 Keisuke Watanabe
Keisuke Watanabe Shunichiro Kuramitsu
Shunichiro Kuramitsu Avery D. Posey Jr.
Avery D. Posey Jr. Carl H. June1,2,4*
Carl H. June1,2,4*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 26 October 2018
Sec. Cancer Immunity and Immunotherapy
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02486
This article is part of the Research Topic CAR-T Cell Therapies for Non-Hematopoietic Malignancies: Taking Off The Training Wheels View all 12 articles
A major obstacle for chimeric antigen receptor (CAR) T cell therapy in solid tumors is the lack of truly tumor-specific target antigens, which translates to the targeting of tumor-associated antigens (TAAs) overexpressed on tumors but shared with normal organs, raising safety concerns. In addition, expression of TAAs in solid tumors is particularly heterogeneous. In this regard, it is critical to deeply understand the sensitivity of CAR T cells, especially against low-density targets and the possible therapeutic window of antigen density targeted by CAR T cells. In this review, we discuss the recent findings of mechanisms of antigen recognition through CAR, including immunological synapse formation, and the impact of target antigen density for induction of distinct T cell functions. We also discuss rational strategies to adjust and expand the therapeutic window for effective and safe targeting of solid tumors by CAR T cell platforms.
Chimeric antigen receptor T cell (CAR T cell) therapy has shown significant efficacy in hematological malignancies (1–3). Recently the U.S. FDA approved two types of CD19-targeting CAR T cells, tisagenlecleucel (Kymriah™–Novartis) in leukemia (August 2017) and lymphoma (May 2018) and axicabtagene ciloleucel (Yescarta™–Kite) in lymphoma (October 2017). The compelling success of CD19-specific CAR T cell therapies propels the development of CARs that can induce similar efficacy in solid tumors; however, the process is faced with multiple challenges that must be addressed to achieve sufficient efficacy.
Among the many challenges of CAR T cell therapy in solid tumors, a major obstacle is the lack of truly tumor-specific target antigens, which forces cellular immunologists to target tumor-associated antigens (TAAs) overexpressed on tumors but also expressed on normal tissues and organs, raising safety concerns. For instance, fatal cytokine release syndrome (CRS) has been reported from the targeting of human epidermal growth factor receptor 2 (HER2) with CAR T cells due to the recognition of low-levels of HER2 expressed on the normal cells of lung epithelium (4). Also, carbonic anhydrase IX-specific CAR T cells in renal cell cancer induced liver toxicities (5) and carcinoembryonic antigen (CEA)-specific transgenic T cell receptor (TCR) T cells induced severe colitis in colon cancer patients (6). In addition, the tumor microenvironment (TME) of solid tumors is particularly immunosuppressive, which prevents effective anti-tumor immune responses. The immunosuppressive TME contains multiple components including physical barriers, such as a dense extracellular matrix; dysfunctional epithelial cells; metabolic checkpoints, such as hypoxia and immunological barriers, such as immunosuppressive cytokines/molecules and immunosuppressive immune cells. To target such tumors effectively, multiple factors impacting efficacy and toxicity must be simultaneously addressed.
In this regard, it is critical to deeply understand CAR T cell biology and multiple factors that can affect the therapeutic window of CAR T cell therapies. In this review, we discuss the recent findings of mechanisms of antigen recognition through CARs, including immunological synapse (IS) formation, impact of target antigen density for induction of distinct T cell functions, and the kinetics of target cell killing. We also discuss rational strategies to adjust and expand the therapeutic window for effective and safe targeting of solid tumors by CAR T cell platforms.
While basic mechanisms by which T cells interact with targets through T cell receptors have been intensively investigated, those of CAR-target interactions are less well understood. As CARs consist of combined parts of the TCR complex and antibodies, it will be valuable to discuss the similarities of CARs to endogenous, unmodified TCR T cells and define distinct differences of CARs to better understanding CAR T cell biology (Table 1).
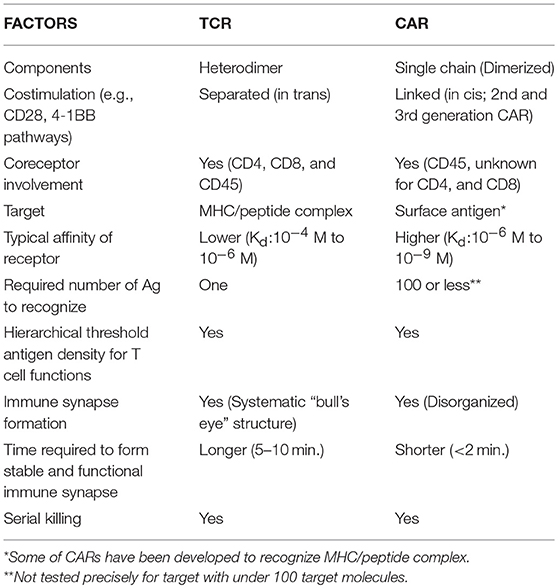
Table 1. Comparison of CAR and TCR T cell biological factors.
The TCR is a heterodimer of two subunits: a TCRα subunit and a TCRβ subunit. Each subunit contains a variable region domain (V) and a constant region domain (C), which is followed by a transmembrane region. Each V domain contains three complementarity-determining regions (CDRs), which interact with peptide presented on the major histocompatibility complex (MHC). The TCR itself does not possess signaling domains, requiring intracellular signaling to be initiated by the CD3 complex. The CD3 complex consists of three dimers, CD3ζε and CD3δε heterodimers and CD3 ζζ homodimer (7). The CD3γ/δ/ε subunits each consist of a single extracellular immunoglobulin (Ig) domain and an immunoreceptor tyrosine-based activation motif (ITAM), whereas CD3ζ has a short extracellular domain (ECD) and three ITAMs (8, 9). TCR and CD3 subunits form a complex on the T cell surface (TCR-CD3 complex).
CARs are synthetic chimeric proteins that are introduced into T cells to redirect antigenic specificity and enhance cellular functionality (10). CARs typically consist of a single-chain variable fragment (scFv) from a mAb, an extracellular spacer region (termed hinge), a transmembrane domain, CD3ζ signaling domain, and usually one or two costimulatory domain(s) for second-generation or third-generation CARs, respectively (11–14). Atypical constructions of CARs utilize receptor ligands or peptides as the extracellular antigen-recognition domain, such as zetakine CARs—e.g., interleukin-13 receptor alpha 2 (IL13Rα2) zetakine CARs (15). CARs endow T cells with the benefit of directly binding surface antigens via scFv (antibody recognition) in an MHC-independent manner, which allows activity from the same CAR molecule in both CD4 and CD8 T cells and reactivity against patient tumors regardless of histocompatibility. CARs can transmit signals through CD3ζ and costimulatory domains simultaneously to the T cell, which can induce a stoichiometric and potentially ideal activation of T cells.
T cell activation is mediated through highly organized and dynamic interaction of TCRs with MHC-peptide complexes, referred to as an IS. A matured IS is an aggregation of TCR-based signalosomes that induce T cell responses. The IS is defined by three concentric rings of clustered molecules (Figure 1A). The inner circle of an IS is termed the central supramolecular activation cluster (cSMAC), where TCR signaling takes place. The cSMAC contains most of the TCR-MHC-peptide complexes, CD28, PKC-θ, and Lck, whereas peripheral SMAC (pSMAC) contains proteins involved in cell adhesion, such as integrin LFA-1, cytoskeletal linker talin, and ICAM1. Large molecules, such as CD43 and CD45, are excluded from the pSMAC and make up the distal SMAC (dSMAC). Inhibitory and costimulatory molecules, such as PD-1, CTLA-4, and ICOS also are aggregated at the region of IS and play crucial roles in the regulation of T cell activation (16).
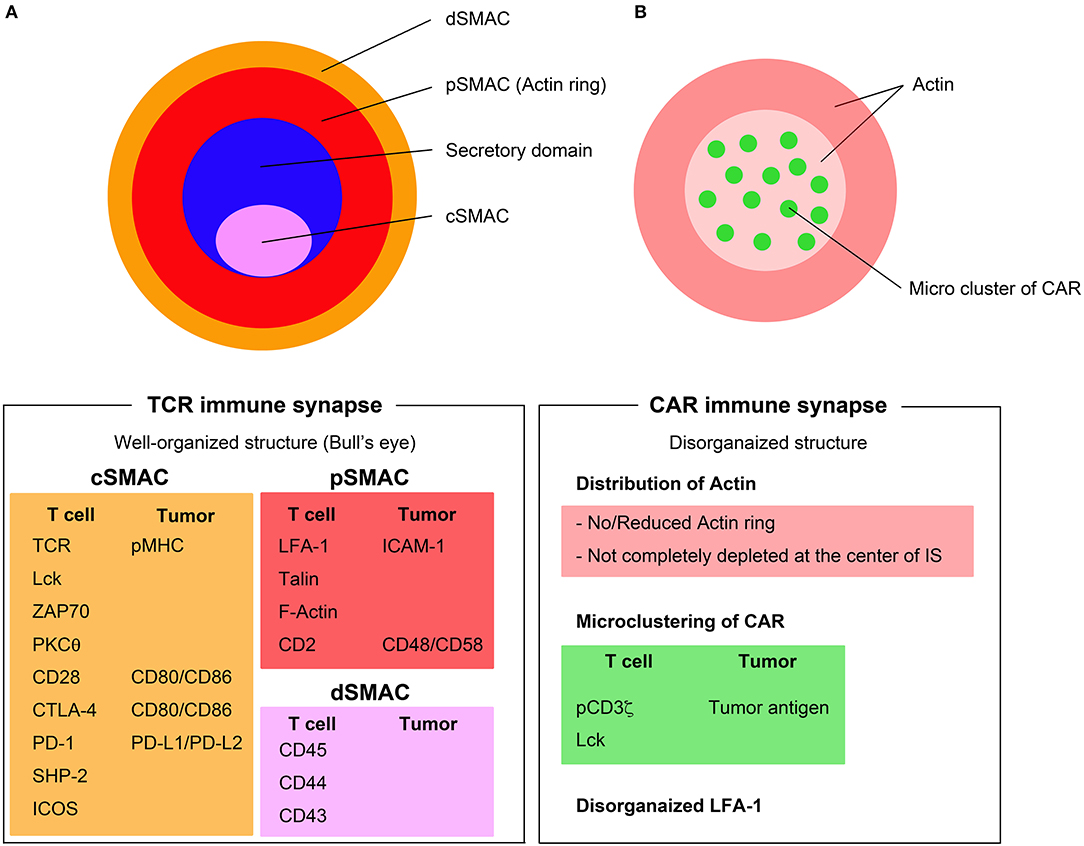
Figure 1. Immunological synapse formation through TCRs and CARs. (A) TCR immune synapse shows a well-organized bull's eye structure including the central supramolecular activation complex (cSMAC) (pink), the peripheral SMAC (pSMAC) (red), and distal SMAC (dSMAC) (orange). (B) CAR immune synapse (right) displays disorganized structure with no/reduced actin ring and microclusters of CAR/tumor antigen in a disorganized pattern. Major components of the TCR and CAR immune synapse are listed below the figures.
Secretion of lytic granules occurs within a variant of IS (namely secretory synapse) between cytotoxic T lymphocytes (CTLs) and target cells. The secretary synapse has two separate and distinct domains in cSMAC: one is a signaling domain, which contains the signaling proteins, and another is a secretory domain for exocytosis of cytokines, perforins, and granzymes. Stinchcombe et al. demonstrated that the transient polarization and docking of the centrosome to the plasma membrane, which is controlled by Lck signaling, has an important role in the mechanism of directing this secretion (17–19).
The intracellular signaling downstream of CARs and the mechanisms of the IS formed by CARs have not been extensively studied. It has been demonstrated that CAR clustering, ZAP70 recruitment to IS, and exclusion of CD45 outside of IS occurs between CD19-specific CAR T cells and target cells that is similar to TCR activation. Downstream signaling molecules of the TCR, such as CD3ζ, LAT, Lck, and ZAP70 are phosphorylated after CD19-CAR T cell activation by autologous CD19+ B cells (20). In this study, third-generation CAR T cells had a significantly higher phosphorylation status on downstream TCR signaling molecules, and another study demonstrated that third-generation CARs, specifically those incorporating CD28 and 4-1BB costimulatory domains, induced a stronger PI3K/Akt activation when compared to second-generation CAR T cells upon in vitro exposure to antigen (21).
The formation of CAR IS has characteristics unlike the structure of TCR IS. The CAR IS does not present a systematic bull's eye structure, which is a characteristic feature of TCR IS. Organization of the actin ring in CAR IS is poor and actin may not be not completely diminished at the center of CAR IS (22). LFA-1 is disorganized and CAR-tumor antigen complexes form microclusters that are randomly distributed at the CAR IS (23) (Figure 1B). While TCR IS requires 5–10 min to form the bull's eye structure, the CAR IS might not need to form these stable structures because the disorganized multifocal pattern of CAR IS is sufficient to rapidly induce significant proximal signaling, which occurs within a short period of time (<2 min). Another important part of IS biology is the delivery of cytotoxic granules, including perforin and granzymes, to the IS mediated by microtubule organizing center (MTOC) (24). The rapid but short duration of proximal signaling of CAR IS also induces rapid MTOC migration to the IS and accelerates the delivery of granules (23). Although the mechanisms of CAR IS have gradually been revealed, it is still unclear whether the differences in CAR IS structure correlate with the efficacy of CAR T cells.
Soluble forms of CAR ligands, such as CD30, mesothelin, and CEA, that exist in monomeric forms cannot trigger CAR signaling (25–27), which is reasonable since they will not induce CAR dimerization. However, CAR T cells can potentially recognize soluble ligands that can exist in oligomeric forms, such as TGF-β, even without cell-cell interaction. Chang et al. recently demonstrated that TGF-β captured by an anti-TGF-β CAR could induce an IS, mimic actin-dependent CAR dimerization, and trigger T cell signaling (28). They also showed that the CAR response to the soluble ligands can be tuned by adjusting the extracellular spacers and the intracellular signaling domains of CARs. These findings reveal mechanisms by which the structures of CARs influence signaling and can also lead to strategies of engineering CAR T cells to overcome tumor immunosuppression by converting TGF-β from a potent immunosuppressive cytokine to a CAR T cell activator.
It has been demonstrated through fluorescence microscopy that, under optimal conditions, as few as one peptide-MHC complex is sufficient to trigger T-cell activation, IL-2, and TNF-α secretion (29, 30), while a contradictory report suggested that four peptide-MHC complexes are the minimum required amount of agonists for half-maximal activation and calcium flux of CD4+ T cells (31). This high sensitivity of TCR signaling may reflect the unique role of the TCR, which requires the detection of a very rare foreign peptide presented on MHC in the presence of thousands of presented self-peptides. Orchestrated assembly of the receptor complex system may provide such high sensitivity while retaining specificity. The co-receptors CD4 and CD8 also participate in the binding and proximal signaling upon TCR interaction with peptide-MHC. For instance, CD4 acts to reduce the amount of peptide-MHC required from over 30 molecules/target cells to just one molecule (29). Interestingly, TCRs have a hierarchical threshold of antigen density for induction of cell lysis, proliferation, and cytokine production (32), where less antigen density is required for cell lysis than for cytokine production. This phenomenon is observed in the single cell levels but not as a T cell population (33).
To address the question of thresholds for CAR activation, Watanabe et al. investigated the density of CD20 required to activate CD20-specific CAR T cells (CD28 co-stimulation domain) with target cells expressing ~200–250,000 CD20 molecules per cell (34). Target cells expressing the lowest density of CD20 within the set of the target cells (~200 molecules/cell) could induce lysis by CAR T cells. This data was consistent with a previous report that CAR targeting a tumor-specific glycoepitope of murine OTS8 that could lyse target cells with similarly low density (~200 molecules/cell) of target antigen (35). This study also demonstrated that the CAR format is more sensitive than bi-specific T cell engagers (BiTEs) constructed with the same scFv.
Watanabe et al. also demonstrated that the target antigen density that is required to induce T cell proliferation and cytokine production was higher than that required to induce CAR mediated lysis: CD20-specific CAR T cells could lyse target cells with 200 molecules/cell, but cytokine production and T cell proliferation required a higher density of CD20, nearly 5,000 molecules/cell. In addition, recently Walker et al. investigated target antigen density required to activate ALK-specific CAR T cells with the 4-1BB co-stimulatory domain using Nalm6 cells expressing various densities of ALK (~18,000–450,000 molecules/cell) (36). In this model, production of IFN-γ, IL-2, and TNF-α showed a sharp threshold dependence on tumor antigen density and there was a significantly higher threshold for IL-2 production compared to IFN-γ production. IL-2 production required 60,000 molecules/cell and IFN-γ production required 30,000 molecules/cell to induce a half maximal response. CAR T cells could lyse target cells with the lowest ALK expression in this target cell panel (~18,000 molecules/cell); however, another panel of target cells with much lower ALK expression will be required to determine the absolute minimal density required for lysis. Liu et al. demonstrated that affinity-tuned anti-HER2 CARs consisting of scFvs derived from high affinity antibodies could degranulate when targeting very low HER2-expressing cells, where the expression was below detection capabilities by flow cytometric analysis (37). Although the number of target molecules was not determined in this study, this result suggests that CARs have a considerably lower threshold of antigen expression for target cell lysis; this threshold may be CAR- or scFv-dependent. There is a one-log difference in the threshold of IFN-γ production between the reports of Watanabe et al. and Walker et al. a discrepancy that might result from differences in the CAR constructs (e.g., affinity of scFv, hinge, co-stimulatory domain) or from the density of CAR expression, two features that could be utilized to more precisely enable control of CAR T cell activation.
These results suggest that CAR T cells can recognize target cells with considerably low levels of target antigen and that they have hierarchical T cell signaling thresholds for cell lysis, proliferation and individual cytokine production. It is likely that each T cell subset or each single T cell has a distinct threshold to be activated, as shown in TCR T cells, but this has not yet been investigated for CAR T cells.
Endogenous T cells and NK cells can sequentially lyse multiple target cells (serial killing), which is likely to be necessary for tumor eradication (38, 39); however, the ability of CAR T cell to mediate serial killing and the kinetics of target cell lysis had not been fully demonstrated until recently. Davenport and colleagues tested the functions of cytotoxic T lymphocytes (CTLs) activated through either the endogenous TCR or an ectopically expressed CAR using a novel transgenic mouse model in which individual T cells co-expressed two antigen receptors (OT-I TCR and anti-HER2 CAR) (40). The authors clearly demonstrated that CAR T cells were serial killers using time-lapse video microscopy; approximately 22% of CAR T cells sequentially delivered a lethal hit to two or three tumor cells and the frequency of serial killing through the CAR was comparable to that through the TCR. Using kinetic analysis of tumor cell killing via a real-time impedance-based assay, the authors showed that CTLs elicited equivalent killing kinetics of target cells regardless of whether recognition occurred through the TCR or CAR for the first 20 h, but CAR-mediated lysis slowed its cytolytic kinetics compared to TCR-mediated lysis after 20 h. This difference in sustained lysis kinetics may be explained by CAR downmodulation after stimulation upon the antigen recognition, which can be ameliorated through TCR-based expression of CAR (41).
T cell activation is regulated by the interaction between the TCR and MHC-peptide complex and the major factors that have influence on the sensitivity of activation are target antigen density and TCR affinity. One of the major immunosuppressive mechanisms in the cancer microenvironment is failed antigen recognition due to low-affinity TCR and cancer associated peptide-MHC complex interactions (42, 43). TCR affinities to self-derived peptides, such as cancer antigens, are lower than TCR affinities to pathogen-derived antigens (44). Therefore, it is generally more difficult to isolate T cells that have sufficient sensitivity to TAAs than to identify pathogen-derived antigen-specific T cells from patients, and this is the first hurdle of adoptive cell therapies (ACTs) (45).
High TCR affinity is, on the other hand, accompanied by autoimmune responses, which sometimes leads to serious adverse events when patients are treated with ACTs. Although the affinity of TCRs is known to be μM range (Kd:10−4M−10−6M), Zhong et al. reported that T cell antitumor activity and autoimmunity are closely coupled but plateau at a defined TCR affinity of 5–10 μM, which suggests that ACT utilizing supra-physiologic, high-affinity TCRs does not improve efficacy (46, 47).
It has also been reported that a small number of peptide-MHC complexes can achieve a high TCR occupancy since a single complex can serially engage and trigger hundreds of TCRs (48–50). Altogether, this suggests a model where the ideal affinity of TCR should provide interaction sufficiently long enough to transduce proximal signaling but appropriately short to detach and allow as many TCRs to encounter MHC-peptide complexes as possible.
The influence of the scFv affinity on CAR T cell functional response is still incompletely understood. In general, CARs constructed with scFvs possess higher affinity (in the nM range, Kd:10−6 M−10−9 M) compared to native TCR affinities. Since most TAAs are highly expressed on tumors and at lower levels on normal tissues, it is essential to consider the threshold of the stimulation to yield optimal specificity of CAR-redirected T cell activation since there is a risk that increasing the affinity of CARs will lead to serious adverse effects due to on-target, off-tumor recognition (37, 51). The high affinity of the 4D5 (trastuzumab) scFv may be responsible for the fatal pulmonary toxicity and CRS that was attributed to anti-HER2 CAR reactivity against low level HER2 expression in the normal lung (4). As mentioned before, one strategy to increase the therapeutic index for TAA such as HER2 is affinity tuning of the scFv to generate HER2-specific CAR T cells unable to degranulate in response to normal human primary cells with low level HER2 expression (37).
Similar to native T cells, CAR T cells can also kill multiple target cells in a sequential fashion. However, tumor cells can be eliminated more rapidly when stimulated through CARs than through TCRs because CARs can dissociate from dying tumor cells more rapidly than TCRs (40). Hence, increasing the affinity of CAR T cells may reduce or prevent serial killing, promote T cell exhaustion, and decrease the generation and persistence of central memory and effector phenotype T cells (52), or increase the loss of T cells through activation-induced cell death (53).
A critical part of adoptive T cell therapy is the selection of the target antigen, in order to deliver sufficient efficacy and minimize toxicity. Some CARs targeting tumor-specific antigens have been developed pre-clinically, including CARs targeting aberrantly glycosylated oncogenes, such as the Tn glycoform of MUC1 (54), and tumor-specific activating forms of integrin (55), and clinically, such as CARs targeting the tumor-specific transcriptional variants EGFRvIII in glioblastoma (56). In the absence of more cancer-specific targets, CAR T cell therapies will most likely continue to target TAAs for solid tumors that also exhibit expression on normal tissues. Indeed, most of ongoing clinical trials of CAR T cell therapies for solid tumors are targeting such TAAs (57).
It is critical to know whether normal tissues express the antigen and its expression levels in order to predict potential toxicities. Several public databases of antigen expression on the normal tissues are available based on gene expression (RNAseq or microarrays) or immunohistochemistry (IHC). However, such technologies contain limitations and pitfalls. For gene expression analysis, antigens expressed by very rare but critical cells may be underestimated. In addition, it may not be possible to distinguish if expressed genes are derived from tissues or from infiltrating cells. For instance, some databases suggest that human intestine is CD4 positive; however, this expression likely represents infiltrated CD4 T cells rather than intestinal tissues themselves. The accuracy of IHC staining in public databases largely depends on the quality of the antibody, its affinity and the epitope for the antigen. For instance, the cancer-specific Tn glycoform of MUC1 recognized by the high-affinity antibody developed by (58) is extremely rare and unlikely to be identified in public IHC databases. Instead, researchers are more likely discover staining for antibodies developed against the normal glycoform of the antigen; in this case, the broad epithelial expression of normally-glycosylated MUC1 would credential it as an unsafe target for CAR T cells. Similar arguments could be made for the lack of cancer-specific splice variants in public IHC databases, such as EGFRvIII vs. EGFR expression. In addition, false positives and false negatives are problems not yet resolved and the sensitivity of IHC for low-expressing antigens may not be sufficient to select CAR targets for solid tumors. These limitations and pitfalls are well discussed in a previous review (59). New technologies, such as single cell RNA sequencing, may provide more accurate expression profiles that enable researchers to better predict efficacy and toxicity of novel CAR T cells.
“Therapeutic window” is a term originally from pharmaceutical toxicology and defined as a range of doses between efficacy and toxicity, achieving the highest therapeutic benefit without resulting in unacceptable toxicity; it is the range between the minimum effective dose (MED) and the maximum tolerated dose (MTD) (Figure 2A). Although the pharmacokinetics of engineered replicating cells are largely different from that of drugs, applying the concept of the therapeutic window to the field of ACTs will be valuable for optimizing the therapies. In CAR T cell therapies, targeting antigens expressed exclusively on tumor cells or antigens that are expressed only on non-critical tissues widens the therapeutic window as direct toxicity on vital tissues would not occur (Figure 2B). On the other hand, targeting antigens that are expressed in critical normal tissues/cells narrows the window by decreasing MTD (Figure 2C).
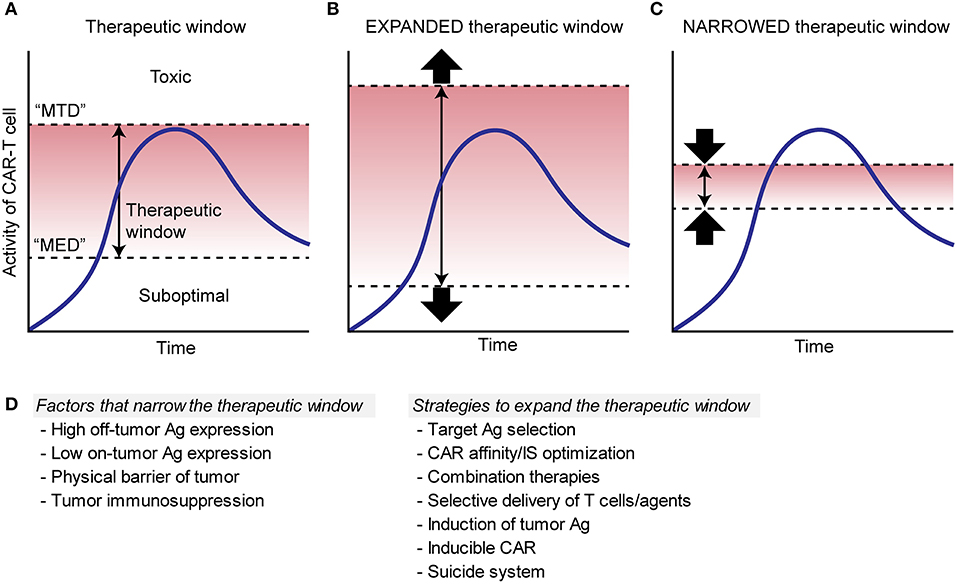
Figure 2. Schema to illustrate therapeutic window of CAR T cell therapies. (A). Therapeutic window (red area) is determined as a range between the minimum effective dose (“MED”) and the maximum tolerated dose (“MTD”), where the therapy can achieve the highest therapeutic benefit without resulting in unacceptable toxicity. Blue lines show the kinetics of CAR T cells. (B). Red area shows an expanded therapeutic window with decreased “MED” and/or increased “MTD,” which increase a chance for the CAR T cells to induce sufficient efficacy without toxicity. (C). Red area shows a narrowed therapeutic window with increased “MED” and/or decreased “MTD,” which can cause toxicity and/or suboptimal efficacy. (D). Factors that can narrow the therapeutic window and possible strategies to expand the window are listed.
Determination of the therapeutic window cannot be resolved solely based on the profile of antigen expression. For instance, even in the case of a large differential in antigen expression by tumors and normal tissues, where the antigen is expressed at higher density in tumors, tumors may still be more resistant to CAR T cells than normal tissue due to inherent immunosuppression within the TME that does not exist within normal tissue. In this case, inhibition of T cell infiltration or induction of T cell hypofunction by the tumor would narrow the therapeutic window of CAR T cells by increasing the MED.
Given that truly tumor-specific target surface antigens have as of yet been rarely found, TAAs with shared expression of normal organs may be our only reasonable targets for the foreseeable future; therefore, strategies to expand the therapeutic window of CAR T cell therapy are necessary for the treatment of solid tumors. The possible approaches to expand therapeutic window include: (1) optimizing CAR affinity and sensing, (2) optimizing immunological synapse formation, (3) combination therapies, (4) local delivery of CAR T cells and therapeutic agents, (5) induction of target antigen expression, or (6) other modifications (Figure 2D).
Although increasing CAR affinity enables recognition of antigens independent of target density (60), that action may cause serious adverse effects, namely on-target, off-tumor toxicity, and reduce the capability of sequential target tumor killing. Therefore, it is important to address rational strategies to determine ideal CAR affinity. The construction of affinity-tuned scFvs using light-chain exchange technology is one of the most feasible methods to measure the optimal affinities of CARs. With this method, Drent et al. identified that CD38-CAR T cells with ~1000-fold lower affinity to the original antibody that exhibited optimal proliferation, cytokine production, and cytotoxicity in vitro and in vivo, but spared normal tissue, compared with high-affinity CAR T cells (61).
Increasing the affinity of scFvs beyond a defined threshold (Kd <10−8 M) does not necessarily induce improved T cell activation (37, 62, 63). For instance, μM affinity CAR T cells exhibited superior cytokine production, expansion and antitumor efficacy, and less systemic off-tumor toxicity compared to nM affinity CAR T cells (64). These reports suggest that CAR T cells with affinities above a defined threshold are not necessarily required, or rather it may be important to generate CARs with varied affinities for the same epitope, and to identify the lowest affinity at which those epitope-specific CAR T cells can exhibit maximal cytolytic, proliferative, and safety potential.
Apart from changing scFv affinity, regulating the level of surface CAR expression is an important factor to induce ideal CAR signaling. CAR T cell function is governed by CAR density as well as target antigen density, where low expression of either can result in limited functionality and sensitivity of CAR T cells (36). On the other hand, continuous signaling (tonic signaling) through CAR can occur depending on the CAR structure and high CAR density, which can induce inferior antitumor effects and T cell engraftment in vivo by increasing T cell differentiation, exhaustion and activation induced cell death (AICD) (65, 66). Modifying CAR density while maintaining expression under the threshold required for tonic signaling induction will be required to induce sufficient anti-tumor efficacy and to keep safety potential for each target antigen and CAR construct. Another approach is combinatorial antigen recognition through two different antigens on tumor cells. Split-signaling CAR T cells have been engineered such that CAR-1 drives only the activation signal (signal 1) of CD3ζ and CAR-2 drives only co-stimulation (signal 2) through co-stimulatory molecules, such as CD28 and 4-1BB. Thereby, CAR T cells can be optimally activated only upon recognition of two separate required antigens simultaneously (“AND” logic gated CAR). This approach has been tested in CAR T cells with split signals utilizing anti-HER2 and anti-MUC1 CARs (67) or anti-CD19 and anti-PSMA CARs (68) in pre-clinical models. Conversely, CAR T cells can be modified so that CARs can drive full signaling upon recognizing either of two different antigens by expressing two CARs or a single CAR with tandem antigen binding domains (“OR” logic gated CAR) (69–71). Engineering T cells with “AND” logic CARs enables more specific and safer targeting and those with “OR” logic CARs potentially overcomes low target antigen expression and tumor escape by target antigen loss.
The use of adapter molecules to develop a “universal” CAR can be another attractive platform to overcome tumor heterogeneity in antigen expression and to make CAR T cell activities more conditional. Urbanska et al. described a biotin-binding immune receptor composed of an extracellular-modified avidin linked to an intracellular T cell signaling domain which can recognize tumor cells pre-treated with antigen-specific molecules such as mAb, scFv, or other tumor-specific ligands (72). Tamada et al. similarly described anti-FITC CAR which can target those pre-treated with FITC conjugated mAbs, and they demonstrated that anti-FITC CAR T cell activity can be attenuated by injecting FITC-labeled non-specific IgG Ab in a preclinical model (73). This platform enables flexible multiple tumor antigen targeting, which potentially prevents target antigen loss and ease off-tumor toxicity by dividing off-tumor toxicities. This system also enables control of CAR T cell activities by adjusting doses of adopter molecules, or more actively, by quenching CAR by adding the excess amount of non-specific tagged molecules. However, it is still unclear whether each adaptor will induce equal activity when ligated to the acceptor CAR molecule. For instance, it has been revealed that the length and composition of the CAR hinge influences the activity of the T cells and the epitope of the antibody also influences this (whether it is distal or proximal to the target cell membrane) (74–76). It will be needed to address these factors so that they each provide an optimal CAR signal. Another potential problem of this platform to translate to the clinic will be the immunogenicity of the adapter molecules.
As discussed above, the CAR IS is more disorganized when compared to the well-organized bull's eye structure of TCR IS and is characterized by a multifocal pattern of Lck arrangement, decreased actin rings, and diffuse LFA-1 distribution. The remarkable capabilities of CAR IS are virtually instant induction of proximal signaling and rapid delivery of cytotoxic granules mediated by a faster migration of MTOC to the CAR IS compared to the TCR IS. These superiorities enable CAR T cells to dissociate quickly from destructed tumor cells and to mediate efficient serial killing.
Recently, several studies have reported important findings on how CAR design affects IS formation. Xiong et al. examined the quality of the CAR IS using CD19-specific CAR constructed with either CD28 or 4-1BB co-stimulatory domains and determined that CD28 plus 4-1BB-based third generation CARs are superior to CD28 based second generation CARs, as measured by IS structure, signaling and function (22). In comparison to bi-specific CAR T cells, CARs specific for two glioma-associated antigens, HER2 and IL13Rα2, exhibited significantly higher F-actin accumulation and increased polarization of the MTOC.
The structural characteristics of CAR IS are now in the beginning of elucidation. Although further studies are essential to reveal the correlation between CAR IS structure and the anti-tumor efficacy, modulation of CAR IS would be a great option of CAR T cell therapy if it is possible to increase the efficacy. There have been several attempts to improve the efficacy of CAR T cells by modifying CAR IS through immunomodulatory drugs (IMiDS), such as lenalidomide—a synthetic derivative of thalidomide. Lenalidomide improves CAR efficacy by increasing actin accumulation at the IS and is a promising combinatorial treatment for enhanced CAR activity (77, 78).
Combinatorial approaches may serve as a promising strategy to drive CAR T cell therapy toward solid tumors by overcoming tumor heterogeneity and expanding the therapeutic window. Oncolytic viruses (OVs) are promising agents for the treatment of solid tumors, and an oncolytic herpes virus expressing GM-CSF has been FDA-approved for the therapy of advanced melanoma based on therapeutic benefit in a clinical study (79). OVs can be programmed to specifically target, replicate in, and lyse cancer cells, while sparing normal cells. The release of virus progeny results in an exponential increase of the virus inoculum, which can cause direct tumor lysis while providing danger signals necessary to awaken the immune system (80). Furthermore, OVs can be genetically modified to express therapeutic transgenes selectively in the TME. Their ability to revert tumor immunosuppression while locally expressing therapeutic transgenes provides a rational strategy for combination with CAR T cell therapies. Indeed, we and other researchers reported enhanced CAR T cell efficacy by combining OVs expressing either cytokines (81), chemokines (82), an anti-PD-L1 minibody (83), a BiTE (84), or the combination of them against solid tumors in pre-clinical mouse models. We have shown that an oncolytic adenovirus expressing IL-2 and TNF-α enhanced the efficacy of mesothelin-redirected CAR T cells, which was associated with enhanced T cell infiltration to the tumor bed and reduced metastases (81). Murine TNF-α and murine IL-2 delivered by adenovirus could increase the efficacy of mesothelin-redirected CAR T cells in immunocompetent mice engrafted with highly immunosuppressive syngeneic LSL-KrasG12D/+;LSL-Trp53R172H/+;Pdx-1-Cre (KPC) mice derived-pancreatic ductal adenocarcinoma (PDA) tumor, whereas multiple injections of anti-mesothelin-CAR T cell monotherapy failed to suppress tumor growth. This combination approach enhanced the efficacy of CAR T cells and did not induce off-tumor toxicity.
Other combinatorial approaches include combination with agonistic antibodies specific for the 4-1BB costimulatory receptor (85), which can directly activate CAR T cells and also can reduce host immunosuppressive immune cells, such as Tregs or MDSCs.
Although efficient trafficking of CAR T cells to cerebrospinal fluid in patients with central nervous system (CNS) involved acute lymphoblastic leukemia has been reported (1), the response of primary or metastatic solid tumors in the CNS may be limited by the accessibility of CAR T cells. On the other hand, enhancing the strength of systemically administrated CAR T cells can raise safety concerns, as reported in HER2-redirected CAR T cell therapy. Direct administration of CAR T cells into the tumor bed is an optional route of drug delivery. Priceman et al. demonstrated that intraventricular delivery of HER2-CAR T cells shows antitumor activity against brain-metastatic breast cancer in orthotopic xenograft models, whereas intravenous delivery of HER2-CAR T cells achieved only partial antitumor responses in mice even at 10-fold higher doses compared with local or regional delivery to the brain (86). In confirmation of this administration route, intraventricular administration of IL13Rα2-targeting CAR T cells induced regression of all intracranial and spinal tumors in a patient with recurrent multifocal glioblastoma (87). We have tested intratumoral administration of mRNA-transduced anti-c-Met CAR T cells in patients with metastatic breast cancer (88) in a clinical trial and confirmed feasibility of this approach for clinical use.
Another approach is to engineer CAR-T cells to work only or dominantly in the tumor site. Han et al. developed the “masked CAR” system, which consists of a masking peptide that blocks the antigen-binding site and a protease-sensitive linker. The authors demonstrated that proteases commonly active in the TME (and presumably inactive in normal tissue) can cleave the linker and disengage the masking peptide, which enables CAR T cells to recognize target antigens only at the tumor site (89).
To overcome the immunosuppressive TME, local delivery of cytokines and chemokines, such as IL-18, IL-12, and the combination of CCL19 and IL-7 (90–92) or checkpoint blocking agents (93) by CAR T cells within the TME may help to overcome impediments to T cell infiltration and functionality. These approaches demonstrate enhanced therapeutic efficacy, while avoiding systemic adverse events in pre-clinical models.
As discussed here, target antigen density can govern the efficacy of CAR T cell therapy. In addition, the loss or down-regulation of target antigen is a major cause of tumor escape (94). Induction or re-induction of antigen expression on target cells may be an attractive approach to expand the therapeutic window. It has been reported that a sublethal dose of radiation can induce the expression of TAAs, such as mesothelin and CEA, on tumor cells (95). Epigenetic control may also modulate target antigen expression; in that vein, an anti-methylation drug, azacytidine (5-AZA), can re-induce CD20 expression on lymphoma cells after treatment, including after treatment with CD20-targeting mAb rituximab (96).
Equipping CAR T cells with a suicide system, such as inducible caspase-9 (iCas9) (97, 98) or co-expression of truncated EGFR (99), will enhance the safety of CAR T cells. These systems can induce depletion of CAR T cells by administrating agents that trigger cell-intrinsic apoptosis or cell-extrinsic antibody-mediated depletion of the therapeutic cells. Transfection of T cells with mRNA encoding CAR enables transient expression of CAR (100) and is a technology that is suitable for early phase clinical trials if new antigens are targeted and dose-limiting toxicity may be predicted. Another approach for remote-controlled safety is an inducible CAR system, including a TET-inducible system (101), which enables drug-inducible control of CAR expression.
Lastly, the synthetic Notch (synNotch) system (102, 103) is another attractive platform for diverse and flexible modification of CAR T cells. SynNotch receptors can allow the addition of custom response programs to T cells upon antigen recognition. For instance, synNotch can drive tailored cytokine secretion, biased T cell differentiation, or local delivery of therapeutic payloads, such as antibodies, upon the recognition of the antigen. In addition, synNotch can be utilized to develop sophisticated antigen recognition by CAR T cells based on the Boolean “AND” logic gating.
The treatment of solid tumors by CAR T cells is complex and multifactorial with a narrower therapeutic window than the targeting of CD19 for the treatment of B cell leukemia and non-Hodgkin lymphoma. Despite a growing list of clinical studies, remarkable responses have been rarely achieved with the exception of a case in glioblastoma with intraventricular delivery of the IL13Rα2 CAR. In this setting, establishment of strategies to expand the therapeutic window is critical. As outlined here, there are several promising approaches to achieve this in the preclinical setting and some of them are currently under investigation in clinical trials. The results of these and future clinical trials will elucidate a more refined path forward for solid tumor treatments.
There remain a lot of unknowns on tumor biology, the TME and CAR T cell biology. Fortunately, powerful tools to address these questions, such as emerging technologies in bioinformatics, mass spectrometry proteomics, mass cytometry, and single cell RNA sequencing, will allow us to access highly multiplexed and precise information on tumors, components of TME and immune cells. Moreover, maturation of technologies in gene-editing, such as the CRISPR/Cas9 system or synthetic biology, such as the synNotch system, will enable flexible design and engineering of T cells. Combining these technologies may lead to breakthroughs for CAR T cell therapies for the treatment of solid tumors.
Finally, several cases with unexpected severe toxicities have been reported when new CAR T cell therapies were first administrated to patients. Unfortunately, current technologies do not allow us to predict all the toxicities in the clinical setting; thus, only clinical trials can currently reveal information on safety and efficacy profiles of adoptive immunotherapies. Continuous development and refinement of preclinical models that can predict toxicity as well as the careful and rational planning and implementation of clinical trials will be crucial for further development of CAR T cell therapies.
KW and SK conceptualized, wrote, and edited the manuscript. ADP and CHJ provided feedback and edited the manuscript.
ADP and CHJ report intellectual property related to chimeric antigen receptor T cells licensed to Novartis. CHJ is a founder and has equity in Tmunity Therapeutics and receives commercial research grants from Novartis. CHJ is a consultant/advisory board member for Tmunity, Immune Design, and Celldex Therapeutics.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Financial support for this work was provided in part by grants from the NCI [P01 CA214278 (CHJ) and R01 CA226983 (CHJ)], the Department of Veterans Affairs [BLR&D IK2-BX-004183 (ADP)], Tmunity Therapeutics (ADP and CHJ), and CureSearch for Children's Cancer (ADP). ADP and CHJ are members of the Parker Institute for Cancer Immunotherapy, which supports the University of Pennsylvania's Cancer Immunotherapy Program.
1. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. (2013) 368:1509–18. doi: 10.1056/NEJMoa1215134
2. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
3. Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. (2015) 33:540–9. doi: 10.1200/JCO.2014.56.2025
4. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. (2010) 18:843–51. doi: 10.1038/mt.2010.24
5. Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. (2006) 24:e20–22. doi: 10.1200/JCO.2006.05.9964
6. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. (2011) 19:620–6. doi: 10.1038/mt.2010.272
7. Birnbaum ME, Berry R, Hsiao YS, Chen Z, Shingu-Vazquez MA, Yu X, et al. Molecular architecture of the alphabeta T cell receptor-CD3 complex. Proc Natl Acad Sci USA. (2014) 111:17576–81. doi: 10.1073/pnas.1420936111
8. Hatada MH, Lu X, Laird ER, Green J, Morgenstern JP, Lou M, et al. Molecular basis for interaction of the protein tyrosine kinase ZAP-70 with the T-cell receptor. Nature (1995) 377:32–8. doi: 10.1038/377032a0
9. Kersh EN, Shaw AS, Allen PM. Fidelity of T cell activation through multistep T cell receptor zeta phosphorylation. Science (1998) 281:572–5. doi: 10.1126/science.281.5376.572
10. Lim WA, June CH. The principles of engineering immune cells to treat cancer. Cell (2017) 168:724–40. doi: 10.1016/j.cell.2017.01.016
11. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA. (1989) 86:10024–8. doi: 10.1073/pnas.86.24.10024
12. Finney HM, Lawson AD, Bebbington CR, Weir AN. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol. (1998) 161:2791–7.
13. Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia (2004) 18:676–84. doi: 10.1038/sj.leu.2403302
14. Guedan S, Posey AD Jr, Shaw C, Wing A, Da T, Patel PR, et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight (2018) 3:96976. doi: 10.1172/jci.insight.96976
15. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. (2015) 21:4062–72. doi: 10.1158/1078-0432.CCR-15-0428
16. Yokosuka T, Kobayashi W, Takamatsu M, Sakata-Sogawa K, Zeng H, Hashimoto-Tane A, et al. Spatiotemporal basis of CTLA-4 costimulatory molecule-mediated negative regulation of T cell activation. Immunity (2010) 33:326–39. doi: 10.1016/j.immuni.2010.09.006
17. Stinchcombe JC, Bossi G, Booth S, Griffiths GM. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity (2001) 15:751–61. doi: 10.1016/S1074-7613(01)00234-5
18. Stinchcombe JC, Majorovits E, Bossi G, Fuller S, Griffiths GM. Centrosome polarization delivers secretory granules to the immunological synapse. Nature (2006) 443:462–5. doi: 10.1038/nature05071
19. Tsun A, Qureshi I, Stinchcombe JC, Jenkins MR, de la Roche M, Kleczkowska J, et al. Centrosome docking at the immunological synapse is controlled by Lck signaling. J Cell Biol. (2011) 192:663–74. doi: 10.1083/jcb.201008140
20. Karlsson H, Svensson E, Gigg C, Jarvius M, Olsson-Stromberg U, Savoldo B, et al. Evaluation of intracellular signaling downstream chimeric antigen receptors. PLoS ONE (2015) 10:e0144787. doi: 10.1371/journal.pone.0144787
21. Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. (2010) 18:413–20. doi: 10.1038/mt.2009.210
22. Xiong W, Chen Y, Kang X, Chen Z, Zheng P, Hsu YH, et al. Immunological synapse predicts effectiveness of chimeric antigen receptor cells. Mol Ther. (2018) 26:963–75. doi: 10.1016/j.ymthe.2018.01.020
23. Davenport AJ, Cross RS, Watson KA, Liao Y, Shi W, Prince HM, et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc Natl Acad Sci USA. (2018) 115:E2068–76. doi: 10.1073/pnas.1716266115
24. Mukherjee M, Mace EM, Carisey AF, Ahmed N, Orange JS. Quantitative imaging approaches to study the CAR immunological synapse. Mol Ther. (2017) 25:1757–68. doi: 10.1016/j.ymthe.2017.06.003
25. Hombach A, Heuser C, Sircar R, Tillmann T, Diehl V, Pohl C, et al. An anti-CD30 chimeric receptor that mediates CD3-zeta-independent T-cell activation against Hodgkin's lymphoma cells in the presence of soluble CD30. Cancer Res. (1998) 58:1116–9.
26. Nolan KF, Yun CO, Akamatsu Y, Murphy JC, Leung SO, Beecham EJ, et al. Bypassing immunization: optimized design of “designer T cells” against carcinoembryonic antigen (CEA)-expressing tumors, and lack of suppression by soluble CEA. Clin Cancer Res. (1999) 5:3928–41.
27. Lanitis E, Poussin M, Hagemann IS, Coukos G, Sandaltzopoulos R, Scholler N, et al. Redirected antitumor activity of primary human lymphocytes transduced with a fully human anti-mesothelin chimeric receptor. Mol Ther. (2012) 20:633–43. doi: 10.1038/mt.2011.256
28. Chang ZL, Lorenzini MH, Chen X, Tran U, Bangayan NJ, Chen YY. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat Chem Biol. (2018) 14:317–24. doi: 10.1038/nchembio.2565
29. Irvine DJ, Purbhoo MA, Krogsgaard M, Davis MM. Direct observation of ligand recognition by T cells. Nature (2002) 419:845–9. doi: 10.1038/nature01076
30. Huang J, Brameshuber M, Zeng X, Xie J, Li QJ, Chien YH, et al. A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4(+) T cells. Immunity (2013) 39:846–57. doi: 10.1016/j.immuni.2013.08.036
31. Manz BN, Jackson BL, Petit RS, Dustin ML, Groves J. T-cell triggering thresholds are modulated by the number of antigen within individual T-cell receptor clusters. Proc Natl Acad Sci USA. (2011) 108:9089–94. doi: 10.1073/pnas.1018771108
32. Au-Yeung BB, Zikherman J, Mueller JL, Ashouri JF, Matloubian M, Cheng DA, et al. A sharp T-cell antigen receptor signaling threshold for T-cell proliferation. Proc Natl Acad Sci USA. (2014) 111:E3679–88. doi: 10.1073/pnas.1413726111
33. Itoh Y, Germain RN. Single cell analysis reveals regulated hierarchical T cell antigen receptor signaling thresholds and intraclonal heterogeneity for individual cytokine responses of CD4+ T cells. J Exp Med. (1997) 186:757–66. doi: 10.1084/jem.186.5.757
34. Watanabe K, Terakura S, Martens AC, van Meerten T, Uchiyama S, Imai M, et al. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 zeta chimeric antigen receptor-modified effector CD8+ T cells. J Immunol. (2015) 194:911–20. doi: 10.4049/jimmunol.1402346
35. Stone JD, Aggen DH, Schietinger A, Schreiber H, Kranz DM. A sensitivity scale for targeting T cells with chimeric antigen receptors (CARs) and bispecific T-cell Engagers (BiTEs). Oncoimmunology (2012) 1:863–73. doi: 10.4161/onci.20592
36. Walker AJ, Majzner RG, Zhang L, Wanhainen K, Long AH, Nguyen SM, et al. Tumor antigen and receptor densities regulate efficacy of a chimeric antigen receptor targeting anaplastic lymphoma kinase. Mol Ther. (2017) 25:2189–201. doi: 10.1016/j.ymthe.2017.06.008
37. Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. (2015) 75:3596–607. doi: 10.1158/0008-5472.CAN-15-0159
38. Isaaz S, Baetz K, Olsen K, Podack E, Griffiths GM. Serial killing by cytotoxic T lymphocytes: T cell receptor triggers degranulation, re-filling of the lytic granules and secretion of lytic proteins via a non-granule pathway. Eur J Immunol. (1995) 25:1071–9. doi: 10.1002/eji.1830250432
39. Choi PJ, Mitchison TJ. Imaging burst kinetics and spatial coordination during serial killing by single natural killer cells. Proc Natl Acad Sci USA. (2013) 110:6488–93. doi: 10.1073/pnas.1221312110
40. Davenport AJ, Jenkins MR, Cross RS, Yong CS, Prince HM, Ritchie DS, et al. CAR-T cells inflict sequential killing of multiple tumor target cells. Cancer Immunol Res. (2015) 3:483–94. doi: 10.1158/2326-6066.CIR-15-0048
41. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature (2017) 543:113–7. doi: 10.1038/nature21405
42. Purbhoo MA, Sutton DH, Brewer JE, Mullings RE, Hill ME, Mahon TM, et al. Quantifying and imaging NY-ESO-1/LAGE-1-derived epitopes on tumor cells using high affinity T cell receptors. J Immunol. (2006) 176:7308–16. doi: 10.4049/jimmunol.176.12.7308
43. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood (2013) 122:863–71. doi: 10.1182/blood-2013-03-490565
44. Aleksic M, Liddy N, Molloy PE, Pumphrey N, Vuidepot A, Chang KM, et al. Different affinity windows for virus and cancer-specific T-cell receptors: implications for therapeutic strategies. Eur J Immunol. (2012) 42:3174–9. doi: 10.1002/eji.201242606
45. Johnson LA, Heemskerk B, Powell DJ Jr, Cohen CJ, Morgan RA, Dudley ME, et al. Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. J Immunol. (2006) 177:6548–59. doi: 10.4049/jimmunol.177.9.6548
46. Schmid DA, Irving MB, Posevitz V, Hebeisen M, Posevitz-Fejfar A, Sarria JC, et al. Evidence for a TCR affinity threshold delimiting maximal CD8 T cell function. J Immunol. (2010) 184:4936–46. doi: 10.4049/jimmunol.1000173
47. Zhong S, Malecek K, Johnson LA, Yu Z, Vega-Saenz de Miera E, Darvishian F, et al. T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc Natl Acad Sci USA. (2013) 110:6973–8. doi: 10.1073/pnas.1221609110
48. Valitutti S, Muller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature (1995) 375:148–51. doi: 10.1038/375148a0
49. Stone JD, Chervin AS, Kranz DM. T-cell receptor binding affinities and kinetics: impact on T-cell activity and specificity. Immunology (2009) 126:165–76. doi: 10.1111/j.1365-2567.2008.03015.x
50. Edwards LJ, Evavold BD. T cell recognition of weak ligands: roles of signaling, receptor number, and affinity. Immunol Res. (2011) 50:39–48. doi: 10.1007/s12026-011-8204-3
51. Richman SA, Nunez-Cruz S, Moghimi B, Li LZ, Gershenson ZT, Mourelatos Z, et al. High-affinity GD2-specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol Res. (2018) 6:36–46. doi: 10.1158/2326-6066.CIR-17-0211
52. Caserta S, Kleczkowska J, Mondino A, Zamoyska R. Reduced functional avidity promotes central and effector memory CD4 T cell responses to tumor-associated antigens. J Immunol. (2010) 185:6545–54. doi: 10.4049/jimmunol.1001867
53. Engels B, Chervin AS, Sant AJ, Kranz DM, Schreiber H. Long-term persistence of CD4(+) but rapid disappearance of CD8(+) T cells expressing an MHC class I-restricted TCR of nanomolar affinity. Mol Ther. (2012) 20:652–60. doi: 10.1038/mt.2011.286
54. Posey AD Jr, Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, et al. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity (2016) 44:1444–54. doi: 10.1016/j.immuni.2016.05.014
55. Hosen N, Matsunaga Y, Hasegawa K, Matsuno H, Nakamura Y, Makita M, et al. The activated conformation of integrin beta7 is a novel multiple myeloma-specific target for CAR T cell therapy. Nat Med. (2017) 23:1436–43. doi: 10.1038/nm.4431
56. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017). 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
57. Castellarin M, Watanabe K, June CH, Kloss CC, Posey AD Jr. Driving cars to the clinic for solid tumors. Gene Ther. (2018) 25:165–75. doi: 10.1038/s41434-018-0007-x
58. Sorensen AL, Reis CA, Tarp MA, Mandel U, Ramachandran K, Sankaranarayanan V, et al. Chemoenzymatically synthesized multimeric Tn/STn MUC1 glycopeptides elicit cancer-specific anti-MUC1 antibody responses and override tolerance. Glycobiology (2006) 16:96–107. doi: 10.1093/glycob/cwj044
59. Hinrichs CS, Restifo NP. Reassessing target antigens for adoptive T-cell therapy. Nat Biotechnol. (2013) 31:999–1008. doi: 10.1038/nbt.2725
60. Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. (2015) 75:3505–18. doi: 10.1158/0008-5472.CAN-15-0139
61. Drent E, Themeli M, Poels R, de Jong-Korlaar R, Yuan H, de Bruijn J, et al. A rational strategy for reducing on-target off-tumor effects of CD38-chimeric antigen receptors by affinity optimization. Mol Ther. (2017) 25:1946–58. doi: 10.1016/j.ymthe.2017.04.024
62. Chmielewski M, Hombach A, Heuser C, Adams GP, Abken H. T cell activation by antibody-like immunoreceptors: increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J Immunol. (2004) 173:7647–53. doi: 10.4049/jimmunol.173.12.7647
63. Turatti F, Figini M, Balladore E, Alberti P, Casalini P, Marks JD, et al. Redirected activity of human antitumor chimeric immune receptors is governed by antigen and receptor expression levels and affinity of interaction. J Immunother. (2007) 30:684–93. doi: 10.1097/CJI.0b013e3180de5d90
64. Park S, Shevlin E, Vedvyas Y, Zaman M, Park S, Hsu YS, et al. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci Rep. (2017) 7:14366. doi: 10.1038/s41598-017-14749-3
65. Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res. (2015) 3:356–67. doi: 10.1158/2326-6066.CIR-14-0186
66. Gomes-Silva D, Mukherjee M, Srinivasan M, Krenciute G, Dakhova O, Zheng Y, et al. Tonic 4-1BB costimulation in chimeric antigen receptors impedes T cell survival and is vector-dependent. Cell Rep. (2017) 21:17–26. doi: 10.1016/j.celrep.2017.09.015
67. Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira AC, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. (2012) 32:1059–70. doi: 10.1007/s10875-012-9689-9
68. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. (2013) 31:71–5. doi: 10.1038/nbt.2459
69. Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, et al. TanCAR: a novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol Ther Nucleic Acids (2013) 2:e105. doi: 10.1038/mtna.2013.32
70. Hegde M, Corder A, Chow KK, Mukherjee M, Ashoori A, Kew Y, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther. (2013) 21:2087–101. doi: 10.1038/mt.2013.185
71. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Invest. (2016) 126:3036–52. doi: 10.1172/JCI83416
72. Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, et al. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. (2012) 72:1844–52. doi: 10.1158/0008-5472.CAN-11-3890
73. Tamada K, Geng D, Sakoda Y, Bansal N, Srivastava R, Li Z, et al. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin Cancer Res. (2012) 18:6436–45. doi: 10.1158/1078-0432.CCR-12-1449
74. Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L, et al. Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol. (2008) 180:4901–9. doi: 10.4049/jimmunol.180.7.4901
75. Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res. (2013) 19:3153–64. doi: 10.1158/1078-0432.CCR-13-0330
76. Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. (2015) 3:125–35. doi: 10.1158/2326-6066.CIR-14-0127
77. Kuramitsu S, Ohno M, Ohka F, Shiina S, Yamamichi A, Kato A, et al. Lenalidomide enhances the function of chimeric antigen receptor T cells against the epidermal growth factor receptor variant III by enhancing immune synapses. Cancer Gene Ther. (2015) 22:487–95. doi: 10.1038/cgt.2015.47
78. Wang X, Walter M, Urak R, Weng L, Huynh C, Lim L, et al. Lenalidomide enhances the function of CS1 chimeric antigen receptor-redirected T cells against multiple myeloma. Clin Cancer Res. (2018) 24:106–19. doi: 10.1158/1078-0432.CCR-17-0344
79. Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. (2015) 33:2780–8. doi: 10.1200/JCO.2014.58.3377
80. Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer (2014) 14:559–67. doi: 10.1038/nrc3770
81. Watanabe K, Luo Y, Da T, Guedan S, Ruella M, Scholler J, et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight (2018) 3:99573. doi: 10.1172/jci.insight.99573
82. Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. (2014) 74:5195–205. doi: 10.1158/0008-5472.CAN-14-0697
83. Rosewell Shaw A, Porter CE, Watanabe N, Tanoue K, Sikora A, Gottschalk S, et al. Adenovirotherapy delivering cytokine and checkpoint inhibitor augments CAR T cells against metastatic head and neck cancer. Mol Ther. (2017) 25:2440–51. doi: 10.1016/j.ymthe.2017.09.010
84. Wing A, Fajardo CA, Posey AD Jr, Shaw C, Da T, Young RM, et al. Improving CART-cell therapy of solid tumors with oncolytic virus-driven production of a bispecific T-cell engager. Cancer Immunol Res. (2018) 6:605–16. doi: 10.1158/2326-6066.CIR-17-0314
85. Mardiana S, John LB, Henderson MA, Slaney CY, von Scheidt B, Giuffrida L, et al. A multifunctional role for adjuvant anti-4-1BB therapy in augmenting antitumor response by chimeric antigen receptor T cells. Cancer Res. (2017) 77:1296–309. doi: 10.1158/0008-5472.CAN-16-1831
86. Priceman SJ, Tilakawardane D, Jeang B, Aguilar B, Murad JP, Park AK, et al. Regional delivery of chimeric antigen receptor-engineered T cells effectively targets HER2(+) breast cancer metastasis to the brain. Clin Cancer Res. (2018) 24:95–105. doi: 10.1158/1078-0432.CCR-17-2041
87. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
88. Tchou J, Zhao Y, Levine BL, Zhang PJ, Davis MM, Melenhorst JJ, et al. Safety and efficacy of intratumoral injections of Chimeric Antigen Receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol Res. (2017) 5:1152–61. doi: 10.1158/2326-6066.CIR-17-0189
89. Han X, Bryson PD, Zhao Y, Cinay GE, Li S, Guo Y, et al. Masked chimeric antigen receptor for tumor-specific activation. Mol Ther. (2017) 25:274–84. doi: 10.1016/j.ymthe.2016.10.011
90. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood (2012) 119:4133–41. doi: 10.1182/blood-2011-12-400044
91. Hu B, Ren J, Luo Y, Keith B, Young RM, Scholler J, et al. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep. (2017) 20:3025–33. doi: 10.1016/j.celrep.2017.09.002
92. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol. (2018) 36:346–51. doi: 10.1038/nbt.4086
93. Suarez ER, Chang de K, Sun J, Sui J, Freeman GJ, Signoretti, S, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget (2016) 7:34341–55. doi: 10.18632/oncotarget.9114
94. Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. (2015) 5:1282–95. doi: 10.1158/2159-8290.CD-15-1020
95. Garnett CT, Palena C, Chakraborty M, Tsang KY, Schlom J, Hodge JW. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res. (2004) 64:7985–94. doi: 10.1158/0008-5472.CAN-04-1525
96. Hiraga J, Tomita A, Sugimoto T, Shimada K, Ito M, Nakamura S, et al. Down-regulation of CD20 expression in B-cell lymphoma cells after treatment with rituximab-containing combination chemotherapies: its prevalence and clinical significance. Blood (2009) 113:4885–93. doi: 10.1182/blood-2008-08-175208
97. Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia (2010) 24:1160–70. doi: 10.1038/leu.2010.75
98. Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. (2011) 365:1673–83. doi: 10.1056/NEJMoa1106152
99. Paszkiewicz PJ, Frassle SP, Srivastava S, Sommermeyer D, Hudecek M, Drexler I, et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J Clin Invest. (2016) 126:4262–72. doi: 10.1172/JCI84813
100. Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. (2014) 2:112–20. doi: 10.1158/2326-6066.CIR-13-0170
101. Sakemura R, Terakura S, Watanabe K, Julamanee J, Takagi E, Miyao K, et al. A tet-on inducible system for controlling CD19-chimeric antigen receptor expression upon drug administration. Cancer Immunol Res. (2016) 4:658–68. doi: 10.1158/2326-6066.CIR-16-0043
102. Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, Thomson M, et al. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell (2016) 164:780–91. doi: 10.1016/j.cell.2016.01.012
Keywords: T cell biology, chimeric antigen receptors, immune synapse formation, immunotherapy, cancer immunology
Citation: Watanabe K, Kuramitsu S, Posey AD Jr and June CH (2018) Expanding the Therapeutic Window for CAR T Cell Therapy in Solid Tumors: The Knowns and Unknowns of CAR T Cell Biology. Front. Immunol. 9:2486. doi: 10.3389/fimmu.2018.02486
Received: 01 August 2018; Accepted: 08 October 2018;
Published: 26 October 2018.
Edited by:
Salem Chouaib, Institut Gustave Roussy, FranceReviewed by:
Maksim Mamonkin, Baylor College of Medicine, United StatesCopyright © 2018 Watanabe, Kuramitsu, Posey and June. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Keisuke Watanabe, a2Vpc3VrZXdAcGVubm1lZGljaW5lLnVwZW5uLmVkdQ==
Carl H. June, Y2p1bmVAdXBlbm4uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.