. (2013) 16:13–46.
59. McCarthy KM, Francis SA, McCormack JM, Lai J, Rogers RA, Skare IB, et al. Inducible expression of claudin-1-myc but not occludin-VSV-G results in aberrant tight junction strand formation in MDCK cells. J Cell Sci. (2000) 113(Pt 19):3387–98.
60. Stevenson BR, Siliciano JD, Mooseker MS, Goodenough DA. Identification of ZO-1: a high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J Cell Biol. (1986) 103:755–66. doi: 10.1083/jcb.103.3.755
61. Gumbiner B, Lowenkopf T, Apatira D. Identification of a 160-kDa polypeptide that binds to the tight junction protein ZO-1. Proc Natl Acad Sci USA. (1991) 88:3460–4. doi: 10.1073/pnas.88.8.3460
62. Haskins J, Gu L, Wittchen ES, Hibbard J, Stevenson BR. ZO-3, a novel member of the MAGUK protein family found at the tight junction, interacts with ZO-1 and occludin. J Cell Biol. (1998) 141:199–208. doi: 10.1083/jcb.141.1.199
63. Takano K, Kojima T, Sawada N, Himi T. Role of tight junctions in signal transduction: an update. EXCLI J. (2014) 13:1145–62.
64. Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol. (2009) 1:a002584. doi: 10.1101/cshperspect.a002584
65. Turner JR, Buschmann MM, Romero-Calvo I, Sailer A, Shen L. The role of molecular remodeling in differential regulation of tight junction permeability. Semin Cell Dev Biol. (2014) 36:204–12. doi: 10.1016/j.semcdb.2014.09.022
66. Furuse M, Izumi Y, Oda Y, Higashi T, Iwamoto N. Molecular organization of tricellular tight junctions. Tissue Barriers (2014) 2:e28960. doi: 10.4161/tisb.28960
67. Meyle J, Gultig K, Rascher G, Wolburg H. Transepithelial electrical resistance and tight junctions of human gingival keratinocytes. J Periodontal Res. (1999) 34:214–22. doi: 10.1111/j.1600-0765.1999.tb02244.x
68. Gumbiner B, Simons K. A functional assay for proteins involved in establishing an epithelial occluding barrier: identification of a uvomorulin-like polypeptide. J Cell Biol. (1986) 102:457–68. doi: 10.1083/jcb.102.2.457
69. Staddon JM, Herrenknecht K, Smales C, Rubin LL. Evidence that tyrosine phosphorylation may increase tight junction permeability. J Cell Sci. (1995) 108(Pt 2):609–19.
70. Claude P. Morphological factors influencing transepithelial permeability: a model for the resistance of the zonula occludens. J Membr Biol. (1978) 39:219–32. doi: 10.1007/BF01870332
71. Groeger S, Doman E, Chakraborty T, Meyle J. Effects of Porphyromonas gingivalis infection on human gingival epithelial barrier function in vitro. Eur J Oral Sci. (2010) 118:582–9. doi: 10.1111/j.1600-0722.2010.00782.x
72. Palant CE, Duffey ME, Mookerjee BK, Ho S, Bentzel CJ. Ca2+ regulation of tight-junction permeability and structure in Necturus gallbladder. Am J Physiol. (1983) 245:C203–12. doi: 10.1152/ajpcell.1983.245.3.C203
73. Huibregtse JM, Beaudenon SL. Mechanism of HPV E6 proteins in cellular transformation. Semin Cancer Biol. (1996) 7:317–26.
74. Groeger S, Michel J, Meyle J. Establishment and characterization of immortalized human gingival keratinocyte cell lines. J Periodontal Res. (2008) 43:604–14. doi: 10.1111/j.1600-0765.2007.01019.x
75. Shi Y, Ratnayake DB, Okamoto K, Abe N, Yamamoto K, Nakayama K. Genetic analyses of proteolysis, hemoglobin binding, and hemagglutination of Porphyromonas gingivalis. Construction of mutants with a combination of rgpA, rgpB, kgp, and hagA. J Biol Chem. (1999) 274:17955–60.
76. Bruewer M, Utech M, Ivanov AI, Hopkins AM, Parkos CA, Nusrat A. Interferon-gamma induces internalization of epithelial tight junction proteins via a macropinocytosis-like process. FASEB J. (2005) 19:923–33. doi: 10.1096/fj.04-3260com
77. Guo W, Wang P, Liu ZH, Ye P. Analysis of differential expression of tight junction proteins in cultured oral epithelial cells altered by Porphyromonas gingivalis, Porphyromonas gingivalis lipopolysaccharide, and extracellular adenosine triphosphate. Int J Oral Sci. (2018) 10:e8. doi: 10.1038/ijos.2017.51
78. Provost E, Rimm DL. Controversies at the cytoplasmic face of the cadherin-based adhesion complex. Curr Opin Cell Biol. (1999) 11:567–72. doi: 10.1016/S0955-0674(99)00015-0
79. Baum B, Georgiou M. Dynamics of adherens junctions in epithelial establishment, maintenance, and remodeling. J Cell Biol. (2011) 192:907–17. doi: 10.1083/jcb.201009141
80. Imamura Y, Itoh M, Maeno Y, Tsukita S, Nagafuchi A. Functional domains of alpha-catenin required for the strong state of cadherin-based cell adhesion. J Cell Biol. (1999) 144:1311–22. doi: 10.1083/jcb.144.6.1311
81. Abe K, Takeichi M. EPLIN mediates linkage of the cadherin catenin complex to F-actin and stabilizes the circumferential actin belt. Proc Natl Acad Sci USA. (2008) 105:13–9. doi: 10.1073/pnas.0710504105
82. Mandai K, Nakanishi H, Satoh A, Obaishi H, Wada M, Nishioka H, et al. A novel actin filament-binding protein with one PDZ domain localized at cadherin-based cell-to-cell adherens junction. J Cell Biol. (1997) 139:517–28. doi: 10.1083/jcb.139.2.517
83. Kussel-Andermann P, El-Amraoui A, Safieddine S, Nouaille S, Perfettini I, Lecuit M, et al. Vezatin, a novel transmembrane protein, bridges myosin VIIA to the cadherin-catenins complex. EMBO J. (2000) 19:6020–9. doi: 10.1093/emboj/19.22.6020
84. Coopman P, Djiane A. Adherens junction and E-cadherin complex regulation by epithelial polarity. Cell Mol Life Sci. (2016) 73:3535–53. doi: 10.1007/s00018-016-2260-8
85. Nakagawa I, Inaba H, Yamamura T, Kato T, Kawai S, Ooshima T, et al. Invasion of epithelial cells and proteolysis of cellular focal adhesion components by distinct types of Porphyromonas gingivalis fimbriae. Infect Immun. (2006) 74:3773–82. doi: 10.1128/IAI.01902-05
86. Yilmaz O, Verbeke P, Lamont RJ, Ojcius DM. Intercellular spreading of Porphyromonas gingivalis infection in primary gingival epithelial cells. Infect Immun. (2006) 74:703–10. doi: 10.1128/IAI.74.1.703-710.2006
87. Potempa J, Banbula A, Travis J. Role of bacterial proteinases in matrix destruction and modulation of host responses. Periodontol (2000) 24:153–92. doi: 10.1034/j.1600-0757.2000.2240108.x
88. Bedi GS, Williams T. Purification and characterization of a collagen-degrading protease from Porphyromonas gingivalis. J Biol Chem. (1994) 269:599–606.
89. Chen Z, Casiano CA, Fletcher HM. Protease-active extracellular protein preparations from Porphyromonas gingivalis W83 induce N-cadherin proteolysis, loss of cell adhesion, and apoptosis in human epithelial cells. J Periodontol. (2001) 72:641–50. doi: 10.1902/jop.2001.72.5.641
90. Sheets SM, Potempa J, Travis J, Fletcher HM, Casiano CA. Gingipains from Porphyromonas gingivalis W83 synergistically disrupt endothelial cell adhesion and can induce caspase-independent apoptosis. Infect Immun. (2006) 74:5667–78. doi: 10.1128/IAI.01140-05
91. Hintermann E, Haake SK, Christen U, Sharabi A, Quaranta V. Discrete proteolysis of focal contact and adherens junction components in Porphyromonas gingivalis-infected oral keratinocytes: a strategy for cell adhesion and migration disabling. Infect Immun. (2002) 70:5846–56. doi: 10.1128/IAI.70.10.5846-5856.2002
92. Amano A. Molecular interaction of Porphyromonas gingivalis with host cells: implication for the microbial pathogenesis of periodontal disease. J Periodontol. (2003) 74:90–6. doi: 10.1902/jop.2003.74.1.90
93. Nakagawa I, Amano A, Kuboniwa M, Nakamura T, Kawabata S, Hamada S. Functional differences among FimA variants of Porphyromonas gingivalis and their effects on adhesion to and invasion of human epithelial cells. Infect Immun. (2002) 70:277–85. doi: 10.1128/IAI.70.1.277-285.2002
94. Tsuda K, Amano A, Umebayashi K, Inaba H, Nakagawa I, Nakanishi Y, et al. Molecular dissection of internalization of Porphyromonas gingivalis by cells using fluorescent beads coated with bacterial membrane vesicle. Cell Struct Funct. (2005) 30:81–91. doi: 10.1247/csf.30.81
95. Huttenlocher A, Ginsberg MH, Horwitz AF. Modulation of cell migration by integrin-mediated cytoskeletal linkages and ligand-binding affinity. J Cell Biol. (1996) 134:1551–62. doi: 10.1083/jcb.134.6.1551
96. Cox EA, Sastry SK, Huttenlocher A. Integrin-mediated adhesion regulates cell polarity and membrane protrusion through the Rho family of GTPases. Mol Biol Cell (2001) 12:265–77. doi: 10.1091/mbc.12.2.265
97. Kato T, Kawai S, Nakano K, Inaba H, Kuboniwa M, Nakagawa I, et al. Virulence of Porphyromonas gingivalis is altered by substitution of fimbria gene with different genotype. Cell Microbiol. (2007) 9:753–65. doi: 10.1111/j.1462-5822.2006.00825.x
98. Lamont RJ, Yilmaz O. In or out: the invasiveness of oral bacteria. Periodontol (2002) 30:61–9. doi: 10.1034/j.1600-0757.2002.03006.x
99. Amano A. Disruption of epithelial barrier and impairment of cellular function by Porphyromonas gingivalis. Front Biosci. (2007) 12:3965–74. doi: 10.2741/2363
100. Mifsud EJ, Tan AC, Jackson DC. TLR agonists as modulators of the innate immune response and their potential as agents against infectious disease. Front Immunol. (2014) 5:79. doi: 10.3389/fimmu.2014.00079
101. Sasai M, Yamamoto M. Pathogen recognition receptors: ligands and signaling pathways by Toll-like receptors. Int Rev Immunol. (2013) 32:116–33. doi: 10.3109/08830185.2013.774391
102. Akira S, Sato S. Toll-like receptors and their signaling mechanisms. Scand J Infect Dis. (2003) 35:555–62. doi: 10.1080/00365540310015683
103. Kawai T, Akira S. TLR signaling. Semin Immunol. (2007) 19:24–32. doi: 10.1016/j.smim.2006.12.004
104. Cario E, Brown D, McKee M, Lynch-Devaney K, Gerken G, Podolsky DK. Commensal-associated molecular patterns induce selective toll-like receptor-trafficking from apical membrane to cytoplasmic compartments in polarized intestinal epithelium. Am J Pathol. (2002) 160:165–73. doi: 10.1016/S0002-9440(10)64360-X
105. Zhang D, Zhang G, Hayden MS, Greenblatt MB, Bussey C, Flavell RA, et al. A toll-like receptor that prevents infection by uropathogenic bacteria. Science (2004) 303:1522–6. doi: 10.1126/science.1094351
106. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science (1998) 282:2085–8. doi: 10.1126/science.282.5396.2085
107. Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, et al. Cutting edge: role of toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol. (2002) 169:10–4. doi: 10.4049/jimmunol.169.1.10
108. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature (2001) 413:732–8. doi: 10.1038/35099560
109. Bauer S, Kirschning CJ, Hacker H, Redecke V, Hausmann S, Akira S, et al. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc Natl Acad Sci USA. (2001) 98:9237–42. doi: 10.1073/pnas.161293498
110. Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature (2001) 410:1099–103. doi: 10.1038/35074106
111. Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. (2002) 3:196–200. doi: 10.1038/ni758
112. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A Toll-like receptor recognizes bacterial DNA. Nature (2000) 408:740–5. doi: 10.1038/35047123
113. Jurk M, Heil F, Vollmer J, Schetter C, Krieg AM, Wagner H, et al. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat Immunol. (2002) 3:499. doi: 10.1038/ni0602-499
114. Fitzgerald KA, Palsson-McDermott EM, Bowie AG, Jefferies CA, Mansell AS, Brady G, et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature (2001) 413:78–83. doi: 10.1038/35092578
115. Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, et al. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J Exp Med. (2003) 198:1043–55. doi: 10.1084/jem.20031023
116. Horng T, Barton GM, Medzhitov R. TIRAP: an adapter molecule in the Toll signaling pathway. Nat Immunol. (2001) 2:835–41. doi: 10.1038/ni0901-835
117. Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, et al. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell (1998) 2:253–8. doi: 10.1016/S1097-2765(00)80136-7
118. Oshiumi H, Matsumoto M, Funami K, Akazawa T. Seya T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat Immunol. (2003) 4:161–7. doi: 10.1038/ni886
119. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science (2003) 301:640–3. doi: 10.1126/science.1087262
120. Akira S, Yamamoto M, Takeda K. Role of adapters in Toll-like receptor signalling. Biochem Soc Trans. (2003) 31(Pt 3):637–42. doi: 10.1042/bst0310637
121. Gohda J, Matsumura T, Inoue J. Cutting edge: TNFR-associated factor (TRAF) 6 is essential for MyD88-dependent pathway but not toll/IL-1 receptor domain-containing adaptor-inducing IFN-beta (TRIF)-dependent pathway in TLR signaling. J Immunol. (2004) 173:2913–7. doi: 10.4049/jimmunol.173.5.2913
122. Kondo T, Kawai T, Akira S. Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol. (2012) 33:449–58. doi: 10.1016/j.it.2012.05.002
123. Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, et al. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat Immunol. (2004) 5:503–7. doi: 10.1038/ni1061
124. Chen HC, Zhan X, Tran KK, Shen H. Selectively targeting the toll-like receptor 9 (TLR9)–IRF 7 signaling pathway by polymer blend particles. Biomaterials (2013) 34:6464–72. doi: 10.1016/j.biomaterials.2013.05.016
125. Beklen A, Hukkanen M, Richardson R, Konttinen YT. Immunohistochemical localization of Toll-like receptors 1–10 in periodontitis. Oral Microbiol Immunol. (2008) 23:425–31. doi: 10.1111/j.1399-302X.2008.00448.x
126. Semlali A, Witoled C, Alanazi M, Rouabhia M. Whole cigarette smoke increased the expression of TLRs, HBDs, and proinflammory cytokines by human gingival epithelial cells through different signaling pathways. PLoS ONE (2012) 7:e52614. doi: 10.1371/journal.pone.0052614
127. Uehara A, Fujimoto Y, Fukase K, Takada H. Various human epithelial cells express functional Toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol Immunol. (2007) 44:3100–11. doi: 10.1016/j.molimm.2007.02.007
128. Promsudthi A, Poomsawat S, Limsricharoen W. The role of Toll-like receptor 2 and 4 in gingival tissues of chronic periodontitis subjects with type 2 diabetes. J Periodontal Res. (2014) 49:346–54. doi: 10.1111/jre.12112
129. Sugawara Y, Uehara A, Fujimoto Y, Kusumoto S, Fukase K, Shibata K, et al. Toll-like receptors, NOD1, and NOD2 in oral epithelial cells. J Dent Res. (2006) 85:524–9. doi: 10.1177/154405910608500609
130. Uehara A, Sugawara S, Tamai R, Takada H. Contrasting responses of human gingival and colonic epithelial cells to lipopolysaccharides, lipoteichoic acids and peptidoglycans in the presence of soluble CD14. Med Microbiol Immunol. (2001) 189:185–92. doi: 10.1007/s004300100063
131. Uehara A, Sugawara Y, Kurata S, Fujimoto Y, Fukase K, Kusumoto S, et al. Chemically synthesized pathogen-associated molecular patterns increase the expression of peptidoglycan recognition proteins via toll-like receptors, NOD1 and NOD2 in human oral epithelial cells. Cell Microbiol. (2005) 7:675–86. doi: 10.1111/j.1462-5822.2004.00500.x
132. Shaddox L, Wiedey J, Bimstein E, Magnuson I, Clare-Salzler M, Aukhil I, et al. Hyper-responsive phenotype in localized aggressive periodontitis. J Dent Res. (2010) 89:143–8. doi: 10.1177/0022034509353397
133. Shaddox LM, Mullersman AF, Huang H, Wallet SM, Langaee T, Aukhil I. Epigenetic regulation of inflammation in localized aggressive periodontitis. Clin Epigenetics (2017) 9:94. doi: 10.1186/s13148-017-0385-8
134. Jin SH, Guan XY, Liang WH, Bai GH, Liu JG. TLR4 polymorphism and periodontitis susceptibility: a meta-analysis. Medicine (2016) 95:e4845. doi: 10.1097/MD.0000000000004845
135. Zhou Q, Wang C, Wang X, Wu X, Zhu Z, Liu B, et al. Association between TLR4 (+896A/G and +1196C/T) polymorphisms and gastric cancer risk: an updated meta-analysis. PLoS ONE (2014) 9:e109605. doi: 10.1371/journal.pone.0109605
136. Saxena M, Yeretssian G. NOD-like receptors: master regulators of inflammation and cancer. Front Immunol. (2014) 5:327. doi: 10.3389/fimmu.2014.00327
137. Becker CE, O'Neill LA. Inflammasomes in inflammatory disorders: the role of TLRs and their interactions with NLRs. Semin Immunopathol. (2007) 29:239–48. doi: 10.1007/s00281-007-0081-4
138. Martinon F, Tschopp J. NLRs join TLRs as innate sensors of pathogens. Trends Immunol. (2005) 26:447–54. doi: 10.1016/j.it.2005.06.004
139. Ting JP, Kastner DL, Hoffman HM. CATERPILLERs, pyrin and hereditary immunological disorders. Nat Rev Immunol. (2006) 6:183–95. doi: 10.1038/nri1788
140. Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science (2003) 300:1584–7. doi: 10.1126/science.1084677
141. Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn's disease. J Biol Chem. (2003) 278:5509–12. doi: 10.1074/jbc.C200673200
142. Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol. (2003) 4:702–7. doi: 10.1038/ni945
143. Inohara C, McDonald C, Nunez G. NOD-LRR proteins: role in host-microbial interactions and inflammatory disease. Annu Rev Biochem. (2005) 74:355–83. doi: 10.1146/annurev.biochem.74.082803.133347
144. Strober W, Murray PJ, Kitani A, Watanabe T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol. (2006) 6:9–20. doi: 10.1038/nri1747
145. Uehara A, Takada H. Synergism between TLRs and NOD1/2 in oral epithelial cells. J Dent Res. (2008) 87:682–6. doi: 10.1177/154405910808700709
146. Hirao K, Yumoto H, Takahashi K, Mukai K, Nakanishi T, Matsuo T. Roles of TLR2, TLR4, NOD2, and NOD1 in pulp fibroblasts. J Dent Res. (2009) 88:762–7. doi: 10.1177/0022034509341779
147. Lee YY, Chan CH, Hung SL, Chen YC, Lee YH, Yang SF. Up-regulation of nucleotide-binding oligomerization domain 1 in inflamed human dental pulp. J Endod. (2011) 37:1370–5. doi: 10.1016/j.joen.2011.06.008
148. Zhou S, Yu P, Guan L, Xing A, Liu S, Xiong Q, et al. NOD1 expression elicited by iE-DAP in first trimester human trophoblast cells and its potential role in infection-associated inflammation. Eur J Obstet Gynecol Reprod Biol. (2013) 170:318–23. doi: 10.1016/j.ejogrb.2013.04.011
149. Gao Y, Jiang W, Qian Y, Zhou Q, Jiang H, Wang X, et al. NOD1 agonist iE-DAP reverses effects of cigarette smoke extract on NOD1 signal pathway in human oral mucosal epithelial cells. Int J Clin Exp Med. (2015) 8:12519–28.
150. Gieseler F, Ungefroren H, Settmacher U, Hollenberg MD, Kaufmann R. Proteinase-activated receptors (PARs) - focus on receptor-receptor-interactions and their physiological and pathophysiological impact. Cell Commun Signal. (2013) 11:86. doi: 10.1186/1478-811X-11-86
151. Bunnett NW. Protease-activated receptors: how proteases signal to cells to cause inflammation and pain. Semin Thromb Hemost. (2006) 32(Suppl. 1):39–48. doi: 10.1055/s-2006-939553
152. Knecht W, Cottrell GS, Amadesi S, Mohlin J, Skaregarde A, Gedda K, et al. Trypsin IV or mesotrypsin and p23 cleave protease-activated receptors 1 and 2 to induce inflammation and hyperalgesia. J Biol Chem. (2007) 282:26089–100. doi: 10.1074/jbc.M703840200
153. Lourbakos A, Chinni C, Thompson P, Potempa J, Travis J, Mackie EJ, et al. Cleavage and activation of proteinase-activated receptor-2 on human neutrophils by gingipain-R from Porphyromonas gingivalis. FEBS Lett. (1998) 435:45–8. doi: 10.1016/S0014-5793(98)01036-9
154. Coughlin SR, Camerer E. PARticipation in inflammation. J Clin Invest. (2003) 111:25–7. doi: 10.1172/JCI17564
155. Camerer E, Huang W, Coughlin SR. Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proc Natl Acad Sci USA. (2000) 97:5255–60. doi: 10.1073/pnas.97.10.5255
156. Cottrell GS, Amadesi S, Grady EF, Bunnett NW. Trypsin IV, a novel agonist of protease-activated receptors 2 and 4. J Biol Chem. (2004) 279:13532–9. doi: 10.1074/jbc.M312090200
157. Ishihara H, Connolly AJ, Zeng D, Kahn ML, Zheng YW, Timmons C, et al. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature (1997) 386:502–6. doi: 10.1038/386502a0
158. Sambrano GR, Huang W, Faruqi T, Mahrus S, Craik C, Coughlin SR. Cathepsin G activates protease-activated receptor-4 in human platelets. J Biol Chem. (2000) 275:6819–23. doi: 10.1074/jbc.275.10.6819
159. Zhang D, Li S, Hu L, Sheng L, Chen L. Modulation of protease-activated receptor expression by Porphyromonas gingivalis in human gingival epithelial cells. BMC Oral Health (2015) 15:128. doi: 10.1186/s12903-015-0105-8
160. Chung WO, An JY, Yin L, Hacker BM, Rohani MG, Dommisch H, et al. Interplay of protease-activated receptors and NOD pattern recognition receptors in epithelial innate immune responses to bacteria. Immunol Lett. (2010) 131:113–9. doi: 10.1016/j.imlet.2010.02.006
161. Skeldon A, Saleh M. The inflammasomes: molecular effectors of host resistance against bacterial, viral, parasitic, and fungal infections. Front Microbiol. (2011) 2:15. doi: 10.3389/fmicb.2011.00015
162. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
163. Said-Sadier N, Ojcius DM. Alarmins, inflammasomes and immunity. Biomed J. (2012) 35:437–49. doi: 10.4103/2319-4170.104408
164. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
165. Yilmaz O, Lee KL. The inflammasome and danger molecule signaling: at the crossroads of inflammation and pathogen persistence in the oral cavity. Periodontol (2015) 69:83–95. doi: 10.1111/prd.12084
166. Akhter A, Caution K, Abu Khweek A, Tazi M, Abdulrahman BA, Abdelaziz DH, et al. Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity (2012) 37:35–47. doi: 10.1016/j.immuni.2012.05.001
167. van de Veerdonk FL, Netea MG, Dinarello CA, Joosten LA. Inflammasome activation and IL-1beta and IL-18 processing during infection. Trends Immunol. (2011) 32:110–6. doi: 10.1016/j.it.2011.01.003
168. Bui FQ, Johnson L, Roberts J, Hung SC, Lee J, Atanasova KR, et al. Fusobacterium nucleatum infection of gingival epithelial cells leads to NLRP3 inflammasome-dependent secretion of IL-1beta and the danger signals ASC and HMGB1. Cell Microbiol. (2016) 18:970–81. doi: 10.1111/cmi.12560
169. Johnson L, Atanasova KR, Bui PQ, Lee J, Hung SC, Yilmaz O, et al. Porphyromonas gingivalis attenuates ATP-mediated inflammasome activation and HMGB1 release through expression of a nucleoside-diphosphate kinase. Microbes Infect. (2015) 17:369–77. doi: 10.1016/j.micinf.2015.03.010
170. Creagh EM. Caspase crosstalk: integration of apoptotic and innate immune signalling pathways. Trends Immunol. (2014) 35:631–40. doi: 10.1016/j.it.2014.10.004
171. Eldridge MJ, Shenoy AR. Antimicrobial inflammasomes: unified signalling against diverse bacterial pathogens. Curr Opin Microbiol. (2015) 23:32–41. doi: 10.1016/j.mib.2014.10.008
172. Bauernfeind F, Ablasser A, Bartok E, Kim S, Schmid-Burgk J, Cavlar T, et al. Inflammasomes: current understanding and open questions. Cell Mol Life Sci. (2011) 68:765–83. doi: 10.1007/s00018-010-0567-4
173. Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, et al. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol. (2006) 176:3877–83. doi: 10.4049/jimmunol.176.7.3877
174. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science (2011) 334:1573–7. doi: 10.1126/science.1208347
175. Perregaux DG, McNiff P, Laliberte R, Conklyn M, Gabel CA. ATP acts as an agonist to promote stimulus-induced secretion of IL-1 beta and IL-18 in human blood. J Immunol. (2000) 165:4615–23. doi: 10.4049/jimmunol.165.8.4615
176. Sanz JM, Di Virgilio F. Kinetics and mechanism of ATP-dependent IL-1 beta release from microglial cells. J Immunol. (2000) 164:4893–8.
177. Yilmaz O, Sater AA, Yao L, Koutouzis T, Pettengill M, Ojcius DM. ATP-dependent activation of an inflammasome in primary gingival epithelial cells infected by Porphyromonas gingivalis. Cell Microbiol. (2010) 12:188–98. doi: 10.1111/j.1462-5822.2009.01390.x
178. Lee SJ, Choi BK. Involvement of NLRP10 in IL-1alpha induction of oral epithelial cells by periodontal pathogens. Innate Immun. (2017) 23:569–77. doi: 10.1177/1753425917722610
179. Belibasakis GN, Thurnheer T, Bostanci N. Interleukin-8 responses of multi-layer gingival epithelia to subgingival biofilms: role of the “red complex” species. PLoS ONE (2013) 8:e81581. doi: 10.1371/journal.pone.0081581
180. Bostanci N, Bao K, Wahlander A, Grossmann J, Thurnheer T, Belibasakis GN. Secretome of gingival epithelium in response to subgingival biofilms. Mol Oral Microbiol. (2015) 30:323–35. doi: 10.1111/omi.12096
181. Feliciani C, Gupta AK, Sauder DN. Keratinocytes and cytokine/growth factors. Crit Rev Oral Biol Med. (1996) 7:300–18. doi: 10.1177/10454411960070040101
182. Dinarello CA. The IL-1 family and inflammatory diseases. Clin Exp Rheumatol. (2002) 20(5 Suppl. 27):S1–13.
183. Ramage G, Lappin DF, Millhouse E, Malcolm J, Jose A, Yang J, et al. The epithelial cell response to health and disease associated oral biofilm models. J Periodontal Res. (2017) 52:325–33. doi: 10.1111/jre.12395
184. Campbell LM, Maxwell PJ, Waugh DJ. Rationale and means to target pro-inflammatory interleukin-8 (CXCL8) signaling in cancer. Pharmaceuticals (2013) 6:929–59. doi: 10.3390/ph6080929
185. St John MA, Li Y, Zhou X, Denny P, Ho CM, Montemagno C, et al. Interleukin 6 and interleukin 8 as potential biomarkers for oral cavity and oropharyngeal squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. (2004) 130:929–35. doi: 10.1001/archotol.130.8.929
186. Goutoudi P, Diza E, Arvanitidou M. Effect of periodontal therapy on crevicular fluid interleukin-6 and interleukin-8 levels in chronic periodontitis. Int J Dent. (2012) 2012:362905. doi: 10.1155/2012/362905
187. Schueller K, Riva A, Pfeiffer S, Berry D, Somoza V. Members of the oral microbiota are associated with IL-8 release by gingival epithelial cells in healthy individuals. Front Microbiol. (2017) 8:416. doi: 10.3389/fmicb.2017.00416
188. Fujimura Y, Hotokezaka H, Ohara N, Naito M, Sakai E, Yoshimura M, et al. The hemoglobin receptor protein of porphyromonas gingivalis inhibits receptor activator NF-kappaB ligand-induced osteoclastogenesis from bone marrow macrophages. Infect Immun. (2006) 74:2544–51. doi: 10.1128/IAI.74.5.2544-2551.2006
189. Fujita Y, Nakayama M, Naito M, Yamachika E, Inoue T, Nakayama K, et al. Hemoglobin receptor protein from Porphyromonas gingivalis induces interleukin-8 production in human gingival epithelial cells through stimulation of the mitogen-activated protein kinase and NF-kappaB signal transduction pathways. Infect Immun. (2014) 82:202–11. doi: 10.1128/IAI.01140-12
190. Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel “alarmin”? PLoS ONE (2008) 3:e3331. doi: 10.1371/journal.pone.0003331
191. Paul WE, Zhu J. How are T(H)2-type immune responses initiated and amplified? Nat Rev Immunol. (2010) 10:225–35. doi: 10.1038/nri2735
192. Tada H, Matsuyama T, Nishioka T, Hagiwara M, Kiyoura Y, Shimauchi H, et al. Porphyromonas gingivalis gingipain-dependently enhances IL-33 production in human gingival epithelial cells. PLoS ONE (2016) 11:e0152794. doi: 10.1371/journal.pone.0152794
193. Kadomatsu T, Endo M, Miyata K, Oike Y. Diverse roles of ANGPTL2 in physiology and pathophysiology. Trends Endocrinol Metab. (2014) 25:245–54. doi: 10.1016/j.tem.2014.03.012
194. Kim I, Moon SO, Koh KN, Kim H, Uhm CS, Kwak HJ, et al. Molecular cloning, expression, and characterization of angiopoietin-related protein. angiopoietin-related protein induces endothelial cell sprouting. J Biol Chem. (1999) 274:26523–8. doi: 10.1074/jbc.274.37.26523
195. Santulli G. Angiopoietin-like proteins: a comprehensive look. Front Endocrinol. (2014) 5:4. doi: 10.3389/fendo.2014.00004
196. Kubota Y, Oike Y, Satoh S, Tabata Y, Niikura Y, Morisada T, et al. Cooperative interaction of Angiopoietin-like proteins 1 and 2 in zebrafish vascular development. Proc Natl Acad Sci USA. (2005) 102:13502–7. doi: 10.1073/pnas.0501902102
197. Ohno T, Yamamoto G, Hayashi JI, Nishida E, Goto H, Sasaki Y, et al. Angiopoietin-like protein 2 regulates Porphyromonas gingivalis lipopolysaccharide-induced inflammatory response in human gingival epithelial cells. PLoS ONE (2017) 12:e0184825. doi: 10.1371/journal.pone.0184825
198. Imai H, Fujita T, Kajiya M, Ouhara K, Yoshimoto T, Matsuda S, et al. Mobilization of TLR4 into lipid rafts by aggregatibacter actinomycetemcomitans in gingival epithelial cells. Cell Physiol Biochem. (2016) 39:1777–86. doi: 10.1159/000447877
199. Armitage GC. Comparison of the microbiological features of chronic and aggressive periodontitis. Periodontol (2010) 53:70–88. doi: 10.1111/j.1600-0757.2010.00357.x
200. Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol. (2010) 8:481–90. doi: 10.1038/nrmicro2337
201. Taubman MA, Valverde P, Han X, Kawai T. Immune response: the key to bone resorption in periodontal disease. J Periodontol. (2005) 76(11 Suppl.):2033–41. doi: 10.1902/jop.2005.76.11-S.2033
202. Lally ET, Golub EE, Kieba IR, Taichman NS, Rosenbloom J, Rosenbloom JC, et al. Analysis of the Actinobacillus actinomycetemcomitans leukotoxin gene. Delineation of unique features and comparison to homologous toxins. J Biol Chem. (1989) 264:15451–6.
203. Mayer MP, Bueno LC, Hansen EJ, DiRienzo JM. Identification of a cytolethal distending toxin gene locus and features of a virulence-associated region in Actinobacillus actinomycetemcomitans. Infect Immun. (1999) 67:1227–37.
204. Feng Z, Weinberg A. Role of bacteria in health and disease of periodontal tissues. Periodontol (2006) 40:50–76. doi: 10.1111/j.1600-0757.2005.00148.x
205. Pahumunto N, Chotjumlong P, Makeudom A, Krisanaprakornkit S, Dahlen G, Teanpaisan R. Pro-inflammatory cytokine responses in human gingival epithelial cells after stimulation with cell wall extract of Aggregatibacter actinomycetemcomitans subtypes. Anaerobe (2017) 48:103–9. doi: 10.1016/j.anaerobe.2017.08.001
206. Simonson LG, Goodman CH, Bial JJ, Morton HE. Quantitative relationship of Treponema denticola to severity of periodontal disease. Infect Immun. (1988) 56:726–8.
207. Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL Jr. Microbial complexes in subgingival plaque. J Clin Periodontol. (1998) 25:134–44.
208. Dashper SG, Seers CA, Tan KH, Reynolds EC. Virulence factors of the oral spirochete Treponema denticola. J Dent Res. (2011) 90:691–703. doi: 10.1177/0022034510385242
209. Brissette CA, Pham TT, Coats SR, Darveau RP, Lukehart SA. Treponema denticola does not induce production of common innate immune mediators from primary gingival epithelial cells. Oral Microbiol Immunol. (2008) 23:474–81. doi: 10.1111/j.1399-302X.2008.00452.x
210. Jo AR, Baek KJ, Shin JE, Choi Y. Mechanisms of IL-8 suppression by Treponema denticola in gingival epithelial cells. Immunol Cell Biol. (2014) 92:139–47. doi: 10.1038/icb.2013.80
211. Ruby J, Martin M, Passineau MJ, Godovikova V, Fenno JC, Wu H. Activation of the innate immune system by Treponema denticola periplasmic flagella through toll-like receptor 2. Infect Immun. (2018) 86: e00573–17. doi: 10.1128/IAI.00251-18
212. Gu C, Wu L, Li X. IL-17 family: cytokines, receptors and signaling. Cytokine (2013) 64:477–85. doi: 10.1016/j.cyto.2013.07.022
213. Yao Z, Painter SL, Fanslow WC, Ulrich D, Macduff BM, Spriggs MK, et al. Human IL-17: a novel cytokine derived from T cells. J Immunol. (1995) 155:5483–6.
214. Adibrad M, Deyhimi P, Ganjalikhani Hakemi M, Behfarnia P, Shahabuei M, Rafiee L. Signs of the presence of Th17 cells in chronic periodontal disease. J Periodontal Res. (2012) 47:525–31. doi: 10.1111/j.1600-0765.2011.01464.x
215. Beklen A, Ainola M, Hukkanen M, Gurgan C, Sorsa T, Konttinen YT. MMPs, IL-1, and TNF are regulated by IL-17 in periodontitis. J Dent Res. (2007) 86:347–51. doi: 10.1177/154405910708600409
216. Cardoso CR, Garlet GP, Crippa GE, Rosa AL, Junior WM, Rossi MA, et al. Evidence of the presence of T helper type 17 cells in chronic lesions of human periodontal disease. Oral Microbiol Immunol. (2009) 24:1–6. doi: 10.1111/j.1399-302X.2008.00463.x
217. Konermann A, Beyer M, Deschner J, Allam JP, Novak N, Winter J, et al. Human periodontal ligament cells facilitate leukocyte recruitment and are influenced in their immunomodulatory function by Th17 cytokine release. Cell Immunol. (2012) 272:137–43. doi: 10.1016/j.cellimm.2011.10.020
218. Allam JP, Duan Y, Heinemann F, Winter J, Gotz W, Deschner J, et al. IL-23-producing CD68(+) macrophage-like cells predominate within an IL-17-polarized infiltrate in chronic periodontitis lesions. J Clin Periodontol. (2011) 38:879–86. doi: 10.1111/j.1600-051X.2011.01752.x
219. Buduneli N, Buduneli E, Kutukculer N. Interleukin-17, RANKL, and osteoprotegerin levels in gingival crevicular fluid from smoking and non-smoking patients with chronic periodontitis during initial periodontal treatment. J Periodontol. (2009) 80:1274–80. doi: 10.1902/jop.2009.090106
220. Vernal R, Dutzan N, Chaparro A, Puente J, Antonieta Valenzuela M, Gamonal J. Levels of interleukin-17 in gingival crevicular fluid and in supernatants of cellular cultures of gingival tissue from patients with chronic periodontitis. J Clin Periodontol. (2005) 32:383–9. doi: 10.1111/j.1600-051X.2005.00684.x
221. Awang RA, Lappin DF, MacPherson A, Riggio M, Robertson D, Hodge P, et al. Clinical associations between IL-17 family cytokines and periodontitis and potential differential roles for IL-17A and IL-17E in periodontal immunity. Inflamm Res. (2014) 63:1001–12. doi: 10.1007/s00011-014-0776-7
222. Lemke G, Rothlin CV. Immunobiology of the TAM receptors. Nat Rev Immunol. (2008) 8:327–36. doi: 10.1038/nri2303
223. Lemke G, Burstyn-Cohen T. TAM receptors and the clearance of apoptotic cells. Ann NY Acad Sci. (2010) 1209:23–9. doi: 10.1111/j.1749-6632.2010.05744.x
224. Nassar M, Tabib Y, Capucha T, Mizraji G, Nir T, Pevsner-Fischer M, et al. GAS6 is a key homeostatic immunological regulator of host-commensal interactions in the oral mucosa. Proc Natl Acad Sci USA. (2017) 114:E337–46. doi: 10.1073/pnas.1614926114
225. Nassar M, Tabib Y, Capucha T, Mizraji G, Nir T, Saba F, et al. Multiple regulatory levels of growth arrest-specific 6 in mucosal immunity against an oral pathogen. Front Immunol. (2018) 9:1374. doi: 10.3389/fimmu.2018.01374
226. Tezal M, Sullivan MA, Reid ME, Marshall JR, Hyland A, Loree T, et al. Chronic periodontitis and the risk of tongue cancer. Arch Otolaryngol Head Neck Surg. (2007) 133:450–4. doi: 10.1001/archotol.133.5.450
227. Tezal M, Grossi SG, Genco RJ. Is periodontitis associated with oral neoplasms? J Periodontol. (2005) 76:406–10. doi: 10.1902/jop.2005.76.3.406
228. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. (2002) 8:793–800. doi: 10.1038/nm730
229. Wu P, Wu D, Li L, Chai Y, Huang J. PD-L1 and survival in solid tumors: a meta-analysis. PLoS ONE (2015) 10:e0131403. doi: 10.1371/journal.pone.0131403
230. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. (2013) 63:11–30. doi: 10.3322/caac.21166
231. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
232. Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. (1999) 5:1365–9. doi: 10.1038/70932
233. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. (2000) 192:1027–34. doi: 10.1084/jem.192.7.1027
234. Wang S, Chen L. T lymphocyte co-signaling pathways of the B7-CD28 family. Cell Mol Immunol. (2004) 1:37–42.
235. Subudhi SK, Zhou P, Yerian LM, Chin RK, Lo JC, Anders RA, et al. Local expression of B7-H1 promotes organ-specific autoimmunity and transplant rejection. J Clin Invest. (2004) 113:694–700. doi: 10.1172/JCI19210
236. Azuma T, Yao S, Zhu G, Flies AS, Flies SJ, Chen L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood (2008) 111:3635–43. doi: 10.1182/blood-2007-11-123141
237. Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. (2013) 73:3591–603. doi: 10.1158/0008-5472.CAN-12-4100
238. Groeger S, Domann E, Gonzales JR, Chakraborty T, Meyle J. B7-H1 and B7-DC receptors of oral squamous carcinoma cells are upregulated by Porphyromonas gingivalis. Immunobiology (2011) 216:1302–10. doi: 10.1016/j.imbio.2011.05.005
239. Hirai M, Kitahara H, Kobayashi Y, Kato K, Bou-Gharios G, Nakamura H, et al. Regulation of PD-L1 expression in a high-grade invasive human oral squamous cell carcinoma microenvironment. Int J Oncol. (2017) 50:41–8. doi: 10.3892/ijo.2016.3785
240. Groeger S, Howaldt HP, Raifer H, Gattenloehner S, Chakraborty T, Meyle J. Oral squamous carcinoma cells express B7-H1 and B7-DC receptors in vivo. Pathol Oncol Res. (2017) 23:99–110. doi: 10.1007/s12253-016-0100-7
241. Groeger S, Jarzina F, Mamat U, Meyle J. Induction of B7-H1 receptor by bacterial cells fractions of Porphyromonas gingivalis on human oral epithelial cells. Immunobiology (2017) 222:137–147. doi: 10.1016/j.imbio.2016.10.011
242. Groeger S, Jarzina F, Domann E, Meyle J. Porphyromonas gingivalis activates NFkappaB and MAPK pathways in human oral epithelial cells. BMC Immunol. (2017) 18:1. doi: 10.1186/s12865-016-0185-5
243. Li S, Lee YC, Li Q, Chen CJ, Hsu WL, Lou PJ, et al. Oral lesions, chronic diseases and the risk of head and neck cancer. Oral Oncol. (2015) 51:1082–7. doi: 10.1016/j.oraloncology.2015.10.014
244. Komiya Y, Shimomura Y, Higurashi T, Sugi Y, Arimoto J, Umezawa S, et al. Patients with colorectal cancer have identical strains of Fusobacterium nucleatum in their colorectal cancer and oral cavity. Gut (2018). doi: 10.1136/gutjnl-2018-316661. [Epub ahead of print].
245. Hoppe T, Kraus D, Novak N, Probstmeier R, Frentzen M, Wenghoefer M, et al. Oral pathogens change proliferation properties of oral tumor cells by affecting gene expression of human defensins. Tumour Biol. (2016) 37:13789–98. doi: 10.1007/s13277-016-5281-x
246. Geng F, Liu J, Guo Y, Li C, Wang H, Wang H, et al. Persistent exposure to Porphyromonas gingivalis promotes proliferative and invasion capabilities, and tumorigenic properties of human immortalized oral epithelial cells. Front Cell Infect Microbiol. (2017) 7:57. doi: 10.3389/fcimb.2017.00057
247. Binder Gallimidi A, Fischman S, Revach B, Bulvik R, Maliutina A, Rubinstein AM, et al. Periodontal pathogens Porphyromonas gingivalis and Fusobacterium nucleatum promote tumor progression in an oral-specific chemical carcinogenesis model. Oncotarget (2015) 6:22613–23. doi: 10.18632/oncotarget.4209
248. Bolos V, Peinado H, Perez-Moreno MA, Fraga MF, Esteller M, Cano A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci. (2003) 116(Pt 3):499–511. doi: 10.1242/jcs.00224
249. Lee J, Roberts JS, Atanasova KR, Chowdhury N, Han K, Yilmaz O. Human primary epithelial cells acquire an epithelial-mesenchymal-transition phenotype during long-term infection by the oral opportunistic pathogen, Porphyromonas gingivalis. Front Cell Infect Microbiol. (2017) 7:493. doi: 10.3389/fcimb.2017.00493
250. Abdulkareem AA, Shelton RM, Landini G, Cooper PR, Milward MR. Potential role of periodontal pathogens in compromising epithelial barrier function by inducing epithelial-mesenchymal transition. J Periodontal Res. (2018) 53:565–74. doi: 10.1111/jre.12546
251. Meyle J, Chapple I. Molecular aspects of the pathogenesis of periodontitis. Periodontol (2015) 69:7–17. doi: 10.1111/prd.12104
252. Paster BJ, Boches SK, Galvin JL, Ericson RE, Lau CN, Levanos VA, et al. Bacterial diversity in human subgingival plaque. J Bacteriol. (2001) 183:3770–83. doi: 10.1128/JB.183.12.3770-3783.2001
253. van Winkelhoff AJ, Loos BG, van der Reijden WA, van der Velden U. Porphyromonas gingivalis, Bacteroides forsythus and other putative periodontal pathogens in subjects with and without periodontal destruction. J Clin Periodontol. (2002) 29:1023–8. doi: 10.1034/j.1600-051X.2002.291107.x
254. Tatakis DN, Kumar PS. Etiology and pathogenesis of periodontal diseases. Dent Clin North Am. (2005) 49:491–516. doi: 10.1016/j.cden.2005.03.001
255. Ji S, Kim Y, Min BM, Han SH, Choi Y. Innate immune responses of gingival epithelial cells to nonperiodontopathic and periodontopathic bacteria. J Periodontal Res. (2007) 42:503–10. doi: 10.1111/j.1600-0765.2007.00974.x
256. Guo W, Wang P, Liu Z, Yang P, Ye P. The activation of pyrin domain-containing-3 inflammasome depends on lipopolysaccharide from Porphyromonas gingivalis and extracellular adenosine triphosphate in cultured oral epithelial cells. BMC Oral Health (2015) 15:133. doi: 10.1186/s12903-015-0115-6
257. Moore WE, Moore LV. The bacteria of periodontal diseases. Periodontol (1994) 5:66–77. doi: 10.1111/j.1600-0757.1994.tb00019.x
258. Kumagai Y, Yagishita H, Yajima A, Okamoto T, Konishi K. Molecular mechanism for connective tissue destruction by dipeptidyl aminopeptidase IV produced by the periodontal pathogen Porphyromonas gingivalis. Infect Immun. (2005) 73:2655–64. doi: 10.1128/IAI.73.5.2655-2664.2005
259. Holt SC, Kesavalu L, Walker S, Genco CA. Virulence factors of Porphyromonas gingivalis. Periodontol (1999) 20:168–238. doi: 10.1111/j.1600-0757.1999.tb00162.x
260. Birkedal-Hansen H, Taylor RE, Zambon JJ, Barwa PK, Neiders ME. Characterization of collagenolytic activity from strains of Bacteroides gingivalis. J Periodontal Res. (1988) 23:258–64. doi: 10.1111/j.1600-0765.1988.tb01369.x
261. Soolari AS, Champagne C, Punzi JS, Amar S, Van Dyke TE. Serum modulation of neutrophil response to Porphyromonas gingivalis LPS in periodontal disease. J Int Acad Periodontol. (1999) 1:101–9.
262. Kesavalu L, Ebersole JL, Machen RL, Holt SC. Porphyromonas gingivalis virulence in mice: induction of immunity to bacterial components. Infect Immun. (1992) 60:1455–64.
263. Amano A, Nakagawa I, Okahashi N, Hamada N. Variations of Porphyromonas gingivalis fimbriae in relation to microbial pathogenesis. J Periodontal Res. (2004) 39:136–42. doi: 10.1111/j.1600-0765.2004.00719.x
264. Jotwani R, Cutler CW. Fimbriated Porphyromonas gingivalis is more efficient than fimbria-deficient P. gingivalis in entering human dendritic cells in vitro and induces an inflammatory Th1 effector response. Infect Immun. (2004) 72:1725–32.
265. Preshaw PM, Schifferle RE, Walters JD. Porphyromonas gingivalis lipopolysaccharide delays human polymorphonuclear leukocyte apoptosis in vitro. J Periodontal Res. (1999) 34:197–202. doi: 10.1111/j.1600-0765.1999.tb02242.x
266. Hamada N, Watanabe K, Sasakawa C, Yoshikawa M, Yoshimura F, Umemoto T. Construction and characterization of a fimA mutant of Porphyromonas gingivalis. Infect Immun. (1994) 62:1696–704.
267. Grenier D. Inactivation of human serum bactericidal activity by a trypsinlike protease isolated from Porphyromonas gingivalis. Infect Immun. (1992) 60:1854–7.
268. Njoroge T, Genco RJ, Sojar HT, Hamada N, Genco CA. A role for fimbriae in Porphyromonas gingivalis invasion of oral epithelial cells. Infect Immun. (1997) 65:1980–4.
269. Lamont RJ, Chan A, Belton CM, Izutsu KT, Vasel D, Weinberg A. Porphyromonas gingivalis invasion of gingival epithelial cells. Infect Immun. (1995) 63:3878–85.
270. Yilmaz O, Watanabe K, Lamont RJ. Involvement of integrins in fimbriae-mediated binding and invasion by Porphyromonas gingivalis. Cell Microbiol. (2002) 4:305–14. doi: 10.1046/j.1462-5822.2002.00192.x
271. Watanabe K, Yamaji Y, Umemoto T. Correlation between cell-adherent activity and surface structure in Porphyromonas gingivalis. Oral Microbiol Immunol. (1992) 7:357–63. doi: 10.1111/j.1399-302X.1992.tb00636.x
272. Shah HN, Gharbia SE, Kowlessur D, Wilkie E, Brocklehurst K. Isolation and characterization of gingivain, a cysteine proteinase from Porphyromonas gingivalis strain W83. Biochem Soc Trans. (1990) 18:578–9. doi: 10.1042/bst0180578
Keywords: oral epithelial cells, differentiation, receptors, cytokines, immuno-modulation, infection, cancer
Citation: Groeger S and Meyle J (2019) Oral Mucosal Epithelial Cells. Front. Immunol. 10:208. doi: 10.3389/fimmu.2019.00208
Received: 03 September 2018; Accepted: 23 January 2019;
Published: 14 February 2019.
Edited by:
Asaf Wilensky, Hadassah Medical Center, IsraelReviewed by:
Denis Francis Kinane, Geneva University Hospitals (HUG), SwitzerlandCopyright © 2019 Groeger and Meyle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sabine Groeger, c2FiaW5lLmUuZ3JvZWdlckBkZW50aXN0Lm1lZC51bmktZ2llc3Nlbi5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.