 Claudia Afferni1
Claudia Afferni1 Carla Buccione2Sara Andreone2
Carla Buccione2Sara Andreone2 Maria Rosaria Galdiero3,4
Maria Rosaria Galdiero3,4 Gilda Varricchi3,4
Gilda Varricchi3,4 Gianni Marone3,4,5
Gianni Marone3,4,5 Fabrizio Mattei2*
Fabrizio Mattei2* Giovanna Schiavoni2*
Giovanna Schiavoni2*- 1National Center for Drug Research and Evaluation, Istituto Superiore di Sanità, Rome, Italy
- 2Department of Oncology and Molecular Medicine, Istituto Superiore di Sanità, Rome, Italy
- 3Department of Translational Medical Sciences and Center for Basic and Clinical Immunology Research (CISI), University of Naples Federico II, Naples, Italy
- 4WAO Center of Excellence, Naples, Italy
- 5Institute of Experimental Endocrinology and Oncology “Gaetano Salvatore”, National Research Council, Naples, Italy
Interleukin-33 (IL-33) is a IL-1 family member of cytokines exerting pleiotropic activities. In the steady-state, IL-33 is expressed in the nucleus of epithelial, endothelial, and fibroblast-like cells acting as a nuclear protein. In response to tissue damage, infections or necrosis IL-33 is released in the extracellular space, where it functions as an alarmin for the immune system. Its specific receptor ST2 is expressed by a variety of immune cell types, resulting in the stimulation of a wide range of immune reactions. Recent evidences suggest that different IL-33 isoforms exist, in virtue of proteolytic cleavage or alternative mRNA splicing, with potentially different biological activity and functions. Although initially studied in the context of allergy, infection, and inflammation, over the past decade IL-33 has gained much attention in cancer immunology. Increasing evidences indicate that IL-33 may have opposing functions, promoting, or dampening tumor immunity, depending on the tumor type, site of expression, and local concentration. In this review we will cover the biological functions of IL-33 on various immune cell subsets (e.g., T cells, NK, Treg cells, ILC2, eosinophils, neutrophils, basophils, mast cells, DCs, and macrophages) that affect anti-tumor immune responses in experimental and clinical cancers. We will also discuss the possible implications of diverse IL-33 mutations and isoforms in the anti-tumor activity of the cytokine and as possible clinical biomarkers.
Introduction to IL-33 Biology
Interleukin-33 (IL-33) is a cytokine member of IL-1 family, including IL-1α, IL-1β, IL-18, and IL-1Ra that are related to each other by receptor structure and signal transduction pathways. These cytokines share a conserved structure of β-trefoil fold comprised of 12 anti-parallel β-strands that are arranged in a three-fold symmetric pattern. The β-barrel core motif is packed by various amounts of helices in each cytokine structure (1). IL-33 was initially described in 2003 by Girard's group as a nuclear protein abundantly expressed in high endothelial venules (HEVs), specialized blood vessels that mediate the entry of lymphocytes into lymphoid organs and therefore named “nuclear factor from high endothelial venules” (NF-HEV) (2).
It is now known that IL-33 is a chromatin-associated nuclear cytokine in vivo through chromatin-binding motif within its N-terminal nuclear domain, suggesting that nuclear localization and binding to histones are important for IL-33 function and regulation (3). Nuclear IL-33 can function as a transcriptional repressor when overexpressed in transfected cells, although there is still no direct evidence that endogenous nuclear IL-33 regulates gene or protein expression (4). IL-33 is constitutively expressed in different human and mouse tissues in the steady-state, including epithelial, endothelial, fibroblast-like cells, and myofibroblasts and its expression can be increased during inflammation (2, 5). After cell stress or necrosis, IL-33 is released into the extracellular space and functions as an endogenous danger signal that alerts the immune system of tissue damage during trauma or infection. Indeed, IL-33 is considered an “alarmin” able to activate different actors of the innate immune system, mediating a variety of immune reactions including anti-cancer immune responses (6). Here, we will review the biological role of IL-33 affecting immune responses with particular emphasis on anti-tumor immunity.
IL-33 Isoforms
Similar to IL-1β and IL-18, IL-33 is synthesized in a full-length form (amino acids 1–270) that is found in the nucleus, in the cytosol and outside the cell. As IL-1β and IL-18, IL-33 is cleaved intracellularly by the enzyme caspase-1 before release outside the cell. This process requires the NLRP3 inflammasome, which can be activated in response to endogenous and exogenous danger signals. This NLRP3 inflammasome leads to Caspase-1 activation and, in turn, to IL-33 processing and release (7). When cells undergo necrosis or injury, full-length IL-33 is released in the extracellular space where it is cleaved by inflammatory proteases. During apoptosis, a process that does not trigger inflammation in vivo, IL-33 is cleaved and inactivated by endogenous caspases (8–10). Processing by apoptotic caspases is an important regulatory mechanism that limits or suppresses the pro-inflammatory properties of IL-33 during homeostatic cell turnover. Another regulatory mechanism limiting IL-33 activity is oxidation. Extracellular IL-33 is susceptible to cysteine oxidation that leads to the formation of disulphide bridges, resulting in conformational changes that inhibit the binding to ST2 receptor, thus rapidly inactivating IL-33 following allergen exposure (11).
Recent studies have demonstrated the existence of several human full-length active mRNA splice variants dependent on both the cell type expressing IL-33 and the pathological condition and triggered by diverse stimulations during immune responses (12–14). Of note, inflammatory proteases from neutrophils (proteinase 3, elastase, and cathepsin G) (15), mast cells (chymase, tryptase, and granzyme B) 16, and environmental allergens (17) can process full-length IL-33 into shorter mature forms (18–21 kDa) whose biological activity is 10- to 30-fold more potent than the full-length form (see Figure 1). The mature form does not translocate into the nucleus because it lacks the nuclear localization signal found in full-length IL-33 (18, 19). Proteolytic cleavage of IL-33 was shown to induce allergic inflammation in vivo (17) highlighting a novel mechanism by which inflammatory and environmental proteases can amplify allergic inflammation. Of interest, isoform variants as well as cleavage by endogenous and exogenous proteases has been described also for other epithelial-derived cytokines, such as thymic stromal lymphopoietin (TSLP), resulting in pleiotropic functions in health and disease (20). Although both isoforms are biologically active the relative importance of full length and mature IL-33 forms in vivo remains unclear (2, 21). In a mouse model of lung delivery of recombinant adenoviruses encoding IL-33 isoforms the full-length IL-33 induced inflammation in an ST2-independent fashion, but not pulmonary eosinophilia, goblet cell hyperplasia, or Th2 skewing, whereas mature IL-33 induced ST2-dependent Th2-associated effects. Both isoforms had similar effects on gene expression, suggesting that the different effects are due to differential utilization of the ST2 receptor (22). In addition, in a mouse model of DNA cancer vaccine, delivery of either full-length or mature IL-33 as an immunoadjuvant induced potent Th1 and cytotoxic T cell (CTL)-associated anti-tumor immunity and complete regression of established TC-1 tumor in mice. Interestingly, the full-length IL-33 was more potent than mature IL-33 in expanding the humoral immune response (23).
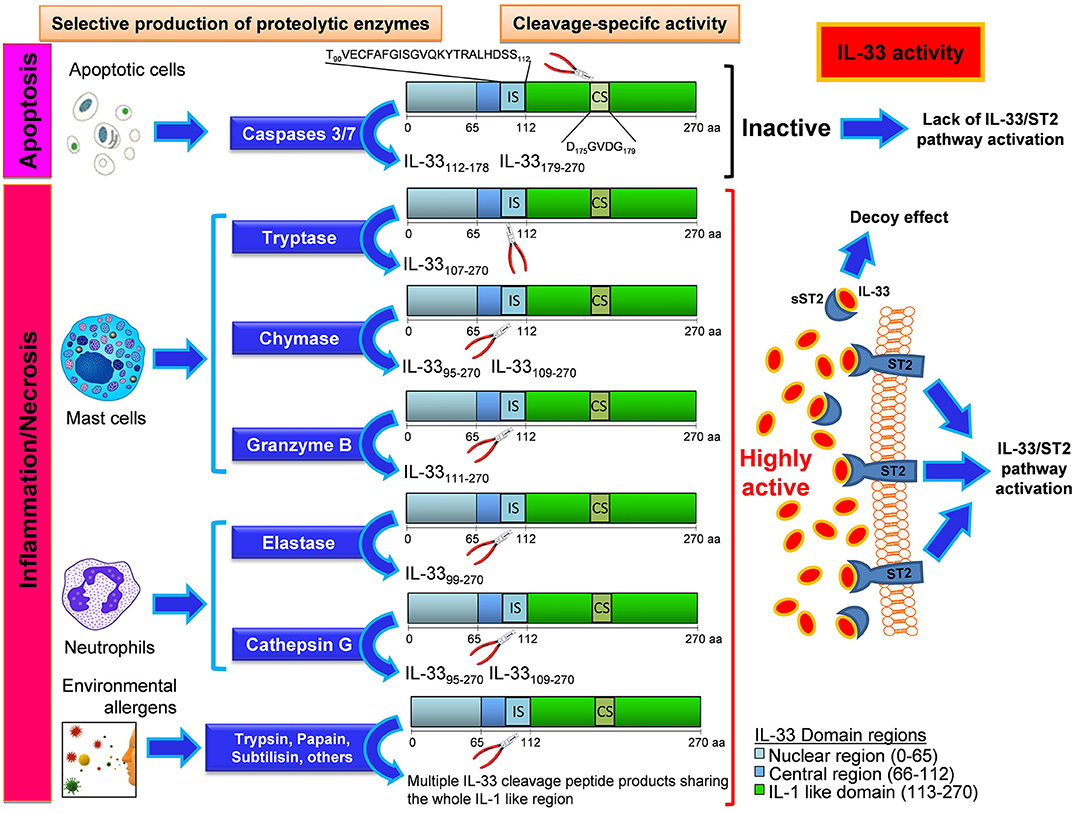
Figure 1. Mechanisms and effects of the enzyme-specific IL-33 cleavage. Biological events such as apoptotic stress, Inflammation, and necrosis can differentially generate various IL-33 protein variants with high biological activity or no activity depending on the enzyme produced by the cells. Apoptotic cells enable the production and release of caspases 3 and 7, that cleaves IL-33 in the caspase site (CS) generating inactive fragments of IL-33 by disruption of some IL-1 like functional domains close to CS. The insurgence of inflammation or necrosis process leads to the local recruitment of mast cells and neutrophils, the main effectors of inflammatory processes. When these cells reach the inflammed site, they produce, and release enzymes that cleave the IL-33 protein at the Inflammatory site (IS) inside the central region. These cleavage-specific enzymes allow the production and release of highly active forms of IL-33, capable to stably bind the ST2 receptor (ST2) on a plethora of ST2-expressing cells. In other cases this active IL-33 does bind to the soluble form of its receptor (sST2), which acts as a decoy receptor. In this latter event the effect of IL-33 will be suppressed by the formation of an sST2/IL-33 decoy complex. When no apoptotic nor inflammatory enzymes are produced, an uncleaved form of IL-33 is released, with a very low biological activity compared to that showed by IL-33 cleavage products originated by neutrophil and mast cell-derived enzymatic cutting. When the IL-33 is cleaved by certain environmental allergens, their enzymatic activity at the IS site gives rise to multiple peptide products sharing the whole IL-1 like region of IL-33, that does retain the ability to bind ST2. The IL-33 cleavage products herein shown are all equipped with the ST2 binding sequence (inside the IL-1 like region). The secondary fragments lacking the ST2 binding sequence and generated during the cleavage reaction are not depicted and have no effect on IL-33/ST2 binding.
The IL-33/ST2 Axis
IL-33 exert its cytokine activity through binding to its primary specific receptor ST2, which is dependent on the co-receptor, IL-1 receptor accessory protein (IL-1RAcP), and the adaptor protein MyD88 for signaling (24). The crystal structure of IL-33 with ST2 has revealed that surface charge complementarity is crucial for specific binding (25). The gene that encodes for ST2 produces its transmembrane receptor but also produces a soluble form of ST2 (sST2), which acts as a binding decoy for IL-33 and thus down-modulates IL-33 activity during inflammatory responses, such as in experimental allergic asthma (26) and collagen-induced arthritis (27). Most hematopoietic cells express ST2. ILC2s, some Treg cells, and mast cells are the primary tissue-resident cells that constitutively express high levels of ST2, implying that these cells are initial targets of IL-33 (3). Non-hematopoietic cells, including endothelial cells, epithelial cells, and fibroblasts, are reported to express ST2 and respond to IL-33, although the in vivo consequences of signaling in these populations are less well-characterized.
In hematopoietic cells, IL-33 acts primarily on immune cells associated with type 2 and regulatory immune responses, including ILC2s, Th2 cells, eosinophils, mast cells, and basophils, as well as subsets of dendritic cells, myeloid-derived suppressor cells, and Tregs (28). However, it is now clear that the action of IL-33 is not limited to the activation of type-2 immune responses. Indeed, recent studies have revealed important roles of IL-33 in the activation of immune cells involved in type-1 immunity, such as Th1 cells, NK cells, CD8+ T cells, neutrophils, macrophages, B cells, and NKT cells (19, 29, 30). This pleiotropic nature of IL-33 (Figure 2) is likely to explain why IL-33 has been implicated in a wide variety of non-allergic diseases, including infectious diseases (fungal, helminth, protozoa, bacterial, and viral infection), cardiovascular diseases, chronic obstructive pulmonary disease (COPD), fibrotic diseases, musculoskeletal diseases, inflammatory bowel diseases, diseases of the central nervous system (Alzheimer), graft vs. host disease (GVHD), obesity, diabetes, and cancer (3).
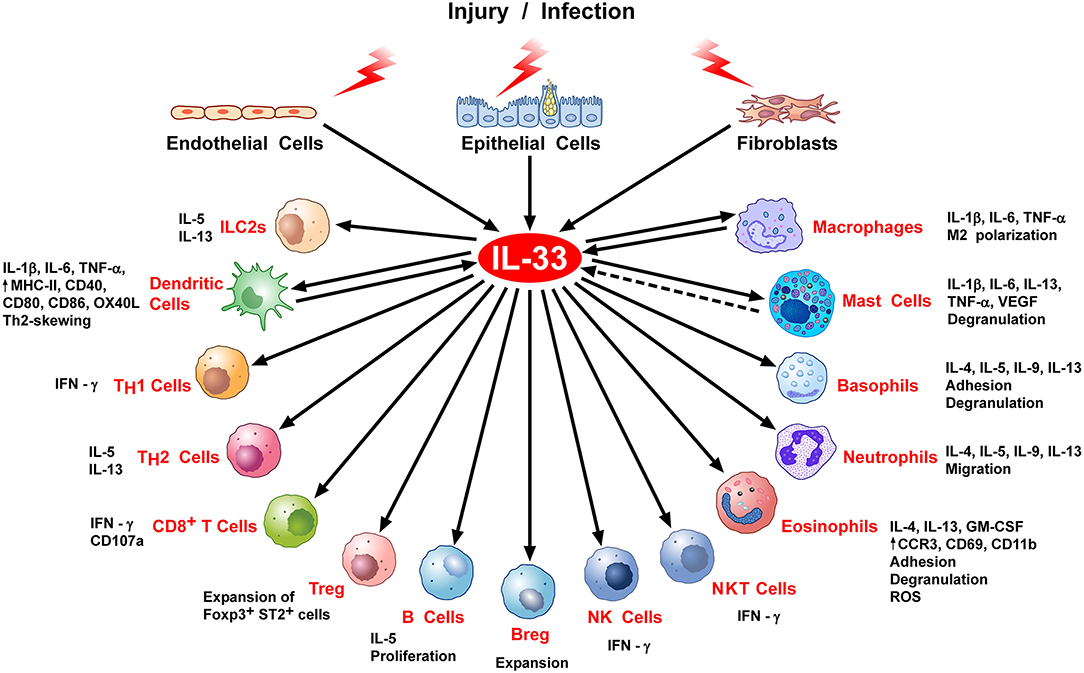
Figure 2. Stimulation of immune cells by IL-33. IL-33 is secreted by epithelial, endothelial, or fibroblasts in response to various stimuli, including infection or cell stress causing injury. IL-33 exerts its biological activities through binding to its specific receptor ST2 expressed by most hematopoietic cells. IL-33/ST2 can stimulate a variety of immune cell reactions, including: an atypical Th2-type of immune response through production of IL-5 and IL-13 by ILC2 and Th2 cells; IFNγ production by NK, NKT, and Th1 cells; CD107a exposure and IFNγ production by activated CD8 T cells; in vivo expansion of ST2+ Treg cells B and Breg cells; release of inflammatory cytokines (e.g., IL-1β, IL-6, TNFα) by DC, macrophages and mast cells; M2 macrophage polarization; degranulation of mast cells, basophils, and eosinophils; neutrophil migration; phenotypic, and functional activation of DC and eosinophils. There is some evidence that activated DCs, macrophages, and perhaps mast cells can produce IL-33.
Immune Cell Targets of IL-33 Affecting Tumor-Immune Responses
CD4+ Th Cells
Naïve CD4+ T helper cells constitutively express ST2 and stimulation with IL-33 skews their differentiation toward a Th2 phenotype. CD4+ T cells are needed in the effector phase of a protective antitumor immune response against tumors lacking MHC class II (31). However, human CD4+ T cells can suppress tumors expressing adequate levels of MHC class II and self-antigens on their surface, through secretion of IFN-γ or direct tumor killing (32). Interestingly, Villareal et al. demonstrated that IL-33 can be an effective adjuvant when combined with an HPV16 E6/E7-encoded DNA vaccine, enhancing both antigen specific CD4+ and CD8+ IFN-γ+ T cells, and antigen specific IgG concentration in the serum, leading to regression of established TC-1 tumor in mice (23). In accordance with this study, Mousa Komai-Koma et al. showed that IL-33 may promote CD4+ T helper 1 (Th1) differentiation by a mechanism depending on IL-12 and ST2. IL-33 and IL-12 synergistically increase both ST2 and IL-12R expression in early activated CD4+ T cells. These data indicate that IL-33 promotes Th1 cell development, while it is ineffective on mature Th1 cells (33). A possible explanation for such differences is that ST2 expression is induced only in early-TCR activated naïve CD4+ T cells and is then gradually inhibited when Th1 cells fully mature. Although the signaling pathway by which IL-33 enhances Th1 polarization is still unknown, it is likely that IL-33 inducing Th1 or Th2response depends on the cytokine milieu, in particular the balance of IL-12 and IL-4 levels in vivo (33). Of note, IL-33 also promotes the differentiation of IL-9-producing Th cells (34) which exert potent antitumor immune responses in vivo (35, 36).
CD4+ Treg Cells
ST2/IL-33 signaling is known to expand suppressive CD4+ Foxp3+ GATA3+ Treg cells in vivo and in vitro (37). IL-33-expanded Tregs express ST2 and can be found in several immune and non-immune tissues exerting potent suppressor function in a variety of pathological conditions, such as autoimmunity, inflammation, transplantation, and allergy (38). ST2+ Treg expansion can be mediated by IL-33 signaling in DCs, through production of IL-2, which selectively expands ST2+ Tregs (39). In the intestine, particularly rich in ST2+ Treg cells, IL-33 signaling stimulates transforming growth factor (TGF)-β1-mediated differentiation of Treg cells and provides a signal for Treg-cell accumulation and maintenance in inflamed tissues (40). In ApcMin/+ mice, epithelial-derived IL-33 promoted the expansion of ST2+ Treg cells in the colon correlating with increased tumor burden (41, 42). A similar observation was recently reported in the CT26 adenocarcinoma model, where rIL-33 administration to tumor-bearing mice promoted, while IL-33 blockade reduced, the expansion of ST2+ Treg cells in tumor tissue and spleen (43). Moreover, IL-33 blockade reduced accumulation of Treg cells in tumor microenvironment and inhibited tumor growth in a preclinical model of human non-small-cell lung cancer (NSCLC) xenografts (44). In contrast, some studies have reported inhibitory effects of IL-33/ST2 on Treg cells expansion. In a melanoma mouse model, IL-33 was shown to inhibit Treg infiltration in the tumor microenvironment indirectly, through stimulation of MDSCs, which had reduced capacity to induce the differentiation or expansion of Treg cells in vitro (45). A recent study using reciprocal bone marrow chimeras in a mouse model of sporadic colon cancer, genetic ablation of ST2 in both hematopoietic and non-hematopoietic compartments leads to increased tumor-infiltrating ST2+ Foxp3+ Tregs and enhanced colon tumor development (46).
CD8+ T Cells
Unlike CD4+ T cells, only effector CD8+ T cells or polarized Tc1 cells, but not naïve and early activated CD8+ T cells, express ST2 (47). High expression of ST2 in CD8+ T cells cultured in Tc1 polarizing conditions is regulated by T-bet, a master transcription regulator of Th1 effector functions. Moreover, it was shown that IL-12 and IL-33 synergistically increased T-bet and Blimp1, transcription factors critical for effector fate of CD8+ T cell (47). Recent studies from transplantable solid tumor models have indicated a direct role of exogenous IL-33 in promoting antitumor CD8+ T cell immunity using either IL-33 transgenic mice (48), IL-33 DNA as vaccine adjuvant (23), or IL-33 expressing tumor cells (49). Systemic administration of rIL-33 in melanoma tumor bearing mice, promoted expansion, increased tumor infiltration and effector function of antigen-specific CD8+ IFN-γ+ T cells by both a direct or DCs-mediated effect (50). In the aggressive C1498 acute myeloid leukemia (AML) model, IL-33 treatment significantly increased the percentage of effector memory liver CD8+ T cells leading to delayed leukemia development and improved overall survival (51). This finding suggest a role of exogenous IL-33 in promoting rapid expansion of the effector memory CD8+ T cell pool, consistent with the results from solid tumor models (23, 48). In this study the increased CD8+ T cells activation level up-regulates PD-1/PD-L1 expression in vivo, therefore combination of PD-1 blockade and IL-33 treatment further improves survival of leukemia-bearing mice (51).
Type-2 Innate Lymphoid Cells
Innate lymphoid cells (ILCs), belonging to the family of innate cells, are characterized by classic lymphoid cell morphology, but lack lineage-specific markers and somatically rearranged antigen receptors. Based on the expression of transcription factors, phenotypic markers, and effector cytokine production profiles, ILCs have been divided into three distinct subclasses: group 1 ILCs, group 2 ILCs, and group 3 ILCs (52). ILC are derived from a common lymphoid progenitor and possess a wide range of cell surface markers, many of which have only recently been elucidated (53). ILC2, originally identified in the mouse and human mesenteric lymph nodes as lineage marker negative, c-kit+, Sca-1+, IL-7Ra+, and ST2+ cells (54), were also found in lung, skin, and gut, while only a small number of circulating ILC2s can be detected in blood (55). They are involved in tissue repair (56), anti-helminth immunity (57), and allergic inflammation (58).These cells are dependent on transcription factor GATA-binding protein 3 for their development and maintenance (59). Activation of ILC2s by alarmins (IL-25, IL-33, and TSLP) secreted by epithelial cells upon cellular stress and tissue damage (55, 60), produce IL-5, IL-13 (54), IL-4, IL-6, IL-9, and amphiregulin which induce Th2 differentiation (61). This group of innate cells was often observed to infiltrate tumors in humans, but their role seems more frequently associated with cancer progression than restriction. Clinical studies suggested that increased numbers of ILC2s in peripheral blood of patients with gastric cancer, could contribute by cytokines they secrete to the immunosuppressive environment maintained by CD4+ T helper 2 (Th2), myeloid-derived suppressor cells (MDSC), and macrophages (62). ILC2s might also induce immune suppression via secretion of amphiregulin (63), which enhances Treg activity in vivo and can thereby inhibit antitumor immune responses induced by DC vaccination (64). The anti-tumoral activity of ILC2 was described for the first time by Ikutani et al. in a mouse model of lung metastatic melanoma. Following tumor induction, administration of rIL-33 induced the development of IL-5-producing ILC2, which recruited and maintained eosinophils responsible for tumor cell death and tumor metastasis prevention (65). Overexpression of IL-33 in several tumor cell lines induced high numbers of ILC2s, when transplanted in mice, with potent anti-tumoral activity. The latter study suggests that local production of IL-33 induces ILC2 to release CXCR2 ligands able to sustain the expression of CXCR2 on tumor cells and induce their apoptosis (66).
NK and NKT Cells
IL-33 directly activates both human (19) and mouse (30) NKT and NK cells inducing IFN-γ production via cooperation with IL-12, thus contributing to establish Th-1 immunity. During viral infection, IL-33/ST2 axis amplifies the expansion of NK cells and enhances host defense (67, 68). The role of IL-33 on NKT in cancer immunity is unknown. In contrast, a number of reports have analyzed the effects of IL-33 on NK cell expansion and/or activation in tumor-bearing mice. In mouse experimental metastasis models of B16 melanoma and Lewis lung carcinoma, transgenic expression of IL-33 in the host promoted the recruitment of cytotoxic NK cells to the pulmonary site that inhibited metastasis formation (48). In vitro, IL-33 directly activated NK cell cytotoxicity, stimulated NF-κB and up-regulated CD69 expression (48). Furthermore, B16 and 4T1 tumor cells overexpressing IL-33 implanted into syngeneic mice induced IFN-γ+ NK cells in tumor tissue that mediated IL-33 anti-tumoral effect (49). Similarly, increased frequencies of CD107a+IFN-γ+ NK cells were observed following exogenous administration of IL-33 in spleens and tumors of B16 melanoma-bearing mice (69). In contrast with these reports, previous studies in 4T1 breast cancer model showed that IL-33/ST2 signaling impairs NK cell activation. ST2-deficient mice bearing 4T1 tumors exhibited increased numbers of activated NK cells (IFN-γ+ CD27high CD11bhigh, CD69+ KLRG−) and NK cytotoxic activity, with respect to wild-type (WT) counterparts. In vivo depletion of NK cells accelerated 4T1 tumor growth in ST2−/− mice (70). Moreover, exogenous administration of IL-33 to WT 4T1 tumor-bearing mice decreased NK cell activation and cytotoxicity and promoted tumor progression (71), thus suggesting a detrimental role for IL-33 in NK cell-dependent anti-tumor responses. These contrasting results suggest that IL-33 may exert opposing effects on NK cells within the tumor microenvironment depending on the levels of IL-33 expressed and on the primary target cells.
Macrophages
Several lines of evidence indicate that IL-33 amplifies the expression of M2 markers on macrophages in vitro and in vivo, thus promoting the suppressor function of tumor-associated macrophages (TAMs). Blockade of IL-33 abrogates the polarization of TAMs into (alternatively activated) M2 macrophages in a model of human non-small-cell lung cancer (44). Expression of IL-33 was found to stimulate the recruitment of M2-like macrophages into the cancer microenvironment in mouse models of colon (41, 72–74) and breast cancer (71) correlating with tumor progression. Of note, IL-33 stimulated macrophages to produce prostaglandin E2, which supported colon cancer stemness (73). Furthermore, in mouse tumor xenografts IL-33 was shown to promote metastasis through recruitment of M2-like TAMs (75). Recently, it was shown in the mouse monocyte/macrophage line RAW264.7 that IL-33 directly induces MMP-9 expression, which facilitates tumor progression, invasion, and angiogenesis (76). These observations indicate that IL-33/ST2 signaling on macrophages promotes M2 polarization, immunosuppression, and tumor progression.
Dendritic Cells
Although dendritic cells (DCs) express low levels of ST2 on their cell surfaces, they respond to IL-33 by up-regulating MHC-II, CD40, CD80, CD86, OX40L, CCR7, and by increasing production of several cytokines (IL-4, IL-5, IL-13, TNF-α, and IL-1β) and chemokines (CCL17 and CCL22) (77–80). In addition, IL-33-activated DCs promote an atypical Th2-type of immune response inducing IL-5- and IL-13-producing CD4+ T cells in vitro and in vivo (77, 79), which can be further amplified during allergic inflammatory response via ST2 (78, 81). In vivo, IL-33 exposure induces DC recruitment and activation in the lung (78, 79). IL-33 promotes the expansion of DCs from bone marrow (BM), by stimulating the secretion of basophils-derived GM-CSF. However, such IL-33 differentiated BM-DCs expressed low levels of MHC-II but high PD-L1 and PD-L2 immune checkpoints on the surface, and displayed reduced capacity to prime naïve T cells (82). In support of this potential tolerogenic effect, IL-33 has been shown to promote IL-2 secretion by murine DCs, thus supporting the in vitro and in vivo expansion of ST2-expressing Treg cells (39). In mice bearing 4T1 breast cancer IL-33 administration increased the percentage of splenic CD11c+ DCs expressing IL-10 (71). In contrast, in a murine AML model, systemic IL-33 administration promoted DC activation and “licensing” for cross-priming of tumor-reactive CD8+ T cells (51). Likewise, in EG7 lymphoma, B16, and inducible BrafV600EPTEN melanoma models exogenous IL-33 activated myeloid DCs within the tumor microenvironment increasing antigen cross-presentation and restoring anti-tumor T cell activity in a ST2, MyD88, and STAT1-dependent manner (50). On the whole, these results suggest that IL-33 depending on the context can stimulate DC antigen presentation and, thus, anti-tumor immune responses, or induce tolerogenic features, thus supporting tumor growth.
Myeloid-Derived Suppressor Cells
Myeloid-derived suppressor cells (MDSCs) are closely related to granulocytes and monocytes but differ from them in that they are absent in healthy individuals but expand under pathological conditions, such as cancer, exerting potent immune suppressive role (83). Several reports described the ability of IL-33 to expand MDSCs in vivo during tumorigenesis. In 4T1 breast cancer model, IL-33 has been reported to promote MDSC expansion (71). Exogenous administration of IL-33 increased intratumoral and systemic accumulation of CD11b+ Gr-1+ MDSCs that expressed TGF-β1 and IL-13α1R with higher incidence of monocytic vs. granulocytic MDSCs. Fittingly, absence of IL-33/ST2 signaling reduced the accumulation, proliferation, and immunosuppressive ability of MDSCs in tumor-bearing mice (71, 84). Moreover, IL-33 upregulated in MDSCs the expression and activity of arginase-1 in vitro and activated NF-κB and MAPK signaling in vivo, which augment their immunosuppressive ability (84). Conversely, in vitro IL-33 was shown to negatively regulate MDSC development from BM progenitor cells inhibiting the differentiation of granulocytic MDSCs (G-MDSCs), but not of monocytic MDSCs (M-MDSCs). In addition, IL-33-treated BM-derived MDSCs exhibited diminished immunosuppressive capacity, reduced inhibition on T-cell proliferation and IFN-γ production, and diminished production of ROS (45). In B16 melanoma mouse models, IL-33 administration was shown to decrease MDSCs accumulation in the spleen and tumor microenvironment (45, 69). These evidences indicate that IL-33 may promote or halt MDSCs expansion depending on the tumor type.
Neutrophils
IL-33 directly acts on murine neutrophils in the lungs (85). Indeed, IL-33-treated neutrophils produced IL-4, IL-5, IL-9, and IL-13 and displayed a distinct gene expression profile in contrast to resting and lipopolysaccharide (LPS)-treated neutrophils. These neutrophils were found in the lungs of ovalbumin (OVA)-induced mouse model of asthma. Adoptive transfer of IL-33-driven neutrophils significantly worsened the severity of the disease in this model (86). In vitro, IL-33 promoted IL-4, IL-5, IL-9, and IL-13 expression in murine neutrophils in time- and dose- dependent manner. IL-33-induced neutrophils expressed high levels of CXCR1, CCR1, IL-1R2, and CXCR2 mRNAs compared with LPS-induced neutrophils. In a mouse model of choriomeningitis virus-induced hepatitis, IL-33 promoted neutrophil recruitment in the liver and dampened liver injury by limiting T-cell activation. Liver neutrophils displayed an immunosuppressive phenotype, characterized by high levels of arginase-1, iNOS and IL-10 (87). Thus, IL-33 may promote inflammatory or regulatory neutrophils, depending on the pathological condition. Little is known about the effects of IL-33 on neutrophils in tumor immunity. In a mouse model of ectopic CT26 colon carcinoma, systemic chemotherapy with irinotecan induced intestinal mucositis, associated with the induction of IL-33, and increased neutrophil accumulation in the intestine. Supernatants from intestine explants treated with irinotecan enhanced migration of neutrophils in vitro in an IL-33/CXCL1/2/CXCR2-dependent manner. Importantly, IL-33 blockade reduced mucositis and enabled prolonged irinotecan treatment of ectopic colon carcinoma leading to a beneficial outcome of the chemotherapy. These results suggest that inhibition of the IL-33/ST2 pathway may represent a novel approach to limit mucositis and improve the effectiveness of chemotherapy (88).
Eosinophils
Administration of IL-33 in mice causes massive tissue infiltration of eosinophils and elevations of typical type 2 cytokines such as IL-5, IL-9, and IL-13, contributing to allergy and fibrosis (89). These responses also occurred in RAG knockout mice, suggesting that innate cells, particularly IL-5 producing ILC2s, were the direct target of IL-33 (54). However, IL-33 is now known to act directly on eosinophils leading to upregulation of CCR3, CD69, and CD11b, production of chemokines (CCL17, CXCL2, CXCL3, and CXCL10), and cytokines (IL-6, IL-13, GM-CSF) (90). IL-33 sustained eosinophil survival in vivo in a ST2-dependent manner and via autocrinous production of GM-CSF. Moreover, IL-33 and GM-CSF promoted the production of IL-4 and IL-13 by eosinophils, which in turn favored macrophage polarization toward M2 phenotype (91). In a murine model of melanoma, in vivo expansion of innate IL-5–producing ILC2 cells following systemic IL-33 injection played an important role for eosinophil recruitment and metastasis control. Innate IL-5-producing cells were increased in response to tumor invasion, and their regulation of eosinophils is critical to halt tumor metastasis (65). We reported that in transplantable B16 melanoma models, IL-33 restricted tumor growth and inhibited lung metastasis through recruitment and activation of eosinophils. Indeed, ST2-deficient mice presented an increased metastatic load and reduced lung eosinophilia compared to wild type mice. Depletion of eosinophils completely abolished the anti-tumor effects of IL-33 administration (69). Kim et al. also reported substantial expansion of intratumoral eosinophils in mice transplanted with IL-33-expressing tumor cells (EL4, CT26, and B16); however, their role in IL-33-induced anti-tumor effects was not addressed (66). In our study, IL-33 expanded eosinophils expressed T cell-attracting chemokines and induced NK and CD8+ T cell-recruitment at the subcutaneous tumor site, but not at the lung metastatic site. In addition, IL-33 activated eosinophils in vitro enhancing CD11b, CD69, and granzyme B expression and activating cytotoxic functions against melanoma cells (69). These findings suggest that depending on the tumor site, eosinophils may play an accessory role, supporting the recruitment of tumor-reactive CD8+ T cells (92), or a direct cytotoxic effect against tumor cells. Indeed, IL-33 can potently activate human eosinophils, enhancing adhesion, promoting survival, and inducing ROS production and degranulation (93, 94). Of note, degranulation of eosinophils has been shown in vivo in proximity of tumors (95, 96) and after in vitro stimulation, resulting in efficient killing of target mouse (97) and human (98) tumor cells, highlighting the tumoricidal properties of eosinophils (99).
Mast Cells
IL-33 activates its receptor complex (ST2: IL-1RAcP) on human mast cells (100) and basophils (19). IL-33 synergizes with IgE- and non-IgE-dependent stimuli to release cytokines from human mast cells (101). Similarly, IL-33 augments substance P-induced vascular endothelial growth factor (VEGF) production from human mast cell lines (102). The latter findings are clinically relevant because VEGFs produced by human mast cells (103) and basophils (104) play an important role in chronic inflammation and in tumor growth (105). We have demonstrated that IL-33 up-regulated the Fcγ receptor type IIa and synergistically enhanced immune complex-triggered activation of human mast cells (106). Collectively, these findings demonstrate that IL-33 can synergistically potentiate the immunologic and non-immunologic release of mediators from human and rodent mast cells. Single cell analysis demonstrated that IL-33 increased both the number of degranulating and chemokine-producing mast cells and the magnitude of individual mast cell response (107). The relevance of IL-33-mediated mast cell response has been found also in vivo in several pathological conditions, including cancer. Mast cells and basophils are known to infiltrate several types of tumors but, due to the wide range of mediators they release, it is difficult to define their specific pro- or anti-tumoral activity (103). Mast cell activation by IL-33 may occur in a number of tumor types. In skin cancers, mast cells accumulated with IL-33 expressing fibroblasts in UV-exposed murine skin samples (108). In the ApcMin/+ mouse model, IL-33 deficiency reduced tumor burden (109, 110) and decreased mast cell density in polyps as well as suppressed the gene expression of mast cell-derived proteases and cytokines that promote angiogenesis, Treg function, and MDSC recruitment within the tumor microenvironment (111–113).
Basophils
Human basophils constitutively express ST2 which is induced by IL-3 (114, 115). Although IL-33 alone failed to directly induce degranulation of human basophils, it exerted priming effects. It enhanced degranulation and IL-4 production in response to IgE cross-linking (114). By contrast, IL-33 alone activated unprimed murine basophils in vitro (116). Recently, Rivellese and collaborators, using highly purified human basophils, elegantly demonstrated that IL-33 alone induces the release of low but detectable amounts of IL-4 (117). IL-33 synergistically potentiated IL-4 production induced by IL-3 or anti-IgE. Interestingly, IL-33 did not induce basophil degranulation, as evaluated by the membrane expression of CD63, but significantly enhanced IgE-mediated histamine release. Collectively, these findings indicate that IL-33 can activate human basophils presumably through the engagement of ST2. Furthermore, IL-33 can enhance IL-3- and anti-IgE-mediated basophil degranulation, histamine secretion, and cytokine production. The role of basophils in anti-cancer immunity is poorly characterized and the effects of IL-33 in stimulating or regulating basophils responses in cancer are unknown. In mouse models of melanoma, Sektioglu et al. reported that intratumoral basophils enhanced CD8+ T cell infiltration via production of the chemokines CCL3 and CCL4, contributing to tumor rejection following Treg cell depletion (118). Recently, low circulating eosinophils and basophils were associated with poor prognosis in CRC patients (119). Moreover, basophils in tumor-draining lymph nodes of both mice and pancreatic ductal adenocarcinoma patients correlated with Th2 responses in tumors and poor prognosis (120).
Role of IL-33 in Cancer: Evidences from Experimental Tumor Models
Accumulating evidences indicate that the axis IL-33/ST2 plays a role in tumor immunity. However, both pro-tumoral and anti-tumoral functions have been reported and the current literature suggest that IL-33 may differently affect tumor immunity depending on the tumor type, immune cell targets and on cooperating microenvironmental factors (121).
Breast Cancer
The majority of reports point to a pro-tumoral role of IL-33 in breast cancer models. In mice, the IL-33/ST2 axis was shown to promote the growth and lung and liver metastases of 4T1 mammary tumors facilitating the intratumoral accumulation of immunosuppressive myeloid (Gr-1+ TGF-β1+ MDSC, IL-10-expressing CD11c+ DCs, and alternatively activated M2 macrophages), ILC2, and Treg cells, while dampening the expansion of activated NK cells (70, 71, 84). Lukic group demonstrated that IL-33/ST2 axis promotes the expression of pro-angiogenic VEGF in tumor cells and attenuates tumor necrosis, thus facilitating mammary tumor growth (122). In contrast, another study reported that transgenic overexpression of IL-33 in 4T1 breast cancer cells reduces tumor growth and metastasis in vivo (49). In vitro, IL-33 acts as a critical tumor promoter during epithelial cell transformation and sustains breast cancer tumorigenesis (123). Recently, it has been reported that IL-33 overexpression in human ER-positive breast cancer cells results in resistance to tamoxifen-induced tumor growth inhibition, by promoting cancer stem cell properties (124). These findings indicate that in breast cancer IL-33/ST2 exert both intrinsic and extrinsic pro-tumoral function favoring tumorigenesis and stemness and reducing anti-tumor immunity.
Colorectal Cancer
A number of evidences from mouse models point to an important role of the IL-33/ST2 axis in promoting colorectal cancer (CRC) tumorigenesis, progression and malignancy (125). In the ApcMin/+ mouse model for human familial adenomatous polyposis, abrogation of the IL-33/ST2 axis by knockout of IL-33 (109) or ST2 (41, 110) inhibits proliferation, induces apoptosis, and suppresses angiogenesis, thus decreasing tumor number and size. Accordingly, overexpression of IL-33 in MC-38 mouse CRC cells results in increased in vitro proliferation and enhanced tumor growth and liver metastasis after orthotopic transplant in syngeneic mice through tumor-derived IL-33-induced recruitment of CD11b+ GR1+ and CD11b+ F4/80+ myeloid cells and angiogenesis (126). In addition, expression of IL-33 in human CRC cells promoted their growth and metastasis in vivo and reduced the survival of recipient nude mice (127). Likewise, IL-33/ST2 signaling in CRC tissues promoted the malignant growth and metastatic spread of CRC through modification of the tumor microenvironment (72). In a recent study, it was shown that IL-33 overexpression or exogenous administration of IL-33 to human or murine colon cancer cells enhanced cell growth in vivo and promoted colon cancer cell stemness through an immune-associated mechanism (73). In contrast, some studies have reported an anti-tumoral function of IL-33 in CRC models. Abrogation of ST2 signaling in CT26 mouse adenocarcinoma cells enhanced tumor development after subcutaneous transplant into syngeneic BALB/c mice (74). In the azoxymethane (AOM)/dextran sodium sulfate (DSS) model, IL-33-deficient mice were shown to be highly susceptible to cancer-associated colitis showing increased tumor number, size, and grade, due to a protective function of IL-33 in regulating an IgA-microbiota axis in the intestine (128). Fittingly, in a mouse model of sporadic colon cancer tumorigenesis in the absence of preexisting inflammation lack of IL-33 signaling enhanced colon tumorigenesis, while IL-33 treatment reduced tumor growth in the transplantable MC38 model, via IFN-γ-mediated antitumor immune response (46). These evidences suggest that in the absence of preexisting chronic or acute inflammation the homeostatic release of IL-33 by dying colon epithelial cells protects against the initiation and development of sporadic colon cancer.
Lung Cancer
The IL-33/ST2 axis plays a critical role in various inflammatory lung diseases, including asthma and fibrosis (129). However, few studies have investigated the contribution of this pathway to lung cancer. Akimoto et al. reported that ST2 expression in human and murine lung cancer is inversely correlated with metastatic potential (130). Exposure to IL-33 enhanced oncotic cell death of ST2+ low-metastatic Lewis lung carcinoma cells, but not of ST2− high-metastatic cells, thus suggesting that IL-33 enhances lung cancer progression by selecting for malignant cells. Moreover, in vitro stimulation with IL-33 promoted the migration and invasiveness of human lung A549 cells (131) and enhanced the growth and metastasis of primary non-small-cell lung cancer (NSCLC) cells after xenotransplant into immunodeficient mice (132). Vice versa, IL-33 blockade efficiently inhibited tumor growth of patient-derived NSCLC xenografts, abrogating M2 polarization of macrophages, and reducing the accumulation of Treg cells within the tumor microenvironment (44). In mouse lung tumor models stable transfection of IL-33 gene into tumor cells inhibited tumor growth and metastatic spread in vivo (49, 133). Of note, IL-33 expressing metastatic A9 lung cancer cells, a TC1-derived cell line with spontaneous down-regulation of MHC-I, restored MHC-I expression and immune recognition in mice (133). Although these evidences indicate that IL-33/ST2 expression in lung tissue or cancer cells fuels tumor growth, the contribution of IL-33 to anti-tumor adaptive immune responses against this cancer remains to be established.
Melanoma
Accumulating evidences indicate that in melanoma models IL-33 exert anti-tumoral and anti-metastatic effects by conditioning the local immune environment. Systemic injections of IL-33 significantly inhibit tumor growth in mice bearing subcutaneous B16.F10 melanomas (50, 69) and in BRAFV600EPTEN-inducible melanoma model (50). Dominguez et al. reported that IL-33 both directly stimulated CD8+ T cell expansion and IFN-γ production and activated myeloid dendritic cells (mDCs) increasing antigen cross-presentation. Of note, combination therapy with rIL-33 and agonistic anti-CD40 antibodies demonstrated synergistic anti-tumoral activity in this model (50). Using subcutaneous and experimental metastasis B16 melanoma models, we recently demonstrated that IL-33 inhibits melanoma tumor growth and pulmonary metastasis in vivo in an eosinophil-dependent manner (69). In a previous study, it was shown that IL-33 transgenic mice inhibit tumor metastasis in the B16 melanoma model (48). Likewise, transgenic expression of a secretable mature form of IL-33 by plasmid DNA in B16 tumor cells reduced tumor growth and metastasis in vivo (49). In these models, both NK and CD8+ T cells were required for the anti-tumoral effect of IL-33. Instead, Kim et al. showed that subcutaneous injection of adenoviral vector-transfected B16 melanoma cells engineered to secrete IL-33 did not develop palpable tumors in mice, in a CD8+ T- and NK-independent manner, through expansion of CXCR2 ligands-secreting intratumoral ILC2 (66). Ex vivo explanted IL-33-expressing B16 tumors exhibited higher reactive oxygen species (ROS) levels and increased CXCR2 expression and apoptosis thus suggesting a role for IL-33 in creating a hypoxic tumor microenvironment supporting ILC2-induced apoptosis (66). The explanation for such diverse immune-mediated anti-tumor mechanisms needs further investigations, although it is plausible that different local concentrations of IL-33 within the tumor microenvironment may stimulate different responses.
Other Tumors
IL-33 has been reported to exert an anti-tumoral role in other tumor models. In a human papilloma virus (HPV)-associated model for cancer immunotherapy IL-33 was shown to act as a potent vaccine adjuvant augmenting Th1 and CD8+ T-cell responses, inducing anti-tumor immunity in vivo (23). Enforced expression of IL-33 in a large collection of murine tumor cell lines, including CT26 colon carcinoma, EL4 lymphoma, B16 melanoma, Lewis lung carcinoma (LLC), A9, and 4T1 lung cancer, results in reduced tumor growth in vivo (49, 66, 133). In a murine acute myeloid leukemia (AML) model administration of IL-33 significantly inhibited leukemia growth and improved mice survival rate in a CD8+ T cell dependent manner (51). Notably, combination of PD-1 checkpoint blockade with IL-33 further prolonged mice survival, and induced complete leukemia regression in 50% of animals. The latter report represents the first evidence for combining IL-33 with immunotherapy targeting immune checkpoint inhibitors. Recently, administration of rhIL-33 was shown to expand Vγ9 T cells improving the therapeutic response to phosphoantigen in preventing tumor growth in a humanized mouse lymphoma model (134). Pro-tumoral effects were observed in several other types of experimental cancers (135). In an organotypic culture model, carcinoma-associated fibroblasts (CAFs) were found to express high levels of IL-33 which promoted cancer invasive behavior of head and neck squamous cell carcinoma (HNSCC) cells (136). In gastric cancer, IL-33 exerted a pro-tumorigenic function inducing cancer cell invasion by stimulating the secretion of MMP-3 and IL-6 via ST2-ERK1/2 pathway (137) and activation of the JNK pathway (138) conferring chemotherapy resistance in vitro. Finally, in mouse models of cholangiocarcinoma (CCA), a malignant neoplasm of the biliary-duct system, administration of IL-33 was found to increase biliary tumorigenesis through an increase of IL-6 expression in tumor tissue (139) and to enhance cholangiocyte proliferation, by increasing the numbers of IL-13-producing ILC2s (140). The effects of IL-33 in experimental cancers are summarized in Table 1.
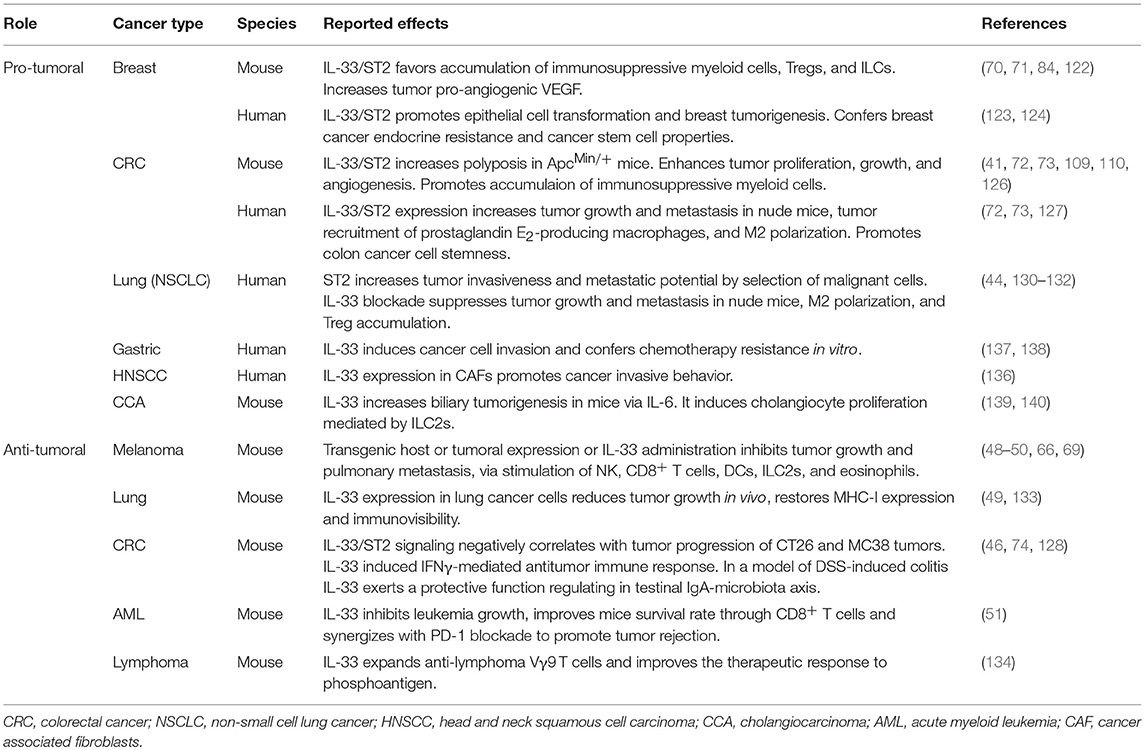
Table 1. Role of IL-33 in experimental tumors.
IL-33/ST2 as a Biomarker Predictive of Cancer Progression and Patients Survival
The recent literature regarding IL-33 involvement in tumorigenesis is controversial, since IL-33 seems to have dual, pro-inflammatory or protective, roles depending on the cellular and cytokine context. Some studies have shown a positive correlation between IL-33 expression in tumor tissue and a favorable prognosis in cancer patients. For example expression levels of IL-33 and ST2 were significantly down-regulated in both adenocarcinoma and squamous cell carcinoma of lung tissues when compared to adjacent normal lung controls (141). Furthermore, plasma IL-33 levels were elevated during the early stage of lung cancer and decreased with advanced cancer stages, probably due to lung volume reduction containing bronchial epithelium and vascular endothelium as sources of IL-33 (142). The observed decreases indicate that the expression levels of IL-33 are inversely associated with lung cancer progression (143). Of interest, in hepatocellular carcinoma resected tissues expression of IL-33 by intratumoral effector memory CD62L−KLRG1+CD107a+ CD8+ T cells was shown to be a prognostic marker for increased patients survival (144). Serum levels of IL-33 were significantly higher in patients with breast cancer compared to patients with benign breast diseases, so the local expression of IL-33 may be a marker for differentiating malignant from normal/benign tissues (145, 146). IL-33 expression in adjacent tissues also tends to be higher compared to normal tissues, suggesting that adjacent non-cancerous tissues may be similarly relevant to cancers in terms of anti-tumor immunity. Local IL-33 expression may also increase intratumoral accumulation of immunosuppressive lymphoid cells in patients with breast cancer (145, 146). Plasma sST2 levels and nuclear IL-33 expression were found to be increased in endothelial cells in bone marrow biopsies from patients with myeloproliferative neoplasms, whereas low/undetectable levels were found in healthy donors (147). It has been proposed that IL-33 induces the production of cytokines and growth factors that promote myelopoiesis and facilitates the development of leukemia by inducing and/or enhancing the proliferation of hematopoietic progenitors in the bone marrow microenvironment from patients with chronic myeloid leukemia (147).
In other clinical conditions, increased IL-33 expression was inversely correlated with the overall survival of cancer patients. Serum IL-33 levels were increased in renal cell carcinoma (RCC) patients compared to healthy volunteers (148). Such over-expression was associated with advanced tumor-lymph node-metastasis (TNM) stage, resulting in reduced survival and increased risk of recurrence in patients. Mechanistically, it has been shown that IL-33 enhances RCC cell growth in vivo and prevents chemotherapy-induced tumor apoptosis in vitro via JNK signaling activation in tumor cells (149). Similarly, both IL-33 and ST2 were up-regulated in ovarian tumors compared to normal ovary and ovarian benign tumors, and the expression levels were further increased in tumor tissues at the metastatic site (150). It has been proposed that IL-33/ST2 axis promotes ovarian cancer migration and metastasis through regulation of ERK and JNK signaling pathways (151). Furthermore, most head and neck squamous cell carcinoma (HNSCC) cases with a low invasion pattern grading score (IPGS) showed low or no expression of IL-33, whereas most HNSCC cases with high IPGS displayed abundant expression of IL-33 in CAFs and in cancer cells. This observation suggests a paracrine effect of IL-33 as a result of the crosstalk between tumor cells and surrounding stromal cells. Hence, CAFs overexpressing IL-33 promote the induction of epithelial-to-mesenchymal trans-differentiation, eventually leading to tumor progression and poor prognosis (136). Higher IL-33 expression was described in glioma tissue compared to normal brain tissues at both transcriptional and translational levels (152, 153). It has been proposed that IL-33 stimulates cell migration through the expression of matrix metalloproteinases (MMP2/MMP9) via the ST2/ NF-κB pathway, thereby promoting cell invasion and tumor growth (154). Higher expression of IL-33 and total ST2 (ST2L and sST2) have also been reported in colorectal cancer (CRC) tissues compared to adjacent normal tissues (127). This observation could be explained considering that inflammatory cytokines are important components of the CRC microenvironment and colon cancer progression is closely related to chronic inflammation. Increased IL-33 expression is observed in poorly differentiated human CRC cells, which is associated with poor survival in patients with metastatic colon cancer (73).
The relation between IL-33 and sST2 serum levels and the survival of patients suffering from liver cirrhosis (LC) and hepatocellular carcinoma (HCC) has not been determined yet. No significant difference in IL-33 serum levels was found in HCC compared to LC. IL-33 levels did not correlate with overall survival or liver function parameters, whereas sST2 levels were significantly elevated in LC and HCC patients, compared to healthy subjects, and were associated with overall survival of HCC. Therefore, its function remains to be clarified (155). Similarly, IL-33 protein levels were significantly lower in gastric cancer tissues than adjacent tissues. These levels were associated to the depth of tumor invasion and the morphology of the tumor, suggesting that IL-33 is involved in the process of inflammatory reaction in the development of gastric cancer, while it is not significantly associated with the overall survival of these patients (156).
Analysis of IL-33 Mutations and Isoforms in Cancer
IL-33 gene undergoes a certain number of somatic mutations during tumorigenesis. Depending on the exact location in the gene (and protein), the mutation may be putative of some key functional aberrancies that influence the IL-33 global functional properties (Figure 3). In general, IL-33 mutations occurred with very low frequencies in all tumors examined (0.072–1.391%). However, some mutations may putatively have a key impact on the functionality of IL-33. IL-33 protein is equipped with three main regions (Nuclear, Central, IL-1-like) with three specific binding and cleavage sequences in each of these domains (Figure 3). For example, there are 15 unique mutations in skin cancer patients, with a missense mutation (M52I at nucleotide 6250538 of the chromosome 9) targeting the chromatin binding site (R1). We hypothesize that this amino acid change potentially breaks the ability of IL-33 to bind DNA and thus to exert its regulatory functions. There are additionally 11 unique somatic missense mutations all affecting the IL-1-like domain of the protein (but outside the R3 sequence), that can putatively compromise the IL-33 binding ability to its specific receptor ST2 (3, 157). Another tumor that displays a mutation affecting the ability of IL-33 to bind the chromatin is the uterine cancer with the R48H amino acid change (Figure 3). Interestingly, the mutations E121K, H221Y, S225F, and H246Y, all occurring in the IL-1-like domain, are classified as deleterious in terms of IL-33 functional changes for skin cancers as evidenced by the cBioPortal database. Some of these mutations, like the amino acid change H221Y, are also shared with other tumors such as bladder cancer, thus denoting their significance. There is only one somatic missense alteration (G176S) occurring inside the R3 region, shared by colon and blood cancer patients (Figure 3). These mutations can putatively disrupt the cleavage site for caspase 3 and 7, thus increasing the IL-33 resistance to these proteases with key consequences in skin tumor survival and progression. Mutations in the cleavage site for inflammatory proteases (R2) occur only in colon and uterine tumors, with the A95V amino acid change (colon) and two mutations in uterine tumor (missense V101L and truncation S111).
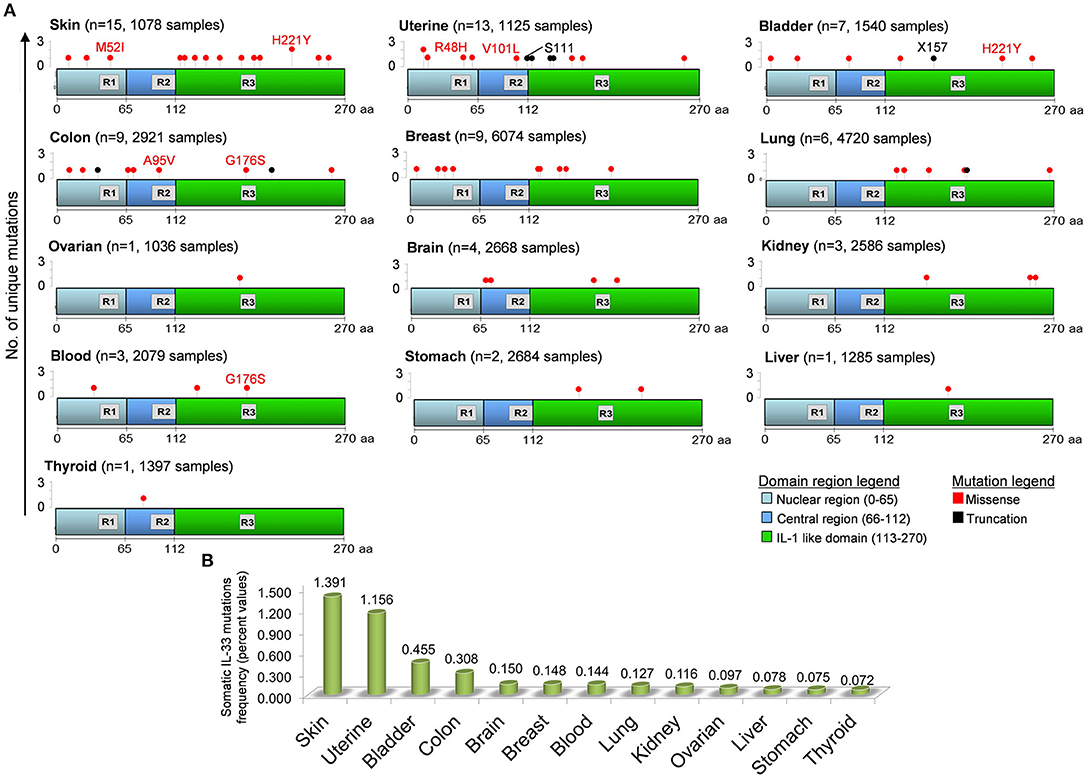
Figure 3. Distribution of unique somatic IL-33 gene mutations in tumors and in the domain regions of the IL-33 protein. (A) IL-33 gene mutations in each tumor have been obtained by inferring the publicly available cBioPortal database for analysis of large-scale cancer genomics data sets (http://www.cbioportal.org/). For each tumor type the number of mutations (n) and the total number of sample patients is depicted. The IL-33 gene mutation positions are aligned to the three principal regions of the IL-33 protein. R1, Chromatin binding domain (M45XLRSGXXI53); R2, Cleavage site for inflammatory Proteases (T90VECFAFGISGVQKYTRALHDSS112); R3, Cleavage site for caspase 3 and caspase 7 (D175GVDG179). Some missense (red text) and truncation (black text) mutations are depicted. (B) Somatic IL-33 gene mutation frequencies calculated by data analyzed in (A) and indicated for each tumor type.
As mentioned above, the IL-33 gene is equipped with a splicing system generating three different variants of the IL-33 protein (Figure 4A). The first isoform variant is composed by 7 exons, whereas the variants 2 and 3 contain 6 and 4 exons, respectively. The role and the extent of expression of each isoform in cancer is still a field of debate. Nevertheless, public databases such as IsoExpresso are now available, containing expression data of isoforms associated to thousands of human genes involved in cancer progression (158). We then extracted data about the expression of IL-33 isoform variants in different types of tumors and the normal counterparts (Figure 4B). The IsoExpresso database revealed a differential expression across the lesions and normal tissues or across the isoform variants of IL-33. Thyroid, liver, breast, and bladder cancers display a change in isoform expression when compared to normal counterparts. Indeed, variants 2 and 3 are highly expressed in these tumors compared to expression levels observed in normal tissues (Figure 4B). Of note, bladder cancer presents a splice site mutation (X157, Figure 3), and the presence of this mutation may probably be associated to the observed expression shift of the IL-33 isoform variants in this type of cancer (Figure 4). Indeed, in the tumor lesions the mainly expressed isoforms are variant 2 and 3, whereas the normal tissues express the variant 1 only (Figure 4B). This strongly suggests that this mutation affects the disappearing of IL-33 exon 4 (Figure 4A). In contrast, uterine tumor has been associated with a splice mutation (X115, close to the S111 truncation, Figure 3) not affecting the expression of IL-33 splice variants (Figure 4B).
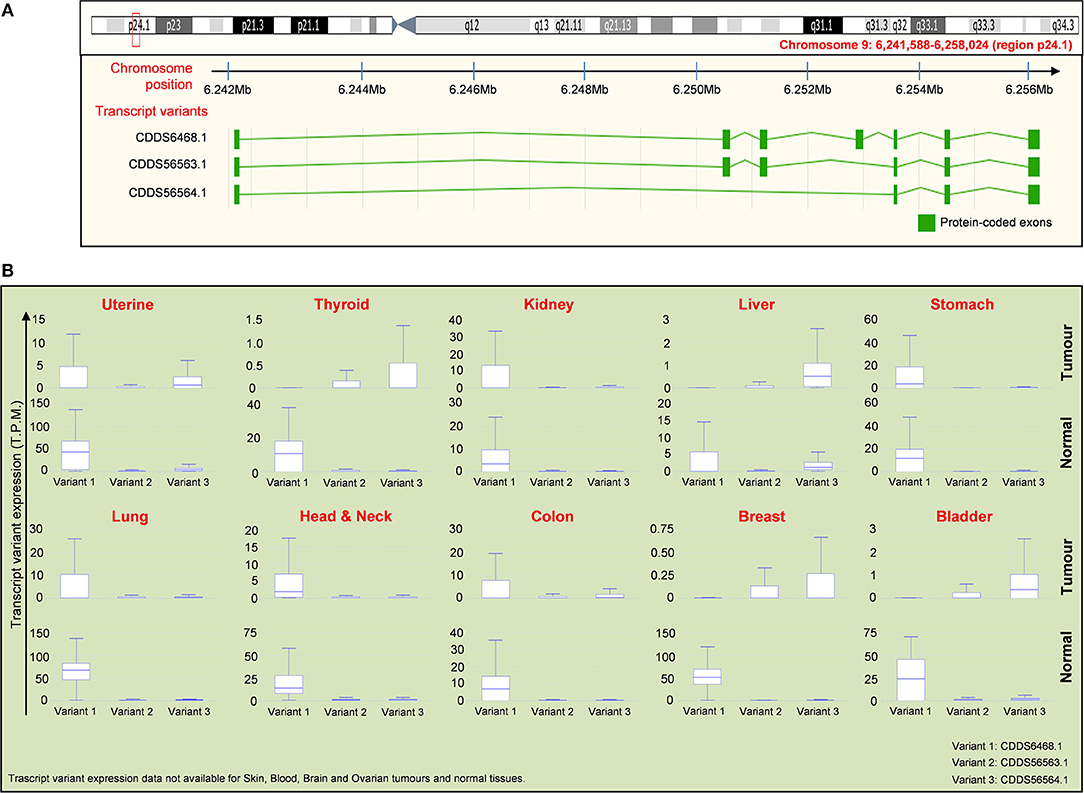
Figure 4. Distribution of IL-33 RNA transcript variants in tumor samples compared to normal tissues. (A) Schematic representation of the chromosome location (red box and text) of the IL-33 gene (upper panel) with the location of exons present in each indicated transcript variant (lower panel). Data are obtained by ENSEMBL genome browser (https://www.ensembl.org/). There are 3 isoforms of IL-33 gene, which are: NM_001199640 (CDDS56563.1), NM_001199641 (CDDS56564.1), NM_033439 (CDDS6468.1). (B) Expression of the IL-33 transcript variants indicated in (A) for each tumor compared to the normal tissues. Expression data (T.P.M, transcript per million) are obtained by interrogating the public database for Isoform expression resource analysis (Isoexpresso, http://wiki.tgilab.org/ISOexpresso/main.php). IL-33 transcript variant expression data are not available for skin, blood, brain, and ovarian cancers/normal tissues within Isoexpresso database. The predominant forms for brain and ovarian tumors are variants 1 whereas the predominant form expressed in skin tumors is the transcript variant 3 (resource: Mammalian Transcriptomic Database, http://mtd.cbi.ac.cn/).
Concluding Remarks
The role of IL-33 in tumorigenesis and cancer immunity remains controversial. The current literature indicate that IL-33 expression can be regulated during the progression of distinct types of cancers and that IL-33/ST2 signaling within the tumor microenvironment may differently contribute to tumorigenesis, promoting antitumor responses or mediating tumor growth or metastasis, depending on the nature of the malignant tissue. Further studies are needed to elucidate the role of this pathway at specific time points during cancer development as a possible diagnostic/prognostic marker for patients and to clarify whether IL-33/ST2 blockade may represent a valid approach for adjuvant therapies of established IL-33-dependent tumors.
It has been suggested that IL-33 has opposing effects in tumor immunity depending on its local concentration (121). In this respect, the tumor histotype may crucially determine the amount of IL-33 expressed. In addition, the presence of different IL-33 isoforms may play a crucial role. Thus, it may be possible that under steady-state conditions the homeostatic release of IL-33 by apoptotic epithelial cells may be regulated by caspase cleavage, thus limiting excessive inflammation. We envisage that, as in several chronic inflammatory diseases, also in tumors IL-33 could exist in several different forms as a result of post-translational processes, including intracellular and extracellular modification, or as a result of mRNA alternative splicing. There is compelling evidence that different forms and cleavage products of IL-33 can exert dissimilar biological activities. Unfortunately, the identification and the concentrations of the different forms of IL-33 in different tumors is largely unknown. Future studies should characterize the different forms of IL-33 in human and experimental tumors. Moreover, the mutation level of IL-33 gene observed in human solid tumors (Figure 3) may be associated with different isoforms and distinct stimulated antitumor immune responses. Lastly, since the new frontier of cancer immunotherapy is the employment of immune checkpoint inhibitors (i.e., against PD-1, PD-L1, CTLA-4) further studies are needed to clarify whether IL-33 conditioning may increase the therapeutic response to checkpoint blockade in cancer patients.
Author Contributions
GS, CA, and FM have conceived and designed the review. CA, CB, SA, MG, GV, GM, FM, and GS contributed intellectually and to the writing of the submitted version of the manuscript.
Funding
This work was supported in part by grants from AIRC IG 14297 to GS; Italian Ministry of Health RF-2011-02347120 to FM; Regione Campania CISI-Lab Project, CRèME Project, and TIMING Project to GM.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank all colleagues that have indirectly supported this work with ideas, discussion and encouragement. Special thanks to Fabrizio Fiorbianco for artwork.
References
1. Krumm B, Xiang Y, Deng J. Structural biology of the IL-1 superfamily: key cytokines in the regulation of immune and inflammatory responses. Protein Sci. (2014) 23:526–38. doi: 10.1002/pro.2441
2. Cayrol C, Girard J-P. Interleukin-33 (IL-33): a nuclear cytokine from the IL-1 family. Immunol Rev. (2018) 281:154–68. doi: 10.1111/imr.12619
3. Liew FY, Girard J-P, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. (2016) 16:676–89. doi: 10.1038/nri.2016.95
4. Carriere V, Roussel L, Ortega N, Lacorre D-A, Americh L, Aguilar L, et al. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci USA. (2007) 104:282–7. doi: 10.1073/pnas.0606854104
5. Moussion C, Ortega N, Girard J-P. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘Alarmin’? PLoS ONE (2008) 3:e3331. doi: 10.1371/journal.pone.0003331
6. Gajardo Carrasco T, Morales RA, Perez F, Terraza C, Yanez L, Campos-Mora M, et al. Alarmin' immunologists: IL-33 as a putative target for modulating T cell-dependent responses. Front Immunol. (2015) 6:232. doi: 10.3389/fimmu.2015.00232
7. Cassel SL, Joly S, Sutterwala FS. The NLRP3 inflammasome: a sensor of immune danger signals. Semin Immunol. (2009) 21:194–8. doi: 10.1016/j.smim.2009.05.002
8. Ali S, Nguyen DQ, Falk W, Martin MU. Caspase 3 inactivates biologically active full length interleukin-33 as a classical cytokine but does not prohibit nuclear translocation. Biochem Biophys Res Commun. (2010) 391:1512–6. doi: 10.1016/j.bbrc.2009.12.107
9. Cayrol C, Girard J-P. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci USA. (2009) 106:9021–6. doi: 10.1073/pnas.0812690106
10. Lüthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity (2009) 31:84–98. doi: 10.1016/j.immuni.2009.05.007
11. Cohen ES, Scott IC, Majithiya JB, Rapley L, Kemp BP, England E, et al. Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat Commun. (2015) 6:8327. doi: 10.1038/ncomms9327
12. Gordon ED, Simpson LJ, Rios CL, Ringel L, Lachowicz-Scroggins ME, Peters MC, et al. Alternative splicing of interleukin-33 and type 2 inflammation in asthma. Proc Natl Acad Sci USA. (2016) 113:8765–70. doi: 10.1073/pnas.1601914113
13. Hong J, Bae S, Jhun H, Lee S, Choi J, Kang T, et al. Identification of constitutively active interleukin 33 (IL-33) splice variant. J Biol Chem. (2011) 286:20078–86. doi: 10.1074/jbc.M111.219089
14. Tsuda H, Komine M, Karakawa M, Etoh T, Tominaga S-I, Ohtsuki M. Novel splice variants of IL-33: differential expression in normal and transformed cells. J Invest Dermatol. (2012) 132:2661–4. doi: 10.1038/jid.2012.180
15. Lefrançais E, Roga S, Gautier V, Gonzalez-de-Peredo A, Monsarrat B, Girard J-P, et al. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci USA. (2012) 109:1673–8. doi: 10.1073/pnas.1115884109
16. Lefrançais E, Duval A, Mirey E, Roga S, Espinosa E, Cayrol C, et al. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc Natl Acad Sci USA. (2014) 111:15502–7. doi: 10.1073/pnas.1410700111
17. Cayrol C, Duval A, Schmitt P, Roga S, Camus M, Stella A, et al. Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat Immunol. (2018) 19:375–85. doi: 10.1038/s41590-018-0067-5
18. Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage–CD25+CD44hi lymphoid cells mediate innate type-2 immunity and allergic inflammation in the lungs. J Immunol. (2012) 188:1503–13. doi: 10.4049/jimmunol.1102832
19. Smithgall MD, Comeau MR, Park Yoon B-R, Kaufman D, Armitage R, Smith DE. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK Cells. Int Immunol. (2008) 20:1019–30. doi: 10.1093/intimm/dxn060
20. Varricchi G, Pecoraro A, Marone G, Criscuolo G, Spadaro G, Genovese A, et al. Thymic stromal lymphopoietin isoforms, inflammatory disorders, and cancer. Front Immunol. (2018) 9:1595. doi: 10.3389/fimmu.2018.01595
21. Villarreal DO, Weiner DB. IL-33 isoforms: their future as vaccine adjuvants? Expert Rev Vaccines (2015) 14:489–92. doi: 10.1586/14760584.2015.1011135
22. Luzina IG, Pickering EM, Kopach P, Kang PH, Lockatell V, Todd NW, et al. Full-length IL-33 promotes inflammation but not Th2 response in vivo in an ST2-independent fashion. J Immunol. (2012) 189:403–10. doi: 10.4049/jimmunol.1200259
23. Villarreal DO, Wise MC, Walters JN, Reuschel EL, Choi MJ, Obeng-Adjei N, et al. Alarmin IL-33 acts as an immunoadjuvant to enhance antigen-specific tumor immunity. Cancer Res. (2014) 74:1789–800. doi: 10.1158/0008-5472.CAN-13-2729
24. Martin MU. Special aspects of interleukin-33 and the IL-33 receptor complex. Semin Immunol. (2013) 25:449–57. doi: 10.1016/j.smim.2013.10.006
25. Liu X, Hammel M, He Y, Tainer JA, Jeng US, Zhang L, et al. Structural insights into the interaction of IL-33 with its receptors. Proc Natl Acad Sci USA. (2013) 110:14918–23. doi: 10.1073/pnas.1308651110
26. Hayakawa H, Hayakawa M, Kume A, Tominaga S. Soluble ST2 blocks interleukin-33 signaling in allergic airway inflammation. J Biol Chem. (2007) 282:26369–80. doi: 10.1074/jbc.M704916200
27. Leung BP, Xu D, Culshaw S, McInnes IB, Liew FY. A novel therapy of murine collagen-induced arthritis with soluble T1/ST2. J Immunol. (2004) 173:145–50. doi: 10.4049/jimmunol.173.1.145
28. Cayrol C, Girard J-P. IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol. (2014) 31:31–7. doi: 10.1016/j.coi.2014.09.004
29. Bonilla WV, Fröhlich A, Senn K, Kallert S, Fernandez M, Johnson S, et al. The alarmin interleukin-33 drives protective antiviral CD8+ T cell responses. Science (2012) 335:984–9. doi: 10.1126/science.1215418
30. Bourgeois E, Van Linh P, Samson M, Diem Sv, Barra A, Roga Sp, et al. The pro-Th2 cytokine IL-33 directly interacts with invariant NKT and NK cells to induce IFN-gamma production. Eur J Immunol. (2009) 39:1046–55. doi: 10.1002/eji.200838575
31. Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon EK, et al. Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. J Exp Med. (2010) 207:651–67. doi: 10.1084/jem.20091921
32. Hombach A, Kohler H, Rappl G, Abken H. Human CD4+ T cells lyse target cells via granzyme/perforin upon circumvention of MHC class II restriction by an antibody-like immunoreceptor. J Immunol. (2006) 177:5668–75. doi: 10.4049/jimmunol.177.8.5668
33. Komai-Koma M, Wang E, Kurowska-Stolarska M, Li D, McSharry C, Xu D. Interleukin-33 promoting Th1 lymphocyte differentiation dependents on IL-12. Immunobiology (2016) 221:412–7. doi: 10.1016/j.imbio.2015.11.013
34. Blom L, Poulsen BC, Jensen BM, Hansen A, Poulsen LK. IL-33 induces IL-9 production in human CD4+ T cells and basophils. PLoS ONE (2011) 6:e21695. doi: 10.1371/journal.pone.0021695
35. Lu Y, Hong S, Li H, Park J, Hong B, Wang L, et al. Th9 cells promote antitumor immune responses in vivo. J Clin Invest. (2012) 122:4160–71. doi: 10.1172/JCI65459
36. Ramadan A, Griesenauer B, Adom D, Kapur R, Hanenberg H, Liu C, et al. Specifically differentiated T cell subset promotes tumor immunity over fatal immunity. J Exp Med. (2017) 214:3577–96. doi: 10.1084/jem.20170041
37. Peine M, Marek RM, Löhning M. IL-33 in T cell differentiation, function, and immune homeostasis. Trends Immunol. (2016) 37:321–33. doi: 10.1016/j.it.2016.03.007
38. Griesenauer B, Paczesny S. The ST2/IL-33 axis in immune cells during inflammatory diseases. Front Immunol. (2017) 8:475. doi: 10.3389/fimmu.2017.00475
39. Matta BM, Lott JM, Mathews LR, Liu Q, Rosborough BR, Blazar BR, et al. IL-33 is an unconventional alarmin that stimulates IL-2 secretion by dendritic cells to selectively expand IL-33R/ST2(+) regulatory T cells. J Immunol. (2014) 193:4010–20. doi: 10.4049/jimmunol.1400481
40. Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, et al. The alarmin IL-33 promotes regulatory T cell function in the intestine. Nature (2014) 513:564–8. doi: 10.1038/nature13577
41. He Z, Chen L, Souto FO, Canasto-Chibuque C, Bongers G, Deshpande M, et al. Epithelial-derived IL-33 promotes intestinal tumorigenesis in Apc(Min/+) mice. Sci Rep. (2017) 7:5520. doi: 10.1038/s41598-017-05716-z
42. Meinicke H, Bremser A, Brack M, Akeus P, Pearson C, Bullers S, et al. Tumour-associated changes in intestinal epithelial cells cause local accumulation of KLRG1+ GATA3+ regulatory T cells in mice. Immunology (2017) 152:74–88. doi: 10.1111/imm.12750
43. Zhou Y, Ji Y, Wang H, Zhang H, Zhou H. IL-33 promotes the development of colorectal cancer through inducing tumor-infiltrating ST2L+ regulatory T Cells in mice. Technol Cancer Res Treat. (2018) 17:1533033818780091. doi: 10.1177/1533033818780091
44. Wang K, Shan S, Yang Z, Gu X, Wang Y, Wang C, et al. IL-33 blockade suppresses tumor growth of human lung cancer through direct and indirect pathways in a preclinical model. Oncotarget (2017) 8:68571–82. doi: 10.18632/oncotarget.19786
45. Lim Hui X, Choi S, Cho D, Kim Tae S. IL-33 inhibits the differentiation and immunosuppressive activity of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. Immunol Cell Biol. (2017) 95:99–107. doi: 10.1038/icb.2016.72
46. Eissmann MF, Dijkstra C, Wouters MA, Baloyan D, Mouradov D, Nguyen PM, et al. Interleukin 33 signaling restrains sporadic colon cancer in an interferon-γ-dependent manner. Cancer Immunol Res. (2018) 6:409–21. doi: 10.1158/2326-6066.CIR-17-0218
47. Yang Q, Li G, Zhu Y, Liu L, Chen E, Turnquist H, et al. IL-33 synergizes with TCR and IL-12 signaling to promote the effector function of CD8+ T cells. Eur J Immunol. (2011) 41:3351–60. doi: 10.1002/eji.201141629
48. Gao K, Li X, Zhang L, Bai L, Dong W, Shi G, et al. Transgenic expression of IL-33 activates CD8+ T cells and NK cells and inhibits tumor growth and metastasis in mice. Cancer Lett. (2013) 335:463–71. doi: 10.1016/j.canlet.2013.03.002
49. Gao X, Wang X, Yang Q, Zhao X, Wen W, Li G, et al. Tumoral expression of IL-33 inhibits tumor growth and modifies the tumor microenvironment through CD8+ T and NK cells. J Immunol. (2015) 194:438–45. doi: 10.4049/jimmunol.1401344
50. Dominguez D, Ye C, Geng Z, Chen S, Fan J, Qin L, et al. Exogenous IL-33 restores dendritic cell activation and maturation in established cancer. J Immunol. (2017) 198:1365–75. doi: 10.4049/jimmunol.1501399
51. Qin L, Dominguez D, Chen S, Fan J, Long A, Zhang M, et al. Exogenous IL-33 overcomes T cell tolerance in murine acute myeloid leukemia. Oncotarget (2016) 7:61069–80. doi: 10.18632/oncotarget.11179
52. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol. (2013) 13:145–9. doi: 10.1038/nri3365
53. Harly C, Cam M, Kaye J, Bhandoola A. Development and differentiation of early innate lymphoid progenitors. J Exp Med. (2018) 215:249–62. doi: 10.1084/jem.20170832
54. Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature (2010) 463:540–4. doi: 10.1038/nature08636
55. Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. (2011) 12:1055–62. doi: 10.1038/ni.2104
56. Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DM, Artis D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci USA. (2015) 112:10762–7. doi: 10.1073/pnas.1509070112
57. Fallon PG, Ballantyne SJ, Mangan NE, Barlow JL, Dasvarma A, Hewett DR, et al. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J Exp Med. (2006) 203:1105–16. doi: 10.1084/jem.20051615
58. Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity (2014) 40:425–35. doi: 10.1016/j.immuni.2014.01.011
59. Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity (2012) 37:634–48. doi: 10.1016/j.immuni.2012.06.020
60. Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, et al. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med. (2013) 5:170ra16. doi: 10.1126/scitranslmed.3005374
61. Dancescu M, Rubio-Trujillo M, Biron G, Bron D, Delespesse G, Sarfati M. Interleukin 4 protects chronic lymphocytic leukemic B cells from death by apoptosis and upregulates Bcl-2 expression. J Exp Med. (1992) 176:1319–26. doi: 10.1084/jem.176.5.1319
62. Bie Q, Zhang P, Su Z, Zheng D, Ying X, Wu Y, et al. Polarization of ILC2s in peripheral blood might contribute to immunosuppressive microenvironment in patients with gastric cancer. J Immunol Res. (2014) 2014:923135. doi: 10.1155/2014/923135
63. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. (2011) 12:1045–54. doi: 10.1038/ni.2131
64. Zaiss DM, van Loosdregt J, Gorlani A, Bekker CP, Grone A, Sibilia M, et al. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity (2013) 38:275–84. doi: 10.1016/j.immuni.2012.09.023
65. Ikutani M, Yanagibashi T, Ogasawara M, Tsuneyama K, Yamamoto S, Hattori Y, et al. Identification of innate IL-5-producing cells and their role in lung eosinophil regulation and antitumor immunity. J Immunol. (2012) 188:703–13. doi: 10.4049/jimmunol.1101270
66. Kim J, Kim W, Moon UJ, Kim HJ, Choi HJ, Sin JI, et al. Intratumorally establishing type 2 innate lymphoid cells blocks tumor growth. J Immunol. (2016) 196:2410–23. doi: 10.4049/jimmunol.1501730
67. Liang Y, Jie Z, Hou L, Yi P, Wang W, Kwota Z, et al. IL-33 promotes innate IFN-g production and modulates dendritic cell response in LCMV-induced hepatitis in mice. Eur J Immunol. (2015) 45:3052–63. doi: 10.1002/eji.201545696
68. Nabekura T, Girard J-P, Lanier LL. IL-33 receptor ST2 amplifies the expansion of natural killer cells and enhances host defense during mouse cytomegalovirus infection. J Immunol. (2015) 194:5948–52. doi: 10.4049/jimmunol.1500424
69. Lucarini V, Ziccheddu G, Macchia I, La Sorsa V, Peschiaroli F, Buccione C, et al. IL-33 restricts tumor growth and inhibits pulmonary metastasis in melanoma-bearing mice through eosinophils. Oncoimmunology (2017) 6:e1317420. doi: 10.1080/2162402X.2017.1317420
70. Jovanovic I, Radosavljevic G, Mitrovic M, Lisnic Juranic V, McKenzie ANJ, Arsenijevic N, et al. ST2 deletion enhances innate and acquired immunity to murine mammary carcinoma. Eur J Immunol. (2011) 41:1902–12. doi: 10.1002/eji.201141417
71. Jovanovic IP, Pejnovic NN, Radosavljevic GD, Pantic JM, Milovanovic MZ, Arsenijevic NN, et al. Interleukin-33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int J Cancer (2014) 134:1669–82. doi: 10.1002/ijc.28481
72. Akimoto M, Maruyama R, Takamaru H, Ochiya T, Takenaga K. Soluble IL-33 receptor sST2 inhibits colorectal cancer malignant growth by modifying the tumour microenvironment. Nat Commun. (2016) 7:13589. doi: 10.1038/ncomms13589
73. Fang M, Li Y, Huang K, Qi S, Zhang J, Zgodzinski W, et al. IL33 Promotes colon cancer cell stemness via JNK activation and macrophage recruitment. Cancer Res. (2017) 77:2735–45. doi: 10.1158/0008-5472.CAN-16-1602
74. O'Donnell C, Mahmoud A, Keane J, Murphy C, White D, Carey S, et al. An antitumorigenic role for the IL-33 receptor, ST2L, in colon cancer. Br J Cancer (2016) 114:37–43. doi: 10.1038/bjc.2015.433
75. Yang Y, Andersson P, Hosaka K, Zhang Y, Cao R, Iwamoto H, et al. The PDGF-BB-SOX7 axis-modulated IL-33 in pericytes and stromal cells promotes metastasis through tumour-associated macrophages. Nat Commun. (2016) 7:11385. doi: 10.1038/ncomms11385
76. Ariyoshi W, Okinaga T, Chaweewannakorn W, Akifusa S, Nisihara T. Mechanisms involved in enhancement of matrix metalloproteinase-9 expression in macrophages by interleukin-33. J Cell Physiol. (2017) 232:3481–95. doi: 10.1002/jcp.25809
77. Rank MA, Kobayashi T, Kozaki H, Bartemes KR, Squillace DL, Kita H. IL-33-activated dendritic cells induce an atypical TH2-type response. J Allergy Clin Immunol. (2009) 123:1047–54. doi: 10.1016/j.jaci.2009.02.026
78. Gabriele L, Schiavoni G, Mattei F, Sanchez M, Sestili P, Butteroni C, et al. Novel allergic asthma model demonstrates ST2-dependent dendritic cell targeting by cypress pollen. J Allergy Clin Immunol. (2013) 132:686–95. doi: 10.1016/j.jaci.2013.02.037
79. Besnard A-G, Togbe D, Guillou N, Erard F, Quesniaux V, Ryffel B. IL-33-activated dendritic cells are critical for allergic airway inflammation. Eur J Immunol. (2011) 41:1675–86. doi: 10.1002/eji.201041033
80. Kurokawa M, Matsukura S, Kawaguchi M, Ieki K, Suzuki S, Watanabe S, et al. Interleukin-33-activated dendritic cells induce the production of thymus and activation-regulated chemokine and macrophage-derived chemokine. Int Arch Allergy Immunol. (2013) 161 (Suppl. 2):52–7. doi: 10.1159/000350363
81. Su Z, Lin J, Lu F, Zhang X, Zhang L, Gandhi NB, et al. Potential autocrine regulation of interleukin-33/ST2 signaling of dendritic cells in allergic inflammation. Mucosal Immunol. (2013) 6:921–30. doi: 10.1038/mi.2012.130
82. Mayuzumi N, Matsushima H, Takashima A. IL-33 promotes DC development in BM culture by triggering GM-CSF production. Eur J Immunol. (2009) 39:3331–42. doi: 10.1002/eji.200939472
83. Tcyganov E, Mastio J, Chen E, Gabrilovich DI. Plasticity of myeloid-derived suppressor cells in cancer. Curr Opin Immunol. (2018) 51:76–82. doi: 10.1016/j.coi.2018.03.009
84. Xiao P, Wan X, Cui B, Liu Y, Qiu C, Rong J, et al. Interleukin 33 in tumor microenvironment is crucial for the accumulation and function of myeloid-derived suppressor cells. Oncoimmunology (2015) 5:e1063772. doi: 10.1080/2162402X.2015.1063772
85. Foley SC, Hamid Q. Images in allergy and immunology: neutrophils in asthma. J Allergy Clin Immunol. (2007) 119:1282–6. doi: 10.1016/j.jaci.2007.02.006
86. Sun B, Zhu L, Tao Y, Sun H-X, Li Y, Wang P, et al. Characterization and allergic role of IL-33-induced neutrophil polarization. Cell Mol Immunol. (2018) 15:782–93. doi: 10.1038/cmi.2017.163
87. Liang Y, Yi P, Yuan DMK, Jie Z, Kwota Z, Soong L, et al. IL-33 induces immunosuppressive neutrophils via a type 2 innate lymphoid cell/IL-13/STAT6 axis and protects the liver against injury in LCMV infection-induced viral hepatitis. Cell Mol Immunol. (2018). doi: 10.1038/cmi.2017.147. [Epub ahead of print].
88. Guabiraba R, Besnard AG, Menezes GB, Secher T, Jabir MS, Amaral SS, et al. IL-33 targeting attenuates intestinal mucositis and enhances effective tumour chemotherapy in mice. Mucosal Immunol. (2014) 7:1079–93. doi: 10.1038/mi.2013.124
89. Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N, et al. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol. (2008) 20:791–800. doi: 10.1093/intimm/dxn037
90. Johnston LK, Bryce PJ. Understanding interleukin 33 and its roles in eosinophil development. Front Med (Lausanne) (2017) 4:51. doi: 10.3389/fmed.2017.00051
91. Willebrand R, Voehringer D. IL-33-Induced cytokine secretion and survival of mouse eosinophils is promoted by autocrine GM-CSF. PLoS ONE (2016) 11:e0163751. doi: 10.1371/journal.pone.0163751
92. Carretero R, Sektioglu IM, Garbi N, Salgado OC, Beckhove P, Hammerling GJ. Eosinophils orchestrate cancer rejection by normalizing tumor vessels and enhancing infiltration of CD8(+) T cells. Nat Immunol. (2015) 16:609–17. doi: 10.1038/ni.3159
93. Cherry WB, Yoon J, Bartemes KR, Iijima K, Kita H. A novel IL-1 family cytokine, IL-33, potently activates human eosinophils. J Allergy Clin Immunol. (2008) 121:1484. doi: 10.1016/j.jaci.2008.04.005
94. Suzukawa M, Koketsu R, Iikura M, Nakae S, Matsumoto K, Nagase H, et al. Interleukin-33 enhances adhesion, CD11b expression and survival in human eosinophils. Lab Invest. (2008) 88:1245–53. doi: 10.1038/labinvest.2008.82
95. Cormier SA, Taranova AG, Bedient C, Nguyen T, Protheroe C, Pero R, et al. Pivotal advance: eosinophil infiltration of solid tumors is an early and persistent inflammatory host response. J Leukoc Biol. (2006) 79:1131–9. doi: 10.1189/jlb.0106027
96. Caruso RA, Parisi A, Quattrocchi E, Scardigno M, Branca G, Parisi C, et al. Ultrastructural descriptions of heterotypic aggregation between eosinophils and tumor cells in human gastric carcinomas. Ultrastruct Pathol. (2011) 35:145–9. doi: 10.3109/01913123.2011.578233
97. Costain DJ, Guha AK, Liwski RS, Lee TDG. Murine hypodense eosinophils induce tumour cell apoptosis by a granzyme B-dependent mechanism. Cancer Immunol Immunother. (2001) 50:293–9. doi: 10.1007/PL00006690
98. Legrand F, Driss V, Delbeke M, Loiseau S, Hermann E, Dombrowicz D, et al. Human eosinophils exert TNF-alpha and granzyme A-mediated tumoricidal activity toward colon carcinoma cells. J Immunol. (2010) 185:7443–51. doi: 10.4049/jimmunol.1000446
99. Varricchi G, Galdiero MR, Loffredo S, Lucarini V, Marone G, Mattei F, et al. Eosinophils: the unsung heroes in cancer? Oncoimmunology (2018) 7:e1393134. doi: 10.1080/2162402X.2017.1393134
100. Iikura M, Suto H, Kajiwara N, Oboki K, Ohno T, Okayama Y, et al. IL-33 can promote survival, adhesion and cytokine production in human mast cells. Lab Invest. (2007) 87:971–8. doi: 10.1038/labinvest.3700663
101. Silver MR, Margulis A, Wood N, Goldman SJ, Kasaian M, Chaudhary D. IL-33 synergizes with IgE-dependent and IgE-independent agents to promote mast cell and basophil activation. Inflamm Res. (2010) 59:207–18. doi: 10.1007/s00011-009-0088-5
102. Theoharides TC, Zhang B, Kempuraj D, Tagen M, Vasiadi M, Angelidou A, et al. IL-33 augments substance P-induced VEGF secretion from human mast cells and is increased in psoriatic skin. Proc Natl Acad Sci USA. (2010) 107:4448–53. doi: 10.1073/pnas.1000803107
103. Varricchi G, Galdiero MR, Loffredo S, Marone G, Iannone R, Marone G, et al. Are mast cells MASTers in cancer? Front Immunol. (2017) 8:424. doi: 10.3389/fimmu.2017.00424
104. de Paulis A, Prevete N, Fiorentino I, Rossi FW, Staibano S, Montuori N, et al. Expression and functions of the vascular endothelial growth factors and their receptors in human basophils. J Immunol. (2006) 177:7322–31. doi: 10.4049/jimmunol.177.10.7322
105. Marone G, Varricchi G, Loffredo S, Granata F. Mast cells and basophils in inflammatory and tumor angiogenesis and lymphangiogenesis. Eur J Immunol. (2016) 778:146–51. doi: 10.1016/j.ejphar.2015.03.088
106. Rivellese F, Suurmond J, Habets K, Dorjée Annemarie L, Ramamoorthi N, Townsend Michael J, et al. Ability of interleukin-33- and immune complex-triggered activation of human mast cells to down-regulate monocyte-mediated immune responses. Arthritis Rheumatol. (2015) 67:2343–53. doi: 10.1002/art.39192
107. Joulia R, L'Faqihi F-E, Valitutti S, Espinosa E. IL-33 fine tunes mast cell degranulation and chemokine production at the single-cell level. J Allergy Clin Immunol. (2017) 140:497–509.e10. doi: 10.1016/j.jaci.2016.09.049
108. Byrne SN, Beaugie C, O'Sullivan C, Leighton S, Halliday GM. The immune-modulating cytokine and endogenous alarmin interleukin-33 is upregulated in skin exposed to inflammatory UVB radiation. Am J Pathol. (2011) 179:211–22. doi: 10.1016/j.ajpath.2011.03.010
109. Maywald RL, Doerner SK, Pastorelli L, De Salvo C, Benton SM, Dawson EP, et al. IL-33 activates tumor stroma to promote intestinal polyposis. Proc Natl Acad Sci USA. (2015) 112:E2487–96. doi: 10.1073/pnas.1422445112
110. Mertz KD, Mager LF, Wasmer MH. The IL-33/ST2 pathway contributes to intestinal tumorigenesis in humans and mice. Oncoimmunology (2015) 26:e1062966. doi: 10.1080/2162402X.2015.1062966
111. Gounaris E, Erdman SE, Restaino C, Gurish MF, Friend DS, Gounari F, et al. Mast cells are an essential hematopoietic component for polyp development. Proc Natl Acad Sci USA. (2007) 104:19977. doi: 10.1073/pnas.0704620104
112. Blatner NR, Bonertz A, Beckhove P, Cheon EC, Krantz SB, Strouch M, et al. In colorectal cancer mast cells contribute to systemic regulatory T-cell dysfunction. Proc Natl Acad Sci USA. (2010) 107:6430–5. doi: 10.1073/pnas.0913683107
113. Cheon EC, Khazaie K, Khan MW, Strouch MJ, Krantz SB, Phillips J, et al. Mast cell 5-lipoxygenase activity promotes intestinal polyposis in APCΔ468 mice. Cancer Res. (2011) 71:1627–36. doi: 10.1158/0008-5472.CAN-10-1923
114. Suzukawa M, Iikura M, Koketsu R, Nagase H, Tamura C, Komiya A, et al. An IL-1 cytokine member, IL-33, induces human basophil activation via its ST2 receptor. J Immunol. (2008) 181:5981–9. doi: 10.4049/jimmunol.181.9.5981
115. Pecaric-Petkovic T, Didichenko SA, Kaempfer S, Spiegl N, Dahinden CA. Human basophils and eosinophils are the direct target leukocytes of the novel IL-1 family member IL-33. Blood (2009) 113:1526–34. doi: 10.1182/blood-2008-05-157818
116. Schneider E, Petit-Bertron A-F, Bricard R, Levasseur Ml, Ramadan A, Girard J-P, et al. IL-33 activates unprimed murine basophils directly in vitro and induces their in vivo expansion indirectly by promoting hematopoietic growth factor production. J Immunol. (2009) 183:3591–7. doi: 10.4049/jimmunol.0900328
117. Rivellese F, Suurmond J, de Paulis A, Marone G, Huizinga Tom WJ, Toes René EM. IgE and IL-33-mediated triggering of human basophils inhibits TLR4-induced monocyte activation. Eur J Immunol. (2014) 44:3045–55. doi: 10.1002/eji.201444731
118. Sektioglu IM, Carretero R, Bulbuc N, Bald T, Tüting T, Rudensky AY, et al. Basophils promote tumor rejection via chemotaxis and infiltration of CD8+ T cells. Cancer Res. (2017) 77:291–302. doi: 10.1158/0008-5472.CAN-16-0993
119. Wei Y, Zhang X, Wang G, Zhou Y, Luo M, Wang S, et al. The impacts of pretreatment circulating eosinophils and basophils on prognosis of stage I-III colorectal cancer. Asia Pac J Clin Oncol. (2018). 14:e243–51. doi: 10.1111/ajco.12871
120. De Monte L, Wörmann S, Brunetto E, Heltai S, Magliacane G, Reni M, et al. Basophil recruitment into tumor-draining lymph nodes correlates with Th2 inflammation and reduced survival in pancreatic cancer patients. Cancer Res. (2016) 76:1792–803. doi: 10.1158/0008-5472.CAN-15-1801-T
121. Lu B, Yang M, Wang Q. Interleukin-33 in tumorigenesis, tumor immune evasion, and cancer immunotherapy. J Mol Med (Berl). (2016) 94:535–43. doi: 10.1007/s00109-016-1397-0
122. Milosavljevic MZ, Jovanovic IP, Pejnovic NN, Mitrovic SLJ, Arsenijevic NN, Simovic Markovic BJ, et al. Deletion of IL-33R attenuates VEGF expression and enhances necrosis in mammary carcinoma. Oncotarget (2016) 7:18106–15. doi: 10.18632/oncotarget.7635
123. Kim JY, Lim SC, Kim G, Yun HJ, Ahn SG, Choi HS. Interleukin-33/ST2 axis promotes epithelial cell transformation and breast tumorigenesis via upregulation of COT activity. Oncogene (2015) 34:4928–38. doi: 10.1038/onc.2014.418
124. Hu H, Sun J, Wang C, Bu X, Liu X-p, Mao YP, et al. IL-33 facilitates endocrine resistance of breast cancer by inducing cancer stem cell properties. Biochem Biophys Res Commun. (2017) 485:643–50. doi: 10.1016/j.bbrc.2017.02.080
125. Akimoto M, Takenaga K. Role of the IL-33/ST2L axis in colorectal cancer progression. Cell Immunol. (2018). doi: 10.1016/j.cellimm.2017.12.014. [Epub ahead of print].
126. Zhang Y, Davis C, Shah S, Hughes D, Ryan JC, Altomare D, et al. IL-33 promotes growth and liver metastasis of colorectal cancer in mice by remodeling the tumor microenvironment and inducing angiogenesis. Mol Carcinogen. (2017) 56:272–87. doi: 10.1002/mc.22491
127. Liu X, Zhu L, Lu X, Bian H, Wu X, Yang W, et al. IL-33/ST2 pathway contributes to metastasis of human colorectal cancer. Biochem Biophys Res Commun. (2014) 453:486–92. doi: 10.1016/j.bbrc.2014.09.106
128. Malik A, Sharma D, Zhu Q, Karki R, Guy CS, Vogel P, et al. IL-33 regulates the IgA-microbiota axis to restrain IL-1a-dependent colitis and tumorigenesis. J Clin Invest. (2016) 126:4469–81. doi: 10.1172/JCI88625
129. Drake LY, Kita H. IL-33: Biological properties, functions and roles in airway disease. Immunol Rev. (2017) 278:173–84. doi: 10.1111/imr.12552
130. Akimoto M, Hayashi JI, Nakae S, Saito H, Takenaga K. Interleukin-33 enhances programmed oncosis of ST2L-positive low-metastatic cells in the tumour microenvironment of lung cancer. Cell Death Dis. (2016) 7:e2057. doi: 10.1038/cddis.2015.418
131. Yang Z, Gao X, Wang J, Xu L, Zheng Y, Xu Y. Interleukin-33 enhanced the migration and invasiveness of human lung cancer cells. Onco Targets Ther. (2018) 11:843–9. doi: 10.2147/OTT.S155905
132. Wang C, Chen Z, Bu X, Han Y, Shan S, Ren T, et al. IL-33 signaling fuels outgrowth and metastasis of human lung cancer. Biochem Biophys Res Commun. (2016) 479:461–8. doi: 10.1016/j.bbrc.2016.09.081
133. Saranchova I, Han J, Huang H, Fenninger F, Choi KB, Munro L, et al. Discovery of a metastatic immune escape mechanism initiated by the loss of expression of the tumour biomarker interleukin-33. Sci Rep. (2016) 6:30555. doi: 10.1038/srep30555
134. Duault C, Betous D, Bezombes C, Roga S, Cayrol C, Girard J-P, et al. IL-33-expanded human Vγ9Vδ2 cells have anti-lymphoma effect in a mouse tumor model. Eur J Immunol. (2017) 47:2137–41. doi: 10.1002/eji.201747093
135. Wasmer M-H, Krebs P. The role of IL-33-dependent inflammation in the tumor microenvironment. Front Immunol. (2017) 7:682. doi: 10.3389/fimmu.2016.00682
136. Chen S-F, Nieh S, Jao S-W, Wu M-Z, Liu C-L, Chang Y-C, et al. The paracrine effect of cancerassociated fibroblast-induced interleukin-33 regulates the invasiveness of head and neck squamous cell carcinoma. J Pathol. (2013) 231:180–9. doi: 10.1002/path.4226
137. Yu X, Hu Z, Shen X, Dong L, Zhou W, Hu W. IL-33 promotes gastric cancer cell invasion and migration via ST2-ERK1/2 pathway. Dig Dis Sci. (2015) 60:1265–72. doi: 10.1007/s10620-014-3463-1
138. Ye X, Zhao Y, Weng G, Chen Y, Wei X, Shao J, et al. IL-33-induced JNK pathway activation confers gastric cancer chemotherapy resistance. Oncol Rep. (2015) 33:2746–52. doi: 10.3892/or.2015.3898
139. Yamada D, Rizvi S, Razumilava N, Bronk SF, Davila JI, Champion MD, et al. IL-33 facilitates oncogene induced cholangiocarcinoma in mice by an IL-6 sensitive mechanism. Hepatology (2015) 61:1627–42. doi: 10.1002/hep.27687
140. Li J, Razumilava N, Gores GJ, Walters S, Mizuochi T, Mourya R, et al. Biliary repair and carcinogenesis are mediated by IL-33-dependent cholangiocyte proliferation. J Clin Invest. (2014) 124:3241–51. doi: 10.1172/JCI73742
141. Yang M, Feng Y, Yue C, Xu B, Chen L, Jiang J, et al. Lower expression level of IL-33 is associated with poor prognosis of pulmonary adenocarcinoma. PLoS ONE (2018) 13:e0193428. doi: 10.1371/journal.pone.0193428
142. Kim MS, Kim E, Heo J-S, Bae D-J, Lee J-UW, Lee T-H, et al. Circulating IL-33 level is associated with the progression of lung cancer. Lung Cancer (2015) 90:346–51. doi: 10.1016/j.lungcan.2015.08.011
143. Hsu Y-L, Hung J-Y, Lee Y-L, Chen F-W, Chang K-F, Chang W-A, et al. Identification of novel gene expression signature in lung adenocarcinoma by using next-generation sequencing data and bioinformatics analysis. Oncotarget (2017) 8:104831–54. doi: 10.18632/oncotarget.21022
144. Brunner SM, Rubner C, Kesselring R, Martin M, Griesshammer E, Ruemmele P, et al. Tumor-infiltrating, interleukin-33-producing effector-memory CD8+ T cells in resected hepatocellular carcinoma prolong patient survival. Hepatology (2015) 61:1957–67. doi: 10.1002/hep.27728
145. Jafarzadeh A, Minaee K, Farsinejad A-R, Nemati M, Khosravimashizi A, Daneshvar H, et al. Evaluation of the circulating levels of IL-12 and IL-33 in patients with breast cancer: influences of the tumor stages and cytokine gene polymorphisms. Iran J Basic Med Sci. (2015) 18:1189–98. doi: 10.22038/IJBMS.2015.6271
146. Liu J, Shen J-X, Hu J-L, Huang W-H, Zhang G-J. Significance of interleukin-33 and its related cytokines in patients with breast cancers. Front Immunol. (2014) 5:141. doi: 10.3389/fimmu.2014.00141
147. Levescot A, Flamant S, Basbous S, Jacomet F, Féraud O, Anne Bourgeois E, et al. BCR-ABL-induced deregulation of the IL-33/ST2 pathway in CD34(+) progenitors from chronic myeloid leukemia patients. Cancer Res. (2014) 74:2669–76. doi: 10.1158/0008-5472.CAN-13-2797
148. Wang Z, Xu L, Chang Y, Zhou L, Fu H, Zhang W, et al. IL-33 is associated with unfavorable postoperative survival of patients with clear-cell renal cell carcinoma. Tumor Biol. (2016) 37:11127–34. doi: 10.1007/s13277-016-4879-3
149. Wu C, Wu Y, Cheng C, Hong Z, Shi Z, Lin S, et al. Interleukin-33 predicts poor prognosis and promotes renal cell carcinoma cell growth through its receptor ST2 and the JNK signaling pathway. Cell Physiol Biochem. (2018) 47:191–200. doi: 10.1159/000489766
150. Saied EM, El-Etreby NM. The role and prognostic value of inducible nitric oxide synthase (iNOS) and interleukin-33 (IL-33) in serous and mucinous epithelial ovarian tumours. Ann Diagn Pathol. (2017) 27:62–8. doi: 10.1016/j.anndiagpath.2017.01.006
151. Tong X, Barbour M, Hou K, Gao C, Cao S, Zheng J, et al. Interleukin-33 predicts poor prognosis and promotes ovarian cancer cell growth and metastasis through regulating ERK and JNK signaling pathways. Mol Oncol. (2016) 10:113–25. doi: 10.1016/j.molonc.2015.06.004
152. Gramatzki D, Frei K, Cathomas G, Moch H, Weller M, Mertz KD. Interleukin-33 in human gliomas: expression and prognostic significance. Oncol Lett. (2016) 12:445–52. doi: 10.3892/ol.2016.4626
153. Zhang J, Wang P, Ji W, Ding Y, Lu X. Overexpression of interleukin-33 is associated with poor prognosis of patients with glioma. Int J Neurosci. (2017) 127:210–7. doi: 10.1080/00207454.2016.1175441
154. Zhang J-F, Wang P, Yan Y-J, Li Y, Guan M-W, Yu J-J, et al. IL-33 enhances glioma cell migration and invasion by upregulation of MMP2 and MMP9 via the ST2-NF-κB pathway. Oncol Rep. (2017) 38:2033–42. doi: 10.3892/or.2017.5926
155. Bergis D, Kassis V, Ranglack A, Koeberle V, Piiper A, Kronenberger B, et al. High serum levels of the interleukin-33 receptor soluble ST2 as a negative prognostic factor in hepatocellular carcinoma. Transl Oncol. (2013) 6:311–8. doi: 10.1593/tlo.12418
156. Hu W, Li X, Li Q, Tan Y, Xu B, Xie Q, et al. Interleukin-33 expression does not correlate with survival of gastric cancer patients. Pathol Oncol Res. (2017) 23:615–9. doi: 10.1007/s12253-016-0167-1
157. De la Fuente M, MacDonald TT, Hermoso MA. The IL-33/ST2 axis: role in health and disease. Cytokine Growth Factor Rev. (2015) 26:615–23. doi: 10.1016/j.cytogfr.2015.07.017
Keywords: IL-33, cancer, immune cell subsets, tumor immunology, IL-33 isoforms
Citation: Afferni C, Buccione C, Andreone S, Galdiero MR, Varricchi G, Marone G, Mattei F and Schiavoni G (2018) The Pleiotropic Immunomodulatory Functions of IL-33 and Its Implications in Tumor Immunity. Front. Immunol. 9:2601. doi: 10.3389/fimmu.2018.02601
Received: 24 July 2018; Accepted: 22 October 2018;
Published: 13 November 2018.
Edited by:
Hui-Rong Jiang, University of Strathclyde, United KingdomReviewed by:
Xiao-Qing Wei, Cardiff University, United KingdomPadraic Fallon, Trinity College, Dublin, Ireland
Copyright © 2018 Afferni, Buccione, Andreone, Galdiero, Varricchi, Marone, Mattei and Schiavoni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabrizio Mattei, ZmFicml6aW8ubWF0dGVpQGlzcy5pdA==
Giovanna Schiavoni, Z2lvdmFubmEuc2NoaWF2b25pQGlzcy5pdA==