 Ourania S. Kotsiou
Ourania S. Kotsiou Konstantinos I. Gourgoulianis
Konstantinos I. Gourgoulianis Sotirios G. Zarogiannis
Sotirios G. Zarogiannis- 1Department of Respiratory Medicine, Faculty of Medicine, School of Health Sciences, University of Thessaly, BIOPOLIS, Larissa, Greece
- 2Department of Physiology, Faculty of Medicine, School of Health Sciences, University of Thessaly, BIOPOLIS, Larissa, Greece
Interleukin 33 (IL-33) is highly expressed in barrier sites, acting via the suppression of tumorigenicity 2 receptor (ST2). IL-33/ST2 axis has long been known to play a pivotal role in immunity and cell homeostasis by promoting wound healing and tissue repair. However, it is also involved in the loss of balance between extensive inflammation and tissue regeneration lead to remodeling, the hallmark of fibrosis. The aim of the current review is to critically evaluate the available evidence regarding the role of the IL-33/ST2 axis in organ fibrosis. The role of the axis in tissue remodeling is better understood considering its crucial role reported in organ development and regeneration. Generally, the IL-33/ST2 signaling pathway has mainly anti-inflammatory/anti-proliferative effects; however, chronic tissue injury is responsible for pro-fibrogenetic responses. Regarding pulmonary fibrosis mature IL-33 enhances pro-fibrogenic type 2 cytokine production in an ST2- and macrophage-dependent manner, while full-length IL-33 is also implicated in the pulmonary fibrotic process in an ST2-independent, Th2-independent fashion. In liver fibrosis, evidence indicate that when acute and massive liver damage occurs, the release of IL-33 might act as an activator of tissue-protective mechanisms, while in cases of chronic injury IL-33 plays the role of a hepatic fibrotic factor. IL-33 signaling has also been involved in the pathogenesis of acute and chronic pancreatitis. Moreover, IL-33 could be used as an early marker for ulcer-associated activated fibroblasts and myofibroblast trans-differentiation; thus one cannot rule out its potential role in inflammatory bowel disease-associated fibrosis. Similarly, the upregulation of the IL-33/ST2 axismay contribute to tubular cell injury and fibrosis via epithelial to mesenchymal transition (EMT) of various cell types in the kidneys. Of note, IL-33 exerts a cardioprotective role via ST2 signaling, while soluble ST2 has been demonstrated as a marker of myocardial fibrosis. Finally, IL-33 is a crucial cytokine in skin pathology responsible for abnormal fibroblast proliferation, leukocyte infiltration and morphologic differentiation of human endothelial cells. Overall, emerging data support a novel contribution of the IL-33/ST2 pathway in tissue fibrosis and highlight the significant role of the Th2 pattern of immune response in the pathophysiology of organ fibrosis.
Introduction
Interleukin (IL) 33 was first described in 1999 as a protein (DV27) overexpressed in vasospastic cerebral arteries in a canine subarachnoid hemorrhage model (1). Later, in 2003, it was characterized at the molecular level as a nuclear factor abundantly expressed in human high endothelial venule cells (2). The mechanisms mediating its effects were elucidated by Schmitz et al., as well as Dinarello, that identified IL-33 as a cytokine of the IL-1 superfamily (3, 4). Its name was given due to its adherence to the style of the IL-1 superfamily nomenclature, indicating that IL-33 was not merely a functional copy of IL-1α and IL-1β proteins (3, 4). The role of IL-33 as an alarmin was established participating in tissue homeostasis, signaling via the IL-1 receptor-related suppression of tumorigenicity 2 receptor (ST2), and inducing T helper type 2 (Th2) immune responses (3, 4).
Normally human IL-33 is mainly expressed and stored in the nucleus of endothelial and epithelial cells (5). IL-33 is a dual-function cytokine: the full-length IL-33 protein (flIL-33) serves as an intranuclear gene regulator, and the mature IL-33 (mIL-33) serves as an extracellular cytokine upon release from damaged or necrotic cells (6). IL-33 is passively and rapidly released from damaged cells, as a tissue-barrier component in response to stimuli or cell injury. However, it can also be actively secreted by immune cells. The abundant basal IL-33 expression in tissues can be further increased during inflammation (7). Notably, it has been documented that the fraction of IL-33 produced by tissues rather than that provided by immune cells, is necessary for Th2-induced airway inflammation (8). An inflammatory microenvironment may exacerbate disease-associated functions of IL-33 through the generation of highly active mature forms (9). Neutrophil serine proteases such as cathepsin G and elastase which secreted during inflammation have been shown to regulate IL-33 activity, by processing flIL-33 and generate biologically highly active mature forms of IL-33, in vivo (10). Furthermore, serine proteases secreted by activated mast cells (chymase and tryptase) generate mIL-33 with potent activity on lymphoid cell type 2 ILC2s (11). On the contrary, it is still unknown whether and which endogenous proteases have a similar capacity (7). It has only been reported an endogenous calcium-dependent caspase which is called calpain that mediates pro-IL-33 cleavage and mIL-33 production. Calpain is secreted when the cells are severely damaged by external stimulation such as inflammatory stimuli; and subsequently, the level of intracellular calcium ion is raised by an influx of extracellular ion or a release from an intracellular store (7, 12). Although both flIL-33 and mIL-33 can bind to and signal through ST2, mIL-33 exhibit 10-fold higher affinity and bioactivity than flIL-33 (6, 10).
Conversely, the ST2 receptor is predominantly expressed by immune cells involved in innate immunity, including mast cells, ILC2s, macrophages, dendritic cells (DCs), eosinophils, basophils, natural killer cells (NK cells). Furthermore, ST2 is expressed by cells participating in adaptive immunity such as CD4 +, CD8 + T cells, and T-regulatory cells (Tregs) (13, 14).
In humans, there are three ST2 isoforms. IL-33 signals via the ST2L receptor which has a membrane-bound domain, an extracellular segment composed of three linked immunoglobulin-like motifs, and a cytosolic Toll/interleukin-1 receptor domain. The soluble ST2 (sST2) isoform lacks the transmembrane and cytoplasmic domains and includes a unique nine amino-acid C-terminal sequence, constitutes a decoy receptor that does not signal. The ST2V isoform which is characterized by the absence of an immunoglobulin-like motif and alternative splicing of the C-terminal portion of ST2 is thought to be a form which is primarily found in gastrointestinal tissues (15, 16).
The IL-33/ST2 axis has been widely studied in respiratory, digestive, urogenital, heart and liver pathologies and the abundance of literature suggests a pivotal role of this pathway in the pathogenesis of an increasing number of diseases (Table 1). Emerging data have shown that IL-33/ST2 axis is involved in a variety of biological processes such as the development and regulation of immune responses, restoration of normal tissue homeostasis by promoting wound healing and repair. However, the IL-33/ST2 signaling pathway is involved in the loss of balance between extensive inflammation and tissue regeneration lead to remodeling that constitutes the hallmark of fibrosis (14, 72).
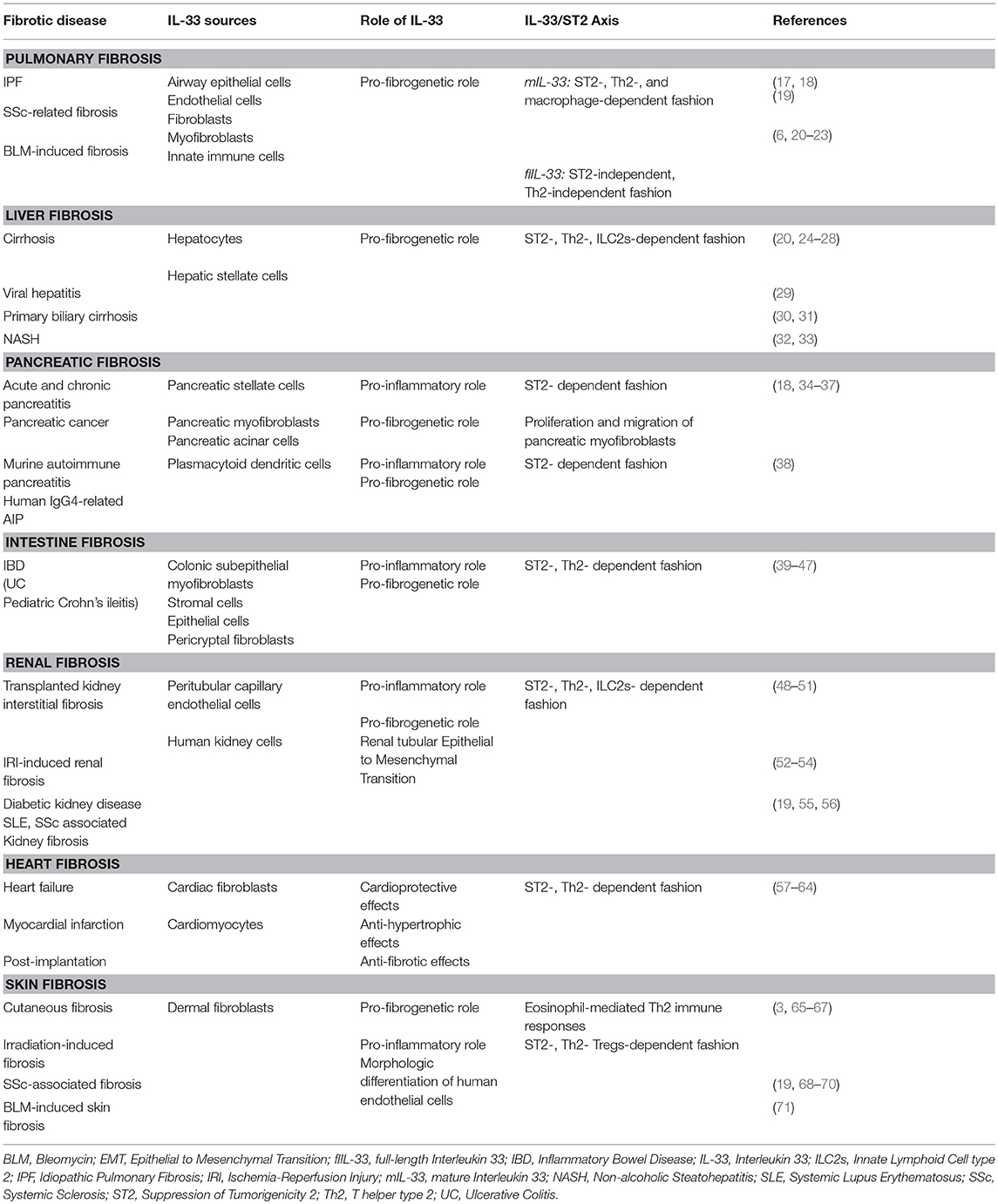
Table 1. The main roles of IL-33 in organ fibrosis.
Despite the burden of human organ fibrosis, there are still many things unknown regarding the underlying mechanisms. On this note, it has been supported that the IL-33/ST2 axis exerts anti-inflammatory and anti-proliferative effects in many diseases; however, it has also been shown that results in fibrotic effects in others. Although several lines of evidence demonstrate that there is a potential role of IL-33/ST2 in remodeling and differentiation processes in, there is still room for better understanding. The aim of this review is to critically evaluate the available evidence regarding the role of the IL-33/ST2 axis in organ fibrosis.
IL33/ST2 Axis Participates in the TH2-Mediated Inflammatory Response and Exacerbates Tissue Remodeling
Acute wounds initiate an early inflammatory response, directing the subsequent phases of healing (73). However, the triggers of inflammation and wound repair upon injury are still unclear (73). IL-33 has been found to participate in the early inflammatory process as other members of the IL-1 family do (73). More specifically, increased mRNA and protein expression after scratching in vivo have been demonstrated; suggesting a role for IL-33 in wound healing (74). Furthermore, ST2 receptor signaling improves wound closure by promoting the transition of macrophages from an inflammatory to a non-inflammatory state during healing, supporting epidermal closure, angiogenesis, and reduced scarring (73). During wound healing extracellular mIL-33 interacts with the ST2L receptor and the complex of IL-33/ST2L activates myeloid differentiation primary response 88 (MyD88) intracellular cascades that drive production of type 2 cytokines (such as IL-13) from polarized Th2 cells (73). Besides, it has been suggested that proteolytically uncleaved, flIL-33, which remains predominantly intracellular and intranuclear, promotes inflammation in an ST2-independent fashion through regulation of gene expression (75).
The key role of the IL-33/ST2 axis in tissue remodeling is better understood considering the crucial role of immune responses during tissue regeneration. Of note, increased production of epithelial IL-33 could lead to accumulation of innate type 2 cells during the alveolar period of lung development; that is when the lung is maximally remodeled (76). IL-33 promotes ILC2s function that enhance tissue healing, remodeling, and homeostasis in the post-partum period. More convincingly, studies in adult murine models showed that IL-33 activates lung ILC2s and renders them resistant to interferon-γ (IFN-γ)-mediated suppression of IL-5 and IL-13 production (22). Th2 cytokines such as IL-13 are crucial mediators of inflammation and remodeling (22). In addition, IL-33 activates alternatively activated M2 macrophages that control tissue remodeling during lung postnatal branching morphogenesis (77).
In other words, type 2 immunity influences lung development and/or remodeling while the spontaneous activation of type 2 cells in the embryonic period have been found to require extracellular IL-33 and ST2 signaling (76, 78). Meanwhile, IL-33 contributes to the development of type 2 immune environment in lungs at a young age as it lowers the threshold for innate immune responses to allergens (76).
IL-33/ST2 Axis in Pulmonary Fibrosis
Pulmonary fibrosis is a non-neoplastic pulmonary disease that is primarily caused by an uncontrolled wound-healing response. Idiopathic pulmonary fibrosis (IPF) is a highly lethal pathological entity of unknown etiology that is characterized by inflammation, fibroblast accumulation, and excessive collagen deposition. Importantly, IL-33 mRNA and protein levels have been found significantly increased in the bronchoalveolar lavage (BAL) fluids of patients with IPF (17) and systemic sclerosis (SSc)-related fibrosis as compared to healthy controls (19). Furthermore, the expression of IL-33 mRNA was also enhanced in IPF lung tissue (23).
IL-33 is also elevated in the bleomycin (BLM)-induced murine model of lung injury and fibrosis (20, 21, 23). The lungs of BLM-treated mice showed a substantial accumulation of IL-33-positive cells (20, 21, 23). Specifically, it has been documented that IL-33 and BLM result in synergistic effects on pulmonary fibrosis in vivo (6). In detail, mIL-33 production is induced in macrophages by BLM (6, 20, 21, 23). Subsequently, IL-33 enhances the polarization of macrophages toward an M2 phenotype (6, 79, 80). It is well established that the pro-fibrogenic activity of IL-33 is mainly attributed to its involvement in M2 macrophage polarization, as macrophages with alternative activation, rather than classical activation, serve to accelerate pulmonary fibrosis (6, 79, 80). A clear link between biologic splice variants of IL-33 (mIL-33) and a Th2 innate immune response has been demonstrated (80). Mature forms of IL-33 have been reported to drive production of extremely high levels of Th2 cytokines such as IL-13 (80). Furthermore, M2 macrophages are polarized by IL-13, and they further promote a Th2 reaction through the IL-13 and the transforming growth factor β (TGF-β) production, vice versa (20–23).
Several studies have demonstrated the importance of Th2 cells in fibrosis since IL-4, IL-5, and IL-13 have been causally linked to fibrosis (80). In detail, IPF fibroblasts are hyper-responsive to cytokines such as IL-13, at the same time, fibroblasts and innate immune cells are important sources of IL-33 (80). Enhanced production of TGF-β and IL-13 are essential for the development of pulmonary fibrosis by inducing myofibroblast differentiation and stimulating the production of extracellular matrix components, such as collagen (79, 80). Therefore, these data support that precise control of alveolar TGF-β activation and IL-13 are essential for alveolar homeostasis (79).
Previous work has shown that deficiency of the Akt2 isoform resulted in M2 macrophages polarization producing IL-13 and TGF-β and in the expansion of IL-13 recruiting ILC2s (6). Thus, Akt2 regulates pulmonary fibrosis by up-regulating the pro-fibrotic TGF-β and IL-13 production by macrophages (81). It is likely that the induction of IL-13 precedes and is essential for the subsequent enhanced M2 macrophage polarization by IL-33 (6). Moreover, it has been shown that in response to IL-33 treatment, Akt2−/−macrophages displayed decreased production of IL-13 and TGF-β1 and attenuated phosphorylation of transcription factor Forkhead box O3a (FoxO3a) to stop acting as a trigger for apoptosis (81). Inhibition of Akt2 marked as a potential strategy for treating IPF (81).
Interestingly, BLM can also induce fIL-33 secretion from airway epithelial cells and alveolar macrophages in an ST2-independent, Th2-independent fashion, likely through cytokine regulation of several non-Th2 cytokines, such as TGF-β, IL-6, and monocyte chemoattractant protein-1 (MCP-1) and possibly by engaging heat shock protein (HSP) 70 (6). flIL-33 can then be processed into various mature forms of IL-33 by neutrophil proteases. mIL-33 subsequently stimulate macrophages and ILC2s to produce IL-13 (75). On the other hand, this response may be mediated only by nuclear-located fIIL-33 affecting gene expression. Collectively, these data suggest that flIL-33 is also potentially implicated in the pulmonary fibrotic process (6, 75).
ST2 is mainly expressed in endothelial and type II alveolar epithelial cells as well as innate immune cells such as macrophages and ILC2s in the lungs. sST2 levels were also found increased in serum and BAL of acute exacerbation of IPF and BLM-induced lung fibrosis, respectively (82). ST2 mRNA expression has been reported to be increased in the BLM-induced lung fibrosis model in vivo, as well as in a human lung fibroblast and a human type II alveolar epithelial cell line, possibly reflecting the development of a type 2 pattern of inflammatory process in the fibrotic lung tissue (82, 83).
Li et al. reported that ST2 deficiency or administration of an anti-IL-33 antibody were able to attenuate bone marrow (BM)-induced pulmonary fibrosis (6). Similarly, intranasal administration of a lentivirus for epithelial over-expression of sST2 has been reported to attenuate pulmonary fibrotic change by inhibiting the expression of pro-inflammatory and pro-fibrotic mediators, such as IL-13, IL-33, and TGF-β1 along with improved survival rates in BLM-treated mice (20).
Increased numbers of lung ILC2s have also been implicated in BLM-induced fibrosis in the mouse (6). Especially, BM-derived ST2-expressive ILC2s have been recently reported to be recruited to the fibrotic lung through the IL-33/ST2 pathway and contributed to fibroblast activation perhaps via transforming TGF-β (84). Furthermore, adoptive ILC2 transfer into recipient mice enhances lung fibrosis, whereas blocking IL-33 or ST2 deficiency diminish fibrosis. The increase in IL-33 responsive ILC2s is not unique to animal models of pulmonary fibrosis since they are also increased in SSc, correlating with the extent of fibrosis. ILC2s are also present in the IPF lung tissue and BAL, wherein they are associated with upregulated expression of lung IL-33.
Even in cases of asthma, experimental results showed that direct murine airway exposure to IL-33 could induce local fibrotic changes. IL-33/ST2 axis is thought to at least partially mediate the fibroblast function and local expression of matrix metalloproteinases (MMPs) and their inhibitors as well as other fibrosis-related proteins (85). Additionally, IL-33 is probably related to prostaglandin E2 (PGE2) production, stimulating mast cells to produce a large quantity of PGE2 demonstrating potent anti-fibrotic activity in the IPF lung (17, 86). IL-33 can regulate deposition of extracellular matrix (ECM) and promote the process of pulmonary fibrosis by inducing the imbalance between MMP9 and tissue inhibitor of MMP9 (TIMP-1) (17, 86).
Finally, fibulin-1 (Fbln1), an important ECM component involved in a matrix organization and wound repair, has been found to predict disease progression in IPF patients (87). Genetically inhibited Fbln1 has been associated with reduced levels of pro-inflammatory cytokines such as IL-33 and pulmonary inflammatory cells in a murine COPD model (87).
Hence, IL-33 is thought to be a novel cytokine that promotes the initiation and progression of pulmonary fibrosis by recruiting and directing inflammatory cell function and enhancing pro-fibrogenic cytokine production in an ST2- and macrophage-dependent manner (6, 79, 80). However, fIL-33 secretion from airway epithelial cells and alveolar macrophages acts in an ST2-independent, Th2-independent fashion in the fibrotic process. The over-expression of sST2 decoy receptor or any other exogenous inhibition of IL-33/ST2L signaling result in markedly lower levels of IL-33 and other pro-inflammatory and pro-fibrotic mediators thus attenuate the fibrotic process.
IL-33/ST2 Axis in Liver Fibrosis
Liver fibrosis is a reversible wound healing response to acute or chronic hepatocellular injury from various etiologies, including viral infection, cholestasis, metabolic diseases, and alcohol abuse. It has been suggested that damage associated molecular patterns (DAMPs) act as molecular links between hepatocyte death and liver fibrogenesis (27). Evidence indicates that when acute and massive liver damage occurs, the release of IL-33 by injured hepatocytes might act as an activator of tissue-protective mechanisms, while in cases of chronic injury IL-33 plays the role of a hepatic fibrotic factor (27). For instance, melatonin which exerts cytoprotective effects via inhibition of oxidative stress and apoptosis in liver ischemia-reperfusion injury (IRI) has been proposed to inhibit liver fibrosis through suppressing necroptotic DAMPs signaling cascades, such as the IL-33 signaling pathway (88).
IL-33/ST2 axis has been implicated in several hepatic diseases, such as cirrhosis, virus infection, fatty liver disease and toxic liver damage leading further to liver fibrosis (24, 27–29). Indeed, IL-33 has shown potential liver fibrosis promoting effect (24, 32). It has been found that in murine and human fibrotic livers, IL-33 levels as well as the mRNA expression of both IL-33 and ST2, are higher as compared to healthy liver (24, 28). Their expression is significantly increased along with the severity of fibrosis, especially in cirrhotic livers (33). Likewise in patients with primary biliary cirrhosis, an autoimmune liver disease that could result in liver failure, and hepatoma carcinoma, the serum IL-33 levels were positively correlated with disease severity (30, 31).
The role of ST2 has also been highlighted in liver fibrosis. It has been documented that liver injury, inflammatory cell infiltration, and fibrosis are reduced in the absence of the receptor ST2L (24). Furthermore, it has been found that ST2L deficient mice did not increase collagen production when challenged with carbon tetrachloride, an organic compound with pro-fibrotic effects (24, 28). Similarly, the absence of ST2L prevented liver inflammation both in the acute and chronic phases, with attenuated activation of mitogen-activated protein kinase (MEK)\extracellular signal-regulated kinase(ERK)/p38- mitogen-activated protein kinase (MAPK) signaling cascade(25, 32).
Besides, sST2 has been regarded as a circulating biomarker to reflect IL-33 activation and fibrosis in patients with liver diseases. In fact, sST2 serum levels differ between hepatitis B virus-infected patients were dependent on the severity of hepatic fibrosis (89). Especially, the plasma levels of sST2 were found to be associated with mortality in patients with HBV-related acute-on-chronic liver failure (89). Notably, in a murine liver fibrosis model, the antibody blockade of sST2 enhances the severity of fibrosis (89).
It has been reported that the major source of IL-33 in fibrotic livers is the hepatic stellate cells (HSCs) that have also been suggested to be the leading producers of ECM proteins (25, 27, 32). Injury-associated immunological processes supporting trans-differentiation of quiescent HSC to fibrogenic myofibroblasts in the course of liver injury are particularly important in fibrosis (90). Moreover, an ST2 expression has been observed on the membrane of HSCs (32). However, other data derived from murine and human studies demonstrated that hepatocytes are the primary sources of IL-33 both in the fibrotic liver and in healthy liver (27, 32). More specifically, IL-33 release as a DAMP upon hepatocyte damage may have a direct effect on HSCs that increases secretion of cytokines and production of collagen (27, 32). Besides, another study showed that activation of HSCs was decreased in ST2-deficient liver fibrosis mice (24). IL-33-mediated Th2 immune response promotes HSCs proliferation, TGF-β synthesis, and fibrogenesis (27). As in lung tissue, Th2 pro-fibrotic cytokines production such as IL-4, IL-5, and IL-13 are known to play a critical role in liver fibrosis (27). On the other hand, Th1 cytokines lead to a rapid and intense inflammatory response while causing little fibrosis.
Interestingly, vector-encoded overexpression of IL-33 was sufficient to induce fibrosis in the liver without administration of chemicals, demonstrating the pro-fibrotic role of IL-33, predominantly exerted through the IL-13 induction (27). In particular, IL-13 could initiate activation and differentiation of HSCs by enhancing TGF-β signaling through IL-4Rα and signal transducer and activator of transcription 6 (STAT6) in HSCs, promoting liver fibrosis (28). Some other data suggest that IL-13, rather than TGF-β, primarily activates HSCs in liver fibrosis (32). Furthermore, the IL-33/ST2/IL-13 pathway is thought to be Galectin-3 (Gal-3) dependent (91–93). Gal-3 has been found to attenuate steatosis while promoting liver injury, inflammation and fibrosis in an obesogenic mouse model of non-alcoholic steatohepatitis (NASH) (91–93). Therefore, Gal-3 inhibitors have been suggested to protect against fibrotic disorders (91, 92).
More convincingly, the stimulation of in vitro activated HSCs with recombinant IL-33 (rIL-33) induced the MAPK pathways that were found to be mediated by ERK, Jun N-terminal kinase (JNK) and p38 protein kinases (32). Moreover, HSCs activated by rIL-33 in vitro, released IL-6, TGF-β, and resulted in the stimulation of α-smooth muscle actin (a-SMA) and collagen expression (32). These data suggest a direct fibrogenic role of IL-33 in HSCs, which is potentially synergistic with its effects on ILC2s (26, 28). Hence, another mechanism proposed to be involved in liver fibrosis is through the activation of ILC2s via the ST2 signaling pathway, resulting again in a release of several Th2 cytokines (30, 33, 94). IL-33 induces the activation and expansion of ILC2s to express IL-13 and IL-5, which subsequently causes M2 macrophage and eosinophil accumulation and regulates ST2+ Tregs homeostasis in liver adipose tissue through attenuating adipose tissue inflammation (95–97). In fact, the upregulation of IL-33 was positively correlated with an increase of ILC2s (32, 98). Activated ILC2-derived IL-13 initiated activation and differentiation of HSCs via the IL-4Rα-STAT6 transcription factor-dependent pathway, as previously described (28).
Non-alcoholic Fatty Liver Disease (NAFLD) which comprises simple steatosis, NASH, cirrhosis and possibly liver carcinoma, is potentially related to a severe form of the fibrotic liver disease; however, how fat deposition renders hepatocytes susceptibility to inflammatory, lipid and oxidative stress mediators is still unidentified. In obesity, immune cells infiltrating the visceral adipose tissue mediate chronic low-grade inflammation that plays a critical role in the pathogenesis of NAFLD (99). It has been demonstrated that administration of rIL-33 aggravates liver fibrosis in an ST2-dependent manner during experimental NAFLD, which is further shown by a substantial reduction of experimentally-induced liver fibrosis in mice lacking IL-33 (28, 33).
In NASH, through secreted cytokines, intrahepatic innate and adaptive immune cells sustain chronic inflammation and induce trans-differentiation of HSCs into myofibroblasts, which are critical cells for liver fibrosis (90). Remarkably, IL-33 treatment has been proposed to attenuate diet-induced hepatic steatosis on the one hand, but aggravate hepatic fibrosis in an ST2-dependent manner on the other hand (33). These findings provide evidence for a dual role of the IL-33/ST2 axis in diet-induced NASH in mice. Similarly, additional results were recently obtained showing that injury-induced endogenous IL-33 release is sufficient to cause inflammation and fibrosis in the bile duct ligated mouse model, which is not further enhanced by rIL-33 (32). More contradictory data reported that IL-33 deficiency in mice does not lessen liver fibrosis during diet-induced steatohepatitis (100).
Hence, in liver fibrosis, evidence indicate that when acute and massive liver damage occurs, the release of IL-33 might act as an activator of tissue-protective mechanisms, while in cases of chronic injury IL-33 shows a significant liver fibrosis promoting effect in an ST2-, Th2- dependent fashion across the entire spectrum of liver pathology.
IL-33/ST2 Axis in Ancreatic Fibrosis
Pancreatic fibrosis is one of the characteristic histopathological findings in cases of chronic pancreatitis. The fibrosis develops as a result of abnormal activation of stromal cells and deposition of ECM proteins. Identification of essential regulators of pancreatic fibrosis, mainly the pancreatic stellate cells (PSCs), has contributed significantly to the understanding of the cellular and molecular basis of these pathogenic processes (101). There is accumulating evidence that PSCs play a key role in the development of pancreatic fibrosis in chronic pancreatitis and pancreatic cancer (34, 102). Additionally, IL-33 is a novel factor involved in the pathogenesis of chronic pancreatitis and possibly pancreatic cancer. In addition, IL-33 has been found to exacerbate acute pancreatic (AP) inflammation in mice (35).
IL-33 is expressed in the nucleus of activated PSCs. Baseline IL-33 expression was reported to be low in quiescent rat PSCs but increased upon cellular activation with mediators such as IL-1b, tumor necrosis factor a (TNF-a), lipopolysaccharide (LPS), platelet-derived growth factor (PDGF)-BB (36). In detail, IL-1b induces IL-33 expression via activation of the nuclear factor (NF)-kβ and ERK pathways and partially through p38 MAPK, whereas PDGF-BB induces IL-33 expression primarily via activating the ERK signaling pathway (18).
It has been recently proposed that IL-33 induction is associated with the transformation to an α-SMA positive PSCs myofibroblastic phenotype. However, treatment of PSCs with rIL-33 did not stimulate any specific phenotype, while a reduction of IL-33 expression resulted in decreased proliferation of PSCs in response to PDGF-BB. Pancreatic myofibroblasts responded to IL-33 by the expression of pro-inflammatory mediators, and increased proliferation and migration, thus playing a crucial role in the progression of pancreatic fibrosis (103). Vice versa, pancreatic myofibroblasts express and secrete modest levels of IL-33 mRNA and protein, respectively. Expression of the ST2 was detected in PSCs and pancreatic myofibroblasts (36).
Moreover, Watanabe et al. found that IL-33 secretion by pancreatic acinar cells under the influence of type I IFN plays a significant role in the development of pancreatic fibrosis occurring in a model of conventional pancreatitis (38). Furthermore, substance P released by pancreatic acinar cells was shown to synergize IL-33 and augment mast cell activation that subsequently regulates the release of several inflammatory mediators in the initiation and progression of AP (35). Remarkably, a triangular link between the cytokine IL-33, pancreatic acinar cells, and mast cells in the development and progression of AP exists (35, 37). Recently Leema G et al. investigated the protective effects of scopoletin, a coumarin compound with anti-inflammatory activities on AP and associated lung injury in mice and found an anti-inflammatory effect by down-regulating substance P signaling via Nf-κB pathway (104).
Besides, activation of plasmacytoid dendritic cells (pDCs) producing IFN-α and IL-33 plays a pivotal role in the chronic fibro-inflammatory responses underlying murine autoimmune pancreatitis (AIP) and human IgG4-related AIP (38).
Watanabe et al. also suggested the possibility that microbe-associated molecular patterns act as pDC activators in AIP, indicating that this form of pancreatic inflammation is initiated and/or driven by gut bacterial components (38). However, further studies defining the gut microbiome in AIP, as well as the demonstration that gut bacteria are translocated into the circulation and can thus contact pancreatic cells, will be required to fully establish this concept (38).
Therefore, IL-33 is considered to be a novel factor implicated in the pathogenesis of acute and chronic pancreatitis and potentially in tissue fibrosis. Specifically, the expression of the ST2 in PSCs, pancreatic myofibroblasts, and pDCs implying a role for IL-33 signaling within the pancreas in an autocrine, and/or paracrine fashion.
IL-33/ST2 Axis in Intestinal Fibrosis
IL-33/ST2 axis seems to represent an important mediator in intestinal fibrosis. Normal epithelium and stroma of the intestine express large amounts of IL-33 and ST2 during the homeostatic turnover of the intestinal mucosa (39). It has been demonstrated that intestinal baseline IL-33 expression was present in pericryptal fibroblasts and was increased during infection (105). A role for IL-33/ST2 signaling in the differentiation of stem cells in organoid culture was also elucidated (105).
IL-33 has been associated with areas of compromised barrier function and plays a critical role in maintaining normal gut homeostasis (44). Uncontrolled IL-33 expansion potentially leads to barrier dysfunction of epithelium, chronic relapsing inflammation, and fibrotic lesions (41–43). Additionally, IL-33 induces enteric glia to secrete glial cell-line derived neurotrophic factor family ligands (GFLs) that play an essential role in intestinal epithelial barrier homeostasis by maintaining tight junctions and negatively regulating local inflammatory response (106, 107). Moreover, IL-33 influences the enteric nervous system to induce intestine hypermotility t to expel invading parasites from the intestine, while is an important regulator of the gut microbiome (108, 109).
Within the gut mucosa, colonic subepithelial myofibroblasts SEMFs are primary sources of IL33, particularly in ulcerated lesions from patients with ulcerative colitis (UC) and induce potent Th2 immune responses (45, 46). The localization of IL-33-producing SEMFs in mucosal ulcerations suggests a significant role of the cytokine in wound healing. Actually, it could be used as an early marker for ulcer-associated activated fibroblasts and myofibroblasts trans-differentiation. Overall, one cannot rule out the potential role of IL-33 in gut-associated fibrosis, particularly in the setting of turnover of chronic tissue damage and repair, characteristics of inflammatory bowel disease (IBD) (45, 46).
The IL-33 expression is enhanced specifically in inflamed mucosa in UC, while exogenous IL-33 treatment in mice, modulated higher colonic mucin release (45, 46). Moreover, IL-33 mRNA levels have been associated with UC disease activity (18, 44, 47). Sponheim et al. found that a feature of IBD-associated mRNA IL-33 expression is the accumulation of both fibroblasts and myofibroblasts in UC lesions. In ulcerations, the fibroblast marker HSP47, platelet-derived growth factor receptor (PDGFR) β, and in part the SEMFs marker α-SMA were expressed (46). Epidermal growth factor has also been demonstrated to contribute to increased IL-33 production and ST2 expression (110).
Epithelial-IL-33 was also increased in pediatric Crohn's ileitis and strongly associated with clinical and histopathological findings, ileal eosinophilia, and complicated fibrostenotic disease (40). Furthermore, neutralization of IL-33 interferes with the massive influx of eosinophils into the gut mucosa and potently decreases fibrogenic gene expression and fibrosis (111).
sST2 constitutes a marker of IBD severity, found to be significantly increased in both the gut mucosa and the serum in both patients and experimental models of IBD. However, in IBD patients, ST2L mRNA expression remained similar to that of healthy controls (14, 44).
IL-33 promotes ILC2s in the gut to produce the growth factor amphiregulin (AREG) that binds to the epidermal growth factor receptor (EGFR) which are responsible for tissue repair during inflammation and restoration of mucosal integrity (42, 112). Disruption of the AREG/EGFR signaling pathway is involved in human patients and murine models of IBD (113, 114). On the other hand, IL-33 stimulation of ILC2s could be beneficial in intestinal inflammation outside the setting of infection for instance in cases of helminth intestinal infection (115). ATP released by parasite-infected cells stimulates local mast cells to produce IL-33, which then activates IL-13-producing ILC2s necessary for helminth expulsion (115).
The role of the IL-33 is not limited to Th2 responses but could also amplify Th1-mediated inflammation (41). IL-33 is an activator of Tregs, activation of which appears to be a compensatory mechanism for intestinal inflammation (112, 116). Colonic Tregs preferentially express ST2. Signaling through ST2/IL-33 promotes both Treg accumulation and maintenance in the gut, enhancing their protective function (117).
The role of IL-33 in colitis as well as in colon cancer is controversial, being very dependent on the timing of alarmin activation or expression relative to the damaging insult (41, 118). Regarding the colon cancer, on the one hand, it has been supported that IL-33 promotes an IFN-γ-mediated immune protective mechanism that helps guard against the development of sporadic colon cancer that links to inflammation (39). IL-33 inhibits colon cancer growth by suppressing cellular proliferation and promoting apoptosis (119). Interestingly, IL-33 has been found to potently induce a neutralizing ant-IL-13 receptor that plays an important protective role in cases of mucosal damage (120–122). Conversely, Zhang et al., found that tumor-derived IL-33 following the activation of tumor cells by pro-inflammatory cytokines such as IL-13, modulates the tumor microenvironment to potently promote colon carcinogenesis and liver metastasis in murine models (123). Furthermore, IL-33 stimulation of human colonic SEMFs has been found to induce the expression of growth factors associated with intestinal tumor progression and extracellular matrix components (124). Similarly, experimental studies in a murine model showed that overexpression of IL-33 promoted the expansion of ST2+Tregs, increased Th2 cytokine milieu, and induced M2 macrophages in the gut, thereby increasing tumor development (125). Furthermore, lower expression of ST2L has been reported in human colon tumors. ST2L expression was negatively associated with the higher tumor grade (126). Inhibition of the IL-33/ST2 pathway may limit mucositis and thus improve the effectiveness of chemotherapy (127).
It is ambiguous if IL-33 is a consequence of intestinal inflammation or if IL-33 is a critical instigator in promoting an inflammatory response. Taken together, there is clear evidence that IL-33/ST2 axis participates in maintaining normal gut homeostasis, particularly in promoting mucosal wound healing and repair. When deregulated, this important ligand-binding pair can also play a critical role in the progression of chronic inflammation and fibrosis, leading to gastrointestinal-related disorders such as IBD as well as colon cancer.
IL-33/ST2 Axis in Renal Fibrosis
Renal fibrosis is characterized by progressive connective tissue expansion through kidney parenchyma, leading to detrimental renal function deterioration (128, 129). Almost all the cell types in the kidneys participate in the pathogenesis of renal fibrosis, illustrating the complexity of this process (48, 51, 52). Specifically, a study by Manetti et al. found that nuclear IL-33 expression in the fibrotic kidneys of patients with SSc was absent in the endothelial cells of peritubular capillaries while ST2 has been expressed abundantly in kidney glomeruli, tubules, and peritubular capillaries (19). Conversely, in control human kidney (HK) specimens, IL-33 was found to be constitutively expressed in peritubular capillary endothelial cells (49). The levels of IL-33 and sST2 were relevant to the progressive deterioration of kidney function while a significant correlation between the serum level of sST2 and disease severity has been shown (50).
Epithelial to Mesenchymal Transition (EMT) of podocytes, tubular epithelial cells, and circulating fibrocytes which are transformed to mesenchymal fibroblasts migrating to adjacent interstitial parenchyma constitutes the principal mechanism of renal fibrosis (129, 130). Other key events in tubulointerstitial fibrosis that also take place in the glomeruli after injury are glomerular infiltration of inflammatory cells and myofibroblastic activation of the mesangial cells (129, 131–133).
Emerging evidence supported that the renal tubular EMT is a remarkable process in the pathogenesis of renal interstitial fibrosis, mediated by IL-33 (50). IL-33 has been found to participate in transplanted kidney interstitial fibrosis promoting EMT of HK-2 cells in a dose- and time-dependent manner, via the activation of the p38 MAPK signaling pathway (129). These data suggest that therapies are targeting a reduction in IL-33 levels or on the downregulation of the p38 MAPK signaling axis may be an effective strategy in the prevention of kidney interstitial fibrosis (129).
IL-33 is also a marker of IRI that contributes to innate immune cell recruitment and development of renal graft damage associated with renal transplantation in humans (53). Furthermore, it plays a significant role in the pathogenesis of IRI-induced renal fibrosis through regulating myeloid fibroblast accumulation, inflammatory cell infiltration, and cytokine and chemokine expression (52, 53). Th2- cytokines, including IL-4, IL-5, and IL-13, all induced by IL-33, play an essential role in the renal fibrotic disease (52, 53).
Experimental data revealed that IL-33-treated IRI mice had increased levels of IL-4 and IL-13 in serum and renal tissue as well as more ILC2s, Tregs, and anti-inflammatory M2 macrophages, as compared to control-treated IRI mice (54). Furthermore, it has been reported that depletion of ILC2s substantially abolished the protective effect of IL-33 on renal IRI (54). Conversely, adoptive transfer of ex vivo-expanded ILC2 prevented renal injury in mice subjected to IRI. Besides, treatment of mice with IL-33 or ILC2 after IRI had a protective effect associated with induction of M2 macrophages in kidney and required the ILC2 production of amphiregulin (54). Interestingly, mice treated with sST2 exhibited less severe renal dysfunction and fibrosis in response to IRI compared with vehicle-treated mice (54). Furthermore, inhibition of IL-33 suppressed BM-derived fibroblast accumulation and myofibroblast formation in the kidneys after IRI stress, which was associated with less expression of ECM proteins (54). Hence, IL-33 signaling in ILC2s plays a critical role in the pathogenesis of IRI–induced renal fibrosis and treatment with IL-33 inhibitor reduced pro-inflammatory cytokine and chemokine levels in the kidneys of mice following IRI insult (54).
Furthermore, it is increasingly recognized that episodes of acute kidney injury (AKI) increase the susceptibility of chronic kidney disease (CKD) and end-stage renal disease (ESRD) that are characterized by organ fibrogenesis (134, 135). It has been found that the administration of rIL-33 exacerbated cisplatin-induced AKI by acting as a pro-inflammatory cytokine (49). In a cisplatin-induced mouse AKI model, IL-33 was reported to promote AKI through CD4+ T cell-mediated production of chemokine (C-X-C motif) ligand (CXCL) 1, which could exacerbate the renal damage. In addition, high expression levels of IL-33 have been observed in LPS-induced acute glomerular injury (49, 136).
Additionally, IL-33 released from necrotic cells has been implicated in autophagy, which can balance increased apoptosis secondary to contrast-induced nephropathy in diabetic kidney disease (55). Another study also reported that IL-33 contributes to kidney fibrosis associated with systemic lupus erythematosus (SLE).
Thus, emerging data indicate that the upregulation of the IL-33/ST2 signaling pathway may promote tubular cell injury and fibrosis predominantly via EMT in the kidneys (56).
IL-33/ST2 Axis in Heart Fibrosis
Heart failure (HF) and cardiac fibrosis are associated with IL-33 mainly in cases of a mechanical strain of cardiac fibroblasts (57, 64). IL-33 demonstrates anti-hypertrophic and anti-fibrotic effects on cardiomyocytes, transduced by ST2L. In 2007, Sanada et al. first documented that IL-33 prevents cardiomyocyte apoptosis, reduces infarct size, fibrosis, and apoptosis through induction of anti-apoptotic proteins after ischemia-reperfusion in rats and improves cardiac function and survival after myocardial infarction (57). IL-33 correlated with the expression kinetics of the anti-apoptotic gene B-cell lymphoma 2 (Bcl-2), which is in agreement with its anti-apoptotic role (58). Thereafter, multiple experimental studies have also illustrated that IL-33 attenuates cardiac fibrosis induced by the increased cardiovascular load, showing that IL-33 directly inhibits pro-fibrotic activities of cardiac fibroblasts (58, 59, 137). Treatment of rat cardiac fibroblasts with IL-33 was also found to impair the migratory activity of fibroblasts or their precursors into the stressed myocardium (57, 138). IL-33 levels were found to be significantly elevated upon a cyclic stretch of cardiac fibroblasts in vitro, and the administration of IL-33 was shown to inhibit myocyte amino acid incorporation and growth thus protecting against cardiac hypertrophy (57). IL-33 protected cardiomyocytes from hypoxia-induced apoptosis in vitro, and this effect was partially inhibited by sST2, highlighting the critical role of IL-33 in regulating cardiac myocyte function and its protective role in cardiac fibrotic diseases (58).
Others have reported that ablation of IL-33 gene caused exaggerated cardiac remodeling in both ischemic and non-ischemic HF, It leads to cardiomyocyte hypertrophy and cardiac fibrosis upon mechanical stress, impaired cardiac function, and survival (60). Furthermore, it has been recently shown that IL-33 acts by reducing a form of erythrocyte superoxide dismutase (eSOD) production, thus eSOD is found decreased in chronic HF (61). eSOD is a protective enzyme against oxidative stress in chronic HF (61). Alternatively, the in vitro administration of IL-33 significantly decreased cardiac interstitial fibrosis in wild-type mice underwent transaortic constriction surgery to increase cardiovascular load (57).
Of note, the aforementioned IL-33 benefits were absent in mice with deletion of the ST2 gene, so these data indicate that IL-33 exerts its cardioprotective role only through the ST2 receptor signaling. Moreover, the microRNA-587b has been proposed to ameliorate cell apoptosis, inflammatory reaction of myocarditis, and fibrosis through inhibition of the IL-33/ST2 pathway by suppressing IL-33 (139).
In contrast, sST2 disrupts the cardioprotective effects of IL-33 by sequestering its availability for binding with the transmembrane receptor ST2L. sST2 has been demonstrated as a marker of myocardial fibrosis and HF progression. Both cardiac fibroblasts and cardiomyocytes express IL-33 and sST2, and expression levels are increased as a response to myocardial stress (15). This issue is supported from a clinical perspective, given that sST2 concentrations have repeatedly been found high in patients with acute myocardial infarction and acute HF and correlate with parameters of infarct magnitude, cardiac dysfunction, hemodynamic impairment, and neurohormonal derangement (15). Based on the above sST2 is thought to be a biomarker for poor outcome in patients with cardiovascular disease (140–142). Moreover, cardiogenic shock and increased C-reactive protein levels are associated with higher sST2 levels. The PRIDE (Pro-Brain Natriuretic Peptide Investigation of Dyspnea in the Emergency Department) study highlighted the potential applications of sST2 in acute HF (143). Thereafter, several studies that followed emphasized on its diagnostic and prognostic utility (144–146). sST2 may also identify patients who benefit most from cardiac resynchronization therapy defibrillators (147), titration of beta blockers (62) and angiotensin-converting enzyme inhibitors (148). Weir et al. showed that sST2 could predict functional recovery and left ventricle remodeling during the post-infarction period (149). The sST2 levels were positively correlated with the degree of cardiac fibrosis (150). Along these lines, it has been recently observed that left ventricular assist device (LVAD) resulted in a significant drop in sST2 levels with normalization within 3 months post-implantation, thus lessened heart fibrosis and inflammation (151).
According to the above-mentioned studies, recently, a highly sensitive ELISA for sST2 (Presage ST2) as well as a rapid quantitative lateral flow immunoassay for measurement of sST2 in human plasma has been developed, allowing for point-of-care testing (142). The first one was approved by regulatory agencies both in Europe and the United States for prognostication in HF. The second one (Aspect-PLUS ST2 test, Critical Diagnostics, San Diego, CA, USA) has received regulatory approval in Europe, but it has yet to be approved by the Food and Drug Administration in the United States (142). The American College of Cardiology Foundation/American Heart Association (ACCF/AHA) guidelines of 2013 have incorporated sST2 as a relevant marker of fibrosis. They recommend it for additive risk stratification in patients with acutely decompensated HF (level of evidence A) or chronic HF (level of evidence B) (63).
Additionally, the metabolic activity of epicardial adipose tissue has been recently associated with a decrease in the IL-33 levels, thus was closely related to the development of cardiac fibrosis at 1-year post-myocardial infraction (150).
Moreover, Gal-3 has been associated with left ventricular remodeling along with an increased risk of incident HF and mortality (152). It has been reported that Gal-3 promotes myocardial fibrosis, whereas myocardial fibrosis and hypertrophy are prevented through interaction between IL-33 and sST2 (57).
Hence, IL-33 demonstrates cardioprotective, anti-hypertrophic and anti-fibrotic effects on cardiomyocytes, transduced by ST2L, and disturbing by sST2.
IL-33/ST2 Axis in Skin Fibrosis
IL-33 is released by dermal fibroblasts (3). It has been documented that the IL-33/ST2 signaling is associated with abnormal fibroblast proliferation, leukocyte infiltration and morphologic differentiation of human endothelial cells, resulting in increased endothelial permeability, consistently with increased angiogenesis and ECM deposition in vivo (65).
IL-33 has been reported to be a crucial signaling cytokine in skin pathology by inducing IL-13-dependent cutaneous fibrosis mechanism, required both eosinophils and recombination-activating gene (RAG)-dependent lymphocytes. Eosinophils contribute to tissue remodeling and fibrosis (65). It is known that in skin diseases, eosinophil expresses a broad spectrum of Th2 cytokines such as IL-4, IL-5, IL-13, and C-C motif chemokine-11 (CCL11/eotaxin) (66). The different cytokine expression patterns suggest distinct functional roles of eosinophils in various diseases that might be related to host defense, immunomodulation, fibrosis, and/or tumor development (67).
More convincingly, in cutaneous fibrosis, the injection of rIL-33 induces collagen production via ST2-dependent recruitment of BM-derived eosinophils that further secrete IL-13 in response to IL-33 stimulation (22, 65). IL-33 has also been proposed as a critical molecule operating in eosinophil-mediated fibrosis in the high-dose-per fraction irradiated skin (66). In detail, vascular endothelial cells damaged by high-dose radiation secrete IL-33, which may stimulate fibrotic responses via eosinophil recruitment and eosinophil-mediated Th2 immune responses.
Rankin et al. demonstrated that IL-33 induces cutaneous fibrosis and intense inflammation that are associated with large numbers of CD3+ cells and F4/80+ myeloid cells, except infiltrating eosinophils (22). Additionally, IL-33 was also shown to induce several others cytokines such as IL-4, IL-5, tissue inhibitor of metalloproteinases 1 (TIMP1), MMP12, and MMP13 gene expression; however, it did not induce the expression of TGF-β1 which participated with varying degrees in fibrogenesis (22).
As far as SSc is concerned the most severe variant is characterized by aggressive skin fibrosis (68). These patients experience a low health-related quality of life that is directly related to the extent of the dermal fibrosis (68). Serum levels of IL-33 are elevated in SSc, even more pronounced in diffuse cutaneous SSc than in limited cutaneous SSc; thus these levels are positively correlated with the total fibrotic skin score. In other words, serum IL-33 levels are likely to reflect the degree of endothelial damage in patients with SSc (68).
IL-33 might mediate very early pathogenic events of SSc through recruitment and stimulation of ST2-expressing cells (immune cells and fibroblast/myofibroblast) (68). Other studies reported that increased circulating levels of IL-33 in SSc correlate with early disease stage and microvascular involvement being a serum marker for vascular abnormalities in SSc (69). IL-33 induces migration of Th2 lymphocytes and enhances Th2 cytokine production. Remarkably, owing to the Th2-type signature, there are elevated levels of both IL-4 and IL-13 in SSc patient's sera (67), while SSc patients exhibit substantial Th2 cytokine production in cultures of CD4+ T lymphocytes isolated from their affected skin (153). It is likely that the signaling mechanism in the dermal fibroblasts mediated by IL-13 is STAT6 (67, 154, 155).
A recent study demonstrated that the rs7044343 polymorphism of the IL-33 gene was associated with susceptibility to SSc in a Turkish population. No similar studies were found in the literature (156).
Conversely, sST2 constitutes a potential marker for disease progression in limited cutaneous SSc with disease duration over 9 years. On the contrary, sST2 was not elevated in healthy controls or SSc patients with early skin involvement or disease duration shorter than 9 years (157). Furthermore, sST2 serum levels were lowered by iloprost (prostacyclin) treatment (157). The question remains why sST2 is elevated in limited cutaneous SSc and not in diffuse cutaneous SSc patients. Diffuse cutaneous SSc and limited cutaneous SSc may be two pathophysiologically different diseases rather than two subtypes of one disease showing significant differences of organ involvement, disease progression and significantly different chemokine levels between two entities (158, 159). Accordingly, sST2 could be a marker for pathological alterations and higher sST2 in limited cutaneous SSc could be partially beneficial by blocking the high inflammatory capacity of IL-33 by neutralizing its bioactivity.
In addition, evidence is presented to support the high tissue-localized expression of IL-33 in patients with SSc, as well as IL-33-dependant skin-localized Tregs trans-differentiation into Th2-like cells, combined with expression of the ST2 receptor on Tregs. In other words, IL-33 might be an important stimulator of tissue-localized loss of normal Tregs function (70). Moreover, friend leukemia virus integration 1 (Fli1) -a predisposing factor of SSc- haploinsufficiency increases Th2- and Th17-like Tregs proportions in BLM-induced pro-fibrotic skin condition, in which IL-33-producing dermal fibroblasts contribute to Th2-like Tregs trans-differentiation (71).
Consequently, IL-33 is a crucial cytokine in skin pathology responsible for abnormal fibroblast proliferation, leukocyte infiltration and morphologic differentiation of human endothelial cells, leading to fibrotic skin conditions.
Conclusion
In this review, we spotlight the distinctive contribution of IL-33/ST2 signaling in organ fibrosis as well as the significant role of the Th2 pattern of immune response in the pathophysiology of organ fibrosis. The IL-33/ST2 axis widely participates in the fibrotic process of many vital organs, demonstrating clear direct effects on wound healing and remodeling. Generally, the IL-33/ST2 signaling pathway has mainly anti-inflammatory/anti-proliferative effects. However, chronic tissue injury is responsible for pro-inflammatory/pro-fibrogenetic responses. At the basal level, both flIL-33 and mIL-33 forms have been reported to contribute to fibrogenesis. The axis influences the capacity of various cells to trans-differentiate into extracellular matrix-secreting activated myofibroblasts which constitute the main cell population of fibrosis, in an organ-specific underlying mechanism. Furthermore, the IL-33/ST2 axis is involved in angiogenesis, production of matrix components, ECM deposition. Importantly, elevated levels of IL-33 and/or sST2 constitute markers of dysfunction and severity in many fibrotic diseases. IL-33/ST2 axis seems to be a promising therapeutic target in fibrosis constitutes, therefore, a critical area for further investigation.
Author Contributions
OK took part in decision on structure and content of the review, performing literature, search, and writing the review. KG revised the draft critically; gave final approval of the submitted version. SZ took part in decision on structure and content of the review, contributed to writing the review and gave thorough feedback throughout the process, and accepting the final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AP, acute pancreatitis; ASCF, American College of Cardiology Foundation; AHA, American Heart Association; Acute Kidney Injury; ALI, Acute Lung Injury; AP-1, Activator Protein 1; a-SMA, α-Smooth Muscle Actin; BAL, Bronchoalveolar Lavage; BLM, Bleomycin; BM, Bone Marrow; CKD, Chronic Kidney Disease; CXCL, Chemokine (C-X-C motif) Ligand; DAMPs, Damage Associated Molecular Patterns; ECM, Extracellular Matrix; EGFR, Epidermal Growth Factor Receptor; EMT, Epithelial to Mesenchymal Transition; ERK, Extracellular Signal-Regulated Kinase, eSOD, Erythrocyte Superoxide Dismutase; ESRD, End-Stage Renal Disease; Fbln1, fibulin-1; Fli1, Friend Leukemia Virus Integration; flIL-33, full-length Interleukin 33; FoxO3a, Forkhead box O3a; Gal-3, Galectin-3; GFLs, glial cell-line derived neurotrophic factor family ligands; HF, Heart Failure; HK, Human Kidney; HSCs, Hepatic Stellate Cells; HSP, Heat Shock Protein; IBD, Inflammatory Bowel Disease; IL, Interleukin; IL-13Ra1, IL-13 Receptor subunit alpha 1; ILC2s, Innate Lymphoid Cell type 2; IFN-γ, Interferon-γ; IPF, Idiopathic Pulmonary Fibrosis; IRI, Ischemia-Reperfusion Injury; JNK, Jun N-terminal kinase; LPS, Lipopolysaccharide; LVAD, Left Ventricular Assist Device; MAPK, Mitogen-Activated Protein Kinase; MCP-1, Monocyte Chemoattractant Protein-1; MEK, Mitogen-Activated Protein Kinase\ERK kinase; mIL-33, mature Interleukin 33; MMP, Matrix Metalloproteinase; MyD88, Myeloid Differentiation primary response 88; NAFLD, Non-alcoholic Fatty Liver Disease; NK cells, Natural Killers cells; NASH, Non-alcoholic Steatohepatitis; NF, Nuclear Factor; PDGF, Platelet-Derived Growth Factor; PDGFR, Platelet-Derived Growth Factor Receptor; pDCs, plasmacytoid Dendritic Cells; PGE2, Prostaglandin E2; PRIDE, Pro-Brain Natriuretic Peptide Investigation of Dyspnea in the Emergency Department; PSCs, Pancreatic Stellate Cells; RAG, Recombination-Activating Gene; SEMFs, Subepithelial Myofibroblasts; SLE, Systemic Lupus Erythematosus; SSc, Systemic Sclerosis; STAT6, Signal Transducer and Activator of Transcription 6; ST2, Suppression of Tumorigenicity 2; sST2, soluble ST2; ST2L, transmembrane ST2; TGF-β, Transforming Growth Factor β; Th1, T helper type 1; Th2, T helper type 2; TIMP1, Tissue Inhibitor of Metalloproteinases 1; TNF-a, Tumor Necrosis Factor a; Tregs, T-regulatory cells; rIL-33, recombinant Interlrukin-33; UC, Ulcerative Colitis.
References
1. Onda H, Kasuya H, Takakura K, Hori T, Imaizumi T, Takeuchi T, et al. Identification of genes differentially expressed in canine vasospastic cerebral arteries after subarachnoid hemorrhage. J Cerebr Blood Flow Met. (1999) 19:1279–88. doi: 10.1097/00004647-199911000-00013
2. Baekkevold ES, Roussigne M, Yamanaka T, Johansen FE, Jahnsen FL, Amalric F, et al. Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Am J Pathol. (2003) 163:69–79. doi: 10.1016/S0002-9440(10)63631-0
3. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. (2005) 23:479–90. doi: 10.1016/j.immuni.2005.09.015
4. Dinarello CA. An IL-1 family member requires caspase-1 processing and signals through the ST2 receptor. Immunity (2005) 23:461–2. doi: 10.1016/j.immuni.2005.10.004
5. Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS ONE (2008) 3:e3331. doi: 10.1371/journal.pone.0003331
6. Li D, Guabiraba R, Besnard AG, Komai-Koma M, Jabir MS, Zhang L, et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol. (2014) 134:1422.e11–32. doi: 10.1016/j.jaci.2014.05.011
7. Drake LY, Kita H. L-33: biological properties, functions, and roles in airway disease. Immunol Rev. (2017) 278:173–84. doi: 10.1111/imr.12552
8. Nakanishi W, Yamaguchi S, Matsuda A, Suzukawa M, Shibui A, Nambu A, et al. IL-33, but not IL-25, is crucial for the development of house dust mite antigen-induced allergic rhinitis. PLoS ONE (2013) 8:e78099. doi: 10.1371/journal.pone.0078099
9. Lefrançais E, Cayrol C. Mechanisms of IL-33 processing and secretion: differences and similarities between IL-1 family members. Eur Cytokine Netw. (2012) 23:120–7. doi: 10.1684/ecn.2012.0320
10. Lefrancais E, Roga S, Gautier V, Gonzalez-de-Peredo A, Monsarrat B, Girard JP, et al. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci USA. (2012) 109:1673–8. doi: 10.1073/pnas.1115884109
11. Lefrançais E, Duval A, Mirey E, Roga S, Espinosa E, Cayrol C, et al. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc Natl Acad Sci USA. (2014) 111:15502–7. doi: 10.1073/pnas.1410700111
12. Hayakawa M, Hayakawa H, Matsuyama Y, Tamemoto H, Okazaki H, Tominaga S. Mature interleukin-33 is produced by calpain-mediated cleavage in vivo. Biochem Biophys Res Commun. (2009) 387:218–22. doi: 10.1016/j.bbrc.2009.07.018
13. Gajardo Carrasco T, Morales RA, Pérez F, Terraza C, Yáñez L, Campos-Mora M, et al. Alarmin' immunologists: IL-33 as a putative target for modulating T cell-dependent responses. Front Immunol. (2015) 6:232. doi: 10.3389/fimmu.2015.00232
14. Griesenauer B, Paczesny S. The ST2/IL-33 axis in immune cells during inflammatory diseases. Front Immunol. (2017) 8:475. doi: 10.3389/fimmu.2017.00475
15. Pascual-Figal DA, Januzzi JL. The biology of ST2: the international ST2 consensus panel. Am J Cardiol. (2015) 115:3B–7. doi: 10.1016/j.amjcard.2015.01.034
16. Mueller T, Jaffe AS. Soluble ST2–analytical considerations. Am J Cardiol. (2015) 115:8B−21. doi: 10.1016/j.amjcard.2015.01.035
17. Lee JU, Chang HS, Lee HJ, Jung CA, Bae DJ, Song HJ, et al. Upregulation of interleukin-33 and thymic stromal lymphopoietin levels in the lungs of idiopathic pulmonary fibrosis. BMC Pulm Med. (2017) 17:39. doi: 10.1186/s12890-017-0380-z
18. Gao Q, Li Y, Li M. The potential role of IL-33/ST2 signaling in fibrotic diseases. J Leukoc Biol. (2015) 98:15–22. doi: 10.1189/jlb.3RU0115-012R
19. Manetti M, Ibba-Manneschi L, Liakouli V, Guiducci S, Milia AF, Benelli G, et al. The IL1-like cytokine IL33 and its receptor ST2 are abnormally expressed in the affected skin and visceral organs of patients with systemic sclerosis. Ann Rheum Dis. (2010) 69:598–605. doi: 10.1136/ard.2009.119321
20. Gao Q, Li Y, Pan X, Yuan X, Peng X, Li M. Lentivirus expressing soluble ST2 alleviates bleomycin-induced pulmonary fibrosis in mice. Int Immunopharmacol. (2016) 30:188–93. doi: 10.1016/j.intimp.2015.11.015
21. Xu J, Zheng J, Song P, Zhou Y, Guan S. IL33/ST2 pathway in a bleomycin induced pulmonary fibrosis model. Mol Med Rep. (2016) 14:1704–8. doi: 10.3892/mmr.2016.5446
22. Rankin AL, Mumm JB, Murphy E, Turner S, Yu N, McClanahan TK, et al. IL-33 induces IL-13-dependent cutaneous fibrosis. J Immunol. (2010) 184:1526–35. doi: 10.4049/jimmunol.0903306
23. Luzina IG, Kopach P, Lockatell V, Kang PH, Nagarsekar A, Burke AP. Interleukin-33 potentiates bleomycin-induced lung injury. Interleukin-33 potentiates bleomycin-induced lung injury. Am. J. Respir. Cell. Mol. Biol. (2013) 49:999–1008. doi: 10.1165/rcmb.2013-0093OC
24. Sun Z, Chang B, Gao M, Zhang J, Zou Z. IL-33-ST2 axis in liver disease: progression and challenge. Mediators Inflamm. (2017) 2017:5314213. doi: 10.1155/2017/5314213.
25. Weiskirchen R, Tacke F. Interleukin-33 in the pathogenesis of liver fibrosis: alarming ILC2 and hepatic stellate cells. Cell Mol Immunol. (2017) 14:143–5. doi: 10.1038/cmi.2016.62
26. Hammerich L, Tacke F. Interleukins in chronic liver disease: lessons learned from experimental mouse models. Clin Exp Gastroenterol. (2014) 7:297–306. doi: 10.2147/CEG.S43737. eCollection 2014
27. Marvie P, Lisbonne M, L'Helgoualc'h A, Rauch M, Turlin B, Preisser L, et al. Interleukin-33 overexpression is associated with liver fibrosis in mice and humans. J Cell Mol Med. (2010) 14:1726–39. doi: 10.1111/j.1582-4934.2009.00801.x
28. McHedlidze T, Waldner M, Zopf S, Walker J, Rankin AL, Schuchmann M, et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity (2013) 39:357–71. doi: 10.1016/j.immuni.2013.07.018
29. Wang J, Cai Y, Ji H, Feng J, Ayana DA, Niu J, et al. Serum IL-33 levels are associated with liver damage in patients with chronic hepatitis B. J Interferon Cytokine Res. (2012) 32:248–53. doi: 10.1089/jir.2011.0109
30. Sun Y, Zhang JY, Lv S, Wang H, Gong M, Du N, et al. Interleukin-33 promotes disease progression in patients with primary biliary cirrhosis. Tohoku J Exp Med. (2014) 234:255–61. doi: 10.1620/tjem.234.255
31. Li J, Razumilava N, Gores GJ, Walters S, Mizuochi T, Mourya R, et al. Biliary repair and carcinogenesis are mediated by IL-33-dependent cholangiocyte proliferation. J Clin Invest. (2014) 124:3241–51. doi: 10.1172/JCI73742
32. Tan Z, Liu Q, Jiang R, Lv L, Shoto SS, Maillet I, et al. Interleukin-33 drives hepatic fibrosis through activation of hepatic stellate cells. Cell. Mol. Immunol. (2017) 15:388–98. doi: 10.1038/cmi.2016.63
33. Gao Y, Liu Y, Yang M, Guo X, Zhang M, Li H, et al. IL-33 treatment attenuated diet-induced hepatic steatosis but aggravated hepatic fibrosis. Oncotarget (2016) 7:33649–61. doi: 10.18632/oncotarget.9259
34. Pang TCY, Wilson JS, Apte MV. Pancreatic stellate cells: what's new? Curr Opin Gastroenterol. (2017) 33:366–73. doi: 10.1097/MOG.0000000000000378
35. Kempuraj D, Twait EC, Williard DE, Yuan Z, Meyerholz DK, Samuel I. The novel cytokine interleukin-33 activates acinar cell proinflammatory pathways and induces acute pancreatic inflammation in mice. PLoS ONE (2013) 8:e56866. doi: 10.1371/journal.pone.0056866
36. Masamune A, Watanabe T, Kikuta K, Satoh K, Kanno A, Shimosegawa T. Nuclear expression of interleukin-33 in pancreatic stellate cells. Am J Physiol Gastrointest Liver Physiol. (2010) 299:G821–32. doi: 10.1152/ajpgi.00178.2010
37. Dib M, Zhao X, Wang X, Andersson R. Role of mast cells in the development of pancreatitis-induced multiple organ dysfunction. Br J Surg. (2002) 89:172–8. doi: 10.1046/j.0007-1323.2001.01991.x
38. Watanabe T, Yamashita K, Arai Y, Minaga K, Kamata K, Nagai T, et al. Chronic fibro-inflammatory responses in autoimmune pancreatitis depend on IFN-α and IL-33 produced by plasmacytoid dendritic cells. J Immunol. (2017) 198:3886–96. doi: 10.4049/jimmunol.1700060
39. Eissmann MF, Dijkstra C, Wouters MA, Baloyan D, Mouradov D, Nguyen PM, et al. Interleukin 33 signaling restrains sporadic colon cancer in an interferon-γ-dependent manner. Cancer Immunol. Res. (2018) 6:409–21. doi: 10.1158/2326-6066.CIR-17-0218
40. Masterson JC, Capocelli KE, Hosford L, Biette K, McNamee EN, de Zoeten EF, et al. Eosinophils and IL-33 perpetuate chronic inflammation and fibrosis in a pediatric population with Stricturing Crohn's Ileitis. Inflamm Bowel Dis (2015) 21:2429–40. doi: 10.1097/MIB.0000000000000512
41. Lopetuso LR, Scaldaferri F, Pizarro TT. Opposing functions of classic and novel IL-1 family members in gut health and disease. Front Immunol. (2013) 4:181. doi: 10.3389/fimmu.2013.00181
42. Monticelli A, Osborne LC, Noti M, Tran SV, Zaiss DMW, Artis D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin–EGFR interactions. Proc Natl Acad Sci USA. (2015) 112:10762–67. doi: 10.1073/pnas.1509070112
43. Pastorelli L, De Salvo C, Cominelli MA, Vecchi M, Pizarro TT. Novel cytokine signaling pathways in inflammatory bowel disease: insight into the dichotomous functions of IL-33 during chronic intestinal inflammation. Therap Adv Gastroenterol. (2011) 4:311–23. doi: 10.1177/1756283X11410770
44. Beltran CJ, Nunez LE, Diaz-Jimenez D, Farfan N, Candia E, Heine C, et al. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflamm Bowel Dis. (2010) 16:1097–107. doi: 10.1002/ibd.21175
45. Kobori A, Yagi Y, Imaeda H, Ban H, Bamba S, Tsujikawa T, et al. Interleukin-33 expression is specifically enhanced in inflamed mucosa of ulcerative colitis. J Gastroenterol. (2010) 45:999–1007. doi: 10.1007/s00535-010-0245-1
46. Sponheim J, Pollheimer J, Olsen T, Balogh J, Hammarström C, Loos T, et al. Inflammatory bowel disease-associated interleukin-33 is preferentially expressed in ulceration-associated myofibroblasts. Am J Pathol. (2010) 177:2804–15. doi: 10.2353/ajpath.2010.100378
47. Seidelin JB, Bjerrum JT, Coskun M, Widjaya B, Vainer B, Nielsen OH. IL-33 is upregulated in colonocytes of ulcerative colitis. Immunol. Lett. (2010) 128:80–5. doi: 10.1016/j.imlet.2009.11.001
48. Humphreys BD. Mechanisms of renal fibrosis. Annu Rev Physiol. (2018) 80:309–26. doi: 10.1146/annurev-physiol-022516-034227
49. Akcay A, Nguyen Q, He Z, Turkmen K, Won D, Hernando AA, et al. IL-33 exacerbates acute kidney injury. J Am Soc Nephrol. (2011) 22:2057–67. doi: 10.1681/ASN.2010091011
50. Chen WY, Chang YJ, Su CH, Tsai TH, Chen SD, Hsing CH, et al. Upregulation of Interleukin-33 in obstructive renal injury. Biochem Biophys Res Commun. (2016) 473:1026–32. doi: 10.1016/j.bbrc.2016.04.010
51. Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. (2011) 7:684–96. doi: 10.1038/nrneph.2011.149
52. Liang H, Xu F, Wen XJ, Liu HZ, Wang HB, Zhong JY, et al. Interleukin-33 signaling contributes to renal fibrosis following ischemia reperfusion. Eur J Pharmacol. (2017) 812:18–27. doi: 10.1016/j.ejphar.2017.06.031
53. Ferhat M, Robin A, Giraud S, Sena S, Goujon JM, Touchard G, et al. Endogenous IL-33 contributes to kidney ischemia-reperfusion injury as an alarmin. J Am Soc Nephrol. (2018) 29:1272–88. doi: 10.1681/ASN.2017060650.
54. Cao Q, Wang Y, Niu Z, Wang C, Wang R, Zhang Z, et al. Potentiating tissue-resident type 2 innate lymphoid cells by IL-33 to prevent renal ischemia-reperfusion injury. J Am Soc Nephrol. (2018) 29:961–76. doi: 10.1681/ASN.2017070774
55. Demirtas L, Turkmen K, Kandemir FM, Ozkaraca M, Kucukler S, Gürbüzel M, et al. The possible role of interleukin-33 as a new player in the pathogenesis of contrast-induced nephropathy in diabetic rats. Ren Fail. (2016) 38:952–60. doi: 10.3109/0886022X.2016.1165034
56. Chen WY, Li LC, Yang YL. Emerging roles of IL-33/ST2 axis in renal diseases. Int J Mol Sci. (2017) 18:783. doi: 10.3390/ijms18040783
57. Sanada S, Hakuno D, Higgins LJ, Schreiter ER, McKenzie AN, Lee RT. IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J Clin Invest. (2007) 117:1538–49. doi: 10.1172/JCI30634
58. Seki K, Sanada S, Kudinova AY, Steinhauser ML, Handa V, Gannon J, et al. Interleukin-33 prevents apoptosis and improves survival after experimental myocardial infarction through ST2 signaling. Circ Heart Fail. (2009) 2:684–91. doi: 10.1161/CIRCHEARTFAILURE.109.873240
59. Sánchez-Más J, Lax A, Asensio-López Mdel C, Fernandez-Del Palacio MJ, Caballero L, Santarelli G, et al. Modulation of IL-33/ST2 system in postinfarction heart failure: correlation with cardiac remodelling markers. Eur J Clin Invest. (2014) 44:643–51. doi: 10.1111/eci.12282
60. Veeraveedu PT, Sanada S, Okuda K, Fu HY, Matsuzaki T, Araki R, et al. Ablation of IL-33 gene exacerbate myocardial remodeling in mice with heart failure induced by mechanical stress. Biochem Pharmacol. (2017) 138:73–80. doi: 10.1016/j.bcp.2017.04.022
61. Zhang HF, Xie SL, Chen YX, Mai JT, Wang JF, Zhu WL, et al. Altered serum levels of IL-33 in patients with advanced systolic chronic heart failure: correlation with oxidative stress. J Transl Med. (2012) 10:120. doi: 10.1186/1479-5876-10-120
62. Gaggin HK, Motiwala S, Bhardwaj A, Parks KA, Januzzi JL Jr. Soluble concentrations of the interleukin receptor family member ST2 and beta-blocker therapy in chronic heart failure. Circ Heart Fail. (2013) 6:1206–13. doi: 10.1161/CIRCHEARTFAILURE.113.000457
63. Yancy CW, Jessup M, Chair V, Bozkurt B, Butler J, Casey DE, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. (2013) 128:e240–327. doi: 10.1016/j.jacc.2013.05.019
64. Kakkar R, Hei H, Dobner S, Lee RT. Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J Biol Chem. (2012) 287:6941–8. doi: 10.1074/jbc.M111.298703
65. Artlett CM. The IL-1 family of cytokines. Do they have a role in scleroderma fibrosis? Immunol Lett. (2018) 195:30–7. doi: 10.1016/j.imlet.2017.11.012
66. Lee EJ, Kim JW, Yoo H, Kwak W, Choi WH, Cho S, et al. Single high-dose irradiation aggravates eosinophil-mediated fibrosis through IL-33 secreted from impaired vessels in the skin compared to fractionated irradiation. Biochem Biophys Res Commun. (2015) 464:20–6. doi: 10.1016/j.bbrc.2015.05.081
67. O'Reilly S. Role of interleukin-13 in fibrosis, particularly systemic sclerosis. Biofactors (2013) 39:593–6. doi: 10.1002/biof.1117
68. Yanaba K, Yoshizaki A, Asano Y, Kadono T, Sato S. Serum IL-33 levels are raised in patients with systemic sclerosis: association with extent of skin sclerosis and severity of pulmonary fibrosis. Clin Rheumatol. (2011) 30:825–30. doi: 10.1007/s10067-011-1686-5
69. Duan L, Chen J, Gong F, Shi G. The role of IL-33 in rheumatic diseases. Clin Dev Immunol. (2013) 2013:924363. doi: 10.1155/2013/924363
70. MacDonald KG, Dawson NA, Huang Q, Dunne JV, Levings MK, Broady R. Regulatory T cells produce profibrotic cytokines in the skin of patients with systemic sclerosis. J Allergy Clin Immunol. (2015) 135:946.e9. doi: 10.1016/j.jaci.2014.12.1932
71. Saigusa R, Asano Y, Taniguchi T, Hirabayashi M, Nakamura K, Miura S, et al. Fli1-haploinsufficient dermal fibroblasts promote skin-localized transdifferentiation of Th2-like regulatory T cells. Arthritis Res Ther. (2018) 20:23. doi: 10.1186/s13075-018-1521-3
72. Ueha S, Shand FHW, Matsushima K. Cellular and molecular mechanisms of chronic inflammation-associated organ fibrosis. Front Immunol. (2012) 3:71. doi: 10.3389/fimmu.2012.00071
73. Lee JS, Seppanen E, Patel J, Rodero MP, Khosrotehrani K. ST2 receptor invalidation maintains wound inflammation, delays healing and increases fibrosis. Exp Dermatol. (2016) 25:71–4. doi: 10.1111/exd.12833
74. Yin H, Li X, Hu S, Liu T, Yuan B, Gu H, et al. IL-33 accelerates cutaneous wound healing involved in upregulation of alternatively activated macrophages. Mol. Immunol. (2013) 56:347–53. doi: 10.1016/j.molimm.2013.05.225
75. Luzina IG, Pickering EM, Kopach P, Kang PH, Lockatell V, Todd NW, et al. Full-length IL-33 promotes inflammation but not Th2 response in vivo in an ST2-independent fashion. J Immunol. (2012) 189:403–10. doi: 10.4049/jimmunol.1200259
76. de Kleer IM, Kool M, de Bruijn MJ, Willart M, van Moorleghem J, Schuijs MJ, et al. Immunity (2016) 45:1285–98. doi: 10.1016/j.immuni.2016.10.031.
77. Kudo F, Ikutani M, Seki Y, Otsubo T, Kawamura YI, Dohi T, et al. Interferon-γ constrains cytokine production of group 2 innate lymphoid cells. Immunology (2016) 147:21–9. doi: 10.1111/imm.12537
78. Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol. (2009) 183:6469–77. doi: 10.4049/jimmunol.0901575
79. John AE, Wilson MR, Habgood A, Porte J, Tatler AL, Stavrou A, et al. Loss of epithelial Gq and G11 signaling inhibits TGFβ production but promotes IL-33-mediated macrophage polarization and emphysema. Sci Signal. (2016) 9:ra104. doi: 10.1126/scisignal.aad5568
80. Cayrol C, Girard JP. Interleukin-33 (IL-33): a nuclear cytokine from the IL-1 family. Immunol Rev. (2018) 281:154–68. doi: 10.1111/imr.12619
81. Arranz AC, Doxaki E, Vergadi Y, Martinez de la Torre K, Vaporidi ED, Lagoudaki E, et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc. Natl. Acad. Sci. USA. (2012) 109:9517–22. doi: 10.1073/pnas.1119038109
82. Tajima S, Oshikawa K, Tominaga S, Sugiyama Y. The increase in serum soluble ST2 protein upon acute exacerbation of idiopathic pulmonary fibrosis. Chest (2003) 124: 1206–14. doi: 10.1378/chest.124.4.1206
83. Tajima S, Bando M, Ohno S, Sugiyama Y, Oshikawa K, Tominaga S, et al. ST2 gene induced by type 2 helper T cell (Th2) and proinflammatory cytokine stimuli may modulate lung injury and fibrosis. Exp Lung Res. (2007) 33:81–97. doi: 10.1080/01902140701198583
84. Zhao Y, Gonzalez De Los Santos F, Wu Z, Liu T, Phan SH. An ST2-dependent role of bone marrow derived group 2 innate lymphoid cells in pulmonary fibrosis. J. Pathol. (2018) 245:399–409. doi: 10.1002/path.5092
85. An G, Zhang X, Wang W, Huang Q, Li Y, Shan S, et al. The effects of interleukin-33 on airways collagen deposition and matrix metalloproteinase expression in a murine surrogate of asthma. Proc Natl Acad Sci USA. (2018) 109:9517–22. doi: 10.1111/imm.12911
86. Park GY, Christman JW. Involvement of cyclooxygenase-2 and prostaglandins in the molecular pathogenesis of inflammatory lung diseases. Am J Physiol Lung Cell Mol Physiol. (2006) 290:L797–805. doi: 10.1152/ajplung.00513.2005
87. Liu G, Cooley MA, Jarnicki AG, Hsu AC, Nair PM, Haw TJ, et al. Fibulin-1 regulates the pathogenesis of tissue remodeling in respiratory diseases. JCI Insight (2016) 1:e86380. doi: 10.1172/jci.insight.86380
88. Kim SH, Lee SM. Cytoprotective effects of melatonin against necrosis and apoptosis induced by ischemia/reperfusion injury in rat liver. J Pineal Res. (2008) 44:165–71. doi: 10.1111/j.1600-079X.2007.00504.x
89. Oztas E, Kuzu UB, Zengin NI, Kalkan IH, Onder FO, Yildiz H, et al. Can serum ST2 levels be used as a marker of fibrosis in chronic hepatitis B infection? Medicine (2015) 94:e1889. doi: 10.1097/MD.0000000000001889
90. Friedman SL. Evolving challenges in hepatic fibrosis. Nat Rev Gastroenterol Hepatol. (2010) 7:425–36. doi: 10.1038/nrgastro.2010.97
91. de Oliveira SA, de Freitas Souza BS, SáBarreto EP, Kaneto CM, Neto HA, et al. Reduction of galectin-3 expression and liver fibrosis after cell therapy in a mouse model of cirrhosis. Cytotherapy (2012) 14:339–49. doi: 10.3109/14653249.2011.637668
92. Traber PG, Chou H, Zomer E, Hong F, Klyosov A, Fiel MI, et al. Regression of fibrosis and reversal of cirrhosis in rats by galectin inhibitors in thioacetamide-induced liver disease. PLoS ONE (2013) 8:e75361. doi: 10.1371/journal.pone.0075361
93. Jeftic I, Jovicic N, Pantic J, Arsenijevic N, Lukic ML, Pejnovic N. Galectin-3 ablation enhances liver steatosis, but attenuates inflammation and IL-33-dependent fibrosis in obesogenic mouse model of nonalcoholic steatohepatitis. Mol Med. (2015) 21:453–65. doi: 10.2119/molmed.2014.00178
94. Forkel M, Berglin L, Kekäläinen E, Carlsson A, Svedin E, Michaëlsson J, et al. Composition and functionality of the intrahepatic innate lymphoid cell-compartment in human nonfibrotic and fibrotic livers. Eur J Immunol. (2017) 47:1280–94. doi: 10.1002/eji.201646890
95. Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med. (2013) 210:535–49. doi: 10.1084/jem.20121964
96. Han JM, Wu D, Denroche HC, Yao Y, Verchere CB, Levings M. IL-33 reverses an obesity-induced deficit in visceral adipose tissue ST2+ T regulatory cells and ameliorates adipose tissue inflammation and insulin resistance. J Immunol. (2015) 194:4777–83. doi: 10.4049/jimmunol.1500020
97. Vasanthakumar A, Moro K, Xin A, Liao Y, Gloury R, Kawamoto S, et al. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat Immunol. (2015) 16:276–85. doi: 10.1038/ni.3085
98. Jeffery HC, McDowell P, Lutz P, Wawman RE, Roberts S, Bagnall C, et al. Human intrahepatic ILC2 are IL-13 positive amphiregulin positive and their frequency correlates with model of end stage liver disease score. PLoS ONE (2017) 12:e0188649. doi: 10.1371/journal.pone.0188649
99. Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. (2011) 121:2111–7. doi: 10.1172/JCI57132
100. Vasseur P, Dion S, Filliol A, Genet V, Lucas-Clerc C, Jean-Philippe G, et al. Endogenous IL-33 has no effect on the progression of fibrosis during experimental steatohepatitis. Oncotarget (2017) 25:848563–74. doi: 10.18632/oncotarget.18335
101. Ferdek PE, Jakubowska MA. Biology of pancreatic stellate cells—more than just pancreatic cancer. Pflugers Arch. (2017) 469:1039–50. doi: 10.1007/s00424-017-1968-0
102. Schmieder A, Multhoff G, Radons J. Interleukin-33 acts as a pro-inflammatory cytokine and modulates its receptor gene expression in highly metastatic human pancreatic carcinoma cells. Cytokine (2012) 60:514–21. doi: 10.1016/j.cyto.2012.06.286
103. Nishida A, Andoh A, Imaeda H, Inatomi O, Shiomi H, Fujiyam AY. Expression of interleukin 1-like cytokine interleukin 33 and its receptor complex (ST2L and IL1RAcP) in human pancreatic myofibroblasts. Gut (2010) 59:531–41. doi: 10.1136/gut.2009.193599
104. Leema G, Tamizhselvi R. Protective effect of scopoletin against cerulein-induced acute pancreatitis and associated lung injury in mice. Pancreas (2018) 47:577–85. doi: 10.1097/MPA.0000000000001034
105. Mahapatro M, Foersch S, Hefele M, He GW, Giner-Ventura E, McHedlidze T, et al. Programming of intestinal epithelial differentiation by IL-33 derived from pericryptal fibroblasts in response to systemic infection. Cell Rep. (2016) 15:1743–56. doi: 10.1016/j.celrep.2016.04.049
106. Zhang DK, He FQ, Li TK, Pang XH, Cui DJ, Xie Q, et al. Glial-derived neurotrophic factor regulates intestinal epithelial barrier function and inflammation and is therapeutic for murine colitis. J Pathol. (2010) 222:213–22. doi: 10.1002/path.2749
107. Powell N, Walker MM, Talley NJ. The mucosal immune system: master regulator of bidirectional gut-brain communications. Nat. Rev. Gastroenterol. Hepatol. (2017) 14:143–59. doi: 10.1038/nrgastro.2016.191
108. Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. (2016) 16:676–89. doi: 10.1038/nri.2016.95
109. Malik A, Sharma D, Zhu Q, Karki R, Guy CS, Vogel P, et al. IL-33 regulates the IgA-microbiota axis to restrain IL-1α-dependent colitis and tumorigenesis. J Clin Invest. (2016) 126:4469–81. doi: 10.1172/JCI88625
110. Islam MS, Horiguchi K, Iino S, Kaji N, Mikawa S, Hori M, et al. Epidermal growth factor is a critical regulator of the cytokine IL-33 in intestinal epithelial cells. Br J Pharmacol. (2016) 173:2532–42. doi: 10.1111/bph.13535
111. De Salvo C, Wang XM, Pastorelli L, Mattioli B, Omenetti S, Buela KA, et al. IL-33 drives eosinophil infiltration and pathogenic type 2 helper T-cell immune responses leading to chronic experimental ileitis. Am J Pathol. (2016) 186:885–98. doi: 10.1016/j.ajpath.2015.11.028
112. Hodzic Z, Schill EM, Bolock AM, Good M. IL-33 and the intestine: the good, the bad, and the inflammatory. Cytokine (2017) 100:1–10. doi: 10.1016/j.cyto.2017.06.017
113. Sipos F, Muzes G, Valcz G, Galamb O, Toth K, Leiszter K, et al. Regeneration associated growth factor receptor and epithelial marker expression in lymphoid aggregates of ulcerative colitis. Scand J Gastroenterol. (2010) 45:440–8. doi: 10.3109/00365521003624144
114. Yan F, Cao H, Cover TL, Washington MK, Shi Y, Liu L, et al. Colon-specific delivery of a probiotic-derived soluble protein ameliorates intestinal inflammation in mice through an EGFR-dependent mechanism. J Clin Invest. (2011) 121:2242–53. doi: 10.1172/JCI44031
115. Shimokawa C, Kanaya T, Hachisuka M, Ishiwata K, Hisaeda H, Kurashima Y, et al. Mast cells are crucial for induction of Group 2 innate lymphoid cells and clearance of helminth infections. Immunity (2017) 46:863.e4–74. doi: 10.1016/j.immuni.2017.04.017
116. Fernandes C, Wanderley CWS, Silva CMS, Muniz HA, Teixeira MA, Souza NRP, et al. Role of regulatory T cells in irinotecan-induced intestinal mucositis. Eur J Pharm Sci. (2018) 115:158–66. doi: 10.1016/j.ejps.2018.01.006
117. Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature (2014) 513:564–8. doi: 10.1038/nature13577
118. Judd LM, Chalinor HV, Walduck A, Pavlic DI, Däbritz J, Dubeykovskaya Z, et al. TFF2 deficiency exacerbates weight loss and alters immune cell and cytokine profiles in DSS colitis, and this cannot be rescued by wild-type bone marrow. Am J Physiol Gastrointest Liver Physiol. (2015) 308:G12–24. doi: 10.1152/ajpgi.00172.2014
119. Chen X, Lu K, Timko NJ, Weir DM, Zhu Z, Qin C, et al. IL-33 notably inhibits the growth of colon cancer cells. Oncol Lett. (2018) 16:769–74. doi: 10.3892/ol.2018.8728
120. Heller F, Fuss IJ, Nieuwenhuis EE, Blumberg RS, Strober W. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity (2002) 17:629–38. doi: 10.1016/S1074-7613(02)00453-3
121. Kawashima R, Kawamura YI, Kato R, Mizutani N, Toyama-Sorimachi N, Dohi T. IL-13 receptor alpha2 promotes epithelial cell regeneration from radiation-induced small intestinal injury in mice. Gastroenterology (2006) 131:130–41. doi: 10.1053/j.gastro.2006.04.022
122. Sezaki T, Hirata Y, Hagiwara T, Kawamura YI, Okamura T, Takanashi R, et al. Disruption of the TWEAK/Fn14 pathway prevents 5-fluorouracil-induced diarrhea in mice. World J Gastroenterol. (2017) 23:2294–307. doi: 10.3748/wjg.v23.i13.2294
123. Zhang Y, Davis C, Shah S, Hughes D, Ryan JC, Altomare D, et al. L-33 promotes growth and liver metastasis of colorectal cancer in mice by remodeling the tumor microenvironment and inducing angiogenesis. Mol Carcinog (2017) 56:272–87. doi: 10.1002/mc.22491
124. Maywald RL, Doerner SK, Pastorelli L, De Salvo C, Benton SM, Dawson EP, et al. IL-33 activates tumor stroma to promote intestinal polyposis. Proc Natl Acad Sci USA. (2015) 112:E2487–96. doi: 10.1073/pnas.1422445112
125. He Z, Chen L, Souto FO, Canasto-Chibuque C, Bongers G, Deshpande M, et al. Epithelial-derived IL-33 promotes intestinal tumorigenesis in Apc Min/+ mice. Sci Rep. (2017) 7:5520. doi: 10.1038/s41598-017-05716-z
126. O'Donnell C, Mahmoud A, Keane J, Murphy C, White D, Carey S, et al. An antitumorigenic role for the IL-33 receptor, ST2L, in colon cancer. Oncol Lett. (2018) 16:769–774. doi: 10.1038/bjc.2015.433
127. Guabiraba R, Besnard AG, Menezes GB, Secher T, Jabir MS, Amaral SS, et al. IL-33 targeting attenuates intestinal mucositis and enhances effective tumor chemotherapy in mice. Mucosal Immunol. (2014) 7:1079–93. doi: 10.1038/mi.2013.124
128. Rabb H, Griffin MD, McKay DB, Swaminathan S, Pickkers P, Rosner MH, et al. Inflammation in AKI: current understanding, key questions, and knowledge gaps. J Am Soc Nephrol. (2016) 27:371–9. doi: 10.1681/ASN.2015030261
129. Xu Z, Zhao C, Wang Z, Tao J, Han Z, Zhang W, et al. Interleukin-33 levels are elevated in chronic allograft dysfunction of kidney transplant recipients and promotes epithelial to mesenchymal transition of human kidney (HK-2) cells. Gene (2017) 644:113–21. doi: 10.1016/j.gene.2017.11.010
130. Stone RC, Pastar I, Ojeh N, Chen V, Liu S, Garzon KI, et al. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. (2016) 365:495–506. doi: 10.1007/s00441-016-2464-0.
131. Li Y, Kang YS, Dai C, Kiss LP, Wen X, Liu Y. Epithelial-to-mesenchymal transition is a potential pathway leading to podocyte dysfunction and proteinuria. Am J Pathol. (2008) 172:299–308. doi: 10.2353/ajpath.2008.070057
132. Yamaguchi Y, Iwano M, Suzuki D, Nakatani K, Kimura K, Harada K, et al. Epithelial-mesenchymal transition as an explanation for podocyte depletion in diabetic nephropathy. Am J Kidney Dis. (2009) 54:653–64. doi: 10.1053/j.ajkd.2009.05.009
133. Kang YS, Li Y, Dai C, Kiss LP, Wu C, Liu Y. Inhibition of integrin-linked kinase blocks podocyte epithelial-mesenchymal transition and ameliorates proteinuria. Kidney Int. (2010) 78:363–73. doi: 10.1038/ki.2010.137
134. Mutsaers HA, Stribos EG, Glorieux G, Vanholder R, Olinga P. Chronic kidney disease and fibrosis: the role of uremic retention solutes. Front Med. (2015) 2:60. doi: 10.3389/fmed.2015.00060
135. Ó hAinmhire E, Humphreys BD. Fibrotic changes mediating acute kidney injury to chronic kidney disease transition. Nephron (2018) 137:264–7. doi: 10.1159/000474960
136. Lee SJ, Borsting E, Declèves AE, Singh P, Cunard R. Podocytes express IL-6 and lipocalin 2/neutrophil gelatinase-associated lipocalin in lipopolysaccharide-induced acute glomerular injury. Nephron Exp Nephrol. (2012) 121:e86–96. doi: 10.1159/000345151
137. Millar NL, O'Donnell C, McInnes IB, Brint E. Wounds that heal and wounds that don't - The role of the IL-33/ST2 pathway in tissue repair and tumorigenesis. Semin Cell Dev Biol. (2017) 61:41–50. doi: 10.1016/j.semcdb.2016.08.007
138. Zhu J, Carver W. Effects of interleukin-33 on cardiac fibroblast gene expression and activity. Cytokine (2012) 58:368–79. doi: 10.1016/j.cyto.2012.02.008
139. Wang EW, Jia XS, Ruan CW, Ge ZR. miR-487b mitigates chronic heart failure through inhibition of the IL-33/ST2 signaling pathway. Oncotarget (2017) 8:51688–702. doi: 10.18632/oncotarget.18393
140. Weinberg EO, Shimpo M, Hurwitz S, Tominaga S, Rouleau JL, Lee RT. Identification of serum soluble ST2 receptor as a novel heart failure biomarker. Circulation (2003) 107:721–6.
141. Sabatine MS, Morrow DA, Higgins LJ, MacGillivray C, Guo W, Bode C, et al. Complementary roles for biomarkers of biomechanical strain ST2 and N-terminal prohormone B-type natriuretic peptide in patients with ST-elevation myocardial infarction. Circulation (2008) 117:1936–44. doi: 10.1161/CIRCULATIONAHA.107.728022
142. McCarthy CP, Januzzi JL Jr. Soluble ST2 in heart failure. Heart Fail Clin (2018) 14:41–8. doi: 10.1016/j.hfc.2017.08.005
143. Januzzi JL Jr Peacock WF, Maisel AS, Chae CU, Jesse RL, Baggish AL, et al. Measurement of the interleukin family member ST2 in patients with acute dyspnea: results from the PRIDE (Pro-Brain Natriuretic Peptide Investigation of Dyspnea in the Emergency Department) study. J Am Coll Cardiol. (2007) 50:607–13. doi: 10.1016/j.jacc.2007.05.014
144. Ky B, French B, McCloskey K, Rame JE, McIntosh E, Shahi P, et al. High-sensitivity ST2 for prediction of adverse outcomes in chronic heart failure. Circ Heart Fail. (2011) 4:180–7. doi: 10.1161/CIRCHEARTFAILURE.110.958223
145. Dieplinger B, Mueller T. Soluble ST2 in heart failure. Clin Chim Acta. (2015) 443:57–70. doi: 10.1016/j.cca.2014.09.021
146. Aimo A, Vergaro G, Ripoli A, Bayes-Genis A, Pascual Figal DA, de Boer RA, et al. Meta-analysis of soluble suppression of tumorigenicity-2 and prognosis in acute heart failure. JACC Heart Fail. (2017) 5:287–96. doi: 10.1016/j.jchf.2016.12.016
147. Skali H, Gerwien R, Meyer TE, Snider JV, Solomon SD, Stolen CM. Soluble ST2 and risk of arrhythmias, heart failure, or death in patients with mildly symptomatic heart failure: results from MADIT-CRT. J Cardiovasc Transl Res. (2016) 9:421–8. doi: 10.1007/s12265-016-9713-1
148. Anand IS, Rector TS, Kuskowski M, Snider J, Cohn JN. Prognostic value of soluble ST2 in the valsartan heart failure trial. Circ Heart Fail. (2014) 7:418–26. doi: 10.1161/CIRCHEARTFAILURE.113.001036
149. Weir RA, Miller AM, Murphy GE, Clements S, Steedman T, Connell JM, et al. Serum soluble ST2: a potential novel mediator in left ventricular and infarct remodeling after acute myocardial infarction. J Am Coll Cardiol. (2010) 55:243–50. doi: 10.1016/j.jacc.2009.08.047
150. Gruzdeva O, Uchasova E, Dyleva Y, Borodkina D, Akbasheva O, Belik E, et al. Relationships between epicardial adipose tissue thickness and adipo-fibrokine indicator profiles post-myocardial infarction. Cardiovasc Diabetol. (2018) 17:40. doi: 10.1186/s12933-018-0679-y
151. Tseng CCS, Huibers MMH, Gaykema LH, Siera-de Koning E, Ramjankhan FZ, Maisel AS, et al. Soluble ST2 in end-stage heart failure, before and after support with a left ventricular assist device. Eur J Clin Invest. (2018) 48:e12886. doi: 10.1111/eci.12886
152. Shah RV, Januzzi JL Jr. Soluble ST2 and galectin-3 in heart failure. Clin Lab Med. (2014) 34, 87–97. doi: 10.1016/j.cll.2013.11.009
153. Komai-Koma M, Xu D, Li Y, McKenzie AN, McInnes IB, Liew FY. IL-33 is a chemoattractant for human Th2 cells. Eur J Immunol. (2007) 37:2779–86. doi: 10.1002/eji.200737547
154. Aoudjehane L, Pissaia AJr, Scatton O, Podevin P, Massault PP, Chouzenoux S, et al. Interleukin-4 induces the activation and collagen production of cultured human intrahepatic fibroblasts via the STAT-6 pathway. Lab Invest. (2008) 88:973–85. doi: 10.1038/labinvest.2008.61
155. Fuschiotti P, Larregina AT, Ho J, Feghali-Bostwick C, Medsger TA Jr. Interleukin-13-producing CD81 T cells mediate dermal fibrosis in patients with systemic sclerosis. Arthritis Rheum. (2013) 65:236–46. doi: 10.1002/art.37706
156. Koca SS, Pehlivan Y, Kara M, Alibaz-Oner F, Oztuzcu S, Yilmaz N, et al. The IL-33 gene is related to increased susceptibility to systemic sclerosis. Rheumatol Int. (2016) 36:579–84. doi: 10.1007/s00296-015-3417-8
157. Wagner A, Köhm M, Nordin A, Svenungsson E, Pfeilschifter JM, Radeke HH. Increased serum levels of the IL-33 neutralizing sST2 in limited cutaneous systemic sclerosis. Scand J Immunol. (2015) 82:269–74. doi: 10.1111/sji.12317
158. Hachulla E, Launay D. Diagnosis and classification of systemic sclerosis. Clinic Rev Allerg Immunol. (2011) 40:78–83. doi: 10.1007/s12016-010-8198-y
Keywords: epithelial cells, inflammation, Interleukin-33, myofibroblasts, organ fibrosis, ST2, tissue remodeling
Citation: Kotsiou OS, Gourgoulianis KI and Zarogiannis SG (2018) IL-33/ST2 Axis in Organ Fibrosis. Front. Immunol. 9:2432. doi: 10.3389/fimmu.2018.02432
Received: 04 June 2018; Accepted: 02 October 2018;
Published: 24 October 2018.
Edited by:
Fang-Ping Huang, University of Hong Kong, Hong KongReviewed by:
Remo Castro Russo, Universidade Federal de Minas Gerais, BrazilMarcela A. Hermoso, Universidad de Chile, Chile
Copyright © 2018 Kotsiou, Gourgoulianis and Zarogiannis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ourania S. Kotsiou, cmFuaWFrb3RzaW91QGdtYWlsLmNvbQ==