 Andrea M. Chambers1†
Andrea M. Chambers1† Sandro Matosevic
Sandro Matosevic- 1Department of Industrial and Physical Pharmacy, Purdue University, West Lafayette, IN, United States
- 2Center for Cancer Research, Purdue University, West Lafayette, IN, United States
Energy metabolism is key to the promotion of tumor growth, development, and metastasis. At the same time, cellular metabolism also mediates immune cell survival, proliferation and cytotoxic responses within the tumor microenvironment. The ability of natural killer cells to eradicate tumors relies on their ability to functionally persist for the duration of their anti-tumor effector activity. However, a tumor's altered metabolic requirements lead to compromised functional responses of cytokine-activated natural killer cells, which result in decreased effectiveness of adoptive cell-based immunotherapies. Tumors exert these immunosuppressive effects through a number of mechanisms, a key driver of which is hypoxia. Hypoxia also fuels the generation of adenosine from the cancer-associated ectoenzymes CD39 and CD73. Adenosine's immunosuppression manifests in decreased proliferation and impaired anti-tumor function, with adenosinergic signaling emerging as an immunometabolic checkpoint blockade target. Understanding such immunometabolic suppression is critical in directing the engineering of a new generation of natural killer cell-based immunotherapies that have the ability to more effectively target difficult-to-treat solid tumors.
Introduction
Warburg metabolism, alongside glutamine turnover, is a hallmark of cancer, associated with elevated metabolism of glucose to produce ATP and sustain rapid cancer cell growth (1). Such metabolic reprogramming from glycolysis to oxidative phosphorylation results in the activation of a number of signaling pathways, upregulation of oncogenes and transcriptional factors, and inactivation of tumor suppressor genes. Similarly, natural killer (NK) cells undergo tumor-induced metabolic reprogramming, when they switch from a basal to an activated state. Such reprogramming leads to altered cytotoxic function, and can result in impaired effectiveness of immunotherapies relying on adoptively-transferred NK cells in vivo. The deregulated metabolism in the tumor microenvironment (TME) exerts an immunosuppressive effect on NK cells through a combination of mechanisms (Table 1). Some of the most representative features of an immunosuppressive TME are environmental heterogeneity to include deep hypoxic cores which lead to the generation of suppressive metabolites, such as adenosine, and the secretion of metabolic by-products. Though adoptive immunotherapy of both unmodified (9) and genetically-engineered NK cells (14) has been extensively demonstrated, the implication of immunometabolism is only starting to be investigated. Most of the insights into immunometabolic suppression of NK cell function currently originate from a limited number of focused studies. Until now, the interest has largely been on improving the cytotoxic activity of NK cells and enhancing target recognition.
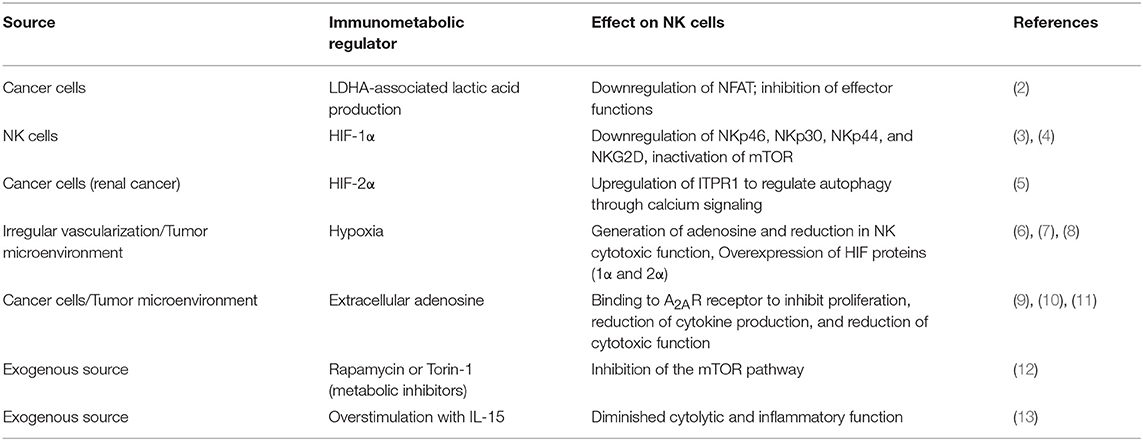
Table 1. Sources of immunometabolic inhibition of NK cell functions.
The use of natural killer cells in adoptive immunotherapy has traditionally relied on equipping these cells with synthetic machinery—such as genetically-engineered chimeric antigen receptors—to improve target recognition (14, 15). It is becoming increasingly apparent that next-generation immunotherapies, for difficult-to-treat solid tumors, will require the consideration of the emerging role of immunometabolism on immune function. Here, we discuss the current state-of-the-art of the field and the approaches aimed at targeting immunometabolic suppression to improve adoptive NK cell immunotherapy.
Natural Killer Cell Subsets
The function of NK cells is regulated by the interplay of a number of activating and inhibitory receptors. NK cells are considered to be the most well-known subset of innate lymphoid cells, which also includes tissue inducer cells and non-cytotoxic innate lymphoid cells (ILCs). Though both human and mouse innate lymphoid cells include conventional NK cells, non-cytotoxic innate lymphoid cell groups 1, 2, and 3 (ILC1, ILC2, and ILC3) as well as intraepithelial innate lymphoid cell group 1 (ieILC1), these cells differ in their expression of surface markers. For instance, murine ILCs do not express CD56 and express NK1.1 instead of NKp44 (16).
Phenotypically, human NK cells are defined by the expression of CD56 and the absence of CD3. NK cells are then additionally split into two main subsets based on the expression of CD56 and CD16: CD56brightCD16± and CD56dimCD16bright(+). These subsets differ in tissue distribution, and are dependent on specific homing properties and the cells' in situ maturation. CD56dimCD16bright cell represent about 90% of all NK cells, and are predominant in peripheral blood. On the other hand, CD56brightCD16±, found mostly in lymphoid organs, can be subdivided into CD16− (which represent about 30–50% of CD56bright cells), and CD16dim (50–70% of CD56bright) subsets. The less common CD56dimCD16− and CD56−CD16+ cells have also been described, but the function of these cells is not well-known (17). Over 90% of peripheral blood NK cells are also killer immunologlobulin-like receptor (KIR)+.
Distribution and trafficking of NK cells in tissues has been extensively described (18). Tissue-resident NK cells express CD69, which blood-derived NK cells lack (19). They also differ in expression of chemokine receptors and adhesion molecules: Tissue resident NK cells tend to express CXCR6 and CCR5 and the integrins CD49a and CD103, while blood-derived NK cells express CXCR3, CXCR4, CCR7, CD62L (L-selectin), and lack CD49a (20).
Murine NK cells differ from human NK cells in a few notable aspects. While human NK cells express KIRs, mouse NK cells are characterized by expression of the C-type lectin-like family of receptors, Ly49s. Mouse NK cells, additionally, lack expression of CD56, which is a hallmark of human NK cells.
Murine NK cells are primarily defined based on their expression of CD27 and CD11b. In adult mice, CD11blow cells are primarily found in the bone marrow, lymph nodes and the liver, while the CD11bhigh subset is located in peripheral blood, the spleen and lungs. Among these, the CD11bhighCD27high subset is the most highly cytotoxic and expresses higher amounts of cytokines (21). Correlations have been made in terms of functionality between CD11blowCD27high and CD11bhighCD27low NK cells in mice with CD56bright and CD56dim in humans, respectively (22). The intratumoral infiltration of these subsets also differs, with CD27+CD11b+ the prevalent subset found in fibrosarcoma (23). Mouse NK cells also express NK1.1, CD16 and CD122 and are regulated by different activating and inhibitory receptors (24).
Immunometabolic Cytokine Activation of NK Cells
Insights into the metabolism of natural killer cells mostly come from studies using murine cells, though a rapidly increasing body of work is contributing to our expanding knowledge of human NK cells. Glycolytic fueling in tumors reduces glucose availability to surrounding immune cells, leading to their metabolic reprogramming (25). In NK cells, regulation of metabolic response by up-regulation of glucose uptake and glycolysis is mediated by mTOR, specifically mTORC1 (26). mTORC1 activation requires sufficient intracellular nutrients and energy. mTOR is also essential for regulating the production of granzyme B and perforin, and can most potently be activated with high-concentrations of IL-15 during early infection, though other cytokines (IL-2, IL-12, IL-18) are also implicated (27). IL-15 activates mTORC1 via PI3K, PDPK1, and AKT (28). While NK cells do not exhibit increased glycolysis during short-term activation, extended stimulation with high-dose IL-15 over multiple days was shown to lead to up-regulation of metabolism, enhancing glycolysis (29). mTORC1 also enhances glycolysis by promoting transcription factor HIFα and mitochondrial biogenesis through PPARγ co-activator 1α (PGC1α) and yin and yang 1 (YY1) (30). Recently, Srebp, otherwise implicated in de novo lipid synthesis, has been shown to regulate functional responses and NK cell effector function, in supporting glycolysis and oxidative phosphorylation by the use of the citrate–malate shuttle, through its targets Acly and Slc25a1 (31). High rates of glycolysis in tumors exert inhibitory effects on tumor-infiltrating NK cells also via cancer-associated lactate dehydrogenase-A (LDHA). LDHA fuels the conversion of excess pyruvate and NADH into lactate and NAD+, thus supporting tumor glycolysis. Brand et al. (2) recently reported that LDHA-associated lactic acid production leads to impaired NK cell activity through downregulation of nuclear factor of activated T cells (NFAT) in T and NK cells.
In response to diminishing glucose supplies, NK cells are thought to undergo metabolic reprogramming by foregoing IL-15 and mTOR dependency, and instead becoming driven by activating receptors (e.g., Ly49H in mice, KIR in humans) (32). However, the metabolic reprogramming of failed NK metabolism in a tumor setting has not been investigated in detail. This reprogramming is likely to be activation-dependent: cytokine-stimulated NK cells can produce IFN-γ independent of glycolysis or mitochondrial oxidative phosphorylation, while activating-receptor stimulated NK cells require oxidative phosphorylation (29).
Though scarce, evidence is also emerging that metabolic signatures also differ among human NK cell subsets, albeit from limited in vitro studies. In addition to greater activation of mTORC1, cytokine-activated CD56bright cells are thought to have higher rates of glucose uptake compared with CD56dim cells, associated with their higher expression of IFNγ (33). While CD56dim cells are more cytotoxically active, they also have a lower biosynthetic burden and are likely to have lower metabolic requirements (33).
Because cytokines are a critical feature of adoptive immunotherapies with NK cells, understanding their activation-specific metabolic requirements to engage in anti-cancer cytotoxic functions is critical to the implementation of NK cells as viable immunotherapies.
Hypoxia-Induced Metabolic Reprogramming of NK Cells
Oxygen availability is dependent on the metabolic requirements and functional status of each organ. During immunological responses in inflammatory and tumor microenvironments, NK cells operate at varying concentrations of oxygen, often reaching regions of severely low oxygen (hypoxia). Hypoxia is, as such, considered an adverse prognostic factor, particularly for solid tumors (34), and has been documented as being a feature of multiple pathologies (35). While oxygen concentrations in human tissues range from as low as 1.3% in bone marrow (36) to 13% in arterial blood (37), the existence of a hypoxic niche at sites of inflammation and in pathological environments has been evidenced by measured oxygen concentrations that are ~3–8 times lower than in corresponding physioxic tissues (38).
Tumor microenvironments are characterized by cycling hypoxia (39), throughout which they are exposed to bursts of varying concentrations of oxygen due to irregular vascularization and blood supply of tumor tissues. Both acute and chronic hypoxia result in DNA damage (40), replication arrest (41), radioresistance (42), epithelial to mesenchymal transition (43), angiogenesis and metastasis (44), and immune resistance (45).
Key NK cell responses to hypoxia are regulated by overexpression of HIF-1α and HIF-2α (6, 7), although inconclusive mechanisms have been proposed for their exact effect on NK cells. HIF-α can signal through and is stabilized via both oxygen-dependent and oxygen-independent mechanisms (46). These transcription factors direct responses of NK cells to low oxygen, influencing trophoblast lineage commitment and promoting development of the invasive trophoblast lineage during fetal development (47).
The overexpression of HIF-1α in hypoxic environments, which is partially dependent on mTOR signaling, leads to the downregulation of NK activating receptors NKp46, NKp30, NKp44, and NKG2D (3). Recent work has shown that deletion of HIF-1α in NK cells renders them hyporesponsive in both hypoxia and normoxia. Under long term hypoxia, tumor-associated NK cells from HIF-1α KO mice did not show a reduction of soluble vascular endothelial growth factor receptor 1 (sVEGFR1) expression, likely compensated by HIF-2α. These cells were also found to be less present in hypoxic zones, suggesting HIF-1α has an effect on intratumoral infiltration of NK cells and, collectively, increasing the bioavailability of VEGF (48). Others have also hypothesized that during long-term hypoxia, the HIF-1α protein accumulates due to the inactivation of its degradation pathway. This buildup of HIF-1α enforces a negative feedback loop to inactivate mTOR and cause diminished cellular translation and deleterious downstream effects (4).
Additionally, hypoxia was shown to cause autophagy-induced degradation of granzyme B released from activated NK cells in the tumor microenvironment (49, 50). The hypoxia-induced onset of autophagy has been described mechanistically for a number of cancers, including renal cancer (51), glioblastoma (52), bladder cancer (53), lung cancer (54) and acute myeloid leukemia.(55) Mechanistic insights in renal cancer have implicated HIF-2α to transcriptionally upregulate inositol triphosphate receptor 1 (ITPR1), a regulator of autophagy through calcium signaling (5).
The strength of the antitumor response of NK cells is largely dependent on the expression of NKG2D ligands on NK cells, and corresponding MICA/B receptors on tumor cells (56). These receptors are highly present in tumor environments in response to cellular stresses, and the MICA/B receptors assist the NK cells in locating the cancer cells for elimination through NKG2D receptor activation. Shedding of MIC receptors—alongside CD16 shedding— has been widely described to contribute to immune evasion and deficiency of adoptive NK cell transfers (57–59). This shedding has shown to be induced by hypoxia through altered nitric oxide (NO) signaling (60), and could be rescued through exogenous induction of NO signaling (61). It bears mentioning that shedding of CD16 from NK cells also occurs in response to various activation stimuli, including cytokines, cross-linking with activating receptors or target cell stimulation, ultimately resulting in decreased intracellular cytokine production and impaired CD107a degranulation (62). Without CD16 shedding, NK cell viability is not sustained, and shedding is thought to possibly prevent activation-induced cell death and modulate NK cell effector functions through the ability to engage multiple target cells.
Because of the broad and potent role of cytokines in activation of NK cells, it is no surprise that studies have investigated the implication of cytokine stimulation in hypoxic environments. Recent reports have indicated that hypoxia-induced loss of NK cell cytotoxicity could be rescued by treatment with high doses (1,000 IU/mL) IL-2 (63) for 14–16 h. While IL-2 treatment abrogated impairment of NK-mediated cytotoxicity against K562 cells in vitro under both normoxic (20% oxygen) and hypoxic (0% oxygen) conditions, it is still unclear how such observations translate in vivo against solid tumor targets. Similarly to IL-2, IL-15 induces potent cytotoxic responses on NK cells. Interestingly, short-term hypoxia (6 h, 1% O2) enhanced the cytotoxic response of NK cells that were simultaneously stimulated with IL-15 (50 ng/mL) against K562 cells in vitro, and induced upregulation of HIF-1α-induced glycolytic gene expression (8). This did not, however, result in changes in glycolytic flux compared to non-IL-15-stimulated NK cells, showing that other factors may also be involved in this response during short-term hypoxia. The synergy between IL-15 and short-term hypoxia has also resulted in the downregulation of the glycolytic/pentose phosphate pathway-linked gene TKTL1, suggesting a possible switch to Warburg metabolism in response to IL-15 and short-term hypoxia. Transcriptional microarray data collected from NK cells under hypoxia and IL-5 activation (6 h) have also been reported (64).
Effects of Adenosinergic Metabolism on NK Cells
Adenosine, a purine ribonucleoside, is involved in a well-characterized network of pathways in many cell types, including immune cells. While adenosine signaling occurs both intra and extracellularly, its roles diverge. Intracellularly, adenosine is involved in energy homeostasis, nucleic acid metabolism, angiogenesis, and the methionine cycle (65–67), exerting a protective effect on cells and tissues (68). Extracellular adenosine, on the other hand, is involved in intercellular signaling. Elevated extracellular concentrations of adenosine in tumors, which can be as much as 100-fold higher than in normal tissues, are known to contribute to immune evasion (69, 70). This immune evasion manifests through a combination of reduced proliferation, inhibition of cytotoxic activity, downregulation of activating receptors, and reduced secretion of cytotoxic cytokines (71). As a result, adenosinergic signaling has emerged as a negative feedback loop that regulates local and systemic anti-tumor response (72). Much is known about adenosine signaling in cells, and with deepening knowledge of its effects on NK cells (73) as a target for adoptive immunotherapy, it has become of increasing therapeutic interest (74).
The physiologic functions of adenosine are largely mediated by four types of G-protein-coupled adenosine receptors, A1, A2A, A2B, and A3, where the A2A adenosine receptor (A2AR) is the subtype that is most frequently expressed on immune cells (75). Extracellular accumulation of adenosine in the tumor microenvironment leads to immunosuppression, particularly through A2AR on infiltrating immune cells, including NK cells (76–78). Indeed, effector functions of NK cells were shown to be susceptible to A2AR stimulation in a number of studies. Among the suppressed immune functions due to extracellular adenosine are inhibited proliferation (79), a reduced production of cytokines from IL-2-stimulated NK cells (11), and a reduced cytotoxic effector function against cancer cells (10). A2A receptor expression on tumor-associated myeloid cells was also shown to inhibit the cytotoxic function of NK cells in primary and metastatic tumor microenvironments (80), indicating that the broad effect of the A2 receptor in tumors is not limited to self-expression on NK cells. Unlike signaling through A2A, agonism of the A1 or, to a lesser extent, the A3 receptor stimulates NK cytotoxicity (77), presumably through a mechanism that involves a decrease in intracellular cAMP (81, 82). A3 agonism was also associated with higher serum levels of IL-12, a known stimulator of NK cell cytotoxicity (82). Using a different A3 receptor agonist, iodobenzyl methylcarboxamidoadenosine, that blocks the adenosine response, inhibition of proliferation, IFN-γ production, and cytotoxicity of NK cells was shown. However, through a mechanism involving the A3 adenosine receptor, and in the presence of adenosine upon stimulation with IFN-α, IFN-γ production from NK cells increased compared to that observed in the absence of adenosine (83). More recently, targeting A2AR via both A2AR inhibition or in A2AR-deficient mice resulted in improved tumor control and inhibition of tumor progression through elimination of the A2AR-induced suppression of NK cell maturation by promoting the accumulation of highly cytotoxic CD56dim NK cells (84). Ongoing pre-clinical studies are also addressing the co-inhibition of the A2AR receptor with the A2B receptor on immune cells in conjunction with chemotherapy (85); however, the A2B receptor has a lower expression on NK cells compared to myeloid-derived and dendritic cells.
Strategies to Modulate Immunometabolic Suppression in the Tumor Microenvironment
Recent evidence that metabolic functions regulate NK cell activation (Table 2) has fueled interest in the preclinical development of strategies that address metabolic reprogramming of NK cells in the context of immunotherapy. While several therapeutic strategies aim to target metabolism of immune cells, approaches aimed at specifically targeting NK cell function are still emerging.
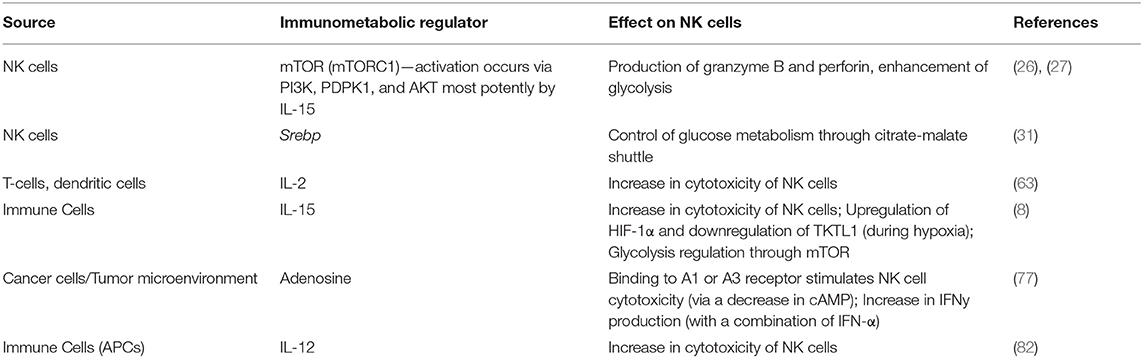
Table 2. Sources of immunometabolic regulation of NK cell functions.
Cytokine Stimulation
Several studies are investigating cytokine stimulation to induce activation signals and enhance in vivo persistence of adoptively-transferred NK cells. Among these, IL-15 has been the most utilized, due to its recognized role in NK cell development, homeostasis and activation (86). Recently, the functions of IL-15 have been linked to mammalian target of rapamycin (mTOR) (87), which is known to regulate NK cell metabolism through glycolysis. There are various clinical trials currently investigating several IL-15 dosing regimens for improving NK cell metabolism for adoptive transfer. Though IL-15-stimulated NK cells were shown to eradicate solid tumors (88), recent findings have indicated that continuous treatment with IL-15 may actually result in exhaustion of NK cells (13), ultimately leading to cells with markedly diminished cytolytic and inflammatory function. As it stands, our understanding of optimal IL-15 dosing, timing, or the overall effects of its use in the immunotherapy of adoptively-transferred NK cells is incomplete.
Metabolic Inhibition
The mTOR inhibitor rapamycin has been used in clinical immunotherapy for a number of years to suppress the metabolic activity of pathogenic cells (89). The metabolic activity of NK cells is regulated by mTOR. In NK cells, mTOR is activated primarily, though not exclusively, by IL-15 (90), rationalizing the use of IL-15 in adoptive immunotherapy. Consequently, stunting metabolic activity for cancer treatment has raised concerns that using IL-15 could lead to associated negative effects on glucose metabolism of immune cells, including NK cells (12). Recently, Yang et al. (91) described the independent roles of mTORC1 and mTORC2 in the development of NK cells: while the loss of either did not disrupt the cytolytic function of NK cells, deletion of mTORC1 function disrupted NK cell homeostasis, and the absence of mTORC2 stunted the terminal maturation of NK cells. Results like these have shown that the use of metabolic inhibitors, such as rapamycin or Torin-1, should be taken into consideration to make sure there are not any secondary and potentially suppressive effects on the immune cell effector function, since manipulation of mTOR signaling can affect NK cell development and repopulation.
Targeting Adenosinergic Signaling With Adoptive NK Cell Immunotherapy
With recent advances in immunotherapy using genetically-engineered immune cells, an emerging therapeutic approach has been to combine an A2A receptor blockade with adoptive cell therapy. Targeting the A2A receptor with a small molecule inhibitor, such as SCH58261, or with A2AR knockdown in cells using shRNA, triggers the enhancement of the cytotoxic function of anti-HER2 CAR-T cells either alone or in combination with anti-PD-1 therapy when transferred adoptively to tumor-bearing C57BL/6 mice (92). Unlike CAR-T cells, however, no such studies using adoptively-transferred NK cells exist yet.
Accumulation of extracellular adenosine in tumors is associated with activity of the CD39-CD73 enzymatic cascade (93). Hypoxia in solid tumors promotes the release of nucleotides including AMP and ATP, which fuel the enzymatic activity of the ectonucleotidases CD39 and CD73 (Figure 1), inducing the conversion of AMP → ATP → extracellular adenosine, with CD73 catalyzing the dephosphorylation of extracellular 5′AMP to adenosine as the final step in this pathway (94). HIF-1 was implicated as regulating the activity of CD73 (95). Though CD73 is expressed on several cell types including NK cells, it is highly upregulated in various cancers (91, 96). Antibody therapy with anti-CD73 antibodies or CD73 shRNA (97, 98) was shown to be effective in inhibiting tumor growth and metastasis. This type of treatment has recently found its way into the clinic and is known as Medimmune's Oleclumab (anti-CD73 antibody), which is currently in Phase I clinical trials (99). Therapeutic inhibition of CD73 was also shown to improve antitumor efficacy of anti-PD-1 and anti-CTLA-4 checkpoint inhibitors in preclinical models of various solid tumors (100). A limited expression of CD73 is observed on NK cells, typically in the region of 1%. NK cells were reported to be able to enhance their expression of CD73 upon co-culture with mesenchymal stem cells (MSCs) (101). CD73 expression conferred by MSCs results in catalytically active ectoenzyme, and NK cells that are capable of producing higher amounts of adenosine via the conversion of 5′-AMP. Further studies have implicated the CD56brightCD16− NK subset as most active in producing adenosine via involvement of CD38—by inhibiting CD38 activity, adenosine production was reduced (102).
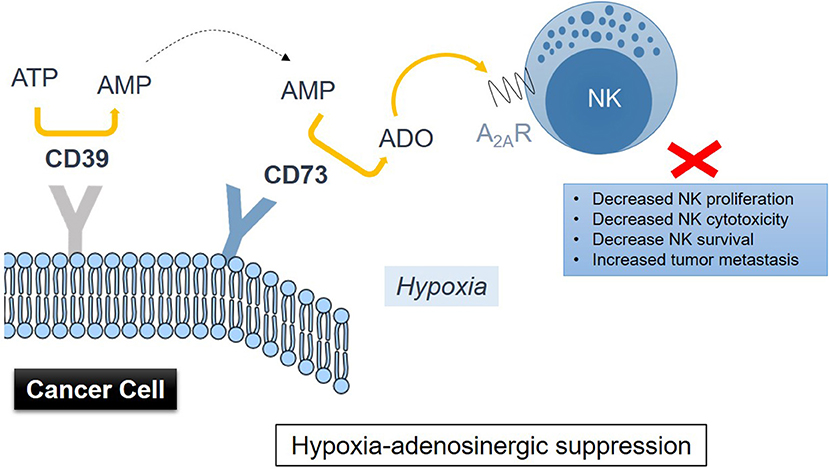
Figure 1. Drawing representation of immunometabolic suppression of NK cell functions induced by adenosine metabolism in the TME involving CD39 and CD73 on cancer cells via signaling through the adenosine A2A receptor on NK cells.
Mechanisms that guide CD73-induced promotion of tumor growth and immune resistance include not only the inhibition of NK cell cytotoxicity, but also induction of the internalization of CD73 expressed on cancer cells (103). For that reason, a number of anti-CD73 clones have been indicated to promote internalization of CD73 with limited effect on its enzymatic activity. While CD73-targeting antibody therapy can potentiate the anti-tumor activity of NK cells via ADCC, a recent study showed that blockade of CD73 with anti-CD73 clone 7G2 in conjunction with anti-CD39 clone A1 enhanced NK cell cytotoxicity in vitro via a mechanism that includes adenosinergic metabolism and is independent of ADCC (104). Our own work showed that a CD73 antibody blockade enhances the killing potential of CAR-engineered NK-92 cells, a widely-used NK cell line that does not express CD16, via mechanisms that implicate extracellular adenosine in the absence of ADCC. NK cells were also implicated in the metastatic control of LWT1 melanoma tumors in vivo when C57BL/6 mice were targeted by anti-CD73 antibody therapy in conjunction with inhibition of A2AR signaling (105). Alongside NK cells, tumor control was optimal when CD8+ T cells, interferon-γ and perforin were also present. Most of the in vivo studies targeting CD73 use mouse clones of these antibodies and are performed in C57BL/6 mice. Therefore, further insights are needed on understanding the effect of targeting the CD73/adenosinergic pathway with human-directed antibody clones in association with adoptive transfer of immune cells into in vivo models that bear human immune components.
Adenosine signaling was also studied in NK-92 cells Hong et al. (106) observed that upon exposure to acute myeloid leukemia-derived exosomes, NK-92 cells, which also express the A2AR receptor, increased their expression of adenosine, inosine and hypoxanthine, ultimately contributing to autocrine inhibition of NK-92 cytotoxic function via upregulation of A2AR function (106).
Adenosine is metabolically unstable, becoming rapidly converted to inosine via the activity of the enzyme adenosine deaminase (ADA). For that reason, most studies on the effect of adenosine on NK cells use its metabolically stable analog, 2-chloroadenosine, thereby bypassing catalytic removal of rapidly-accumulating extracellular adenosine. Extreme dysfunction in purinergic metabolism can lead to adenosine deaminase-severe combined immunodeficiency (ADA-SCID), which causes the accumulation of adenosine due to ADA loss of function, ultimately resulting in partial or complete lymphopenia (107).
Though direct studies on the effects of purine metabolism, particularly that of adenosine, on the functions and phenotypes of NK cells are limited, insights are starting to emerge. McCarthy et al. (108) observed that the generation of purine nucleotides, including adenosine, through glycolysis drives the expression of MICA ligands on cells, which are then targeted by NKG2D receptors on NK cells. Restricting early glycolytic pathway intermediates abrogated the expression of MICA, showing that cell proliferation was not shown to be a prerequisite for MICA expression.
Conclusion
An emerging body of work is starting to highlight the critical role of immunometabolism in innate immunity and the development of emerging cancer immunotherapies. Among the immunosuppressive features of tumors is hypoxia, which results in the generation of adenosine, a metabolite that is highly suppressive to NK cell cytotoxicity and proliferation. These immunometabolic changes are specific to NK cells and occur under various cytokine stimulation programs. While mTOR has been recognized as a key driver of metabolic reprogramming in NK cells, the mechanisms by which mTOR regulates the metabolic system and NK cell effector responses in the tumor microenvironment are still largely unknown. Moreover, the role of mTOR-mediated regulation of protein translation during NK cell effector responses has not been studied in depth. Limited knowledge also exists on how these metabolic changes occur in phenotypically distinct NK subsets, particularly licensed cells. For instance, metabolic responses of licensed and unlicensed NK cells are not known, and neither are the metabolic programs of NK cells that display exhausted functional phenotypes. This dearth of knowledge precludes our utilization of NK cells as effective adoptive NK immunotherapies; however, rapid advances are fueling remarkable discoveries in the field. Future immunotherapies with adoptively-transferred NK cells are expected to employ modalities that reverse or avoid immunometabolic suppression of NK cell function in their design to ultimately lead to the improved targeting of solid tumors.
Author Contributions
SM researched the literature and wrote the initial draft of the manuscript. AC and KL performed the literature review, generated the tables and prepared an edited version of the manuscript. AC, KL, and SM critically revised the manuscript. AC, KL, and SM wrote the manuscript and edited the figure and tables.
Funding
The authors gratefully acknowledge the Flow Cytometry Shared Resource Grant and support from the Purdue University Center for Cancer Research, P30CA023168. This publication was made possible, in part, with support from the Indiana Clinical and Translational Sciences Institute funded, in part by Award Number UL1TR001108 from the National Institutes of Health, National Center for Advancing Translational Sciences, Clinical, and Translational Sciences Award. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Liberti MV, Locasale JW. The warburg effect: how does it benefit cancer cells? Trends Biochem Sci. (2016) 41:211–8. doi: 10.1016/j.tibs.2015.12.001
2. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metabol. (2016) 24:657–71. doi: 10.1016/j.cmet.2016.08.011
3. Balsamo M, Manzini C, Pietra G, Raggi F, Blengio F, Mingari MC, et al. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur J Immunol. (2013) 43:2756–64. doi: 10.1002/eji.201343448
4. Knaup KX, Jozefowski K, Schmidt R, Bernhardt WM, Weidemann A, Juergensen JS, et al. Mutual regulation of hypoxia-inducible factor and mammalian target of rapamycin as a function of oxygen availability. Mol Cancer Res. (2009) 7:88–98. doi: 10.1158/1541-7786.MCR-08-0288
5. Messai Y, Noman MZ, Hasmim M, Escudier B, Chouaib S. HIF-2α/ITPR1 axis: a new saboteur of NK-mediated lysis. Oncoimmunology (2015) 4:e985951. doi: 10.4161/2162402X.2014.985951
6. Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol. (2009) 9:609–17. doi: 10.1038/nri2607
7. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell (2012) 148:399–408. doi: 10.1016/j.cell.2012.01.021
8. Velásquez SY, Killian D, Schulte J, Sticht C, Thiel M, Lindner HA. Short-term hypoxia synergizes with interleukin 15 priming in driving glycolytic gene transcription and supports human natural killer cell activities. J Biol Chem. (2016) 291:12960–77. doi: 10.1074/jbc.M116.721753
9. Davis ZB, Felices M, Verneris MR, Miller JS. Natural killer cell adoptive transfer therapy: exploiting the first line of defense against cancer. Cancer J. (2015) 21:486–91. doi: 10.1097/PPO.0000000000000156
10. Raskovalova T, Huang X, Sitkovsky M, Zacharia LC, Jackson EK, Gorelik E. Gs protein-coupled adenosine receptor signaling and lytic function of activated NK cells. J Immunol. (2005) 175:4383–91. doi: 10.4049/jimmunol.175.7.4383
11. Raskovalova T, Lokshin A, Huang X, Jackson EK, Gorelik E. Adenosine-mediated inhibition of cytotoxic activity and cytokine production by IL-2/NKp46-activated NK cells: involvement of protein kinase A isozyme I (PKA I). Immunol Res. (2006) 36:91–9. doi: 10.1385/IR:36:1:91
12. Baeyens A, Pérol L, Fourcade G, Cagnard N, Carpentier W, Woytschak J, et al. Limitations of IL-2 and rapamycin in immunotherapy of type 1 diabetes. Diabetes (2013) 62:3120–31. doi: 10.2337/db13-0214
13. Felices M, Lenvik AJ, McElmurry R, Chu S, Hinderlie P, Bendzick L, et al. Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight (2018) 3:96219. doi: 10.1172/jci.insight.96219
14. Matosevic S. Viral and nonviral engineering of natural killer cells as emerging adoptive cancer immunotherapies. J Immunol Res. (2018) 2018:4054815. doi: 10.1155/2018/4054815
15. Hu Y, Tian Z, Zhang C. Chimeric antigen receptor (CAR)-transduced natural killer cells in tumor immunotherapy. Acta Pharmacol Sin. (2017) 39:167–76. doi: 10.1038/aps.2017.125
16. Artis D, Spits H. The biology of innate lymphoid cells. Nature (2015) 517:293–301. doi: 10.1038/nature14189
17. Mavilio D, Lombardo G, Benjamin J, Kim D, Follman D, Marcenaro E, et al. Characterization of CD56–/CD16+ natural killer (NK) cells: a highly dysfunctional NK subset expanded in HIV-infected viremic individuals. PNAS (2005) 102:2886–91. doi: 10.1073/pnas.0409872102
18. Carrega P, Ferlazzo G. Natural killer cell distribution and trafficking in human tissues. Front Immunol. (2012) 3:347. doi: 10.3389/fimmu.2012.00347
19. Lugthart G, Melsen JE, Vervat C, van Ostaijen-Ten Dam MM, Corver WE, Roelen DL, et al. Human lymphoid tissues harbor a distinct CD69+CXCR6+ NK Cell population. J Immunol. (2016) 197:78–84. doi: 10.4049/jimmunol.1502603
20. Melsen JE, Lugthart G, Lankester AC, Schilham MW. Human circulating and tissue-resident CD56bright natural killer cell populations. Front Immunol. (2016) 7:262. doi: 10.3389/fimmu.2016.00262
21. Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, Walzer T. Maturation of mouse NK cells is a 4-stage developmental program. Blood (2009) 113:5488–96. doi: 10.1182/blood-2008-10-187179
22. Sungur CM, Murphy WJ. Utilization of mouse models to decipher natural killer cell biology and potential clinical applications. Hematology (2013) 2013:227–33. doi: 10.1182/asheducation-2013.1.227
23. Hayakawa Y, Sato-Matsushita M, Takeda K, Iwakura Y, Tahara H, Irimura T. Early activation and interferon-γ production of tumor-infiltrating mature CD27 high natural killer cells. Cancer Sci. (2011) 102:1967–71. doi: 10.1111/j.1349-7006.2011.02042.x
24. Smith HR, Idris AH, Yokoyama WM. Murine natural killer cell activation receptors. Immunol Rev. (2001) 181:115–25. doi: 10.1034/j.1600-065X.2001.1810109.x
25. O'Neill LAJ, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
26. Marçais A, Cherfils-Vicini J, Viant C, Degouve S, Viel S, Fenis A, et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat Immunol. (2014) 15:749–57. doi: 10.1038/ni.2936
27. Nandagopal N, Ali AK, Komal AK, Lee S-H. The Critical Role of IL-15–PI3K–mTOR Pathway in natural killer cell effector functions. Front Immunol. (2014) 5:187. doi: 10.3389/fimmu.2014.00187
28. Ali AK, Nandagopal N, Lee S-H. IL-15–PI3K–AKT–mTOR: A critical pathway in the life journey of natural killer cells. Front Immunol. (2015) 6:355. doi: 10.3389/fimmu.2015.00355
29. Keppel MP, Topcagic N, Mah AY, Vogel TP, Cooper MA. Activation-specific metabolic requirements for NK cell IFN-γ production. J Immunol. (2015) 194:1954–62. doi: 10.4049/jimmunol.1402099
30. Weichhart T, Hengstschläger M, Linke M. Regulation of innate immune cell function by mTOR. Nat Rev Immunol. (2015) 15:599–614. doi: 10.1038/nri3901
31. Assmann N, O'Brien KL, Donnelly RP, Dyck L, Zaiatz-Bittencourt V, Loftus RM, et al. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat Immunol. (2017) 18:1197–206. doi: 10.1038/ni.3838
32. Sun JC, Ma A, Lanier LL. Cutting edge: IL-15-independent NK cell response to mouse cytomegalovirus infection. J Immunol. (2009) 183:2911–4. doi: 10.4049/jimmunol.0901872
33. Keating SE, Zaiatz-Bittencourt V, Loftus RM, Keane C, Brennan K, Finlay DK, et al. Metabolic reprogramming supports IFN-γ production by CD56bright NK cells. J Immunol. (2016) 196:2552–60. doi: 10.4049/jimmunol.1501783
34. Harris AL. Hypoxia–a key regulatory factor in tumour growth. Nat Rev Cancer (2002) 2:38–47. doi: 10.1038/nrc704
35. Noman MZ, Messai Y, Carré T, Akalay I, Méron M, Janji B, et al. Microenvironmental hypoxia orchestrating the cell stroma cross talk, tumor progression and antitumor response. Crit Rev Immunol. (2011) 31:357–77. doi: 10.1615/CritRevImmunol.v31.i5.10
36. Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM, et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature (2014) 508:269–73. doi: 10.1038/nature13034
37. Carreau A, Hafny-Rahbi BE, Matejuk A, Grillon C, Kieda C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med. (2011) 15:1239–53. doi: 10.1111/j.1582-4934.2011.01258.x
38. Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (2015) 3:83–92. doi: 10.2147/HP.S93413
39. Kimura H, Braun RD, Ong ET, Hsu R, Secomb TW, Papahadjopoulos D, et al. Fluctuations in red cell flux in tumor microvessels can lead to transient hypoxia and reoxygenation in tumor parenchyma. Cancer Res. (1996) 56:5522–8.
40. Riffle S, Pandey RN, Albert M, Hegde RS. Linking hypoxia, DNA damage and proliferation in multicellular tumor spheroids. BMC Cancer (2017) 17:338. doi: 10.1186/s12885-017-3319-0
41. Hammond EM, Green SL, Giaccia AJ. Comparison of hypoxia-induced replication arrest with hydroxyurea and aphidicolin-induced arrest. Mutat Res. (2003) 532:205–13. doi: 10.1016/j.mrfmmm.2003.08.017
42. Horsman MR, Nordsmark M, Khalil AA, Hill SA, Chaplin DJ, Siemann DW, et al. Reducing acute and chronic hypoxia in tumours by combining nicotinamide with carbogen breathing. Acta Oncologica (1994) 33:371–6. doi: 10.3109/02841869409098431
43. Hill RP, Marie-Egyptienne DT, Hedley DW. Cancer stem cells, hypoxia and metastasis. Semin Radiat Oncol. (2009) 19:106–11. doi: 10.1016/j.semradonc.2008.12.002
44. Rofstad EK, Gaustad J-V, Egeland TAM, Mathiesen B, Galappathi K. Tumors exposed to acute cyclic hypoxic stress show enhanced angiogenesis, perfusion and metastatic dissemination. Int J Cancer (2010) 127:1535–46. doi: 10.1002/ijc.25176
45. Noman MZ, Hasmim M, Messai Y, Terry S, Kieda C, Janji B, et al. Hypoxia: a key player in antitumor immune response. A review in the theme: cellular responses to hypoxia. Am J Physiol Cell Physiol. (2015) 309:C569–79. doi: 10.1152/ajpcell.00207.2015
46. Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity (2014) 41:518–28. doi: 10.1016/j.immuni.2014.09.008
47. Chakraborty D, Rumi MAK, Soares M. NK cells, hypoxia and trophoblast cell differentiation. Cell Cycle (2012) 11:2427–30. doi: 10.4161/cc.20542
48. Krzywinska E, Kantari-Mimoun C, Kerdiles Y, Sobecki M, Isagawa T, Gotthardt D, et al. Loss of HIF-1α in natural killer cells inhibits tumour growth by stimulating non-productive angiogenesis. Nat Commun. (2017) 8:1597. doi: 10.1038/s41467-017-01599-w
49. Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K, et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci USA. (2013) 110:17450–5. doi: 10.1073/pnas.1304790110
50. Viry E, Baginska J, Berchem G, Noman MZ, Medves S, Chouaib S, et al. Autophagic degradation of GZMB/granzyme B: a new mechanism of hypoxic tumor cell escape from natural killer cell-mediated lysis. Autophagy (2014) 10:173–5. doi: 10.4161/auto.26924
51. Messai Y, Noman MZ, Hasmim M, Janji B, Tittarelli A, Boutet M, et al. ITPR1 protects renal cancer cells against natural killer cells by inducing autophagy. Cancer Res. (2014) 74:6820–32. doi: 10.1158/0008-5472.CAN-14-0303
52. Jawhari S, Ratinaud M-H, Verdier M. Glioblastoma, hypoxia and autophagy: a survival-prone ‘ménage-à-trois.' Cell Death Dis. (2016) 7:e2434. doi: 10.1038/cddis.2016.318
53. Yang X, Yin H, Zhang Y, Li X, Tong H, Zeng Y, et al. Hypoxia-induced autophagy promotes gemcitabine resistance in human bladder cancer cells through hypoxia-inducible factor 1α activation. Int J Oncol. (2018) 53:215–24. doi: 10.3892/ijo.2018.4376
54. Wu HM, Jiang ZF, Ding PS, Shao LJ, Liu RY. Hypoxia-induced autophagy mediates cisplatin resistance in lung cancer cells. Sci Rep. (2015) 5:12291–91. doi: 10.1038/srep12291
55. Zhang SP, Niu YN, Yuan N, Zhang AH, Chao D, Xu QP, et al. Role of autophagy in acute myeloid leukemia therapy. Chin J Cancer. (2013) 32:130–5. doi: 10.5732/cjc.012.10073
56. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science (1999) 285:727–9.
57. Salih HR, Rammensee H-G, Steinle A. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J Immunol. (2002) 169:4098–102. doi: 10.4049/jimmunol.169.8.4098
58. Holdenrieder S, Stieber P, Peterfi A, Nagel D, Steinle A, Salih HR. Soluble MICA in malignant diseases. Int J Cancer (2006) 118:684–7. doi: 10.1002/ijc.21382
59. Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res. (2011) 17:6287–97. doi: 10.1158/1078-0432.CCR-11-1347
60. Barsoum IB, Hamilton TK, Li X, Cotechini T, Miles EA, Siemens DR, et al. Hypoxia induces escape from innate immunity in cancer cells via increased expression of ADAM10: role of nitric oxide. Cancer Res. (2011) 71:7433–41. doi: 10.1158/0008-5472.CAN-11-2104
61. Siemens DR, Hu N, Sheikhi AK, Chung E, Frederiksen LJ, Pross H, et al. Hypoxia increases tumor cell shedding of MHC class I chain-related molecule: role of nitric oxide. Cancer Res. (2008) 68:4746–53. doi: 10.1158/0008-5472.CAN-08-0054
62. Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D, et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood (2013) 121:3599–608. doi: 10.1182/blood-2012-04-425397
63. Sarkar S, Germeraad WTV, Rouschop KMA, Steeghs EMP, van Gelder M, Bos GMJ, et al. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS ONE (2013) 8:e64835. doi: 10.1371/journal.pone.0064835
64. Figueiredo AS, Killian D, Schulte J, Sticht C, Lindner HA. Whole transcriptome data of primary human NK cells under hypoxia and interleukin 15 priming: A 2×2 factorial design experiment. Data in Brief (2017) 14:77–83. doi: 10.1016/j.dib.2017.07.018
65. Xu Y, Wang Y, Yan S, Zhou Y, Yang Q, Pan Y, et al. Intracellular adenosine regulates epigenetic programming in endothelial cells to promote angiogenesis. EMBO Mol Med. (2017) 9:1263–78. doi: 10.15252/emmm.201607066
66. Fredholm BB, Johansson S, Wang Y-Q. Adenosine and the regulation of metabolism and body temperature. Adv Pharmacol. (2011) 61:77–94. doi: 10.1016/B978-0-12-385526-8.00003-5
67. Hjemdahl P, Fredholm BB. Cyclic AMP-dependent and independent inhibition of lipolysis by adenosine and decreased pH. Acta Physiol Scand. (1976) 96:170–9. doi: 10.1111/j.1748-1716.1976.tb10186.x
68. Linden J. Adenosine in tissue protection and tissue regeneration. Mol Pharmacol. (2005) 67:1385–7. doi: 10.1124/mol.105.011783
69. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MKK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci USA. (2006) 103:13132–7. doi: 10.1073/pnas.0605251103
70. Young A, Mittal D, Stagg J, Smyth MJ. Targeting cancer-derived adenosine:new therapeutic approaches. Cancer Discov. (2014) 4:879–88. doi: 10.1158/2159-8290.CD-14-0341
71. Whiteside TL. Targeting adenosine in cancer immunotherapy: a review of recent progress. Expert Rev Anticancer Ther. (2017) 17:527–35. doi: 10.1080/14737140.2017.1316197
72. Muller-Haegele S, Muller L, Whiteside TL. Immunoregulatory activity of adenosine and its role in human cancer progression. Expert Rev Clin Immunol. (2014) 10:897–914. doi: 10.1586/1744666X.2014.915739
73. Chambers AM, Wang J, Lupo KB, Yu H, Atallah Lanman NM, Matosevic S. Adenosinergic signaling alters natural killer cell functional responses. Front Immunol. (2018) 9:2533. doi: 10.3389/fimmu.2018.02533
74. Wang J, Matosevic S. Adenosinergic signaling as a target for natural killer cell immunotherapy. J Mol Med. (2018) 96:903–13. doi: 10.1007/s00109-018-1679-9
75. Huang S, Apasov S, Koshiba M, Sitkovsky M. Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood (1997) 90:1600–10.
76. Ohta A. A Metabolic Immune checkpoint: adenosine in tumor microenvironment. Front Immunol. (2016) 7:109. doi: 10.3389/fimmu.2016.00109
77. Priebe T, Platsoucas CD, Nelson JA. Adenosine receptors and modulation of natural killer cell activity by purine nucleosides. Cancer Res. (1990) 50:4328–31.
78. Williams BA, Manzer A, Blay J, Hoskin DW. Adenosine acts through a novel extracellular receptor to inhibit granule exocytosis by natural killer cells. Biochem Biophys Res Commun. (1997) 231:264–9. doi: 10.1006/bbrc.1997.6077
79. Miller JS, Cervenka T, Lund J, Okazaki IJ, Moss J. Purine metabolites suppress proliferation of human NK cells through a lineage-specific purine receptor. J Immunol. (1999) 162:7376–82.
80. Cekic C, Day Y-J, Sag D, Linden J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. (2014) 74:7250–9. doi: 10.1158/0008-5472.CAN-13-3583
81. Goto T, Herberman RB, Maluish A, Strong DM. Cyclic AMP as a mediator of prostaglandin E-induced suppression of human natural killer cell activity. J Immunol. (1983) 130:1350–5.
82. Harish A, Hohana G, Fishman P, Arnon O, Bar-Yehuda S. A3 adenosine receptor agonist potentiates natural killer cell activity. Int J Oncol. (2003) 23:1245–9. doi: 10.3892/ijo.23.4.1245
83. Jeffe F, Stegmann KA, Broelsch F, Manns MP, Cornberg M, Wedemeyer H. Adenosine and IFN-{alpha} synergistically increase IFN-gamma production of human NK cells. J Leukoc Biol. (2009) 85:452–61. doi: 10.1189/jlb.0108046
84. Young A, Ngiow SF, Gao Y, Patch A-M, Barkauskas DS, Messaoudene M, et al. A2AR Adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res. (2018) 78:1003–16. doi: 10.1158/0008-5472.CAN-17-2826
85. Schindler U, Seitz L, Ashok D, Piovesan D, Tan J, DiRenzo D, et al. AB928, a dual antagonist of the A2aR and A2bR adenosine receptors, leads to greater immune activation and reduced tumor growth when combined with chemotherapy. Eur J Cancer (2018) 92:S14–5. doi: 10.1016/j.ejca.2018.01.037
86. Rautela J, Huntington ND. IL-15 signaling in NK cell cancer immunotherapy. Curr Opin Immunol. (2017) 44:1–6. doi: 10.1016/j.coi.2016.10.004
87. Mao Y, Hoef V van, Zhang X, Wennerberg E, Lorent J, Witt K, et al. IL-15 activates mTOR and primes stress-activated gene expression leading to prolonged antitumor capacity of NK cells. Blood (2016) 128:1475–89. doi: 10.1182/blood-2016-02-698027
88. Liu RB, Engels B, Arina A, Schreiber K, Hyjek E, Schietinger A, et al. Densely granulated murine NK cells eradicate large solid tumors. Cancer Res. (2012) 72:1964–74. doi: 10.1158/0008-5472.CAN-11-3208
89. Mineharu Y, Kamran N, Lowenstein PR, Castro MG. Blockade of mTOR signaling via rapamycin combined with immunotherapy augments anti-glioma cytotoxic and memory T cells' functions. Mol Cancer Ther. (2014) 13:3024–36. doi: 10.1158/1535-7163.MCT-14-0400
90. Viel S, Besson L, Marotel M, Walzer T, Marçais A. Regulation of mTOR, metabolic fitness, and effector functions by cytokines in natural killer cells. Cancers (2017) 9:E132. doi: 10.3390/cancers9100132
91. Yang C, Tsaih S-W, Lemke A, Flister MJ, Thakar MS, Malarkannan S. mTORC1 and mTORC2 differentially promote natural killer cell development. eLife 7:e35619. doi: 10.7554/eLife.35619
92. Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest. (2017) 127:929–41. doi: 10.1172/JCI89455
93. Antonioli L, Pacher P, Vizi ES, Haskó G. CD39 and CD73 in immunity and inflammation. Trends Mol Med. (2013) 19:355–67. doi: 10.1016/j.molmed.2013.03.005
94. Antonioli L, Blandizzi C, Malavasi F, Ferrari D, Haskó G. Anti-CD73 immunotherapy: a viable way to reprogram the tumor microenvironment. Oncoimmunology (2016) 5:e1216292. doi: 10.1080/2162402X.2016.1216292
95. Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, et al. Ecto-5'-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. (2002) 110:993–1002. doi: 10.1172/JCI15337
96. Beavis PA, Stagg J, Darcy PK, Smyth MJ. CD73: a potent suppressor of antitumor immune responses. Trends Immunol. (2012) 33:231–7. doi: 10.1016/j.it.2012.02.009
97. Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, et al. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci USA. (2010) 107:1547–52. doi: 10.1073/pnas.0908801107
98. Jin D, Fan J, Wang L, Thompson LF, Liu A, Daniel BJ, et al. CD73 on tumor cells impairs anti-tumor T cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res. (2010) 70:2245–55. doi: 10.1158/0008-5472.CAN-09-3109
99. Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, McGlinchey KA, et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology (2016) 5:e1208875. doi: 10.1080/2162402X.2016.1208875
100. Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res. (2013) 19:5626–35. doi: 10.1158/1078-0432.CCR-13-0545
101. Chatterjee D, Tufa DM, Baehre H, Hass R, Schmidt RE, Jacobs R. Natural killer cells acquire CD73 expression upon exposure to mesenchymal stem cells. Blood (2014) 123:594–5. doi: 10.1182/blood-2013-09-524827
102. Morandi F, Horenstein AL, Chillemi A, Quarona V, Chiesa S, Imperatori A, et al. CD56brightCD16– NK cells produce adenosine through a CD38-mediated pathway and act as regulatory cells inhibiting autologous CD4+ T cell proliferation. J Immunol. (2015) 195:965–72. doi: 10.4049/jimmunol.1500591
103. Terp MG, Olesen KA, Arnspang EC, Lund RR, Lagerholm BC, Ditzel HJ, et al. Anti-human CD73 monoclonal antibody inhibits metastasis formation in human breast cancer by inducing clustering and internalization of CD73 expressed on the surface of cancer cells. J Immunol. (2013) 191:4165–73. doi: 10.4049/jimmunol.1301274
104. Häusler SF, Del Barrio IM, Diessner J, Stein RG, Strohschein J, Hönig A, et al. Anti-CD39 and anti-CD73 antibodies A1 and 7G2 improve targeted therapy in ovarian cancer by blocking adenosine-dependent immune evasion. Am J Transl Res. (2014) 6:129–39. Available online at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3902223/
105. Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, et al. Co-inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell (2016) 30:391–403. doi: 10.1016/j.ccell.2016.06.025
106. Hong C-S, Sharma P, Yerneni SS, Simms P, Jackson EK, Whiteside TL, et al. Circulating exosomes carrying an immunosuppressive cargo interfere with cellular immunotherapy in acute myeloid leukemia. Sci Rep. (2017) 7:14684. doi: 10.1038/s41598-017-14661-w
107. Sauer AV, Brigida I, Carriglio N, Hernandez RJ, Scaramuzza S, Clavenna D, et al. Alterations in the adenosine metabolism and CD39/CD73 adenosinergic machinery cause loss of Treg cell function and autoimmunity in ADA-deficient SCID. Blood (2012) 119:1428–39. doi: 10.1182/blood-2011-07-366781
Keywords: immunometabolism, natural killer cells, immunotherapy, tumor microenvironment, adenosine
Citation: Chambers AM, Lupo KB and Matosevic S (2018) Tumor Microenvironment-Induced Immunometabolic Reprogramming of Natural Killer Cells. Front. Immunol. 9:2517. doi: 10.3389/fimmu.2018.02517
Received: 30 May 2018; Accepted: 12 October 2018;
Published: 08 November 2018.
Edited by:
Kate Stringaris, National Institutes of Health (NIH), United StatesReviewed by:
Anna Karolina Kozlowska, Poznan University of Medical Sciences, PolandChristine Susanne Falk, Hannover Medical School, Germany
Copyright © 2018 Chambers, Lupo and Matosevic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandro Matosevic, c2FuZHJvQHB1cmR1ZS5lZHU=
†These authors have contributed equally to this work