 Melanie Salvermoser
Melanie Salvermoser Daniela Begandt
Daniela Begandt Ronen Alon
Ronen Alon Barbara Walzog
Barbara Walzog
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 16 November 2018
Sec. Inflammation
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02680
This article is part of the Research Topic Leukocyte Trafficking in Homeostasis and Disease View all 12 articles
Cell migration is indispensable for various biological processes including angiogenesis, wound healing, and immunity. In general, there are two different migration modes described, the mesenchymal migration mode and the amoeboid migration mode. Neutrophils rapidly migrate toward the sites of injury, infection, and inflammation using the amoeboid migration mode which is characterized by cell polarization and a high migration velocity. During site-directed trafficking of neutrophils from the blood stream into the inflamed tissue, neutrophils must first withstand shear stress while migrating on the 2-dimensional endothelial surface. Subsequently, they have to cross different physical barriers during the extravasation process including the squeezing through the compact endothelial monolayer that comprises the blood vessel, the underlining basement membrane and then the 3-dimensional meshwork of extracellular matrix (ECM) proteins in the tissue. Therefore, neutrophils have to rapidly switch between distinct migration modes such as intraluminal crawling, transmigration, and interstitial migration to pass these different confinements and mechanical barriers. The nucleus is the largest and stiffest organelle in every cell and is therefore the key cellular element involved in cellular migration through variable confinements. This review highlights the importance of nuclear deformation during neutrophil crossing of such confinements, with a focus on transendothelial migration and interstitial migration. We discuss the key molecular components involved in the nuclear shape changes that underlie neutrophil motility and squeezing through cellular and ECM barriers. Understanding the precise molecular mechanisms that orchestrate these distinct neutrophil migration modes introduces an opportunity to develop new therapeutic concepts for controlling pathological neutrophil-driven inflammation.
Neutrophils are important players in innate immunity as they represent the first immune cells arriving at site of tissue injury or infection. Besides their ability to control local infections, neutrophils are critically involved in tissue remodeling including wound healing, angiogenesis, and tumor metastasis (1–6). During acute inflammation, neutrophils are recruited from the blood stream into the inflamed tissue following a well-defined multi-step recruitment cascade. This cascade is initiated by neutrophil capturing via specific adhesion receptors on inflamed blood vessels, followed by fast and slow rolling, arrest, adhesion strengthening, intraluminal crawling, and protrusion through endothelial junctions in search for exit cues (7). These intravascular events are then followed by neutrophil squeezing through junctions ending in successful transendothelial migration (TEM), followed by abluminal crawling of the neutrophil in between the endothelial layer and its associated pericyte sheet, and interstitial migration to the final destination at the site of inflammation (8). An indispensable prerequisite for efficient neutrophil recruitment is their ability to migrate in different microenvironments. Following 2-dimensional (2D) crawling on the inflamed endothelium in search of potential exit sites, neutrophils protrude and transmigrate through the endothelial monolayer establishing sub-endothelial crawling which involves simultaneous engagement of the endothelial layer and the subjacent basement membrane (BM). Soon thereafter the neutrophil begins to crawl on and navigate in between individual pericytes on its way to the interstitial space where they migrate in a 3D collagen-rich environment toward the site of inflammation (Figure 1) (9–11). During these processes, neutrophil migration is characterized by rapid shape changes underlying polarization into a lamellipodium and a uropod (12, 13). In this review, we describe the diverse environmental conditions which dictate the different migration modes neutrophils employ with a stress on the molecular mechanisms of nuclear deformation events critical for neutrophil squeezing through different cellular (i.e., endothelial and pericytic), and extracellular barriers at sites of inflammation.
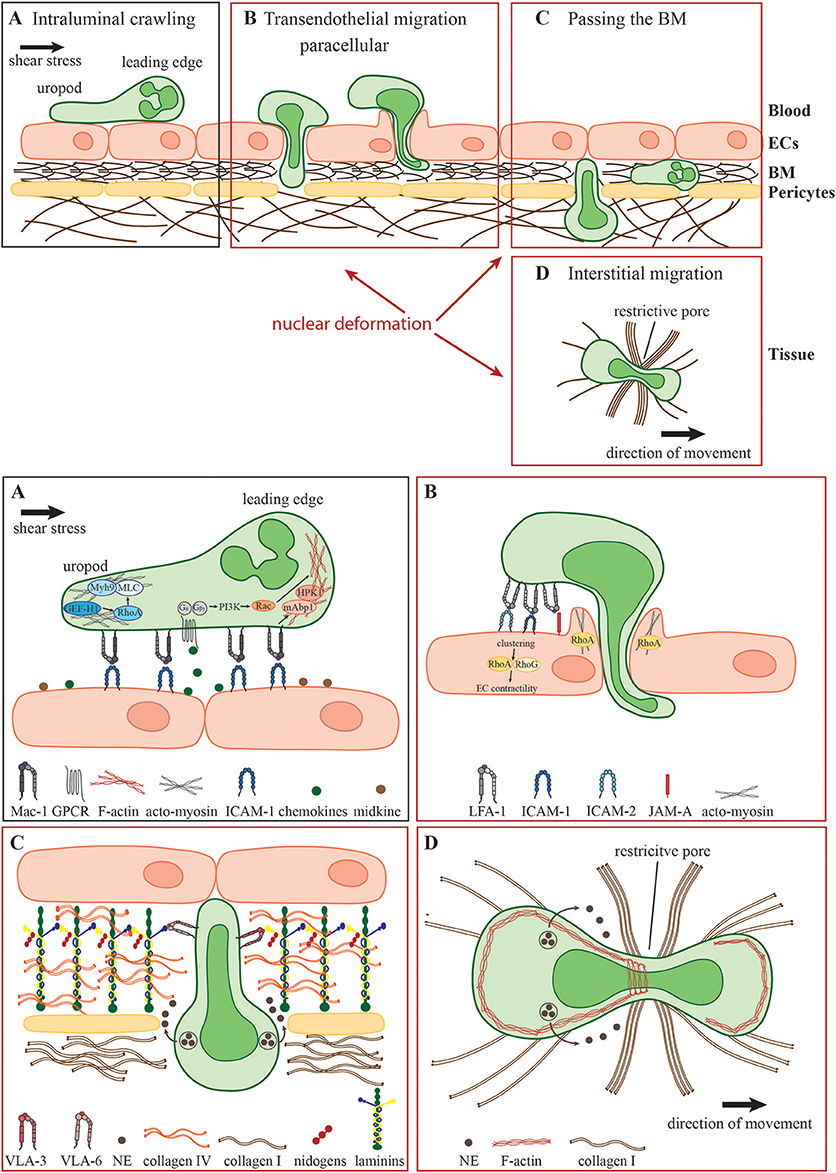
Figure 1. Neutrophil migration in different environmental conditions. During the acute inflammatory response, (A) recently arrested neutrophils migrate along the inflamed endothelium (intraluminal crawling) toward potential exit sites. Intraluminal crawling is mainly mediated by the interaction of Mac-1 on neutrophils with ICAM-1 on inflamed ECs. Binding of chemokines and other chemoattractants to their respective GPCRs results in the dissociation of the Gα and Gβγ subunits with subsequent intracellular signaling inducing cell polarization with an F-actin-rich leading edge and an acto-myosin-rich uropod. (B) Neutrophils protrude and transmigrate through the endothelial monolayer via the paracellular route. Here, LFA-1- and Mac-1-engagement of endothelial ligands including ICAM-1, ICAM-2, and JAM-A activates endothelial Rho-GTPases e.g., RhoA, RhoG, and NMII leading to EC contractility and plasma leakage restriction. Neutrophils must use their own Rho GTPases to squeeze their nuclei through paracellular endothelial junctions triggering gap formation. (C) Following transmigration, neutrophils pass the subjacent basement membrane (BM) and often also crawl on adjacent pericytes embedded in the BM. The main components of the BM are laminins, collagen type IV and nidogens. Neutrophils penetrate this meshwork through LER which can be enlarged by the secretion of elastase (NE) and by nuclear squeezing, both potentially coordinated by the neutrophil integrins VLA-3 and VLA-6 and their interactions with BM collagens and laminins. (D) In the inflamed tissue, neutrophils migrate within a 3D collagen I-rich environment toward the site of inflammation (interstitial migration). Here, along with NE secretion, neutrophils deform and push forward their nuclei to pass through restrictive barriers in the meshwork of collagen fibers, most probably by dynamic interactions of their actin cytoskeleton with the nuclear lamina.
An important characteristic of neutrophils is their high flexibility to adapt their mode of migration rapidly to the environmental conditions. Intraluminal crawling occurs either under low hemodynamic shear stress conditions in postcapillary venules during acute inflammation or under high shear stress conditions in inflamed arteries e.g., during the development of a chronic disease such as atherosclerosis (14). How neutrophils resist shear stress has been reviewed in detail elsewhere (15). Briefly, the process of intraluminal crawling involves specific β2 integrin-mediated shear resistant adhesive interactions of neutrophils with endothelial cells (ECs) (Figure 1A). The key integrin ligands on inflamed ECs that enable efficient intravascular neutrophil crawling are ICAMs (16). During this mode of 2D migration β2 integrins anchor neutrophils to the adhesive substratum enabling force transmission from the actin cytoskeleton to the environment (17–20). Additional molecules that facilitate intravascular crawling are the cytokine midkine and the serine protease Cathepsin G (CatG) (21, 22). Weckbach et al. demonstrated that the genetic absence of midkine abrogates neutrophil adhesion and extravasation in TNF α-stimulated mouse cremaster muscle venules arguing for a pro-adhesive role of midkine probably by binding to the neutrophil LDL-receptor-related protein-1 (LRP-1) (6, 21, 23). In contrast, CatG displayed by ECs has been found to be exclusively important for neutrophil adhesion to arteries under high flow conditions (22). Integrin-dependent neutrophil adhesion and crawling require the binding of chemokines presented by inflamed blood vessels with respective G-protein-coupled receptors (GPCR) on neutrophils eliciting intracellular signaling that triggers integrin adhesiveness, as well as shape changes and polarization (24). Upon GPCR engagement, primarily CXCR2 (25), the G-protein dissociates into distinct Gαi and Gβγ subunits, which regulate the activity of different molecules such as ion channels, adenylyl cyclase and phosphatidylinositol 3-kinase (PI3K) (26, 27). Activation of the PI3K leads to the recruitment of small guanosine triphosphatases (GTPases) of the Rho family including Rac, Cdc42, and RhoA. Neutrophil GPCRs can also activate these different Rho GTPases via PI3K-independent pathways (28). The appropriate subcellular and spatiotemporal regulation of these signaling molecules mediate cell polarization into an F-actin-rich lamellipodium and a myosin-rich trailing edge—a prerequisite of the amoeboid migration mode (29–31). The non-muscle myosin class II (NMII) protein complex is fundamentally important to maintain cell polarization by linking and translocating F-actin filaments (32, 33). Recently, Zehrer et al. demonstrated the important role of Myh9, the heavy chain of NMIIa in neutrophils for their proper 2D migration (crawling), TEM, and 3D migration. Myh9 was found to be critical for the retraction of the uropod and the consolidation of the leading edge ensuring proper neutrophil polarization and migration (34). Notably, integrin-mediated neutrophil crawling can occur with, against and perpendicular to the direction of blood flow ensuring optimal scanning capacity of the endothelial surface for appropriate extravasation sites (35). Neutrophil crawling to and protrusion through endothelial junctions are regulated by specific cytoskeletal adaptors such as the Rho-GTPase specific guanine exchange factor (GEF) Vav1, the mammalian actin binding protein 1 (mAbp1), the hematopoietic progenitor kinase 1 (HPK1), and GEF-H1 (36–39). It has been shown that CXCL2-stimulated neutrophils use Vav1 for their shear stress-induced perpendicular crawling as the genetic absence of Vav1 results in migration exclusively in the direction of blood flow. Under artificial shear free conditions, the migration behavior of neutrophils is intact in the genetic absence of Vav1 pointing toward the specialized role of Vav1 for Rho activities orchestrating integrin-mediated neutrophil crawling under shear flow (36). The same is true for mAbp1 and its interacting protein HPK1 indicating that these two proteins are additionally required for neutrophil crawling under shear flow (37, 38). Recently, Fine et al. demonstrated that spreading and mechanotactic migration are impaired in the genetic absence of GEF-H1, a specific RhoA GEF (29, 39). These data indicate that neutrophils possess tightly regulated molecular mechanisms that allow their integrin-mediated migration on inflamed vessels under shear stress conditions, a critical checkpoint in their extravasation into inflamed tissues. Once reaching potential exit sites, primarily paracellular endothelial junctions, neutrophils traverse the endothelial monolayer via sequential steps of protrusion through these junction, formation of large pseudopodia in the subendothelial compartments and squeezing of their multi-lobular nuclei through adjacent ECs (Figure 1B). The ECs lining the blood vessel are connected by elaborated endothelial junctions composed of variable tight junctions, adherens junctions and gap junctions (40). In general, neutrophils take almost exclusively the paracellular route for their TEM (41). Neutrophil adhesion triggers also EC signaling events believed to facilitate the disassembly of the endothelial junctions enabling neutrophils to send their protrusions through the endothelial monolayer in search for exit signals, primarily chemokines highly enriched within the endothelial BM (42). Engagements of neutrophil integrins with different endothelial ligands like ICAM-1, ICAM-2, and JAM-A initiates the formation of “docking structures” (43) or “transmigratory cups” (44) consisting of pseudopod-like, F-actin-rich endothelial membrane extensions surrounding the leukocyte (45). Subsequent activation of endothelial Rho-GTPases like RhoA, and RhoG, and NMII triggers EC contractility temporally linked to gap formation (46–48). However, recent works have proposed an alternative mechanism whereby endothelial RhoA and acto-myosin contractility are not required for gap formation by transmigrating neutrophils. Instead, gap formation is dictated by neutrophil protrusion and nucleus squeezing through the paracellular endothelial junctions and at rare instances also through transcellular pores, which generate large displacements of the highly elastic endothelial stress fibers and collapse of thin actin filaments interlaced in between these actin bundles (49, 50). One of these works suggested that RhoA activation in endothelial cells is essential for restricting plasma leakage through the gaps generated by squeezing neutrophils (49). Collectively these studies suggested that neutrophils rather than endothelial cells control their TEM dynamics and that nuclear squeezing determines both the gap size generated by transmigrating leukocytes and the speed of TEM (50).
After successful TEM, neutrophils have to pass the perivascular BM predominantly consisting of laminins (isoform 411 and 511), collagen type IV, heparan sulfate proteoglycans, and nidogens (51–56) as well as embedded pericytes adjacent to the blood vessels (Figure 1C) (57, 58). The venular BM exhibits low-expression regions (LER) of laminins and collagen IV which are enriched between pericytes and are favored exit sites for neutrophils to overcome the BM (59, 60). However, the exact mechanism how neutrophils penetrate the BM is still highly debated. Neutrophils contain specific proteases including matrix metalloproteases and the serine protease neutrophil elastase (NE) and use these proteases to degrade the BM and squeeze through LERs (61, 62). Indeed, elastase-deficient neutrophils can normally cross inflamed endothelium but fail to penetrate the BM (62). In addition, the binding of the neutrophil integrins VLA-3 (α3β1) and VLA-6 (α6β1) to the BM is thought to facilitate neutrophil remodeling of the BM enlargement of LER and interstitial migration at sites of inflammation (59, 63, 64). Interestingly, VLA-3, VLA-6, and NE are located in intracellular vesicles which need to be translocated to the cell surface for efficient neutrophil transmigration through the BM, implicating these integrins as potential scavengers of elastase that restricts its proteolytic activity to BM regions enriched with VLA-3 and VLA-6 binding collagens and laminins (65, 66). Recently, Kurz et al. showed that the mammalian sterile 20-like kinase 1 (Mst1) is critically involved in this unique mobilization of VLA-3, VLA-6, and NE to the neutrophil surface (67). Accordingly, Mst1-deficient neutrophils that successfully extravasate through the venular wall get stuck between the endothelial monolayer and the BM and fail to pass the BM. Neutrophil crossing of the endothelial BM is also tightly associated with neutrophil crawling along venular pericytes (59, 68). This type of 2D migration is also ICAM-1-dependent, and during the onset of inflammation pericytes upregulate this ligand for neutrophil LFA-1 and Mac-1 (68). Whether these neutrophil integrins also rely on stimulatory chemokines co-elevated on inflamed pericytes for crawling, a migration mode that takes place in the absence of shear forces is unknown. It is likely, however, that the GTPase machineries discussed above as critical for β2 integrin-mediated neutrophil crawling on inflamed endothelial cells under shear flow- are not identical to those involved in neutrophil crawling on inflamed pericytes.
In the inflamed tissue, neutrophils migrate in 3D collagen-rich environments toward their final destinations at the site of inflammation (Figure 1D). Of note, the microenvironment in which the neutrophils migrate differs both mechanically and biochemically between different organs (69). However, the extracellular matrix as the non-cellular component of all tissues consists predominantly of type I collagen, elastin, proteoglycans, and non-collageneous glycoproteins (70). Here, type I collagen assembles into mechanically stable fibrils providing physical stability of the connective tissue (71). In vivo this fibrillary collagen meshwork exhibits interfibrillar spaces ranging from 2 to 30 μm as shown for mouse cremaster tissue (71, 72). Neutrophils migrate within this confined tissue in a low-adhesive and largely β2 integrin-independent manner. Furthermore, integrin-deficient as well as talin-deficient neutrophils show intact migration in 3D environments compared to control cells, ruling out contributions from either β1 and β3 integrins to this mode of neutrophil motility (17, 73). These data indicate that the traction forces needed for successful 3D migration are transmitted to the environment without integrin-dependent anchoring of the cell to the surface, the prevalent mechanism for neutrophil migration in 2D environments (17, 74). However, the exact mechanism how neutrophils translate their intracellular actomyosin-driven forces to the traction forces critical for their locomotion inside various collagenous 3D environments is still not entirely understood.
In order to study the underlying mechanism experimentally, 3D collagen gels are widely employed. These gels mimic different meshwork architectures with different pore sizes, dependent on the collagen concentration. A collagen concentration of 1.5 mg/mL yields a low-density meshwork with pore cross sections of 10–12 μm2 and a high-density collagen matrix with a collagen concentration of 3.0 mg/mL exhibits pore cross sections ranging between 2 and 3 μm2 (17, 72). As the exact structure of collagen gels cannot be experimentally controlled, various microchannels were recently developed to closely mimic parameters including pore sizes and micro-geometry to improve the analysis of interstitial migration (75, 76). During migration in such confined 3D environments, neutrophils need to pass physical restrictions much smaller than their nucleus similar to the situation in the tissue or 3D collagen gels. Nevertheless, while microchannels are rigid, dense 3D collagen polymers are not only more elastic but can be also locally degraded by neutrophil proteases. Thus, neutrophil passage through microchannels and collagen barriers involve similar but not identical requirements of nucleus deformation.
During cell migration through different mechanical constrictions the dynamic interaction of the nucleus with the actin cytoskeleton is required to ensure proper positioning of the nucleus and nuclear deformation to successfully squeeze the cell through these constrictions (77). Indeed, nucleus deformation is the rate-limiting step for cells to pass through different constrictions smaller than the nucleus (78–81). The neutrophil nucleus is composed of 2–6 nuclear lobes with a diameter of 2 μm connected by a segment with a size of ~ 0.5 μm (82, 83). Nuclear deformation follows three different phases while the cell squeezes through physical barriers, namely the initiation phase, the deformation phase and the remodeling phase (Figure 2A) (78, 84). When the cell reaches the constriction, the nucleus is the first organelle pushing against the constriction (50, 84). During the deformation phase the nucleus elongates into an hour-glass shaped nuclear morphology while squeezing through the constriction. After passing the constriction, the rear of the nucleus pushes forward to refold into its original spherical morphology. The nucleus is mechanically stabilized by a thin, elastic shell encoded by three genes, LMNA, encoding lamins A/C, and LMNB1 and LMNB2, encoding lamin B1 and lamin B2, respectively (85). The rigidity of the nuclei is determined by the relative levels of their A and B lamins (86, 87). The nuclear lamina of neutrophils is much softer than the lamina of most tissue resident cells due to their negligible content of lamin A/C (Figures 2B,C) (88). The neutrophil nucleus is further adjusted for rapid squeezing through small confinements by its unique multi-lobular shape. Expression of a lamin B receptor (LBR) on the inner nuclear membrane is critical for this multi-lobular shape (Figures 2B,C) (89). Interestingly, interference with the multi-lobular shape of the nucleus by LBR knockdown keeping lamin A content low bears minimal effects on nuclear squeezing via rigid pores (90). Notably, bone marrow neutrophil precursors regulate both their nuclear shape and lamin A/C content during maturation. The nuclei of immature neutrophils are stiff and circular as they express higher levels of lamin A/C and lack LBR. Upon full maturation neutrophils adapt their nuclear shape and rigidity to optimize their squeezing through bone marrow sinusoids (89). Similarly, naïve T cells temporally upregulate their lamin A/C expression during TCR activation and remain stationary until they downregulate lamin A/C expression and regain nuclear deformability as they become migratory (91). Thus, nuclear shape and deformability are adapted to the squeezing needs of particular cells.
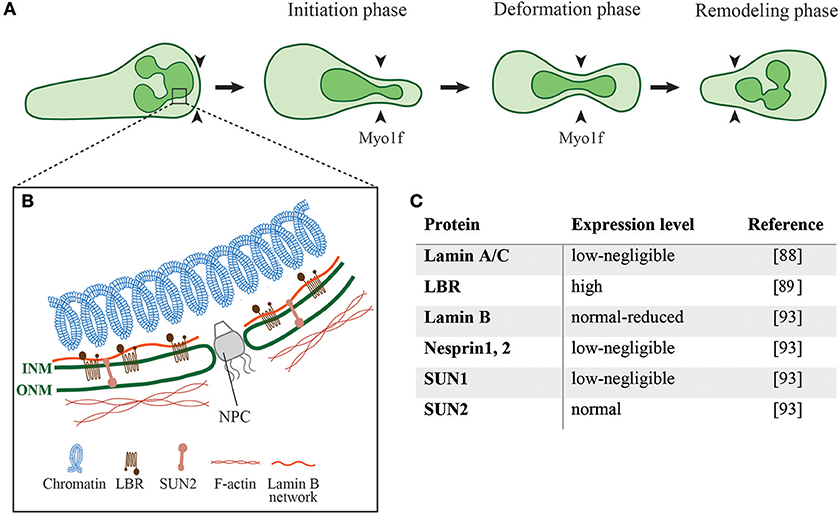
Figure 2. Different phases of nuclear deformation and structural components of the neutrophil nucleus. (A) While neutrophils squeeze through restrictive barriers (indicated by black arrowheads) smaller than their nucleus, the individual nuclear lobes undergo different phases of deformation. During the initiation phase neutrophils use one of its preexistent lobes to penetrate the barrier. This is followed by pushing, deformation, and elongation of the lobe and its neighbor lobes. Myo1f is critically required for nuclear pushing and deformation during this squeezing process. (B) Schematics of the structural proteins that regulate the shape and mechanical properties of a neutrophil nucleus as well as its crosstalk with the neutrophil cytoskeleton. ONM, outer nuclear membrane; INM, inner nuclear membrane; NPC, nuclear pore complex; LBR, Lamin B receptor; (C) Expression levels of different nuclear proteins in neutrophils.
In contrast to epithelial and mesenchymal cells, which keep their stiff nuclei at their rear, motile leukocytes readily translocate their nuclei to their pseudopodia and do so irrespectively of barrier rigidity (50). In a recent study on granulocyte-like differentiated HL-60 cells, we found that this property of the neutrophil nucleus is conserved and is independent of the barrier rigidity the neutrophil is squeezed through. Nevertheless, when the stiffness of the nucleus was elevated by overexpression of lamin A, and when the neutrophil was embedded in a dense collagen matrix the nucleus could no longer translocate to the neutrophil pseudopodia (92). Thus, the exceptional ability of neutrophils to squeeze through mechanically rigid barriers such as collagen-rich interstitial spaces, or the BMs of blood vessels and epithelial barriers likely depends on both low lamin A content, nuclear lamina deformability, and high LBR expression (90). These requirements are dispensable, however, for neutrophil squeezing through the much softer endothelial junctions and consequently for TEM, because of the higher elasticity of the endothelial cytoskeleton than the elasticity of individual collagen fibers within collagen-rich interstitial spaces and the BMs of blood vessels and epithelial barriers.
The nuclear translocation to the neutrophil's pseudopodia, a shared feature among all motile leukocytes which appears to facilitate their squeezing may be regulated by specific interactions of the nuclear cytoskeleton (nucleoskeleton) and the perinuclear actin filaments. The neutrophil nucleoskeleton is deficient of several linker of the nucleoskeleton and cytoskeleton (LINC) complex proteins, including nesprin1, 2, and SUN1 (Figures 2B,C) (93) which are implicated in force transmission in adherent cells (94). Mature neutrophils are possibly devoid of these nuclear-cytoskeletal interactions as part of their highly motile nature and preference of chemotactic cues over integrin-dependent adhesions specialized to transduce forces to the nucleus (95). The nuclei of neutrophils can be also pushed to the leading edge by actomyosin machineries that orchestrate nuclear positioning and squeezing and bridge the neutrophil's uropod with the microtubule-organizing center (MTOC) at the back of the squeezed nucleus (96).
In addition to the unique shape and deformability of the neutrophil nucleus, neutrophil migration to sites of inflammation critically depends on the unconventional class I myosin Myosin 1f (Myo1f), found to facilitate nuclear deformation (84). This recent work suggests that the deformation of the nucleus is almost completely absent in Myo1f-deficient neutrophils compared to control cells resulting in diminished in vitro neutrophil migration within 3D collagen gels and impaired in vivo trafficking toward sites of lesions. Whereas, class II myosins are involved in the generation of contractility forces, class I myosins exist as monomers and link membranes to the actin cytoskeleton (97) potentially implicating these myosins in nuclear deformation critical for neutrophil squeezing. How precisely this unique myosin communicates with the nucleus and its closely associated microtubules remains an open question for future investigations.
Neutrophil migration to sites of inflammation is indispensable for innate immunity as neutrophils are the predominant immune cells combating pathogens. Efficient neutrophil migration critically relies on the exceptionally dynamic deformation of the nucleus of neutrophils. Accordingly, neutrophils obtained from mice lacking LBR expression show hyposegmentation of the nucleus associated with a decreased nuclear deformability and impaired neutrophil responses (98). The same is true for patients suffering from Pelger-Huet anomaly (PHA), a mutation in the human LBR leading to hyposegmentation of the neutrophil nucleus (99). Thus, impaired nuclear deformability can hamper neutrophil migration and function in inflammation. The improvement of our current knowledge of the molecular mechanisms underlying nuclear deformation events critical for neutrophil crossing through distinct mechanical barriers may therefore help to identify novel therapeutic targets for the treatment of neutrophil-driven acute and chronic inflammatory pathologies as well as for the manipulation of neutrophil crosstalks with tumor cells.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The review is based on work supported by grants from the Deutsche Forschungsgemeinschaft (SFB 914, project A02 [BW]). RA is supported by the Israel Science Foundation, the Flight Attendant Medical Research Institute Foundation (FAMRI), U.S.A., as well as a research grant from Carol A. Milett.
1. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. (2013) 13:159–75. doi: 10.1038/nri3399
2. Theilgaard-Mönch K, Knudsen S, Follin P, Borregaard N. The transcriptional activation program of human neutrophils in skin lesions supports their important role in wound healing. J Immunol. (2004) 172:7684–93. doi: 10.4049/jimmunol.172.12.7684
3. Wculek SK, Malanchi I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature (2015) 528:413–7. doi: 10.1038/nature16140
4. Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature (2015) 522:345–8. doi: 10.1038/nature14282
5. Sindrilaru A, Peters T, Schymeinsky J, Oreshkova T, Wang H, Gompf A, et al. Wound healing defect of Vav3-/- mice due to impaired {beta}2-integrin-dependent macrophage phagocytosis of apoptotic neutrophils. Blood (2009) 113:5266–76. doi: 10.1182/blood-2008-07-166702
6. Weckbach LT, Groesser L, Borgolte J, Pagel JI, Pogoda F, Schymeinsky J, et al. Midkine acts as proangiogenic cytokine in hypoxia-induced angiogenesis. Am J Physiol Heart Circ Physiol. (2012) 303:H429–38. doi: 10.1152/ajpheart.00934.2011
7. Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity (2014) 41:694–707. doi: 10.1016/j.immuni.2014.10.008
8. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. (2007) 7:678–89. doi: 10.1038/nri2156
9. Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med. (2006) 203:2569–75. doi: 10.1084/jem.20060925
10. Voisin MB, Woodfin A, Nourshargh S. Monocytes and neutrophils exhibit both distinct and common mechanisms in penetrating the vascular basement membrane in vivo. Arterioscler Thromb Vasc Biol. (2009) 29:1193–9. doi: 10.1161/ATVBAHA.109.187450
11. Stark K, Eckart A, Haidari S, Tirniceriu A, Lorenz M, von Brühl ML, et al. Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and ‘instruct’ them with pattern-recognition and motility programs. Nat Immunol. (2013) 14:41–51. doi: 10.1038/ni.2477
12. Wolf K, Müller R, Borgmann S, Bröcker EB, Friedl P. Amoeboid shape change and contact guidance: T-lymphocyte crawling through fibrillar collagen is independent of matrix remodeling by MMPs and other proteases. Blood (2003) 102:3262–9. doi: 10.1182/blood-2002-12-3791
13. Friedl P, Borgmann S, Bröcker EB. Amoeboid leukocyte crawling through extracellular matrix: lessons from the Dictyostelium paradigm of cell movement. J Leukoc Biol. (2001) 70:491–509. doi: 10.3410/f.1003679.37654
14. Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev. (2011) 91:327–87. doi: 10.1152/physrev.00047.2009
15. Begandt D, Thome S, Sperandio M, Walzog B. How neutrophils resist shear stress at blood vessel walls: molecular mechanisms, subcellular structures, and cell-cell interactions. J Leukoc Biol. (2017) 102:699–709. doi: 10.1189/jlb.3MR0117-026RR
16. Staunton DE, Marlin SD, Stratowa C, Dustin ML, Springer TA. Primary structure of ICAM-1 demonstrates interaction between members of the immunoglobulin and integrin supergene families. Cell (1988) 52:925–33. doi: 10.1016/0092-8674(88)90434-5
17. Lämmermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Söldner R, Hirsch K, et al. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature (2008) 453:51–5. doi: 10.1038/nature06887
18. Malawista SE, de Boisfleury Chevance A. Random locomotion and chemotaxis of human blood polymorphonuclear leukocytes (PMN) in the presence of EDTA: PMN in close quarters require neither leukocyte integrins nor external divalent cations. Proc Natl Acad Sci USA. (1997) 94:11577–82. doi: 10.1073/pnas.94.21.11577
19. Shulman Z, Shinder V, Klein E, Grabovsky V, Yeger O, Geron E, et al. Lymphocyte crawling and transendothelial migration require chemokine triggering of high-affinity LFA-1 integrin. Immunity (2009) 30:384–96. doi: 10.1016/j.immuni.2008.12.020
20. Renkawitz J, Schumann K, Weber M, Lämmermann T, Pflicke H, Piel M, et al. Adaptive force transmission in amoeboid cell migration. Nat Cell Biol. (2009) 11:1438–43. doi: 10.1038/ncb1992
21. Weckbach LT, Gola A, Winkelmann M, Jakob SM, Groesser L, Borgolte J, et al. The cytokine midkine supports neutrophil trafficking during acute inflammation by promoting adhesion via beta2 integrins (CD11/CD18). Blood (2014) 123:1887–96. doi: 10.1182/blood-2013-06-510875
22. Ortega-Gomez A, Salvermoser M, Rossaint J, Pick R, Brauner J, Lemnitzer P, et al. Cathepsin G controls arterial but not venular myeloid cell recruitment. Circulation (2016) 134:1176–88. doi: 10.1161/CIRCULATIONAHA.116.024790
23. Weckbach LT, Preissner KT, Deindl E. The role of midkine in arteriogenesis, involving mechanosensing, endothelial cell proliferation, and vasodilation. Int J Mol Sci. (2018) 19:E2559. doi: 10.3390/ijms19092559
24. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. (2014) 32:659–702. doi: 10.1146/annurev-immunol-032713-120145
25. Smith ML, Olson TS, Ley K. CXCR2- and E-selectin-induced neutrophil arrest during inflammation in vivo. J Exp Med. (2004) 200:935–9. doi: 10.1084/jem.20040424
26. Mocsai A, Walzog B, Lowell CA. Intracellular signaling during neutrophil recruitment. Cardiovasc Res. (2015) 107:373–85. doi: 10.1093/cvr/cvv159
27. Surve CR, Lehmann D, Smrcka AV. A chemical biology approach demonstrates G protein betagamma subunits are sufficient to mediate directional neutrophil chemotaxis. J Biol Chem. (2014) 289:17791–801. doi: 10.1074/jbc.M114.576827
28. Laudanna C, Bolomini-Vittori M. Integrin activation in the immune system. Wiley Interdiscip Rev Syst Biol Med. (2009) 1:116–27. doi: 10.1002/wsbm.9
29. Xu J, Wang F, Van Keymeulen A, Herzmark P, Straight A, Kelly K, et al. Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell (2003) 114:201–14. doi: 10.1016/S0092-8674(03)00555-5
30. Pestonjamasp KN, Forster C, Sun C, Gardiner EM, Bohl B, Weiner O, et al. Rac1 links leading edge and uropod events through Rho and myosin activation during chemotaxis. Blood (2006) 108:2814–20. doi: 10.1182/blood-2006-01-010363
31. Niggli V. Rho-kinase in human neutrophils: a role in signalling for myosin light chain phosphorylation and cell migration. FEBS Lett. (1999) 445:69–72. doi: 10.1016/S0014-5793(99)00098-8
32. Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. (2009) 10:778–90. doi: 10.1038/nrm2786
33. Maupin P, Phillips CL, Adelstein RS, Pollard TD. Differential localization of myosin-II isozymes in human cultured cells and blood cells. J Cell Sci. (1994) 107 (Pt 11):3077–90. doi: 10.1002/cm.970310203
34. Zehrer A, Pick R, Salvermoser M, Boda A, Miller M, Stark K, et al. A fundamental role of Myh9 for neutrophil migration in innate immunity. J Immunol. (2018) 201:1748–64. doi: 10.4049/jimmunol.1701400
35. Sumagin R, Prizant H, Lomakina E, Waugh RE, Sarelius IH. LFA-1 and Mac-1 define characteristically different intralumenal crawling and emigration patterns for monocytes and neutrophils in situ. J Immunol. (2010) 185:7057–66. doi: 10.4049/jimmunol.1001638
36. Phillipson M, Heit B, Parsons SA, Petri B, Mullaly SC, Colarusso P, et al. Vav1 is essential for mechanotactic crawling and migration of neutrophils out of the inflamed microvasculature. J Immunol. (2009) 182:6870–8. doi: 10.4049/jimmunol.0803414
37. Hepper I, Schymeinsky J, Weckbach LT, Jakob SM, Frommhold D, Sixt M, et al. The mammalian actin-binding protein 1 is critical for spreading and intraluminal crawling of neutrophils under flow conditions. J Immunol. (2012) 188:4590–601. doi: 10.4049/jimmunol.1100878
38. Jakob SM, Pick R, Brechtefeld D, Nussbaum C, Kiefer F, Sperandio M, et al. Hematopoietic progenitor kinase 1 (HPK1) is required for LFA-1-mediated neutrophil recruitment during the acute inflammatory response. Blood (2013) 121:4184–94. doi: 10.1182/blood-2012-08-451385
39. Fine N, Dimitriou ID, Rullo J, Sandí MJ, Petri B, Haitsma J, et al. GEF-H1 is necessary for neutrophil shear stress-induced migration during inflammation. J Cell Biol. (2016) 215:107–19. doi: 10.1083/jcb.201603109
40. Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev. (2004) 84:869–901. doi: 10.1152/physrev.00035.2003
41. Nourshargh S, Hordijk PL, Sixt M. Breaching multiple barriers: leukocyte motility through venular walls and the interstitium. Nat Rev Mol Cell Biol. (2010) 11:366–78. doi: 10.1038/nrm2889
42. Bao X, Moseman EA, Saito H, Petryniak B, Thiriot A, Hatakeyama S, et al. Endothelial heparan sulfate controls chemokine presentation in recruitment of lymphocytes and dendritic cells to lymph nodes. Immunity (2010) 33:817–29. doi: 10.1016/j.immuni.2010.10.018
43. Barreiro O, Yanez-Mo M, Serrador JM, Montoya MC, Vicente-Manzanares M, Tejedor R, et al. Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J Cell Biol. (2002) 157:1233–45. doi: 10.1083/jcb.200112126
44. Carman CV, Springer TA. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol. (2004) 167:377–88. doi: 10.1083/jcb.200404129
45. Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol. (2015) 15:692–704. doi: 10.1038/nri3908
46. Stroka KM, Aranda-Espinoza H. Endothelial cell substrate stiffness influences neutrophil transmigration via myosin light chain kinase-dependent cell contraction. Blood (2011) 118:1632–40. doi: 10.1182/blood-2010-11-321125
47. Saito H, Minamiya Y, Saito S, Ogawa J. Endothelial rho and rho kinase regulate neutrophil migration via endothelial myosin light chain phosphorylation. J Leukoc Biol. (2002) 72:829–36. doi: 10.1189/jlb.72.4.829
48. van Buul JD, Allingham MJ, Samson T, Meller J, Boulter E, García-Mata R, et al. RhoG regulates endothelial apical cup assembly downstream from ICAM1 engagement and is involved in leukocyte trans-endothelial migration. J Cell Biol. (2007) 178:1279–93. doi: 10.1083/jcb.200612053
49. Heemskerk N, Schimmel L, Oort C, van Rijssel J, Yin T, Ma B, et al. F-actin-rich contractile endothelial pores prevent vascular leakage during leukocyte diapedesis through local RhoA signalling. Nat. Commun. (2016) 7:10493. doi: 10.1038/ncomms10493
50. Barzilai S, Yadav SK, Morrell S, Roncato F, Klein E, Stoler-Barak L, et al. Leukocytes breach endothelial barriers by insertion of nuclear lobes and disassembly of endothelial actin filaments. Cell Rep. (2017) 18:685–99. doi: 10.1016/j.celrep.2016.12.076
51. Hallmann R, Zhang X, Di Russo J, Li L, Song J, Hannocks MJ, et al. The regulation of immune cell trafficking by the extracellular matrix. Curr Opin Cell Biol. (2015) 36:54–61. doi: 10.1016/j.ceb.2015.06.006
52. Sorokin L. The impact of the extracellular matrix on inflammation. Nat Rev Immunol. (2010) 10:712–23. doi: 10.1038/nri2852
53. Bader BL, Smyth N, Nedbal S, Miosge N, Baranowsky A, Mokkapati S, et al. Compound genetic ablation of nidogen 1 and 2 causes basement membrane defects and perinatal lethality in mice. Mol Cell Biol. (2005) 25:6846–56. doi: 10.1128/MCB.25.15.6846-6856.2005
54. Frieser M, Nöckel H, Pausch F, Röder C, Hahn A, Deutzmann R, et al. Cloning of the mouse laminin alpha 4 cDNA. Expression in a subset of endothelium. Eur J Biochem. (1997) 246:727–35. doi: 10.1111/j.1432-1033.1997.t01-1-00727.x
55. Kenne E, Soehnlein O, Genové G, Rotzius P, Eriksson EE, Lindbom L. Immune cell recruitment to inflammatory loci is impaired in mice deficient in basement membrane protein laminin alpha4. J Leukoc Biol. (2010) 88:523–8. doi: 10.1189/jlb.0110043
56. Wu C, Ivars F, Anderson P, Hallmann R, Vestweber D, Nilsson P, et al. Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat Med. (2009) 15:519–27. doi: 10.1038/nm.1957
57. Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. (2005) 97:512–23. doi: 10.1161/01.RES.0000182903.16652.d7
58. Andreeva ER, Pugach IM, Gordon D, Orekhov AN. Continuous subendothelial network formed by pericyte-like cells in human vascular bed. Tissue Cell (1998) 30:127–35. doi: 10.1016/S0040-8166(98)80014-1
59. Wang S, Voisin MB, Larbi KY, Dangerfield J, Scheiermann C, Tran M, et al. Venular basement membranes contain specific matrix protein low expression regions that act as exit points for emigrating neutrophils. J Exp Med. (2006) 203:1519–32. doi: 10.1084/jem.20051210
60. Voisin MB, Pröbstl D, Nourshargh S. Venular basement membranes ubiquitously express matrix protein low-expression regions: characterization in multiple tissues and remodeling during inflammation. Am J Pathol. (2010) 176:482–95. doi: 10.2353/ajpath.2010.090510
61. Reichel CA, Rehberg M, Bihari P, Moser CM, Linder S, Khandoga A, et al. Gelatinases mediate neutrophil recruitment in vivo: evidence for stimulus specificity and a critical role in collagen IV remodeling. J Leukoc Biol. (2008) 83:864–74. doi: 10.1189/jlb.1007666
62. Young RE, Thompson RD, Larbi KY, La M, Roberts CE, Shapiro SD, et al. Neutrophil elastase (NE)-deficient mice demonstrate a nonredundant role for NE in neutrophil migration, generation of proinflammatory mediators, and phagocytosis in response to zymosan particles in vivo. J Immunol. (2004) 172:4493–502. doi: 10.4049/jimmunol.172.7.4493
63. Hyun YM, Sumagin R, Sarangi PP, Lomakina E, Overstreet MG, Baker CM, et al. Uropod elongation is a common final step in leukocyte extravasation through inflamed vessels. J Exp Med. (2012) 209:1349–62. doi: 10.1084/jem.20111426
64. Dangerfield JP, Wang S, Nourshargh S. Blockade of alpha6 integrin inhibits IL-1beta- but not TNF-alpha-induced neutrophil transmigration in vivo. J Leukoc Biol. (2005) 77:159–65. doi: 10.1189/jlb.0704421
65. Wang S, Dangerfield JP, Young RE, Nourshargh S. PECAM-1, alpha6 integrins and neutrophil elastase cooperate in mediating neutrophil transmigration. J Cell Sci. (2005) 118(Pt 9):2067–76. doi: 10.1242/jcs.02340
66. Uriarte SM, Powell DW, Luerman GC, Merchant ML, Cummins TD, Jog NR, et al. Comparison of proteins expressed on secretory vesicle membranes and plasma membranes of human neutrophils. J Immunol. (2008) 180:5575–81. doi: 10.4049/jimmunol.180.8.5575
67. Kurz AR, Pruenster M, Rohwedder I, Ramadass M, Schäfer K, Harrison U, et al. MST1-dependent vesicle trafficking regulates neutrophil transmigration through the vascular basement membrane. J Clin Invest. (2016) 126:4125–39. doi: 10.1172/JCI87043
68. Proebstl D, Voisin MB, Woodfin A, Whiteford J, D'Acquisto F, Jones GE, et al. Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J Exp Med. (2012) 209:1219–34. doi: 10.1084/jem.20111622
69. De Bruyn PP. The amoeboid movement of the mammalian leukocyte in tissue culture. Anat Rec. (1946) 95:177–91. doi: 10.1002/ar.1090950209
70. Daley WP, Peters SB, Larsen M. Extracellular matrix dynamics in development and regenerative medicine. J Cell Sci. (2008) 121(Pt 3):255–64. doi: 10.1242/jcs.006064
71. Wolf K, Alexander S, Schacht V, Coussens LM, von Andrian UH, van Rheenen J, et al. Collagen-based cell migration models in vitro and in vivo. Semin Cell Dev Biol. (2009) 20:931–41. doi: 10.1016/j.semcdb.2009.08.005
72. Wolf K, Te Lindert M, Krause M, Alexander S, Te Riet J, Willis AL, et al. Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol. (2013) 201:1069–84. doi: 10.1083/jcb.201210152
73. Woolf E, Grigorova I, Sagiv A, Grabovsky V, Feigelson SW, Shulman Z, et al. Lymph node chemokines promote sustained T lymphocyte motility without triggering stable integrin adhesiveness in the absence of shear forces. Nat Immunol. (2007) 8:1076–85. doi: 10.1038/ni1499
74. Renkawitz J, Sixt M. Mechanisms of force generation and force transmission during interstitial leukocyte migration. EMBO Rep. (2010) 11:744–50. doi: 10.1038/embor.2010.147
75. Ambravaneswaran V, Wong IY, Aranyosi AJ, Toner M, Irimia D. Directional decisions during neutrophil chemotaxis inside bifurcating channels. Integr Biol. (2010) 2:639–47. doi: 10.1039/c0ib00011f
76. Heuzé ML, Collin O, Terriac E, Lennon-Duménil AM, Piel M. Cell migration in confinement: a micro-channel-based assay. Methods Mol Biol. (2011) 769:415–34. doi: 10.1007/978-1-61779-207-6_28
77. Maniotis AJ, Chen CS, Ingber DE. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc Natl Acad Sci USA. (1997) 94:849–54. doi: 10.1073/pnas.94.3.849
78. Friedl P, Wolf K, Lammerding J. Nuclear mechanics during cell migration. Curr Opin Cell Biol. (2011) 23:55–64. doi: 10.1016/j.ceb.2010.10.015
79. Davidson PM, Denais C, Bakshi MC, Lammerding J. Nuclear deformability constitutes a rate-limiting step during cell migration in 3-D environments. Cell Mol Bioeng. (2014) 7:293–306. doi: 10.1007/s12195-014-0342-y
80. Denais C, Lammerding J. Nuclear mechanics in cancer. Adv Exp Med Biol. (2014) 773:435–70. doi: 10.1007/978-1-4899-8032-8_20
81. Thiam HR, Vargas P, Carpi N, Crespo CL, Raab M, Terriac E, et al. Perinuclear Arp2/3-driven actin polymerization enables nuclear deformation to facilitate cell migration through complex environments. Nat Commun. (2016) 7:10997. doi: 10.1038/ncomms10997
82. Campbell MS, Lovell MA, Gorbsky GJ. Stability of nuclear segments in human neutrophils and evidence against a role for microfilaments or microtubules in their genesis during differentiation of HL60 myelocytes. J Leukoc Biol. (1995) 58:659–66. doi: 10.1002/jlb.58.6.659
83. Aquiles Sanchez J, Karni RJ, Wangh LJ. Fluorescent in situ hybridization (FISH) analysis of the relationship between chromosome location and nuclear morphology in human neutrophils. Chromosoma (1997) 106:168–77. doi: 10.1007/s004120050236
84. Salvermoser M, Pick R, Weckbach LT, Zehrer A, Löhr P, Drechsler M, et al. Myosin 1f is specifically required for neutrophil migration in 3D environments during acute inflammation. Blood (2018) 131:1887–98. doi: 10.1182/blood-2017-10-811851
85. Funkhouser CM, Sknepnek R, Shimi T, Goldman AE, Goldman RD, Olvera de la Cruz M. Mechanical model of blebbing in nuclear lamin meshworks. Proc Natl Acad Sci USA. (2013) 110:3248–53. doi: 10.1073/pnas.1300215110
86. Lammerding J, Fong LG, Ji JY, Reue K, Stewart CL, Young SG, et al. Lamins A and C but not lamin B1 regulate nuclear mechanics. J Biol Chem. (2006) 281:25768–80. doi: 10.1074/jbc.M513511200
87. Shin JW, Spinler KR, Swift J, Chasis JA, Mohandas N, Discher DE. Lamins regulate cell trafficking and lineage maturation of adult human hematopoietic cells. Proc Natl Acad Sci USA. (2013) 110:18892–7. doi: 10.1073/pnas.1304996110
88. Olins AL, Zwerger M, Herrmann H, Zentgraf H, Simon AJ, Monestier M, et al. The human granulocyte nucleus: unusual nuclear envelope and heterochromatin composition. Eur J Cell Biol. (2008) 87:279–90. doi: 10.1016/j.ejcb.2008.02.007
89. Zwerger M, Herrmann H, Gaines P, Olins AL, Olins DE. Granulocytic nuclear differentiation of lamin B receptor-deficient mouse EPRO cells. Exp Hematol. (2008) 36:977–87. doi: 10.1016/j.exphem.2008.03.003
90. Rowat AC, Jaalouk DE, Zwerger M, Ung WL, Eydelnant IA, Olins DE, et al. Nuclear envelope composition determines the ability of neutrophil-type cells to passage through micron-scale constrictions. J Biol Chem. (2013) 288:8610–8. doi: 10.1074/jbc.M112.441535
91. González-Granado JM, Silvestre-Roig C, Rocha-Perugini V, Trigueros-Motos L, Cibrián D, Morlino G, et al. Nuclear envelope lamin-A couples actin dynamics with immunological synapse architecture and T cell activation. Sci Signal. (2014) 7:ra37. doi: 10.1126/scisignal.2004872
92. Yadav SK, Feigelson SW, Roncato F, Antman-Passig M, Shefi O, Lammerding J, et al. Frontline science: elevated nuclear lamin A is permissive for granulocyte transendothelial migration but not for motility through collagen I barriers. J Leukoc Biol. (2018) 104:239–51. doi: 10.1002/JLB.3HI1217-488R
93. Olins AL, Hoang TV, Zwerger M, Herrmann H, Zentgraf H, Noegel AA, et al. The LINC-less granulocyte nucleus. Eur J Cell Biol. (2009) 88:203–14. doi: 10.1016/j.ejcb.2008.10.001
94. Crisp M, Liu Q, Roux K, Rattner JB, Shanahan C, Burke B, et al. Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol. (2006) 172:41–53. doi: 10.1083/jcb.200509124
95. Graham DM, Andersen T, Sharek L, Uzer G, Rothenberg K, Hoffman BD, et al. Enucleated cells reveal differential roles of the nucleus in cell migration, polarity, and mechanotransduction. J Cell Biol. (2018) 217:895–914. doi: 10.1083/jcb.201706097
96. Hind LE, Vincent WJ, Huttenlocher A. Leading from the back: the role of the uropod in neutrophil polarization and migration. Dev Cell (2016) 38:161–9. doi: 10.1016/j.devcel.2016.06.031
97. Kalhammer G, Bahler M. Unconventional myosins. Essays Biochem. (2000) 35:33–42. doi: 10.1042/bse0350033
98. Gaines P, Tien CW, Olins AL, Olins DE, Shultz LD, Carney L, et al. Mouse neutrophils lacking lamin B-receptor expression exhibit aberrant development and lack critical functional responses. Exp Hematol. (2008) 36:965–76. doi: 10.1016/j.exphem.2008.04.006
Keywords: inflammation, neutrophil, migration, nuclear deformation, myosin1f
Citation: Salvermoser M, Begandt D, Alon R and Walzog B (2018) Nuclear Deformation During Neutrophil Migration at Sites of Inflammation. Front. Immunol. 9:2680. doi: 10.3389/fimmu.2018.02680
Received: 06 September 2018; Accepted: 30 October 2018;
Published: 16 November 2018.
Edited by:
Andres Hidalgo, Centro Nacional de Investigaciones Cardiovasculares (CNIC), SpainReviewed by:
Toshiyuki Murai, Osaka University, JapanCopyright © 2018 Salvermoser, Begandt, Alon and Walzog. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Barbara Walzog, d2Fsem9nQGxyei51bmktbXVlbmNoZW4uZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.