 Kohei Sugihara
Kohei Sugihara Tina L. Morhardt1,2
Tina L. Morhardt1,2 Nobuhiko Kamada
Nobuhiko Kamada- 1Division of Gastroenterology, Department of Internal Medicine, University of Michigan, Ann Arbor, MI, United States
- 2Division of Pediatric Gastroenterology, Hepatology and Nutrition, Department of Internal Medicine, University of Michigan, Ann Arbor, MI, United States
Inflammatory bowel disease (IBD) is a chronic and relapsing inflammatory disease of the gastrointestinal tract. Although the precise etiology of IBD remains incompletely understood, accumulating evidence suggests that various environmental factors, including dietary nutrients, contribute to its pathogenesis. Dietary nutrients are known to have an impact on host physiology and diseases. The interactions between dietary nutrients and intestinal immunity are complex. Dietary nutrients directly regulate the immuno-modulatory function of gut-resident immune cells. Likewise, dietary nutrients shape the composition of the gut microbiota. Therefore, a well-balanced diet is crucial for good health. In contrast, the relationships among dietary nutrients, host immunity and/or the gut microbiota may be perturbed in the context of IBD. Genetic predispositions and gut dysbiosis may affect the utilization of dietary nutrients. Moreover, the metabolism of nutrients in host cells and the gut microbiota may be altered by intestinal inflammation, thereby increasing or decreasing the demand for certain nutrients necessary for the maintenance of immune and microbial homeostasis. Herein, we review the current knowledge of the role dietary nutrients play in the development and the treatment of IBD, focusing on the interplay among dietary nutrients, the gut microbiota and host immune cells. We also discuss alterations in the nutritional metabolism of the gut microbiota and host cells in IBD that can influence the outcome of nutritional intervention. A better understanding of the diet-host-microbiota interactions may lead to new therapeutic approaches for the treatment of IBD.
Introduction
Inflammatory bowel disease (IBD) is a chronic and relapsing inflammatory condition of the gastrointestinal tract. Crohn's disease (CD) and ulcerative colitis (UC) are the two principal types of IBD. The prevalence of IBD has been increasing worldwide with the highest incidence found in Western countries (1). Although the precise etiology of IBD remains unclear, interactions between genetic and environmental factors are associated with its pathogenesis (2, 3). Advances in next-generation gene sequencing technology have led to the identification of over 160 genetic polymorphisms associated with the risk for IBD (4). Since the number of IBD patients in developing countries has rapidly increased in the past several decades, in concert with industrialization and westernization of lifestyle, it is unlikely that susceptibility genes are the primary driver of rising rates of IBD (5). Therefore, it is likely that environmental exposure is the most significant risk factor in IBD.
Among environmental factors, accumulating evidence suggests that dietary nutrients contribute to the pathogenesis of IBD (6, 7). Specifically, diets rich in fat and protein, common in the western world and countries with similar lifestyles, have been identified as risk factors for the development of IBD. Hence, nutritional intervention, which aims to reduce the intake of potential nutritional hazards, is a treatment option in IBD that induces and extends disease remission (8). Moreover, some dietary nutrients can potentiate the host immune system and intestinal barrier function, which in turn protect the host from disease. Thus, providing beneficial nutrients, while limiting nutritional hazards, is a key strategy for successful dietary therapies designed for the treatment of IBD. In addition to affecting host immunity and intestinal barrier function, dietary nutrients have an impact on the composition and function of the gut microbiota. The altered gut microbiota can, in turn, influence host physiology and disease. Furthermore, the metabolism of host immune and non-immune cells, as well as that of the gut microbiota, are known to change during inflammation. Hence, the demand for certain nutrients by the host and/or the microbiota may be changed in IBD (9–11). A more thorough understating of the complex interplay among dietary nutrients, host immunity, and the gut microbiota is required to increase the effectiveness of dietary interventions used to treat IBD. Herein, we review the current knowledge of the role of dietary nutrients in the development and the treatment of IBD.
Dietary Amino Acids and IBD
Dietary amino acids act as key regulatory factors in cellular and microbial metabolic pathways. They also play important roles in gut homeostasis. Intestinal inflammation affects several metabolic pathways and disturbances in amino acid metabolism are observed in IBD patients. Amino acid metabolic profiles in the blood, urine, feces, and intestinal tissues are also altered in IBD patients and correlate with the severity of disease (Table 1). Additionally, metagenomic studies have revealed that amino acid biosynthesis genes are downregulated and amino acid transporter genes are upregulated in the gut microbiome of IBD patients, indicating that the gut microbiota lessens the production of amino acids and increases the rate of their utilization (11, 25). In addition to bacteria, host immune cells also utilize amino acids differently during inflammation. For instance, certain amino acids are critical for T cell effector function as well as the proliferation of macrophages (26, 27). Thus, the demand for certain amino acids by host cells and the gut microbiota may increase as a result of inflammation.
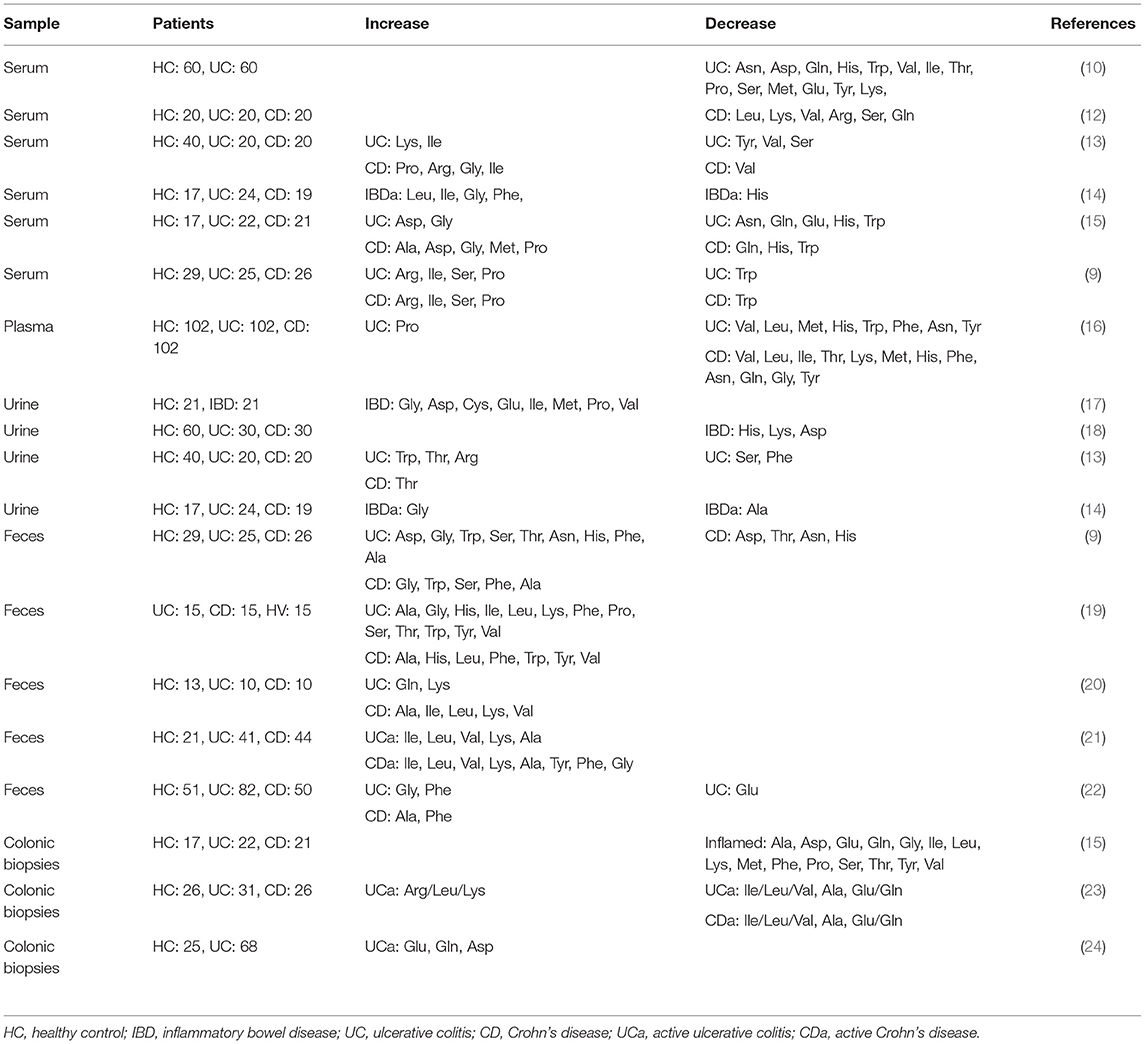
Table 1. Comparison of amino acid levels between healthy subjects and IBD patients.
Tryptophan
Tryptophan is an essential amino acid and a common constituent of protein-based foods, such as fish, meat, and cheese. It is utilized in the synthesis of nicotinamide derivatives, indole derivatives and serotonin (28). Tryptophan metabolites, such as kynurenine, indole-3-aldehyde, and indole-3-acetic acid, can act as ligands for the aryl hydrocarbon receptor (AhR), a critical regulator of immunity and inflammation involved in adaptive immunity and intestinal barrier function (Figure 1) (29, 30). In a murine model of colitis, mice fed a tryptophan-deficient diet showed exacerbation of colitis accompanied by increased weight loss and reduced levels of antimicrobial peptides (31). Conversely, dietary supplementation with tryptophan and tryptophan metabolites ameliorated intestinal inflammation in experimental colitis (31–35).
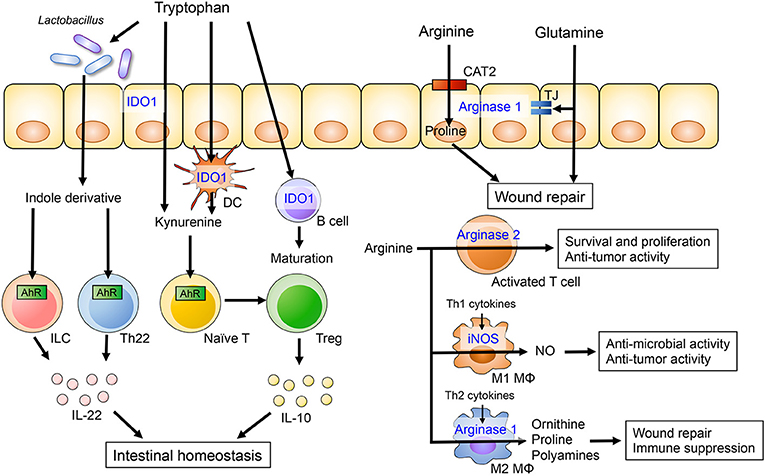
Figure 1. The role of dietary amino acids in intestinal homeostasis. Dietary tryptophan is metabolized to kynurenine or indole derivatives by host cells or the gut microbiota, respectively. Kynurenine promotes the differentiation of Treg and induces IL-10 production by Treg cells through AhR. Lactobacillus species are capable of catabolizing tryptophan into indole derivatives that are ligands for the AhR. Activation of AhR in gut-resident T cells and ILC enhances production of IL-22, which in turn potentiates mucosal barrier integrity. Arginine and glutamine metabolism in intestinal epithelial cells is associated with epithelial barrier and intestinal wound repair. Arginine also plays an important role of the immune system. Arginine is catabolized by iNOS in M1 macrophages and by arginase II in M2 macrophages. The arginine metabolisms regulate anti-microbial and -tumor activity and immune suppression in M1 and M2 macrophages, respectively.
Indoleamine 2,3 dioxygenase-1 (IDO1) is ubiquitously expressed in epithelial cells, dendritic cells and macrophages. IDO1 is the first step in the kynurenine pathway, a major route for tryptophan catabolism (28). IDO1 regulates the differentiation and maturation of adaptive immune cells (36). Kynurenine is an initial metabolite of IDO1-mediated tryptophan catabolism and the kynurenine/tryptophan ratio is a surrogate marker of IDO1 activity. A recent clinical study has shown that serum tryptophan levels are lower and the kynurenine/tryptophan ratio is elevated in IBD patients compared to healthy controls (37). Additionally, IDO1 mRNA expression in colonic tissues is significantly higher in IBD and correlates with disease severity, suggesting the kynurenine pathway is upregulated in IBD. The expression of IDO1 is also higher in murine models of experimental colitis and has been shown to regulate inflammatory response (38). Since kynurenine is known to have anti-inflammatory properties, elevated levels of IDO1 in IBD may be a counter reaction to inflammation. Indeed, in the 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis model, disease severity is exacerbated in IDO1 deficient mice or mice receiving an IDO inhibitor (38, 39). IDO1 also attenuates intestinal inflammation in other models of colitis, including graft vs. host disease (40) and the Citrobacter rodentium infection model (41). Kynurenine promotes the differentiation of regulatory T cell (Treg) and induces IL-10 production by Treg cells through AhR activation (30, 42). Likewise, kynurenine-mediated AhR activation increases expression of the IL-10 receptor in intestinal epithelial cells (35). IL-10 signaling regulates mucosal wound repair through WNT1-inducible signaling pathway protein 1, suggesting that kynurenine plays a role in mucosal wound repair (43). Additionally, kynurenine supplementation ameliorates body weight loss, intestinal permeability and histology in the model of dextran sodium sulfate (DSS)-induced colitis (35).
Peripheral immune activation can affect the systemic metabolism of tryptophan and emotional behavior. Activation of T cells in Pdcd1−/− mice, which lack the inhibitory programmed cell death protein 1 (PD-1), has been shown to affect the blood metabolic profile (44). Specifically, it results in an increase of amino acid uptake and intracellular accumulation of free amino acids, resulting in the reduction of amino acid levels (especially tryptophan) in the blood. Tryptophan is essential for the synthesis of the neurotransmitter serotonin, which regulates many aspects of animal behavior, including anxiety, aggression and fear (45). Brain serotonin levels are lower in Pdcd1−/− mice. Accordingly, these animals are prone to anxiety and exhibit enhanced fear responses. Collectively, this evidence suggests that activation of T cells causes a systemic metabolic shift, resulting in abnormal emotional behavior. Importantly, dietary supplementation of tryptophan ameliorates these behavioral abnormalities. Anxiety and depression are more common in patients with IBD and symptoms stemming from these conditions tend to be more severe during periods of active disease (46). Thus, tryptophan is an important nutrient that plays a role in the regulation of inflammation and maintenance of mental health.
In addition to tryptophan metabolism in the host, tryptophan metabolites generated by the gut microbiota also contribute to the regulation of mucosal homeostasis (47). A genome-wide association study has found that caspase recruitment domain-containing protein 9 (CARD9), an adaptor protein involved in apoptosis and antifungal immunity, is a susceptibility gene for IBD (4). CARD9 deficient mice produce reduced amounts of IL-22, a cytokine with an important role in maintaining mucosal immunity and integrity (48), and are more susceptible to colitis (49). Interestingly, the gut microbiota of CARD9 deficient mice lack certain bacteria, such as Lactobacillus reuteri and Allobaculum, that are capable of catabolizing tryptophan into indole derivatives (49). Hence, dysbiotic microbiotas in CARD9 deficient mice do not generate indole derivatives, which promote mucosal IL-22 production through AhR activation (49). Consistently, reduced levels of AhR ligands are also typically observed in the microbiotas found in IBD patients, particularly in those with the CARD9 risk alleles associated with IBD (49). Thus, gut dysbiosis in the context of IBD may lead to compromised tryptophan catabolism, which in turn influences IL-22-mediated mucosal protection.
Arginine
Arginine is a semi-essential amino acid and a substrate for 4 enzymes including arginases, nitric oxide synthases (NOS), arginine-glycine amidinotransferase and arginine decarboxylase (50). Arginases have two isoforms, arginase I and arginase II, both of which metabolize L-arginine to NO and citrulline. Arginase I is highly expressed in the liver, whereas arginase II is expressed in the brain, kidneys, mammary glands and intestine. nitric oxide synthases (NOS) catalyzes the synthesis of NO from arginine. There are three NOS isoenzymes: neuronal nitric oxide synthase (nNOS), inducible nitric oxide synthase (iNOS), and endothelial nitric oxide synthase (eNOS). Arginine uptake into cells is mainly mediated by the cationic amino acid transporter (CAT) family of proteins encoded by the solute carrier (SLC).
Alterations in arginine metabolism have been reported in animal models of colitis as well as IBD patients, and arginine supplementation has been shown to ameliorate experimental colitis (51–53). A prospective cohort study has demonstrated that colonic arginine levels are decreased in UC patients and they correlate with disease severity (51). Altered levels of arginine in the colonic tissue are associated with increased mRNA expression of the arginine metabolic enzymes arginase II and iNOS. Although previous animal and clinical studies have revealed contradictory findings regarding arginine transporter expression during inflammation, arginine transport is critical for the maintenance of gut homeostasis (51–53). Wounding of intestinal epithelial cells has been shown to alter the expression of genes related to arginine transport and metabolism in vitro (52). CAT2, unlike CAT1, mediates intestinal wound repair and cell migration (54). Therefore, a CAT2 deficiency renders mice susceptible to DSS-induced colitis (53). Arginine catabolism also promotes wound repair. L-arginine metabolites, including proline and ornithine, are associated with restitution of intestinal epithelial integrity. Hence, supplementation of arginine during the recovery phase of DSS-induced colitis alleviates intestinal inflammation and promotes the migration of colonic epithelial cells (52). Thus, arginine metabolism plays a crucial role in the resolution of inflammation and, therefore, arginine supplementation could be used to promote mucosal healing in IBD.
Arginine has also been suggested to be crucial for immune cell activation and function. Arginase II is up-regulated in activated T cell, resulting in enhanced CD4+ and CD8+ T cell survival and anti-tumor activity (55). Depletion of extracellular arginine has been found to impair aerobic glycolysis, T cell proliferation and cytokine production (56–58). Additionally, the deletion of argininosuccinate 1 (ASS1), an enzyme utilized in the de novo synthesis of arginine, blunts Th1 and Th17 cell polarization even in the presence of extracellular arginine (59). Similarly, arginine metabolism may play a role in the regulation of macrophage functions. It has been reported that macrophages can undergo polarization toward the M1 or M2 phenotype based on environmental polarization cues. Stimulation with LPS plus IFN-γ induces M1 polarization, whereas Th2 cytokines, such as IL-4, IL-10, and IL-13, induce M2 polarization (60, 61). Interestingly, these two types of macrophages catabolize arginine differently. M1 macrophages express iNOS, which converts arginine to NO and citrulline. Both of these arginine metabolites are involved in the elimination of intracellular pathogens and tumor cells (62). In contrast, M2 macrophages express arginase I, which regulates the synthesis of proline and polyamines from ornithine, and is critical for wound healing and immune suppression (63). While a number of studies have demonstrated the importance of arginine metabolism in immune cells, the role of arginine in intestinal immunity is poorly understood. Thus, more research is necessary to understand how arginine metabolism shapes intestinal immune response during inflammation.
Glutamine
Glutamine is considered a conditionally essential amino acid because its consumption is increased during conditions of catabolic stress, including trauma, sepsis and post-surgery recovery (64). Glutamine is the most important fuel for enterocytes and immune cells, and has beneficial effects on clinical outcomes (65). Activation of lymphocytes results in a metabolic reprogramming that switches energy metabolism from oxidative phosphorylation to aerobic glycolysis and glutaminolysis, the process by which cells convert glutamine into TCA cycle metabolites (66, 67). Thus, glutamine metabolism and the demand for glutamine during inflammation may be increased in certain cells. Indeed, glutamine levels in the colonic tissue of IBD patients in remission are decreased compared to control subjects. This difference is even more considerable during active disease (15, 68). In addition to the colonic tissue, blood glutamine levels are also diminished in IBD patients and experimental animal models of colitis (10, 69). Oral or rectal supplementation of glutamine attenuates intestinal inflammation, intestinal fibrosis, and colitis-associated colon tumorigenesis in various models of colitis (69–73). Glutamine treatment significantly improves histology results, reduces oxidative stress and cytokine production via downregulation of the NF-κB and STAT signaling pathways (72). Glutamine also regulates epithelial integrity. Glutamine has been implicated in the preservation of gut barrier function, maintenance of epithelial tight junction integrity (74) and modulating paracellular permeability (75). In contrast, glutamine deprivation or inhibition of glutamine synthase significantly increase paracellular permeability and decrease the expression of tight junction proteins (76, 77). Glutamine is also associated with the proliferation of Lgr5-positive intestinal stem cell and subsequent crypt expansion. In intestinal organoid culture, the deprivation of glutamine suppresses epithelial proliferation (78). Replenishment of culture medium with supplementation of glutamine rescues the proliferation of epithelial cells via the activation of mammalian target of rapamycin (mTOR) (78). However, glutamine deprivation does not affect the proportions of Paneth and goblet cells, indicating that certain amino acids may support the differentiation and proliferation of intestinal epithelial cells in a lineage-specific manner. Although glutamine supplementation has been shown to be beneficial in murine models of experimental colitis, the utility of this amino acid in IBD patients remains poorly understood. Clinical studies showed that glutamine-supplemented enteral nutrition and total parenteral nutrition did not improve therapeutic outcomes for pediatric CD, adult CD, and adult UC patients (79, 80).
Other Amino Acids
Metabolome analyses have shown that the levels of other amino acids, such as histidine, glycine, and threonine, are altered in the serum and intestinal tissues of IBD patients (10, 13, 15). These amino acids have also been reported to be beneficial to IBD patients and in models of experimental colitis. Histidine is a natural amino acid and is one of conditionally essential amino acids. Histidine acts as a scavenger of hydroxyl radicals and singlet oxygen (81), and hence can suppress oxidative stress in intestinal epithelial cells (82). Moreover, dietary histidine ameliorates intestinal inflammation in the IL-10-deficient cell transfer model of colitis through the inhibition of NF-κB activation, following the down-regulation of pro-inflammatory cytokine production in macrophages (83). In IBD patients, the levels of histidine in the blood and intestinal tissues are markedly decreased compared to control subjects (10, 15). A recent clinical study has shown that the plasma histidine level is a predictor of relapse risk in UC patients (84). Glycine has been reported to be protective against various kinds of organ injuries as well as intestinal inflammation. Dietary glycine improves histology and dampens the expression of pro-inflammatory cytokines in TNBS- and DSS-induced colitis (85). Threonine is an essential amino acid with roles in maintaining intestinal homeostasis. It is important for mucosal barrier function, which prevents luminal bacteria from penetrating the mucosal tissue (86). Defects in intestinal mucosal barrier function are observed in IBD patients and experimental colitis, enabling gut bacteria to penetrate the intestinal mucosa (87, 88). Mucins contain high levels of threonine, serine and proline (89). Consequently, mucin synthesis requires robust amounts of these amino acids and restriction of dietary threonine impairs intestinal mucin synthesis (89). In contrast, dietary supplementation of threonine, serine, proline and cysteine ameliorates intestinal damage in DSS-treated rats, presumably by improving mucosal barrier function (90).
Intestinal inflammation changes the metabolic requirements of host cells and the gut microbiota. The up-regulation of amino acid metabolic pathways is one of the major inflammation-related metabolic shifts in the host and the microbiota (11, 44, 55). In other words, host cells and resident microbes have an increased demand for certain amino acids in the context of IBD. Thus, dietary supplementation of these sought-after amino acids is a key strategy for the treatment of IBD. However, it is noteworthy to mention that other amino acids may fuel pro-inflammatory responses in IBD and restriction of certain amino acids can attenuate intestinal inflammation. Amino acid deficiency is sensed by a serine/threonine-protein kinase called general control non-derepressible 2 (GCN2) (91). GCN2 suppresses pathogenic Th17 responses by inhibiting ROS-mediated activation of the inflammasome (92). Therefore, when GCN2 is deleted in a tissue-specific manner from intestinal epithelial cells and CD11c+ antigen-presenting cells, the animals experienced more severe weight loss and intestinal inflammation, indicating that both intestinal epithelial cells and antigen-presenting cells mediate the protective effects of GCN2 during colitis. Notably, amino acid starvation, induced by the consumption of a low-protein diet or a leucine-deficient diet, can suppress DSS-induced colitis through the activation of GCN2 (92). Since deprivation of other amino acids, such as tryptophan, tends to enhance intestinal inflammation (31), it is likely that each amino acid plays a distinct role in the promotion and/or suppression of intestinal inflammation. Thus, a more thorough understanding of the specific roles different amino acids play in IBD and supplying the optimal amount of anti-inflammatory amino acids while limiting the consumption of pro-inflammatory amino acids can lead to the development of effective amino acid-based dietary interventions.
Carbohydrate
Carbohydrates are an important source of nutrients for both host cells and the gut bacteria. However, host cells largely lack the capacity to digest complex poly-saccharides (93). Rather, dietary fibers and other carbohydrate polymers are degraded and fermented by the gut microbiota into mono-saccharides as well as various by-products. Recent accumulating evidence indicates these gut microbial byproducts can modulate the host immune system and intestinal barrier function, thus playing a crucial role in intestinal homeostasis (94). Therefore, the intake of indigestive carbohydrates (dietary fibers) benefits the host. On the other hand, gut dysbiosis may compromise the metabolic activities of the gut microbiota, resulting in impaired generation of the protective microbial byproducts. Furthermore, high intake of undigested and fermentable carbohydrates, such as mono- and di-saccharides, induces various gastrointestinal symptoms. In this section, we summarize the roles of dietary fiber and fermentable carbohydrates in intestinal homeostasis and the management of IBD symptoms.
Fibers
Dietary fibers are complex carbohydrates consisting of both soluble and insoluble components. Dietary fibers are not digested or absorbed by host cells, as mammalian cells largely lack the enzymes necessary to degrade them. Instead, dietary fibers are subjected to bacterial fermentation in the gastrointestinal tract. Although a wide range of bacteria ferment dietary fibers, each bacterium has a substrate preference based on its enzymatic activity. For example, the human gut symbionts Bacteroides thetaiotaomicron and B. ovatus can degrade a wide variety of glycans, whereas some Bacteroides species are restricted to one or only a few types (95). Thus, dietary intervention can remodel the gut microbial composition by customizing the content of dietary fibers. Also, bacterial byproducts generated through the fermentation of dietary fibers have various effects on host immune cells and intestinal barrier function. Short-chain fatty acids (SCFAs), such as acetate, propionate and butyrate, are the major end products of microbial fermentation of dietary fibers (Figure 2) (93). Dietary fiber-derived SCFAs are key energy substrates utilized by colonocytes (96). Additionally, SCFAs act as signaling molecules via G-protein-coupled receptors (GPRs), such as GPR41, GPR43, and GPR109a. SCFAs can regulate the differentiation of immune cells and immune responses through GPRs. SCFAs have been shown to promote anti-inflammatory responses through the activation of GPRs, as mice deficient in GPR43 and GPR109a develop more severe DSS-induced colitis (97, 98). Furthermore, GPR109a signaling promotes anti-inflammatory properties in colonic macrophages and dendritic cells as well as inducing the differentiation of regulatory type T cells, such as Treg cells and IL-10-producing T cells (97). Since SCFAs are generated by the gut microbiota through fermentation of dietary fibers, germ-free (GF) mice display lower levels of colonic Foxp3+ Treg cells (99). Butyrate is known to regulate histone acetylation, which in turn up-regulates the expression of Foxp3. Transcriptional factor Foxp3 is a lineage specification factor of Tregs that has been shown to prevent the development of colitis in a T cell transfer model of colitis (100). Collectively, SCFAs are important immunomodulatory molecules that exert numerous beneficial effects on host metabolism and immunity.
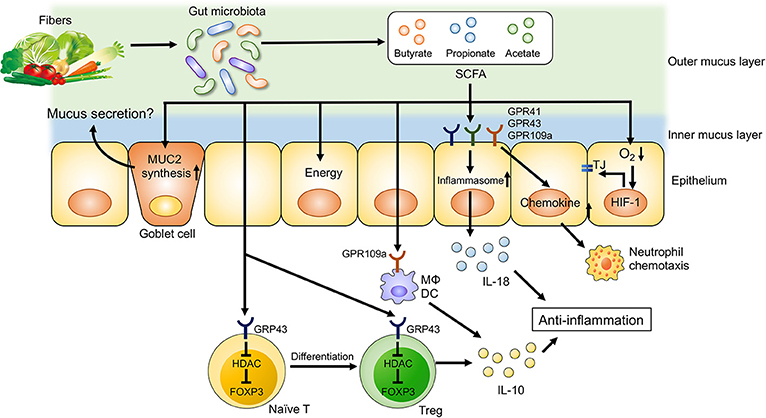
Figure 2. Dietary fiber-derived SCFAs regulate intestinal homeostasis. Dietary fiber-derived SCFAs serve as energy substrates for colonocytes. Likewise, SCFAs regulate intestinal barrier function and immune system through GPCRs signaling. SCFAs promote the differentiation into Treg cells and the production of IL-10 from Treg cells through GPR43. SCFA facilitate inflammasome activation in colonic epithelial cells through GPR43, resulting in an IL-18 production that is critical for anti-inflammation and epithelial repair. SCFAs also regulate intestinal barrier function via enhancing the expression of tight junction proteins and the synthesis of MUC2.
In addition to their regulatory function, SCFAs and, by extension, dietary fibers are also important for mucosal barrier function. Butyrate enhances intestinal epithelial barrier function via hypoxia inducible factor-1 (HIF-1) that regulates the integrity of epithelial tight junctions (101, 102). Likewise, butyrate treatment in mice facilitates the assembly of tight junction proteins and increases the synthesis of MUC2 protein, the main component of intestinal mucus (103, 104). Thus, a lack of dietary fibers may compromise epithelial integrity and mucus production due to insufficient SCFA generation, resulting in impaired intestinal barrier function (100, 101). In contrast, butyrate is known to suppress the proliferation of intestinal stem cells. Butyrate inhibits histone acetylation and enhances the promoter activity for the negative cell-cycle regulator Foxo3 in intestinal stem cells (105). Increased expression of Foxo3 in intestinal stem cells results in delayed wound repair after mucosal injuries (105). Notably, a recent report has unveiled that consumption of a low fiber diet leads to the disruption of intestinal barrier function through a mechanism that is independent of SCFAs. In the absence of dietary fibers, some commensal bacteria, such as Akkermansia muciniphila, utilize host mucus glycans to meet their energy needs (106). As a result, these mucolytic bacteria become the predominant species within the gut microbiota (106). Importantly, a bloom of mucolytic bacteria results in the degradation of the colonic mucus layer that renders the host susceptible to enteric pathogens, such as C. rodentium (106).
Gut dysbiosis observed in IBD patients is primarily characterized by reduced bacterial diversity, the enrichment of the phylum of Proteobacteria and a lower abundance of Firmicutes and Bacteroidetes phyla (107). IBD-associated gut dysbiosis is accompanied by functional changes in the gut microbiota that affect its ability to ferment dietary fibers. The abundance of butyrate-producing bacteria, such as Roseburia hominis and Faecalibacterium prausnitzii, is decreased in IBD patients (11, 108, 109) and, therefore, fecal SCFAs levels are lower in IBD patients compared to controls (20). On the other hand, recent clinical trials did not demonstrate overt therapeutic benefits associated with butyrate enemas in UC patients (110, 111). These results suggest that IBD patients might have functional impairments involving butyrate utilization in addition to its impaired generation, likely due to gut dysbiosis. For instance, several studies have reported that butyrate oxidation and the expression of genes involved in butyrate oxidation are diminished in the intestinal mucosa of IBD patients (112–115), indicating that butyrate cannot be used properly in the inflamed gut. Moreover, butyrate uptake by colonocytes is impaired in IBD patients. Although butyrate uptake is mediated by monocarboxylate transporter 1 (MCT1), the transcription of Mct1 is downregulated upon stimulation with IFN-γ and TNF-α, thereby reducing the uptake of butyrate (116). Consistently, expression levels of Mct1 mRNA negatively correlate with the degree of intestinal inflammation in IBD (116).
Although numerous animal studies have reported that dietary fiber supplementation attenuates intestinal inflammation (117–119), only limited evidence is available from clinical trials involving IBD patients (120). Prebiotic fibers might promote the growth of protective members of the gut microbiota. However, there is no study that provides statistically significant evidence for the efficacy of prebiotic fibers as treatment for IBD (121). It implies that fiber treatment is not effective for active IBD. Regarding this notion, it has been reported that intestinal inflammation down-regulates carbohydrate metabolism in the gut microbiota (11, 25). Thus, it is possible that gut microbes are not able to utilize supplied fibers efficiently in the inflamed gut. Consistently, consumption of a high fiber diet does not attenuate colitis, while pretreatment with the same diet can prevent the development of colitis (119). Thus, metabolic activities of the microbiota, which might be altered as a result of disease, may determine the usefulness of administrating prebiotic fibers to IBD patients. In addition to disease status, the initial composition of the microbiota may influence the efficacy of dietary fiber treatment. For example, dietary fiber can improve postprandial glucose metabolism in healthy individuals, but they are divided into responders and non-responders (122). The abundance of Prevotella, a bacterial genus capable of digesting fibers, is higher in responders compared to non-responders. In other words, a high fiber diet is not beneficial in individuals who do not have sufficient numbers of fiber-degrading bacteria in their gut microbiota. Taken together, personalized nutritional management will likely be a key component of any successful therapeutic approach for IBD since intestinal inflammation results in both compositional and functional changes of the gut microbiota.
Elimination of Fermentable Carbohydrate
Several dietary therapies that eliminate specific carbohydrates have been developed and evaluated for induction of remission, maintenance and improvement of functional symptoms in IBD. Symptoms of IBD commonly include abdominal pain, discomfort, rectal bleeding, and a change in stool consistency and frequency during active periods. However, similar gastrointestinal symptoms can also be observed during periods of remission, even in the absence of clinical inflammation (123). These functional-like gastrointestinal symptoms are negatively correlated with the quality of life (QOL) experienced by IBD patients (124). Naturally, a more effective control of functional-like gastrointestinal symptoms is needed to improve the QOL of IBD patients. Dietary therapies for these functional-like gastrointestinal symptoms have recently been developed. These therapies include a low Fermentable Oligo-saccharides, Di-saccharides, Mono-saccharides And Polyols (FODMAP) diet and a Specific Carbohydrate Diet (SCD) (8). We will review the effects of a low-FODMAP diet on IBD symptoms, because there is emerging clinical evidence suggesting the usefulness of this approach.
A diet low in FODMAP is used to manage symptoms in patients with irritable bowel syndrome (IBS) (125). A randomized, double-blind, placebo-controlled, cross-over study has revealed that the administration of fructans, unlike galacto-oligosaccharides or sorbitol, exacerbates functional-like gastrointestinal symptoms in quiescent IBD (126). A recent meta-analysis has shown that a low FODMAP diet can reduce gastrointestinal symptoms in patients with quiescent IBD (127). Although the precise mechanism by which FODMAPs promotes functional-like gastrointestinal symptoms is unclear, FODMAP intake is associated with increased luminal water content and colonic gas production (125). Whereas, most carbohydrates are completely digested and absorbed in the small intestine, some carbohydrates are not. Unabsorbed carbohydrates, such as fructose, polyols, and lactose, alter intestinal osmolality and increase water content in the small intestine. Other unabsorbed carbohydrates, including fructans and galacto-oligosaccharides, are fermented by the bacteria in the colon, leading to gas production. High levels of luminal water and gas result in luminal distension, thereby causing functional-like gastrointestinal symptoms. In an animal study, a high FODMAP diet increased intestinal permeability and altered the composition of microbial communities, leading to increased luminal levels of LPS (128). Inhibition of Toll-like receptor (TLR) activation by LPS ameliorates intestinal barrier function and visceral nociception. Moreover, since fecal LPS is higher in IBS patients compared to healthy subjects, a low-FODMAP diet improves IBS symptoms by reducing the levels of fecal LPS. Dietary intervention is an exciting topic in the current IBD research. Further studies are needed to unravel the mechanism of action and the long-term effects of a low FODMAP diet.
Fatty Acids
The Western diet is characterized by high intake of fats. There is evidence that links increased fat intake to the pathogenesis of IBD. Dietary fat comprises saturated fatty acids (SFAs), monounsaturated fatty acids (MUFAs), and polyunsaturated fatty acids (PUFAs). Most fatty acids, except for linoleic acid and alpha-linolenic acid which are essential fatty acids, can synthesize in the body. Fatty acids are important energy sources and membrane constituents, and their biological activities influence both the immune system and the gut microbiota. A number of studies have revealed that high-fat diets change the composition of the gut microbiota and impair intestinal barrier function, resulting in intestinal and systemic inflammation (129–131). Furthermore, consumption of a diet containing high levels of fat and sugar promotes the colonization of adherent-invasive E. coli (AIEC), an IBD-associated pathobiont (132). A recent systematic literature review of epidemiological data has shown that high dietary intakes of total fats, SFAs, omega-6 (n-6) and PUFAs are associated with an increased risk of both CD and UC (133). Similar to amino acids and carbohydrates, abnormal fatty acid profiles and metabolism are observed in the blood and intestinal mucosa of IBD patients (134). On the other hand, dietary supplementation with specific fatty acids can improve perturbed fatty acids profiles and intestinal inflammation (135). We summarize the roles of fatty acids in the pathogenesis and treatment of IBD.
Saturated Fatty Acids (SFAs)
SFAs, mainly myristic acid, palmitic acid, and stearic acid, are found in animal fat-containing products, such as meat, butter, whole milk, and other dairy products. Although a connection between saturated fats and the risk of IBD has been uncovered by small case–control studies, prospective cohorts have not identified a statistical association, suggesting the relationship is more complex. In animal studies, a diet high in SFAs induces colonic inflammation, including increased expression of pro-inflammatory cytokines and enhanced intestinal permeability in a microbiota- or TLR4-dependent manner (129–131). Similarly, a diet high in SFAs exacerbates intestinal inflammation in experimental models of colitis (130, 136). A milk-derived diet high in saturated fats, unlike a diet high in polyunsaturated fats, promotes the expansion of a sulphite-reducing pathobiont, Bilophila wadsworthia, and aggravates Th1-mediated colitis in IL-10 deficient mice (136). Milk-derived fat promotes taurine conjugation of hepatic bile acids, thereby leading to the accumulation of sulfur that aids the growth of B. wadsworthia.
In a murine model of DSS-induced colitis, the level of a saturated fat 1-stearoyl-sn-glycero-3-phosphorylcholine (stearoyl-LPC) in the blood is increased compared to control mice, whereas the unsaturated fat 1-oleoyl-sn-glycero-3-phosphorylcholine (oleoyl-LPC) is decreased following DSS treatment. Thus, intestinal inflammation shifts the balance between saturated LPCs and monounsaturated LPCs (135). The perturbation of the lipid serum profile is caused by decreased expression of stearoyl-CoA desaturase 1 (SCD1) in the liver. SCD1 is an enzyme found in the endoplasmic reticulum. It is responsible for the biosynthesis of oleic acid (18:1) and palmitoleic acid (16:1) from stearic acid (18:0), and palmitic acid (16:0), respectively (137). Saturated LPCs, unlike their monounsaturated counterparts, increase IL-1β production in human monocytes (138). Consistently, SCD1-deficient mice are more susceptible to DSS and exacerbated colitis can be ameliorated by means of oleic acid supplementation (135).
Monounsaturated Fatty Acids (MUFAs)
MUFAs are classified as fatty acids containing a single double bond. Oleic acid, the most common omega-9 MUFA, is found in various vegetable oils, such as olive oil and canola oil. The levels of MUFAs, including oleic acid, are significantly reduced in IBD patients compared to control subjects, both systemically (blood) and locally (intestinal mucosa) (139, 140). Epidemiologically, the Mediterranean diet containing high levels of MUFAs, especially oleic acid, is well-recognized for its beneficial effects on metabolic syndrome and cardiovascular health (141). The effects of MUFAs on IBD remain unresolved. There are conflicting reports regarding the therapeutic potential of MUFAs in IBD. A large prospective cohort study has demonstrated that dietary oleic acid intake is inversely associated with UC development (142). In contrast, other studies have shown that a high intake of MUFAs increases the risk for the development of UC and CD. Several animal studies have consistently demonstrated that supplementation of oleic acid or olive oil attenuates intestinal inflammation in DSS-induced colitis (135, 143).
Polyunsaturated Fatty Acids (PUFAs)
PUFAs are classified into omega-3 (n-3) and n-6 families. These two families are thought to have different effects on inflammation. Conventionally, n-6 PUFAs are considered to be pro-inflammatory. Linoleic acid, the major PUFA in the diet, is converted to arachidonic acid, a precursor of inflammatory mediators such as prostaglandins and leukotrienes (134). N-6 PUFAs are incorporated into phospholipids found in cellular membranes. Multiple members of the phospholipase family of enzymes, such as phospholipase A2 (PLA2), hydrolyze n-6 PUFAs during inflammation. Arachidonic acid is a primary target for PLA2 and is converted to inflammatory mediators involved in pro-inflammatory cell signaling events. The levels of PLA2 are elevated in the mucosal biopsies of CD and UC patients (144), resulting in increased hydrolysis of arachidonic acid and the subsequent generation of inflammatory mediators. In contrast, n-3 PUFAs appear to be critical for the suppression of inflammation. α-linolenic acid, an essential n-3 PUFA, is a precursor of eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), both of which are contained in fish oils. N-3 PUFAs compete with n-6 PUFAs in the substrate pool of the lipoxygenase pathway, inhibiting the production of inflammatory mediators downstream of n-6 PUFAs (134). Likewise, pro-resolving mediators are generated from n-3 fatty acids via several enzymatic reactions (145). Resolvin E1 (RvE1) is an anti-inflammatory lipid mediator derived from EPA through aspirin-acetylated cyclooxygenase-2 (COX2) and microbial cytochrome P450 (146, 147). RvE1 inhibits leukocyte infiltration and IL-12 production by dendritic cells and macrophages (148, 149). Consistent with the anti-inflammatory effects on immune cells, the administration of RvE1 attenuates TNBS- and DSS-induced colitis in mice (146, 149). Moreover, n-3 PUFAs can directly inhibit the TLR4 and NF-κB signaling pathways, leading to the down-regulation of pro-inflammatory genes, such as COX2 and TNF-α (150, 151). Thus, an appropriate balance of n-6/n-3 PUFAs is important for the regulation of inflammation.
A Western diet typically contains high levels of n-6 PUFAs and low levels of n-3 PUFAs (152). The skewed ratio between the intake of n-6 PUFAs and n-3 PUFAs is considered a risk factor for IBD (133). Although previous studies have examined the effect of n-3 fatty acid supplementation in prevention and treatment of IBD, the outcomes were inconsistent in both human IBD and animal models of colitis. In the animal studies, supplementation with n-3 fatty acids found in fish oil attenuated intestinal inflammation (153, 154), whereas another study showed that n-3 fatty acids exacerbated colitis (155). This disparity may have been caused by various confounding factors, such as control diet, administration period and sensitivity to oxidation. Another report demonstrated that fat-1 transgenic mice are protected against DSS-induced colitis (156). Since fat-1 transgenic mice have increased endogenous levels of n-3 PUFAs, the protective phenotype observed in this mouse strain may be due to n-3 PUFAs (156). In human IBD, a systematic review does not support the notion that supplementation of n-3 fatty acids can induce and maintain remission of IBD (157). The latest Cochrane review has reached a similar conclusion (158). Interestingly, several studies have demonstrated that different genotypes may be associated with the variable response to nutritional intervention with n-3 PUFAs. For example, genetic polymorphisms of TNF-α and PPARα have been associated with an altered response to nutritional intervention with n-3 PUFAs (159, 160). In IBD, genetic polymorphisms, such as CYP4F3 and Caspase 9+93C/T, modify the association between dietary fatty acid intake and risk of IBD (161, 162). Since fatty acid utilization may vary depending on the genetic background, personalized nutritional management is likely required to maximize the efficacy of n-3 PUFAs supplementation in IBD.
Phytochemicals
Phytochemicals are a wide range of natural compounds found in plants. Accumulating evidence suggest that phytochemicals have beneficial effect on several chronic diseases (163). A number of polyphenols, including flavonoids, phenolic acids, stilbenes, and lignans, have been shown to influence the gut microbiota and intestinal immunity. For example, curcumin, a hydrophobic polyphenol derived from Curcuma longa, displays a wide variety of biological functions, such as anti-inflammatory, anti-cancer, and anti-oxidant activities (164). Curcumin suppresses cytokine production by macrophages and intestinal epithelial cells via the inhibition of NF-κB activation (165, 166), and thus administration of curcumin ameliorates DSS- and TNBS-induced colitis (167, 168). Polyphenols including curcumin are not efficiently absorbed from the small intestine, and therefore polyphenol complexes contained in diet have limited bioavailability (169). Given this evidence, a large portion of unabsorbed polyphenols are likely transported to the large intestine where they interact with the colonic gut microbiota. Of note, the gut resident bacteria are capable of catabolizing polyphenols and degrading them into small fragments (170). For instance, curcumin is converted into tetrahydrocurcumin by curcumin-converting enzyme purified from E. coli (171). Curcumin-derived tetrahydrocurcumin can prevent colitis and colitis-associated colon cancer in mice (167, 172). Recent epidemiological study reported a potential association of polyphenol in CD (173). Moreover, clinical trials show that curcumin supplementation is effective for the induction and maintenance of remission in UC patients (174, 175).
Food Additives
Recent widespread use of processed food increases consumption of food additives. Emulsifiers are complex molecules that are incorporated into most processed foods to enhance texture and stability. It has been implied that the increased consumption of emulsifier is associated with IBD pathogenesis (176). In recent studies, the consumption of dietary emulsifier disrupted host-microbiota interaction and promoted intestinal inflammation and colon carcinogenesis (177, 178). Although precise mechanism remains incompletely understood, dietary emulsifiers foster the blooms of mucolytic bacteria, such as Ruminococcus gnavus and Akkermansia muciniphila (177). Importantly, the negative impact of dietary emulsifiers is not observed in GF mice, suggesting that the compositional and functional modulation of the gut microbiota by emulsifiers play a key role in their adverse effect (177). Other food additives, for example artificial sweeteners, also induce gut dysbiosis (179, 180). The consumption of artificial sweeteners promotes the expansion of Proteobacteria and increases the infiltration of bacteria into the ileal lamina propria in CD-like ileitis model mice (180). Moreover, dietary phosphate, the main component of many food additives, enhances intestinal inflammation through the activation of NF-κB in macrophages (181). These studies suggest that many food additives may be associated with the risk of IBD. In fact, epidemiological and clinical studies reported that processed meats and beverages are risk factors for the onset and relapse of IBD (182, 183). However, it is difficult to ascertain whether food additives are risk factors for developing IBD, because most food frequency questionnaires cannot fully evaluate the consumption of food additives.
Conclusion and Future Direction
The relationship between dietary nutrients and intestinal homeostasis is complex. It is influenced by a myriad of interactions between host immunity, the intestinal epithelium and the gut microbiota. Moreover, intestinal inflammation changes cellular and microbial metabolisms, adding another layer of complexity to an already complex system (Table 2). The demand for certain nutrients necessary for the maintenance of intestinal homeostasis is likely altered in IBD. The scarcity or overabundance of some nutrients can disturb intestinal homeostasis even further, thus exacerbating the disease. Genetic polymorphisms also influence the efficacy of nutritional intervention. Thus, an in-depth knowledge of a patient's genetic predispositions, gut microbial compositions, disease activity and type (i.e., ileal, colonic, etc.) will be required to maximize the effectiveness of nutritional intervention approaches in IBD.
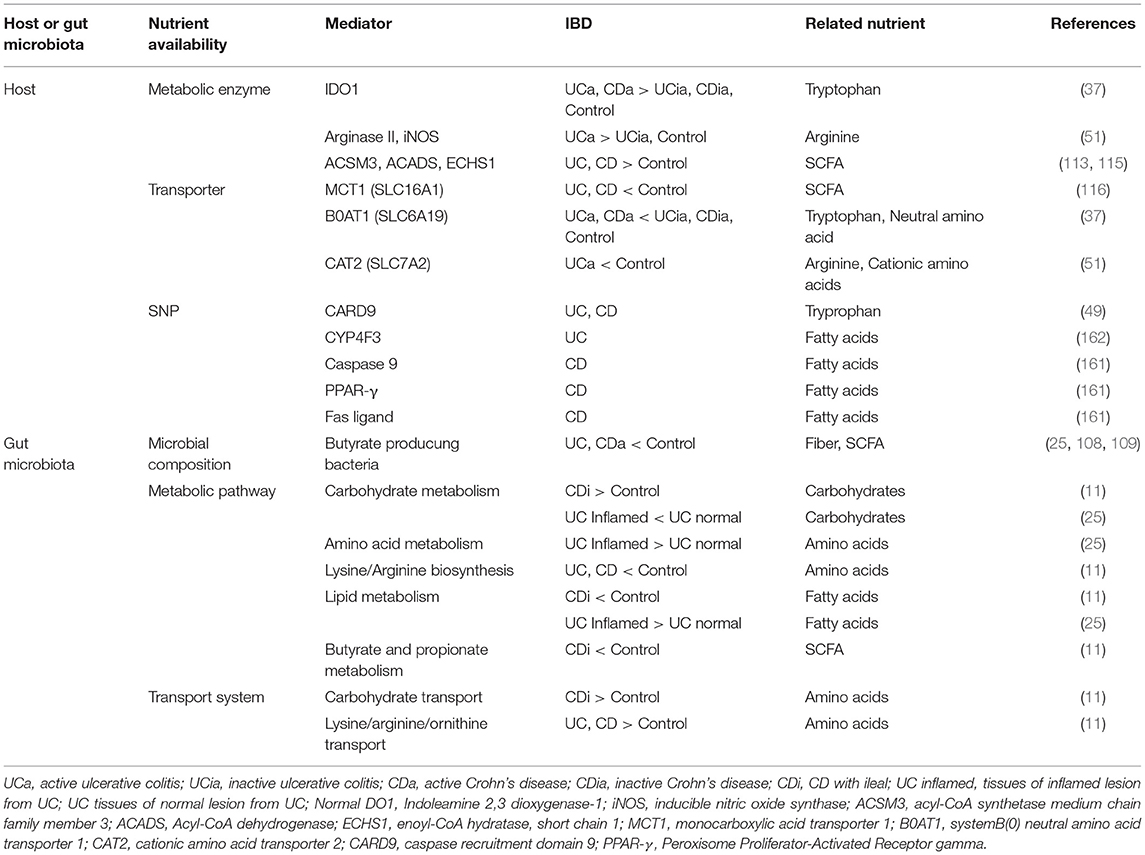
Table 2. Changes of dietary nutrient availavility in the host and gut microbiota of IBD.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank P. Kuffa for critical reading of the manuscript. This work was supported by the Kenneth Rainin Foundation, National Institute of Health DK110146 and DK108901, Crohn's and Colitis Foundation of America (to NK), T32 DK094775 (to TM), and the Uehara Memorial Foundation Postdoctoral Fellowship Award (to KS).
References
1. Cosnes J, Gowerrousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology (2011) 140:1785–94. doi: 10.1053/j.gastro.2011.01.055
2. Ananthakrishnan AN. Epidemiology and risk factors for IBD. Nat Rev Gastroenterol Hepatol. (2015) 12:205–17. doi: 10.1038/nrgastro.2015.34
3. Loddo I, Romano C. Inflammatory bowel disease: genetics, epigenetics, and pathogenesis. Front Immunol. (2015) 6:6–11. doi: 10.3389/fimmu.2015.00551
4. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature (2012) 491:119–24. doi: 10.1038/nature11582
5. Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology (2012) 142:46–54.e42. doi: 10.1053/j.gastro.2011.10.001
6. Lee D, Albenberg L, Compher C, Baldassano R, Piccoli D, Lewis JD, et al. Diet in the pathogenesis and treatment of inflammatory bowel diseases. Gastroenterology (2015) 148:1087–106. doi: 10.1053/j.gastro.2015.01.007
7. Spooren CEGM, Pierik MJ, Zeegers MP, Feskens EJM, Masclee AAM, Jonkers DMAE. Review article: the association of diet with onset and relapse in patients with inflammatory bowel disease. Aliment Pharmacol Ther. (2013) 38:1172–87. doi: 10.1111/apt.12501
8. Sigall-Boneh R, Levine A, Lomer M, Wierdsma N, Allan P, Fiorino G, et al. Research gaps in diet and nutrition in inflammatory bowel disease. a topical review by D-ECCO Working Group (Dietitians of ECCO). J Crohns Colitis. (2017) 1407–19. doi: 10.1093/ecco-jcc/jjx109
9. Kolho KL, Pessia A, Jaakkola T, de Vos WM, Velagapudi V. Faecal and serum metabolomics in paediatric inflammatory bowel disease. J Crohns Colitis. (2017) 11:321–34. doi: 10.1093/ecco-jcc/jjw158
10. Kohashi M, Nishiumi S, Ooi M, Yoshie T, Matsubara A, Suzuki M, et al. A novel gas chromatography mass spectrometry-based serum diagnostic and assessment approach to ulcerative colitis. J Crohns Colitis. (2014) 8:1010–21. doi: 10.1016/j.crohns.2014.01.024
11. Morgan X, Tickle T, Sokol H, Gevers D, Devaney K, Ward D. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. (2012) 13:R79. doi: 10.1186/gb-2012-13-9-r79
12. Scoville EA, Allaman MM, Brown CT, Motley AK, Horst SN, Williams CS, et al. Alterations in lipid, amino acid, and energy metabolism distinguish Crohn's disease from ulcerative colitis and control subjects by serum metabolomic profiling. Metabolomics (2018) 14:1–12. doi: 10.1007/s11306-017-1311-y
13. Schicho R, Shaykhutdinov R, Ngo J, Nazyrova A, Schneider C, Panaccione R, et al. Quantitative metabolomic profiling of serum, plasma, and urine by 1H NMR spectroscopy discriminates between patients with inflammatory bowel disease and healthy individuals. J Proteome Res. (2012) 11:3344–57. doi: 10.1021/pr300139q
14. Dawiskiba T, Deja S, Mulak A, Zabek A, Jawien E, Pawełka D, et al. Serum and urine metabolomic fngerprinting in diagnostics of inflammatory bowel diseases. World J Gastroenterol. (2014) 20:163–74. doi: 10.3748/wjg.v20.i1.163
15. Ooi M, Nishiumi S, Yoshie T, Shiomi Y, Kohashi M, Fukunaga K, et al. GC/MS-based profiling of amino acids and TCA cycle-related molecules in ulcerative colitis. Inflamm Res. (2011) 60:831–40. doi: 10.1007/s00011-011-0340-7
16. Hisamatsu T, Okamoto S, Hashimoto M, Muramatsu T, Andou A, Uo M, et al. Novel, objective, multivariate biomarkers composed of plasma amino acid profiles for the diagnosis and assessment of inflammatory bowel disease. PLoS ONE (2012) 7:e31131. doi: 10.1371/journal.pone.0031131
17. Martin FP, Su MM, Xie GX, Guiraud SP, Kussmann M, Godin JP, et al. Urinary metabolic insights into host-gut microbial interactions in healthy and IBD children. World J Gastroenterol. (2017) 23:3643–54. doi: 10.3748/wjg.v23.i20.3643
18. Stephens NS, Siffledeen J, Su X, Murdoch TB, Fedorak RN, Slupsky CM. Urinary NMR metabolomic profiles discriminate inflammatory bowel disease from healthy. J Crohns Colitis. (2013) 7:e42–8. doi: 10.1016/j.crohns.2012.04.019
19. Bosch S, Struys EA, Van Gaal N, Bakkali A, Jansen EW, Diederen K, et al. Fecal amino acid analysis can discriminate de novo treatment-naïve pediatric inflammatory bowel disease from controls. J Pediatr Gastroenterol Nutr. (2018) 66:773–8. doi: 10.1097/MPG.0000000000001812
20. Marchesi JR, Holmes E, Khan F, Kochhar S, Scanlan P, Shanahan F, et al. Rapid and noninvasive metabonomic characterization of inflammatory bowel disease. J Proteome Res. (2007) 6:546–51. doi: 10.1021/pr060470d
21. Bjerrum JT, Wang Y, Hao F, Coskun M, Ludwig C, Günther U, et al. Metabonomics of human fecal extracts characterize ulcerative colitis, Crohn's disease and healthy individuals. Metabolomics (2014) 11:122–33. doi: 10.1007/s11306-014-0677-3
22. Santoru ML, Piras C, Murgia A, Palmas V, Camboni T, Liggi S, et al. Cross sectional evaluation of the gut-microbiome metabolome axis in an Italian cohort of IBD patients. Sci Rep. (2017) 25:9523. doi: 10.1038/s41598-017-10034-5
23. Balasubramanian K, Kumar S, Singh R, Sharma U, Ahuja V, Makharia G, et al. Metabolism of the colonic mucosa in patients with inflammatory bowel diseases: an in vitro proton magnetic resonance spectroscopy study. Magn Reson Imag. (2009) 27:79–86. doi: 10.1016/j.mri.2008.05.014
24. Bjerrum JT, Nielsen OH, Hao F, Tang H, Nicholson JK, Wang Y, et al. Metabonomics in ulcerative colitis: diagnostics, biomarker identification, and insight into the pathophysiology. J Proteome Res. (2010) 9:954–62. doi: 10.1021/pr9008223
25. Davenport M, Poles J, Leung JM, Wolff MJ, Abidi WM, Ullman T, et al. Metabolic alterations to the mucosal microbiota in inflammatory bowel disease. Inflamm Bowel Dis. (2014) 20:723–31. doi: 10.1097/MIB.0000000000000011
26. Ren W, Liu G, Yin J, Tan B, Wu G, Bazer FW, et al. Amino-acid transporters in T-cell activation and differentiation. Cell Death Dis. (2017) 8:1–9. doi: 10.1038/cddis.2016.222
27. Langston PK, Shibata M, Horng T. Metabolism supports macrophage activation. Front Immunol. (2017) 8:61. doi: 10.3389/fimmu.2017.00061
28. Palego L, Betti L, Rossi A, Giannaccini G. Tryptophan biochemistry: structural, nutritional, metabolic, and medical aspects in humans. J Amino Acids (2016) 2016:1–13. doi: 10.1155/2016/8952520
29. Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol. (2012) 13:144–52. doi: 10.1038/ni.2187
30. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between Kynurenine and the Aryl Hydrocarbon receptor can generate regulatory T cells. J Immunol. (2010) 185:3190–8. doi: 10.4049/jimmunol.0903670
31. Hashimoto T, Perlot T, Rehman A, Trichereau J, Ishiguro H, Paolino M, et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature (2012) 487:477–81. doi: 10.1038/nature11228
32. Islam J, Sato S, Watanabe K, Watanabe T, Ardiansyah, Hirahara K, et al. Dietary tryptophan alleviates dextran sodium sulfate-induced colitis through aryl hydrocarbon receptor in mice. J Nutr Biochem. (2017) 42:43–50. doi: 10.1016/j.jnutbio.2016.12.019
33. Kim CJ, Kovacs-Nolan JA, Yang C, Archbold T, Fan MZ, Mine Y. L-Tryptophan exhibits therapeutic function in a porcine model of dextran sodium sulfate (DSS)-induced colitis. J Nutr Biochem. (2010) 21:468–75. doi: 10.1016/j.jnutbio.2009.01.019
34. Bettenworth D, Nowacki TM, Ross M, Kyme P, Schwammbach D, Kerstiens L, et al. Nicotinamide treatment ameliorates the course of experimental colitis mediated by enhanced neutrophil-specific antibacterial clearance. Mol Nutr Food Res. (2014) 58:1474–90. doi: 10.1002/mnfr.201300818
35. Lanis JM, Alexeev EE, Curtis VF, Kitzenberg DA, Kao DJ, Battista KD, et al. Tryptophan metabolite activation of the aryl hydrocarbon receptor regulates IL-10 receptor expression on intestinal epithelia. Mucosal Immunol. (2017) 10:1133–44. doi: 10.1038/mi.2016.133
36. El-Zaatari M, Bass AJ, Bowlby R, Zhang M, Syu LJ, Yang Y, et al. Indoleamine 2,3-Dioxygenase 1, increased in human gastric pre-neoplasia, promotes inflammation and metaplasia in mice and is associated with type II hypersensitivity/autoimmunity. Gastroenterology (2018) 154:140–53.e17. doi: 10.1053/j.gastro.2017.09.002
37. Nikolaus S, Schulte B, Al-Massad N, Thieme F, Schulte DM, Bethge J, et al. Increased Tryptophan metabolism is associated with activity of inflammatory bowel diseases. Gastroenterology (2017) 153:1504–16.e2. doi: 10.1053/j.gastro.2017.08.028
38. Gurtner GJ, Newberry RD, Schloemann SR, McDonald KG, Stenson WF. Inhibition of Indoleamine 2,3-Dioxygenase augments trinitrobenzene sulfonic acid colitis in mice. Gastroenterology (2003) 125:1762–73. doi: 10.1053/j.gastro.2003.08.031
39. Takamatsu M, Hirata A, Ohtaki H, Hoshi M, Hatano Y, Tomita H, et al. IDO1 plays an immunosuppressive role in 2,4,6-Trinitrobenzene sulfate-induced colitis in mice. J Immunol. (2013) 191:3057–64. doi: 10.4049/jimmunol.1203306
40. Jasperson LK, Bucher C, Panoskaltsis-Mortari A, Taylor PA, Mellor AL, Munn DH, et al. Indoleamine 2,3-dioxygenase is a critical regulator of acute graft-versus-host disease lethality. Blood (2008) 111:3257–65. doi: 10.1182/blood-2007-06-096081
41. Harrington L, Srikanth CV, Antony R, Rhee SJ, Mellor AL, Hai NS, et al. Deficiency of indoleamine 2,3-dioxygenase enhances commensal-induced antibody responses and protects against Citrobacter rodentium-induced colitis. Infect Immun. (2008) 76:3045–53. doi: 10.1128/IAI.00193-08
42. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature (2008) 453:65–71. doi: 10.1038/nature06880
43. Quiros M, Nishio H, Neumann PA, Siuda D, Brazil JC, Azcutia V, et al. Macrophage-derived IL-10 mediates mucosal repair by epithelial WISP-1 signaling. J Clin Invest. (2017) 127:3510–20. doi: 10.1172/JCI90229
44. Miyajima M, Zhang B, Sugiura Y, Sonomura K, Guerrini MM, Tsutsui Y, et al. Metabolic shift induced by systemic activation of T cells in PD-1-deficient mice perturbs brain monoamines and emotional behavior. Nat Immunol. (2017) 18:1342–52. doi: 10.1038/ni.3867
45. Bauer EP. Serotonin in fear conditioning processes. Behav Brain Res. (2015) 277:68–77. doi: 10.1016/j.bbr.2014.07.028
46. Selinger CP, Bannaga A. Inflammatory bowel disease and anxiety: links, risks, and challenges faced. Clin Exp Gastroenterol. (2015) 8:111–7. doi: 10.2147/CEG.S57982
47. Zelante T, Iannitti RG, Cunha C, DeLuca A, Giovannini G, Pieraccini G, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity (2013) 39:372–85. doi: 10.1016/j.immuni.2013.08.003
48. Dudakov JA, Hanash AM, van den Brink MRM. Interleukin-22: immunobiology and pathology. Annu Rev Immunol. (2015) 33:747–85. doi: 10.1146/annurev-immunol-032414-112123
49. Lamas B, Richard ML, Leducq V, Pham HP, Michel ML, Da Costa G, et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med. (2016) 22:598–605. doi: 10.1038/nm.4102
50. Patel VB, Preedy VR, Rajendram R. L -Arginine in Clinical Nutrition. New York, NY: Humana Press (2017).
51. Coburn LA, Horst SN, Allaman MM, Brown CT, Williams CS, Hodges ME, et al. L -Arginine availability and metabolism is altered in ulcerative colitis. Inflamm Bowel Dis. (2016) 22:1847–58. doi: 10.1097/MIB.0000000000000790
52. Coburn LA, Gong X, Singh K, Asim M, Scull BP, Allaman MM, et al. L-arginine supplementation improves responses to injury and inflammation in dextran sulfate sodium colitis. PLoS ONE (2012) 7:e33546. doi: 10.1371/journal.pone.0033546
53. Singh K, Coburn LA, Barry DP, Asim M, Scull BP, Allaman MM, et al. Deletion of cationic amino acid transporter 2 exacerbates dextran sulfate sodium colitis and leads to an IL-17-predominant T cell response. AJP Gastrointest Liver Physiol. (2013) 305:G225–40. doi: 10.1152/ajpgi.00091.2013
54. Singh K, Coburn LA, Barry DP, Boucher J-L, Chaturvedi R, Wilson KT. L-arginine uptake by cationic amino acid transporter 2 is essential for colonic epithelial cell restitution. AJP Gastrointest Liver Physiol. (2012) 302:G1061–73. doi: 10.1152/ajpgi.00544.2011
55. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-Arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell (2016) 167:829–42.e13. doi: 10.1016/j.cell.2016.09.031
56. Fletcher M, Ramirez ME, Sierra RA, Raber P, Thevenot P, Al-Khami AA, et al. L-Arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res. (2015) 75:275–83. doi: 10.1158/0008-5472.CAN-14-1491
57. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell responses. Cancer Res. (2004) 64:5839–49. doi: 10.1158/0008-5472.CAN-04-0465
58. Rodriguez PC, Zea AH, DeSalvo J, Culotta KS, Zabaleta J, Quiceno DG, et al. L-Arginine consumption by macrophages modulates the expression of CD3 chain in T lymphocytes. J Immunol. (2003) 171:1232–9. doi: 10.4049/jimmunol.171.3.1232
59. Tarasenko TN, Gomez-Rodriguez J, McGuire PJ. Impaired T cell function in argininosuccinate synthetase deficiency. J Leukoc Biol. (2015) 97:273–8. doi: 10.1189/jlb.1AB0714-365R
60. Hesse M, Modolell M, La Flamme AC, Schito M, Fuentes JM, Cheever AW, et al. Differential regulation of nitric oxide Synthase-2 and Arginase-1 by Type 1/Type 2 cytokines in vivo: granulomatous pathology is shaped by the pattern of L-Arginine metabolism. J Immunol. (2001) 167:6533–44. doi: 10.4049/jimmunol.167.11.6533
61. Munder M, Eichmann K, Modolell M. Alternative metabolic states in murine macrophages reflected by the nitric oxide synthase/arginase balance: competitive regulation by CD4+ T cells correlates with Th1/Th2 phenotype. J Immunol. (1998) 160:5347–54.
62. MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. (1997) 15:323–50. doi: 10.1146/annurev.immunol.15.1.323
63. Campbell L, Saville CR, Murray PJ, Cruickshank SM, Hardman MJ. Local arginase 1 activity is required for cutaneous wound healing. J Invest Dermatol. (2013) 133:2461–70. doi: 10.1038/jid.2013.164
64. Hankard R, Mauras N, Hammond D, Haymond M, Darmaun D. Is glutamine a “conditionally essential” amino acid in Duchenne muscular dystrophy? Clin Nutr. (1999) 18:365–9. doi: 10.1054/clnu.1999.0054
65. Sayles C, Hickerson SC, Bhat RR, Hall J, Garey KW, Trivedi MV. Oral glutamine in preventing treatment-related mucositis in adult patients with cancer: a systematic review. Nutr Clin Pract. (2016) 31:171–9. doi: 10.1177/0884533615611857
66. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol. (2010) 185:1037–44. doi: 10.4049/jimmunol.0903586
67. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35:871–82. doi: 10.1016/j.immuni.2011.09.021
68. Sido B, Seel C, Hochlehnert A, Breitkreutz R, Droge W. Low intestinal glutamine level and low glutaminase activity in Crohn's disease: a rational for glutamine supplementation? Dig Dis Sci. (2006) 51:2170–9. doi: 10.1007/s10620-006-9473-x
69. Shiomi Y, Nishiumi S, Ooi M, Hatano N, Shinohara M, Yoshie T, et al. GCMS-based metabolomic study in mice with colitis induced by dextran sulfate sodium. Inflamm Bowel Dis. (2011) 17:2261–74. doi: 10.1002/ibd.21616
70. Vicario M, Amat C, Rivero M, Moretó M, Pelegrí C. Dietary glutamine affects mucosal functions in rats with mild DSS-induced colitis. J Nutr. (2007) 137:1931–7. doi: 10.1093/jn/137.8.1931
71. Crespo I, San-Miguel B, Prause C, Marroni N, Cuevas MJ, González-Gallego J, et al. Glutamine treatment attenuates endoplasmic reticulum stress and apoptosis in TNBS-induced colitis. PLoS ONE (2012) 7:e50407. doi: 10.1371/journal.pone.0050407
72. Kretzmann NA, Fillmann H, Mauriz JL, Marroni CA, Marroni N, González-Gallego J, et al. Effects of glutamine on proinflammatory gene expression and activation of nuclear factor Kappa B and signal transducers and activators of transcription in TNBS-induced colitis. Inflamm Bowel Dis. (2008) 14:1504–13. doi: 10.1002/ibd.20543
73. Tian Y, Wang K, Wang Z, Li N, Ji G. Chemopreventive effect of dietary glutamine on colitis-associated colon tumorigenesis in mice. Carcinogenesis (2013) 34:1593–600. doi: 10.1093/carcin/bgt088
74. Suzuki T. Regulation of intestinal epithelial permeability by tight junctions. Cell Mol Life Sci. (2013) 70:631–59. doi: 10.1007/s00018-012-1070-x
75. Seth A. L-Glutamine ameliorates acetaldehyde-induced increase in paracellular permeability in Caco-2 cell monolayer. AJP Gastrointest Liver Physiol. (2004) 287:G510–7. doi: 10.1152/ajpgi.00058.2004
76. Li N. Glutamine regulates Caco-2 cell tight junction proteins. AJP Gastrointest Liver Physiol. (2004) 287:G726–33. doi: 10.1152/ajpgi.00012.2004
77. Li N, Neu J. Glutamine deprivation alters intestinal tight junctions via a PI3-K/Akt mediated pathway in Caco-2 cells 1,2. J Nutr. (2009) 139:710–4. doi: 10.3945/jn.108.101485
78. Moore SR, Guedes MM, Costa TB, Vallance J, Maier EA, Betz KJ, et al. Glutamine and alanyl-glutamine promote crypt expansion and mTOR signaling in murine enteroids. Am J Physiol Liver Physiol. (2015) 15:831–9. doi: 10.1152/ajpgi.00422.2014
79. Ockenga J, Borchert K, Stüber E, Lochs H, Manns MP, Bischoff SC. Glutamine-enriched total parenteral nutrition in patients with inflammatory bowel disease. Eur J Clin Nutr. (2005) 59:1302–9. doi: 10.1038/sj.ejcn.1602243
80. Akobeng AK, Miller V, Stanton J, Elbadri AM, Thomas AG. Double-blind randomized controlled trial of glutamine-enriched polymeric diet in the treatment of active Crohn's disease. J Pediatr Gastroenterol Nutr. (2000) 30:78–84. doi: 10.1097/00005176-200001000-00022
81. Wade AM, Tucker HN. Antioxidant characteristics of L-histidine. J Nutr Biochem. (1998) 9:308–15. doi: 10.1016/S0955-2863(98)00022-9
82. Son DO, Satsu H, Shimizu M. Histidine inhibits oxidative stress- and TNF-α-induced interleukin-8 secretion in intestinal epithelial cells. FEBS Lett. (2005) 579:4671–7. doi: 10.1016/j.febslet.2005.07.038
83. Andou A, Hisamatsu T, Okamoto S, Chinen H, Kamada N, Kobayashi T, et al. Dietary histidine ameliorates murine colitis by inhibition of proinflammatory cytokine production from macrophages. Gastroenterology (2009) 136:564–74.e2. doi: 10.1053/j.gastro.2008.09.062
84. Hisamatsu T, Ono N, Imaizumi A, Mori M, Suzuki H, Uo M, et al. Decreased plasma histidine level predicts risk of relapse in patients with ulcerative colitis in remission. PLoS ONE (2015) 10:e0140716. doi: 10.1371/journal.pone.0140716
85. Tsune I, Ikejima K, Hirose M, Yoshikawa M, Enomoto N, Takei Y, et al. Dietary glycine prevents chemical-induced experimental colitis in the rat. Gastroenterology (2003) 125:775–85. doi: 10.1016/S0016-5085(03)01067-9
86. Johansson MEV, Hansson GC. Immunological aspects of intestinal mucus and mucins. Nat Rev Immunol. (2016) 16:639–49. doi: 10.1038/nri.2016.88
87. Johansson MEV, Gustafsson JK, Sjöberg KE, Petersson J, Holm L, Sjövall H, et al. Bacteria penetrate the inner mucus layer before inflammation in the dextran sulfate colitis model. PLoS ONE (2010) 5:e12238. doi: 10.1371/journal.pone.0012238
88. Johansson MEV, Gustafsson JK, Holmen-Larsson J, Jabbar KS, Xia L, Xu H, et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut (2014) 63:281–91. doi: 10.1136/gutjnl-2012-303207
89. Faure M, Moënnoz D, Montigon F, Mettraux C, Breuillé D, Ballèvre O. Dietary threonine restriction specifically reduces intestinal mucin synthesis in rats. J Nutr. (2005) 135:486–91. doi: 10.1093/jn/135.3.486
90. Faure M, Mettraux C, Moennoz D, Godin J-P, Vuichoud J, Rochat F, et al. Specific amino acids increase mucin synthesis and microbiota in dextran sulfate sodium-treated rats. J Nutr. (2006) 136:1558–64. doi: 10.1093/jn/136.6.1558
91. Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. (2016) 17:1374–95. doi: 10.15252/embr.201642195
92. Ravindran R, Loebbermann J, Nakaya HI, Khan N, Ma H, Gama L, et al. The amino acid sensor GCN2 controls gut inflammation by inhibiting inflammasome activation. Nature (2016) 531:523–7. doi: 10.1038/nature17186
93. Holscher HD. Dietary fiber and prebiotics and the gastrointestinal microbiota. Gut Microbes (2017) 8:172–84. doi: 10.1080/19490976.2017.1290756
94. Thorburn AN, Macia L, Mackay CR. Diet, Metabolites, and “Western-Lifestyle” Inflammatory Diseases. Immunity (2014) 40:833–42. doi: 10.1016/j.immuni.2014.05.014
95. Salyers AA, West SE, Vercellotti JR, Wilkins TD. Fermentation of mucins and plant polysaccharides by anaerobic bacteria from the Fermentation of Mucins and Plant Polysaccharides by Anaerobic Bacteria from the Human Colon. Appl Environ Microbiol. (1977) 34:529–33.
96. Marsman KE, McBurney MI. Dietary fiber increases oxidative metabolism in colonocytes but not in distal small intestinal enterocytes isolated from rats. J Nutr. (1995) 125:273–82.
97. Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity (2014) 40:128–39. doi: 10.1016/j.immuni.2013.12.007
98. Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Di Yu, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature (2009) 461:1282–6. doi: 10.1038/nature08530
99. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. The microbial metabolites, short-chain fatty acids, regulate colonic treg cell homeostasis. Science (2013) 341:569–73. doi: 10.1126/science.1241165
100. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature (2013) 504:446–50. doi: 10.1038/nature12721
101. Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, et al. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe (2015) 17:662–71. doi: 10.1016/j.chom.2015.03.005
102. Saeedi BJ, Kao DJ, Kitzenberg DA, Dobrinskikh E, Schwisow KD, Masterson JC, et al. HIF-dependent regulation of claudin-1 is central to intestinal epithelial tight junction integrity. Mol Biol Cell (2015) 26:2252–62. doi: 10.1091/mbc.E14-07-1194
103. Peng L, Li Z-R, Green RS, Holzman IR, Lin J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J Nutr. (2009) 139:1619–25. doi: 10.3945/jn.109.104638
104. Burger-van Paassen N, Vincent A, Puiman PJ, van der Sluis M, Bouma J, Boehm G, et al. The regulation of intestinal mucin MUC2 expression by short-chain fatty acids: implications for epithelial protection. Biochem J. (2009) 420:211–9. doi: 10.1042/BJ20082222
105. Kaiko GE, Ryu SH, Koues OI, Collins PL, Solnica-Krezel L, Pearce EJ, et al. The colonic crypt protects stem cells from microbiota-derived metabolites. Cell (2016) 165:1708–20. doi: 10.1016/j.cell.2016.05.018
106. Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, et al. A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell (2016) 167:1339–53.e21. doi: 10.1016/j.cell.2016.10.043
107. Nagao-Kitamoto H, Kamada N. Host-microbial Cross-talk in inflammatory bowel disease. Immune Netw. (2017) 17:1–12. doi: 10.4110/in.2017.17.1.1
108. Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, et al. A decrease of the butyrate-producing species roseburia hominis and faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut (2014) 63:1275–83. doi: 10.1136/gutjnl-2013-304833
109. Wang W, Chen L, Zhou R, Wang X, Song L, Huang S, et al. Increased proportions of Bifidobacterium and the Lactobacillus group and loss of butyrate-producing bacteria in inflammatory bowel disease. J Clin Microbiol. (2014) 52:398–406. doi: 10.1128/JCM.01500-13
110. Soergel KH, Lashner BA, Hanauer SB, Vanagunas A, Sellin JH, Weinberg D, et al. Short chain fatty acid rectal irrigation for left-sided ulcerative colitis: a randomised, placebo controlled trial. Gut (1997) 40:485–91.
111. Scheppach W, Sommer H, Kirchner T, Paganelli GM, Bartram P, Christl S, et al. Effect of butyrate enemas on the colonic mucosa in distal ulcerative colitis. Gastroenterology (1992) 103:51–6. doi: 10.1016/0016-5085(92)91094-K
112. Chapman MA, Grahn MF, Boyle MA, Hutton M, Rogers J, Williams NS. Butyrate oxidation is impaired in the colonic mucosa of sufferers of quiescent ulcerative colitis. Gut (1994) 35:73–6. doi: 10.1136/gut.35.1.73
113. De Preter V, Arijs I, Windey K, Vanhove W, Vermeire S, Schuit F, et al. Impaired butyrate oxidation in ulcerative colitis is due to decreased butyrate uptake and a defect in the oxidation pathway. Inflamm Bowel Dis. (2012) 18:1127–36. doi: 10.1002/ibd.21894
114. Duffy MM, Regan MC, Ravichandran P, O'Keane C, Harrington MG, Fitzpatrick JM, et al. Mucosal metabolism in ulcerative colitis and Crohn's disease. Dis Colon Rectum (1998) 41:1399–405. doi: 10.1007/BF02237056
115. De Preter V, Rutgeerts P, Schuit F, Verbeke K, Arijs I. Impaired expression of genes involved in the butyrate oxidation pathway in crohn's disease patients. Inflamm Bowel Dis. (2013) 19:43–4. doi: 10.1002/ibd.22970
116. Thibault R, De Coppet P, Daly K, Bourreille A, Cuff M, Bonnet C, et al. Down-regulation of the monocarboxylate Transporter 1 is involved in butyrate deficiency during intestinal inflammation. Gastroenterology (2007) 133:1916–27. doi: 10.1053/j.gastro.2007.08.041
117. Hartog A, Belle FN, Bastiaans J, De Graaff P, Garssen J, Harthoorn LF, et al. A potential role for regulatory T-cells in the amelioration of DSS induced colitis by dietary non-digestible polysaccharides. J Nutr Biochem. (2015) 26:227–33. doi: 10.1016/j.jnutbio.2014.10.011
118. Wang H, Shi P, Zuo L, Dong J, Zhao J, Liu Q, et al. Dietary non-digestible polysaccharides ameliorate intestinal epithelial barrier dysfunction in IL-10 knockout mice. J Crohns Colitis (2016) 10:1076–86. doi: 10.1093/ecco-jcc/jjw065
119. Silveira ALM, Ferreira AVM, de Oliveira MC, Rachid MA, da Cunha Sousa LF, dos Santos Martins F, et al. Preventive rather than therapeutic treatment with high fiber diet attenuates clinical and inflammatory markers of acute and chronic DSS-induced colitis in mice. Eur J Nutr. (2017) 56:179–91. doi: 10.1007/s00394-015-1068-x
120. Wedlake L, Slack N, Andreyev HJN, Whelan K. Fiber in the treatment and maintenance of inflammatory bowel disease: a systematic review of randomized controlled trials. Inflamm Bowel Dis. (2014) 20:576–86. doi: 10.1097/01.MIB.0000437984.92565.31
121. Benjamin JL, Hedin CRH, Koutsoumpas A, Ng SC, McCarthy NE, Hart AL, et al. Randomised, double-blind, placebo-controlled trial of fructo-oligosaccharides in active Crohn's disease. Gut (2011) 60:923–9. doi: 10.1136/gut.2010.232025
122. Kovatcheva-Datchary P, Nilsson A, Akrami R, Lee YS, De Vadder F, Arora T, et al. Dietary fiber-induced improvement in glucose metabolism is associated with increased abundance of prevotella. Cell Metab. (2015) 22:971–82. doi: 10.1016/j.cmet.2015.10.001
123. Halpin SJ, Ford AC. Prevalence of symptoms meeting criteria for irritable bowel syndrome in inflammatory bowel disease: systematic review and meta-analysis. Am J Gastroenterol. (2012) 107:1474–82. doi: 10.1038/ajg.2012.260
124. Simrén M, Axelsson J, Gillberg R, Abrahamsson H, Svedlund J, Björnsson ES. Quality of life in inflammatory bowel disease in remission: the impact of IBS-like symptoms and associated psychological factors. Am J Gastroenterol. (2002) 97:389–96. doi: 10.1111/j.1572-0241.2002.05475.x
125. Staudacher HM, Whelan K. The low FODMAP diet: recent advances in understanding its mechanisms and efficacy in IBS. Gut (2017) 66:1517–27. doi: 10.1136/gutjnl-2017-313750
126. Cox SR, Prince AC, Myers CE, Irving PM, Lindsay JO, Lomer MC, et al. Fermentable Carbohydrates [FODMAPs] exacerbate functional gastrointestinal symptoms in patients with inflammatory bowel disease: a randomised, double-blind, placebo-controlled, cross-over, re-challenge trial. J Crohns Colitis (2017)1420–9. doi: 10.1093/ecco-jcc/jjx073
127. Zhan YL, Zhan YA, Dai SX. Is a low FODMAP diet beneficial for patients with inflammatory bowel disease? A meta-analysis and systematic review. Clin Nutr. (2017) 37:123–9. doi: 10.1016/j.clnu.2017.05.019
128. Zhou SY, Gillilland M, Wu X, Leelasinjaroen P, Zhang G, Zhou H, et al. FODMAP diet modulates visceral nociception by lipopolysaccharide-mediated intestinal inflammation and barrier dysfunction. J Clin Invest. (2018) 128:267–80. doi: 10.1172/JCI92390
129. Kim KA, Gu W, Lee IA, Joh EH, Kim DH. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS ONE (2012) 7:e47713. doi: 10.1371/journal.pone.0047713
130. Gulhane M, Murray L, Lourie R, Tong H, Sheng YH, Wang R, et al. High fat diets induce colonic epithelial cell stress and inflammation that is reversed by IL-22. Sci Rep. (2016) 6:1–17. doi: 10.1038/srep28990
131. Cani PD, Bibiloni R, Knauf C, Neyrinck AM, Delzenne NM. Changes in gut microbiota control metabolic diet–induced obesity and diabetes in mice. Diabetes (2008) 57:1470–81. doi: 10.2337/db07-1403.
132. Martinez-Medina M, Denizot J, Dreux N, Robin F, Billard E, Bonnet R, et al. Western diet induces dysbiosis with increased e coli in CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut (2014) 63:116–24. doi: 10.1136/gutjnl-2012-304119
133. Hou JK, Abraham B, El-Serag H. Dietary intake and risk of developing inflammatory bowel disease: A systematic review of the literature. Am J Gastroenterol. (2011) 106:563–73. doi: 10.1038/ajg.2011.44
134. Shores DR, Binion DG, Freeman BA, Baker PRS. New insights into the role of fatty acids in the pathogenesis and resolution of inflammatory bowel disease. Inflamm Bowel Dis. (2011) 17:2192–204. doi: 10.1002/ibd.21560
135. Chen C, Shah YM, Morimura K, Krausz KW, Miyazaki M, Richardson TA, et al. Metabolomics reveals that hepatic Stearoyl-CoA Desaturase 1 downregulation exacerbates inflammation and acute colitis. Cell Metab. (2008) 7:135–47. doi: 10.1016/j.cmet.2007.12.003
136. Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/-mice. Nature (2012) 487:104–8. doi: 10.1038/nature11225
137. Miyazaki M, Kim YC, Gray-Keller MP, Attie AD, Ntambi JM. The biosynthesis of hepatic cholesterol esters and triglycerides is impaired in mice with a disruption of the gene for stearoyl-CoA desaturase 1. J Biol Chem. (2000) 275:30132–8. doi: 10.1074/jbc.M005488200
138. Liu-Wu Y, Hurt-Camejo E, Wiklund O. Lysophosphatidylcholine induces the production of IL-1beta by human monocytes. Atherosclerosis (1998) 137:351–7.
139. Fernández-Bañares F, Esteve-Comas M, Mañé J, Navarro E, Bertrán X, Cabré E, et al. Changes in mucosal fatty acid profile in inflammatory bowel disease and in experimental colitis: a common response to bowel inflammation. Clin Nutr. (1997) 16:177–83. doi: 10.1016/S0261-5614(97)80003-9
140. Esteve-Comas M, Nunez MC, Fernandez-Banares F, Abad-Lacruz A, Gil A, Cabre E, et al. Abnormal plasma polyunsaturated fatty acid pattern in non-active inflammatory bowel disease. Gut (1993) 34:1370–3. doi: 10.1136/gut.34.10.1370
141. Serra-Majem L, Roman B, Estruch R. Scientific evidence of interventions using the Mediterranean diet: a systematic review. Nutr Rev. (2006) 64:S27–47. doi: 10.1301/nr.2006.feb.S27
142. De Silva PSA, Luben R, Shrestha SS, Khaw KT, Hart AR. Dietary arachidonic and oleic acid intake in ulcerative colitis etiology: a prospective cohort study using 7-day food diaries. Eur J Gastroenterol Hepatol. (2014) 26:11–8. doi: 10.1097/MEG.0b013e328365c372
143. Camuesco D, Gálvez J. Dietary olive oil supplemented with fish oil, rich in EPA and DHA (n-3) polyunsaturated fatty acids, attenuates colonic inflammation in rats with DSS-induced colitis. J Nutr. (2005) 135:687–94. doi: 10.1093/jn/135.4.687
144. Morita H, Makanishi K, Dohi T, Yasugi E, Oshima M. Phospholipid turnover in the inflammed intestinal mucosa: arachidonic ccid-rich phosphatidyl/plasmenyl-ethanolamine in the mucosa in inflammatory bowel disease. J Gastroenterol. (1999) 34:46–53.
145. Serhan CN, Chiang N. Endogenous pro-resolving and anti-inflammatory lipid mediators: a new pharmacologic genus. Br J Pharmacol. (2009) 153:S200–15. doi: 10.1038/sj.bjp.0707489
146. Arita M, Yoshida M, Hong S, Tjonahen E, Glickman JN, Petasis NA, et al. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc Natl Acad Sci USA. (2005) 102:7671–6. doi: 10.1073/pnas.0409271102
147. Haas-Stapleton EJ, Lu Y, Hong S, Arita M, Favoreto S, Nigam S, et al. Candida albicans modulates host defense by biosynthesizing the pro-resolving mediator resolvin E1. PLoS ONE (2007) 2:e1316. doi: 10.1371/journal.pone.0001316
148. Arita M, Bianchini F, Aliberti J, Sher A, Chiang N, Hong S, et al. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J Exp Med. (2005) 201:713–22. doi: 10.1084/jem.20042031
149. Ishida T, Yoshida M, Arita M, Nishitani Y, Nishiumi S, Masuda A, et al. Resolvin E1, an endogenous lipid mediator derived from eicosapentaenoic acid, prevents dextran sulfate sodium-induced colitis. Inflamm Bowel Dis. (2010) 16:87–95. doi: 10.1002/ibd.21029
150. Lee JY, Plakidas A, Lee WH, Heikkinen A, Chanmugam P, Bray G, et al. Differential modulation of Toll-like receptors by fatty acids. J Lipid Res. (2003) 44:479–86. doi: 10.1194/jlr.M200361-JLR200
151. Novak TE, Babcock TA, Jho DH, Helton WS, Espat NJ. NF-κB inhibition by ω-3 fatty acids modulates LPS-stimulated macrophage TNF-α transcription. Am J Physiol. (2003) 284:L84–9. doi: 10.1152/ajplung.00077.2002
152. Simopoulos AP. The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp Biol Med. (2008) 233:674–88. doi: 10.3181/0711-MR-311
153. Jia Q, Ivanov I, Zlatev ZZ, Alaniz RC, Eeks BR, Calla Ay ES, et al. Dietary fish oil and curcumin combine to modulate colonic cytokinetics and gene expression in dextran sodium sulphate-treated mice. Br J Nutr. (2011) 106:519–29. doi: 10.1017/S0007114511000390
154. Hassan A, Ibrahim A, Mbodji K, Coëffier M, Ziegler F, Bounoure F, et al. An α-linolenic acid-rich formula reduces oxidative stress and inflammation by regulating NF-κB in rats with TNBS-induced colitis. J Nutr. (2010) 140:1714–21. doi: 10.3945/jn.109.119768
155. Matsunaga H, Hokari R, Kurihara C, Okada Y, Takebayashi K, Okudaira K, et al. Omega-3 fatty acids exacerbate DSS-induced colitis through decreased adiponectin in colonic subepithelial myofibroblasts. Inflamm Bowel Dis. (2008) 14:1348–57. doi: 10.1002/ibd.20491
156. Hudert CA, Weylandt KH, Lu Y, Wang J, Hong S, Dignass A, et al. Transgenic mice rich in endogenous omega-3 fatty acids are protected from colitis. Proc Natl Acad Sci USA. (2006) 103:11276–81. doi: 10.1073/pnas.0601280103
157. Turner D, Shah PS, Steinhart AH, Zlotkin S, Griffiths AM. Maintenance of remission in inflammatory bowel disease using omega-3 fatty acids (fish oil): A systematic review and meta-analyses. Inflamm Bowel Dis. (2011) 17:336–45. doi: 10.1002/ibd.21374
158. Turner D, Zlotkin SH, Shah PS, Griffiths AM. Omega 3 fatty acids (fish oil) for maintenance of remission in Crohn ' s disease (Review). Cochrane Libr. (2014) CD006320. doi: 10.1002/14651858.CD006320.pub2
159. Fontaine-bisson B, Wolever TMS, Chiasson J, Rabasa-Lhoret R, Maheux P, Josse RG, et al. Genetic polymorphisms of tumor necrosis factor-alpha modify the association between dietary polyunsaturated fatty acids and fasting HDL-concentrations and apo A-1 concentrations. Am J Clin Nutr. (2007) 86:768–74. doi: 10.1093/ajcn/86.3.768
160. Paradis A-M, Fontaine-Bisson B, Bossé Y, Robitaille J, Lemieux S, Jacques H, et al. The peroxisome proliferator-activated receptor alpha Leu162Val polymorphism influences the metabolic response to a dietary intervention altering fatty acid proportions in healthy men. Am J Clin Nutr. (2005) 81:523–30. doi: 10.1093/ajcn.81.2.523
161. Ferreira P, Cravo M, Guerreiro CS, Tavares L, Santos PM, Brito M. Fat intake interacts with polymorphisms of Caspase9, FasLigand and PPARgamma apoptotic genes in modulating Crohn's disease activity. Clin Nutr. (2010) 29:819–23. doi: 10.1016/j.clnu.2010.06.008
162. Ananthakrishnan AN, Khalili H, Song M, Higuchi LM, Lochhead P, Richter JM, et al. Genetic polymorphisms in fatty acid metabolism modify the association between dietary n3. Inflamm Bowel Dis. (2017) 23:1898–904. doi: 10.1097/MIB.0000000000001236
163. Zhang YJ, Gan RY, Li S, Zhou Y, Li AN, Xu DP, et al. Antioxidant phytochemicals for the prevention and treatment of chronic diseases. Molecules (2015) 20:21138–56. doi: 10.3390/molecules201219753
164. Maheshwari RK, Singh AK, Gaddipati J, Srimal RC. Multiple biological activities of curcumin: a short review. Life Sci. (2006) 78:2081–7. doi: 10.1016/j.lfs.2005.12.007
165. Wang J, Ghosh SS, Ghosh S. Curcumin improves intestinal barrier function: modulation of intracellular signaling, and organization of tight junctions. Am J Physiol. (2017) 312:438–45. doi: 10.1152/ajpcell.00235.2016
166. Pan MH, Lin-Shiau SY, Lin JK. Comparative studies on the suppression of nitric oxide synthase by curcumin and its hydrogenated metabolites through down-regulation of IκB kinase and NFκB activation in macrophages. Biochem Pharmacol. (2000) 60:1665–76. doi: 10.1016/S0006-2952(00)00489-5
167. Yang J, Zhong X, Kim S, Kim D, Kim HS, Lee J, et al. Comparative effects of curcumin and tetrahydrocurcumin on dextran sulfate sodium-induced colitis and inflammatory signaling in mice. J Cancer Prev. (2018) 23:18–24. doi: 10.15430/JCP.2018.23.1.18
168. Sugimoto K, Hanai H, Tozawa K, Aoshi T, Uchijima M, Nagata T, et al. Curcumin prevents and ameliorates trinitrobenzene sulfonic acid-induced colitis in mice. Gastroenterology (2002) 123:1912–22. doi: 10.1053/gast.2002.37050
169. Manach C. Polyphenols: food sources and bioavailability. Am J Clin Nutr. (2004) 79:727–47. doi: 10.1038/nature05488
170. Cardona F, Andrés-Lacueva C, Tulipani S, Tinahones FJ, Queipo-Ortuño MI. Benefits of polyphenols on gut microbiota and implications in human health. J Nutr Biochem. (2013) 24:1415–22. doi: 10.1016/j.jnutbio.2013.05.001
171. Hassaninasab A, Hashimoto Y, Tomita-Yokotani K, Kobayashi M. Discovery of the curcumin metabolic pathway involving a unique enzyme in an intestinal microorganism. Proc Natl Acad Sci USA. (2011) 108:6615–20. doi: 10.1073/pnas.1016217108
172. Lai CS, Wu JC, Yu SF, Badmaev V, Nagabhushanam K, Ho CT, et al. Tetrahydrocurcumin is more effective than curcumin in preventing azoxymethane-induced colon carcinogenesis. Mol Nutr Food Res. (2011) 55:1819–28. doi: 10.1002/mnfr.201100290
173. Lu Y, Zamora-Ros R, Chan S, Cross AJ, Ward H, Jakszyn P, et al. Dietary polyphenols in the aetiology of crohn's disease and ulcerative colitis-A multicenter European Prospective Cohort Study (EPIC). Inflamm Bowel Dis. (2017) 23:2072–82. doi: 10.1097/MIB.0000000000001108
174. Hanai H, Iida T, Takeuchi K, Watanabe F, Maruyama Y, Andoh A, et al. Curcumin maintenance therapy for ulcerative colitis: randomized, multicenter, double-blind, placebo-controlled trial. Clin Gastroenterol Hepatol. (2006) 4:1502–6. doi: 10.1016/j.cgh.2006.08.008
175. Lang A, Salomon N, Wu JCY, Kopylov U, Lahat A, Har-Noy O, et al. Curcumin in combination with mesalamine induces remission in patients with mild-to-moderate ulcerative colitis in a randomized controlled trial. Clin Gastroenterol Hepatol. (2015) 13:1444–9. doi: 10.1016/j.cgh.2015.02.019
176. Roberts CL, Rushworth SL, Richman E, Rhodes JM. Hypothesis: increased consumption of emulsifiers as an explanation for the rising incidence of Crohn's disease. J Crohns Colitis (2013) 7:338–41. doi: 10.1016/j.crohns.2013.01.004
177. Chassaing B, Koren O, Goodrich JK, Poole AC, Srinivasan S, Ley RE, et al. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature (2015) 519:92–6. doi: 10.1038/nature14232
178. Viennois E, Merlin D, Gewirtz AT, Chassaing B. Dietary emulsifier-induced low-grade inflammation promotes colon carcinogenesis. Cancer Res. (2017) 77:27–40. doi: 10.1158/0008-5472.CAN-16-1359
179. Suez J, Korem T, Zeevi D, Zilberman-Schapira G, Thaiss CA, Maza O, et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature (2014) 514:181–6. doi: 10.1038/nature13793
180. Rodriguez-Palacios A, Harding A, Menghini P, Himmelman C, Retuerto M, Nickerson KP, et al. The artificial sweetener splenda promotes gut proteobacteria, dysbiosis, and myeloperoxidase reactivity in crohn's disease-like ileitis. Inflamm Bowel Dis. (2018) 24:1005–20. doi: 10.1093/ibd/izy060
181. Sugihara K, Masuda M, Nakao M, Abuduli M, Imi Y, Oda N, et al. Dietary phosphate exacerbates intestinal inflammation in experimental colitis. J Clin Biochem Nutr. (2017) 61:91–9. doi: 10.3164/jcbn.16
182. Jowett SL, Seal CJ, Pearce MS, Phillips E, Gregory W, Barton JR, et al. Influence of dietary factors on the clinical course of ulcerative colitis: a prospective cohort study. Gut (2004) 53:1479–84. doi: 10.1136/gut.2003.024828
Keywords: inflammatory bowel disease (IBD), diet, microbiota, immunity, intestinal barrier
Citation: Sugihara K, Morhardt TL and Kamada N (2019) The Role of Dietary Nutrients in Inflammatory Bowel Disease. Front. Immunol. 9:3183. doi: 10.3389/fimmu.2018.03183
Received: 13 June 2018; Accepted: 27 December 2018;
Published: 15 January 2019.
Edited by:
Thomas Thornton MacDonald, Queen Mary University of London, United KingdomReviewed by:
Luca Pastorelli, University of Milan, ItalyTimothy L. Denning, Georgia State University, United States
Jun Kunisawa, NIBIOHN, Japan
Copyright © 2019 Sugihara, Morhardt and Kamada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nobuhiko Kamada, bmthbWFkYUB1bWljaC5lZHU=