
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 12 March 2019
Sec. Microbial Immunology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00096
 Kun Yang1,2†Yingxia He1†
Kun Yang1,2†Yingxia He1† Chae Gyu Park3Young Sun Kang3Pei Zhang4Yanping Han5Yujun Cui5Silvia Bulgheresi6
Chae Gyu Park3Young Sun Kang3Pei Zhang4Yanping Han5Yujun Cui5Silvia Bulgheresi6 Andrey P. Anisimov7*Svetlana V. Dentovskaya7Xiaoling Ying1Lingyu Jiang1Honghui Ding1Olivia Adhiambo Njiri1,8Shusheng Zhang4Guoxing Zheng4Lianxu Xia9Biao Kan9Xin Wang9Huaiqi Jing9Meiying Yan9Wei Li9Yuanzhi Wang2Xiding Xiamu2Gang Chen10Ding Ma11Sara Schesser Bartra12Gregory V. Plano12John D. Klena13Ruifu Yang5*
Andrey P. Anisimov7*Svetlana V. Dentovskaya7Xiaoling Ying1Lingyu Jiang1Honghui Ding1Olivia Adhiambo Njiri1,8Shusheng Zhang4Guoxing Zheng4Lianxu Xia9Biao Kan9Xin Wang9Huaiqi Jing9Meiying Yan9Wei Li9Yuanzhi Wang2Xiding Xiamu2Gang Chen10Ding Ma11Sara Schesser Bartra12Gregory V. Plano12John D. Klena13Ruifu Yang5* Mikael Skurnik14
Mikael Skurnik14 Tie Chen1,2*
Tie Chen1,2*Yersinia pestis, a Gram-negative bacterium and the etiologic agent of plague, has evolved from Yersinia pseudotuberculosis, a cause of a mild enteric disease. However, the molecular and biological mechanisms of how Y. pseudotuberculosis evolved to such a remarkably virulent pathogen, Y. pestis, are not clear. The ability to initiate a rapid bacterial dissemination is a characteristic hallmark of Y. pestis infection. A distinguishing characteristic between the two Yersinia species is that Y. pseudotuberculosis strains possess an O-antigen of lipopolysaccharide (LPS) while Y. pestis has lost the O-antigen during evolution and therefore exposes its core LPS. In this study, we showed that Y. pestis utilizes its core LPS to interact with SIGNR1 (CD209b), a C-type lectin receptor on antigen presenting cells (APCs), leading to bacterial dissemination to lymph nodes, spleen and liver, and the initiation of a systemic infection. We therefore propose that the loss of O-antigen represents a critical step in the evolution of Y. pseudotuberculosis into Y. pestis in terms of hijacking APCs, promoting bacterial dissemination and causing the plague.
Yersinia pestis is the bacterium that causes bubonic, septicemic, and pneumonic forms of plague and that was the cause of the Black Death in Europe during the middle ages. Recent studies have proved that all three suspected plague pandemics (the Justinian, the Black Death and the third pandemic) were caused by this bacterium (1–6). Based on a study (7), the New York Times on October 31, 2010 reported that the plague pathogen responsible for all known plague pandemics in the recorded history of human civilization might have originated in China, but more likely from Eurasia (8, 9). Y. pestis directly evolved from Y. pseudotuberculosis, the cause of a self-limited mesenteric lymphadenitis, within the last 2,600 to 28,000 years (6, 7, 10–12). The apparent question is how did Y. pseudotuberculosis evolve into such a virulent, dangerous and remarkably different pathogen, Y. pestis? Could there still be an ancestral Y. pseudotuberculosis circulating in China that might hold clues to the evolution to Y. pestis?
A distinguishing difference between these pathogens is that Y. pseudotuberculosis contains an O-antigen of lipopolysaccharide (LPS), which was lost by Y. pestis during its evolution (13–15) (Figure 1B). LPS plays a major role in the pathogenicity of Gram-negative bacterial pathogens including pathogenic species of the genera Escherichia, Shigella, Klebsiella, Yersinia, and Salmonella. The presence of LPS promotes toxicity as well as resistance to phagocytosis and serum-dependent (complement-dependent) killing (16–21). LPS generally consists of three structural regions: (i) the lipid A backbone, (ii) an oligosaccharide core (core LPS), and (iii) the somatic O-polysaccharide outer region (also called O-antigen, O-specific antigen, or O-specific side chain) (Figure 1A). Gram-negative bacteria are classified as smooth or rough based on the presence or lack of the O-antigen (O-Ag), respectively. Y. pestis does not contain an O-antigen (14, 15) and therefore the shortened LPS is also referred to as lipooligosaccharide (LOS). Why would Y. pseudotuberculosis sacrifice the production of O-Ag, one of its key virulence factors, during the evolution to Y. pestis?
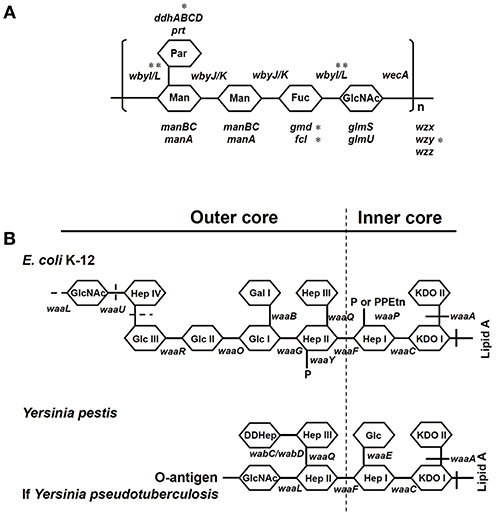
Figure 1. Structures of O-antigen of Y. pseudotuberculosis and core LPS. (A) Genes involved in the biosynthesis of O-antigen of Y. pseudotuberculosis. *The genes were sequenced in this study. (B) Genes involved in the biosynthesis of core LPS are shown at their approximate site of action (solid line). The sites, which are variably substituted or still under investigation, are indicated by dashed lines. Fuc, L-fucose; Gal, galactose; GalNAc, N-Acetyl-galactosamine; Glc, glucose; GlcNAc, N-Acetylglucosamine; Hep, heptose; KDO, 2-keto-3-deoxyoctonate; Man, mannose; P, phosphate; Par, Paratose; PEA, phosphoethanolamine; PPEtn, phosphoethanolamine. It should be noted that E. coli K12 and Y. pestis naturally do not possess an O-antigen.
We have shown that human DC-specific intercellular adhesion molecule-grabbing nonintegrin (hDC-SIGN, CD209a), a C-type lectin receptor on antigen presenting cells (APCs) such as macrophages and dendritic cells (DCs), is a receptor for the core LPS of several Gram-negative bacteria, including E. coli, Haemophilus ducreyi, Neisseria gonorrhoeae, Yersinia spp., and Salmonella typhimurium, promoting bacterial adherence and phagocytosis (22–27). In addition, Y. pseudotuberculosis, via its core LPS-CD209 interaction, may hijack APCs to be disseminated to lymph nodes, spleen and liver (28).
Moreover, hDC-SIGN (CD209a) is a receptor for HIV gp120 that uses DC-SIGN to be captured and trafficked to target cells such as CD4+ T cells (29–31). Mouse DC-SIGN-related protein 1 (SIGNR1, CD209b), expressed on splenic marginal zone, lymph nodes, and peritoneal macrophages, plays a role in lymphocyte migration from the blood into tissues. Here, we show that SIGNR1 is a cellular receptor for Y. pestis and that an exposed core LPS is essential for the APCs/Y. pestis interaction, host dissemination and infection. Therefore, it is possible that the loss of expression of O-antigen during evolution from Y. pseudotuberculosis might have endowed Y. pestis the ability to hijack APCs in rodents in order to spread into lymph nodes and initiate host infections (32, 33).
Y. pseudotuberculosis strains possess an O-antigen, the production of which was lost by Y. pestis during evolution. Based on a recent study on the evolution of Y. pseudotuberculosis (12), we sequenced six O-antigen synthesis genes (Figure 1A) just outside of waaL (Figure 1B) of 39 strains of Y. pseudotuberculosis and eight strains of Y. pestis to investigate the changes in these genes during evolution (12). As shown in a summary (Figure 2), unlike Y. pestis, the genes from the Y. pseudotuberculosis strains showed extreme diversity, which echoes the conclusion of the Cui's study (12). However, there are several new findings; (1) wbyL: Only one Y. pestis strain from the eight analyzed showed a non-synonymous mutation in wbyL compared to Y. pseudotuberculosis. (2) wbyI: The wbyI genes from all eight Y. pestis strains appeared to have lost their potential functions due to the loss of a 62 base pair fragment presents in Y. pseudotuberculosis. (3) gmd: The gmd gene in all eight Y. pestis strains appeared to be non-functional due to an insertion. (4) fcl: Only one strain of the eight Y. pestis strains (Orientalis, the cause of third pandemic of plague) analyzed showed a deletion. The other seven carried a fully functional gene. (5) wzy: Except for one strain (Pestoides F), all the strains analyzed appeared to have lost the function of this gene. (6) ddhB: Except for Pestoides A (0.PE4c) and Microtus (0.PE4i), all lost function due to a frame-shift mutation. It is reported that lack of O-antigen is essential for plasminogen activation and invasiveness of Y. pestis (34). Therefore, loss of genes involved in O-antigen synthesis in Y. pestis affects its function.
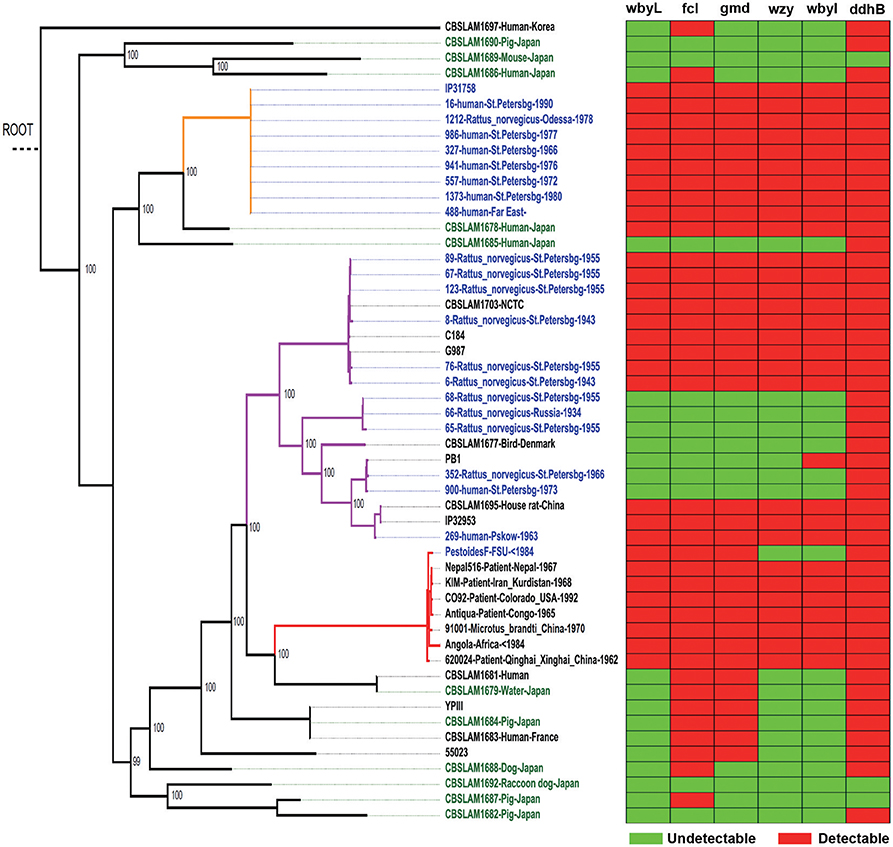
Figure 2. Y. pseudotuberculosis rather than Y. pestis, isolated from China, show the multiple mutations of O-antigen synthesis genes. In the heatmap, rows represent six O-antigen synthesis genes, and columns represent data from Y. pseudotuberculosis and Y. pestis strains. Red cell indicates the gene was detectable in this strain while green cell indicates the gene was undetectable. In the phylogenetic tree, red branches are Y. pestis, and the rest are Y. pseudotuberculosis.
In short, based on the detailed analysis of O-antigen synthesis genes, we showed again that Y. pestis might evolve from one of several specific evolutionary branches of Y. pseudotuberculosis.
Although the O-antigen synthesis genes in Y. pseudotuberculosis are diverse, the core LPS appears to be identical to that of Y. pestis (Figure 1B) (35, 36). Several Gram-negative bacterial strains use their core LPS to interact with human DCs (22–24, 28). Furthermore, another study showed that N-acetylglucosamine (GlcNAc) within the core LPS (Figure 1B) may mediate the interaction with DCs (24). To investigate the hypothesis that Y. pestis might also use its core LPS to interact with mouse macrophages, Y. pestis KIM10-Δail (a natural rough strain with the core LPS exposed), and its two isogenic derivatives; KIM10-Δail-O+ (a smooth strain in which the outer-core LPS is shielded by O-antigen) and KIM10-Δail-Core− (with truncated LPS outer-core, i.e., a deep rough strain) were examined for their ability to invade mouse macrophages. Three corresponding E. coli K-12 strains: CS180 (rough), CS1861 (CS180 expressing an O-antigen, smooth), and CS2429 (deep rough) were used as controls. We have used this set of strains in previous studies that demonstrate that the exposure of the core LPS by several Gram-negative bacteria is essential to initiate contact with human DCs (22–24). Figure 3A shows that Y. pestis KIM10-Δail (rough) and E. coli K12 180 (rough) invade mouse macrophages. In contrast, both deep rough and smooth strains resulted in a reduced level of invasion of mouse macrophages in both assays (Figures 3A,B). All strains used were cultured at 26°C, at which Y. pestis does not produce the F1 capsule that blocks interaction with host cells (37, 38). This result indicates that phagocytosis of these bacteria by mouse macrophages involves the core LPS ligand. The fact that the O-antigen-expressing and the deep rough-Y. pestis still interact with these host cells, although at a reduced level, suggests that besides the core LPS, other components of Y. pestis also mediate interactions with macrophages.
Figure 3. Interaction of Y. pestis with mouse peritoneal macrophages involves its core LPS. (A) Gentamicin protection- and (B) flow cytometry-based assays were used to determine the invasion rate of these sets of E. coli K-12(CS180, CS1861, and CS2429) and Y. pestis KIM10 (Δail, Δail-O+ and Δail-Core−) into purified mouse peritoneal macrophages. (B) CFDA-SE labeled and unlabelled bacteria are indicated by open and filled symbols, respectively. Data are representative of three independent experiments.
In addition, deep rough mutants of Gram-negative bacteria in general are more sensitive to biological killing (39, 40). This fact should be taken into consideration in interpreting the data from the gentamicin survival assay of KIM10-Δail-Core− and CS2429 (Figure 3A).
To determine if the invasion of Y. pestis into mouse macrophages was a result of the interaction between Y. pestis and murine DC-SIGN (mDC-SIGN), five CHO transfectants stably expressing the mouse C-type lectin receptors mDC-SIGN, SIGNR1, SIGNR3, mDEC-205 (CD205), and mLangerin (CD207) (41, 42) (Figure 4A) were infected with Y. pestis KIM10-Δail and CS180. Y. pseudotuberculosis (Y1) grown at 26°C was used as a positive control in this experiment because it invades most epithelial cell lines, including CHO (43), via the invasin-integrin interaction (44, 45). The expression of each C-type lectin is shown in Figure 4B. CHO-SIGNR1, but not other C-type lectin transfectants including CHO-mDC-SIGN, efficiently phagocytized Y. pestis KIM10-Δail and CS180 (Figure 4A). Since macrophages from the mouse peritoneal cavity express SIGNR1 (Figure 8B) (41, 42), it is anticipated that the phagocytosis of Y. pestis by these macrophages involves the SIGNR1-core LPS interaction.
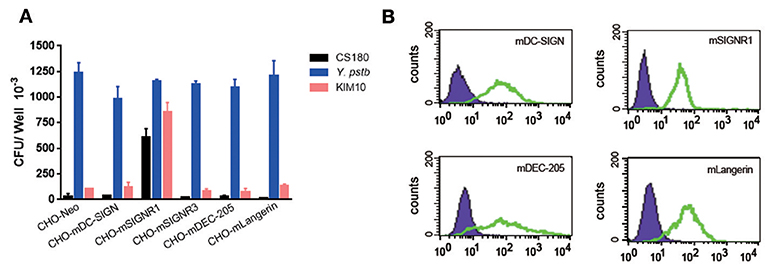
Figure 4. Y. pestis invades CHO-SIGNR1, but not other CHO transfectants. (A) The invasion of three bacteria; E. coli K-12 CS180, Y. pseudotuberculosis (Y1) and Y. pestis KIM10-Δail into several CHO transfectants with the expression of mouse C-type lectins; CHO-mDC-SIGN, CHO-SIGNR1, CHO-SIGNR3, CHO-mDEC-205 (CD205), and CHO-mLangerin (CD207). CHO transfectants were incubated with the bacterial strains for 2.5 h and the extracellular bacteria were killed by 100 μg/ml of gentamicin. The number of intracellular bacteria was determined by counting the recovered CFUs. (B) The expression of level of each transfectant and CHO-Neo was shown in open and filled curve, respectively. Data are representative of three independent experiments.
To confirm that a specific sugar epitope within the core LPS is responsible for interacting with SIGNR1, another set of core LPS mutants from Y. pestis strain D27 (46) was utilized (Figure 1B and Table 1). Consistent with the results obtained for E. coli (22), Salmonella (24), Neisseria gonorrhoeae (23), Neisseria meningitidis (52), H. ducreyi (24), and Y. pseudotuberculosis (28), the rough Y. pestis strain (wild-type) (25), rather than the O-antigen expressing Y. pestis promoted a typical SIGNR1-mediated adherence and phagocytosis (Figure 5). Furthermore, the deletion of the GlcNAc epitope in the waaL mutant (Figure 1B) reduced the ability of Y. pestis to interact with CHO-SIGNR1, indicating again that the core LPS is the ligand. Interestingly, phagocytosis of the Y. pestis mutants by CHO-SIGNR1 was consistent with the idea that the lengthier the core LPS is, the greater the ability to promote phagocytosis, as shown with Salmonella (24).
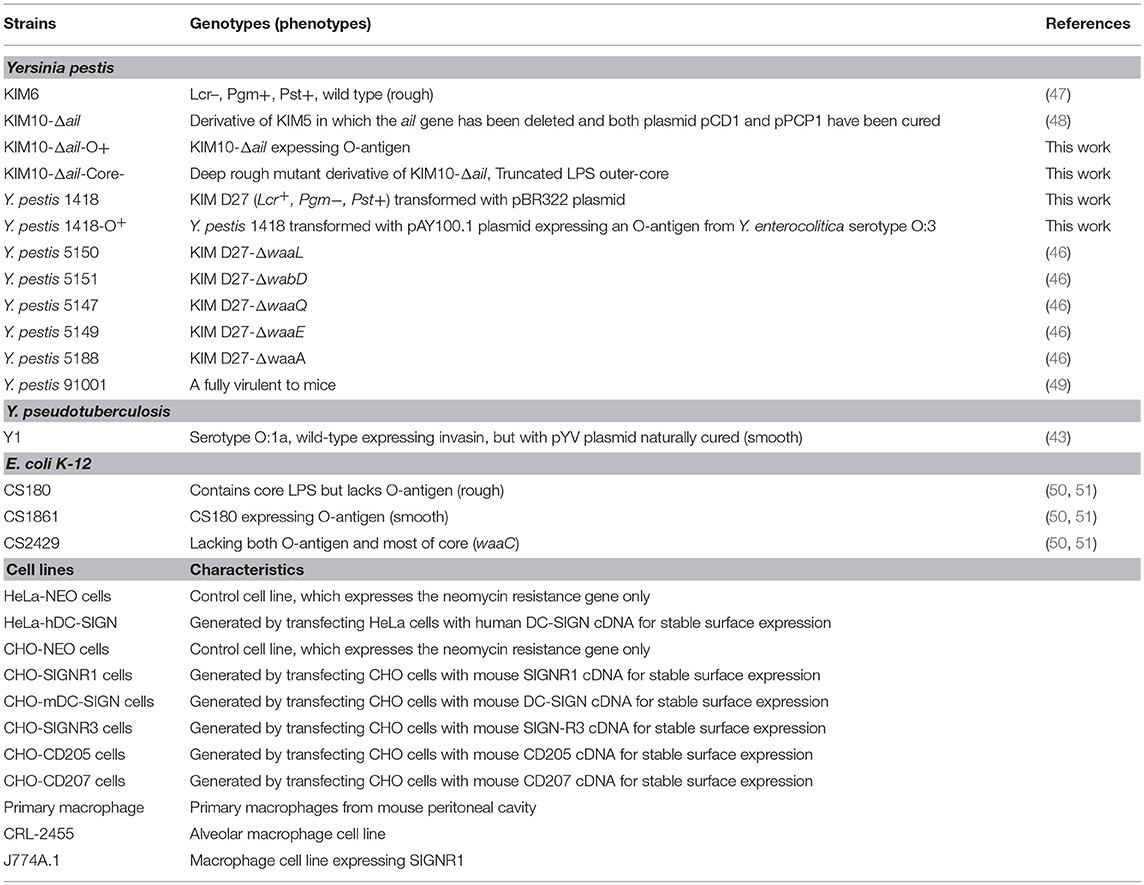
Table 1. Bacterial strains, cell lines used in this study.
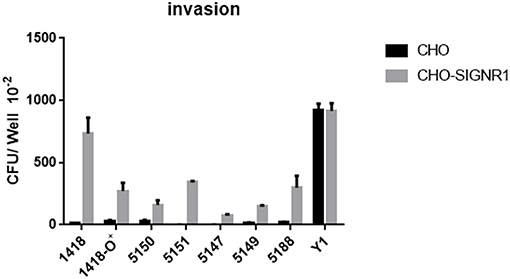
Figure 5. A specific sugar epitope within core LPS may be responsible for interacting with SIGNR1. Y. pestis 1418 (D27, wild type) and its mutants (1418-O+, 5147 D27-Δ-waaQ, 5149 D27-Δ-waaE, 5150 D27-Δ-waaL, 5151 D27-Δ-wabD, and 5188 D27-Δ-waaA) that possess specific sugar epitope within core LPS were used to determine the invasion rate. Y. pseudotuberculosis (Y1) were used as control strains. Data are representative of three independent experiments.
The results from Figures 3–5 lead to three conclusions. First, SIGNR1 is a receptor for Y. pestis. Second, the core LPS of Y. pestis as a ligand is involved in this interaction. Third, GlcNAc is important for the SIGNR1 interaction (24). In fact, the data published 2015 in Immunology and Cell Biology (27), regarding the interaction of human CD207, awarded an editorial comments, entitled “A new cellular target for Yersinia pestis” (53).
Y. pestis invades certain macrophage cell lines, such as the J774A.1 (54, 55). In order to determine if the same ligands are involved in this interaction as was found with primary macrophages (Figure 3A) and CHO-SIGNR1 (Figures 4, 5), the KIM10-Δail and KIM10-Δail-O+ were examined for their ability to invade two macrophage cell lines, J774A.1, and CRL-2455 (Figure 6A). The expression level of SIGNR1 on J774A.1 is shown in Figure 6B, but the expression of this receptor on CRL-2455 cell line was undetectable (Figure 6B).
Figure 6. SIGNR1-mediated phagocytosis depends in part on core LPS. (A) The two sets of bacteria; E. coli K-12 strain (CS180 and CS1861) and Y. pestis KIM10-Δail (Δail and Δail-O+) were tested for whether the SIGNR1-mediated interaction is depended on this core LPS with CHO transfectants and macrophage cell lines J774A.1 and CRL-2455. (B) The expression of SIGNR1 of these macrophage cell lines and control CHO-SIGNR1. Data are representative of three independent experiments. ***P < 0.001.
The interaction of the KIM10-Δail and KIM10-Δail-O+ with J774A.1 resembled the results obtained using primary macrophages. KIM10-Δail invades host cells better than KIM10-Δail-O+, and CS180 is also able to invade J774A.1 cells. However, the CRL-2455 cell line, which does not express SIGNR1, is still able to phagocytose low levels of both KIM10-Δail and KIM10-Δail-O+, but not CS180, indicating that the core LPS does not interact with the CRL-2455 cell line.
In short, these results indicated that other components or mechanisms can also lead to internalization of Y. pestis by macrophages, besides the core LPS-SIGNR1 interaction. Because CS180 invades SIGNR1-expressing macrophage cell line, but not the CRL-2455, it also confirms the core LPS-SIGNR1 interaction with macrophages.
To verify the specificity of the interaction of Y. pestis with SIGNR1, we examined whether the core LPS-SIGNR1 interaction could be inhibited by SIGNR1 antibody, mannan, His-Mermaid and CD66 antibody. Mannan is a well-documented reagent for its ability to block the DC-SIGN-mediated interactions with HIV. His-Mermaid is the recombinant form of Mermaid, a newly identified DC-SIGN-like protein (56) that has previously been shown to inhibit the core-LPS-hDC-SIGN interaction (24). Anti-CD66 antibody was employed as a control antibody. E. coli K12 CS180 and Y. pseudotuberculosis serotype O:1b, mediating a SIGNR1-dependent and -independent interaction, respectively, were again utilized as control strains. Figure 7A shows that the anti-SIGNR1 and mannan inhibit the interaction between Y. pestis or CS180 and CHO-SIGNR1, indicating a specific interaction between SIGNR1-Y. pestis, which promotes the invasion of this bacteria into mouse APCs.
Figure 7. The inhibition of SIGNR1-mediated phagocytosis of Y. pestis by anti-SIGNR1 antibody, mannan, peptides, and oligosaccharides. (A) Y. pestis KIM10-Δail were incubated with CHO-SIGNR1 in the presence or absence of anti-CD66, anti-SIGNR1, mannan and DC-SIGN-like protein (His-Mermaid). The phagocytosis rate of Y. pestis was evaluated by the recovery of bacteria from gentamicin protection. E. coli K12 180 and Y. pseudotuberculosis serotype O:1b were used as control strains to show core LPS-dependent or independent interaction with CHO-SIGNR1. (B,C) Various oligosaccharides and lactoferrin were also tested for their ability to inhibit the interaction of CHO-SIGNR1 and HeLa-hDC-SIGN with Y. pestis KIM10-Δail and Y. pseudotuberculosis sero-type O:1b (Y. pstb). Data are representative of three independent experiments.
Mermaid possesses the ability to inhibit hDC-SIGN-mediated interaction with several Gram-negative bacteria (24), but the inhibition of the Y. pestis-SIGNR1 interaction by Mermaid is limited (Figure 7A), suggesting that hDC-SIGN and SIGNR1 have distinguishing features employed during interactions with core LPS of Y. pestis (41, 42).
Certain oligosaccharides inhibit CS180-HeLa-hDC-SIGN-promoted interaction (24). Figures 7B,C show that although these reagents have very limited abilities to inhibit the SIGNR1-mediated interaction, but the β-D-Gal-(1 → 3)-D-GalNAc (A0167), β-D-Gal-(1 → 6)-D-GlcNAc (A7916) and α-NeuNAc-(2 → 3)-β-D-Gal-(1 → 4)(α-L-Fuc-)-D-GlcNAc (S1782) oligosaccharides inhibit the core-LPS-hDC-SIGN interaction of Y. pestis KIM10-Δail very well (25). The recovery rates of Y1 and KIM10-Δail bacteria in both cell lines are dramatically reduced in the presence of lactoferrin, indicating that these bacteria are killed by this peptide, which is well-known for its ability to kill bacteria (57).
In short, the results also indicated that hDC-SIGN (hCD209a) and SIGNR1 (CD209b) are different in terms of their interactions with Y. pestis.
Yesinia pestis can invade macrophages as well as other APCs during infection in mice (32, 58, 59). To test whether the core LPS-mediated interaction also occurs in vivo, we injected bacterial suspensions directly into the mouse peritoneal cavity. This approach is analogous to our previous study showing that the interaction of mouse CD205 (DEC-205) receptor on alveolar macrophages with the Y. pestis plasminogen activator (Pla) occurs in vivo (26).
After 1.5 h of infection, the intraperitoneal fluids or exudates were collected and placed in gentamicin media to kill the extracellular bacteria. Figure 8A shows that a higher percentage of viable Y. pestis was recovered compared to the O-antigen expressing-Y. pestis, KIM10-Δail-O+. This increased recovery rate of KIM10-Δail is not due to the ability of the O-antigen to protect against complement-mediated killing (16–21), as in the serum killing assay KIM10-Δail-O+ was more resistant than KIM10-Δail (Figure 8C). In short, because KIM10-Δail-O+ was more resistant than KIM10-Δail, the increased recovery of KIM10-Δail suggests that core LPS-mediated phagocytosis of Y. pestis occurs in vivo.
Figure 8. Core LPS-SIGNR1 interaction with macrophages occurs in vivo. Y. pestis (KIM10-Δail and KIM10-Δail-O+) were tested for their ability to invade macrophages in vivo. (A) Bacterial suspensions were inoculated into the mouse peritoneal cavity. After incubation for 1.5 h, macrophages were examined for the rate of internalized bacteria. (B) The SIGNR1 expression of the mouse peritoneal macrophages. (C) KIM10-Δail-O+ was more resistant than KIM10-Δail in serum resistant assay. *P < 0.05, ***P < 0.001. Data are representative of three independent experiments.
Our recently published data showed that Y. pseudotuberculosis uses its core LPS to interact with hDC-SIGN and SIGNR1 receptors, leading to its dissemination (28). We therefore hypothesized that the dissemination of Y. pestis to the lymph node (LN), spleen and liver would also be facilitated by this host-pathogen interaction. Subsequently, if the exposed core LPS of Y. pestis could be shielded that should reduce dissemination.
To achieve a Y. pestis strain producing smooth LPS in vivo, we introduced plasmid pAY100.1 into strain Y. pestis 1418. Plasmid pAY100.1 carries the O-Ag gene cluster of Y. enterocolitica serotype O:3 and produces the O-Ag (25, 27, 60, 61). Y. pestis 1418 (KIM D27) is a conditionally virulent strain, which is able to cause typical plague in mice depending on the route of infection and dose (Figure 9A) (46). As a control, the plasmid vector pBR322 was transformed to Y. pestis 1418, which was described in the previous publications (25–27). Mice were infected via injection into hind paws and sacrificed after 72 h. LN, spleen and liver were then homogenized. The dissemination rates of the bacteria into the different organs were calculated by plating and counting CFUs and fluorescent intensity. Figures 9A,C show that higher numbers of 1418 than 1418-O+ bacteria were isolated from LN, spleen, and liver. It should be recognized that both strains exhibited no differences in growth and adhered to both HeLa and CHO cells (data not shown).
Figure 9. O-antigen expressing Y. pestis have a reduced ability to be disseminated to lymph nodes. (A) Y. pestis 1418 and 1418-O+ were examined for their ability to be disseminated to lymph node, spleen and liver. (B) Bacterial load determined by real time PCR. (C) Dissemination of Y. pestis 1418 and 1418-O+ with pXEN-18 were monitored by the bioluminescence at 72 h post bacterial inoculation into hind paws of mice. (D) Reduction the phagocytosis of Y. pestis 1418 by peritoneal macrophages of SIGNR1 KO mice. (E) Reduction in Y. pestis 1418 dissemination to lymph node in Y. pestis infected SIGNR1 KO mice at 5 hpi as compared to infected WT mice. (F) Survival rates of WT mice and SIGNR1 KO mice after the infection of Y. pestis 1418. For each group, 10 mice were infected with 108 CFU Y. pestis 1418 and observed until 15 days post infection. Log-rank test was performed. The data shown are obtained from the three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
The mice were challenged as described above, but the mice were sacrificed after 8 h infection. The ail gene DNA of Y. pestis in LN, spleen and liver was quantitated by real time PCR. The bacterial load was higher in mice infected with Y. pestis 1418 than in Y. pestis 1418-O+ (Figure 9B), which was consistent with the data of bacterial recovery assay described above.
C57BL/6J mice were subcutaneously inoculated in hind paws with Y. pestis 1418 or 1418-O+ transformed with the pXEN-luxCDABE (pXEN-18) plasmid, and bioluminescent signals were monitored at 0, 48, and 72 h post inoculation (hpi). The bioluminescent scale ranges from most intense (red) to least intensity (violet) (Figure 9C). All images are standardized to the same radiance scale. Bioluminescence was detected in the abdomen and thoracic region of the mice at 48 hpi (data not shown), but the highest level of signal was observed in the region corresponding to liver and spleen at 72 hpi. Signals from the mice infected with Y. pestis 1418-O+ were significantly increased in intensity than those in Y. pestis1418 group, suggesting the dissemination ability of O-antigen expressing Y. pestis was reduced.
To demonstrate the involvement of SIGNR1 in the interaction of Y. pestis and macrophages in vivo, we evaluated the phagocytic and intracellular killing capacity of peritoneal macrophages derived from SIGNR1 knock-out (SIGNR1) mice. The phagocytosis of Y. pestis by peritoneal macrophages (Figure 9D) and host dissemination (Figure 9E) in by SIGNR1 KO mice were significantly reduced, indicating the direct involvement of SIGNR1. However, the SIGNR1 KO mice were shown more susceptible to infection of Y. pestis 1418 (Figure 9F), which is addressed in Discussion.
In summary, the results suggest that Y. pestis could utilize its core LPS to interact with SIGNR1 to enhance the dissemination in host tissues.
To examine whether the reduced dissemination, when the interaction of core LPS-SIGNR1 interaction was blocked by expression of O-antigen, leads to reduction of infection, C57BL/6J mice were challenged with two sets of Y. pestis 1418 and 91001. Survival analyses following subcutaneous injection, the route bubonic plague, revealed that the mice infected with Y. pestis 1418 suffered from a significant survival disadvantage compared to those infected with Y. pestis 1418-O+ (Figure 10A). However, there is no difference with the intravenous inoculation which mimic the septicemic plague (Figure 10B). Y. pestis 91001, a fully virulent strain isolated from China (62), was used to challenge each mouse with 30 CFU via subcutaneous inoculation. When cover the core LPS of Y. pestis 91001 with the expression of O-antigen, the mice infected with Y. pestis 91001-O+ displayed significant survival advantage relative to Y. pestis 91001 (Figure 10C). The results shown above demonstrate that the exposure of core LPS is important for Y. pestis in host dissemination and bacterial infection.
Figure 10. The expression of O-antigen reduces death rates by infection. (A,B) Mice were subcutaneously and intravenously inoculated with Y. pestis 1418/1418-O+. (C) Mice were subcutaneously inoculated with Y. pestis 91001. The rates of mortality were recorded every 12 hpi. *P < 0.05.
Y. pestis, the cause of plagues, has directly evolved from Y. pseudotuberculosis within the last 2,600 to 28,000 years (7, 10, 11). It is well-documented that a distinguishing characteristic between these two Yersinia species is that Y. pseudotuberculosis strains possess an O-antigen (intact LPS expression), which was lost by Y. pestis during evolution. As a result, after entering the skin by an infected flea, Y. pestis, with its core LPS, can directly interact with APCs, leading to phagocytosis of the pathogen (25). The infected APCs consequently serve as a Trojan Horse to deliver the Y. pestis to lymph nodes (33) and initiate the plagues. In this study, we demonstrated that it is the murine SIGNR1 that serves as a receptor for the core LPS of Y. pestis to promote bacterial dissemination and infection.
The current model for the initial stages in the pathogenic process mediated by Y. pestis is reminiscent of how HIV-1 infects hosts. It is well-established that HIV-1 pirates DC-SIGN (CD209), a C-type lectin receptor expressed by APCs, to be captured and transmitted to target cells such as CD4+ lymphocytes (29–31).
The connection between Y. pestis and HIV extends to another receptor, CCR5, which is a co-receptor for HIV. There is evidence that a certain subpopulation of Caucasians is resistant to HIV infection because of their natural deletion of this receptor. It has been suggested that human populations have also been selected by Y. pestis, based on its sensitivity to human CCR5 (63). Therefore, CCR5 knock-out mice were challenged with Y. pestis, but no protection was observed (64, 65).
Besides showing that Y. pestis uses its core LPS to interact with SIGNR1 in vitro, we designed and performed an in vivo interaction assay to determine whether the core LPS-SIGNR1 interaction occurs in vivo between Y. pestis and macrophages. Even so, the fundamental question remains as to whether core LPS-SIGNR1 interaction plays a role in in vivo infections.
The straightforward approach is to test if SIGNR1 knock-out mice would be resistant to plague, as KO mice have been used successfully to identify viral receptors. For example, the CEACAM1 (CD66a) receptor KO mice have an increased resistance to mouse hepatitis viral infection (66), because mouse CEACAM1 is a receptor for mouse hepatitis virus (67). However, there are potential limitations of this approach to study bacterial-host cell interactions. Strangely, there are no credible receptor knock-out models that are more resistant to bacterial infection. The reason might be simple; viral infection is less complicated than bacterial infections. One receptor might not be enough to determine the fate of a bacterial infection, which might contribute to the failure of CCR5-knock-out mice to resist Y. pestis infection (64, 65), even if CCR5 were a receptor for Y. pestis. In addition, SIGNR1 knock-out mice are more susceptible to bacterial infection (68), probably because of the role of this receptor in the complement pathway (69). In short, this strategy could only work if SIGNR1 is the only, or a very prominent, receptor for Y. pestis. Unfortunately, many pathogens do not depend on only one receptor in their interactions with host cells.
The oligosaccharides and small peptide were chosen to analyse their ability to inhibit Y. pestis-C-type lectin interactions for the four following rationales: (1) Oligosaccharides that interfere with the interaction between host cells and Y. pestis have been examined for their therapeutic potential (70, 71). (2) Several Gram-negative bacteria might use their core LPS, consisting of oligosaccharides, to interact with hDC-SIGN, which could be inhibited by oligosaccharides (24). (3) SIGNR1 binds to the capsular polysaccharide of Streptococcus pneumoniae (68, 69, 72). (4) HIV uses gp 120-DC-SIGN interaction to be captured by DCs and transmitted to CD4+ cells (29–31). Therefore, blockage of DC-SIGN-mediated transmission of HIV has been undertaken by many investigators in order to find therapeutic strategies for HIV infection. For example, lactoferrin, a small peptide from milk, and Lewis X components (oligosaccharides) have been shown to prevent DC-mediated HIV-1 transmission by blocking the DC-SIGN-gp120 interaction (73, 74). Interestingly, our data show that some oligosaccharides indeed inhibit the interaction between Y. pestis and hDC-SIGN.
Taken together, this study has demonstrated that SIGNR1 is a cellular receptor for Y. pestis and possibly plays a role in host dissemination and bacterial infection. Since hDC-SIGN and SIGNR1 share a similar ability to interact with core LPS, we speculate that Y. pestis may hijack APCs, through the core LPS-SIGNR1 interaction, to reach the lymph nodes, utilizing a similar mechanism as demonstrated in the HIV-hDC-SIGN interaction. The knowledge acquired from this study may allow us to develop novel strategies to combat this bacterial pathogen by blocking the interaction between Y. pestis and host receptors.
All animal procedures were carried out in strict accordance with the guidelines of Institutional Animal Care and Use Committees (IACUCs) and Institutional Review Board (IRB) of Tongji Hospital, HUST. The handling of the mice and all experimental procedures were specifically approved for this study by the Medical Ethics Committee of Tongji Hospital and were conducted in accordance with the institutional guidelines (IRB ID: TJ-A20141220).
E. coli K12 strain CS180 contains core LPS but lacks O-antigen (75). CS1861 is the strain of CS180 harboring pSS37, a plasmid containing all the genes necessary for the expression of the Shigella dysenteriae 1 O-antigen (50, 51, 75). A deep rough isogenic mutant CS2429 (waaC), lacking both O-antigen and most of core (50, 51, 75), was used to assess the role of LPS in bacterial-macrophage interactions. E. coli strains were cultured on Luria-Bertani medium (LB) supplemented with 1.5% agar at 37°C overnight.
Yersinia pseudotuberculosis (Y1) is a serotype O:1a strain, lacking the virulence plasmid (pYV) and expression of Ail protein. The strain was obtained from the CDC and used as a control strain for invasion (43), since this bacterium invades almost all epithelial cell lines via an invasin-integrin interaction (76).
The Y. pestis strain 1418 used in this study is originated from KIM5 (KIM-D27), whose pgm (pigmentation) 104 kb locus has been deleted (47, 48), and it is therefore classified as an avirulent and non-selected strain. KIM10-Δail is a derivative of KIM5, in which the ail gene has been deleted and its pPCP1 plasmid was also cured (48). The KIM10-Δail used in this study is derivative of KIM5 and was also pYV plasmid-cured strain, selected using a combination of magnesium oxalate and Congo red selection methods (77). KIM10-Δail-O+ that expresses an O-antigen from Y. enterocolitica serotype O:3 (61) is an isogenic derivative of KIM10-Δail. KIM10-Δail-Core− is also an isogenic derivative of KIM10-Δail, in which the outer core LPS has been deleted as described in the construction procedures shown below. Y. pestis core LPS mutants 5147, 5149, 5150, 5151, and 5188 were generously provided by Dr. Skurnik (46). Strains were cultured on GC based-plates (Difco, Sparks, MD) supplemented with 1% hemoglobin (USB Co., Cleveland, OH).
For construction of gmhA-deletion KIM10-Δail, we followed the procedures described by Dr. Darby (78). The inner core structure contains KDO linked to lipid A followed by heptoses to which the outer core hexoses attach (Figure 1). Phosphoheptose isomerase, encoded by gmhA, catalyzes the first step in the GDP-heptose biosynthesis pathway (78). Y. pestis KIM6 gmhA mutants are deep rough mutants as they do not make GDP-heptose that results in truncation of the outer core of LPS (78). To construct a deep rough mutant derivative of KIM10-Δail, the gmhA allelic exchange plasmid pCBD41 was mobilized from E. coli SM10λpir/pCBD41 into KIM10-Δail. pCBD41 (a kind gift of Dr. Greg Darby) contains two gmhA-flanking 900 bp PCR-amplified fragments cloned into suicide vector pCVD442 (78). KIM10-Δail transconjugants were selected on Yersinia selective agar (Difco) containing chloramphenicol and ampicillin. As the suicide vector replication requires the pir gene, which does not present in Yersinia spp., the recovered KIM10-Δail transconjugants should contain the plasmid cointegrated at the gmhA chromosomal locus. The deletion of gmhA was confirmed by PCR assays using the primers; GMHF 5′-GCTTGGATCCCATAATGAAGCTCCTGAGATGTAG and GMHR 5′-AGTGGGTCGACACAGAAGATTGAGGTGATCAAC.
The expression of O-antigen by Y. pestis was lost during evolution from the ancestor, Y. pseudotuberculosis. To express the O-antigen, Y. pestis strain 1418 was transformed with plasmid pAY100.1 that carries all the necessary genes for the expression of the O-ag of Y. enterocolitica serotype O:3 (60, 61). The expression of O-antigen, coded by pAY100.1 plasmid, is not influenced by growth temperature (25–27).
Two mouse macrophage cell lines were purchased from ATCC. The CRL-2455 is an alveolar macrophage cell line. J774A.1 was selected, since this cell line shows its ability to phagocytose Y. pestis when grown at 26°C (54, 55).
Mouse C-type lectin tranfectants, CHO-mDC-SIGN, CHO-SIGNR1, CHO-SIGNR3, CHO-mDEC-205 (CD205), and CHO-mLangerin (CD207) were generated by transfecting CHO cells (purchased from purchased from the Type Culture Collection of the Chinese Academy of Sciences, Shanghai, China) with mouse corresponding C-type lectin cDNA followed by selection for stable surface expression as originally described (41). CHO-Neo is the control cell line, which expresses the neomycin resistance gene only.
HeLa-DC-SIGN cells were generated by transfecting HeLa cells (purchased from ATCC, USA) with human DC-SIGN cDNA followed by selection for stable surface DC-SIGN expression as originally described (79, 80). The cell lines were recently used for identification of core LPS from several Gram-negative bacteria as ligand for DC-SIGN receptor (22–24).
C57BL/6J and BALB/cJ were purchased from Wuhan University Animal Center. SIGNR1 KO mice were kindly provided by The Consortium for Functional Glycomics (CFG, http://www.functionalglycomics.org) and bred in the animal facility of Tongji Hospital. Mice were housed in direct accordance with guidelines drafted by the Animal Care Committees of Tongji Hospital.
Anti-mouse SIGNR1 antibody was purchased from Pharmingen (San Diego, CA). YTH71.3, a rat antibody which recognizes CEACAM1 (CD66a), CEACAM6 (CD66c), and CEACAM3 (CD66d), was purchased from Roche (Indianapolis, IN).
Oligosaccharides: β-D-Gal-(1 → 6)-D-GlcNAc {2-Acetamido-2-deoxy-6-O-(β-D-galactopyranosyl)-D-glucopyranose, A7916}, β-D-Gal-(1 → 4)-D-GlcNAc {N-Acetyl-D-lactosamine A7791}, β-D-Gal-(1 → 3)-D-GalNAc {Galacto-N-biose, A0167}, β-D-GlcNAc-(1 → 3)-β-D-Gal-1 → OMe {(Methyl 3-O-(N-acetyl-β-D-glucosaminyl)-β-D-galactopyranoside, M0775}, α-NeuNAc-(2 → 3)-β-D-Gal-(1 → 4)(α-L-Fuc-)-D-GlcNAc {3'-Sialyl-Lewis-X tetrasaccharide, S1782} and β-D-GlcNAc-(1 → 6)-β-D-Gal-(1 → 4)-D-Glc {β 6'-GlcNAc-lactose, A8297)}, mannan and lactoferrin were purchased from Sigma-Aldrich (St. Louis, MO). Mannan is a ligand antagonist of human mannose receptors. For purpose of easy labeling, the product numbers of each oligosaccharide from Sigma are also included. The background information of each product is listed in Sigma-Aldrich catalog.
Mermaid is a DC-SIGN-like molecule expressed by the marine nematode Laxus oneistus. The carbohydrate recognition domain of Mermaid shares both structural and functional similarity with that of DC-SIGN as described (56). A recombinant form of Mermaid (His-Mermaid) was expressed and purified as described (56).
wbyL, fcl, gmd, wzy, wbyI, and ddhB of 39 Y. pseudotuberculosis strains and 8 Y. pestis strains were sequenced. First, these genes were copied by Polymerase Chain Reaction using high-fidelity DNA polymerase (PCR SuperMix, Transgene Biotech, Beijing). And then the products were sent to perform bidirectional Sanger sequencing. Primers for these O-antigen synthesis genes; wbyL Forward 5′- GTCGGCATTGCTCATTCTATTG- 3′, wbyL Reverse 5′- TCACTGGTTAATCGAACATCCC- 3′, fcl Forward 5′- TGCTGAAATGGTCGCTAGTG-3′, fcl Reverse 5′- AGAGTCGCCATATCCAAATAGC-3′, gmd Forward 5′- AGGTGATGCCGCTATATTAGTG-3′, gmd Reverse 5′- GAGGTCAAGTTCAGTACGATCC-3′, wzy-1 Forward 5′- TCGACTACCTTCTCATTCTTGG-3′, wzy-1 Reverse 5′- TCACGACGAAGAGCCTTTATAG-3′, wzy-2 Forward 5′- GGCCTCTTGTACCAAACTTC-3′, wzy-2 Reverse 5′- TCCGAGAAATAGACAGTTACCC-3′, wbyI Forward 5′- TGTGTCAAGTTAGTCGGATATG-3′, wbyI Reverse 5′- CTTGCGAAGACCATTTCATTAG-3′, ddhB Forward 5′- GGCAGGGCACCTTGGAAG-3′, and ddhB Reverse 5′- CCAGCTCAGCAATCTGTTGAC-3′. Sequencing data was analyzed by BioNumerics Software Version 7 and heatmap was made by R software.
The peritoneal macrophages were selected as our primary cells. After the 6- to 8-week-old female mice were euthanized, intact abdomen was exposed, cleaned with 70% ethanol and opened. Five milliliter of RPMI was injected into intraperitoneal cavity. Mouse abdomen was gently massaged for 3 min and then the lavage fluid was collected. The suspension containing the macrophages was seeded in flasks and placed in a CO2 incubator for 2 h. The cell layers were washed 3 times to remove non-adherent cells. Macrophages were then removed from the plastic surface by incubating with citrate saline and re-seeded for interaction assays or stained with antibodies to check the expression level of receptors.
The assays for adherence and phagocytosis have been described previously (81, 82). Briefly, host cells (CHO, HeLa, and macrophages) were plated in 24 or 96-well plates. Cells were suspended in RPMI with 2% FCS at a concentration of 4 × 105/ml. One half ml each of these cell suspensions was added to 24-well plates and after addition of 50 μl of bacterial suspensions at a concentration of 1 × 107 colony forming units (CFU)/ml, the cells were allowed to incubate for 2.5 h (2 h for alveolar macrophages) at 37°C in the presence of 5% CO2. The cell monolayers were then washed 3 times with PBS. The number of associated bacteria (adherent and internalized) per cell was quantified by washing the cells 3 times with RPMI containing 2% FCS and plating the culture after the cells were lysed by 0.5% saponin (Calbiochem Corp., San Diego, CA).
To determine the internalization of bacteria, gentamicin, which kills extracellular bacteria but cannot penetrate host cells, was added into each well to a final concentration of 100 μg/ml, and the cultures were incubated for 60 min. Cells were washed twice to remove the antibiotics. Then, the cells were suspended in PBS containing 0.5% saponin, diluted and plated on LB and GC as well as Y. pestis plates. The level of internalization of bacteria in these host cells was calculated by determining the CFU recovered from lysed cells.
For the inhibition assay, reagents were added 20 min prior to the addition of bacteria at the following concentrations: anti-SIGNR1 antibody, 5 μg/ml; mannan, 500 μg/ml; DC-SIGN-like protein (Mermaid), 10 μg/ml and anti-CD66, 5 μg/ml. The concentrations used were based on our preliminary data, and were selected based on the fact that at these concentrations, there was no influence on the survival of bacteria and HeLa cells, or the interaction between pEXI and HeLa-CEACAM3 (22–24, 82, 83).
The following method was used to supplement the survival-based phagocytosis assay described previously (23). Briefly, bacteria were suspended in RPMI medium containing 5- and 6-carboxyfluorescein diacetate, succinimidyl ester (CFDA-SE; Molecular Probes, Eugene. OR) for 40 min and washed twice with RPMI to remove the excess dye. Labeled bacteria were added to macrophage cultures for 2 h. Cell cultures were washed twice to remove unbound bacteria. Macrophages plus associated bacteria were fixed with 2% paraformaldehyde. Before flow cytometry, a 1:10 dilution of Trypan blue (0.4%, Sigma, St. Louis, MO) was added to the fixed cell cultures and the mixture was incubated at ambient temperature for 10 min (23) to quench the fluorescence from extracellular labeled bacteria. Trypan blue blocks fluorescence but cannot penetrate host cells, therefore, fluorescence from internalized bacteria will not be influenced by addition of Trypan blue. The rate of bacterial internalization was determined by comparing the intensity fluorescence-positive macrophages with various bacteria. The higher of the fluorescence-intensity shows, the more of bacteria are phagocytosed by macrophages.
One milliliter of bacterial suspensions (OD = 0.1) were injected into 6- to 8-week-old female mouse intraperitoneal cavity. Mouse abdomen was gently massaged for 1 min. After 1.5 h, mice were euthanized and another 4 ml of RPMI with 2% FCS were immediately injected into each mouse peritoneal cavity and the abdomen was gently massaged for another 1 min. The intraperitoneal fluids or exudates were collected, and the numbers of cells were counted for each collection of intraperitoneal lavage. 1 × 106 was seeded onto each well of 24-well plates, containing RPMI with 2% FCS and gentamycin at concentration of 100 μg/ml, and were then incubated for 1.5 h to allow the macrophages adhere to plates and kill the extracellular bacteria. Each well was washed three times with RPMI with 2% FCS to remove non-adhered cells and lysed with saponin, followed the same procedures as in vitro phagocytosis assays.
Six- to eight-week-old female C57BL/6J and SIGNR1 KO mice were used in the following experiments. The dissemination rate was defined as the transport of Y. pestis to LN, spleen, and liver. The infectivity was defined as the mortality after inoculations of pathogens.
The protocol follows a similar assay we previously developed (26, 27). Y. pestis were cultured at 26°C to avoid the expression of OPM capsule and then suspended in PBS at a concentration of OD600 = 1.5 in PBS. Hundred microliters of the Yersinia suspension was injected in hind paws of mice. It should be noted 30 min before inoculation, mice were injected with ampicillin at a final concentration of 50 μg/g of mouse body weight to maintain the plasmid-based expression of O-antigen. (1) For CFU determination, the mice were euthanized and the inguinal lymph nodes were isolated 24 h post-injection. The isolated inguinal lymph nodes were then homogenized and lysed with 0.5% Triton X-100 to release the bacteria prior to plating onto agar plates containing ampicillin. The total isolated CFU of inguinal lymph nodes per mouse were defined as the dissemination rate. (2) For bioluminescence imaging, C57BL/6J mice were inoculated with Y. pestis 1418 and Y. pestis 1418-O+ transformed with the plasmid pXEN-18 which expresses the lux genes. The bioluminescence signal was detected by Night OWL II LB983 imaging system (Berthold Technologies, Bad Wildbad, Germany).
All statistical analyses were completed using Prism software, Version 6 (Graph Pad, San Diego, CA, USA). Statistical significance was assessed using Student's unpaired t-test. Survival assay was analyzed by log-rank test.
TC, CP, YK, AA, RY, JK, and MS contributed conception and design of the study. KY, YXH, YPH, and YJC performed the assays. YXH and YPH performed the statistical analysis. KY and YXH wrote the first draft of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by PHS grants (R01AI 47736) while working at the University of Illinois at Chicago, by grants from the National Natural Science Foundation of China (NSFC81271780 and 81471915) and by two local grants from Tongji Hospital, Tongji Medical College to TC. CP was supported by grants from the National Research Foundation of Korea (NRF-2013R1A1A2058427, NRF-2014R1A4A1008625) and a faculty research grant of Yonsei University College of Medicine for 2014 (6-2014-0062). AA and SD were supported by the Sectoral Scientific Program of the Russian Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing. We would greatly like to thank Drs. Olivier Schwartz at Pasteur Institute, Paris and Joseph Hinnebusch at the Rocky Mountain Laboratories, NIH, USA for their long term supports and helps to initiate the project of C-type lectin and Yersinia spp. interactions. We are indebted to Dr. Ralph Steinman when he was at the Rockefeller University, USA for his insightful advice and generous financial supports for this project. Finally, the PI would specifically like to take this opportunity to thank the Tongji Hospital, HUST, for their supports, allowing this project being continued.
1. Prentice MB, Gilbert T, Cooper A. Was the Black Death caused by Yersinia pestis? Lancet Infect Dis. (2004) 4:72. doi: 10.1016/S1473-3099(04)00923-5
2. Haensch S, Bianucci R, Signoli M, Rajerison M, Schultz M, Kacki S, et al. Distinct clones of Yersinia pestis caused the black death. PLoS Pathog. 6:e1001134. doi: 10.1371/journal.ppat.1001134
3. Bos KI, Schuenemann VJ, Golding GB, Burbano HA, Waglechner N, Coombes BK, et al. A draft genome of Yersinia pestis from victims of the Black Death. Nature. (2011) 478:506–10. doi: 10.1038/nature10549
4. Harbeck M, Seifert L, Hansch S, Wagner DM, Birdsell D, Parise KL, et al. Yersinia pestis DNA from skeletal remains from the 6(th) century AD reveals insights into Justinianic Plague. PLoS Pathog. 9:e1003349. doi: 10.1371/journal.ppat.1003349
5. Wagner DM, Klunk J, Harbeck M, Devault A, Waglechner N, Sahl JW, et al. Yersinia pestis and the plague of Justinian 541-543 AD: a genomic analysis. Lancet Infect Dis. (2014) 14:319–26. doi: 10.1016/S1473-3099(13)70323-2
6. Spyrou MA, Tukhbatova RI, Feldman M, Drath J, Kacki S, Beltran De Heredia J, et al. Historical Y. pestis genomes reveal the European Black Death as the source of ancient and modern plague pandemics. Cell Host Microbe. (2016) 19:874–81. doi: 10.1016/j.chom.2016.05.012
7. Morelli G, Song Y, Mazzoni CJ, Eppinger M, Roumagnac P, Wagner DM, et al. Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity. Nat Genet. (2010) 42:1140–3. doi: 10.1038/ng.705
8. Rasmussen S, Allentoft ME, Nielsen K, Orlando L, Sikora M, Sjogren KG, et al. Early divergent strains of Yersinia pestis in Eurasia 5,000 years ago. Cell. (2015) 163:571–82. doi: 10.1016/j.cell.2015.10.009
9. Valtuena A, Mittnik A, Key FM, Haak W, Allmae R, Belinskij A, et al. The stone age plague and its persistence in Eurasia. Curr Biol. (2017) 27: 3683–91.e8. doi: 10.1016/j.cub.2017.10.025
10. Achtman M, Zurth K, Morelli G, Torrea G, Guiyoule A, Carniel E. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc Natl Acad Sci USA. (1999) 96:14043–8. doi: 10.1073/pnas.96.24.14043
11. Achtman M, Morelli G, Zhu P, Wirth T, Diehl I, Kusecek B, et al. Microevolution and history of the plague bacillus, Yersinia pestis. Proc Natl Acad Sci USA. (2004) 101:17837–42. doi: 10.1073/pnas.0408026101
12. Cui Y, Yu C, Yan Y, Li D, Li Y, Jombart T, et al. Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis. Proc Natl Acad Sci USA. (2013) 110:577–82. doi: 10.1073/pnas.1205750110
13. Minka S, Bruneteau M. Isolation and chemical characterization of type R lipopolysaccharides of a hypovirulent strain of Yersinia pestis. Can J Microbiol. (1998) 44:477–81.
14. Skurnik M, Peippo A, Ervela E. Characterization of the O-antigen gene clusters of Yersinia pseudotuberculosis and the cryptic O-antigen gene cluster of Yersinia pestis shows that the plague bacillus is most closely related to and has evolved from Y. pseudotuberculosis serotype O:1b. Mol Microbiol. (2000) 37:316–30. doi: 10.1046/j.1365-2958.2000.01993.x
15. Prior JL, Parkhill J, Hitchen PG, Mungall KL, Stevens K, Morris HR, et al. The failure of different strains of Yersinia pestis to produce lipopolysaccharide O-antigen under different growth conditions is due to mutations in the O-antigen gene cluster. FEMS Microbiol Lett. (2001) 197:229–33. doi: 10.1111/j.1574-6968.2001.tb10608.x
16. Burns SM, Hull SI. Comparison of loss of serum resistance by defined lipopolysaccharide mutants and an acapsular mutant of uropathogenic Escherichia coli O75:K5. Infect Immun. (1998) 66:4244–53.
17. Cortes G, Borrell N, De Astorza B, Gomez C, Sauleda J, Alberti S. Molecular analysis of the contribution of the capsular polysaccharide and the lipopolysaccharide O side chain to the virulence of Klebsiella pneumoniae in a murine model of pneumonia. Infect Immun. (2002) 70:2583–90. doi: 10.1128/IAI.70.5.2583-2590.2002
18. Morona R, Daniels C, Van Den Bosch L. Genetic modulation of Shigella flexneri 2a lipopolysaccharide O antigen modal chain length reveals that it has been optimized for virulence. Microbiology. (2003) 149:925–39. doi: 10.1099/mic.0.26141-0
19. Murray GL, Attridge SR, Morona R. Regulation of Salmonella typhimurium lipopolysaccharide O antigen chain length is required for virulence; identification of FepE as a second Wzz. Mol Microbiol. (2003) 47:1395–406. doi: 10.1046/j.1365-2958.2003.03383.x
20. Russo TA, Davidson BA, Carlino-Macdonald UB, Helinski JD, Priore RL, Knight PRIII. The effects of Escherichia coli capsule, O-antigen, host neutrophils, and complement in a rat model of Gram-negative pneumonia. FEMS Microbiol Lett. (2003) 226:355–61. doi: 10.1016/S0378-1097(03)00636-0
21. Bengoechea JA, Najdenski H, Skurnik M. Lipopolysaccharide O antigen status of Yersinia enterocolitica O:8 is essential for virulence and absence of O antigen affects the expression of other Yersinia virulence factors. Mol Microbiol. (2004) 52:451–69. doi: 10.1111/j.1365-2958.2004.03987.x
22. Klena J, Zhang P, Schwartz O, Hull S, Chen T. The core lipopolysaccharide of Escherichia coli is a ligand for the dendritic-cell-specific intercellular adhesion molecule nonintegrin CD209 receptor. J Bacteriol. (2005) 187:1710–5. doi: 10.1128/JB.187.5.1710-1715.2005
23. Zhang P, Schwartz O, Pantelic M, Li G, Knazze Q, Nobile C, et al. DC-SIGN (CD209) recognition of Neisseria gonorrhoeae is circumvented by lipooligosaccharide variation. J Leukoc Biol. (2006a) 79:731–8. doi: 10.1189/jlb.0405184
24. Zhang P, Snyder S, Feng P, Azadi P, Zhang S, Bulgheresi S, et al. Role of N-acetylglucosamine within core lipopolysaccharide of several species of Gram-negative bacteria in targeting the DC-SIGN (CD209). J Immunol. (2006b) 177:4002–11. doi: 10.4049/jimmunol.177.6.4002
25. Zhang P, Skurnik M, Zhang SS, Schwartz O, Kalyanasundaram R, Bulgheresi S, et al. Human dendritic cell-specific intercellular adhesion molecule-grabbing nonintegrin (CD209) is a receptor for Yersinia pestis that promotes phagocytosis by dendritic cells. Infect Immun. (2008) 76:2070–9. doi: 10.1128/IAI.01246-07
26. Zhang SS, Park CG, Zhang P, Bartra SS, Plano GV, Klena JD, et al. Plasminogen activator Pla of Yersinia pestis utilizes murine DEC-205 (CD205) as a receptor to promote dissemination. J Biol Chem. (2008) 283:31511–21. doi: 10.1074/jbc.M804646200
27. Yang K, Park CG, Cheong C, Bulgheresi S, Zhang S, Zhang P, et al. Host Langerin (CD207) is a receptor for Yersinia pestis phagocytosis and promotes dissemination. Immunol Cell Biol. (2015) 93:815–24. doi: 10.1038/icb.2015.46
28. He YX, Ye CL, Zhang P, Li Q, Park CG, Yang K, et al. Yersinia pseudotuberculosis exploits CD209 receptors for promoting host dissemination and infection. Infect Immun. (2018) 87:e00654–18. doi: 10.1128/IAI.00654-18
29. Geijtenbeek TB, Kwon DS, Torensma R, Van Vliet SJ, Van Duijnhoven GC, Middel J, et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. (2000) 100:587–97. doi: 10.1016/S0092-8674(00)80694-7
30. Engering A, Van Vliet SJ, Geijtenbeek TB, Van Kooyk Y. Subset of DC-SIGN+ dendritic cells in human blood transmits HIV-1 to T lymphocytes. Blood. (2002) 100:1780–6. doi: 10.1182/blood-2001-12-0179
31. McDonald D, Wu L, Bohks SM, Kewalramani VN, Unutmaz D, Hope TJ. Recruitment of HIV and its receptors to dendritic cell-T cell junctions. Science. (2003) 300:1295–7. doi: 10.1126/science.1084238
32. Viboud GI, Bliska JB. Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu Rev Microbiol. (2005) 59:69–89. doi: 10.1146/annurev.micro.59.030804.121320
33. St John AL, Ang WX, Huang MN, Kunder CA, Chan EW, Gunn MD, et al. S1P-dependent trafficking of intracellular Yersinia pestis through lymph nodes establishes Buboes and systemic infection. Immunity. (2014) 41:440–50. doi: 10.1016/j.immuni.2014.07.013
34. Kukkonen M, Suomalainen M, Kyllonen P, Lahteenmaki K, Lang H, Virkola R, et al. Lack of O-antigen is essential for plasminogen activation by Yersinia pestis and Salmonella enterica. Mol Microbiol. (2004) 51:215–25. doi: 10.1046/j.1365-2958.2003.03817.x
35. Knirel Y, Kondakova A, Bystrova O, Lindner B, Shaikhutdinova R, Dentovskaya S, et al. New features of Yersinia lipopolysaccharide structures as revealed by high-resolution electrospray ionization mass spectrometry. Adv Sci Lett. (2008) 1:192–8. doi: 10.1166/asl.2008.020
36. Dentovskaya SV, Anisimov AP, Kondakova AN, Lindner B, Bystrova OV, Svetoch TE, et al. Functional characterization and biological significance of Yersinia pestis lipopolysaccharide biosynthesis genes. Biochemistry. (2011) 76:808–22. doi: 10.1134/s0006297911070121
37. Du Y, Rosqvist R, Forsberg A. Role of fraction 1 antigen of Yersinia pestis in inhibition of phagocytosis. Infect Immun. (2002) 70:1453–60. doi: 10.1128/IAI.70.3.1453-1460.2002
38. Weeks S, Hill J, Friedlander A, Welkos S. Anti-V antigen antibody protects macrophages from Yersinia pestis -induced cell death and promotes phagocytosis. Microb Pathog. (2002) 32:227–37. doi: 10.1006/mpat.2002.0498
39. Robertson BD, Frosch M, Van Putten JPM. The role of galE in the biosynthesis and function of gonococcal lipopolysaccharide. Mol Microbiol. (1993) 8:891–901. doi: 10.1111/j.1365-2958.1993.tb01635.x
40. Mattsby-Baltzer I, Ahlstrom B, Edebo L, De Man P. Susceptibility of lipopolysaccharide-responsive and -hyporesponsive ItyS Mice to infection with rough mutants of Salmonella typhimurium. Infect Immun. (1996) 64:1321–7.
41. Kang YS, Yamazaki S, Iyoda T, Pack M, Bruening SA, Kim JY, et al. SIGN-R1, a novel C-type lectin expressed by marginal zone macrophages in spleen, mediates uptake of the polysaccharide dextran. Int Immunol. (2003) 15:177–86. doi: 10.1093/intimm/dxg019
42. Takahara K, Yashima Y, Omatsu Y, Yoshida H, Kimura Y, Kang YS, et al. Functional comparison of the mouse DC-SIGN, SIGNR1, SIGNR3 and Langerin, C-type lectins. Int Immunol. (2004) 16:819–29. doi: 10.1093/intimm/dxh084
43. Chen T, Belland RJ, Wilson J, Swanson J. Adherence of pilus- Opa+ gonococci to epithelial cells in vitro involves heparan sulfate. J Exp Med. (1995) 182:511–7. doi: 10.1084/jem.182.2.511
44. Isberg RR, Voorhis DL, Falkow S. Identification of invasin: a protein that allows enteric bacteria to penetrate cultured mammalian cells. Cell. (1987) 50:769–78. doi: 10.1016/0092-8674(87)90335-7
45. Isberg RR, Leong JM. Cultured mammalian cells attach to the invasin protein of Yersinia pseudotuberculosis. Proc Natl Acad Sci USA. (1988) 85:6682–6. doi: 10.1073/pnas.85.18.6682
46. Kiljunen S, Datta N, Dentovskaya SV, Anisimov AP, Knirel YA, Bengoechea JA, et al. Identification of the lipopolysaccharide core of Yersinia pestis and Yersinia pseudotuberculosis as the receptor for bacteriophage phiA1122. J Bacteriol. (2011) 193:4963–72. doi: 10.1128/JB.00339-11
47. Fetherston JD, Schuetze P, Perry RD. Loss of the pigmentation phenotype in Yersinia pestis is due to the spontaneous deletion of 102 kb of chromosomal DNA which is flanked by a repetitive element. Mol Microbiol. (1992) 6:2693–704. doi: 10.1111/j.1365-2958.1992.tb01446.x
48. Plano GV. Role of Ail family proteins in serum resistance of Yersinia pestis. In Proceedings of the 9th International Symposium on Yersinia. Lexington, KY; Washington, DC: American Society for Microbiology (2006). p. 18.
49. Song Y, Tong Z, Wang J, Wang L, Guo Z, Han Y, et al. Complete genome sequence of Yersinia pestis strain 91001, an isolate avirulent to humans. DNA Res. (2004) 11:179–97. doi: 10.1093/dnares/11.3.179
50. Klena JD, Ashford RSII, Schnaitman CA. Role of Escherichia coli K-12 rfa genes and the rfp gene of Shigella dysenteriae 1 in generation of lipopolysaccharide core heterogeneity and attachment of O antigen. J Bacteriol. (1992) 174:7297–307. doi: 10.1128/jb.174.22.7297-7307.1992
51. Klena JD, Schnaitman CA. Function of the rfb gene cluster and the rfe gene in the synthesis of O antigen by Shigella dysenteriae 1. Mol Microbiol. (1993) 9:393–402. doi: 10.1111/j.1365-2958.1993.tb01700.x
52. Steeghs L, Uronen-Hansson U, Van Vliet S, Van Mourik A, Klein N, Van Kooyk Y, et al. Lipopolysaccharide-mediated targeting of Neisseria meningitidis to dendritic cells: binding of lgtB LPS to DC-SIGN. Cell Microbiol. (2006) 8:316–25. doi: 10.1111/j.1462-5822.2005.00623.x
53. Garcia-Vallejo JJ, Van Kooyk Y. A new cellular target for Yersinia pestis. Immunol Cell Biol. (2015) 93:769–70. doi: 10.1038/icb.2015.60
54. Cowan C, Philipovskiy AV, Wulff-Strobel CR, Ye Z, Straley SC. Anti-LcrV antibody inhibits delivery of Yops by Yersinia pestis KIM5 by directly promoting phagocytosis. Infect Immun. (2005) 73:6127–37. doi: 10.1128/IAI.73.9.6127-6137.2005
55. Grabenstein JP, Fukuto HS, Palmer LE, Bliska JB. Characterization of phagosome trafficking and identification of PhoP-regulated genes important for survival of Yersinia pestis in macrophages. Infect Immun. (2006) 74:3727–41. doi: 10.1128/IAI.00255-06
56. Bulgheresi S, Schabussova I, Chen T, Mullin NP, Maizels RM, Ott JA. A new C-type lectin similar to the human immunoreceptor DC-SIGN mediates symbiont acquisition by a marine nematode. Appl Environ Microbiol. (2006) 72:2950–6. doi: 10.1128/AEM.72.4.2950-2956.2006
57. Valenti P, Antonini G. Lactoferrin: an important host defence against microbial and viral attack. Cell Mol Life Sci. (2005) 62:2576–87. doi: 10.1007/s00018-005-5372-0
58. Marketon MM, Depaolo RW, Debord KL, Jabri B, Schneewind O. Plague bacteria target immune cells during infection. Science. (2005) 309:1739–41. doi: 10.1126/science.1114580
59. Zhang Y, Bliska JB. Role of macrophage apoptosis in the pathogenesis of Yersinia. Curr Top Microbiol Immunol. (2005) 289:151–73. doi: 10.1007/3-540-27320-4_7
60. Al-Hendy A, Toivanen P, Skurnik M. Expression cloning of Yersinia enterocolitica O:3 rfb gene cluster in Escherichia coli K12. Microb Pathog. (1991) 10:47–59. doi: 10.1016/0882-4010(91)90065-I
61. Oyston PC, Prior JL, Kiljunen S, Skurnik M, Hill J, Titball RW. Expression of heterologous O-antigen in Yersinia pestis KIM does not affect virulence by the intravenous route. J Med Microbiol. (2003) 52:289–94. doi: 10.1099/jmm.0.05044-0
62. Zhou D, Tong Z, Song Y, Han Y, Pei D, Pang X, et al. Genetics of metabolic variations between Yersinia pestis biovars and the proposal of a new biovar, microtus. J Bacteriol. (2004) 186:5147–52. doi: 10.1128/JB.186.15.5147-5152.2004
63. Hummel S, Schmidt D, Kremeyer B, Herrmann B, Oppermann M. Detection of the CCR5-Δ32 HIV resistance gene in Bronze Age skeletons. Genes Immun. (2005) 6:371–4. doi: 10.1038/sj.gene.6364172
64. Elvin SJ, Williamson ED, Scott JC, Smith JN, Perez De Lema G, Chilla S, et al. Evolutionary genetics: ambiguous role of CCR5 in Y. pestis infection. Nature. (2004) 430:417. doi: 10.1038/nature02822
65. Mecsas J, Franklin G, Kuziel WA, Brubaker RR, Falkow S, Mosier DE. Evolutionary genetics: CCR5 mutation and plague protection. Nature. (2004) 427:606. doi: 10.1038/427606a
66. Hemmila E, Turbide C, Olson M, Jothy S, Holmes KV, Beauchemin N. Ceacam1a-/- mice are completely resistant to infection by murine coronavirus mouse hepatitis virus A59. J Virol. (2004) 78:10156–65. doi: 10.1128/JVI.78.18.10156-10165.2004
67. Tan K, Zelus BD, Meijers R, Liu JH, Bergelson JM, Duke N, et al. Crystal structure of murine sCEACAM1a[1,4]: a coronavirus receptor in the CEA family. Embo J. (2002) 21:2076–86. doi: 10.1093/emboj/21.9.2076
68. Lanoue A, Clatworthy MR, Smith P, Green S, Townsend MJ, Jolin HE, et al. SIGN-R1 contributes to protection against lethal pneumococcal infection in mice. J Exp Med. (2004) 200:1383–93. doi: 10.1084/jem.20040795
69. Kang YS, Do Y, Lee HK, Park SH, Cheong C, Lynch RM, et al. A dominant complement fixation pathway for pneumococcal polysaccharides initiated by SIGN-R1 interacting with C1q. Cell. (2006) 125:47–58. doi: 10.1016/j.cell.2006.01.046
70. Thomas R, Brooks T. Common oligosaccharide moieties inhibit the adherence of typical and atypical respiratory pathogens. J Med Microbiol. (2004) 53:833–40. doi: 10.1099/jmm.0.45643-0
71. Thomas R, Brooks T. Attachment of Yersinia pestis to human respiratory cell lines is inhibited by certain oligosaccharides. J Med Microbiol. (2006) 55:309–15. doi: 10.1099/jmm.0.46102-0
72. Kang YS, Kim JY, Bruening SA, Pack M, Charalambous A, Pritsker A, et al. The C-type lectin SIGN-R1 mediates uptake of the capsular polysaccharide of Streptococcus pneumoniae in the marginal zone of mouse spleen. Proc Natl Acad Sci USA. (2004) 101:215–20. doi: 10.1073/pnas.0307124101
73. Groot F, Geijtenbeek TB, Sanders RW, Baldwin CE, Sanchez-Hernandez M, Floris R, et al. Lactoferrin prevents dendritic cell-mediated human immunodeficiency virus type 1 transmission by blocking the DC-SIGN–gp120 interaction. J Virol. (2005) 79:3009–15. doi: 10.1128/JVI.79.5.3009-3015.2005
74. Naarding MA, Ludwig IS, Groot F, Berkhout B, Geijtenbeek TB, Pollakis G, et al. Lewis X component in human milk binds DC-SIGN and inhibits HIV-1 transfer to CD4+ T lymphocytes. J Clin Invest. (2005) 115:3256–64. doi: 10.1172/JCI25105
75. Schnaitman CA, Klena JD. Genetics of lipopolysaccharide biosynthesis in enteric bacteria. Microbiol Rev. (1993) 57:655–82.
76. Isberg R, Leong J. Multiple beta 1 chain integrins are receptors for invasin, a protein that promotes bacterial penetration into mammalian cells. Cell. (1990) 60:861–71. doi: 10.1016/0092-8674(90)90099-Z
77. Riley G, Toma S. Detection of pathogenic Yersinia enterocolitica by using congo red-magnesium oxalate agar medium. J Clin Microbiol. (1989) 27:213–4.
78. Darby C, Ananth SL, Tan L, Hinnebusch BJ. Identification of gmhA, a Yersinia pestis gene required for flea blockage, by using a Caenorhabditis elegans biofilm system. Infect Immun. (2005) 73:7236–42. doi: 10.1128/IAI.73.11.7236-7242.2005
79. SolFoulon N, Moris A, Nobile C, Boccaccio C, Engering A, Abastado JP, et al. HIV-1 Nef-induced upregulation of DC-SIGN in dendritic cells promotes lymphocyte clustering and viral spread. Immunity. (2002) 16:145–55. doi: 10.1016/S1074-7613(02)00260-1
80. Nobile C, Moris A, Porrot F, Sol-Foulon N, Schwartz O. Inhibition of human immunodeficiency virus type 1 Env-mediated fusion by DC-SIGN. J Virol. (2003) 77:5313–23. doi: 10.1128/JVI.77.9.5313-5323.2003
81. Chen T, Grunert F, Medina-Marino A, Gotschlich E. Several carcinoembryonic antigens (CD66) serve as receptors for gonococcal opacity proteins. J Exp Med. (1997) 185:1557–64. doi: 10.1084/jem.185.9.1557
82. Chen T, Bolland S, Chen I, Parker J, Pantelic M, Grunert F, et al. The CGM1a (CEACAM3/CD66d) mediated phagocytic pathway of Neisseria gonorrhoeae expressing Opacity (Opa) proteins Is also the pathway to cell death. J Biol Chem. (2001) 276:17413–9. doi: 10.1074/jbc.M010609200
Keywords: Yersinia pestis, SIGNR1 (CD209b), macrophages, dendritic cells (DCs), antigen presenting cells (APCs), core lipopolysaccharide/lipooligosaccharides (core LPS/LOS), bacterial dissemination, host-pathogen interactions
Citation: Yang K, He Y, Park CG, Kang YS, Zhang P, Han Y, Cui Y, Bulgheresi S, Anisimov AP, Dentovskaya SV, Ying X, Jiang L, Ding H, Njiri OA, Zhang S, Zheng G, Xia L, Kan B, Wang X, Jing H, Yan M, Li W, Wang Y, Xiamu X, Chen G, Ma D, Bartra SS, Plano GV, Klena JD, Yang R, Skurnik M and Chen T (2019) Yersinia pestis Interacts With SIGNR1 (CD209b) for Promoting Host Dissemination and Infection. Front. Immunol. 10:96. doi: 10.3389/fimmu.2019.00096
Received: 01 July 2018; Accepted: 14 January 2019;
Published: 12 March 2019.
Edited by:
Amy Rasley, Lawrence Livermore National Laboratory, United StatesReviewed by:
Teunis Geijtenbeek, University of Amsterdam, NetherlandsCopyright © 2019 Yang, He, Park, Kang, Zhang, Han, Cui, Bulgheresi, Anisimov, Dentovskaya, Ying, Jiang, Ding, Njiri, Zhang, Zheng, Xia, Kan, Wang, Jing, Yan, Li, Wang, Xiamu, Chen, Ma, Bartra, Plano, Klena, Yang, Skurnik and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrey P. Anisimov, YW5pc2ltb3ZAb2JvbGVuc2sub3Jn; YS1wLWFuaXNpbW92QHlhbmRleC5ydQ==
Ruifu Yang, MTM4MDEwMzQ1NjBAMTYzLmNvbQ==
Tie Chen, Y2hlbnRpZUBodXN0LmVkdS5jbg==; dGllY2hlbjIwMDVAeWFob28uY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.