 Monika C. Brunner-Weinzierl
Monika C. Brunner-Weinzierl Christopher E. Rudd
Christopher E. Rudd
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 27 November 2018
Sec. T Cell Biology
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02737
This article is part of the Research Topic Inhibitory Receptors and Pathways of Lymphocytes View all 15 articles
CTLA-4 is a co-receptor on T-cells that controls peripheral tolerance and the development of autoimmunity. Immune check-point blockade (ICB) uses monoclonal antibodies (MAbs) to block the binding of inhibitory receptors (IRs) to their natural ligands. A humanized antibody to CTLA-4 was first approved clinically followed by the use of antibody blockade against PD-1 and its ligand PD-L1. Effective anti-tumor immunity requires the activation of tumor-specific effector T-cells, the blockade of regulatory cells and the migration of T-cells into the tumor. Here, we review data implicating CTLA-4 and PD-1 in the motility of T-cells with a specific reference to the potential exploitation of these pathways for more effective tumor infiltration and eradication.
T-cells circulate continuously between blood, lymphoid tissues and lymph nodes as a mechanism to encounter and respond to foreign antigen. The movement or motility of T-cells involves integrin and selectin mediated adhesion, increased velocity and arrest, chemotaxis to sites of inflammation, homing back to compartments of initial antigen contact, transmigration to enter tissues and movement inside tissues (Figure 1). Antigen-experienced T-cells extravase into non-lymphoid tissue and travel back via lymphatic vessels. In other instances, i.e., in the lymph nodes where foreign antigen is presented to T-cells by dendritic cells (DCs), integrins such as lymphocyte function-associated antigen 1 (LFA-1) are activated by chemokines and antigen-receptor (T-cell receptor; TCR) ligation to bind to their ligands inter-adhesion molecules (ICAMs) to facilitate the “stop signal” for T-cell-dendritic cell (DC) conjugate formation (Figures 1, 2A). The operations of adhesion and chemokine reactivity from blood to tissue involves multi-step transmigration (6).
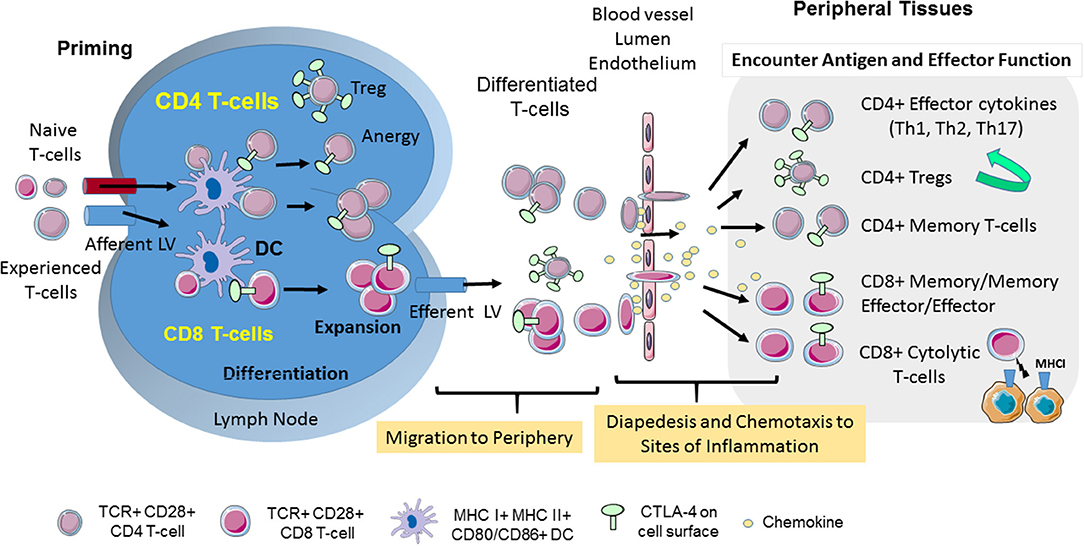
Figure 1. CD28 and CTLA-4-mediated T-cell motility. T-cell response is initiated in secondary lymphoid organs. Naïve and experienced T-cells enter lymph nodes where they encounter antigen presented by DCs. CTLA-4 limits the interaction of CD4+ T-cells with DCs in the reverse-stop signal model involving an increase in T-cell motility, and a raising of the threshold needed to activate T-cells. In the “reverse-stop signal model”, CTLA-4 induces T-cell motility and limits T-cell binding to DCs during antigen-presentation (1, 2). Reverse stop-signaling might also promote the egress of T-cells as mediated by responses to Sphingosine-1-phosphate (S1P) and chemokines. T-cells then migrate from the vasculature to infected tissue via a combination of chemokines and CTLA-4. CTLA-4 can alter motility by up-regulating key chemokine receptors CCR5 and CCR7 and the sensitivity toward the chemokines (3, 4). In the presence of antibody blockade, T-cells accumulate in the blood and remain circulating in the body (3). Upon entry into tissues, different T-cell subsets play important roles in determining the immune response to infection. The scheme was drawn using pictures from Servier Medical Art.
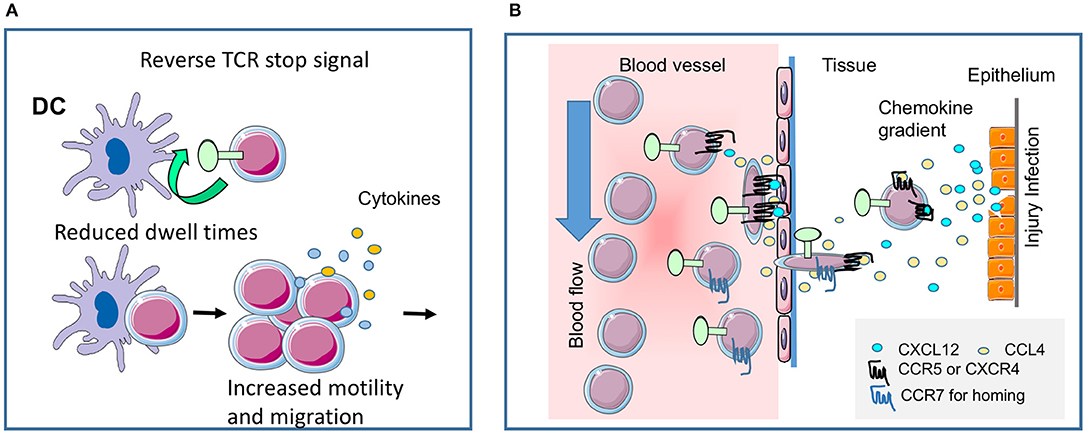
Figure 2. CTLA-4 regulates T-cell motility. (A) Reverse-stop signal model of CTLA-4 (and PD-1). CTLA-4 induces T-cell motility and limits T-cell binding to DCs during antigen-presentation (1, 2). Agonistic CTLA-4 ligation could directly activate the motility of T-cells and thereby interfere with the dwell times of cells with DCs presenting antigenic peptide. PD-1 can function in a similar way (5). (B) CTLA-4 modulates response to chemokines. Chemokine gradients attract T-cells to the site of injury and inflammation. CTLA-4 can alter motility by up-regulating key chemokine receptors CCR5 and CCR7 and the sensitivity toward the chemokines CCL4 (MIP-1β), CXCL12 (SDF1α) and CCL19, but not CXCL9 (MIG) (3). The scheme was drawn using pictures from Servier Medical Art.
Integrin-activation supports activation of chemokine receptors that directs migration of T-cells from blood into tissues or back home into lymph nodes and spleen. The movement of T-cells responds to intrinsic and environmental clues. Chemokines play central roles in inducing the movement of mammalian cells to various niches of the immune system (7, 8). Chemokines effect the motility of CD4 and CD8 T-cells, as well as, suppressor regulatory T-cells (Tregs), although not always in a similar fashion (9, 10) (Figure 1). T-cells in distinct differentiation states such as naïve, effector, or memory T-cells move differently in the same environment to the same clues. Classically, the presence of sensitive CCR7 mediates homing of T-cells to lymph nodes and spleen, while the presence of CXCR5 in follicular T-cells dictates their movement to germinal centers, whereas CXCR3 and CCR5 directs them to the site of injury and inflammation (11). Antigen-experienced T-cells involve movement over long distances were infection might occur, while naïve cells tend to explore the local environment over shorter distances in search of presented antigen (12). Co-receptors such as CD28 and CTLA-4 also modulate these pathways for effective migration.
CD28 plays a central role in providing a second signal needed for T-cell activation (13, 14). Activation signals from the antigen-receptor (TCR) are modified by signals from CD28 and other co-receptors (15–17). CD28 signals via the binding of the lipid kinase phosphatidylinositol- 3-kinase (PI3K) and the adaptor GrB2-SOS complex (18, 19) and p56lck which recruits the protein kinase C (20). It changes the organization of the cytoskeleton (16, 21, 22) and promotes the localization of T-cells to target tissue following antigenic priming (23). With this, it promotes egress from lymphoid tissue and migration to sites of inflammation. Although the downstream pathways that link CD28 to adhesion and migration are not fully understood, loss of CD28 binding to PI3K changes localization to tissues and may favor primed T-cell migration to non-lymphoid tissues (24).
Check-point blockade of cytotoxic T lymphocyte antigen 4 (CTLA-4, CD152) is a major focus in tumor immunotherapy (25, 26). Ipilimumab, a humanized antibody against the inhibitory co-receptor CTLA-4, was the first checkpoint-block mAb to be approved (27). It is thought to act during neo-antigen presentation in lymph nodes and can affect primary and secondary responses to antigen. The loss of CTLA-4 in mice leads to a dramatic lymphoproliferative disorder where animals die within 3–5 weeks of age. Activated CD4 T-cells show an increased localization and infiltration of non-lymphoid and lymphoid organs where they accumulate in lymph nodes, the heart, liver, and pancreas (28–30). Other in vivo models involving antigen-specific T-cell responses combined with CTLA-4 blockade using specific antibodies (31, 32) or reduced CTLA-4 expression (10) support the notion that CTLA-4 can control T-cell infiltration into allo-grafts and tumors.
CTLA-4 dampens T-cell responses via cell intrinsic and extrinsic pathways. Intrinsic events include the inhibition of protein translation, recruitment of phosphatases, activation of ubiquitin ligases, inhibition of cytokine receptor signaling (33–38) and inhibition of lipid microdomain formation on the surface of T-cells (39). CTLA-4 has also been reported to bind to the phosphatases SHP2 and PP2A (34, 40, 41), although the cytoplasmic tail lacks ITIMs for SHP2 binding (42) and PP2A also binds to CD28 (34). Cell extrinsic events include the competition for CD28 in binding to its ligands CD80/86 (43), the removal of CD80/86 (44), the release of suppressive indoleamine (2,3)-dioxygenase (IDO) and the modulation of Treg function (35, 45). Each model has strengths and weaknesses. While competition with CD28 can occur, the induction of autoimmune disease in Ctla-4−/− mice depends on a C-terminal intracellular proline CD28 motif in in vivo co-stimulation (46). Similarly, while CD80/86 can be trans-endocytosed from the surface of DCs by CTLA-4 (44), the level of CD80/86 removal in vivo is low and the ligands can be rapidly re-expressed on presenting cells. Further, whereas the selective deletion of CTLA-4 on FoxP3+ Tregs can delay the onset of disease, mice still die within 2–3 months (35, 45). Moreover, the CTLA-4 YVKM motif binding to PI3K activates pro-survival signals (47, 48) and LFA-1 adhesion (49). Beyond this, the TCR/CD3 mediated stop-signal is decoupled in T-cells from CTLA-4 deficient mice (50) and CTLA-4 has regulatory effects on homeostasis which modulates overall levels of peripheral T-cells (35). It is likely that multiple factors account for the auto-proliferative phenotype in the Ctla-4−/− mice.
PD-1 is a member of the CD28 superfamily which negatively regulates T-cell activation. Blockade of the inhibitory co-receptor PD-1 or its ligand ligand PD-L1 has shown survival rates of 20–30% in treating various types of cancer (27, 51). Negative signals are generated by a cytoplasmic immunoreceptor tyrosine-based switch motif (ITSM) motif that binds to the protein tyrosine phosphatase SHP-2 and which can limit B-cell and T-cell signaling (52, 53). While PD-1-SHP-2 inhibits TCR and/or CD28 signaling (52–54), it is unclear whether PD-1 signals in the same manner in different T-cell subsets. To date, PD-1 has been found to primarily regulate the cytolytic effector function of CD8+ cells (55, 56). Anti-PD-1 immunotherapy also depends on the expression of CD28 (57).
The massive infiltration of organs observed in the Ctla-4−/− provided the first clue that the co-receptor could alter migration of T-cells. Whether this was due to the hyper-activated state of activated Ctla-4−/− CD4 T-cells and/or was related to a direct effect of the co-receptor on mechanisms that affected T-cell motility and/or migration was unclear. An initial clue suggesting that a cell intrinsic pathway might be induced by CTLA-4 was apparent in the observation that T-cells in Ctla-4−/−mice expressing a tailless form of the gene showed alterations in cell migration (58). Further, the acceleration of allograft rejection by CTLA-4 blockade in vivo is associated with more severe mononuclear cell infiltration (59). In addition, depletion of CTLA-4 on T-cell subpopulations in vivo showed that while CTLA-4 on Tregs inhibits the aberrant activation of T-cells, the expression of CTLA-4 on conventional T-cells prevents aberrantly activated T-cells from infiltrating and fatally damaging non-lymphoid tissues (60).
CTLA-4 has been shown to engage mechanisms linked to T-cell movement (1–4, 61) (Figures 1, 2). It was first shown to activate LFA-1 adhesion via increased clustering of integrin receptors (49). YVKM motif binding to PI3K mediates this adhesion (49). This observation suggested that distinct motifs in co-receptor might mediate different intracellular events. Further, it offered the interesting possibility that CTLA-4 could generate both negative and positive signals. Indeed, a precedent was seen in nerve growth factor (NGF) signaling where the binding of PI3K determined whether positive or negative signals leading to apoptosis or cell death were generated (62). The absence of PI3K binding resulted in proapoptotic signaling via the receptor.
One key function of CTLA-4 is to interfere with the ability of T-cells to form stable conjugates with antigen-presenting cells (APCs) (Figure 2A). In the “reverse-stop signal model”, CTLA-4 was found to induce T-cell motility and to limit T-cell binding to DCs during antigen-presentation (1, 2). CTLA-4 ligation with specific antibodies activates the motility of T-cells, while CTLA-4 on T-cells interferes with the dwell times of cells with DCs presenting antigenic peptide. Strikingly, antigen-specific Ctla-4−/− T-cells continue to move even in the presence of antigen (1). Similarly, the expression of CTLA-4 in transformed cell line, Jurkat promotes its motility (63). In terms of cell biology, CTLA-4 ligation induces a polarized morphology typical of motile T-cells, which in turn depends on the mediator's phosphatidylinositol 3-kinase, Vav-1, Cdc42, and myosin light chain kinase (64). From this, we proposed that the ability of CTLA-4 to limit contact times reduced the efficacy of TCR ligation and signaling which in turn raises the threshold needed to activate T-cells (2). Antigen-attracted T-cells competent for CTLA-4 move specifically to sites of inflammation and easily home to lymph nodes in vitro and in vivo, whereas CTLA-4 incompetent T-cells migrate to a lesser extent (3, 60).
It is noteworthy that the effects of CTLA-4 on motility may not operate equally in all T-cells. The reverse-stop effects appear limited to conventional T-cells (Tconvs) (9). It does not operate as efficiently in regulatory T-cells (9), or in anergic T-cells (5). Further, in certain antigen-presentation systems, the blockade of CD80/CD86 itself was as effective as CTLA-4 blockade in promoting the dissociation of T-cells from DCs and increased motility (65). While blockade of CD80/86 will also affect the induction of activation signals from CD28, and indirectly act to terminate T-cell-APC binding, it is also possible that the steric blockade of CTLA-4 with CD80/86 might release T-cells in a manner seen with reverse-stop signaling. Lastly, we also observed that T-cells from Ctla-4−/− mice are unable to arrest when ligated with anti-CD3 (50). The reason for this is unclear but may involve the heightened activation status of T-cells in an inflamed immune environment. It provides a potential explanation for the massive infiltration of all organs of the Ctla-4−/− mice with T-cells. Conversely, the expression of CTLA-4 on conventional T-cells prevents aberrantly activated T-cells from infiltrating and fatally damaging non-lymphoid tissues (60).
In a second pathway of regulation, CTLA-4 can alter motility by up-regulating key chemokine receptors CCR5 and CCR7 and increasing their sensitivity to chemokines CCL4 (MIP-1β), CXCL12 (SDF1α) and CCL19, but not CXCL9 (MIG) (3) (Figure 1, middle; Figure 2B). We have proposed a model for chemotaxis that integrates CD28 and CTLA-4 signals via the G protein-coupled receptor kinase GRK that its phosphorylation of chemokine receptors for de-sensitization and degradation (4). Whereas, CD28 induces GRK to phosphorylate the CCR5 receptor, CTLA-4 engagement inactivates GRK2, leading to delaying or preventing phosphorylation of CCR5, and thereby halts desensitization. In addition, CTLA-4-enhanced specific migration might be partly the consequence of integrin-supported chemotaxis (66, 67), but is also mediated by TCR-mediated PI3K-Akt phosphorylation which synergizes with CD28-mediated migration (4). Antigen-attracted T-cells competent for CTLA-4 move specifically to sites of inflammation and easily home to lymph nodes in vitro and in vivo whereas CTLA-4 incompetent T-cells migrate much less (3, 60). Others have shown that T-cells poorly exit an IFN-treated peritoneal cavity, when before antigen recognition by T-cells anti-CTLA-4 antibodies and anti-hamster antibodies were applied (24). T-cells under this treatment did not move and therefore it is unclear whether the antibody-treatment blocked or crosslinked CTLA-4 and to which degree CTLA-4 operated in trans or without CD28 ligation (4).
Anti-CTLA-4 interference with the interaction between T-cells and DCs (1) laid a precedent for the follow-on finding that PD-1 blockade has similar effects in disrupting T-cell bindings to other cells (5, 68). Antibodies to PD-1 also limit contact times of anergic T-cells (5) and CD8 T-cells (68). In the latter study, PD-L1 was found to localize to the central supramolecular activation cluster, to decrease antiviral CD8 T-cell motility, and promote stable immunological synapse formation. Antibodies to PD-1-PD-L1 restored CD8 T-cell motility in the presence of high viral loads (68).
In this model, anti-PD-1 blockade has shared and distinct properties relative to CTLA-4 blockade. PD-L1 ligation of PD-1 appears to enforce adhesion that is released by anti-PD-1 blockade. PD-1 associated SHP-2 does not appear to negatively regulate adhesion. It is likely that CTLA-4 binding to CD80/86 might also promote adhesion and it blockade might release the T-cell from binding to another cell. However, in addition to this event, anti-CTLA-4 also promotes motility (1, 69). CTLA-4 expressing T-cells simply failed to undergo motility arrest in vivo in the presence of antigen, without the need for antibody blockade (1, 50). Antibody blockade of receptor binding to ligand and the induction of motility are therefore likely to cooperate in disrupting T-cell binding to other cells. In the presence of blocking antibody, the natural expression of CTLA-4 might limit contact of T-cells, while the additional blockade with anti-CTLA-4 ensures the complete release of the weakly adhesive T-cells. In both instances, anti-CTLA-4 and PD-1 limit T-cell binding to DCs during antigen presentation, thereby reducing the efficacy of TCR signaling and raising the threshold needed for the activation of T-cells. This is further complicated by the observation that T-cells from CTLA-4 deficient mice fail to stop in response to anti-CD3 ligation (50). It is unclear whether this feature is due to chronic stimulation that might over-ride the stop signal over time. Overall, the current data indicates that CTLA-4 and PD-1 alters the interaction of T-cells with other cells, including antigen-presenting cells, and consequently, alters the overall motility and migration of T-cells. The exact nature of the regulatory effect may vary depending on the nature of the T-cell, whether CTLA-4 ligation occurs, as well as, the inflammatory conditions in the lymphoid microenvironment.
A prediction from this work has been that CTLA-4 plays a similar role for T-cell entry and movement in tumors. Many tumors express neo-antigens that can be recognized by resident and peripheral T-cells. This aspect might contribute to the synergy seen between anti-CTLA-4 check-point blockade and other modalities of immune intervention (34, 70–73) (Figure 3). As mentioned, CTLA-4 limits dwell times with DCs and potential other tissues (1, 2) and the in vitro and in vivo migration of T-cells is enhanced by CTLA-4 (3, 4). In the presence of antibody blockade, T-cells accumulate in the blood and remain circulating in the body (3). Due to angiogenesis, the enhanced presence of circulating T-cells in the blood may provide an advantage in facilitating tumor access (75). In particular, as vessels at the tumor side are highly branched, irregular, and show a discontinued blood flow (75).
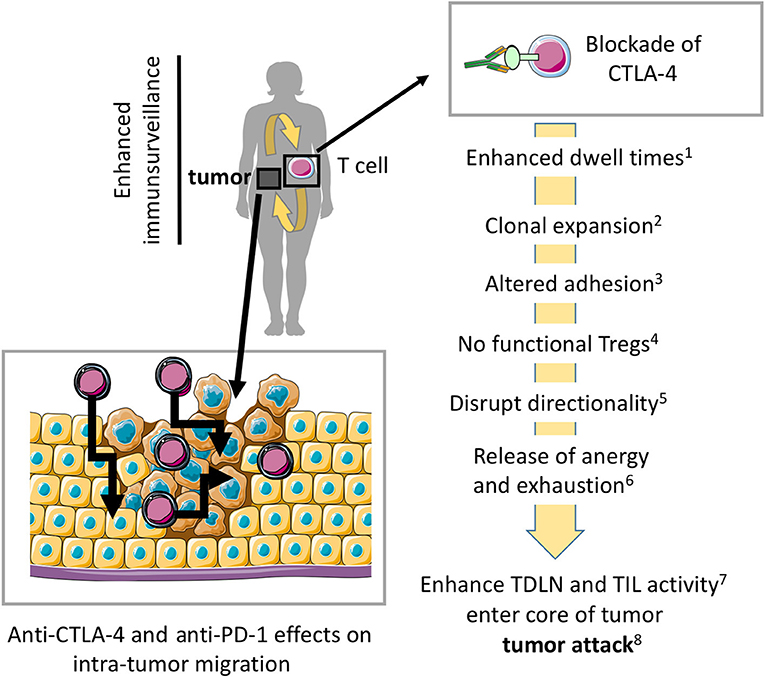
Figure 3. Model where blockade of CTLA-4 and PD-1 enhances migration into tumors and within tumors for more effective tumor rejection. Preventing CTLA-4 engagement, i.e., using anti-CTLA-4-antibodies in vivo modulates the entry and migration of T-cells within tumors for more effective tumor elimination. Anti-CTLA-4 and anti-PD-1 effects on antibodies may also modulate T-cell movement within the tumor mass. (1) (5, 43), (2) (28–30, 74), (3) (49), (4) (35, 45), (5) (3, 4, 32), (6) (65, 69), (7) (10, 31, 32), (8) (25, 26, 37, 38). The scheme was drawn using pictures from Servier Medical Art.
Furthermore, the tumor generates a local immune privileged microenvironment where access by T-cells is limited since integrin-mediated extravasation from blood stream is made difficult as ligands are downregulated at the barrier (75). Some tumors may even grow in immune privileged sides such as the central nervous system. Of note, immune privilege is an active process involving induction of inhibitory mechanisms such as the instructed upregulation of CTLA-4 on T-cells, which can accumulate at the border of the privileged side (76, 77). In addition, T-cells in the tumor microenvironment express CTLA-4 so that blockade releases this localization which enables them to even enter immune privileged microenvironments (32). Therefore, under CTLA-4 blockade using specific antibodies, tumors can be reached by migrating T-cells. However, enhanced motility and migration may also explain immune-related adverse events reported under therapy.
As mentioned, anti-CTLA-4 in tumor models has shown to increase T-cell movement in the tumor (61, 69). In murine breast cancer models, CTLA4 blockade using specific antibodies increased the motility of tumor infiltrating lymphocytes (TILs) in the tumor cavity in vivo (69). The expression of NKG2D then offset this effect by enhancing TILs arrest. In some manner, this combination of anti-CTLA-4 effects on motility combined with stabilization as mediated by NKG2D enhanced tumor eradication. In general, anti-CTLA-4 check-point blockade has been associated with greater tumor entry, although the exact mechanism for this increase in tumor entry has yet to be determined (51). Similarly, in allo-graft models, anti-CTLA-4 blockade increased motility of CD4 effector and Treg cells, it may decrease the motility of CD8 effector T-cells (10). The explanation for these different effects is unclear but may relate to kinetics of CTLA-4 expression on subpopulations and thus, whether it is expressed under anti-CTLA-4 treatment at the cell surface of T-cell helpers and/or CD8 T-cell attackers (47, 74, 78). As CTLA-4 has a much higher affinity to CD80 and CD86 than CD28, the outcome will also be influenced by CD80/86 expression and subsequent ligation in the TDLN and tumor sites (65, 69).
CTLA-4 and PD-1 may have similar effects on T-cell reactivity against tumors; however, the differences in their mode of action may also suggest differences. For example, the more restricted ability of anti-PD-1 to block PD-1 binding to PD-L1 may suggest a more restricted role for T-cells already localized in tumors. Indeed, anti-PD-1 therapy has been reported to have fairly minor effects in promoting an increase in numbers of TILs in tumors such as melanoma. It predominate function on CD8 T-cells may also lead to a restricted effect on this subset. This may operate in conjunction with the effects of anti-PD-1 in restoring functionality to exhausted T-cells (79). By contrast, the combined effects of blockade and direct enhanced motility may be expected to lead to an increase in the migration of T-cells into and within tumors. At the same time, its effect on CD4 and CD8 T-cells might imply a more generalized role on these two major subsets within the T-cell population. Taken together, under CTLA-4 blockade, immune surveillance may be enhanced to sites where T-cells have restricted for tumor entry such as in peri-tumor sites where T-cells can be paralyzed. The synergy of combinational therapy such as CTLA-4 and PD-1 blockade could be due to enhanced motility and a reversal of T-cell exhaustion on different T-cells and in different microenvironments.
Although CTLA-4 impinges on many features of T-cell biology, its effect on tissue and tumor infiltration will be the subject of exciting future work. Antibodies to CTLA-4 may act to facilitate tumor entry and alter the movement in tumors, rates of egress. Further studies will elicit and exploit this feature to facilitate tumor entry for more effective tumor eradication.
MB-W and CR contributed equally to the writing of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The work was supported by Deutsch Forschungsgemeinschaft (DFG) SFB854 B14 and DFG Br1860/8.
1. Schneider H, Downey J, Smith A, Zinselmeyer BH, Rush C, Brewer JM, et al. Reversal of the TCR stop signal by CTLA-4. Science (2006) 313:1972–5. doi: 10.1126/science.1131078
2. Rudd CE. The reverse stop-signal model for CTLA4 function. Nat Rev Immunol. (2008) 8:153–60. doi: 10.1038/nri2253
3. Knieke K, Hoff H, Maszyna F, Kolar P, Schrage A, Hamann A, et al. CD152 (CTLA-4) determines CD4 T-cell migration in vitro and in vivo. PLoS ONE (2009) 4:e5702. doi: 10.1371/journal.pone.0005702
4. Knieke K, Lingel H, Chamaon K, Brunner-Weinzierl MC. Migration of Th1 lymphocytes is regulated by CD152 (CTLA-4)-mediated signaling via PI3 kinase-dependent Akt activation. PLoS ONE (2012) 7:e31391. doi: 10.1371/journal.pone.0031391
5. Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol. (2009) 10:1185–92. doi: 10.1038/ni.1790
6. Andrian UH, von Chambers JD, McEvoy LM, Bargatze RF, Arfors KE, Butcher EC. Two-step model of leukocyte-endothelial cell interaction in inflammation: distinct roles for LECAM-1 and the leukocyte beta 2 integrins in vivo. Proc Nat Acad Sci USA. (1991) 88:7538–42. doi: 10.1073/pnas.88.17.7538
7. Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science (1996) 272:60–6. doi: 10.1126/science.272.5258.60
8. Dustin ML, Bivona TG, Philips MR. Membranes as messengers in T-cell adhesion signaling. Nat Immunol. (2004) 5:363–72. doi: 10.1038/ni1057
9. Lu Y, Schneider H, Rudd CE. Murine regulatory T-cells differ from conventional T-cells in resisting the CTLA-4 reversal of TCR stop-signal. Blood (2012) 120:4560–70. doi: 10.1182/blood-2012-04-421420
10. Miska J, Abdulreda MH, Devarajan P, Lui JB, Suzuki J, Pileggi A, et al. Real-time immune cell interactions in target tissue during autoimmune-induced damage and graft tolerance. J Exp Med. (2014) 211:441–56. doi: 10.1084/jem.20130785
11. Crotty S. Follicular helper CD4 T-cells (TFH). Ann Rev Immunol. (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
12. Krummel MF, Bartumeus F, Gérard A. T-cell migration, search strategies and mechanisms. Nat Rev Immunol. (2016) 16:193–201. doi: 10.1038/nri.2015.16
13. Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nature Rev Immunol. (2001) 1:220–8. doi: 10.1038/35105024
14. Rudd CE, Schneider H. Unifying concepts in CD28, ICOS and CTLA4 co-receptor signalling. Nat Rev Immunol. (2003) 3:544–56. doi: 10.1038/nri1131
15. Rudd CE. Upstream-downstream: CD28 cosignaling pathways and T-cell function. Immunity (1996) 4:527–34. doi: 10.1016/S1074-7613(00)80479-3
16. Zell T, Hunt SW, Mobley JL, Finkelstein LD, Shimizu Y. CD28-mediated up-regulation of beta 1-integrin adhesion involves phosphatidylinositol 3-kinase. J Immunol. (1996) 156:883–6.
17. Kaga S, Ragg S, Rogers KA, Ochi A. Stimulation of CD28 with B7-2 promotes focal adhesion-like cell contacts where Rho family small G proteins accumulate in T-cells. J Immunol. (1998) 160:24–7.
18. Prasad KV, Cai YC, Raab M, Duckworth B, Cantley L, Shoelson SE, et al. T-cell antigen CD28 interacts with the lipid kinase phosphatidylinositol 3-kinase by a cytoplasmic Tyr(P)-Met-Xaa-Met motif. Proc Nat Acad Sci USA. (1994) 91:2834–8. doi: 10.1073/pnas.91.7.2834
19. Schneider H, Cai YC, Prasad KV, Shoelson SE, Rudd CE. T-cell antigen CD28 binds to the GRB-2/SOS complex, regulators of p21ras. Eur J Immunol. (1995) 25:1044–50. doi: 10.1002/eji.1830250428
20. Kong K-F, Yokosuka, T, Canonigo-Balancio, AJ, Isakov N, Saito T, Altman A. A motif in the V3 domain of the kinase PKC-θ determines its localization in the immunological synapse and functions in T-cells via association with CD28. Nat Immunol. (2011) 12:1105–12. doi: 10.1038/ni.2120
21. Raab M, Pfister S, Rudd CE. CD28 signaling via VAV/SLP-76 adaptors: regulation of cytokine transcription independent of TCR ligation. Immunity (2001) 15:921–33. doi: 10.1016/S1074-7613(01)00248-5
22. Wang H, Lim D, Rudd CE. Immunopathologies linked to integrin signalling. Semi Immunopathol. (2010) 32:173–82. doi: 10.1007/s00281-010-0202-3
23. Chang TT, Jabs C, Sobel RA, Kuchroo VK, Sharpe AH. Studies in B7-deficient mice reveal a critical role for B7 costimulation in both induction and effector phases of experimental autoimmune encephalomyelitis. J Exp Med. (1999) 190:733–40. doi: 10.1084/jem.190.5.733
24. Mirenda V, Jarmin SJ, David R, Dyson J, Scott D, Gu Y, et al. Physiologic and aberrant regulation of memory T-cell trafficking by the costimulatory molecule CD28. Blood (2007) 109:2968–77. doi: 10.1182/blood-2006-10-050724
25. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (1996) 271:1734–6. doi: 10.1126/science.271.5256.1734
26. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
27. Page DB, Postow MA, Callahan MK, Allison JP, Wolchok JD. Immune modulation in cancer with antibodies. Annu Rev Med. (2014) 65:185–202. doi: 10.1146/annurev-med-092012-112807
28. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity (1995) 3:541–7. doi: 10.1016/1074-7613(95)90125-6
29. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science (1995) 270:985–8. doi: 10.1126/science.270.5238.985
30. Chambers CA, Krummel MF, Boitel B, Hurwitz A, Sullivan TJ, Fournier S, et al. The role of CTLA-4 in the regulation and initiation of T-cell responses. Immunol Rev. (1996) 153:27–46. doi: 10.1111/j.1600-065X.1996.tb00919.x
31. Schwaiger T, van den Brandt C, Fitzner B, Zaatreh S, Kraatz F, Dummer A, et al. Autoimmune pancreatitis in MRL/Mp mice is a T-cell-mediated disease responsive to cyclosporine A and rapamycin treatment. Gut (2014) 63:494–05. doi: 10.1136/gutjnl-2012-303635
32. Yshii LM, Gebauer CM, Pignolet B, Mauré E, Quériault C, Pierau M, et al. CTLA4 blockade elicits paraneoplastic neurological disease in a mouse model. Brain (2014) 139:2923–34. doi: 10.1093/brain/aww225
33. Marengère LE, Waterhouse P, Duncan GS, Mittrücker HW, Feng GS, Mak TW. Regulation of T-cell receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science (1996) 272:1170–3.
34. Chuang E, Fisher TS, Morgan RW, Robbins MD, Duerr JM, Vander Heiden MG, et al. The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity (2000) 13:313–22. doi: 10.1016/S1074-7613(00)00031-5
35. Kolar P, Knieke K, Hegel JKE, Quandt D, Burmester G-R, Hoff H, et al. CTLA-4 (CD152) controls homeostasis and suppressive capacity of regulatory T-cells in mice. Arthrit Rheumat. (2009) 60:123–32. doi: 10.1002/art.24181
36. Hoff H, Kolar P, Ambach A, Radbruch A, Brunner-Weinzierl MC. CTLA-4 (CD152) inhibits T-cell function by activating the ubiquitin ligase Itch. Mol Immunol. (2010) 47:1875–81. doi: 10.1016/j.molimm.2010.03.017
37. Arra A, Lingel H, Kuropka B, Pick J, Schnoeder T, Fischer T, et al. The differentiation and plasticity of Tc17 cells are regulated by CTLA-4-mediated effects on STATs. Oncoimmunology (2017) 6:e1273300. doi: 10.1080/2162402X.2016.1273300
38. Lingel H, Wissing J, Arra A, Schanze D, Lienenklaus S, Klawonn F, et al. CTLA-4-mediated posttranslational modifications direct cytotoxic T-lymphocyte differentiation. Cell Death Diff. (2017) 24:1739–49. doi: 10.1038/cdd.2017.102
39. Martin M, Schneider H, Azouz A, Rudd CE. Cytotoxic T lymphocyte antigen 4 and CD28 modulate cell surface raft expression in their regulation of T-cell function. J Exp Med. (2001) 194:1675–81. doi: 10.1084/jem.194.11.1675
40. Lee KM, Chuang E, Griffin M, Khattri R, Hong DK, Zhang W, et al. Molecular basis of T-cell inactivation by CTLA-4. Science (1998) 282:2263–6. doi: 10.1126/science.282.5397.2263
41. Baroja ML, Vijayakrishnan L, Bettelli E, Darlington PJ, Chau TA, Ling V, et al. Inhibition of CTLA-4 function by the regulatory subunit of serine/threonine phosphatase 2A. J Immunol. (2002) 168:5070–8. doi: 10.4049/jimmunol.168.10.5070
42. Schneider H, Rudd CE. Tyrosine phosphatase SHP-2 binding to CTLA-4: absence of direct YVKM/YFIP motif recognition. Biochem Biophy Res Commun. (2000) 269:279–83. doi: 10.1006/bbrc.2000.2234
43. Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory pathways in the B7-CD28 ligand-receptor family. Immunity (2016) 44:955–72. doi: 10.1016/j.immuni.2016.05.002
44. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science (2011) 332:600–3. doi: 10.1126/science.1202947
45. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T-cell function. Science (2008) 322:271–5. doi: 10.1126/science.1160062
46. Tai X, van Laethem F, Sharpe AH, Singer A. Induction of autoimmune disease in CTLA-4-/- mice depends on a specific CD28 motif that is required for in vivo costimulation. Proc Nat Acad Sci USA. (2007) 104:13756–61. doi: 10.1073/pnas.0706509104
47. Pandiyan P, Gärtner D, Soezeri O, Radbruch A, Schulze-Osthoff K, Brunner-Weinzierl MC. CD152 (CTLA-4) determines the unequal resistance of Th1 and Th2 cells against activation-induced cell death by a mechanism requiring PI3 kinase function. J Exp Med. (2004) 199:831–42. doi: 10.1084/jem.20031058
48. Schneider H, Valk E, Leung R, Rudd CE. CTLA-4 Activation of phosphatidylinositol 3-Kinase (PI 3-K) and Protein Kinase B (PKB/AKT) sustains T-cell anergy withouT-cell death. PLoS ONE (2008) 3:e3842. doi: 10.1371/journal.pone.0003842
49. Schneider H, Valk E, da Rocha Dias S, Wei B, Rudd CE. CTLA-4 up-regulation of lymphocyte function-associated antigen 1 adhesion and clustering as an alternate basis for coreceptor function. Proc Nat Acad Sci USA. (2005) 102:12861–6. doi: 10.1073/pnas.0505802102
50. Downey J, Smith A, Schneider H, Hogg N, Rudd CE. TCR/CD3 mediated stop-signal is decoupled in T-cells from Ctla4 deficient mice. Immunol Lett. (2008) 115:70–2. doi: 10.1016/j.imlet.2007.09.004
51. Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer (2011) 11:805–12. doi: 10.1038/nrc3153
52. Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Nat Acad Sci USA. (2001) 98:13866–71. doi: 10.1073/pnas.231486598
53. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T-cell stimulation, but only receptor ligation prevents T-cell activation. J Immunol. (2002) 173:945–54. doi: 10.4049/jimmunol.173.2.945
54. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T-cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science (2017) 355:1428–33. doi: 10.1126/science.aaf1292
55. Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. (2013) 14:1212–8. doi: 10.1038/ni.2762
56. Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory pathways in immunotherapy for cancer. Ann Rev Immunol. (2016) 34:539–73. doi: 10.1146/annurev-immunol-032414-112049
57. Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, et al. Rescue of exhausted CD8 T-cells by PD-1-targeted therapies is CD28-dependent. Science (2017) 355:1423–7. doi: 10.1126/science.aaf0683
58. Masteller EL, Chuang E, Mullen AC, Reiner SL, Thompson CB. Structural analysis of CTLA-4 function in vivo. J Immunol. (2000) 164:5319–27. doi: 10.4049/jimmunol.164.10.5319
59. Lin H, Rathmell JC, Gray GS, Thompson CB, Leiden JM, Alegre ML. Cytotoxic T lymphocyte antigen 4 (CTLA4) blockade accelerates the acute rejection of cardiac allografts in CD28-deficient mice: CTLA4 can function independently of CD28. J Exp Med. (1998) 188:199–204. doi: 10.1084/jem.188.1.199
60. Jain N, Nguyen H, Chambers C, Kang J. Dual function of CTLA-4 in regulatory T-cells and conventional T-cells to prevent multiorgan autoimmunity. Proc Nat Acad Sci USA. (2010) 107:1524–8. doi: 10.1073/pnas.0910341107
61. Poirier N, Azimzadeh AM, Zhang T, Dilek N, Mary C, Nguyen B, et al. Inducing CTLA-4-dependent immune regulation by selective CD28 blockade promotes regulatory T-cells in organ transplantation. Sci Transl. Med. (2010) 2:17ra10. doi: 10.1126/scitranslmed.3000116
62. Minshall C, Arkins S, Freund GG, Kelley KW. Requirement for phosphatidylinositol 3'-kinase to protect hemopoietic progenitors against apoptosis depends upon the extracellular survival factor. J Immunol. (1996) 156:939–47.
63. Schneider H, Smith X, Liu H, Bismuth G, Rudd CE. CTLA-4 disrupts ZAP70 microcluster formation with reduced T-cell/APC dwell times and calcium mobilization. Eur J Immunol. (2008) 38:40–7. doi: 10.1002/eji.200737423
64. Wei B, da Rocha Dias S, Wang H, Rudd CE. CTL-associated antigen-4 ligation induces rapid T-cell polarization that depends on phosphatidylinositol 3-kinase, Vav-1, Cdc42, and myosin light chain kinase. J Immunol. (2007) 179:400–8. doi: 10.4049/jimmunol.179.1.400
65. Pentcheva-Hoang T, Simpson TR, Montalvo-Ortiz W, Allison JP. Cytotoxic T lymphocyte antigen-4 blockade enhances antitumor immunity by stimulating melanoma-specific T-cell motility. Cancer Immunol Res. (2014) 2:970–80. doi: 10.1158/2326-6066.CIR-14-0104
66. Schneider H, Valk E, Dias SDR, Wei B, Rudd CE. CTLA-4 regulation of T-cell function via RAP-1-mediated adhesion. Adv Exp Med Biol. (2006) 584:115–26. doi: 10.1007/0-387-34132-3_9
67. Marelli-Berg FM, Fu H, Vianello F, Tokoyoda K, Hamann A. Memory T-cell trafficking: new directions for busy commuters. Immunology (2010) 130:158–65. doi: 10.1111/j.1365-2567.2010.03278.x
68. Zinselmeyer BH, Heydari S, Sacristán C, Nayak D, Cammer M, Herz, J, et al. PD-1 promotes immune exhaustion by inducing antiviral T-cell motility paralysis. J Exp Med. (2013) 210:757–74. doi: 10.1084/jem.20121416
69. Ruocco MG, Pilones KA, Kawashima N, Cammer M, Huang J, Babb JS, et al. Suppressing T-cell motility induced by anti-CTLA-4 monotherapy improves antitumor effects. J Clin Invest. (2012) 122:3718–30. doi: 10.1172/JCI61931
70. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T-cell exhaustion and restore anti-tumor immunity. J Exp Med. (2010) 207:2187–94. doi: 10.1084/jem.20100643
71. Huang R-Y, Eppolito C, Lele S, Shrikant P, Matsuzaki J, Odunsi K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T-cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget (2015) 6:27359–77. doi: 10.18632/oncotarget.4751
72. Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3 — potential mechanisms of action. Nat Rev Immunol. (2015) 15:45–56. doi: 10.1038/nri3790
73. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
74. Maszyna F, Hoff H, Kunkel D, Radbruch A, Brunner-Weinzierl MC. Diversity of clonal T-cell proliferation is mediated by differential expression of CD152 (CTLA-4) on the cell surface of activated individual T lymphocytes. J Immunol. (2003) 171:3459–66. doi: 10.4049/jimmunol.171.7.3459
75. Sanctis F, de Ugel S, Facciponte J, Facciabene A. The dark side of tumor-associated endothelial cells. Semi Immunol. (2018) 35:35–47. doi: 10.1016/j.smim.2018.02.002
76. Gimsa U, ØRen A, Pandiyan P, Teichmann D, Bechmann I, Nitsch R, et al. Astrocytes protect the CNS: antigen-specific T helper cell responses are inhibited by astrocyte-induced upregulation of CTLA-4 (CD152). J Mol Med. (2004) 82:364–72. doi: 10.1007/s00109-004-0531-6
77. Gimsa U, Mitchison NA, Brunner-Weinzierl MC. Immune privilege as an intrinsic CNS property: astrocytes protect the CNS against T-cell-mediated neuroinflammation. Med Inflamm. (2013) 2013:1–11. doi: 10.1155/2013/320519
78. Pandiyan P, Hegel JKE, Krueger M, Quandt D, Brunner-Weinzierl MC. High IFN- production of individual CD8 T lymphocytes is controlled by CD152 (CTLA-4). J Immunol. (2007) 178:2132–40. doi: 10.4049/jimmunol.178.4.2132
Keywords: CTLA4, check-point blockade, cancer, T-cell, motility, migration, PD1, immune surveillance
Citation: Brunner-Weinzierl MC and Rudd CE (2018) CTLA-4 and PD-1 Control of T-Cell Motility and Migration: Implications for Tumor Immunotherapy. Front. Immunol. 9:2737. doi: 10.3389/fimmu.2018.02737
Received: 26 June 2018; Accepted: 06 November 2018;
Published: 27 November 2018.
Edited by:
Alexandre M. Carmo, i3S, Instituto de Investigação e Inovação em Saúde, PortugalReviewed by:
Peter Steinberger, Medizinische Universität Wien, AustriaCopyright © 2018 Brunner-Weinzierl and Rudd. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher E. Rudd, Y2hyaXN0b3BoZXIuZS5ydWRkQHVtb250cmVhbC5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.