 Liya Li
Liya Li Honglin Zhu
Honglin Zhu Xiaoxia Zuo
Xiaoxia Zuo- 1Department of Rheumatology and immunology, Xiangya Hospital, Central South University, Changsha, China
- 2The Institute of Rheumatology and Immunology, Central South University, Changsha, China
Interleukin-33 (IL-33), a member of the IL-1 superfamily, functions as a traditional cytokine and nuclear factor. It is proposed to have an “alarmin” role. IL-33 mediates biological effects by interacting with the ST2 receptor and IL-1 receptor accessory protein, particularly in innate immune cells and T helper 2 cells. Recent articles have described IL-33 as an emerging pro-fibrotic cytokine in the immune system as well as a novel potential target for systemic sclerosis. Here, we review the available information and focus on the pleiotropic expression and pathogenesis of IL-33 in systemic sclerosis, as well as the feasibility of using IL-33 in clinical applications.
Introduction
Systemic sclerosis (scleroderma, SSc) is a heterogeneous autoimmune disease of unknown etiology, clinically characterized with obliterative microvasculopathy, inflammation, and extensive fibrosis of the skin and multiple organ systems and serologically characterized by the presence of circulating specific autoantibodies. SSc has the highest cause-specific mortality among connective tissue diseases (1, 2), and pulmonary artery hypertension and interstitial lung disease (ILD) are the leading causes of death (3, 4). Therapeutic interventions for SSc mainly involve the comprehensive administration of glucocorticoids and immunosuppressants and targeted treatment. To date, no effective medical intervention has been developed to control and reverse the progression of this fibrotic disease (5). Thus, effective and safe targeted therapies for SSc-related fibrosis are urgently needed. In the pathogenesis of SSc, endothelial damage may be a primary event. SSc also exhibits complex interactions during the transition from fibroblasts to myofibroblasts and non-infective inflammation or autoimmunity.
Interleukin (IL)-33 belongs to the IL-1 superfamily and is widely expressed throughout the human body. During cell damage or tissue injury, IL-33 is released into the extracellular space, wherein it produces endogenous danger signals to alert adjacent cells. This function deems IL-33 as an alarmin. IL-33 also functions as a nuclear factor regulating gene transcription in cytokine-expressing or cytokine-responsive cells (6). IL-33 is known to play crucial roles in inflammation. However, recent studies indicated that IL-33 participates in the development and progression of fibrotic diseases and SSc. Here, we review the profibrotic roles of IL-33 and its related mechanisms and discuss its potential application in the treatment of SSc.
Biological Characteristics of IL-33
IL-33, also known as IL-1F11, is a member of the IL-1 superfamily (7) and exhibits dual functionality (8). This cytokine was first identified as a nuclear factor in high endothelial venules in 2003 (9) but was renamed as IL-33 when a study in 2005 demonstrated its role as a specific extracellular ligand for the orphan IL-1 receptor family member ST2 (also known as IL-1RL1, DER4, T1, and FIT-1). ST2 is a member of the Toll-like receptor (TLR)/IL-1 receptor superfamily (10), which has two main isoforms, namely, a short soluble form (sST2) and longer transmembrane form (ST2L), with four isoforms in total, including ST2V and ST2LV (11). The mRNA encoding sST2 is a secretory sequence that is generated by alternative splicing and lacks the sequence encoding the transmembrane domain of ST2L (12).
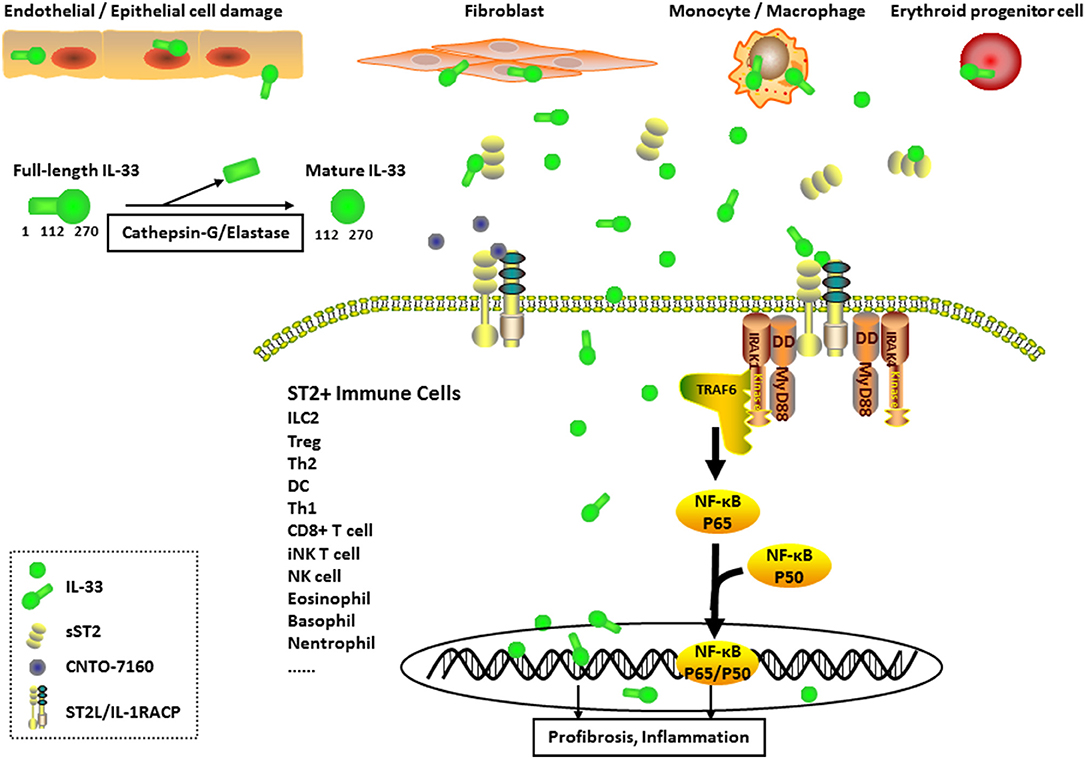
Graphical Abstract. Expression and Pathogenesis of interleukin (IL)-33 in Systemic sclerosis (SSc). IL-33-producing cells (mainly endothelial cells, epithelial cells, fibroblasts, monocytes, macrophages, and erythroid progenitor cells) secrete IL-33 into the extracellular environment. Full-length IL-33 is transformed into mature IL-33 through cleavage by inflammatory proteases (such as cathepsin-G and elastase). The formation of the IL-33/ST2 longer transmembrane form (ST2L)/IL-1 receptor accessory protein complex in ST2+ immune cell membranes results in the activation of nuclear factor-κB transcription factors through the MyD88, IL-1R-associated kinase (IRAK), and tumor necrosis factor receptor associated factor 6 (TRAF6) signaling pathways, leading to the induction of inflammation and profibrosis in pathological cells. IL-33 also functions as a nuclear factor to regulate gene transcription in cytokine-expressing or cytokine-responsive cells. Moreover, sST2 acts as a decoy receptor for IL-33 (full-length IL-33 or mature IL-33), and CNTO-7160 (the first monoclonal anti-ST2 antibody) was designed as a new IL-33 inhibitor. Both of these molecules block the downstream signaling of IL-33.
The IL-33 gene is located on human chromosome 9 (or chromosome 19 in mice) and is transcribed from seven coding exons. The protein is synthesized as a 31-kDa pro-IL-331−−270 (full-length IL-33). Following synthesis, IL-33 is transported into the nucleus as a nuclear factor. Similar to the IL-1 family members IL-1β and IL-18, IL-33 lacks the classic signal sequence necessary for the transport by the endoplasmic reticulum/Golgi secretion pathway (13). Upon natural secretion from pathological cells undergoing necrosis or necroptosis, the full-length IL-33 is cleaved by caspase-3 and caspase-7 to activate apoptotic pathways in the cytoplasm, followed by its release into the extracellular environment (14). Once released into the extracellular matrix, full-length IL-33 is further processed by serine proteases (such as cathepsin-G and elastase) into the 18-kDa IL-33112−−270 (mature IL-33) with increased activity (15, 16), forming a soluble recombinant cytokine in circulation. However, both full-length and mature IL-33 bind to ST2L in ST2+ immune cell membranes and interact with IL-1 receptor accessory protein (IL-1RAcP), eventually leading to the formation of an IL-33/ST2L/IL-1RAcP complex. This complex induces signaling pathways through MyD88, IL-1R-associated kinase (IRAK), and tumor necrosis factor receptor associated factor 6 (TRAF6) and activates the canonical nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) pathways (17).
IL-33 is principally produced by stromal cells, including epithelial cells, endothelial cells, fibroblast-like cells, and myofibroblasts of lymphoid as well as non-lymphoid organs, under both steady state and inflammation conditions (18–20). In erythroid progenitor cells, IL-33 is produced during the maturation of red blood cells and released upon haemolysis (21). Innate immune cells expressing ST2 mainly include dendritic cells (DCs), natural killer (NK) cells, eosinophils, basophils, macrophages, and neutrophils (12). Full-length IL-33 predominantly remains inside the cell and regulates the expression of genes, which induce pulmonary inflammation and fibrosis. In contrast, mature IL-33 promotes asthma as well as allergic and anti-parasitic responses through the ST2 receptor and Th2 mechanisms (22).
Mechanisms such as inactivation by oxidation of cysteine residues, nuclear localization or sequestration, and proteolytic processing, and receptor antagonists as well as sST2 have evolved to regulate the expression and activities of IL-33 (12, 14, 15, 23, 24). sST2 is constitutively expressed in the human serum, wherein it acts as a decoy receptor for IL-33 and is not involved in signaling (25, 26). sST2, induced during tissue damage, may restrict the deleterious effects of increased IL-33 level. A novel mechanism for the rapid inactivation of IL-33 protein released from the cell in vivo was reported, wherein an oxidation-driven conformational change involving the formation of two disulphide bonds was observed, resulting in the elimination of ST2-dependent activity and reduction of inflammation, consistent with the mechanism of many other IL-1 family members (23).
Expression of IL-33 and ST2 in SSC
According to recent studies, increased IL-33 and sST2 levels have been observed in patients with infections, cardiovascular disorders, allergic diseases, and rheumatic diseases such as systemic lupus erythematosus, rheumatoid arthritis (RA), Wegener's granulomatosis, and Behcet's disease (27–31). The serum levels of sST2 and synovial fluid of IL-33 were higher in patients with RA than in healthy controls and patients with osteoarthritis (27). Furthermore, serum sST2 levels were higher in patients with active, newly diagnosed, anti-neutrophil cytoplasmic antibody-associated vasculitis than in patients in remission, indicative of the marker role for sST2 (32). Furthermore, ST2 and IL-33 were highly expressed around ectopic germinal centers in salivary glands from patients with IgG4-related disease, whereas IL-33 was expressed only in epithelial cells in patients with Sjögren's syndrome and controls (33). Interestingly, the exposure of mice in vivo or human skin samples ex vivo to inflammatory doses of ultraviolet B irradiation induced IL-33 expression within the epidermal and dermal skin layers (34). Proteomic analysis used to determine the extracellular and intracellular roles of IL-33 in primary human endothelial cells revealed the induction of inflammation-related protein expression of the exogenous extracellular IL-33, whereas the knockdown of the endogenous nuclear IL-33 expression had no reproducible effect on the endothelial cell proteome (35).
The results described above support that the expression level and biological role of IL-33 are similar to those of ST2. In general, IL-33 expression is upregulated in inflamed tissues following pro-inflammatory stimulation, and the role of IL-33 in cells may vary under different pathophysiological conditions. In SSc, with an exception during tissue inflammation, the authors proposed that IL-33 commonly responds to tissue injury and typically affects rapid tissue repair and regeneration (36–38).
In patients with SSc, serum levels of IL-33 and sST2 were elevated (39) and positively correlated with the extent of skin sclerosis (higher in diffused cutaneous SSc than in limited cutaneous SSc), severity of pulmonary interstitial fibrosis, and vascular involvement in SSc development (40–44). In the lesion skin tissues, IL-33 expression is altered depending on the disease stage. IL-33 is downregulated in most endothelial cells in early SSc but not in late SSc (45). IL-33 produced by activated dermal fibroblasts/myofibroblasts has been implicated in the fibrotic pathology associated with SSc, which is profoundly increased by hypertrophic and mechanical stress (46, 47).
The expression of IL-33 mRNA is reported to increase in the primary pulmonary fibroblasts from patients with SSc-ILD as well as in those from patients with idiopathic pulmonary fibrosis (IPF). The elevated levels of IL-33 in bronchoalveolar lavage fluids may be useful in differentiating IPF from other chronic ILDs (48). In patients with IPF and SSc-ILD, the expression of full-length IL-33 was elevated in the affected lungs, consistent with the observation reported in a bleomycin-induced mouse model. Under the conditions of ST2 gene deficiency, the full-length IL-33 could stimulate the expression of several non-Th2 cytokines and heat shock protein 70. On the other hand, the matured form of IL-33 was unaffected and instead activated Th2 responses (49). In contrast, the expression of the matured form of IL-33 was enhanced but that of the full-length counterpart reduced in the macrophages of bleomycin-induced mouse lung tissues (50). These findings suggest that the full-length IL-33 may serve as a synergistic pro-inflammatory and pro-fibrotic regulator in the lungs.
Pathogenesis of IL-33/ST2 in SSC
Fibrosis, a prominent pathological characteristic of SSc (38), is characterized with a deregulated and uncontrolled repair process. Many molecular and signaling pathways involved in the fibrosis of SSc (51, 52), including transforming growth factor (TGF)-β, TLR4, and interferons (IFNs), are well-studied. TGF-β is responsible for both physiological and pathological matrix remodeling (53) as well as fibroblast-myofibroblast transformation (54). TLR4 induces pro-fibrotic responses by activating NF-κB signaling through MyD88, IRAK, and TRAF6. The TLR/NF-κB signaling pathways enhance the TGF-β-dependent fibrotic process (55, 56). IFNs generally act as negative regulators of collagen synthesis and TGF-β-mediated fibrotic responses, while the mechanism of type I IFN signaling in SSc-promoted fibrosis remains unclear (37, 57). The role of IL-33 in SSc was recently evaluated. In pediatric patients with limited cutaneous SSc, high levels of IL-33 and IFN-γ positively correlated with anti-histone and anti-ssDNA antibodies, indicating that the co-expression of IL-33 and IFN-γ may contribute to the pathogenesis of SSc (58). Subcutaneous injection of IL-33 in mice resulted in the development of cutaneous fibrosis, similar to that observed in patients with SSc, including dermal mast cells and skin-infiltrating neutrophils through the suppression of Th1-mediated contact hypersensitivity responses (59). This observation highlights the important roles of IL-33 in SSc. However, the exact mechanisms require further investigation.
Known as a master regulator of pathological fibrosis, TGF-β may be produced by IL-33-induced cells. During the amplification of the alternatively activated M2 macrophage polarization, the IL-33/ST2 pathway was shown to play a significant role (60). IL-33 polarized M2 macrophages to produce IL-13 and TGF-β1 and induced the expansion of type 2 innate lymphoid cells (ILC2s) for the production of IL-13 in vitro and in vivo. ST2 may protect ILC2s from IL-33 stimulation by reducing the production of IL-5 and IL-13 (61). IL-13 is a well-known profibrotic cytokine downstream of IL-33 in the immune system (51).
IFN-γ may play regulatory roles in physiological processes involving IL-33. In type 2 immune responses, IL-33 and ILC2s are central mediators that promote tissue and metabolic homeostasis, whereas IFN-γ suppresses this pathway and promotes inflammatory responses (62). In vivo, the co-expression of IL-33 and IFN-γ in pulmonary fibroblast culture and lungs resulted in the attenuation of IL-33 protein levels (63). IFN-regulated genes may regulate IL-33 gene expression. In both human monocytes and macrophages from C57BL/6 mice, transcriptional activation of the IL-33 gene stimulated by the acute-phase protein serum amyloid A, a TLR2 ligand, may be regulated by IFN regulatory factor 7 (IRF-7) recruited to the IL-33 promoter. Silencing of IRF-7 expression may result in the abrogation of the expression of IL-33 induced by serum amyloid A (64).
In fibrosis, DCs elevated the expression of IL-33 via TLR/NF-κB signaling pathways in response to allergic inflammation, resulting in an increase in the expression levels of MyD88, NF-κB1, NF-κB2, and RelA accompanied with NF-κB p65 nuclear translocation, possibly through a potential autocrine regulation. These elevations may be blocked with a TLR5 antibody or NF-κB inhibitor quinazoline and thought to be decreased in DCs from MyD88-knockout mice (65). The deficiency in the NF-κB negative feedback regulator A20 in hyperactive mast cells may result in amplified pro-inflammatory responses downstream of IgE/FcεRI, TLRs, IL-1R, and IL-33R (ST2), thereby exacerbating inflammatory disorders (66). In addition, Th2-stimulated (allergen-specific IgG immune complexes and house dust mites) signaling occurs through FcRγ-associated receptors on DCs to upregulate IL-33 production and induce Th2-mediated allergic airway inflammation (67).
In conclusion, IL-33 functions as a pro-fibrogenic cytokine in the development of SSc. IL-33 may enhance the TGF-β-dependent fibrotic process by increasing the production of TGF-β and activate TLR/NF-κB-dependent fibrosis signaling pathways, which are regulated by IFN-γ (Table 1).
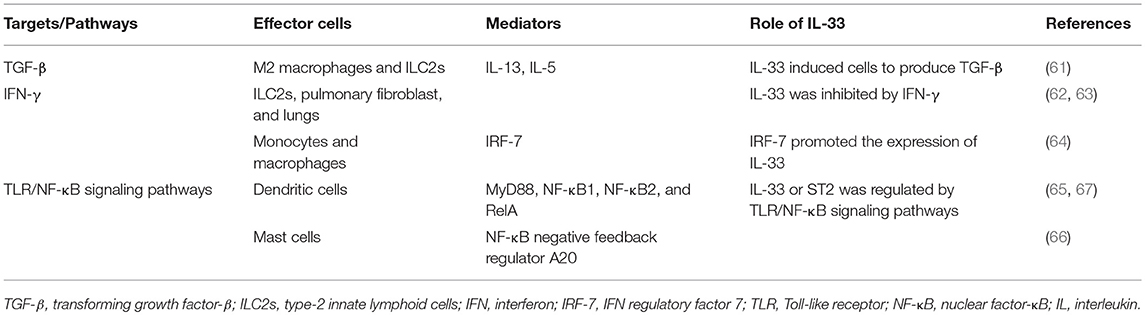
Table 1. Targets/pathways involved in IL-33-dependent fibrosis process.
To determine whether IL-33 is a useful therapeutic target, Locksley et al. described the complexity of using IL-33 and therapeutic strategies for altering IL-33 activities in vivo (68). The framework of IL-33 biology was described as a stepwise process. First, the focal cellular necrosis or other signals induce the release of IL-33 from the nucleus to maintain homeostasis; IL-33 acts on tissue-resident ST2-expressing effector cells such as ILC2s, regulatory T cells (Tregs), and mast cells to create a tissue environment that limits inflammation and promotes a reparative state characterized by tolerance. Second, amplification occurs upon exposure to chronic stimuli such as allergens and repetitive tissue damage, wherein excess extracellular IL-33 leads to multiple self-stimulating cycles of release to promote chronic allergic pathology, fibrosis, and excess stores of IL-33 in the circulation and tissues. The third step is conversion, wherein the activated inflammatory cells and cytokines responsive to the IL-33/ST2 axis play various roles such as killing pathogens, mounting anticancer immune responses, increasing tissue damage, and repressing the type 2-associated immune regulation responses. In patients with SSc, repetitive tissue damage by other pro-fibrotic mediators in fibroblasts and endothelial cells likely suppresses the IL-33 pool increases and regulatory mechanisms. Next, inflammation is amplified, fibrosis occurs, and tissue IL-33 levels increase, ultimately contributing to tissue fibrosis and sclerosis.
Therefore, IL-33 from different sources can be up- or downregulated to exert pleiotropic roles in SSc. Zhao et al. proposed that these apparently contradictory results indicate the presence of an extremely complex process of IL-33 processing and secretion (69). The functional properties of recombinant IL-33 used in previous studies are becoming well-characterized, whereas the cellular sources of IL-33 in natural and stimulated expression remain largely unknown. Additional studies are warranted to explain the differences between in vitro and in vivo results.
Clinical Applications of IL-33 in SSC
Various aspects of the clinical applications of IL-33 have been examined. However, few studies have evaluated these effects in patients with SSc. Thus, information may be obtained from studies of other diseases that may be applicable to SSc.
IL-33-responsive ILC2s may promote the restoration of injured skin, lung, and gut cells (70). During the regeneration of injured muscles, fibro-adipogenic progenitor cells are the only known source of IL-33 in muscles. The low level of IL-33 expression in older, injured muscle reduces the recruitment and proliferation effects of non-increased muscle-resident Tregs; after the administration of IL-33, the Treg population increases and regeneration is enhanced (71, 72). Furthermore, the upstream and downstream regulation of the IL-33 gene may promote the remodeling of tissues such as nerves and tendons (73, 74).
In general, studies of IL-33 in patients with SSc have indicated that IL-33 is a novel and important pro-fibrogenic cytokine and a potential biomarker for monitoring disease activity (40–45). Genetic polymorphisms in the IL-33 gene may be useful for the prediction of the risk of various diseases. The IL-33 rs7044343 CC genotype was suggested to be associated with an increased risk of developing SSc and a decreased risk of developing RA; the T allele may be a susceptibility marker for premature coronary artery disease and central obesity and possibly involved in the regulation of IL-33 production (53, 75–77). The first monoclonal anti-ST2 antibody, CNTO-7160, was recently designed as a new IL-33 inhibitor; this antibody is being evaluated in phase I clinical trials for the treatment of severe asthma and atopic dermatitis, but no data have been published to date (78).
Prospects
The alarmin IL-33 has dual functions of a cytokine and nuclear factor. However, differences in the levels of IL-33 and systemic sST2 indicate intra-individual and inter-individual biological variation, reference changes, and sex-specific differences (79). Moreover, the evaluation of the circulating concentrations of sST2, full-length IL-33, mature IL-33, and complexes of sST2 and IL-33 in the same patients is interesting; measurement of these four analytes and their ratios may increase the understanding of IL-33-related pathophysiology in various diseases (80).
Recent investigations suggested that IL-33 is a novel pro-fibrogenic cytokine in the development of SSc, mainly because it affects the TLR/NF-κB signaling pathways, and TGF-β1 expression is also regulated by IFN-γ. These effects are crucial for the early diagnosis of pulmonary fibrosis. Whether IL-33 is involved in fibroblast activation alone or in combination with other factors is unclear; however, this molecule is likely a potential biomarker and novel therapy target for managing fibrosis in patients with SSc. Furthermore, the inhibitor of IL-33 (CNTO-7160), currently being examined in clinical trials, may possibly be developed as a new therapy for fibrosis in patients with SSc (78).
Author Contributions
LL devised and wrote the manuscript. HZ and XZ revised the manuscript.
Funding
This work was supported by The National Key Research and Development Program of China (2016YFC0903900), grants from National Natural Science Foundation of China (81671622, 81701621) and Independent Innovation Projects of Central South University (2018zzst290).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ST2L, ST2 longer transmembrane form; sST2, soluble ST2; ILC2, type 2 innate lymphoid cell; Treg, regulatory T cell; DC, dendritic cell; Th1, T helper 1 cell; CD8+ T cell, CD8-positive T cell; iNK T cell, invariant natural killer T cell; NK cell, natural killer cell; SSc, systemic sclerosis; IRAK, IL-1R-associated kinase; TRAF6, tumor necrosis factor receptor associated factor 6; IL, interleukin; ILD, interstitial lung disease; IL-1RAcP, IL-1 receptor accessory protein; NF-κB, nuclear factor-κB; MAPK, mitogen-activated protein kinase; IPF, idiopathic pulmonary fibrosis; TGF, transforming growth factor; TLR, Toll-like receptor; IFN, interferon; IRF-7, IFN regulatory factor 7.
References
1. Li L, Zuo X, Luo H, Li Y, You Y, Duan L, et al. Mortality trend of inpatients with connective tissue diseases: 2005-2014. Zhong Nan Da Xue Xue Bao Yi Xue Ban (2017) 42:927–33. doi: 10.11817/j.issn.1672-7347.2017.08.009
2. Elhai M, Meune C, Avouac J, Kahan A, Allanore Y. Trends in mortality in patients with systemic sclerosis over 40 years: a systematic review and meta-analysis of cohort studies. Rheumatology (2012) 51:1017–26. doi: 10.1093/rheumatology/ker269
3. Elhai M, Avouac J, Kahan A, Allanore Y. Systemic sclerosis: recent insights. Joint Bone Spine (2015) 82:148–53. doi: 10.1016/j.jbspin.2014.10.010
4. Tyndall A J, Bannert B, Vonk M, Airo P, Cozzi F, Carreira PE, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. (2010) 69:1809–15. doi: 10.1136/ard.2009.114264
5. Allanore Y, Distler O. Systemic sclerosis in 2014: advances in cohort enrichment shape future of trial design. Nat Rev Rheumatol. (2015) 11:72–4. doi: 10.1038/nrrheum.2014.222
6. Bertheloot D, Latz E. HMGB1, IL-1alpha, IL-33 and S100 proteins: dual-function alarmins. Cell Mol Immunol. (2017) 14:43–64. doi: 10.1038/cmi.2016.34
7. Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity (2013) 39:1003–18. doi: 10.1016/j.immuni.2013.11.010
8. Luheshi NM, Rothwell NJ, Brough D. Dual functionality of interleukin-1 family cytokines: implications for anti-interleukin-1 therapy. Br J Pharmacol. (2009) 157:1318–29. doi: 10.1111/j.1476-5381.2009.00331.x
9. Baekkevold ES, Roussigne M, Yamanaka T, Johansen FE, Jahnsen FL, Amalric F, et al. Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Am J Pathol. (2003) 163:69–79. doi: 10.1016/S0002-9440(10)63631-0
10. O'Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol Rev. (2008) 226:10–8. doi: 10.1111/j.1600-065X.2008.00701.x
11. Yanagisawa K, Takagi T, Tsukamoto T, Tetsuka T, Tominaga S. Presence of a novel primary response gene ST2L, encoding a product highly similar to the interleukin 1 receptor type 1. FEBS Lett. (1993) 318:83–7.
12. Cayrol C, Girard JP. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol Rev. (2018) 281:154–68. doi: 10.1111/imr.12619
13. Arend WP, Palmer G, Gabay C. IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev. (2008) 223:20–38. doi: 10.1111/j.1600-065X.2008.00624.x
14. Luthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity (2009) 31:84–98. doi: 10.1016/j.immuni.2009.05.007
15. Lefrancais E, Roga S, Gautier VA, Gonzalez-de-Peredo Monsarrat B, Girard JP, et al. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci USA. (2012) 109:1673–8. doi: 10.1073/pnas.1115884109
16. Lefrancais E, Cayrol C. Mechanisms of IL-33 processing and secretion: differences and similarities between IL-1 family members. Eur Cytokine Netw. (2012) 23:120–7. doi: 10.1684/ecn.2012.0320
17. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity (2005) 23:479–90. doi: 10.1016/j.immuni.2005.09.015
18. Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS ONE (2008) 3:e3331. doi: 10.1371/journal.pone.0003331
19. Kuchler AM, Pollheimer J, Balogh J, Sponheim J, Manley L, Sorensen DR, et al. Nuclear interleukin-33 is generally expressed in resting endothelium but rapidly lost upon angiogenic or proinflammatory activation. Am J Pathol. (2008) 173:1229–42. doi: 10.2353/ajpath.2008.080014
20. Pichery M, Mirey E, Mercier P, Lefrancais E, Dujardin A, Ortega N, et al. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel Il-33-LacZ gene trap reporter strain. J Immunol. (2012) 188:3488–95. doi: 10.4049/jimmunol.1101977
21. Wei J, Zhao J, Schrott V, Zhang Y, Gladwin M, Bullock G, et al. Red blood cells store and release interleukin-33. J Investig Med. (2015) 63:806–10. doi: 10.1097/JIM.0000000000000213
22. Luzina IG, Kopach P, Lockatell V, Kang PH, Nagarsekar A, Burke AP, et al. Interleukin-33 potentiates bleomycin-induced lung injury. Am J Respir Cell Mol Biol. (2013) 49:999–1008. doi: 10.1165/rcmb.2013-0093OC
23. Cohen ES, Scott IC, Majithiya JB, Rapley L, Kemp BP, England E, et al. Sleeman and T. mustelin: oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat Commun. (2015) 6:8327. doi: 10.1038/ncomms9327
24. Cayrol C, Girard JP. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci USA. (2009) 106:9021–6. doi: 10.1073/pnas.0812690106
25. Ohto-Ozaki H, Kuroiwa K, Mato N, Matsuyama Y, Hayakawa M, Tamemoto H, et al. Characterization of ST2 transgenic mice with resistance to IL-33. Eur J Immunol. (2010) 40:2632–42. doi: 10.1002/eji.200940291
26. Oshikawa K, Kuroiwa K, Tago K, Iwahana H, Yanagisawa K, Ohno S, et al. Elevated soluble ST2 protein levels in sera of patients with asthma with an acute exacerbation. Am J Respir Crit Care Med. (2001) 164:277–81. doi: 10.1164/ajrccm.164.2.2008120
27. Liew FY, Pitman NI, McInnes IB. Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat Rev Immunol. (2010) 10:103–10. doi: 10.1038/nri2692
28. Kageyama Y, Torikai E, Tsujimura K, Kobayashi M. Involvement of IL-33 in the pathogenesis of rheumatoid arthritis: the effect of etanercept on the serum levels of IL-33. Mod Rheumatol. (2012) 22:89–93. doi: 10.1007/s10165-011-0480-1
29. Hong YS, Moon SJ, Joo YB, Jeon CH, Cho ML, Ju JH, et al. Measurement of interleukin-33 (IL-33) and IL-33 receptors (sST2 and ST2L) in patients with rheumatoid arthritis. J Korean Med Sci. (2011) 26:1132–9. doi: 10.3346/jkms.2011.26.9.1132
30. Kuroiwa K, Arai T, Okazaki H, Minota S, Tominaga S. Identification of human ST2 protein in the sera of patients with autoimmune diseases. Biochem Biophys Res Commun. (2001) 284:1104–8. doi: 10.1006/bbrc.2001.5090
31. Wang S, Ding L, Liu SS, Wang C, Leng RX, Chen GM, et al. IL-33: a potential therapeutic target in autoimmune diseases. J Investig Med. (2012) 60:1151–6. doi: 10.2310/JIM.0b013e31826d8fcb
32. Hladinova Z, Hruskova Z, Svobodova B, Malickova K, Lanska V, Konopasek P, et al. Increased levels of soluble ST2 in patients with active newly diagnosed ANCA-associated vasculitis. Mediators Inflamm. (2015) 2015:603750. doi: 10.1155/2015/603750
33. Furukawa S, Moriyama M, Miyake K, Nakashima H, Tanaka A, Maehara T, et al. Interleukin-33 produced by M2 macrophages and other immune cells contributes to Th2 immune reaction of IgG4-related disease. Sci Rep. (2017) 7:42413. doi: 10.1038/srep42413
34. Byrne SN, Beaugie C, O'Sullivan C, Leighton S, Halliday GM. The immune-modulating cytokine and endogenous Alarmin interleukin-33 is upregulated in skin exposed to inflammatory UVB radiation. Am J Pathol. (2011) 179:211–22. doi: 10.1016/j.ajpath.2011.03.010
35. Gautier V, Cayrol C, Farache D, Roga S, Monsarrat B, Burlet-Schiltz O, et al. Extracellular IL-33 cytokine, but not endogenous nuclear IL-33, regulates protein expression in endothelial cells. Sci Rep. (2016) 6:34255. doi: 10.1038/srep34255
36. Pei C, Barbour M, Fairlie-Clarke KJ, Allan D, Mu R, Jiang HR. Emerging role of interleukin-33 in autoimmune diseases. Immunology (2014) 141:9–17. doi: 10.1111/imm.12174
37. Bhattacharyya S, Wei J, Varga J. Understanding fibrosis in systemic sclerosis: shifting paradigms, emerging opportunities. Nat Rev Rheumatol. (2011) 8:42–54. doi: 10.1038/nrrheum.2011.149
38. Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest. (2007) 117:557–67. doi: 10.1172/JCI31139
39. Zhang YJ, Zhang Q, Yang GJ, Tao JH, Wu GC, Huang XL, et al. Elevated serum levels of interleukin-1beta and interleukin-33 in patients with systemic sclerosis in Chinese population. Z Rheumatol. (2016) 77:151–9. doi: 10.1007/s00393-016-0202-3
40. Terras S, Opitz E, Moritz R K, Hoxtermann S, Gambichler T, Kreuter A. Increased serum IL-33 levels may indicate vascular involvement in systemic sclerosis. Ann Rheum Dis. (2013) 72:144–5. doi: 10.1136/annrheumdis-2012-201553
41. Yanaba K, Yoshizaki A, Asano Y, Kadono T, Sato S. Serum IL-33 levels are raised in patients with systemic sclerosis: association with extent of skin sclerosis and severity of pulmonary fibrosis. Clin Rheumatol. (2011) 30:825–30. doi: 10.1007/s10067-011-1686-5
42. Manetti M, Guiducci S, Ceccarelli C, Romano E, Bellando-Randone S, Conforti ML, et al. Increased circulating levels of interleukin 33 in systemic sclerosis correlate with early disease stage and microvascular involvement. Ann Rheum Dis. (2011) 70:1876–8. doi: 10.1136/ard.2010.148247
43. Wagner A, Kohm M, Nordin A, Svenungsson E, Pfeilschifter JM, Radeke HH. Increased serum levels of the IL-33 neutralizing sST2 in limited cutaneous systemic sclerosis. Scand J Immunol. (2015) 82:269–74. doi: 10.1111/sji.12317
44. Mostmans Y, Cutolo M, Giddelo C, Decuman S, Melsens K, Declercq H, et al. The role of endothelial cells in the vasculopathy of systemic sclerosis: a systematic review. Autoimmun Rev. (2017) 16:774–86. doi: 10.1016/j.autrev.2017.05.024
45. Vettori S, Cuomo G, Iudici M, D'Abrosca V, Giacco V, Barra G, et al. Early systemic sclerosis: serum profiling of factors involved in endothelial, T-cell, and fibroblast interplay is marked by elevated interleukin-33 levels. J Clin Immunol. (2014) 34:663–8. doi: 10.1007/s10875-014-0037-0
46. Manetti M, Ibba-Manneschi L, Liakouli V, Guiducci S, Milia AF, Benelli G, et al. The IL1-like cytokine IL33 and its receptor ST2 are abnormally expressed in the affected skin and visceral organs of patients with systemic sclerosis. Ann Rheum Dis. (2010) 69:598–605. doi: 10.1136/ard.2009.119321
47. Kakkar R, Hei H, Dobner S, Lee RT. Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J Biol Chem. (2012) 287:6941–8. doi: 10.1074/jbc.M111.298703
48. Lee JU, Chang HS, Lee HJ, Jung CA, Bae DJ, Song HJ, et al. Upregulation of interleukin-33 and thymic stromal lymphopoietin levels in the lungs of idiopathic pulmonary fibrosis. BMC Pulm Med. (2017) 17:39. doi: 10.1186/s12890-017-0380-z
49. Luzina IG, Pickering EM, Kopach P, Kang PH, Lockatell V, Todd NW, et al. Full-length IL-33 promotes inflammation but not Th2 response in vivo in an ST2-independent fashion. J Immunol. (2012) 189:403–10. doi: 10.4049/jimmunol.1200259
50. Li D, Guabiraba R, Besnard AG, Komai-Koma M, Jabir MS, Zhang L, et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol. (2014) 134:1422–32. e11doi: 10.1016/j.jaci.2014.05.011
51. Greenblatt MB, Aliprantis AO. The immune pathogenesis of scleroderma: context is everything. Curr Rheumatol Rep. (2013) 15:297. doi: 10.1007/s11926-012-0297-8
52. Zuo X, Zhang L, Luo H, Li Y, Zhu H. Systematic approach to understanding the pathogenesis of systemic sclerosis. Clin Genet. (2017) 92:365–71. doi: 10.1111/cge.12946
53. Koca SS, Pehlivan Y, Kara M, Alibaz-Oner F, Oztuzcu S, Yilmaz N, et al. The IL-33 gene is related to increased susceptibility to systemic sclerosis. Rheumatol Int. (2016) 36:579–84. doi: 10.1007/s00296-015-3417-8
54. Varga J, Pasche B. Transforming growth factor beta as a therapeutic target in systemic sclerosis. Nat Rev Rheumatol. (2009) 5:200–6. doi: 10.1038/nrrheum.2009.26
55. Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. (2007) 13:1324–32. doi: 10.1038/nm1663
56. Csak T, Velayudham A, Hritz I, Petrasek J, Levin I, Lippai D, et al. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am J Physiol Gastrointest Liver Physiol. (2011)300:G433–41. doi: 10.1152/ajpgi.00163.2009
57. Kim D, Peck A, Santer D, Patole P, Schwartz SM, Molitor JA, et al. Induction of interferon-alpha by scleroderma sera containing autoantibodies to topoisomerase I: association of higher interferon-alpha activity with lung fibrosis. Arthritis Rheum. (2008) 58:2163–73. doi: 10.1002/art.23486
58. Torok KS, Kurzinski K, Kelsey C, Yabes J, Magee K, Vallejo AN, et al. Peripheral blood cytokine and chemokine profiles in juvenile localized scleroderma: T-helper cell-associated cytokine profiles. Semin Arthritis Rheum. (2015) 45:284–93. doi: 10.1016/j.semarthrit.2015.06.006
59. Rankin AL, Mumm JB, Murphy E, Turner S, Yu N, McClanahan TK, et al. IL-33 induces IL-13-dependent cutaneous fibrosis. J Immunol. (2010) 184:1526–35. doi: 10.4049/jimmunol.0903306
60. Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol. (2009) 183:6469–77. doi: 10.4049/jimmunol.0901575
61. Hayakawa H, Hayakawa M, Tominaga SI. Soluble ST2 suppresses the effect of interleukin-33 on lung type 2 innate lymphoid cells. Biochem Biophys Rep. (2016) 5:401–07. doi: 10.1016/j.bbrep.2016.02.002
62. Molofsky AB, Van Gool F, Liang HE, Van Dyken SJ, Nussbaum JC, Lee J, et al. Interleukin-33 and interferon-gamma counter-regulate group 2 innate lymphoid cell activation during immune perturbation. Immunity (2015) 43:161–74. doi: 10.1016/j.immuni.2015.05.019
63. Kopach P, Lockatell V, Pickering EM, Haskell RE, Anderson RD, Hasday JD, et al. IFN-gamma directly controls IL-33 protein level through a STAT1- and LMP2-dependent mechanism. J Biol Chem. (2014) 289:11829–43. doi: 10.1074/jbc.M113.534396
64. Sun L, Zhu Z, Cheng N, Yan Q, Ye RD. Serum amyloid A induces interleukin-33 expression through an IRF7-dependent pathway. Eur J Immunol. (2014) 44:2153–64. doi: 10.1002/eji.201344310
65. Su Z, Lin J, Lu F, Zhang X, Zhang L, Gandhi NB, et al. Potential autocrine regulation of interleukin-33/ST2 signaling of dendritic cells in allergic inflammation. Mucosal Immunol. (2013) 6:921–30. doi: 10.1038/mi.2012.130
66. Heger K, Fierens K, Vahl J C, Aszodi A, Peschke K, Schenten D, et al. A20-deficient mast cells exacerbate inflammatory responses in vivo. PLoS Biol. (2014) 12:e1001762. doi: 10.1371/journal.pbio.1001762
67. Tjota MY, Hrusch CL, Blaine KM, Williams JW, Barrett NA, Sperling AI. Signaling through FcRgamma-associated receptors on dendritic cells drives IL-33-dependent TH2-type responses. J Allergy Clin Immunol. (2014) 134:706–13. e8doi: 10.1016/j.jaci.2014.06.013
68. Molofsky AB, Savage AK, Locksley nRM. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity (2015) 42:1005–19. doi: 10.1016/j.immuni.2015.06.006
69. Zhao W, Hu Z. The enigmatic processing and secretion of interleukin-33. Cell Mol Immunol (2010) 7:260–2. doi: 10.1038/cmi.2010.3
70. Rak GD, Osborne LC, Siracusa MC, Kim BS, Wang K, Bayat A, et al. IL-33-dependent group 2 innate lymphoid cells promote cutaneous wound healing. J Invest Dermatol. (2016) 136:487–96. doi: 10.1038/JID.2015.406
71. Castiglioni A, Corna G, Rigamonti E, Basso V, Vezzoli M, Monno A, et al. FOXP3+ T cells recruited to sites of sterile skeletal muscle injury regulate the fate of satellite cells and guide effective tissue regeneration. PLoS ONE (2015) 10:e0128094. doi: 10.1371/journal.pone.0128094
72. Kuswanto W, Burzyn D, Panduro M, Wang KK, Jang YC, Wagers AJ, et al. Poor repair of skeletal muscle in aging mice reflects a defect in local, interleukin-33-dependent accumulation of regulatory T cells. Immunity (2016) 44:355–67. doi: 10.1016/j.immuni.2016.01.009
73. Natarajan C, Yao SY, Sriram S. TLR3 agonist poly-IC induces IL-33 and promotes myelin repair. PLoS ONE (2016) 11:e0152163. doi: 10.1371/journal.pone.0152163
74. Millar NL, Gilchrist DS, Akbar M, Reilly JH, Kerr SC, Campbell AL, et al. MicroRNA29a regulates IL-33-mediated tissue remodelling in tendon disease. Nat Commun. (2015) 6:6774. doi: 10.1038/ncomms7774
75. Huang XL, Wu GC, Wang YJ, Yang XK, Yang GJ, Tao JH, et al. Association of interleukin-1 family cytokines single nucleotide polymorphisms with susceptibility to systemic sclerosis: an independent case-control study and a meta-analysis. Immunol Res. (2016) 64:1041–52. doi: 10.1007/s12026-016-8797-7
76. Li C, Mu R, Guo J, Wu X, Tu X, Liu X, et al. Genetic variant in IL33 is associated with susceptibility to rheumatoid arthritis. Arthritis Res Ther. (2014) 16:R105. doi: 10.1186/ar4554
77. Angeles-Martinez J, Posadas-Sanchez R, Llorente L, Alvarez-Leon E, Ramirez-Bello J, Villarreal-Molina T, et al. The rs7044343 polymorphism of the interleukin 33 gene is associated with decreased risk of developing premature coronary artery disease and central obesity, and could be involved in regulating the production of IL-33. PLoS ONE (2017) 12:e0168828. doi: 10.1371/journal.pone.0168828
78. Stri I. Cytokines of the IL-1 family: recognized targets in chronic inflammation underrated in organ transplantations. Clin Sci. (2017) 131:2241–56. doi: 10.1042/CS20170098
79. Mueller T, Zimmermann M, Dieplinger B, Ankersmit HJ, Haltmayer M. Comparison of plasma concentrations of soluble ST2 measured by three different commercially available assays: the MBL ST2 assay, the Presage ST2 Assay, and the R&D ST2 assay. Clin. Chim. (2012) 413:1493–94. doi: 10.1016/j.cca.2012.06.021
Keywords: interleukin-33, ST2, systemic sclerosis, pathogenesis, fibrosis
Citation: Li L, Zhu H and Zuo X (2018) Interleukin-33 in Systemic Sclerosis: Expression and Pathogenesis. Front. Immunol. 9:2663. doi: 10.3389/fimmu.2018.02663
Received: 06 January 2018; Accepted: 29 October 2018;
Published: 15 November 2018.
Edited by:
Fang-Ping Huang, University of Hong Kong, Hong KongReviewed by:
Piergiuseppe De Berardinis, Istituto di Biochimica delle Proteine (IBP), ItalyAngela Bonura, Consiglio Nazionale Delle Ricerche (CNR), Italy
Copyright © 2018 Li, Zhu and Zuo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Honglin Zhu, aG9uZ2xpbnpodUBjc3UuZWR1LmNu
Xiaoxia Zuo, c3VzYW56dW9AY3N1LmVkdS5jbg==