
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 29 January 2019
Sec. Primary Immunodeficiencies
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00073
 Tao Li1†
Tao Li1† Xian Zhou1†Yun Ling2†Ning Jiang3Jingwen Ai1
Xian Zhou1†Yun Ling2†Ning Jiang3Jingwen Ai1 Jing Wu1
Jing Wu1 Jiazhen Chen1Li Chen4Xiaowen Qian5Xuhui Liu2Xiuhong Xi2Lu Xia2
Jiazhen Chen1Li Chen4Xiaowen Qian5Xuhui Liu2Xiuhong Xi2Lu Xia2 Xiaoyong Fan2Shuihua Lu2*
Xiaoyong Fan2Shuihua Lu2* Wen-Hong Zhang1*
Wen-Hong Zhang1*Background: Disseminated Bacillus Calmette-Guérin disease (D-BCG) in children with chronic granulomatous disease (CGD) can be fatal, while its clinical characteristics remain unclear because both diseases are extremely rare. The patients with CGD receive BCG vaccination, because BCG vaccination is usually performed within 24 h after delivery in China.
Methods: We prospectively followed-up Chinese patients with CGD who developed D-BCG to characterize their clinical and genetic characteristics. The diagnoses were based on the patients' clinical, genetic, and microbiological characteristics.
Results: Between September 2009 and September 2016, we identified 23 patients with CGD who developed D-BCG. Their overall 10-year survival rate was 34%. We created a simple dissemination score to evaluate the number of infected organ systems and the survival probabilities after 8 years were 62 and 17% among patients with simple dissemination scores of ≤3 and >3, respectively (p = 0.0424). Survival was not significantly associated with the CGD stimulation index or interferon-γ treatment. Eight patients underwent umbilical cord blood transplantation and 5 of them were successfully treated. The genetic analyses found mutations in CYBB (19 patients), CYBA (1 patient), NCF1 (1 patient), and NCF2 (1 patient). We identified 6 novel highly likely pathogenic mutations, including 4 mutations in CYBB and 2 mutations in NCF1.
Conclusions: D-BCG is a deadly complication of CGD. The extent of BCG spreading is strongly associated with clinical outcomes, and hematopoietic stem cell transplantation may be a therapeutic option for this condition.
Bacille Calmette-Guerin (BCG), an attenuated strain of Mycobacterium bovis, was originally isolated from a cow with tuberculosis. The BCG vaccine was first used to immunize humans in 1921 (1). With protective effect against life-threatening tuberculosis(TB) such as tuberculosis meningitis and disseminated TB in children, BCG vaccination is recommended in countries with a high TB burden (2). It is one of the most widely used of all current vaccines and over 150 countries have universal BCG vaccination program (3). Although more than 14 substrains of BCG have been used as BCG vaccine strains in different parts of the world differ in terms of their genetic and phenotypic properties, commonly administered BCG vaccine strains induce comparable protective immunity against TB (1).
Disseminated Bacillus Calmette-Guérin (D-BCG) disease is the most fatal adverse event after BCG vaccination, and is associated with a mortality rate of 60–80% (4–6). Early studies have indicated that acquired or inherited immunodeficiency can predispose the patient to infection with an attenuated strain of M. bovis BCG (7). Among them, chronic granulomatous disease (CGD) is one of the representative (8). Patients with CGD suffer from a variety of recurrent bacterial and fungal infections (9). It is caused by an inherited defect in the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzyme complex that consists of two membrane-spanning subunits, gp91phox and p22phox, as well as three cytosolic components p47phox, p67phox, and p40phox (10). Mutations within the X-linked gp91 phox gene (CYBB) cause the X-linked form of CGD, while the autosomal recessive forms of CGD are due to mutations in in CYBA, NCF1, NCF2, or NCF4 which encode for p22 phox, p47 phox, p67 phox, and p40 phox, respectively (10).
BCG vaccination is contraindicated for persons with congenital cell-mediated or severe combined immunodeficiency, immunodeficiency syndromes (e.g., HIV/AIDS, known or suspected congenital immunodeficiency, leukemia, lymphoma, or other malignant disease) (2). Because patients with CGD have impaired reactive oxygen species [ROS]-producing phagocyte NADPH oxidase, which may weaken their anti-mycobacterial defense (8), CGD is one of the contraindications of BCG vaccination. However, as BCG is usually vaccinated very early in infancy, there is not enough time to screen for CGD and other vaccination contraindications. Many reports have described minor BCG complications, such as regional lymphadenitis, in CGD cases (11–16), D-BCG associated with CGD is extremely rare (14, 17), as the incidences of D-BCG and CGD are ~2 cases per 1 million vaccinations (18) and 5 cases per 1 million live births (19), respectively. This manuscript describes the genetic and clinical characteristics of 23 Chinese patients with D-BCG associated with CGD.
CGD was diagnosed based on the patient's clinical presentation and confirmed using either the dihydrorhodamine 123 (DHR) oxidation assay or genetic analysis. DHR assay is tested using FACScam(Becton Dickinson, CA, USA) with methods established by Yu et al. (20). We detected CYBB mutation by PCR using synthetic oligonucleotide primers designed to amplify the CYBB gene. For patients with CYBB mutation negative, we performed whole exon sequencing. D-BCG was classified according to the criteria in Table 1. A simple dissemination site-based score (1 point per involved organ system) was used to evaluate the degree of dissemination to the lungs, bones, liver, spleen, kidney, heart or pericardium, gastrointestinal tract, peritoneum, nose, ears, skin, and soft tissues. The scoring was performed by attending physicians based on the results of the clinical examination, imaging, and laboratory testing.

Table 1. The diagnostic criteria and classifications for Bacillus Calmette-Guérin (BCG) disease.
We conducted a prospective observational study to follow up patients who had clinical symptoms of D-BCG and were hospitalized at the Shanghai Public Health Clinical Center of Fudan University from September 1, 2009, to September 1, 2016. All patients and/or their guardians provided written informed consent to be followed-up. This study's observational protocol was approved by the ethics committee of the hospital. The patients were diagnosed and categorized based on the criteria that are listed in Table 1, and their clinical, radiological, pathological, and microbiological characteristics.
All statistical analyses were performed using GraphPad Prism (version 6.0) and IBM SPSS software (version 22.0). Categorical and continuous variables were compared using Fisher's exact test and the Mann–Whitney U-test, respectively. Kaplan–Meier survival curves were compared between the groups using the log-rank test. All tests were two-sided, and differences were considered statistically significant at p < 0.05.
Between September 2009 and September 2016, 78 patients were diagnosed with D-BCG at our center. Among them, 23 patients were diagnosed with CGD (Table 2). The diagnosis of CGD was based on DHR testing in 1 patient, genetic analysis in 6 patients, and a combination of DHR testing and genetic analysis in 16 patients. The diagnoses of D-BCG in CGD patients were definite in 6 patients, highly probable in 9 patients, and probable in 8 patients. All 23 patients were male, and their median age at referral to our hospital was 12.2 months (mean: 25.7 months, range: 2.9–123.7 months). The children were from 23 different families that resided in 10 Chinese provinces (mostly in the southeast region). All patients had undergone BCG vaccination with strain D2PB302 at the left deltoid region within 24 h after birth, based on the national vaccination policy and protocol.
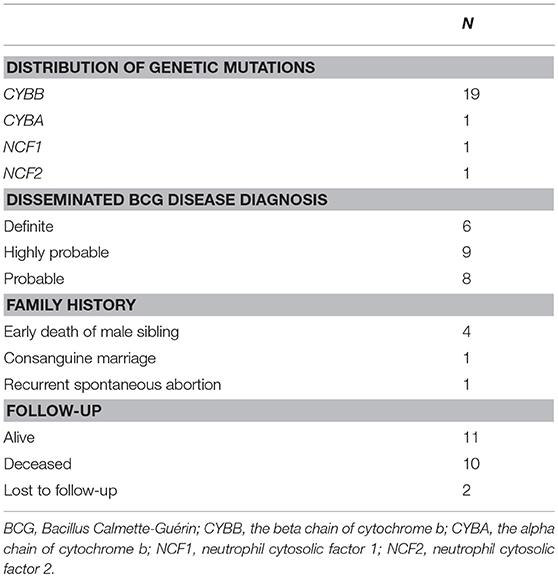
Table 2. Patient characteristics.
The median age at the onset of BCG disease was 3.0 months (mean: 7.6 months, range: 0.3–58.5 months) (Figure 1A). Nineteen of the 23 children (82.6%) had symptoms of BCG disease before the age of 1 year. The median age at the diagnosis of D-BCG was 12.4 months (mean: 24.7 months, range: 2.9–123.1 months) (Figure 1B). The median lag between the onset and diagnosis of BCG disease was 5.4 months (mean: 17.2 months, range: 0.6–121.1 months). Seventeen of the 23 children (73.9%) developed symptoms of BCG disease as the initial symptom of CGD. The median age at the diagnosis of CGD was 12.6 months (mean: 23.9 months, range: 1.3–121.1 months), and 13 of the 23 patients had been diagnosed with D-BCG before developing CGD.
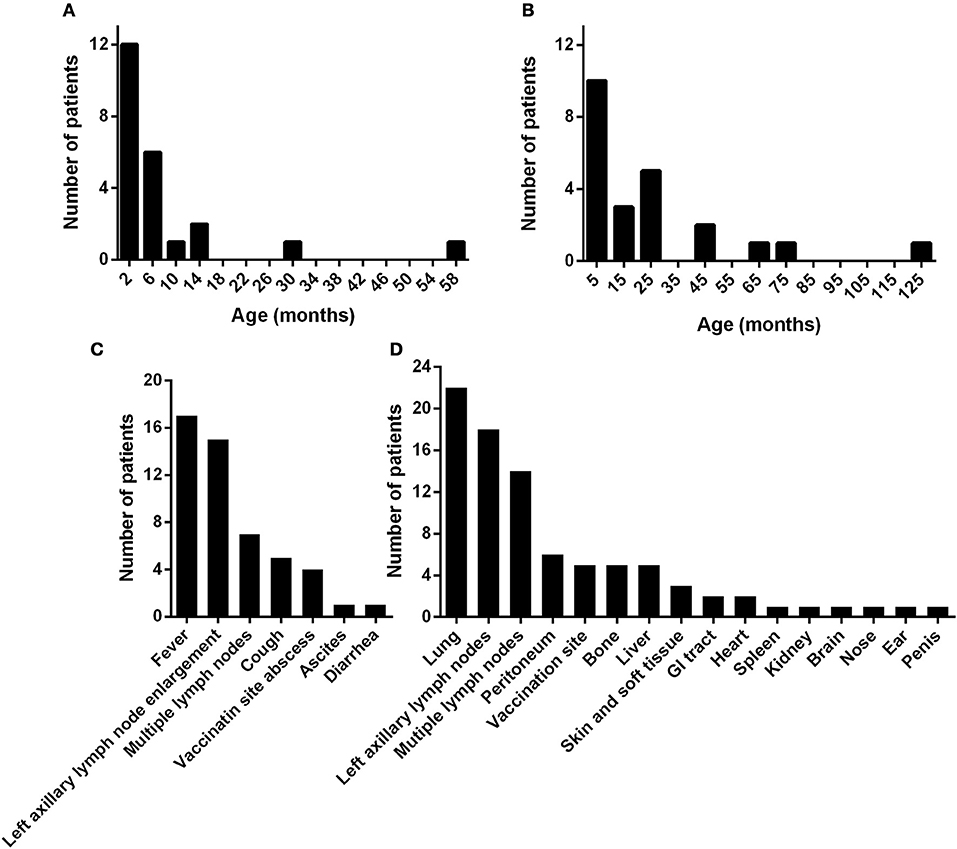
Figure 1. The ages at symptom onset (A) and at the diagnosis of disseminated Bacillus Calmette-Guérin disease (B) in 23 patients with CGD. The (C) initial symptoms, and (D) dissemination sites of Bacillus Calmette-Guérin disease. GI, gastrointestinal.
The local adverse reactions to the BCG vaccine included left axillary lymph node enlargement (n = 15, 65%) and an abscess at the vaccination site (n = 4, 17%). The initial symptoms of D-BCG were fever (n = 17, 74%), multiple lymph node enlargement (n = 7, 30%), cough (n = 5, 22%), ascites (n = 1, 4%), and diarrhea (n = 1, 4%) (Figure 1C). As the infection deteriorated, various organs developed signs of BCG dissemination (Figure 1D). Most patients (n = 22, 96%) had pulmonary involvement, 14 patients (61%) had multiple systematic lymph node enlargement, and abdominal involvement was common (ascites: 6 patients, liver involvement: 5 patients, gastrointestinal tract involvement: 2 patients, splenomegaly: 1 patient, and hydronephrosis: 1 patient). Five patients (22%) had bone lesions, 3 patients had BCG dissemination to the skin or soft tissues beyond the vaccination site, and 2 patients had BCG dissemination to the pericardium. We also observed dissemination to the central nervous system, nose, ears, and penis (1 patient each). The average simple dissemination score was 2.8 affected organ systems (range: 1–6 organ systems).
At the last follow-up (April 21, 2018), 10 patients (44%) had died from complications of D-BCG at a median age of 21.3 months (mean: 35.2 months, range: 6.2–102.0 months), 11 patients (48%) were alive, and 2 patients (9%) had been lost to follow-up. The overall 10-year survival rate was 34% (Figure S1A). The survival probabilities after 8 years were 62% among patients with a dissemination score of ≤3 and 17% among patients with a dissemination score of >3 (p = 0.0424) (Figure 2A).
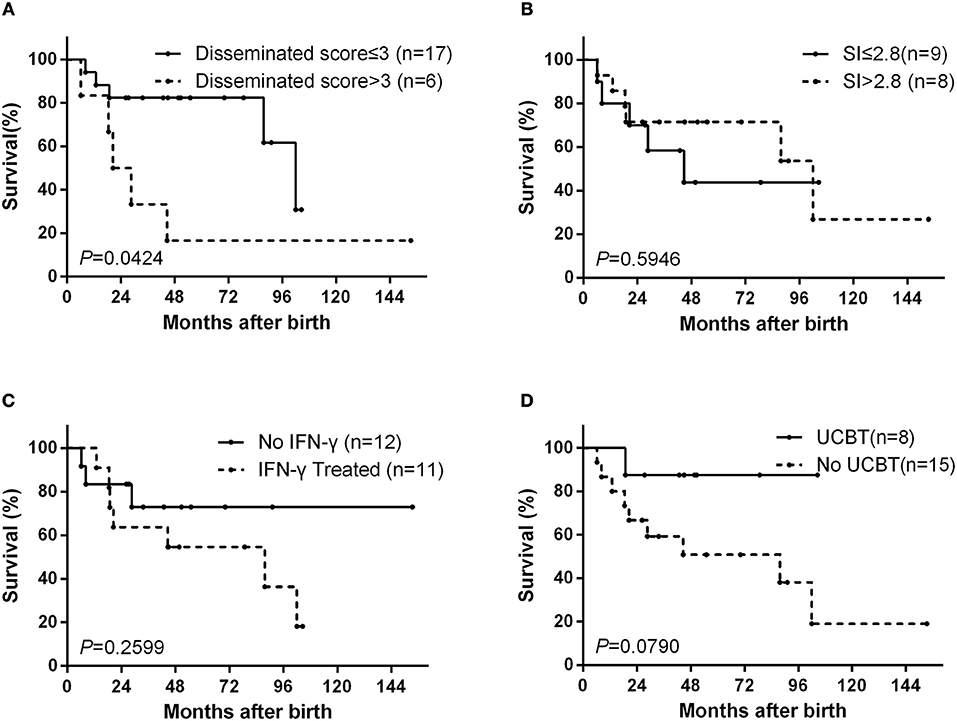
Figure 2. Kaplan–Meier survival curves from the date ot birth. The curves are shown for (A) according to dissemination score, (B) according to the dihydrorhodamine assay stimulation index (SI), (C) according to interferon (IFN)-γ treatment status, and (D) according to umbilical cord blood transplantation (UCBT) status.
Among the 17 patients that underwent DHR testing, the median stimulation index (SI) was 2.79 (mean: 5.31, range: 1.5–18.4). There was no significant difference in the SI values when we compared deceased and surviving patients (Mann–Whitney test, p = 0.9336). The survival probabilities after 8 years were 44% for patients with an SI of ≤2.8 and 54% for patients with an SI of >2.8 (p = 0.5946) (Figure 2B).
In addition to the BCG infection, 14 patients (61%) had other infections that were caused by a virus, fungus, or bacterium. Six patients had ≥2 infections, and 7 patients had disseminated infection caused by chronic active Epstein-Barr virus infection (2 patients), blood culture-confirmed bacterial infections (4 patients), and autopsy-confirmed tuberculosis infection (1 patient). The most frequently isolated organisms were Klebsiella pneumonia and Aspergillus spp., and the lungs were the most frequently involved organs.
After the diagnosis, all patients received anti-BCG treatment, which was generally based on a regimen of isoniazid, rifampin, and ethambutol. Some patients received alternative treatment because of isoniazid resistance (1 patient), adverse reactions (5 patients), or clinical experience. As the subtle changes in the anti-BCG regimen were too complicated to analyze throughout the long-term treatment, we have listed the regimens in Table 3.
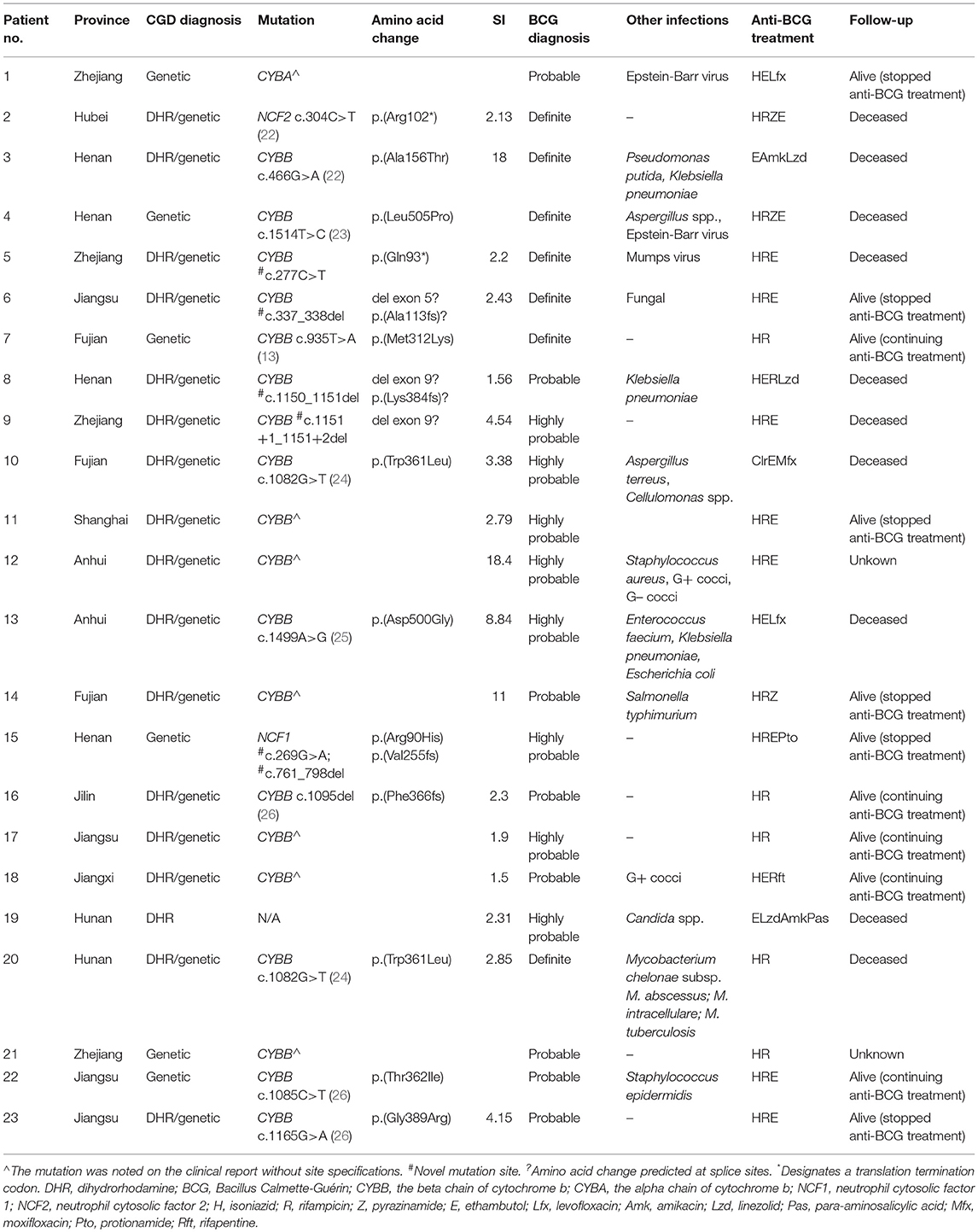
Table 3. The cohort's clinical and genetic characteristics.
Interferon (IFN)-γ was added to the treatment protocol for 11 patients. The use of IFN-γ was not associated with the dissemination score (Fisher's exact test, p = 1.0000), and the survival probabilities after 8 years were not significantly different between patients who did and did not receive IFN-γ treatment (36 vs. 73%, p = 0.2599) (Figure 2C). Eight patients underwent umbilical cord blood transplantation (UCBT), with a success rate of 63% (5 cases). 3 of 5 children who underwent successful UCBT have stopped the anti-BCG treatment. One child died at 4 months after failed UCBT. The other 2 child failed UCBT is continuing the anti-BCG treatment and waiting for the second one. The survival probabilities after 8 years were 88% among patients with UCBT and 38% among patients without UCBT (p = 0.0790) (Figure 2D).
Nineteen patients (82.6%) had X–linked recessive mutations in CYBB gene. One child's mother had a history of recurrent spontaneous abortion and 4 children had deceased brothers who died before the age of 1 year. Additionally, three children were identified with compound heterozygous or hemizygous mutations in other autosomal genes, including CYBA (1 patient), NCF1 (1 patient), and NCF2 (1 patient). A homozygous non-sense mutation (NM_000433.3: c.304C>T) was found in NCF2 of one male, whose parents were consanguineous and carried the heterozygous mutation in this locus. Furthermore, we identified 6 novel highly likely pathogenic mutations, including 4 mutations in CYBB and 2 mutations in NCF1 (Figure 3 and Table 3).
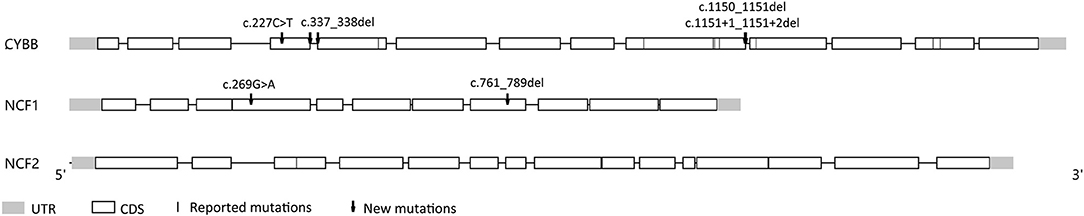
Figure 3. A schematic diagram of the mutations in our cohort.
As both diseases have very low incidence rates, the clinical characteristics of D-BCG with CGD remains unclear. The present study evaluated 23 Chinese cases of D-BCG with CGD, which is, to the best of our knowledge, the largest reported cohort of these patients. The development of antifungal and antibiotic treatments has greatly decreased the mortality rate in conventionally treated CGD cases, and most patients can survive until their thirties (19, 27). However, the prognosis of patients with D-BCG and CGD remains poor. The 10-year survival rate in our cohort was 34%, which is consistent with the findings of previous studies (4, 18). Compared to D-BCG patients with unknown genetic defects, D-BCG patients with CGD required longer anti-BCG treatment (Figure S1B).
In the present study, 5 of 8 patients were successfully treated using UCBT, and 3 of 5 successful UCBT patients have stopped anti-BCG treatment, which indicates that UCBT may be a curative treatment for patients with CGD. Furthermore, compared to conventionally treated CGD, a previous study revealed that hematopoietic stem cell transplantation (HSCT) was associated with lower rates of infection and hospitalization (28). Although our small sample size limited the power of this analysis, we noticed an improved survival rate in this subgroup, and suggest considering early HSCT for patients with D-BCG and CGD, given the deadly effects of BCG dissemination. Unfortunately, the precise timing and conditioning intensity remain unclear for using HSCT to treat primary immunodeficiency (29), and this problem would be further aggravated among patients with BCG infection, given the lack of useful data.
Unlike relatively mild BCG complications that are usually limited to the regional lymph nodes (14, 30), our patients with D-BCG exhibited characteristic BCG dissemination to distant organs. Thus, we created a simple dissemination score to evaluate the number of infected organ systems, and found that it was both easy to measure and may help predict the prognosis in cases of D-BCG with CGD, which is quite bad for patients with score more than 3. In the present study, the BCG dissemination most commonly involved the lungs and systemic lymph nodes, with 22 of 23 cases exhibiting lung involvement and 13 of 23 cases exhibiting concurrent lung and systemic lymph node involvement.
More than 700 different mutations responsible for CGD have been identified (31), and we found 6 new mutations, 4 in CYBB and 2 in NCF1. Although we did not perform functional experiment, the clinical presentation, compromised granulocyte function with evidence of DHR assay and non-synonymous mutations in CGD-related genes were convicing evidence for causative mutations. However, the correlation between the genotype and the clinical presention, especially the occurrence of D-BCG in CGD patients is not clear. For BCG-vaccinated CGD patients, 8–56% of them had BCG lymphadenitis (11, 13, 15, 16, 27), 0–10% had D-BCG (11–13). The present study did not reveal a significant association between D-BCG and the DHR assay's SI value (residual NADPH oxidase function). This result is different from the findings of two previous studies (15, 32), but is consistent with the findings of Köker et al. in Turkey (15). Similar to China, Turkey has a national policy of BCG vaccination, and Köker et al. concluded that the residual NADPH oxidase activity of neutrophils was associated with later and less severe clinical presentation, although it did not affect the occurrence of BCG. Early studies have indicated that CYBB mutations (33) can cause a selectively impaired respiratory burst in macrophages and are associated with mendelian susceptibility to mycobacterial disease. Thus, the residual NADPH oxidase function of macrophages could be a useful predictor of prognosis among patients with both BCG and CGD.
The efficacy of IFN-γ for treating CGD remains controversial (34–37). The most cited study supporting the use of IFN-γ therapy was a randomized double-blind placebo-controlled trial of 128 patients with CGD, and revealed that IFN-γ substantially reduced the frequency of serious infections (34). However, a prospective multicenter study in Italy (37) found that long-term use of IFN-γ did not change the rate of infections per patient-year. In those studies, the IFN-γ was administered prophylactically and the primary endpoints were serious infections, while we evaluated treatment during the active infection period and survival outcomes. Therefore, additional studies are needed to provide better evidence regarding the efficacy of IFN-γ treatment for CGD.
Our study is limited by its observational nature and the small sample size, which limited the power of our statistics analyses. Furthermore, the sample size is too small to perform multivariate survival analysis. However, both D-BCG and CGD are extremely rare diseases, and the 23 cases in our cohort provide valuable information regarding the genetic and clinical characteristics of patients with both D-BCG and CGD.
This study of 23 Chinese patients with D-BCG and CGD confirmed that D-BCG is a severe complication of CGD, and CGD patients is a subgroup of D-BCG patients with relatively poor prognosis. The extent of BCG spreading was strongly associated with clinical outcomes, although these outcomes were not associated with the residual NADPH oxidase function of neutrophils. Furthermore, IFN-γ therapy during active BCG infection was not associated with an improved survival rate. Based on our findings, we suggest that all patients with this fatal disease should be considered for early HSCT.
TL, XZ, YL, SL, and W-HZ: conception or design of the work; TL, XZ, XQ, LX, XL, and XX: data collection and patient care; TL, XZ, NJ, JA, JW, JC, and XF: data analysis and interpretation; TL and XZ: drafting the article. YL, JW, LC, and W-HZ: critical revision of the article; TL, XZ, YL, NJ, JA, JW, JC, LC, XQ, XL, XX, LX, XF, SL, and W-HZ: Final approval of the version to be published.
The present study was partly supported by Shanghai Public Health Clinical Center (SPHCC-KY-GW-2017-08), the Key Technologies Research and Development Program for Infectious Diseases of China (2017ZX10202301-003, 2017ZX10301301-004) and National Natural Science Foundation of China 81700014, 81373064).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.00073/full#supplementary-material
BCG, Bacille Calmette-Guerin; CGD, chronic granulomatous disease; D-BCG, Disseminated Bacillus Calmette-Guérin disease; DHR, dihydrorhodamine 123; HSCT, hematopoietic stem cell transplantation; IFN, interferon; NADPH, nicotinamide adenine dinucleotide phosphate; SI, stimulation index; TB, tuberculosis; UCBT, umbilical cord blood transplantation
1. World Health Organization and WHO Expert Committee on Biological Standardization. WHO Expert Committee on Biological Standardization: Sixty-Second Report. World Health Organization (2013).
2. World Health Organization. BCG vaccines: WHO position paper - February 2018. Wkly Epidemiol Rec. (2018) 93:73–96. doi: 10.1016/j.vaccine.2018.03.009
3. Zwerling A, Behr MA, Verma A, Brewer TF, Menzies D, Pai M. The BCG World Atlas: a database of global BCG vaccination policies and practices. PLoS Med. (2011) 8:e1001012. doi: 10.1371/journal.pmed.1001012
4. Lotte A, Wasz-Hockert O, Poisson N, Engbaek H, Landmann H, Quast U, et al. Second IUATLD study on complications induced by intradermal BCG-vaccination. Bull Int Union Against Tuberculosis Lung Dis. (1988) 63:47–59.
5. Sadeghi-Shanbestari M, Ansarin K, Maljaei SH, Rafeey M, Pezeshki Z. Immunologic aspects of patients with disseminated bacille Calmette-Guerin disease in north-west of Iran. Ital J Pediatr. (2009) 35:42 doi: 10.1186/1824-7288-35-42
6. Paiman SA, Siadati A, Mamishi S, Tabatabaie P, Khotaee G. Disseminated Mycobacterium bovis infection after BCG vaccination. Iran J Allergy Asthma Immunol. (2006) 5:133–7. Available online at: http://ijaai.tums.ac.ir/index.php/ijaai/article/view/146
7. Boisson-Dupuis S, Bustamante J, El-Baghdadi J, Camcioglu Y, Parvaneh N, El Azbaoui S, et al. Inherited and acquired immunodeficiencies underlying tuberculosis in childhood. Immunol Rev. (2015) 264:103–20. doi: 10.1111/imr.12272
8. Deffert C, Cachat J, Krause K. Phagocyte NADPH oxidase, chronic granulomatous disease and mycobacterial infections. Cell Microbiol. (2014) 16:1168–78. doi: 10.1111/cmi.12322
10. Goldblatt D. Recent advances in chronic granulomatous disease. J Infect. (2014) 69:S32–35. doi: 10.1016/j.jinf.2014.07.013
11. Baba LA, Ailal F, El Hafidi N, Hubeau M, Jabot-Hanin F, Benajiba N, et al. Chronic granulomatous disease in morocco: genetic, immunological, and clinical features of 12 patients from 10 Kindreds. J Clin Immunol. (2014) 34:452–8. doi: 10.1007/s10875-014-9997-3
12. Xu H, Tian W, Li S, Zhang L, Liu W, Zhao Y, et al. Clinical and molecular features of 38 children with chronic granulomatous disease in Mainland China. J Clin Immunol. (2014) 34:633–41. doi: 10.1007/s10875-014-0061-0
13. Lee PPW, Chan K, Jiang L, Chen T, Li C, Lee T, et al. Susceptibility to mycobacterial infections in children with x-linked chronic granulomatous disease. Pediatric Infect Dis J. (2008) 27:224–30. doi: 10.1097/INF.0b013e31815b494c
14. Conti F, Lugo-Reyes SO, Blancas GL, He J, Aksu G, Borges DOEJ, et al. Mycobacterial disease in patients with chronic granulomatous disease: a retrospective analysis of 71 cases. J Allergy Clin Immunol. (2016) 138:241–8. doi: 10.1016/j.jaci.2015.11.041
15. Köker MY, Camcioglu Y, van Leeuwen K, Kiliç S, Barlan I, Yilmaz M, et al. Clinical, functional, and genetic characterization of chronic granulomatous disease in 89 Turkish patients. J Allergy Clin Immun. (2013) 132:1156–63. doi: 10.1016/j.jaci.2013.05.039
16. Fattahi F, Badalzadeh M, Sedighipour L, Movahedi M, Fazlollahi MR, Mansouri SD, et al. Inheritance pattern and clinical aspects of 93 Iranian patients with chronic granulomatous disease. J Clin Immunol. (2011) 31:792–801. doi: 10.1007/s10875-011-9567-x
17. Bustamante J, Aksu G, Vogt G, de Beaucoudrey L, Genel F, Chapgier A, et al. BCG-osis and tuberculosis in a child with chronic granulomatous disease. J Allergy Clin Immun. (2007) 120:32–8. doi: 10.1016/j.jaci.2007.04.034
18. Lotte A, Wasz-Höckert O, Poisson N, Dumitrescu N, Verron M, Couvet E. BCG complications. Estimates of the risks among vaccinated subjects and statistical analysis of their main characteristics. Adv Tuberculosis Res. (1983) 21:107–93.
19. Winkelstein JA, Marino MC, Johnston RB Jr, Boyle J, Curnutte J, Gallin JI, et al. Chronic granulomatous disease: report on a national registry of 368 patients. Medicine (2000) 79:155–69. doi: 10.1097/00005792-200005000-00003
20. Yu Y, Zhu W, Wang X. Flow cytometry assay of function of neutrophil in adult and children. Fudan Uni J Med Sci. (2005) 1:101–4. doi: 10.3969/j.issn.1672-8467.2005.01.028
21. Parsons LM, Brosch R, Cole ST, Somoskövi Á, Loder A, Bretzel G, et al. Rapid and simple approach for identification of Mycobacterium tuberculosis complex isolates by PCR-based genomic deletion analysis. J Clin Microbiol. (2002) 40:2339–45. doi: 10.1128/jcm.40.7.2339-2345.2002
22. Bolscher BG, De Boer M, De Klein A, Weening RS, Roos D. Point mutations in the beta-subunit of cytochrome b558 leading to X-linked chronic granulomatous disease. Blood (1991) 77:2482–7.
23. Heyworth PG, Curnutte JT, Rae J, Noack D, Roos D, van Koppen E, et al. Hematologically Important Mutations: X-Linked Chronic Granulomatous Disease (Second Update). Blood Cells Mol Dis. (2001) 27:16–26. doi: 10.1006/bcmd.2000.0347
24. Sun J, Wang Y, Liu D, Yu Y, Wang J, Ying W, et al. Prenatal diagnosis of x-linked chronic granulomatous disease by percutaneous umbilical blood sampling. Scand J Immunol. (2012) 76:512–8. doi: 10.1111/j.1365-3083.2012.02772.x
25. Leusen JH, De Boer M, Bolscher BG, Hilarius PM, Weening RS, Ochs HD, et al. A point mutation in gp91-phox of cytochrome b558 of the human NADPH oxidase leading to defective translocation of the cytosolic proteins p47-phox and p67-phox. J Clin Invest. (1994) 93:2120. doi: 10.1172/JCI117207
26. Roos D, Kuhns DB, Maddalena A, Roesler J, Lopez JA, Ariga T, et al. Hematologically important mutations: X-linked chronic granulomatous disease (third update). Blood Cells Mol Dis. (2010) 45:246–65. doi: 10.1016/j.bcmd.2010.07.012
27. Van den Berg JM, Van Koppen E, Åhlin A, Belohradsky BH, Bernatowska E, Corbeel L, et al. Chronic granulomatous disease: the European experience. PLoS ONE (2009) 4:e5234. doi: 10.1371/journal.pone.0005234
28. Cole T, Pearce MS, Cant AJ, Cale CM, Goldblatt D, Gennery AR. Clinical outcome in children with chronic granulomatous disease managed conservatively or with hematopoietic stem cell transplantation. J Allergy Clin Immun. (2013) 132:1150–5. doi: 10.1016/j.jaci.2013.05.031
29. Güngör T, Teira P, Slatter M, Stussi G, Stepensky P, Moshous D, et al. Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet (2014) 383:436–48. doi: 10.1016/S0140-6736(13)62069-3
30. Casanova J, Jouanguy E, Lamhamedi S, Blanche S, Fischer A. Immunological conditions of children with BCG disseminated infection. Lancet (1995) 346:581. doi: 10.1016/S0140-6736(95)91421-8
31. Wolach B, Gavrieli R, de Boer M, van Leeuwen K, Berger Achituv S, Stauber T, et al. Chronic granulomatous disease: clinical, functional, molecular, and genetic studies. the Israeli experience with 84 patients. Am J Hematol. (2017) 92:28–36. doi: 10.1002/ajh.24573
32. Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. (2010) 363:2600–10. doi: 10.1056/NEJMoa1007097
33. Bustamante J, Arias AA, Vogt G, Picard C, Galicia LB, Prando C, et al. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol. (2011) 12:213–21. doi: 10.1038/ni.1992
34. Gallin JI, Malech HL, Curnutte W, Quie PG, Jaffe HS, Ezkowitz R. A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. N Engl J Med. (1991) 324:509–16. doi: 10.1056/NEJM199102213240801
35. Weening RS, Leitz GJ, Seger RA. Recombinant human interferon-gamma in patients with chronic granulomatous disease—European follow up study. Eur J Pediatr. (1995) 154:295–8.
36. Marciano BE, Wesley R, Ellen S, Anderson VL, Barnhart LA, Darnell D, et al. Long-term interferon-γ therapy for patients with chronic granulomatous disease. Clin Infect Dis. (2004) 39:692–9. doi: 10.1086/422993
Keywords: Bacillus Calmette-Guérin, disseminated BCG disease, BCGosis, vaccination, chronic granulomatous disease, complications
Citation: Li T, Zhou X, Ling Y, Jiang N, Ai J, Wu J, Chen J, Chen L, Qian X, Liu X, Xi X, Xia L, Fan X, Lu S and Zhang W-H (2019) Genetic and Clinical Profiles of Disseminated Bacillus Calmette-Guérin Disease and Chronic Granulomatous Disease in China. Front. Immunol. 10:73. doi: 10.3389/fimmu.2019.00073
Received: 16 October 2018; Accepted: 11 January 2019;
Published: 29 January 2019.
Edited by:
Guzide Aksu, Ege University, TurkeyReviewed by:
Ruben Martinez-Barricarte, Rockefeller University, United StatesCopyright © 2019 Li, Zhou, Ling, Jiang, Ai, Wu, Chen, Chen, Qian, Liu, Xi, Xia, Fan, Lu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuihua Lu, bHVzaHVpaHVhNjZAMTI2LmNvbQ==
Wen-Hong Zhang, emhhbmd3ZW5ob25nQGZ1ZGFuLmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.