 Vandana Kalia1,2*
Vandana Kalia1,2* Surojit Sarkar1,2,3,4*
Surojit Sarkar1,2,3,4*- 1Division of Hematology and Oncology, Department of Pediatrics, University of Washington School of Medicine, Seattle, WA, United States
- 2Ben Towne Center for Childhood Cancer Research, Seattle Children's Research Institute, Seattle, WA, United States
- 3M3D Graduate Program, University of Washington School of Medicine, Seattle, WA, United States
- 4Department of Pathology, University of Washington School of Medicine, Seattle, WA, United States
Interleukin-2 (IL-2) regulates key aspects of CD8 T cell biology–signaling through distinct pathways IL-2 triggers critical metabolic and transcriptional changes that lead to a spectrum of physiological outcomes such as cell survival, proliferation, and effector differentiation. In addition to driving effector differentiation, IL-2 signals are also critical for formation of long-lived CD8 T cell memory. This review discusses a model of rheostatic control of CD8 T cell effector and memory differentiation by IL-2, wherein the timing, duration, dose, and source of IL-2 signals are considered in fine-tuning the balance of key transcriptional regulators of cell fate.
Introduction
Interleukin-2 (IL-2)–the first cytokine to be identified and characterized more than three decades ago—has emerged as a pleiotropic player in a variety of seemingly paradoxical immune functions. Originally discovered for its immunoenhancing role of promoting T cell expansion during mitogenic stimulation, IL-2 is also implicated in activation-induced cell death (AICD). Likewise, IL-2 promotes a variety of effector (cytotoxic CD8, TH1) T cell responses, yet is indispensable for the development, maintenance and function of regulatory T cells (Treg)—the very cells that serve to suppress effector T cell responses. Further adding to the intrigue, even amongst the effector subsets, IL-2 promotes CD8, TH1, and TH2 effector responses, but suppresses inflammatory TH17 responses, and also inhibits the differentiation of follicular helper T (TFH) cells required for B cell germinal center reactions in secondary lymphoid organs. Collectively, these findings support the thesis that IL-2 critically regulates the balance of immunostimulatory and immunosuppressive forces during immune responses to foreign antigens as well as self-antigens during homeostasis. While our understanding of the molecular, transcriptional, and metabolic regulation of CD4 T cell differentiation into TH1, TH2, TH17, TFH, and Treg subsets by IL-2 is abundant [see previous reviews (1–5)], the IL-2-dependent gene regulatory networks that drive effector and memory CD8 T cell differentiation remain to be fully defined. In this review we will focus on IL-2 regulation of CD8 T cell responses; alongside a summary of current literature in the context of CD4 and CD8 T cells, we will also discuss how this niche area is poised for significant advances owing to newer tools such as conditional ablation of IL-2 production and signaling in key subsets of immune cells in the physiologically relevant setting of immunocompetent hosts.
Balancing Primary and Secondary CD8 T Cell Immunity
CD8 T Cell Responses to Acute Infections
A typical CD8 T cell response to primary infection with acute viral or intracellular bacterial pathogens is characterized by three distinct phases—expansion, contraction, and memory. Upon stimulation with cognate antigen in conjunction with costimulatory and inflammatory ligands, naïve cells undergo massive clonal expansion (up to 50,000-fold) and concomitant effector differentiation to generate large numbers of cytotoxic T lymphocytes (CTL), which serve to control the pathogen by migrating to peripheral sites of infection and elaborating cytotoxicity against infected target cells and producing effector cytokines such as IFN-γ and TNF-α (6–11). It is now well-established that the effector CTL pool broadly contains two distinct subsets—(1) short-lived effector cells (SLECs), which are fated to rapidly die after pathogen clearance, and (2) memory precursor effector cells (MPECs) (12–16), which are imprinted with antigen-independent survival capabilities for mediating long-term protection against secondary challenge (17–19). Thus, supporting the concept of memory programming, or imprinting of cardinal memory properties during primary expansion (20–23), several studies have now demonstrated that the balance of MPECs and SLECs can be altered by manipulating the duration of antigen, IL-2 and other inflammatory cytokine signals (14–16, 24). In fact, as discussed later, the heterogeneity of the memory CD8 T cell pool is likely programmed by differential signals accrued during the primary expansion phase. IL-2 signals (paracrine or autocrine) in particular exert crucial roles in effector and memory differentiation and function.
Regulation of Effector CD8 T Cell Responses by IL-2
Optimal T cell activation with cognate peptide-MHC-I and costimulatory ligands result in IL-2 production and induction of IL-2Rα (CD25) expression, which along with IL-2Rβ (CD122, also used for IL-15 signaling), and IL-2Rγ (CD132, also referred to as common γ-chain as it is shared by other cytokines of the γ-chain family such as IL-4, 7, 9, 21) (5), forms the high affinity heterotrimeric receptor for robust IL-2 signal transduction and clonal expansion and effector differentiation (2). Much of the early work on IL-2 regulation of T cell responses relied on reductionist in vitro studies where amount and duration of TCR and IL-2 stimulation can be tightly controlled. These studies established a critical role for IL-2 as a T cell growth factor in driving cell cycle progression and expansion of CD8 T cells following TCR stimulation (25). Similar conclusions were reached following in vivo administration of IL-2, which engendered enhanced effector and memory pools of antigen-specific CD8 T cells (26–29). While these studies demonstrate that CD8 T cell differentiation events are amenable to manipulation by IL-2, physiological relevance of IL-2 in shaping a developing CD8 T cell response was uncovered following the development of Il2 germline-deleted mice. Studies in IL-2 knockout mice are confounded by Treg deficiency and associated spontaneous lymphoproliferative disease (30, 31). Hence, irreconcilably disparate outcomes of reduced or unaltered expansion and effector differentiation were reported in the context of infections and peptide immunization in IL-2 knockout mice (32–35). Nonetheless, bypassing pleiotropic immune effects in straight IL-2 and IL-2Rα (CD25) knockout mice, subsequent studies engaged the strategy of adoptively transferring IL-2- or IL-2Rα-deficient TCR transgenic CD8 T cells into wild-type recipients. In these studies, enumeration of antigen-specific CD8 T cells in an otherwise wild-type milieu using congenic differences without the need for restimulation, clearly established a requirement for IL-2 signals in driving optimal primary expansion of antigen-specific CD8 T cells in secondary lymphoid as well as non-lymphoid tissues (36, 37). IL-2 promotes effector differentiation through STAT-5-mediated Blimp-1-dependent induction of effector molecules (16, 38–42). In this regard, proinflammatory cytokine signals such as IL-12, IFN-γ, and type-1 interferons (IFN-α/β)—commonly referred to as signal 3 for their role in promoting optimal clonal expansion of effector CD8 T cells—are believed to complement IL-2, possibly non-redundantly (43, 44). Such collaboration, particularly between IL-12 and IL-2 has been recently shown to be important for optimal expression of transcription factors T-bet and Blimp-1, which synergize to drive a terminal effector differentiation program in CD8 T cells (45).
Regulation of Memory CD8 T Cell Responses by IL-2
In addition to promoting CD8 T cell expansion and effector differentiation, IL-2 signals are also necessary for memory responses. IL-2Rα upregulation early after TCR stimulation is critical for formation of memory cells with robust secondary expansion capability (46, 47). Subsequent correlations of the duration of IL-2Rα expression with final memory outcome in a physiologically relevant setting—where the natural course of CD8 T cell response was not disturbed—revealed that rapid downregulation of IL-2Rα is equally important for memory development (16). Fate-tracking analyses showed that following an initial burst of IL-2 signals through IL-2Rα, curtailed expression of IL-2Rα and diminished IL-2 signaling is associated with memory fate, whereas prolonged expression of IL-2Rα and stronger IL-2 signaling drives terminal effector differentiation (16). Stronger IL-2 stimulation (100 U/ml) during in vitro priming also drives terminal differentiation compared to weaker signals (10 U/ml) (41). Similar findings have been reported in the DC-peptide immunization models as well as during murine infection with Lymphocytic choriomeningitis virus (LCMV), Listeria monocytogenes (LM), Vaccinia virus (VV), and Vesicular stomatitis virus (VSV) (16, 48). Moreover, constitutive activation of STAT-5 (key signal transducer of common γ-chain cytokines) also causes terminal differentiation (49). Consistent with the pro-proliferative role of IL-2, terminally differentiated effector CD8 T cells (SLECs) that express IL-2Rα for longer duration during an acute infection expand more than their memory-fated counterparts (MPECs) that downregulate the expression of IL-2Rα earlier (15, 16, 50–52). Together, these findings support the notion that metered IL-2 signals are required for optimal protective immunity and present a model of rheostatic control of CD8 T cell fates by IL-2 during acute infections.
All memory cells that survive after clearance of a primary infection are not created equal. Protective CD8 T cell immunity, as we understand it today, consists of collaborative defense against secondary challenge through concerted actions by a complex mixture of memory cells with distinct phenotypes, location, migratory properties, polyfunctionality, antigen-independent longevity, and potential for mounting rapid and robust clonal expansion and effector functions upon secondary challenge (44). As is expected from a spectrum of effector CTLs—that develop in response to varying doses and durations of antigen perceived in a variety of immune contexts, such as dose and duration of cytokines (e.g., IL-2, IFN-I, IL-12, IL-21, TGFβ, etc.), costimulatory signals, CD4 T cell interactions—a veritable spectrum of memory cells exist in a host after antigen clearance. At the risk of oversimplifying the CD8 T cell memory complexity, one can arguably categorize memory cells broadly into two major subsets—lymphoid or central memory (TCM), and non-lymphoid memory, which is further distinguished into tissue-resident memory (TRM), and migratory memory. Defined by their location, central memory cells largely recirculate through secondary lymphoid organs; tissue-resident memory (TRM) cells—true to their name—set up permanent residence at front-lines of pathogen exposure; whereas migratory memory cells comprise a heterogeneous population that is capable of recirculation to peripheral tissues, and may be further distinguished by intravascular staining methodology into the CX3CR1hi effector memory subset (TEM) which does not enter extravascular space, and the less differentiated CX3CR1int memory subset capable of migration into extravascular spaces (53, 54). TRM cells serve effectively as the first line of defense against infections by virtue of their key properties of location at barrier sites and rapid elaboration of effector functions (cytotoxicity against infected target cells and effector cytokine production). Consistent with their ability to recirculate through peripheral tissues, TEM cells retain higher expression of effector molecules, and are believed to aid TRM cells in protecting against secondary challenge along with the extravascular migratory memory cells. In contrast, TCM cells largely downregulate their effector program after antigen clearance, but are capable of rapid upregulation of the effector program upon antigenic rechallenge, also have superior polyfunctionality (ability to coproduce multiple cytokines such as IL-2, IFN-γ, and TNF-α), and expand more vigorously to aid the TRM and migratory cells during secondary challenge.
Developmentally, fate-tracking experiments show that effector CD8 T cells that rapidly downregulate IL-2Rα largely give rise to central memory and effector memory cells. In comparison, effector CTLs, with prolonged IL-2Rα expression, largely give rise to terminal effector and effector memory fates; and curtailed stimulation of these cells by adoptive transfer into infection-controlled recipients (removal of antigen, IL-2, and all other infection-related signals) results in less terminal differentiation, as evidenced by increased proportions of effector memory cells compared to short-lived effector cells. These observations are consistent with a role for increasing IL-2 in driving effector CD8 T cells progressively toward terminal differentiation. It is believed that TRM cells arise from relatively less differentiated memory precursors, which first seed the peripheral sites such as skin and small intestines (55, 56). In situ, the precursors receive microenvironment-specific developmental cues that drive the expression of unique chemokine receptors, integrins, and transcription factors for TRM cell tissue residency and local protection (56–61). Within the tissue, the transforming growth factor β (TGF-β) exerts a critical role in directing the TRM differentiation program in concert with other tissue-specific signals (55, 56, 62, 63). While CD8 TRM cells capable of IL-2 production have been recently reported in skin and liver (64, 65), and IL-2 signals have been shown to be important for maintenance of allergic TH2-type cells in the lungs (66), murine studies directed at understanding whether early IL-2 signals are necessary for TRM seeding of tissue sites, whether prolonged IL-2 signals compromise TRM cells, and how TGF-β signals and other tissue-specific factors work in conjunction with IL-2 signals (synergistically or antagonistically) to drive the differentiation, maintenance, and recall function of TRM cells within the local sites remains to be fully explored. Likewise, whether similar rules of progressive terminal differentiation with increasing IL-2 signals are also active in situations of chronic antigen stimulation—as occurs during persistent viral infections and cancers—remains to be defined.
Autocrine and Paracrine Programming of T Cell Fates
During thymic development, T cell-derived IL-2 is critical for development of Treg cells. (67). During homeostasis, IL-2 is largely produced by CD25int and CD25lo CD4 T cells (activated by self-peptide and foreign peptide MHC-II complexes on DCs) (68), the regulatory TR1 subset in peyer's patches that also produces IL-10 and IFN-γ (69), and to some extent by NK, NKT, and CD8 T cells [evaluated by mRNA (68)]. Recent studies, involving IL-2 ablation in defined immune cells, have shown that T cell-derived IL-2 is critical for maintaining numbers and regulatory function of Treg cells in most secondary lymphoid organs, with the exception of mesenteric lymph nodes where DC-derived IL-2 was also observed to be important (67). During an immune response, activated CD4 T cells produce copious amounts of IL-2 (2), with other IL-2 producers being CD8 T cells (70), DCs (71), NKT cells (72), and mast cells (73). There is evidence that IL-2 may be transpresented by CD25 expressing DCs (74) to deliver high affinity IL-2 signals to CD8 T cells that lack CD25 expression and only express the intermediate affinity β/γ IL-2 receptor heterodimer—analogously to IL-15 transpresentation—thus, suggesting that IL-2 may be delivered in a context-specific manner in vivo depending on the nature and activation status of antigen-presenting DCs.
With highest levels in secondary lymphoid organs, IL-2 is believed to act in an autocrine or paracrine manner to support effector and memory CD8 T cell differentiation. We have previously shown that memory-fated effector CD8 T cells selectively retain the ability for robust IL-2 production in response to antigenic stimulation compared to their terminally differentiated effector counterparts (8, 15, 16, 44). Likewise, polyfunctionality—the capacity for potent IL-2 production along with other effector cytokines such as IFN-γ and TNF-α in response to antigenic restimulation –is a hallmark property of lymphoid central memory CD8 T cells. Querying the functional relevance of autocrine IL-2 production by memory-fated CD8 T cells, studies involving ablation of Il2 in a fraction of antigen-specific CD8 T cells during attenuated LM immunization (75) as well as acute LCMV infection (unpublished observations), demonstrate that the IL-2 needed for development of robust memory CD8 T cells capable of optimal secondary expansion is largely autochthonous. Since CD4 T cells are the major producers of IL-2, it was long presumed that IL-2 serves as the mode of CD4 help for development of protective memory CD8 T cells capable of robust secondary expansion. Thus, largely dismissing CD4 T cell-derived paracrine IL-2 as a mode of help, it is now proposed that CD4 T cells license DCs through the CD40-CD40L axis to induce memory-fated CD8 T cells to produce IL-2 (75). Autocrine IL-2 production through CD27 signals has also been shown to sustain survival of antigen-specific CD8 T cells in virus-infected non-lymphoid tissues (76, 77).
We further employed novel conditional IL-2 gene-deleted mice (78) to investigate whether autocrine IL-2 signals are specifically required during the programming phase of primary responses, or during secondary expansion (unpublished observations). Ablation of Il2 in memory CD8 T cell immediately prior to rechallenge did not result in compromised secondary expansion, but ablation prior to primary infection resulted in defective recall responses. These data suggest that autocrine IL-2 signals during primary CD8 T cell expansion are required to institute a program of optimal secondary expansion. In the context of CD4 help to CD8 T cells, these instructive autocrine IL-2 signals are believed to in part promote the expression of a transcriptional corepressor, Nab2 for blocking TRAIL-mediated apoptosis during secondary expansion (79, 80). Defects in protective CD8 T cell immunity associated with IL-2Rα ablation are rescued by a strong bolus of exogenous IL-2 during primary expansion (47), further supporting the idea that IL-2 exerts an early instructive role. Whether secondary expansion defect associated with lack of autochthonous IL-2 maybe similarly rescued by excessive paracrine IL-2 signals remains unknown. Alternatively, it is possible that there are fundamental differences (quantitative and/or qualitative) between autocrine and paracrine IL-2 signals. In the case of CD4 T cells, autocrine IL-2 production in response to cognate antigen and CD70 signals during late stages of influenza A virus infection has been shown to be critical for upregulation of IL-7Rα (CD127) and survival into memory phase (81). More recently, TFH and TH1 fates have been linked to autocrine and paracrine IL-2 signals, respectively, with different gene expression programs being triggered for lineage determination in IL-2-producing and non-producing CD4 T cells (82). While CD8 T cells that receive differential strength or duration of IL-2 signals have expectedly unique gene expression programs, it remains to be defined how autocrine and paracrine IL-2 signals impact CD8 T cell gene regulation and metabolism.
Fine-Tuning The Regulators of T Cell Fates
Transcriptional and Metabolic Regulation of CD8 T Cell Differentiation
IL-2 couples T cell expansion and effector differentiation through induction of multiple downstream signaling cascades. Expression of pro-differentiation transcription factors, Blimp-1 (16, 38, 40–42) and Id-2 (83), is largely mediated through STAT-5 activation in response to IL-2 stimulation (2) (Figure 1). Reciprocal suppression by IL-2 of transcriptional factors that promote T cell memory such as Bcl-6 (41, 84–88) (which also represses Blimp-1 expression) is believed to further fix the terminal effector differentiation program (45). IL-2 is believed to regulate the expression of Bcl-6 through activation of Akt, which serves to control the activity of Foxo family transcription factors (89), Activation of Akt also alters the expression of proteins involved in CD8 T cell trafficking such as CD62L, CCR7, and S1P1, so as to promote their migration to peripheral sites of infection and inflammation (90–92). In addition to activation of STAT-5 and Akt, which largely promote effector differentiation, IL-2 links effector differentiation with clonal expansion through activation of MAPK signaling and T cell activation, cell cycle progression and survival programs (89). Sustained expression of cMYC through IL-2 drives proliferation by upregulating cyclins and anti-apoptotic molecule B-cell lymphoma 2 (Bcl-2), and by downregulating p21 (93, 94). In addition to cell cycle regulators, Myc also controls key metabolic aspects of T cell activation and proliferation (95). Myc promotes glycolysis and glutaminolysis through upregulation of key enzymatic and transporter proteins (96, 97). In this regard, mTOR also serves as a primary hub to integrate environmental cues from growth factors such as nutrients and IL-2 to promote glycolysis (94, 98, 99), oxidative phosphorylation and anabolic processes such as protein, lipid, and nucleotide biosynthesis necessary to sustain proliferation (96, 97). How effector and memory CD8 T cell fates are defined in vivo through differential metabolic programming by varying IL-2 strength or duration remains to be elucidated.
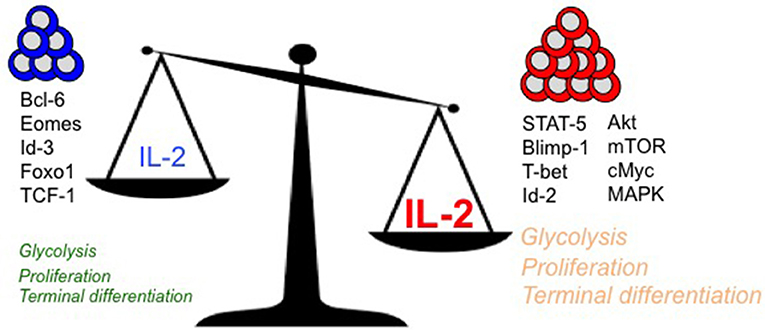
Figure 1. Regulation of key intracellular signaling, transcriptional, and metabolic mediators of terminal effector vs. long-lived polyfunctional memory CD8 T cell fates are presented in the context of differential levels of IL-2 signals.
A Model for Rheostatic Control of T Cell Fates by IL-2
With diverse outcomes of memory and terminal effector differentiation in CD8 T cells that receive short/weak as opposed to prolonged/strong IL-2 signals (as described earlier), it remains to be determined whether rheostatic control of transcriptional and metabolic regulation occurs. It is plausible that curtailed or weak IL-2 signals drive lower levels of STAT-5, Akt and mTOR activity, thus resulting in lesser proliferation, effector differentiation and trafficking to peripheral sites of infection. In contrast, strong and prolonged IL-2 signals may drive stronger STAT-5, Akt, mTOR and MAPK activity, thus leading to augmented proliferation, effector differentiation and migration to peripheral sites of infection, where the microenvironmental niches further reinforce the terminal differentiation programs through induction of receptors for inflammatory cytokines such as IL-12 (100), and inhibition of IL-7 (101) receptor levels. Notably, a role for Tregs has been implicated in regulating the amount of IL-2 signals to memory-fated CD8 T cells by acting as IL-2 sinks (102) during CD8 T cell expansion. During later stages in the absence of antigen (when IL-2 is limiting) also, Tregs continue to curtail T cell stimulation and proliferation to maintain memory CD8 T cell quiescence through CTLA-4 (103) and IL-10 (104) inhibitory mechanisms and possibly through IL-2 restriction (105). IL-2 is also bound to the extracellular matrix through heparan sulfate moieties (106) to presumably increase local concentrations, thus supporting the notion that strong and prolonged IL-2 signals can be achieved in vivo. Effectually, quantal differences in IL-2 signals may lead to differences in signaling thresholds that ultimately result in terminal effector gene expression patterns driven by Blimp-1, T-bet, Id-2, and cMyc, or in memory lineage gene expression patterns characterized by augmented Bcl-6, Eomesodermin and Id-3. Indeed, analogous rheostatic control of CD4 T cell fates by differential levels of IL-2 signaling has been reported in the balance of TH1 and TFH fate determination (107, 108) through reciprocal regulation of T-bet and Bcl-6 by mTORC1-dependent control of the glycolysis gene expression program (109).
Concluding Remarks
Tightly coupled to antigen and costimulation, IL-2 signals follow close suit in T cell activation. In addition to driving expansion and effector differentiation, IL-2 regulates long-term memory outcome as well. Hence, it has been proposed as vaccine adjuvant (110) to augment the size of the memory pool. However, given its rheostatic regulation of terminal effector and memory fates (Figure 1), careful investigation into the dose and duration of IL-2 in a context specific manner is warranted to fine-tune the balance of terminal effector and memory lineages. Hence, based on the clinical need, timely and curtailed IL-2 signals might be exploited to augment memory outcome during vaccination. Alternatively—owing to its ability to induce proliferation and effector differentiation–strong and sustained IL-2 signals might be employed for immunotherapeutic interventions against cancers and chronic infections that rely on activation of a large pool of antigen-specific CD8 T cells. In this quest, IL-2 has gained particular recognition in treating melanomas and renal cell carcinomas (111) by augmenting the tumor-reactive CD8 T cell pool. In the case of gene-modified T cell immunotherapies also—for e.g., when patient T cells are bioengineered to express chimeric antigen receptors or TCRs directed against select tumor antigens–IL-2 is critical for expansion of CAR T cells to sufficient numbers for therapeutic benefit (112). Even in the case of PD-1 checkpoint blockade immunotherapy, IL-2 supplementation has offered combinatorial success with PD-1 blockade in boosting quantitative and functional aspects of exhausted CD8 T cells for enhanced viral control (113). Needless to say, the pleiotropic effects of IL-2 have posed significant hurdles such as off-target side effects of IL-2 administration—e.g., vascular leak syndrome due to activation of endothelial cells, or induction of immune regulation by Tregs. To minimize side effects, novel IL-2 muteins and immune complexes have been developed to selectively target IL-2 to either effector or regulatory T cells (5, 111, 114–116). By enhancing IL-2 binding to the β/γ heterodimer typically expressed on effector CD8 T cells, and thus directing IL-2 away from Tregs—which typically express high levels of IL-2Rα–these immune complexes and muteins provide a means to avoid concomitant induction of Treg suppression observed in case of rIL-2 administration that is counteractive to the desired outcome of effector differentiation. We envisage that detailed molecular dissection of the signal transduction and transcriptional networks downstream of IL-2 signaling vis a vis biological outcomes in individual immune cell-types will guide innovative immunomodulatory strategies designed for distinct clinical mandates. Along this concept, manipulations of the Bcl-6-Blimp-1 and CD27-CD70 axes are being considered with the goal of uncoupling effector differentiation effects of IL-2 from expansion effects (89). Beyond the binary terminal effector or memory outcomes conceived thus far, it is enticing to speculate whether rheostatic regulation of MPEC and SLEC differentiation states by controlling IL-2 signals might be exploited to balance the immediate therapeutic benefits and long-term protective outcomes during adoptive T cell therapy and therapeutic cancer vaccines.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by NIH grant R03AI113635 to VK and by research funds from Seattle Children's Research Institute.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bachmann MF, Oxenius A. Interleukin 2: from immunostimulation to immunoregulation and back again. EMBO Rep. (2007) 8:1142–8. doi: 10.1038/sj.embor.7401099
2. Malek TR. The biology of interleukin-2. Annu Rev Immunol. (2008) 26:453–79. doi: 10.1146/annurev.immunol.26.021607.090357
3. Malek TR, Castro I. Interleukin-2 receptor signaling: at the interface between tolerance and immunity. Immunity (2010) 33:153–65. doi: 10.1016/j.immuni.2010.08.004
4. Liao W, Lin JX, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity (2013) 38:13–25. doi: 10.1016/j.immuni.2013.01.004
5. Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol. (2012) 12:180–90. doi: 10.1038/nri3156
6. Arens R, Schoenberger SP. Plasticity in programming of effector and memory CD8 T-cell formation. Immunol Rev. (2010) 235:190–205. doi: 10.1111/j.0105-2896.2010.00899.x
7. Cui W, Kaech SM. Generation of effector CD8+ T cells and their conversion to memory T cells. Immunol Rev. (2010) 236:151–66. doi: 10.1111/j.1600-065X.2010.00926.x
8. Kalia V, Sarkar S, Ahmed R. CD8 T-cell memory differentiation during acute and chronic viral infections. Adv Exp Med Biol. (2010) 684:79–95. doi: 10.1007/978-1-4419-6451-9_7
9. Williams MA, Bevan MJ. Effector and memory CTL differentiation. Annu Rev Immunol. (2007) 25:171–92. doi: 10.1146/annurev.immunol.25.022106.141548
10. Harty JT, Badovinac VP. Shaping and reshaping CD8+ T-cell memory. Nat Rev Immunol. (2008) 8:107–19. doi: 10.1038/nri2251
11. Lefrancois L, Obar JJ. Once a killer, always a killer: from cytotoxic T cell to memory cell. Immunol Rev. (2010) 235:206–18. doi: 10.1111/j.0105-2896.2010.00895.x
12. Huster KM, Busch V, Schiemann M, Linkemann K, Kerksiek KM, Wagner H, et al. Selective expression of IL-7 receptor on memory T cells identifies early CD40L-dependent generation of distinct CD8+ memory T cell subsets. Proc Natl Acad Sci USA. (2004) 101:5610–5. doi: 10.1073/pnas.0308054101
13. Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. (2003) 4:1191–8. doi: 10.1038/ni1009
14. Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity (2007) 27:281–95. doi: 10.1016/j.immuni.2007.07.010
15. Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med. (2008) 205:625–40. doi: 10.1084/jem.20071641
16. Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R. Prolonged interleukin-2Ralpha expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity (2010) 32:91–103. doi: 10.1016/j.immuni.2009.11.010
17. Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity (2008) 29:848–62. doi: 10.1016/j.immuni.2008.11.002
18. Kaech SM, Hemby S, Kersh E, Ahmed R. Molecular and functional profiling of memory CD8 T cell differentiation. Cell (2002) 111:837–51. doi: 10.1016/S0092-8674(02)01139-X
19. Wherry EJ, Teichgraber V, Becker TC, Masopust D, Kaech SM, Antia R, et al. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. (2003) 4:225–34. doi: 10.1038/ni889
20. Kaech SM, Ahmed R. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat Immunol. (2001) 2:415–22. doi: 10.1038/87720
21. Mercado R, Vijh S, Allen SE, Kerksiek K, Pilip IM, Pamer EG. Early programming of T cell populations responding to bacterial infection. J Immunol. (2000) 165:6833–9. doi: 10.4049/jimmunol.165.12.6833
22. van Stipdonk MJ, Hardenberg G, Bijker MS, Lemmens EE, Droin NM, Green DR, et al. Dynamic programming of CD8+ T lymphocyte responses. Nat Immunol. (2003) 4:361–5. doi: 10.1038/ni912
23. van Stipdonk MJ, Lemmens EE, Schoenberger SP. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat Immunol. (2001) 2:423–9. doi: 10.1038/87730
24. Gerlach C, van Heijst JW, Swart E, Sie D, Armstrong N, Kerkhoven RM, et al. One naive T cell, multiple fates in CD8+ T cell differentiation. J Exp Med. (2010) 207:1235–46. doi: 10.1084/jem.20091175
25. Smith KA. Interleukin-2: inception, impact, and implications. Science (1988) 240:1169–76. doi: 10.1126/science.3131876
26. Cheng LE, Greenberg PD. Selective delivery of augmented IL-2 receptor signals to responding CD8+ T cells increases the size of the acute antiviral response and of the resulting memory T cell pool. J Immunol. (2002) 169:4990–7. doi: 10.4049/jimmunol.169.9.4990
27. Cheng LE, Ohlen C, Nelson BH, Greenberg PD. Enhanced signaling through the IL-2 receptor in CD8+ T cells regulated by antigen recognition results in preferential proliferation and expansion of responding CD8+ T cells rather than promotion of cell death. Proc Natl Acad Sci USA. (2002) 99:3001–6. doi: 10.1073/pnas.052676899
28. Blattman JN, Grayson JM, Wherry EJ, Kaech SM, Smith KA, Ahmed R. Therapeutic use of IL-2 to enhance antiviral T-cell responses in vivo. Nat Med. (2003) 9:540–7. doi: 10.1038/nm866
29. Kim MT, Kurup SP, Starbeck-Miller GR, Harty JT. Manipulating memory CD8 T Cell numbers by timed enhancement of IL-2 signals. J Immunol. (2016) 197:1754–61. doi: 10.4049/jimmunol.1600641
30. Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell (1993) 75:253–61. doi: 10.1016/0092-8674(93)80067-O
31. Suzuki H, Kundig TM, Furlonger C, Wakeham A, Timms E, Matsuyama T, et al. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science (1995) 268:1472–6. doi: 10.1126/science.7770771
32. Kundig TM, Schorle H, Bachmann MF, Hengartner H, Zinkernagel RM, Horak I. Immune responses in interleukin-2-deficient mice. Science (1993) 262:1059–61. doi: 10.1126/science.8235625
33. Kramer S, Mamalaki C, Horak I, Schimpl A, Kioussis D, Hung T. Thymic selection and peptide-induced activation of T cell receptor-transgenic CD8 T cells in interleukin-2-deficient mice. Eur J Immunol. (1994) 24:2317–22. doi: 10.1002/eji.1830241009
34. Cousens LP, Orange JS, Biron CA. Endogenous IL-2 contributes to T cell expansion and IFN-gamma production during lymphocytic choriomeningitis virus infection. J Immunol. (1995) 155:5690–9.
35. Bachmann MF, Schorle H, Kuhn R, Muller W, Hengartner H, Zinkernagel RM, et al. Antiviral immune responses in mice deficient for both interleukin-2 and interleukin-4. J Virol. (1995) 69:4842–6.
36. D'Souza WN, Schluns KS, Masopust D, Lefrancois L. Essential role for IL-2 in the regulation of antiviral extralymphoid CD8 T cell responses. J Immunol. (2002) 168:5566–72. doi: 10.4049/jimmunol.168.11.5566
37. D'Souza WN, Lefrancois L. IL-2 is not required for the initiation of CD8 T cell cycling but sustains expansion. J Immunol. (2003) 171:5727–35. doi: 10.4049/jimmunol.171.11.5727
38. Gong D, Malek TR. Cytokine-dependent Blimp-1 expression in activated T cells inhibits IL-2 production. J Immunol. (2007) 178:242–52. doi: 10.4049/jimmunol.178.1.242
39. Kallies, Xin A, Belz GT, Nutt SL. Blimp-1 transcription factor is required for the differentiation of effector CD8(+) T cells and memory responses. Immunity (2009) 31:283–95. doi: 10.1016/j.immuni.2009.06.021
40. Rutishauser RL, Martins GA, Kalachikov S, Chandele A, Parish IA, Meffre E, et al. Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity (2009) 31:296–308. doi: 10.1016/j.immuni.2009.05.014
41. Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity (2010) 32:79–90. doi: 10.1016/j.immuni.2009.11.012
42. Boulet S, Daudelin JF, Labrecque N. IL-2 induction of Blimp-1 is a key in vivo signal for CD8+ short-lived effector T cell differentiation. J Immunol. (2014) 193:1847–54. doi: 10.4049/jimmunol.1302365
43. Curtsinger JM, Mescher MF. Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol. (2010) 22:333–40. doi: 10.1016/j.coi.2010.02.013
44. Kalia V, Ahmed R, Sarkar S. CD8 T cell memory to pathogens. In: Ratcliff M, editor. Encyclopedia of Immunobiology. Vol. 4. 1st ed. Cambridge, MA: Academic Press; Elsevier. (2016). p. 300–17. doi: 10.1016/B978-0-12-374279-7.14012-3
45. Xin, Masson F, Liao Y, Preston S, Guan T, Gloury R, et al. A molecular threshold for effector CD8(+) T cell differentiation controlled by transcription factors Blimp-1 and T-bet. Nat Immunol. (2016) 17:422–32. doi: 10.1038/ni.3410
46. Mitchell DM, Ravkov EV, Williams MA. Distinct roles for IL-2 and IL-15 in the differentiation and survival of CD8+ effector and memory T cells. J Immunol. (2010) 184:6719–30. doi: 10.4049/jimmunol.0904089
47. Williams MA, Tyznik AJ, Bevan MJ. Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature (2006) 441:890–3. doi: 10.1038/nature04790
48. Obar JJ, Molloy MJ, Jellison ER, Stoklasek TA, Zhang W, Usherwood EJ, et al. CD4+ T cell regulation of CD25 expression controls development of short-lived effector CD8+ T cells in primary and secondary responses. Proc Natl Acad Sci USA. (2010) 107:193–8. doi: 10.1073/pnas.0909945107
49. Hand TW, Cui W, Jung YW, Sefik E, Joshi NS, Chandele A, et al. Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc Natl Acad Sci USA. (2010) 107:16601–6. doi: 10.1073/pnas.1003457107
50. Khan AA, Penny LA, Yuzefpolskiy Y, Sarkar S, Kalia V. MicroRNA-17~92 regulates effector and memory CD8 T-cell fates by modulating proliferation in response to infections. Blood (2013) 121:4473–83. doi: 10.1182/blood-2012-06-435412
51. Buchholz VR, Flossdorf M, Hensel I, Kretschmer L, Weissbrich B, Graf P, et al. Disparate individual fates compose robust CD8+ T cell immunity. Science (2013) 340:630–5. doi: 10.1126/science.1235454
52. Gerlach C, Rohr JC, Perie L, van Rooij N, van Heijst JW, Velds A, et al. Heterogeneous differentiation patterns of individual CD8+ T cells. Science (2013) 340:635–9. doi: 10.1126/science.1235487
53. Gerlach C, Moseman EA, Loughhead SM, Alvarez D, Zwijnenburg AJ, Waanders L, et al. The chemokine receptor CX3CR1 defines three antigen-experienced CD8 T cell subsets with distinct roles in immune surveillance and homeostasis. Immunity (2016) 45:1270–84. doi: 10.1016/j.immuni.2016.10.018
54. Herndler-Brandstetter D, Ishigame H, Shinnakasu R, Plajer V, Stecher C, Zhao J, et al. KLRG1(+) effector CD8(+) T cells lose KLRG1, differentiate into all memory T cell lineages, and convey enhanced protective immunity. Immunity (2018) 48:716–29.e8. doi: 10.1016/j.immuni.2018.03.015
55. Sheridan BS, Pham QM, Lee YT, Cauley LS, Puddington L, Lefrancois L. Oral infection drives a distinct population of intestinal resident memory CD8(+) T cells with enhanced protective function. Immunity (2014) 40:747–57. doi: 10.1016/j.immuni.2014.03.007
56. Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol. (2013) 14:1294–301. doi: 10.1038/ni.2744
57. Campbell DJ, Butcher EC. Intestinal attraction: CCL25 functions in effector lymphocyte recruitment to the small intestine. J Clin Invest. (2002) 110:1079–81. doi: 10.1172/JCI0216946
58. Liu L, Fuhlbrigge RC, Karibian K, Tian T, Kupper TS. Dynamic programming of CD8+ T cell trafficking after live viral immunization. Immunity (2006) 25:511–20. doi: 10.1016/j.immuni.2006.06.019
59. Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med. (2010) 207:553–64. doi: 10.1084/jem.20090858
60. Masopust D, Vezys V, Wherry EJ, Barber DL, Ahmed R. Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J Immunol. (2006) 176:2079–83. doi: 10.4049/jimmunol.176.4.2079
61. Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol. (2013) 14:1285–93. doi: 10.1038/ni.2745
62. Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol. (2012) 188:4866–75. doi: 10.4049/jimmunol.1200402
63. Zhang N, Bevan MJ. Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity (2013) 39:687–96. doi: 10.1016/j.immuni.2013.08.019
64. Seidel JA, Vukmanovic-Stejic M, Muller-Durovic B, Patel N, Fuentes-Duculan J, Henson SM, et al. Skin resident memory CD8(+) T cells are phenotypically and functionally distinct from circulating populations and lack immediate cytotoxic function. Clin Exp Immunol. (2018) 194:79–92. doi: 10.1111/cei.13189
65. Pallett LJ, Davies J, Colbeck EJ, Robertson F, Hansi N, Easom N.J.W, et al. IL-2(high) tissue-resident T cells in the human liver: Sentinels for hepatotropic infection. J Exp Med. (2017) 214:1567–80. doi: 10.1084/jem.20162115
66. Hondowicz BD, An D, Schenkel JM, Kim KS, Steach HR, Krishnamurty AT, et al. Interleukin-2-dependent allergen-specific tissue-resident memory cells drive asthma. Immunity (2016) 44:155–66. doi: 10.1016/j.immuni.2015.11.004
67. Owen DL, Mahmud SA, Vang KB, Kelly RM, Blazar BR, Smith KA, et al. Identification of cellular sources of IL-2 needed for regulatory T cell development and homeostasis. J Immunol. (2018) 200:3926–33. doi: 10.4049/jimmunol.1800097
68. Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med. (2005) 201:723–35. doi: 10.1084/jem.20041982
69. Yamamoto M, Seki Y, Iwai K, Ko I, Martin A, Tsuji N, et al. Ontogeny and localization of the cells produce IL-2 in healthy animals. Cytokine (2013) 61:831–41. doi: 10.1016/j.cyto.2012.11.026
70. Paliard X, de Waal Malefijt R, Yssel H, Blanchard D, Chretien I, Abrams J, et al. Simultaneous production of IL-2, IL-4, and IFN-gamma by activated human CD4+ and CD8+ T cell clones. J Immunol. (1988) 141:849–55.
71. Granucci F, Vizzardelli C, Pavelka N, Feau S, Persico M, Virzi E, et al. Inducible IL-2 production by dendritic cells revealed by global gene expression analysis. Nat Immunol. (2001) 2:882–8. doi: 10.1038/ni0901-882
72. Yui MA, Sharp LL, Havran WL, Rothenberg EV. Preferential activation of an IL-2 regulatory sequence transgene in TCR gamma delta and NKT cells: subset-specific differences in IL-2 regulation. J Immunol. (2004) 172:4691–9. doi: 10.4049/jimmunol.172.8.4691
73. Hershko AY, Suzuki R, Charles N, Alvarez-Errico D, Sargent JL, Laurence A, et al. Mast cell interleukin-2 production contributes to suppression of chronic allergic dermatitis. Immunity (2011) 35:562–71. doi: 10.1016/j.immuni.2011.07.013
74. Wuest SC, Edwan JH, Martin JF, Han S, Perry JS, Cartagena CM, et al. A role for interleukin-2 trans-presentation in dendritic cell-mediated T cell activation in humans, as revealed by daclizumab therapy. Nat Med. (2011) 17:604–9. doi: 10.1038/nm.2365
75. Feau S, Arens R, Togher S, Schoenberger SP. Autocrine IL-2 is required for secondary population expansion of CD8(+) memory T cells. Nat Immunol. (2011) 12:908–13. doi: 10.1038/ni.2079
76. Peperzak V, Xiao Y, Veraar EA, Borst J. CD27 sustains survival of CTLs in virus-infected nonlymphoid tissue in mice by inducing autocrine IL-2 production. J Clin Invest. (2010) 120:168–78. doi: 10.1172/JCI40178
77. Feau S, Garcia Z, Arens R, Yagita H, Borst J, Schoenberger SP. The CD4(+) T-cell help signal is transmitted from APC to CD8(+) T-cells via CD27-CD70 interactions. Nat Commun. (2012) 3:948. doi: 10.1038/ncomms1948
78. Popmihajlov Z, Xu D, Morgan H, Milligan Z, Smith KA. Conditional IL-2 gene deletion: consequences for T cell proliferation. Front Immunol. (2012) 3:e102. doi: 10.3389/fimmu.2012.00102
79. Wolkers MC, Bensinger SJ, Green DR, Schoenberger SP, Janssen EM. Interleukin-2 rescues helpless effector CD8+ T cells by diminishing the susceptibility to TRAIL mediated death. Immunol Lett. (2011) 139:25–32. doi: 10.1016/j.imlet.2011.04.011
80. Wolkers MC, Gerlach C, Arens R, Janssen EM, Fitzgerald P, Schumacher TN, et al. Nab2 regulates secondary CD8+ T-cell responses through control of TRAIL expression. Blood (2012) 119:798–804. doi: 10.1182/blood-2011-08-373910
81. McKinstry KK, Strutt TM, Bautista B, Zhang W, Kuang Y, Cooper AM, et al. Effector CD4 T-cell transition to memory requires late cognate interactions that induce autocrine IL-2. Nat Commun. (2014) 5:5377. doi: 10.1038/ncomms6377
82. DiToro D, Winstead CJ, Pham D, Witte S, Andargachew R, Singer JR, et al. Differential IL-2 expression defines developmental fates of follicular versus nonfollicular helper T cells. Science (2018) 361:eaao2933. doi: 10.1126/science.aao2933
83. Yang CY, Best JA, Knell J, Yang E, Sheridan AD, Jesionek AK, et al. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat Immunol. (2011) 12:1221–9. doi: 10.1038/ni.2158
84. Banerjee, Gordon SM, Intlekofer AM, Paley MA, Mooney EC, Lindsten T, et al. Cutting edge: The transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J Immunol. (2010) 185:4988–92. doi: 10.4049/jimmunol.1002042
85. Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol. (2005) 6:1236–44. doi: 10.1038/ni1268
86. Okabe S, Fukuda T, Ishibashi K, Kojima S, Okada S, Hatano M, et al. BAZF, a novel Bcl6 homolog, functions as a transcriptional repressor. Mol Cell Biol. (1998) 18:4235–44. doi: 10.1128/MCB.18.7.4235
87. Ichii H, Sakamoto A, Hatano M, Okada S, Toyama H, Taki S, et al. Role for Bcl-6 in the generation and maintenance of memory CD8+ T cells. Nat Immunol. (2002) 3:558–63. doi: 10.1038/ni802
88. Ichii H, Sakamoto A, Kuroda Y, Tokuhisa T. Bcl6 acts as an amplifier for the generation and proliferative capacity of central memory CD8+ T cells. J Immunol. (2004) 173:883–91. doi: 10.4049/jimmunol.173.2.883
89. Crompton JG, Sukumar M, Restifo NP. Uncoupling T-cell expansion from effector differentiation in cell-based immunotherapy. Immunol Rev. (2014) 257:264–76. doi: 10.1111/imr.12135
90. Finlay DK, Sinclair LV, Feijoo C, Waugh CM, Hagenbeek TJ, Spits H, et al. Phosphoinositide-dependent kinase 1 controls migration and malignant transformation but not cell growth and proliferation in PTEN-null lymphocytes. J Exp Med. (2009) 206:2441–54. doi: 10.1084/jem.20090219
91. Waugh C, Sinclair L, Finlay D, Bayascas JR, Cantrell D. Phosphoinositide (3,4,5)-triphosphate binding to phosphoinositide-dependent kinase 1 regulates a protein kinase B/Akt signaling threshold that dictates T-cell migration, not proliferation. Mol Cell Biol. (2009) 29:5952–62. doi: 10.1128/MCB.00585-09
92. Finlay D, Cantrell DA. Metabolism, migration and memory in cytotoxic T cells. Nat Rev Immunol. (2011) 11:109–17. doi: 10.1038/nri2888
93. Lord JD, McIntosh BC, Greenberg PD, Nelson BH. The IL-2 receptor promotes lymphocyte proliferation and induction of the c-myc, bcl-2, and bcl-x genes through the trans-activation domain of Stat5. J Immunol. (2000) 164:2533–41. doi: 10.4049/jimmunol.164.5.2533
94. Preston GC, Sinclair LV, Kaskar A, Hukelmann JL, Navarro MN, Ferrero I, et al. Single cell tuning of Myc expression by antigen receptor signal strength and interleukin-2 in T lymphocytes. EMBO J. (2015) 34:2008–24. doi: 10.15252/embj.201490252
95. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35:871–82. doi: 10.1016/j.immuni.2011.09.021
96. Pollizzi KN, Powell JD. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat Rev Immunol. (2014) 14:435–46. doi: 10.1038/nri3701
97. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic instruction of immunity. Cell (2017) 169:570–86. doi: 10.1016/j.cell.2017.04.004
98. Macintyre AN, Finlay D, Preston G, Sinclair LV, Waugh CM, Tamas P, et al. Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is dispensable for T cell metabolism. Immunity (2011) 34:224–36. doi: 10.1016/j.immuni.2011.01.012
99. Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med. (2012) 209:2441–53. doi: 10.1084/jem.20112607
100. Athie-Morales V, Smits HH, Cantrell DA, Hilkens CM. Sustained IL-12 signaling is required for Th1 development. J Immunol. (2004) 172:61–9. doi: 10.4049/jimmunol.172.1.61
101. Xue HH, Kovanen PE, Pise-Masison CA, Berg M, Radovich MF, Brady JN, et al. IL-2 negatively regulates IL-7 receptor alpha chain expression in activated T lymphocytes. Proc Natl Acad Sci USA. (2002) 99:13759–64. doi: 10.1073/pnas.212214999
102. Kastenmuller W, Gasteiger G, Subramanian N, Sparwasser T, Busch DH, Belkaid Y, et al. Regulatory T cells selectively control CD8+ T cell effector pool size via IL-2 restriction. J Immunol. (2011) 187:3186–97. doi: 10.4049/jimmunol.1101649
103. Kalia V, Penny LA, Yuzefpolskiy Y, Baumann FM, Sarkar S. Quiescence of memory CD8(+) T cells is mediated by regulatory T cells through inhibitory receptor CTLA-4. Immunity (2015) 42:1116–29. doi: 10.1016/j.immuni.2015.05.023
104. Laidlaw BJ, Cui W, Amezquita RA, Gray SM, Guan T, Lu Y, et al. Production of IL-10 by CD4(+) regulatory T cells during the resolution of infection promotes the maturation of memory CD8(+) T cells. Nat Immunol. (2015) 16:871–9. doi: 10.1038/ni.3224
105. Smith KA. The molecular mechanisms of regulatory T cell immunosuppression. Front Immunol. (2012) 3:379. doi: 10.3389/fimmu.2012.00379
106. Miller JD, Clabaugh SE, Smith DR, Stevens RB, Wrenshall LE. Interleukin-2 is present in human blood vessels and released in biologically active form by heparanase. Immunol Cell Biol. (2012) 90:159–67. doi: 10.1038/icb.2011.45
107. Oestreich KJ, Mohn SE, Weinmann AS. Molecular mechanisms that control the expression and activity of Bcl-6 in TH1 cells to regulate flexibility with a TFH-like gene profile. Nat Immunol. (2012) 13:405–11. doi: 10.1038/ni.2242
108. Oestreich KJ, Read KA, Gilbertson SE, Hough KP, McDonald PW, Krishnamoorthy V, et al. Bcl-6 directly represses the gene program of the glycolysis pathway. Nat Immunol. (2014) 15:957–64. doi: 10.1038/ni.2985
109. Ray JP, Staron MM, Shyer JA, Ho PC, Marshall HD, Gray SM, et al. The interleukin-2-mTORc1 kinase axis defines the signaling, differentiation, and metabolism of T helper 1 and follicular B helper T cells. Immunity (2015) 43:690–702. doi: 10.1016/j.immuni.2015.08.017
110. Heath AW, Playfair JH. Cytokines as immunological adjuvants. Vaccine (1992) 10:427–34. doi: 10.1016/0264-410X(92)90389-2
111. Boyman O, Kolios AG, Raeber ME. Modulation of T cell responses by IL-2 and IL-2 complexes. Clin Exp Rheumatol. (2015) 33:S54–7. doi: 10.5167/uzh-123113
112. Jensen MC, Riddell SR. Designing chimeric antigen receptors to effectively and safely target tumors. Curr Opin Immunol. (2015) 33:9–15. doi: 10.1016/j.coi.2015.01.002
113. West EE, Jin HT, Rasheed AU, Penaloza-Macmaster P, Ha SJ, Tan WG, et al. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J Clin Invest. (2013) 123:2604–15. doi: 10.1172/JCI67008
114. Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science (2006) 311:1924–7. doi: 10.1126/science.1122927
115. Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, et al. Exploiting a natural conformational switch to engineer an interleukin-2 'superkine'. Nature (2012) 484:529–33. doi: 10.1038/nature10975
Keywords: IL-2, CD8 T cell memory, terminal effectors, autocrine, transcription factors, metabolism
Citation: Kalia V and Sarkar S (2018) Regulation of Effector and Memory CD8 T Cell Differentiation by IL-2—A Balancing Act. Front. Immunol. 9:2987. doi: 10.3389/fimmu.2018.02987
Received: 16 October 2018; Accepted: 04 December 2018;
Published: 20 December 2018.
Edited by:
Michael Vajdy, EpitoGenesis (United States), United StatesReviewed by:
Kimberly Sue Schluns, University of Texas MD Anderson Cancer Center, United StatesCarmen Gerlach, Karolinska Institutet (KI), Sweden
Copyright © 2018 Kalia and Sarkar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vandana Kalia, dmthbGlhQHV3LmVkdQ==
Surojit Sarkar, c2Fya2Fya2FsaWFAZ21haWwuY29t