 Jan Rossaint1
Jan Rossaint1 Alexander Zarbock
Alexander Zarbock- 1Department of Anesthesiology, Intensive Care and Pain Medicine, University Hospital Münster, Münster, Germany
- 2Interdisciplinary Centre for Clinical Research, University Hospital Münster, Münster, Germany
Platelets are most often recognized for their crucial role in the control of acute hemorrhage. However, current research has greatly expanded the appreciation of platelets beyond their contribution to primary hemostasis, indicating that platelets also actively participate in leukocyte recruitment and the regulation of the host defense in response to exogenous pathogens and sterile injury. Early recruitment of leukocytes, especially neutrophils, is the evolutionary stronghold of the innate immune response to successfully control exogenous infections. Platelets have been shown to physically interact with different leukocyte subsets during inflammatory processes. This interaction holds far-reaching implications for the leukocyte recruitment into peripheral tissues as well as the regulation of leukocyte cell autonomous functions, including the formation and liberation of neutrophil extracellular traps. These functions critically depend on the interaction of platelets with leukocytes. The host immune response and leukocyte recruitment must be tightly regulated to avoid excessive tissue and organ damage and to avoid chronification of inflammation. Thus, platelet-leukocyte interactions and the resulting leukocyte activation and recruitment also underlies tight regulation by several inherited feedback mechanisms to limit the extend of vascular inflammation and to protect the host from collateral damage caused by overshooting immune system activation. After the acute inflammatory phase has been overcome the host defense response must eventually be terminated to allow for resolution from inflammation and restoration of tissue and organ function. Besides their essential role for leukocyte recruitment and the initiation and propagation of vascular inflammation, platelets have lately also been implicated in the resolution process. Here, their contribution to phagocyte clearance, T cell recruitment and macrophage reprogramming is also of outmost importance. This review will focus on the role of platelets in leukocyte recruitment during the initiation of the host defense and we will also discuss the participation of platelets in the resolution process after acute inflammation.
Introduction
The adequate regulated recruitment of leukocytes is an indispensable element of the innate immune response (1–3). Neutrophils are the predominant leukocyte subset that is recruited to inflamed tissue by the initial innate immune system response during the onset of inflammation. Their primary function lies in combating and removal of invading pathogens. If defective, reduced neutrophil recruitment and activation can be the cause of severe immune deficiency syndromes (4). Platelets are traditionally well recognized for their important role in primary hemostasis, yet research over the past decade has created a broader understanding of platelets as an essential element of the innate immune system (5, 6). Platelets serve as a major contributor of several pro-inflammatory chemokines and possess a whole inventory of surface receptors and adhesion molecules that enable platelets to bind to leukocytes as well as circulating pathogens, e.g., bacteria (7). Platelets circulate in the blood in a resting, quiescent state under physiological conditions. When platelets are activated, e.g., in the situation of acute vascular inflammation, they may physically directly interact with circulating leukocytes in the blood stream (8–13). The consequences of this interaction are manifold and include leukocyte activation and may enable leukocytes to fulfill their multiple cell-intrinsic functions and immunological task. Furthermore, the interaction of platelets in particular with neutrophils is a prerequisite for neutrophil extravasation and recruitment into inflamed organs in multiple inflammatory scenarios (14). Activated neutrophils can produce and release neutrophil extracellular traps (NETs) (15). NETs are capable of physically entrapping and killing circulating pathogens, e.g., bacteria. The interaction of platelets and neutrophils has been demonstrated to be a prerequisite for NET formation and release under different inflammatory conditions (9, 11, 12, 15, 16).
Beyond their role as auxiliary cells interacting with leukocytes and supporting them in fulfilling their immunological fate, platelets are also capable of direct interaction with circulating pathogens (7). The liver plays a central role in this process. Here, platelets patrol the microvasculature and perform multiple “touch-and-go” maneuvers with sinusoidal Kupffer cells. This interaction is mainly mediated by bond formation between GPIb on platelets and vWF expressed on hepatic Kupffer cells as an element of the innate immune surveillance system of the liver (17). Kupffer cells in the liver act like tissue-resident macrophages and may catch pathogens in the circulation that may have reached the bloodstream, e.g., from the intestines. This process changes platelet behavior in the liver and the interaction of platelets with Kupffer cells becomes permanent with subsequent activation of platelets. Therefore, the activated platelets may initiate the recruitment of circulation neutrophils to eliminate the entrapped pathogens. In this situation, platelets also serve as sentinel cells together with Kupffer cells to guide the innate immune responses elicited by leukocytes. Currently this phenomenon is best described in the liver (18). If platelets may also patrol other cell types apart from Kupffer cells in organs apart from the liver still has to be investigated.
The focus of this review is the role of platelets in leukocyte recruitment during inflammatory processes and during resolution from inflammation. We will emphasize the molecular mechanism regulating the complex formation between platelets and leukocytes and will highlight the functional consequences associated with these processes under different inflammatory conditions.
Platelet Physiology
Platelets do not possess a cellular nucleus and are essentially produced by fragmentation of megakaryocytes in the bone marrow, from where they are released into the circulation in large amounts. They are traditionally well known for their essential functions in primary hemostasis. Subendothelial structures of the extracellular matrix, e.g., collagen fibers and von-Willebrand factor, are usually inaccessible for circulating platelets. If the vessel wall injury leads to exposure of these molecules, platelet adhesion is triggered. The establishment of bonds between adhesion receptors on the cell surface of platelets and their ligands in the exposed extracellular matrix leads to signaling events in platelets and the cells become activated. Consequently, further adhesion molecules on platelets, e.g., integrins, become activated and platelets may release the contents of their intracellular granules including highly active pro-coagulatory mediators (e.g., ADP, thrombin and prostaglandins). The adhesion and activation of single platelets quickly recruits and activates further platelets to the site of vascular injury and leads to the formation of a leak-sealing thrombus. In addition to platelets, leukocytes are recruited and red blood cells are incorporated into the thrombus (19, 20). Apart from stopping blood loss from the injured vessel during traumatic tissue injury, a second objective is to limit and control the possible entry of exogenous pathogens, e.g., bacteria, via the wound surface into the circulation and thus into the organism. This process could potentially lead to local and/or systemically disseminated infections. From this perspective, the formation of an occlusive platelet thrombus resembles not only a barrier preventing blood loss to the outside, but may also serve as a shield to reduce local blood flow in the injured vessel and prevent the dissemination of pathogens from the outside into the organism, which may explain why platelets have evolutionary evolved to a cell type that also executes immunological functions.
A common cell line known as “hematocytes” were abundant in certain invertebrates and very early vertebrates, and are still conserved e.g., in horshoe crabs, a member of the anthropod family which originated about 450 million years ago. Hematocytes combined immunological as well as hemostatic functions primarily found in leukocytes and platelets of today's mammals (7, 21, 22). During evolution and the appearance of mammals, several more specialized hematopoietic cell lines originated from hematocytes, including lymphocytes, monocytes, neutrophils, and eventually also platelets. These cell lines are characterized by the fact that they are actually able to execute less cell-autonomous functions, but with higher specialization. As a matter of fact, the relationship between the hemostatic and the immune system remained very tight during the development of higher organism with a high degree of interconnectivity. The term “immunothrombosis” has lately been proposed to describe the pathophysiological events modulated by immune cells in cooperation with the coagulation system to facilitate the recognition, containment and destruction of exogenous pathogens during vascular inflammation (23).
Platelets possess a wide inventory of cellular adhesion molecules. These molecules have individual functions and enable platelets to act in different hemostatic and inflammatory situations. Furthermore, they contain intracellular granules (α-granules, dense granules and lysosome granules) packed with various pro-coagulant and immune-modulatory mediators that may be released in response to exposure to different activating stimuli (24). Platelets circulate in the blood stream in very high numbers and it does not come as a surprise that the immune system utilizes platelets to serve as cellular sentinels which are needed for broad surveillance of the circulation and detection of pathogens and possible threats.
Platelet Cellular Activation, Adhesion Molecules and Surface Receptors
Initial platelet activation is the key element in platelet function. Platelets can be activated either by binding of soluble platelet agonists, e.g., ADP or thrombin, or by exposure to subendothelial extracellular matrix components, e.g., collagen (25, 26). Ligand binding to platelets leads to the activation of intracellular signaling pathways which cause platelet shape change and cytoskeletal rearrangement, release of platelet granule content and the activation of cell surface adhesion molecules. Platelet granules are a source of many pro-inflammatory and pro-coagulant mediators. These also include chemoattractive cytokines and chemokines and allow platelets to actively fulfill their role in in primary hemostasis and also in inflammatory processes. Interestingly, experimental evidence suggests that the response of platelets to different activating stimuli is actually not uniform. This indicates the existence of a stimulus-dependent platelet response, e.g., degranulation (27, 28). Further in vitro studies revealed more insights of stimulus-dependent release characteristics of platelet granule content. It could be shown that although the composition of released platelet granule-derived mediators following agonist exposition appeared to be mixed in a stochastic manner, the temporal kinetics of platelet granule release clearly followed different stimulus-characteristic patterns (29). These findings are supported by imaging studies using immunofluorescence staining to visualize pair-wise packing of different molecules stored in α-granules. Here, the packaging pattern of platelet granule content also followed a stochastic distribution (30). However, alternative mechanism other that individual platelet granule packing and release might contribute to a stimulus-dependent platelet response, e.g., incomplete granule fusion upon content release and the interaction of individual mediators in a complete signaling network, and this questions remains the topic of current investigations.
Apart from activation by soluble mediators of cellular interaction by direct binding of ligands for platelet cell surface adhesion molecules, platelets are capable of direct interactions with bacteria. For example, platelets may bind and take up Listeria monocytogenes, a facultative intracellular bacterium. In turn, platelets selectively bind to DCs (CD8α+ dendritic cells) for pathogen delivery and presentation initiating an adaptive immune response (31). In another example, platelets have been shown to be necessary for viral clearance by cytotoxic T cells in lymphocytic choriomeningitis virus (LCMV) infections (32, 33). These findings also underline the fact that platelets do not only play a crucial immunological role by interaction with the innate immune system but are also capable of directly affecting the adaptive immune response.
To acknowledge the exact role of platelets in initiating and modulating the immune response to inflammatory stimuli, the function of the main platelet surface adhesion molecules and platelet surface receptors are of great importance. Integrins are a family of surface adhesion molecules which are abundantly expressed an many cell types where they mostly mediate direct cell-matrix and cell-cell interactions (34). Platelets express several integrins, which are the most important class of cell adhesion molecules on platelets. Integrins are formed as a heterodimer consisting of an α- and a β-chain. Integrins reside in an inactive state not capable for ligand binding under resting conditions (low affinity conformation). If activated, conformational change of both the α- and β-subunit occurs and access to the ligand binding site is granted (high affinity conformation) (35). Individual integrins may also possess the ability to change into an intermediate conformation with limited ligand binding affinity. Platelets express mostly integrins of the β1- and β3-subfamily, including αIIbβ3 (GPIIb/IIIa), α2β1 (VLA-2, GPIa/IIa), α5β1 (VLA-5), and α6β1 (VLA-6) (36, 37). Platelet integrins fulfill divergent functions in the interplay of platelets with the subendothelial, extracellular matrix, leukocytes and endothelial cells (36, 37). A second important feature of integrins is their ability to transduce activating signals into the cell in a process called outside-in signaling (38). Thus, following ligand binding to integrins an intracellular signaling cascaded may be triggered inside platelets leading to further cell activation or degranulation.
Furthermore, platelets express additional glycoprotein complexes, including the glycoprotein (GP) Ib-V-IX complex. This molecule cluster serves as the most important binding partner on platelets for von Willebrand factor (vWF) (39). This complex generally mediates the first contact of platelets with structures of the subendothelial, extracellular matrix which is exposed following blood vessel injury. Another important glycoprotein, GPVI, can bind to collagen. Noticeably, platelet activation also leads to an increased surface expression and activation of glycoproteins. The glycoprotein GPIIbIIIa, which is a synonym for the integrin αIIbβ3 is a binding partner for fibronectin, retronectin and vWF (36, 40). GPIIbIIIa is the most abundantly expressed platelet surface adhesion molecule and in its activated conformation binds various ligands, e.g., fibrinogen, vitronectin, fibronectin, vWF or thrombospondin (40). Platelet adhesion molecules are involved in many immunological tasks elicited by platelets. In vitro studies under static conditions demonstrated that β3-integrins on platelets are necessary to mediate firm platelet adhesion to the cell surface of inflamed endothelial cells (41, 42) and both the used of blocking antibodies directed against the integrin αIIbβ3 on platelets and the genomic knockout of this adhesion molecule caused less platelet adhesion to inflamed endothelial cells in vivo (43). Besides this very important role in mediating the contact to endothelial cells the integrin αIIbβ3 is also crucially involved in the initiation and regulation of direct physical interactions of platelets with leukocytes under inflammatory conditions. Here, the integrin αIIbβ3 serves as a binding partner the integrin αLβ2 (Mac-1) on neutrophils via a bridge of soluble fibrinogen (Figure 1) (40). Beyond physical bond formation, the binding of platelet αIIbβ3 to neutrophil Mac-1 also initiates outside-in signaling into neutrophils and is necessary for NET formation and leukocyte recruitment (12, 16).
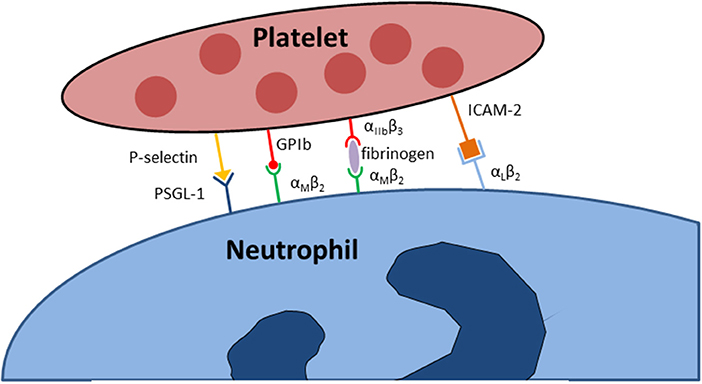
Figure 1. Adhesion molecules implicated in the physical interaction between neutrophils and platelets.
Selectins are adhesion molecular that are abundant on numerous cells types, including endothelial cells, leukocytes, and platelets (44). P-selectin is stored in platelet α-granules in resting platelets. When activated, platelets incorporated P-selectin into the plasma membrane where it becomes available for interaction with its binding partners, e.g., P-selectin glycoprotein ligand-1 (PSGL-1) on neutrophils and monocytes (Figure 1) (7, 45–49). Experimental evidence suggests that the binding of PSGL-1 to P-selectin is necessary to initiate the first interaction between platelets and neutrophils (50, 51). Further adhesion molecules expressed on platelets include several cellular adhesion molecules, including the junctional adhesion molecules (JAM-A, JAM-C), intercellular adhesion molecule (ICAM)-2, and PECAM-1 (platelet endothelial cell adhesion molecule-1) (37). In particular, ICAM-2 on platelets is capable of binding to Mac-1 on neutrophils, but the physiological relevance of these adhesion molecules for the immunological functions of platelets and neutrophils are not fully understood.
Besides adhesion molecules platelets also express different receptors on their cell surface, e.g., complement receptors, pattern recognition receptors (PRRs) of the Toll-like receptor family (TLR1-9) and receptors for detecting immunoglobulins (FcR) (7, 52, 53). These receptors provide platelets with the ability to sense and respond to endogenous pro-coagulant and/or pro-inflammatory mediators, exogenous pathogens and incorporate these signals into cell activation (52). TLRs are a family of evolutionary highly conserved pattern recognition receptors to sense common motifs of exogenous pathogens, termed “pathogen associated molecular patterns” (PAMPs). The detection of PAMPs by TLRs leads to the initiation of an adequate immunological response (54). Functional TLR4 is expressed on platelets (52) and its main function is to recognize lipopolysaccharide (LPS) (55). However, TLR4 binding of LPS does not directly lead to platelet activation and aggregation (52, 56), but merely causes significant platelet priming in the lung and in the liver of mice during LPS-induced vascular inflammation (55). The reasons for this phenomenon are not fully understood to date, but it has been proposed that LPS-induced platelet priming induced the increased production of the pro-inflammatory cytokine TNF-α in the context of bacteremia, and the LPS-binding to TLR4 leads to increased phagocytosis by mononuclear cells (57). Furthermore, it is known that TLR4 activation during systemic inflammation causes the production and release of neutrophil extracellular traps (NETs) helping to catch circulating pathogens from the bloodstream (9). The exact direct or indirect molecular interactions between neutrophils and platelets following platelet TLR4 activation remain poorly defined.
Platelets express various prostaglandin receptors. Prostaglandins are synthesized from membrane-derived phospholipids and are involved in the modulation and regulation of a wide range of physiological processes, e.g., in the cardiovascular, central nervous and immune system. Platelets possess receptors to sense thromboxane, prostacyclin (PGI2), PGD2, and PGE2. Thromboxane A2/prostaglandin H2 (TxA2/PGH2) receptor activation causes the activation of phospholipase A2. This in terms leads to the amplification of platelet activation by autocrine mechanisms. The major inhibitory prostaglandin receptor on platelets are prostacyclin (PGI2) receptors. Prostacyclin is synthesized and released by resting, non-inflamed endothelial cells. PGI2 receptors on platelets sense prostacyclin and suppress platelet activation (58). TxA2 has been shown to be an major regulator of inflammatory processes in vivo and is involved in endothelial cells activation and amplification of inflammation (8, 13, 59). Platelets may synthesize TxA2 by cyclooxygenases, but they are lacking sufficient substrates on their own (60). The main substrate needed for prostaglandin synthesis is arachidonic acid, which is produced by phospholipase A2 by enzymatic hydrolysis of plasma membrane phospholipids. TxA2 production in platelets is significantly increased in the presence of neutrophils. Here, arachidonic acid is shuttled from neutrophils by transcellular metabolism into platelets (13, 61). This process substantially increases platelet TxA2 production. Of note, the binding of P-selectin on platelet to PSGL-1 on neutrophils plays a role in this process, possibly by providing and maintaining the physical proximity between the two cell types (13, 62) (Figure 2).
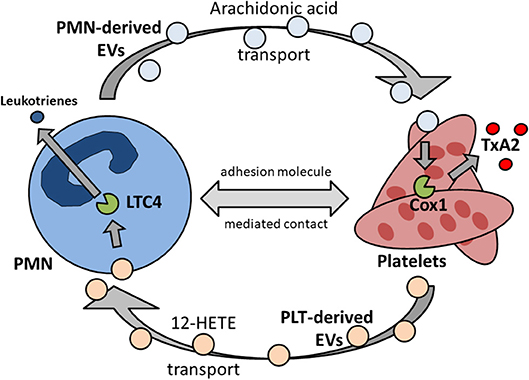
Figure 2. Reciprocal transcellular exchange of substrate and metabolites by extracellular vesicles governs synthesis and release of pro-inflammatory mediators. PMN, polymorphonuclear granulocyte, neutrophil; EV, extracellular vesicles; Cox1, cyclooxygenase 1; TxA2, thromboxane A2; PLT, platelet; 12-HETE, 12-hydroxyeicosatetenoic acid; LTC4, leukotriene 4 synthase.
Platelet-Derived Soluble Mediators
Platelet granules are a storage for various mediators. These include pro-coagulant factors, e.g., ADP and mediators without a direct hemostatic function, e.g., PDGF (platelet-derived growth factor). PDGF plays a role in the regulation of wound healing during and after local inflammation (63). Beyond pro-coagulant factors, platelet granules are also a packed with different pro- and anti-inflammatory mediators, e.g.. transforming growth factor-β (TGF-β) (64). TGF-β, together with IL-10, is one of the most important negative regulatory chemokines of inflammatory processes. Platelets are a major source of TGF-β in the organism, and ITP (idiopathic immune thrombocytopenia) in humans is also characterized by reduced plasma TGF-β levels, which may rise again if low platelet counts during ITP recover to normal values (65, 66). Platelets are also a major source of the chemokines CXCL4 (platelet factor 4), CCL5 (RANTES), and CXCL7 (Neutrophil-activating peptide-2, NAP-2) which may be liberated by activated platelets and are actively involved in neutrophil recruitment and activation (67). Platelet CXCL4 and CCL5 have been shown to be crucial involved in the recruitment and activation of neutrophils during the pathogenesis of acute lung injury (12, 68). Here the two chemokines are deposited on the luminal cell surface of endothelial cells to form a heterodimer and are needed to induce endothelial arrest of intravascular neutrophils (68–71). Furthermore, platelet CXCL4/CCL5 heterodimer binding to neutrophils has been shown to be necessary for neutrophil NET formation and release during the pathogenesis of acute lung injury (12). CXCL7 (NAP7) is a potent CXCR2 agonist and is produced from its precursor molecules CTAP (connective tissue-activating peptide)-III and PBP (platelet basic protein). CXCL7 has also been implicated in the complex formation of neutrophils and platelets under inflammatory conditions in vitro and in vivo (72) and promotes chemotaxis of neutrophils (73). CXCL7 and CXCL4 possess unique structural properties that modulate neutrophil recruitment and processes including chemokine heterodimer formation, glycosaminoglycan (GAG) interactions, and gradient formation. CXCL7 is known to form several biologically active heterodimers with other chemokines, e.g., CXCL7-CXCL1, CXCL7-CXCL4, and CXCL7-CXCL8 heterodimers (74). Interestingly, the binding properties of these heterodimers to GAGs on endothelial cells are substantially different compared to monomeric CXCL7 and this also modulated the receptor binding of the chemokines (74, 75). It was described that the binding of GAGs to monomeric CXCL7 might dynamically modulate the chemokines receptor binding properties and that the GAG-bound monomeric CXCL7 shows less receptor binding affinity than GAG-bound CXCL7 heterodimers (75). CXCL7 liberation by platelet degranulation, interaction with GAGs on the endothelial surface and the resulting gradient formation between free and GAG-bound forms of CXCL7 complexes are also events that contribute to the directed neutrophil recruitment to the site of vascular inflammation (76). CXCL7 also forms tetramers, but to this date nothing is known about the pathophysiological role of these complexes. Interestingly, a negative feedback loop exits to limit the pro-inflammatory action of CXCL7 by CTAP-II (the precursor molecule of CXCL7) inducing the downregulation of CXCR2 on neutrophils. Likewise, also PBP may dampen CXCL7-induced neutrophil activation, degranulation and chemotaxis (77). These studies provide evidence that the release of platelet-derived chemokines may itself lead to the desensitization of chemokine receptors on neutrophils. This may represent an important negative-feedback regulation limiting neutrophil activation.
Platelets are not only a source of chemokines, but the cells themselves also possess chemokine receptors and respond to chemokine stimulation (78). The most prominent chemokine receptors expressed on the cell surface of platelets include CXCR4, CX3CR1, CCR1, CCR3, and CCR4 (79, 80). Several chemokines are known as binding ligands of these receptors and induce platelet activation, including SDF-1 (CXCL12) released by inflamed endothelial cells, TARC (CCL17) and MCD (CCL22) which may be produced by mononuclear cells (81). Besides platelet shape change and activation of cell adhesion molecules, another major consequence of platelet activation by chemokines is the platelet degranulation during which platelet P-selectin stored in platelet granules is integrated into the platelet plasma membrane. The binding of P-selectin of activated platelets to PSGL-1 on neutrophils is an essential step during the formation of physical platelet-neutrophil interactions during inflammatory processes. The formation of platelet-neutrophil complexes is of great importance for neutrophil recruitment and neutrophil function during the pathogenesis of numerous inflammatory diseases (see paragraph below). Current research has demonstrated that platelets are capable of active migration, and activation of CXCR4 on platelets by CXCL12 is critically involved in this process (82–84), but the pathophysiological relevance of this finding during inflammatory diseases has yet to be investigated.
Adenosine Diphosphate Receptors
Adenosine diphosphate (ADP) is a platelet agonist and kept in dense platelet granules in the quiescent state. Upon platelet activation, ADP is set free following platelet degranulation. Extracellular ADP may bind to P2Y1 an P2Y12 receptors. Both receptors are GTP-coupled, platelet-activating receptors. Furthermore, APD may also act by binding to the receptor P2X1, which acts as an ion channel for free calcium ions upon ligand binding and subsequently leads to platelet cytoskeletal rearrangement and induction of platelet shape change. ADP alone is a rather weak platelet agonist. However, it significantly increases the platelet-activating response induced by additional platelet agonists, e.g. thrombin, and leads to the synthesis and liberation of TxA2 from activated platelets, which in turn resembles a strong paracrine platelet agonist. Furthermore, ADP binding to platelets induces the platelet integrin activation (e.g., GPIIbIIIa, integrin αIIbβ3) and platelet aggregation (85). Platelet activation by ADP plays an important pathophysiological role during several inflammatory diseases, including sepsis (86–89).
Platelet-Leukocyte Interactions
Direct physical interactions between platelets and leukocytes are regulated by several distinct molecular interactions but are also enforced by non-biological physical propensities of the vascular system. This is in parts explained by the rheological properties of blood as a mixture of fluids and corpuscular components (90, 91). Here, erythrocytes and larger cells (e.g., leukocytes) stay relatively centered in the middle of the blood flow, whereas platelets are more enriched in the peripheral vicinity of the blood flow closer to the endothelial cell surface lining the inner lumen of the blood vessel. The enrichment of platelets near the vessel wall make encounters with leukocytes in this area more likely. This increases the chance of transient platelet-leukocyte interactions in this area, which might become permanent in case of vascular inflammation with activation of platelets, leukocytes and endothelial cells (91). But also under physiological condition in the absence of inflammation, a small number of transient platelet-neutrophil interactions has been described close to the vascular endothelium (92, 93). The first and probably most important physical interaction between platelets and leukocytes, in particular neutrophils, is established by bond formation between P-selectin on activated platelets and PSGL-1 constitutively expressed on neutrophils (Figure 1). This causes the phenomenon of secondary capturing of free-flowing neutrophils by initial binding of PSGL-1 on these cells to P-selectin expressed by adherent, activated platelets on the vascular endothelial cell surface (94). However, ligand binding to neutrophil PSGL-1 does not only mediate cell-cell interactions, but also induces intracellular signaling. Following PSGL-1 engagement, a cascade of signaling events in neutrophils, including BTK (bruton's tyrosine kinase), Src and MAP kinases, leads to the activation of integrins expressed on neutrophils, e.g., αLβ2 (LFA-1) and αMβ2 (Mac-1) (50, 95–100). LFA-1 is a binding partner of ICAM-2 (intercellular adhesion molecule 2) on platelets, although the exact pathophysiological contribution of this interaction under different inflammatory conditions remains unclear (101–103). Activated Mac-1 on neutrophils is of particular importance for the interaction of neutrophils with platelets, as it is a direct binding ligand for the platelet surface adhesion molecule GPIbα and also indirectly binds to activated platelet GPIIbIIIa through a “bridge” of fibrinogen (Figure 1) (104, 105). The role of GPIIbIIIa binding to Mac-1 in the regulation of neutrophil recruitment and activation has been shown in different inflammatory diseases, e.g., pulmonary inflammation, whereas GPIbα binding to Mac-1 is known to regulate platelet adherence in vitro and is involved in leukocyte recruitment following femoral artery injury in the murine system (105–107). While research over the past decades has revealed several mechanisms by which the platelet and neutrophil may directly and indirectly interact, we are just beginning to understand the specific role and contribution of this phenomenon in different inflammatory diseases. While some diseases models appear to be critically dependent on this interaction, others may not, and even within the same organ system differences may exist in between different inflammatory stimuli (3, 8, 12).
Beyond mediating physical cell-cell interactions, the binding of platelets to neutrophils also modulates and induces cellular immunological functions in neutrophils. A major cellular function of neutrophils is the production and release of ROS (reactive oxygen species). Due to their nature as free radicals, ROS are extremely cell-toxic, and they aid in the destruction of pathogens, e.g., invading bacteria at sites of infection. The complex formation and interaction of platelets and neutrophils induces subsequent integrin-mediated outside-in signaling into neutrophils, which in addition to chemokine stimulation triggers ROS production and release by activated neutrophils. It has been shown that platelet binding to neutrophils increases neutrophil ROS generation efficiency (108, 109). The molecular interaction of P-selectin on platelets and neutrophil PSGL-1 is also of great importance for this process in vitro and in vivo (110, 111). Likewise, pharmacological blockade of ADP binding to its cellular platelet receptor P2Y12 also impaired ROS production in neutrophils (112). As platelet activation by ADP also induces P-selectin mobilization and membrane integration, an implication of the P-selectin/PSGL-1 binding system could be involved in the underlying molecular mechanism. However, exact evidence for this hypothesis is lacking as ADP stimulation of platelets also induces the activation of additional platelet surface adhesion molecules. The second important immunological function by which neutrophils eliminate pathogens is phagocytosis and this process is also affected by platelet-neutrophil interactions. Here, indirect interaction pathways mediated by soluble inflammatory mediators, e.g., prostaglandins and purine nucleotides, play a more important role than direct ligand-receptor interactions (113–116). However, direct cellular interactions also seem to be involved, at least under distinct inflammatory conditions. This was shown by results from study utilizing a periodontitis model where efficient phagocytosis by neutrophils relied on the complex formation of neutrophils and platelets (117), indicating a possible tissue- and stimulus-specificity of platelet-dependency.
The third cell-autonomous immunological feature by which neutrophils may directly engage and kill bacteria is the formation and released of neutrophil extracellular traps (NETs) generated by “NETosis” (15). NETs are essentially decondensed nuclear chromatin, which is decorate with granular proteins from neutrophils and spun into the extracellular space. Although the generation of NETs leaves the neutrophils without a nucleus, the cells are still alive and are capable or cellular functions, e.g. intravascular crawling and transmigration (118). Physically, NETs may act like real-life fishing nets and entangle pathogens circulating in the blood stream. The relevance of this effect has been shown in different models of inflammatory diseases involving the blood-borne distribution of pathogens in the organism (11). Interestingly, NETs are also implicated in the pathogenesis of inflammatory disorders not involving infectious pathogens or stimuli. Here, NET formation has been shown to be a prerequisite for efficient neutrophil recruitment from the vasculature to the site of inflammation and platelet-neutrophil complex formation has been demonstrated to be critically involved in this process (12, 16). The pattern recognition receptor TLR4 is expressed on platelets and may be activated by binding of bacterial products, e.g., LPS. TLR4 activation on platelets leads to neutrophil NET formation and liberation, but the exact mechanism remains unclear (9). Furthermore, the direct physical interaction of platelets and neutrophils by binding of activated GPIIbIIIa to Mac-1 on neutrophils (via a bridge of fibrinogen) also induces NET formation by neutrophils together with simultaneous activation of GPCRs (G-protein coupled receptors) on neutrophils by CXCL4/CCL5 heterodimers released by activated platelets to facilitate neutrophil recruitment during sterile pulmonary inflammation (12). An example for indirect platelet-neutrophil interactions inducing NET release is hBD-1 (human β-defensin 1). Platelets secrete hBD-1 in response to contact with toxin from S. aureus, and hBD-1 has been shown to cause NET release by neutrophils (119).
Platelet Microparticles
Although platelets are small fragments originating from larger cells (megakaryocytes) themselves, they are still capable to generate microparticles with dimensions in the sub-micrometer range (120). Current research has demonstrated that microparticles are associated with multiple physiological and pathophysiological functions (121). Also not restricted to platelets as originating cells, the majority of microparticles in the blood are actually coming from platelets (122). The fact that microparticles may carry certain proteins that are normally not expressed or only expressed in much smaller quantities in their originating cells indicates that microparticles are produced and packed with dedicated proteins in an active process and not just by random cell sequestration (123). Yet the exact regulatory processes guiding these pathways in platelets still have to be investigated. Microparticles may also well interact with and bind to leukocytes, since they inherit the adhesion receptors, e.g., P-selectin and the platelet integrin αIIbβ3, from the platelets cells surface (124–126). However, it is unknown if the platelet integrin αIIbβ3 is activated on the microparticle surface and contributes to adhesion. Microparticles originating from platelets are also capable of binding to other cell types than leukocytes, e.g., endothelial cells. In fact, excessive microparticle binding to the surface of endothelial cells may lead to endothelial cell activation (127).
As a specialized class of microparticles, extracellular vesicles (EVs) are actively released by cells, e.g., neutrophils, following active cell-internal production, packaging and release. EV transport in between neutrophils and platelets has gained attention as it could be demonstrated that intermediate metabolites necessary for sufficient prostaglandin synthesis and release by platelets are shuffled from neutrophils into platelets via specific EV release and uptake (13, 61). Likewise, the transcellular transport vice versa from platelets to neutrophils also plays an important pathophysiological role. Here, neutrophils receive 12-hydroxyeicosatetenoic acid (12-HETE) from platelets to synthesize leukotrienes (128). Interestingly, this interaction also modulates LTC4 synthase activity further downstream in neutrophils, as does the transport vice versa from neutrophils to platelets regulate cyclooxygenase 1 activity in platelets (62, 129). It seems fair to argue that transcellular metabolite exchange between neutrophils and platelets via EVs is a two-way interaction (Figure 2) (130).
The Role of Platelets in the Pathogenesis of Acute Inflammatory Diseases
Acute lung injury is a respiratory disorder characterized by pulmonary leukocyte recruitment and edema formation leading to impaired gas exchange with severe consequences for patients, depending on its severity (131, 132). It may occur in response to different stimuli, e.g., pulmonary bacterial infections, sepsis, and aspiration of gastric content (133). The pathogenesis of pulmonary inflammation and acute lung injury has been demonstrated to rely on platelet-neutrophil complex formation in various disease models. They include transfusion-related acute lung injury (TRALI) (10, 16, 134), LPS-induced lung injury (68, 135), acid-induced lung injury (8), and ventilator-induced lung injury (VILI) (12). Experimental evidence has demonstrated that platelet-neutrophil complexes can be detected in a circulating manner in the blood as well as directly attached to the vessel walls in the lung microcirculation as early as 30 min after exposure to the inflammatory stimulus (8). Here, complex formation involving platelet P-selectin is critically involved and pharmacological blockade of this molecule or cellular depletion of platelets showed a protective effect in reducing immune cell recruitment and limiting the vascular permeability increase. Platelet-neutrophil complex formation also regulated the production of TxA2 and this caused endothelial cell activation and expression of the endothelial cell adhesion molecule ICAM-1 (8). The importance of the direct cellular interaction between platelets and neutrophils in this process was also underlined by a later study demonstrating that platelet-neutrophil complex formation is necessary for the transcellular transport of metabolites from neutrophils into platelets to booster TxA2 production during the host immune response following induction of bacterial pneumonia (13). Pulmonary inflammation may also be induced by non-inflammatory stimuli, e.g., barotrauma during ventilator-induced lung injury (VILI). The interaction of platelets and neutrophils has also been shown to be required for neutrophil recruitment into the lung during VILI by intravascular formation and release of NETs (12). Moreover, Grommes et al. could show that platelet-neutrophil complex formation is also involved in a murine model of LPS-induced lung injury (68).
Massive transfusion of blood products, e.g., during severe hemorrhage following trauma, may cause transfusion-related acute lung injury (TRALI) and is a feared complication in transfusion medicine (134). It has become evident, that the deterioration of gas exchange during TRALI is not a result of an intravascular fluid overload, but essentially involved immunological pathways leading to inflammatory activation of the pulmonary endothelium, immune cell recruitment and increased vascular permeability. Lately, the platelets and platelet-neutrophil interaction shave been shown to be critically involved in the development of TRALI (109). Here, platelets may liberate CD154 in response to TLR-ligand binding (136). Subsequent CD154 binding to CD40 on neutrophils may cause activation of these cells and lead to neutrophil recruitment into the lung (137).
Whether platelet depletion or the attenuation of a platelet-elicited immunological response is associated with improvement of deterioration of the outcome critically relies on the nature of the underlying inflammatory stimulus in a particular model. Whereas the attenuation of the innate host immune response may be beneficial in disease models using aseptic inflammatory stimuli (e.g., LPS inhalation, intratracheal acid instillation or TRALI), the same intervention may substantially worsen the outcome in an infectious model, e.g., following the intratracheal instillation of viable bacteria to induce pulmonary inflammation (13, 138). In addition, it remains unclear if and how platelets may reach other compartments in the lung than the intravascular space, e.g., the lung interstitium or the alveoli. Evidence from first studies suggest that platelets may also be present in the lung as far as in the alveoli (139). There is first evidence that platelets may also be released by megakaryocytes situated outside of the bone marrow, e.g., within the pulmonary microcirculation and it was claimed that this extra-medullar platelet synthesis contributes to a large amount of circulating platelets (140). Yet, the specific contribution to this putative new platelet reservoir in the lungs as well as the functional role of platelets in the different compartments of the lung and their specific contribution to the disease progression here still has to be investigated in more detail.
Platelets are traditionally thought to possess only little motile capabilities, mainly related to rolling, adhesion and aggregation. However, several recent reports have substantially challenged this dogma with the discovery of platelet migration. First reports indicated that human platelets adapt to the application of high shear forces by cellular polarization and flow-directed migration and show migratory behavior toward a SDF-1 (stromal cell-derived factor 1) gradient in vitro (82, 83). First in vivo studies demonstrated that platelet migrate into the extravascular compartment of the lung during allergen-induced airway inflammation (141). Lately, Gärtner et al. showed that platelet migration occurs under inflammatory conditions in mice in vivo and is crucial for bacterial host defense and bundling of bacteria for improved phagocytosis (84). For this process, GPIIb/IIIa, as well as ADP and thromboxane A2 are needed, and it has previously been shown that platelet-TxA2 contributes to the neutrophil recruitment into the lung (13). Interestingly, it was noted that platelets adherent to leukocytes migrate faster than independent platelets, and only a certain percentage of all platelets does migrate (84). However, the relevance of platelet migration in the lung remains unclear to this date.
The liver represents another organ in which platelets and platelet-neutrophil complexes are prominently involved in host defense. The liver is uniquely characterized by the fact that the hepatic microcirculation is placed second in line beyond the intestinal microcirculation, connected by the portal vein. Thus, the liver is also exposed as the first organ that might be passed by invading exogenous bacteria from the intestinal tract (142). The liver is equipped with a unique and specialized immune surveillance system that resides in the livers sinusoidal space where Kupffer cells sense distinct bacterial structures and components. Circulating platelets are in constant temporary contact with Kupffer cells, performing “touch-and-go” maneuvers. Once Kupffer cells become activated following pathogen contact, platelets permanently attach to Kupffer cells by GPIIb-mediated adhesion and attract neutrophils to the liver sinusoids to aid in pathogen clearance (17).
Overwhelming systemic inflammation may occur due to uncontrolled local inflammation and can be potentially life-threatening for host. Interestingly, systemic inflammation and sepsis are often accompanied by transient low platelet counts in the blood, which may rise again after the initial phase of systemic inflammation is overcome (143). One factor contributing to decreased blood platelets counts may be the occurrence of DIC (disseminated intravascular coagulation) consuming platelets. However, emerging evidence also hints to a possible consumption of platelets caused by immunological processes in the circulation during systemic inflammatory disorders (144). This is also supported by results from clinical studies indicating that low circulating platelet counts are often associated with increased circulating microparticles and that circulating platelets from patients with sepsis show increased P-selectin surface expression levels indicating platelet activation (126).
Platelets in the Resolution of Inflammation
Timely resolution of inflammation is important to impede uncontrolled host tissue destruction and organ dysfunction leading to chronic inflammation and fibrosis (145). It is essential that neutrophils are rapidly and efficiently removed from the inflammatory site upon clearance of the invading microorganisms thus avoiding excessive tissue damage (146). Neutrophil apoptosis and consequent engulfment by macrophages is the major route by which the host clears neutrophils. Efficient phagocytosis of apoptotic neutrophils by macrophages not only prevents their secondary necrosis but also turns pro-inflammatory macrophages into cells with an anti-inflammatory, reparative signature (147). Dysfunction in the neutrophil apoptosis machinery is considered critical for the pathogenesis of many chronic human inflammatory diseases, e.g., pulmonary fibrosis after ARDS (148). While various pro-resolving mediators and pathways that govern resolution from inflammation in the lung have been described, the role of platelets in this process remains vaguely investigated. Interestingly, a current report also indicated that delayed neutrophil apoptosis and clearance are also associated with delayed recovery from ischemia/reperfusion-induced acute kidney injury and accelerated renal fibrosis (149).
In the lunge, several studies have provided evidence that platelets do not only act during vascular inflammation within the intravascular compartment, but eventually also appear in the lung alveoli. Platelets have been found to extravasate and accumulate beneath the airways in a model of allergic inflammation (141). Further evidence supported the observation that platelets, eventually coupled to leukocytes, can be detected in the bronchoalveolar lavage fluid after induction of pulmonary inflammation (139). Supporting this finding, platelets could also be found in the BAL of mice after intratracheal instillation of LPS (150). Platelets were long thought to be passive corpuscular blood components that reach their site of action by chance, enforced by their sheer numbers (151). Contradicting this dogma it could recently be demonstrated that platelets are capable of active migration (84). This observation may contribute to the concept that the distribution of platelets is not only restricted to the intravascular compartment, but that platelets also translocate toward the alveolar space, i.e., into the organ tissue. However, previous studies in other organs provided hints that platelets may not only be important for the propagation of vascular inflammation. It could be shown that platelet activating factor (PAF) plays a role in mediating the uptake of urate crystals during the resolution of gouty inflammation (152). Platelets are also a major source for anti-inflammatory mediators of the lipoxin family, e.g., specialized pro-resolving mediators (SPMs) such as resolvins and maresins (153). These lipoxins are produced and released already during the inflammatory onset phase of acute inflammation and their concentrations sharply rise during the convergence toward the resolution phase (154). Interestingly, these lipoxins also promote phagocytic clearance of apoptotic immune cells, e.g., neutrophils, during resolution (155).
Neutrophils and macrophages have traditionally been regarded as dominant cell types during the resolution of inflammation. Regulatory T cells (Tregs) represent a T cell subpopulation with predominantly immune regulatory functions and are mainly acting immunosuppressive. Tregs are a source of the anti-inflammatory cytokines interleukin 10 (IL-10) and transforming growth factor ß (TGF-ß). Yet, it is unknown how exactly platelets, macrophages and Tregs participate in the resolution of pulmonary inflammation. Platelets have been previously described to interact with regulatory T cells under inflammatory conditions. In this context, it could be shown that platelets are needed to control the anti-inflammatory actions of CD4+ regulatory T cells following burn injury trauma in mice (156). In another organ, platelets have also been shown to interact with CD4+ T cells in the liver following ischemic injury and during atherosclerosis (157, 158). Interestingly, platelets are also thought to be capable of inducing CD4+ T cell differentiation by both the release of distinct chemokines and by direct cell-cell contact with T cells. As a consequence, IL-10 production and release by T cells was enhanced (159).
Conclusion
While platelets are traditionally perceived as essential elements of primary hemostasis, the contemporary perception of their pathophysiological role should also clearly include their prominent contribution to inflammatory processes. Current and past research has shed light on their participation in the generation of an adequate immune response. Here, both the direct and indirect interactions with leukocytes, in particular neutrophils, are of outmost importance. Future research will further characterize the detailed, spatio-temporal role of platelets in the pathogenesis of distinct tissue- and stimulus-specific inflammatory situations. Furthermore, platelets may also be involved in the resolution of acute inflammation, a field of research of growing importance. A more detailed understanding of the underlying molecular mechanisms will be the key to the development of targeted therapeutic approaches and interventions to improve the treatment of patients suffering from inflammatory diseases.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the DFG (Deutsche Forschungsgemeinschaft, RO4537/2-1 and RO4537/3-1 to JR, and ZA428/14-1 to AZ) and the Interdisciplinary Center for Clinical Research (IZKF, SEED 12/18 to AM).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. (2007) 7:678–89. doi: 10.1038/nri2156
2. Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nat Med. (2011) 17:1381–90. doi: 10.1038/nm.2514
3. Rossaint J, Zarbock A. Tissue-specific neutrophil recruitment into the lung, liver, and kidney. J Innate Immun. (2013) 5:348–57. doi: 10.1159/000345943
4. Badolato R. Defects of leukocyte migration in primary immunodeficiencies. Eur J Immunol. (2013) 43:1436–40. doi: 10.1002/eji.201243155
5. Zarbock A, Polanowska-Grabowska RK, Ley K. Platelet-neutrophil-interactions: linking hemostasis and inflammation. Blood Rev. (2007) 21:99–111. doi: 10.1016/j.blre.2006.06.001
6. Rossaint J, Zarbock A. Platelets in leucocyte recruitment and function. Cardiovasc Res. (2015) 107:386–95. doi: 10.1093/cvr/cvv048
7. Semple JW, Italiano JEJr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. (2011) 11:264–74. doi: 10.1038/nri2956
8. Zarbock A, Singbartl K, Ley K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J Clin Invest. (2006) 116:3211–9. doi: 10.1172/JCI29499
9. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. doi: 10.1038/nm1565
10. Looney MR, Nguyen JX, Hu Y, Van Ziffle JA, Lowell CA, Matthay MA. Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J Clin Investig. (2009) 119:3450–61. doi: 10.1172/JCI38432
11. McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe (2012) 12:324–33. doi: 10.1016/j.chom.2012.06.011
12. Rossaint J, Herter JM, Van Aken H, Napirei M, Doring Y, Weber C, et al. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood (2014) 123:2573–84. doi: 10.1182/blood-2013-07-516484
13. Rossaint J, Kuhne K, Skupski J, Van Aken H, Looney MR, Hidalgo A, et al. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat Commun. (2016) 7:13464. doi: 10.1038/ncomms13464
14. Mine S, Fujisaki T, Suematsu M, Tanaka Y. Activated platelets and endothelial cell interaction with neutrophils under flow conditions. Intern Med. (2001) 40:1085–92. doi: 10.2169/internalmedicine.40.1085
15. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
16. Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Investig. (2012) 122:2661–71. doi: 10.1172/JCI61303
17. Wong CH, Jenne CN, Petri B, Chrobok NL, Kubes P. Nucleation of platelets with blood-borne pathogens on Kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat Immunol. (2013) 14:785–92. doi: 10.1038/ni.2631
18. McDonald B, Kubes P. Innate immune cell trafficking and function during sterile inflammation of the liver. Gastroenterology (2016) 151:1087–95. doi: 10.1053/j.gastro.2016.09.048
19. Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest. (2005) 115:3378–84. doi: 10.1172/JCI27196
20. Nieswandt B, Varga-Szabo D, Elvers M. Integrins in platelet activation. J Thromb Haemost. (2009) 7(Suppl 1):206–9. doi: 10.1111/j.1538-7836.2009.03370.x
21. Jiravanichpaisal P, Lee BL, Soderhall K. Cell-mediated immunity in arthropods: hematopoiesis, coagulation, melanization and opsonization. Immunobiology (2006) 211:213–36. doi: 10.1016/j.imbio.2005.10.015
22. Buchmann K. Evolution of innate immunity: clues from invertebrates via fish to mammals. Front Immunol. (2014) 5:459. doi: 10.3389/fimmu.2014.00459
23. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. (2013) 13:34–45. doi: 10.1038/nri3345
24. Rendu F, Brohard-Bohn B. The platelet release reaction: granules' constituents, secretion and functions. Platelets (2001) 12:261–73. doi: 10.1080/09537100120068170
25. Watson SP. Platelet activation by extracellular matrix proteins in haemostasis and thrombosis. Curr Pharm Des. (2009) 15:1358–72. doi: 10.2174/138161209787846702
26. De Candia E. Mechanisms of platelet activation by thrombin: a short history. Thromb Res. (2012) 129:250–6. doi: 10.1016/j.thromres.2011.11.001
27. Italiano JEJr, Richardson JL, Patel-Hett S, Battinelli E, Zaslavsky A, Short S, et al. Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood (2008) 111:1227–33. doi: 10.1182/blood-2007-09-113837
28. Rex S, Beaulieu LM, Perlman DH, Vitseva O, Blair PS, McComb ME, et al. Immune versus thrombotic stimulation of platelets differentially regulates signalling pathways, intracellular protein-protein interactions, and alpha-granule release. Thromb Haemost. (2009) 102:97–110. doi: 10.1160/TH08-08-0513
29. Jonnalagadda D, Izu LT, Whiteheart SW. Platelet secretion is kinetically heterogeneous in an agonist-responsive manner. Blood (2012) 120:5209–16. doi: 10.1182/blood-2012-07-445080
30. Kamykowski J, Carlton P, Sehgal S, Storrie B. Quantitative immunofluorescence mapping reveals little functional coclustering of proteins within platelet alpha-granules. Blood (2011) 118:1370–3. doi: 10.1182/blood-2011-01-330910
31. Verschoor A, Neuenhahn M, Navarini AA, Graef P, Plaumann A, Seidlmeier A, et al. A platelet-mediated system for shuttling blood-borne bacteria to CD8alpha+ dendritic cells depends on glycoprotein GPIb and complement C3. Nat Immunol. (2011) 12:1194–201. doi: 10.1038/ni.2140
32. Iannacone M, Sitia G, Isogawa M, Whitmire JK, Marchese P, Chisari FV, et al. Platelets prevent IFN-alpha/beta-induced lethal hemorrhage promoting CTL-dependent clearance of lymphocytic choriomeningitis virus. Proc Natl Acad Sci USA. (2008) 105:629–34. doi: 10.1073/pnas.0711200105
33. Baccala R, Welch MJ, Gonzalez-Quintial R, Walsh KB, Teijaro JR, Nguyen A, et al. Type I interferon is a therapeutic target for virus-induced lethal vascular damage. Proc Natl Acad Sci USA. (2014) 111:8925–30. doi: 10.1073/pnas.1408148111
34. Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. (2007) 25:619–47. doi: 10.1146/annurev.immunol.25.022106.141618
35. Springer TA, Wang JH. The three-dimensional structure of integrins and their ligands, and conformational regulation of cell adhesion. Adv Protein Chem. (2004) 68:29–63. doi: 10.1016/S0065-3233(04)68002-8
36. Ruggeri ZM, Mendolicchio GL. Adhesion mechanisms in platelet function. Circ Res. (2007) 100:1673–85. doi: 10.1161/01.RES.0000267878.97021.ab
37. van Gils JM, Zwaginga JJ, Hordijk PL. Molecular and functional interactions among monocytes, platelets, and endothelial cells and their relevance for cardiovascular diseases. J Leukoc Biol. (2009) 85:195–204. doi: 10.1189/jlb.0708400
38. Luo BH, Springer TA. Integrin structures and conformational signaling. Curr Opin Cell Biol. (2006) 18:579–86. doi: 10.1016/j.ceb.2006.08.005
39. Cruz MA, Diacovo TG, Emsley J, Liddington R, Handin RI. Mapping the glycoprotein Ib-binding site in the von willebrand factor A1 domain. J Biol Chem. (2000) 275:19098–105. doi: 10.1074/jbc.M002292200
40. Bennett JS. Structure and function of the platelet integrin alphaIIbbeta3. J Clin Invest. (2005) 115:3363–9. doi: 10.1172/JCI26989
41. Gawaz M, Neumann FJ, Dickfeld T, Reininger A, Adelsberger H, Gebhardt A, et al. Vitronectin receptor (alpha(v)beta3) mediates platelet adhesion to the luminal aspect of endothelial cells: implications for reperfusion in acute myocardial infarction. Circulation (1997) 96:1809–18. doi: 10.1161/01.CIR.96.6.1809
42. Bombeli T, Schwartz BR, Harlan JM. Adhesion of activated platelets to endothelial cells: evidence for a GPIIbIIIa-dependent bridging mechanism and novel roles for endothelial intercellular adhesion molecule 1 (ICAM-1), alphavbeta3 integrin, and GPIbalpha. J Exp Med. (1998) 187:329–39. doi: 10.1084/jem.187.3.329
43. Massberg S, Enders G, Matos FC, Tomic LI, Leiderer R, Eisenmenger S, et al. Fibrinogen deposition at the postischemic vessel wall promotes platelet adhesion during ischemia-reperfusion in vivo. Blood (1999) 94:3829–38.
44. Zarbock A, Ley K, McEver RP, Hidalgo A. Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood (2011) 118:6743–51. doi: 10.1182/blood-2011-07-343566
45. Hamburger SA, McEver RP. GMP-140 mediates adhesion of stimulated platelets to neutrophils. Blood (1990) 75:550–4.
46. Larsen E, Palabrica T, Sajer S, Gilbert GE, Wagner DD, Furie BC, et al. PADGEM-dependent adhesion of platelets to monocytes and neutrophils is mediated by a lineage-specific carbohydrate, LNF III (CD15). Cell (1990) 63:467–74. doi: 10.1016/0092-8674(90)90443-I
47. Moore KL, Varki A, McEver RP. GMP-140 binds to a glycoprotein receptor on human neutrophils: evidence for a lectin-like interaction. J Cell Biol. (1991) 112:491–9. doi: 10.1083/jcb.112.3.491
48. Moore KL, Patel KD, Bruehl RE, Li F, Johnson DA, Lichenstein HS, et al. P-selectin glycoprotein ligand-1 mediates rolling of human neutrophils on P-selectin. J Cell Biol. (1995) 128:661–71. doi: 10.1083/jcb.128.4.661
49. von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res. (2007) 100:27–40. doi: 10.1161/01.RES.0000252802.25497.b7
50. Evangelista V, Manarini S, Sideri R, Rotondo S, Martelli N, Piccoli A, et al. Platelet/polymorphonuclear leukocyte interaction: P-selectin triggers protein-tyrosine phosphorylation-dependent CD11b/CD18 adhesion: role of PSGL-1 as a signaling molecule. Blood (1999) 93:876–85.
51. Yang J, Furie BC, Furie B. The biology of P-selectin glycoprotein ligand-1: its role as a selectin counterreceptor in leukocyte-endothelial and leukocyte-platelet interaction. Thromb Haemost. (1999) 81:1–7. doi: 10.1055/s-0037-1614407
52. Andonegui G, Kerfoot SM, McNagny K, Ebbert KV, Patel KD, Kubes P. Platelets express functional Toll-like receptor-4. Blood (2005) 106:2417–23. doi: 10.1182/blood-2005-03-0916
53. Semple JW, Freedman J. Platelets and innate immunity. Cell Mol Life Sci. (2010) 67:499–511. doi: 10.1007/s00018-009-0205-1
54. Albiger B, Dahlberg S, Henriques-Normark B, Normark S. Role of the innate immune system in host defence against bacterial infections: focus on the Toll-like receptors. J Intern Med. (2007) 261:511–28. doi: 10.1111/j.1365-2796.2007.01821.x
55. Andonegui G, Bonder CS, Green F, Mullaly SC, Zbytnuik L, Raharjo E, et al. Endothelium-derived Toll-like receptor-4 is the key molecule in LPS-induced neutrophil sequestration into lungs. J Clin Invest. (2003) 111:1011–20. doi: 10.1172/JCI16510
56. Ward JR, Bingle L, Judge HM, Brown SB, Storey RF, Whyte MK, et al. Agonists of toll-like receptor (TLR)2 and TLR4 are unable to modulate platelet activation by adenosine diphosphate and platelet activating factor. Thromb Haemost. (2005) 94:831–8. doi: 10.1160/TH05-01-0009
57. Aslam R, Speck ER, Kim M, Crow AR, Bang KW, Nestel FP, et al. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood (2006) 107:637–41. doi: 10.1182/blood-2005-06-2202
58. Katsuyama M, Sugimoto Y, Namba T, Irie A, Negishi M, Narumiya S, et al. Cloning and expression of a cDNA for the human prostacyclin receptor. FEBS Lett. (1994) 344:74–8. doi: 10.1016/0014-5793(94)00355-6
59. Fang W, Wei J, Han D, Chen X, He G, Wu Q, et al. MC-002 exhibits positive effects against platelets aggregation and endothelial dysfunction through thromboxane A2 inhibition. Thromb Res. (2014) 133:610–5. doi: 10.1016/j.thromres.2014.01.029
60. Evangelista V, Rajtar G, de Gaetano G, White JG, Cerletti C. Platelet activation by fMLP-stimulated polymorphonuclear leukocytes: the activity of cathepsin G is not prevented by antiproteinases. Blood (1991) 77:2379–88.
61. Maugeri N, Evangelista V, Piccardoni P, Dell'Elba G, Celardo A, de Gaetano G, et al. Transcellular metabolism of arachidonic acid: increased platelet thromboxane generation in the presence of activated polymorphonuclear leukocytes. Blood (1992) 80:447–51.
62. Maugeri N, Evangelista V, Celardo A, Dell'Elba G, Martelli N, Piccardoni P, et al. Polymorphonuclear leukocyte-platelet interaction: role of P-selectin in thromboxane B2 and leukotriene C4 cooperative synthesis. Thromb Haemost. (1994) 72:450–6. doi: 10.1055/s-0038-1648888
63. Mazzucco L, Borzini P, Gope R. Platelet-derived factors involved in tissue repair-from signal to function. Transfus Med Rev. (2010) 24:218–34. doi: 10.1016/j.tmrv.2010.03.004
64. Assoian RK, Komoriya A, Meyers CA, Miller DM, Sporn MB. Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization. J Biol Chem. (1983) 258:7155–60.
65. Andersson PO, Stockelberg D, Jacobsson S, Wadenvik H. A transforming growth factor-beta1-mediated bystander immune suppression could be associated with remission of chronic idiopathic thrombocytopenic purpura. Ann Hematol. (2000) 79:507–13. doi: 10.1007/s002770000177
66. Andersson PO, Olsson A, Wadenvik H. Reduced transforming growth factor-beta1 production by mononuclear cells from patients with active chronic idiopathic thrombocytopenic purpura. Br J Haematol. (2002) 116:862–7. doi: 10.1046/j.0007-1048.2002.03345.x
67. Brandt E, Petersen F, Ludwig A, Ehlert JE, Bock L, Flad HD. The beta-thromboglobulins and platelet factor 4: blood platelet-derived CXC chemokines with divergent roles in early neutrophil regulation. J Leukoc Biol. (2000) 67:471–8. doi: 10.1002/jlb.67.4.471
68. Grommes J, Alard JE, Drechsler M, Wantha S, Morgelin M, Kuebler WM, et al. Disruption of platelet-derived chemokine heteromers prevents neutrophil extravasation in acute lung injury. Am J Respir Crit Care Med. (2012) 185:628–36. doi: 10.1164/rccm.201108-1533OC
69. Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, et al. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med. (2003) 9:61–7. doi: 10.1038/nm810
70. von Hundelshausen P, Koenen RR, Sack M, Mause SF, Adriaens W, Proudfoot AE, et al. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood (2005) 105:924–30. doi: 10.1182/blood-2004-06-2475
71. Koenen RR, von Hundelshausen P, Nesmelova IV, Zernecke A, Liehn EA, Sarabi A, et al. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat Med. (2009) 15:97–103. doi: 10.1038/nm.1898
72. Ghasemzadeh M, Kaplan ZS, Alwis I, Schoenwaelder SM, Ashworth KJ, Westein E, et al. The CXCR1/2 ligand NAP-2 promotes directed intravascular leukocyte migration through platelet thrombi. Blood (2013) 121:4555–66. doi: 10.1182/blood-2012-09-459636
73. Walz A, Dewald B, von Tscharner V, Baggiolini M. Effects of the neutrophil-activating peptide NAP-2, platelet basic protein, connective tissue-activating peptide III and platelet factor 4 on human neutrophils. J Exp Med. (1989) 170:1745–50. doi: 10.1084/jem.170.5.1745
74. Brown AJ, Joseph PR, Sawant KV, Rajarathnam K. Chemokine CXCL7 heterodimers: structural insights, CXCR2 receptor function, and glycosaminoglycan interactions. Int J Mol Sci. (2017) 18:748. doi: 10.3390/ijms18040748
75. Brown AJ, Sepuru KM, Rajarathnam K. Structural basis of native CXCL7 monomer binding to CXCR2 receptor N-domain and glycosaminoglycan heparin. Int J Mol Sci. (2017). 18:508. doi: 10.3390/ijms18030508
76. Brown AJ, Sepuru KM, Sawant KV, Rajarathnam K. Platelet-derived chemokine CXCL7 Dimer preferentially exists in the glycosaminoglycan-bound form: implications for neutrophil-platelet crosstalk. Front Immunol. (2017) 8:1248. doi: 10.3389/fimmu.2017.01248
77. Ehlert JE, Ludwig A, Grimm TA, Lindner B, Flad HD, Brandt E. Down-regulation of neutrophil functions by the ELR(+) CXC chemokine platelet basic protein. Blood (2000) 96:2965–72.
78. Gleissner CA, von Hundelshausen P, Ley K. Platelet chemokines in vascular disease. Arterioscler Thromb Vasc Biol. (2008) 28:1920–7. doi: 10.1161/ATVBAHA.108.169417
79. Clemetson KJ, Clemetson JM, Proudfoot AE, Power CA, Baggiolini M, Wells TN. Functional expression of CCR1, CCR3, CCR4, and CXCR4 chemokine receptors on human platelets. Blood (2000) 96:4046–54.
80. Schafer A, Schulz C, Eigenthaler M, Fraccarollo D, Kobsar A, Gawaz M, et al. Novel role of the membrane-bound chemokine fractalkine in platelet activation and adhesion. Blood (2004) 103:407–12. doi: 10.1182/blood-2002-10-3260
81. Gear AR, Camerini D. Platelet chemokines and chemokine receptors: linking hemostasis, inflammation, and host defense. Microcirculation (2003) 10:335–50. doi: 10.1080/mic.10.3-4.335.350
82. Kraemer BF, Borst O, Gehring EM, Schoenberger T, Urban B, Ninci E, et al. PI3 kinase-dependent stimulation of platelet migration by stromal cell-derived factor 1 (SDF-1). J Mol Med. (2010) 88:1277–88. doi: 10.1007/s00109-010-0680-8
83. Kraemer BF, Schmidt C, Urban B, Bigalke B, Schwanitz L, Koch M, et al. High shear flow induces migration of adherent human platelets. Platelets (2011) 22:415–21. doi: 10.3109/09537104.2011.556277
84. Gaertner F, Ahmad Z, Rosenberger G, Fan S, Nicolai L, Busch B, et al. Migrating platelets are mechano-scavengers that collect and bundle bacteria. Cell (2017) 171:1368–82.e23. doi: 10.1016/j.cell.2017.11.001
85. Gachet C. ADP receptors of platelets and their inhibition. Thromb Haemost. (2001) 86:222–32. doi: 10.1055/s-0037-1616220
86. Winning J, Claus RA, Pletz MW, Bauer M, Losche W. Adenosine diphosphate receptor antagonist clopidogrel sulfate attenuates LPS-induced systemic inflammation in a rat model. Shock (2011) 36:317; author reply 317–318. doi: 10.1097/SHK.0b013e318224f66a
87. Totani L, Dell'Elba G, Martelli N, Di Santo A, Piccoli A, Amore C, et al. Prasugrel inhibits platelet-leukocyte interaction and reduces inflammatory markers in a model of endotoxic shock in the mouse. Thromb Haemost. (2012) 107:1130–40. doi: 10.1160/TH11-12-0867
88. Liverani E, Kilpatrick LE, Tsygankov AY, Kunapuli SP. The role of P2Y(1)(2) receptor and activated platelets during inflammation. Curr Drug Targets (2014) 15:720–8. doi: 10.2174/1389450115666140519162133
89. Liverani E, Rico MC, Yaratha L, Tsygankov AY, Kilpatrick LE, Kunapuli SP. LPS-induced systemic inflammation is more severe in P2Y12 null mice. J Leukoc Biol. (2014) 95:313–23. doi: 10.1189/jlb.1012518
90. Goldsmith HL, Spain S. Margination of leukocytes in blood flow through small tubes. Microvasc Res. (1984) 27:204–22. doi: 10.1016/0026-2862(84)90054-2
91. Goldsmith HL, Spain S. Radial distribution of white cells in tube flow. Kroc Found Ser. (1984) 16:131–46.
92. Rinder HM, Bonan JL, Rinder CS, Ault KA, Smith BR. Dynamics of leukocyte-platelet adhesion in whole blood. Blood (1991) 78:1730–7.
93. Peters MJ, Heyderman RS, Hatch DJ, Klein NJ. Investigation of platelet-neutrophil interactions in whole blood by flow cytometry. J Immunol Methods (1997) 209:125–35. doi: 10.1016/S0022-1759(97)00139-7
94. Schmidtke DW, Diamond SL. Direct observation of membrane tethers formed during neutrophil attachment to platelets or P-selectin under physiological flow. J Cell Biol. (2000) 149:719–30. doi: 10.1083/jcb.149.3.719
95. Yeo EL, Sheppard JA, Feuerstein IA. Role of P-selectin and leukocyte activation in polymorphonuclear cell adhesion to surface adherent activated platelets under physiologic shear conditions (an injury vessel wall model). Blood (1994) 83:2498–507.
96. Evangelista V, Manarini S, Rotondo S, Martelli N, Polischuk R, McGregor JL, et al. Platelet/polymorphonuclear leukocyte interaction in dynamic conditions: evidence of adhesion cascade and cross talk between P-selectin and the beta 2 integrin CD11b/CD18. Blood (1996) 88:4183–94.
97. Hidari KI, Weyrich AS, Zimmerman GA, McEver RP. Engagement of P-selectin glycoprotein ligand-1 enhances tyrosine phosphorylation and activates mitogen-activated protein kinases in human neutrophils. J Biol Chem. (1997) 272:28750–6. doi: 10.1074/jbc.272.45.28750
98. Blanks JE, Moll T, Eytner R, Vestweber D. Stimulation of P-selectin glycoprotein ligand-1 on mouse neutrophils activates beta 2-integrin mediated cell attachment to ICAM-1. Eur J Immunol. (1998). 28:433–43.
99. Konstantopoulos K, Neelamegham S, Burns AR, Hentzen E, Kansas GS, Snapp KR, et al. Venous levels of shear support neutrophil-platelet adhesion and neutrophil aggregation in blood via P-selectin and beta2-integrin. Circulation (1998) 98:873–82. doi: 10.1161/01.CIR.98.9.873
100. Evangelista V, Pamuklar Z, Piccoli A, Manarini S, Dell'elba G, Pecce R, et al. Src family kinases mediate neutrophil adhesion to adherent platelets. Blood (2007) 109:2461–9. doi: 10.1182/blood-2006-06-029082
101. Diacovo TG, de Fougerolles AR, Bainton DF, Springer TA. A functional integrin ligand on the surface of platelets: intercellular adhesion molecule-2. J Clin Invest. (1994) 94:1243–51. doi: 10.1172/JCI117442
102. Kuijper PH, Gallardo Tores HI, Lammers JW, Sixma JJ, Koenderman L, Zwaginga JJ. Platelet associated fibrinogen and ICAM-2 induce firm adhesion of neutrophils under flow conditions. Thromb Haemost. (1998) 80:443–8. doi: 10.1055/s-0037-1615227
103. Kirton CM, Nash GB. Activated platelets adherent to an intact endothelial cell monolayer bind flowing neutrophils and enable them to transfer to the endothelial surface. J Lab Clin Med. (2000) 136:303–13. doi: 10.1067/mlc.2000.109406
104. Weber C, Springer TA. Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of Mac-1 with fibrinogen bound to alphaIIbbeta3 and stimulated by platelet-activating factor. J Clin Invest. (1997) 100:2085–93. doi: 10.1172/JCI119742
105. Simon DI, Chen Z, Xu H, Li CQ, Dong J, McIntire LV, et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J Exp Med. (2000) 192:193–204. doi: 10.1084/jem.192.2.193
106. Wang Y, Sakuma M, Chen Z, Ustinov V, Shi C, Croce K, et al. Leukocyte engagement of platelet glycoprotein Ibalpha via the integrin Mac-1 is critical for the biological response to vascular injury. Circulation (2005) 112:2993–3000. doi: 10.1161/CIRCULATIONAHA.105.571315
107. Lo SC, Hung CY, Lin DT, Peng HC, Huang TF. Involvement of platelet glycoprotein Ib in platelet microparticle mediated neutrophil activation. J Biomed Sci. (2006) 13:787–96. doi: 10.1007/s11373-006-9107-5
108. Miedzobrodzki J, Panz T, Plonka PM, Zajac K, Dracz J, Pytel K, et al. Platelets augment respiratory burst in neutrophils activated by selected species of gram-positive or gram-negative bacteria. Folia Histochem Cytobiol. (2008) 46:383–8. doi: 10.2478/v10042-008-0052-1
109. Hidalgo A, Chang J, Jang JE, Peired AJ, Chiang EY, Frenette PS. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat Med. (2009) 15:384–91. doi: 10.1038/nm.1939
110. Suzuki K, Sugimura K, Hasegawa K, Yoshida K, Suzuki A, Ishizuka K, et al. Activated platelets in ulcerative colitis enhance the production of reactive oxygen species by polymorphonuclear leukocytes. Scand J Gastroenterol. (2001) 36:1301–6. doi: 10.1080/003655201317097164
111. Wettero J, Tengvall P, Bengtsson T. Platelets stimulated by IgG-coated surfaces bind and activate neutrophils through a selectin-dependent pathway. Biomaterials (2003) 24:1559–73. doi: 10.1016/S0142-9612(02)00543-4
112. Evangelista V, Manarini S, Dell'Elba G, Martelli N, Napoleone E, Di Santo A, et al. Clopidogrel inhibits platelet-leukocyte adhesion and platelet-dependent leukocyte activation. Thromb Haemost. (2005) 94:568–77. doi: 10.1160/TH05-01-0020
113. Sakamoto H, Ooshima A. Activation of neutrophil phagocytosis of complement coated and IgG coated sheep erythrocytes by platelet release products. Br J Haematol. (1985) 60:173–81. doi: 10.1111/j.1365-2141.1985.tb07398.x
114. Sakamoto H, Yokoya Y, Ooshima A. In vitro control of neutrophilic phagocytosis of IgG-coated SRBC by macromolecules involved in released products from platelets. J Leukoc Biol. (1987) 41:55–62. doi: 10.1002/jlb.41.1.55
115. Miyabe K, Sakamoto N, Wu YH, Mori N, Sakamoto H. Effects of platelet release products on neutrophilic phagocytosis and complement receptors. Thromb Res. (2004) 114:29–36. doi: 10.1016/j.thromres.2004.04.003
116. Wu B, Liu G, Yube K, Ueno M, Tanaka S, Onodera M, et al. Effects of platelet release products on neutrophilic activity in human whole blood. Inflamm Res. (2009) 58:321–8. doi: 10.1007/s00011-009-8230-y
117. Assinger A, Laky M, Schabbauer G, Hirschl AM, Buchberger E, Binder BR, et al. Efficient phagocytosis of periodontopathogens by neutrophils requires plasma factors, platelets and TLR2. J Thromb Haemost. (2011) 9:799–809. doi: 10.1111/j.1538-7836.2011.04193.x
118. Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. (2012) 18:1386–93. doi: 10.1038/nm.2847
119. Kraemer BF, Campbell RA, Schwertz H, Cody MJ, Franks Z, Tolley ND, et al. Novel anti-bacterial activities of beta-defensin 1 in human platelets: suppression of pathogen growth and signaling of neutrophil extracellular trap formation. PLoS Pathogens (2011) 7:e1002355. doi: 10.1371/journal.ppat.1002355
120. Hargett LA, Bauer NN. On the origin of microparticles: from “platelet dust” to mediators of intercellular communication. Pulm Circ. (2013) 3:329–40. doi: 10.4103/2045-8932.114760
121. Italiano JE Jr, Mairuhu AT, Flaumenhaft R. Clinical relevance of microparticles from platelets and megakaryocytes. Curr Opin Hematol. (2010) 17:578–84. doi: 10.1097/MOH.0b013e32833e77ee
122. Horstman LL, Ahn YS. Platelet microparticles: a wide-angle perspective. Crit Rev Oncol Hematol. (1999) 30:111–42. doi: 10.1016/S1040-8428(98)00044-4
123. Garcia BA, Smalley DM, Cho H, Shabanowitz J, Ley K, Hunt DF. The platelet microparticle proteome. J Proteome Res. (2005) 4:1516–21. doi: 10.1021/pr0500760
124. Gawaz M, Dickfeld T, Bogner C, Fateh-Moghadam S, Neumann FJ. Platelet function in septic multiple organ dysfunction syndrome. Intensive Care Med. (1997) 23:379–85. doi: 10.1007/s001340050344
125. Jacoby RC, Owings JT, Holmes J, Battistella FD, Gosselin RC, Paglieroni TG. Platelet activation and function after trauma. J Trauma (2001) 51:639–47. doi: 10.1097/00005373-200110000-00003
126. Ogura H, Kawasaki T, Tanaka H, Koh T, Tanaka R, Ozeki Y, et al. Activated platelets enhance microparticle formation and platelet-leukocyte interaction in severe trauma and sepsis. J Trauma (2001) 50:801–9. doi: 10.1097/00005373-200105000-00005
127. Mause SF, von Hundelshausen P, Zernecke A, Koenen RR, Weber C. Platelet microparticles: a transcellular delivery system for RANTES promoting monocyte recruitment on endothelium. Arterioscler Thromb Vasc Biol. (2005) 25:1512–8. doi: 10.1161/01.ATV.0000170133.43608.37
128. Marcus AJ, Safier LB, Ullman HL, Islam N, Broekman MJ, Falck JR, et al. Platelet-neutrophil interactions. (12S)-hydroxyeicosatetraen-1,20-dioic acid: a new eicosanoid synthesized by unstimulated neutrophils from (12S)-20-dihydroxyeicosatetraenoic acid. J Biol Chem. (1988) 263:2223–9.
129. Laidlaw TM, Kidder MS, Bhattacharyya N, Xing W, Shen S, Milne GL, et al. Cysteinyl leukotriene overproduction in aspirin-exacerbated respiratory disease is driven by platelet-adherent leukocytes. Blood (2012) 119:3790–8. doi: 10.1182/blood-2011-10-384826
130. Page C, Pitchford S. Neutrophil and platelet complexes and their relevance to neutrophil recruitment and activation. Int Immunopharmacol. (2013) 17:1176–84. doi: 10.1016/j.intimp.2013.06.004
131. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. (2000) 342:1334–49. doi: 10.1056/NEJM200005043421806
132. Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol. Med. (2011) 17:293–307. doi: 10.2119/molmed.2010.00138
133. Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. (2012) 122:2731–40. doi: 10.1172/JCI60331
134. Looney MR, Gropper MA, Matthay MA. Transfusion-related acute lung injury: a review. Chest (2004) 126:249–58. doi: 10.1378/chest.126.1.249
135. Kornerup KN, Salmon GP, Pitchford SC, Liu WL, Page CP. Circulating platelet-neutrophil complexes are important for subsequent neutrophil activation and migration. J Appl Physiol. (2010) 109:758–67. doi: 10.1152/japplphysiol.01086.2009
136. Cognasse F, Lafarge S, Chavarin P, Acquart S, Garraud O. Lipopolysaccharide induces sCD40L release through human platelets TLR4, but not TLR2 and TLR9. Intensive Care Med. (2007) 33:382–4. doi: 10.1007/s00134-006-0488-8
137. Khan SY, Kelher MR, Heal JM, Blumberg N, Boshkov LK, Phipps R, et al. Soluble CD40 ligand accumulates in stored blood components, primes neutrophils through CD40, and is a potential cofactor in the development of transfusion-related acute lung injury. Blood (2006) 108:2455–62. doi: 10.1182/blood-2006-04-017251
138. de Stoppelaar SF, van 't Veer C, Claushuis TA, Albersen BJ, Roelofs JJ, van der Poll T. Thrombocytopenia impairs host defense in gram-negative pneumonia derived sepsis. Blood (2014) 124:3781–90. doi: 10.1182/blood-2014-05-573915
139. Ortiz-Munoz G, Mallavia B, Bins A, Headley M, Krummel MF, Looney MR. Aspirin-triggered 15-epi-lipoxin A4 regulates neutrophil-platelet aggregation and attenuates acute lung injury in mice. Blood (2014) 124:2625–34. doi: 10.1182/blood-2014-03-562876
140. Lefrancais E, Ortiz-Munoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature (2017) 544:105–9. doi: 10.1038/nature21706
141. Pitchford SC, Momi S, Baglioni S, Casali L, Giannini S, Rossi R, et al. Allergen induces the migration of platelets to lung tissue in allergic asthma. Am J Respir Crit Care Med. (2008) 177:604–12. doi: 10.1164/rccm.200702-214OC
142. Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol. (2009) 27:147–63. doi: 10.1146/annurev.immunol.021908.132629
143. Vincent JL, Yagushi A, Pradier O. Platelet function in sepsis. Crit Care Med. (2002) 30:S313–7. doi: 10.1097/00003246-200205001-00022
144. Greinacher A, Selleng K. Thrombocytopenia in the intensive care unit patient. Hematology Am Soc Hematol Educ Program (2010) 2010:135–43. doi: 10.1182/asheducation-2010.1.135
145. Levy BD, Serhan CN. Resolution of acute inflammation in the lung. Annu Rev Physiol. (2014) 76:467–92. doi: 10.1146/annurev-physiol-021113-170408
146. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. (2012) 30:459–89. doi: 10.1146/annurev-immunol-020711-074942
147. Kennedy AD, DeLeo FR. Neutrophil apoptosis and the resolution of infection. Immunol Res. (2009) 43:25–61. doi: 10.1007/s12026-008-8049-6
148. Matute-Bello G, Liles WC, Radella F II, Steinberg KP, Ruzinski JT, Jonas M, et al. Neutrophil apoptosis in the acute respiratory distress syndrome. Am J Respir Crit Care Med. (1997) 156:1969–77. doi: 10.1164/ajrccm.156.6.96-12081
149. Cho W, Song JY, Oh SW, Kim MG, Ko YS, Lee HY, et al. Fate of neutrophils during the recovery phase of ischemia/reperfusion induced acute kidney injury. J Korean Med Sci. (2017) 32:1616–25. doi: 10.3346/jkms.2017.32.10.1616
150. Lax S, Rayes J, Wichaiyo S, Haining EJ, Lowe K, Grygielska B, et al. Platelet CLEC-2 protects against lung injury via effects of its ligand podoplanin on inflammatory alveolar macrophages in the mouse. Am J Physiol Lung Cell Mol Physiol. (2017) 313:L1016–29. doi: 10.1152/ajplung.00023.2017
151. Herter JM, Rossaint J, Zarbock A. Platelets in inflammation and immunity. J Thrombosis Haemost. (2014) 12:1764–75. doi: 10.1111/jth.12730
152. Yagnik D. Macrophage derived platelet activating factor implicated in the resolution phase of gouty inflammation. Int J Inflam. (2014) 2014:526496. doi: 10.1155/2014/526496
153. Yadav H, Kor DJ. Platelets in the pathogenesis of acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. (2015) 309:L915–23. doi: 10.1152/ajplung.00266.2015
154. Haworth O, Cernadas M, Yang R, Serhan CN, Levy BD. Resolvin E1 regulates interleukin 23, interferon-gamma and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat Immunol. (2008) 9:873–9. doi: 10.1038/ni.1627
155. Mitchell S, Thomas G, Harvey K, Cottell D, Reville K, Berlasconi G, et al. Lipoxins, aspirin-triggered epi-lipoxins, lipoxin stable analogues, and the resolution of inflammation: stimulation of macrophage phagocytosis of apoptotic neutrophils in vivo. J Am Soc Nephrol. (2002) 13:2497–507. doi: 10.1097/01.ASN.0000032417.73640.72
156. Bergmann CB, Hefele F, Unger M, Huber-Wagner S, Biberthaler P, van Griensven M, et al. Platelets modulate the immune response following trauma by interaction with CD4+ T regulatory cells in a mouse model. Immunol Res. (2016) 64:508–17. doi: 10.1007/s12026-015-8726-1
157. Khandoga A, Hanschen M, Kessler JS, Krombach F. CD4+ T cells contribute to postischemic liver injury in mice by interacting with sinusoidal endothelium and platelets. Hepatology (2006) 43:306–15. doi: 10.1002/hep.21017
158. Li N. CD4+ T cells in atherosclerosis: regulation by platelets. Thromb Haemost. (2013) 109:980–90. doi: 10.1160/TH12-11-0819
Keywords: platelets, leukocytes, neutrophils, inflammation, resolution
Citation: Rossaint J, Margraf A and Zarbock A (2018) Role of Platelets in Leukocyte Recruitment and Resolution of Inflammation. Front. Immunol. 9:2712. doi: 10.3389/fimmu.2018.02712
Received: 28 August 2018; Accepted: 02 November 2018;
Published: 20 November 2018.
Edited by:
Joaquin Teixidó, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Philipp Von Hundelshausen, Ludwig-Maximilians-Universität München, GermanyKrishna Rajarathnam, The University of Texas Medical Branch at Galveston, United States
Copyright © 2018 Rossaint, Margraf and Zarbock. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander Zarbock, emFyYm9ja0B1bmktbXVlbnN0ZXIuZGU=