 Ying Long
Ying Long Weilin Chen
Weilin Chen Qian Du
Qian Du Xiaoxia Zuo
Xiaoxia Zuo Honglin Zhu
Honglin Zhu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 17 October 2018
Sec. Primary Immunodeficiencies
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02383
This article is part of the Research Topic Etiopathogenesis of Systemic Sclerosis: An Update View all 11 articles
Scleroderma (systemic sclerosis, SSc) is a highly heterogeneous rheumatic disease, and uncontrolled fibrosis in visceral organs is the major cause of death in patients. The transforming growth factor-β (TGF-β) and WNT/β-catenin signaling pathways, along with signal transducer and activator of transcription 3 (STAT3), play crucial roles in this fibrotic process. Currently, no therapy is available that effectively arrests or reverses the progression of fibrosis in patients with SSc. Ubiquitination is an important post-translational modification that controls many critical cellular functions. Dysregulated ubiquitination events have been observed in patients with systemic lupus erythematosus, rheumatoid arthritis and fibrotic diseases. Inhibitors targeting the ubiquitination pathway have considerable potential for the treatment of rheumatic diseases. However, very few studies have examined the role and mechanism of ubiquitination in patients with SSc. In this review, we will summarize the molecular mechanisms of ubiquitination in patients with SSc and explore the potential targets for treatment.
Scleroderma (systemic sclerosis, SSc) is a complicated heterogeneous rheumatic disease that is characterized by progressive fibrosis in the skin and multiple other organs. Both environmental and genetic factors contribute to the etiology of SSc and trigger a chronic self-amplifying inflammatory process, leading to vascular alterations, autoimmunity and fibrosis (1). Many molecules and signaling pathways participate in the progression of fibrosis, such as the transforming growth factor-β (TGF-β) and WNT/β-catenin signaling pathways, signal transducer and activator of transcription 3 (STAT3), platelet-derived growth factor (PDGF), endothelin 1, interleukin 6, interleukin 13, autoantibodies, and numerous biologically active substances. Among these pathways, the TGF-β and WNT/β-catenin signaling pathways and STAT3 play key roles. Based on accumulating evidence, post-translational modifications have important regulatory roles in these pathways, such as acetylation, phosphorylation and ubiquitination, suggesting that these modifications are potential targets for the treatment of fibrosis (2–4).
Ubiquitin is a highly evolutionarily conserved protein that modifies other proteins for degradation. Ubiquitination is a process by which target protein is covalently bound to ubiquitin through an enzymatic cascade that is orchestrated sequentially by activating (E1), conjugating (E2) and ligating (E3) enzymes (5). E1 enzymes activate ubiquitin and transfer it onto the E2 conjugating enzyme, and then E3 ligases interact with the ubiquitin-loaded E2 enzyme and substrate protein to mediate the formation of polyubiquitin chains. Subsequently, the polyubiquitin chain is recognized by the 26S proteasome complex and degraded into individual amino acids.
The human genome contains two E1 enzymes, approximately forty E2 enzymes and >600 E3 ligases. The diverse E3 ligases have important roles in the selective recognition of each targeted protein. Additionally, >100 deubiquitinating enzymes (DUBs) have been identified that remove ubiquitin from the substrate proteins (6).
Ubiquitination plays important roles in the proteasomal degradation of proteins, inflammatory signaling, immune responses, autophagy, and T cell activation and differentiation (7). Dysregulation of ubiquitination has been observed in many autoimmune diseases, such as systemic lupus erythematous (8), rheumatoid arthritis (RA) (9) and fibrotic diseases (10). Anti-ubiquitin antibodies are present in 42% of patients with SSc and are associated with anti-histone antibodies. The latter might be positively correlated with the severity of pulmonary fibrosis in patients with SSc (11). According to results from whole-exome sequencing, an E3 ubiquitin ligase-related gene was associated with a higher risk of SSc. Therefore, ubiquitination might play an important role in SSc (12). However, little is known about ubiquitination in the pathology of SSc. Herein, we will summarize the role of ubiquitination in SSc and then discuss the future perspectives for SSc therapy.
Many susceptibility regions have been identified in patients with SSc by genome-wide association studies (GWAS), and the most common and confirmed susceptibility locus is the HLA locus. Recently, non-HLA susceptibility genes have also been identified, and most are correlated with inflammation, T cell differentiation and autoantibodies (13).
The susceptibility genes TNF-α-induced protein 3 (TNFAIP3), TNF receptor-interacting protein (TNIP1), ankyrin repeat and SOCS box-containing 10 (ASB10), and autophagy-related 5 (ATG5) are involved in the ubiquitination-proteasome system (UPS). TNFAIP3 expression (encodes A20) is rapidly induced by TNF-α. TNFAIP3 possesses both E3 ubiquitin ligase and deubiquitinase activities and negatively regulates the inflammatory response by deubiquitinating proteins in the NF-κB pathway, such as IKKg/NEMO, RIP1 and RIP2. TNFAIP3 is associated with diffuse cutaneous SSc, anti-topoisomerase I antibody, lung fibrosis and pulmonary arterial hypertension (14). TNIP1 interacts with A20 and represses the activity of the TLR-induced NF-κB signaling pathway, decreasing the production of proinflammatory cytokines in patients with SSc. Recombinant TNIP1 downregulates inflammatory cytokine-induced collagen synthesis (15). ASB10 belongs to the E3 ubiquitin ligase complex and may be involved in the pathogenesis of SSc and pulmonary vascular complications (12, 16). The protein encoded by the ATG5 gene interacts with ATG12 and forms a complex that functions as an E1-like activating enzyme. ATG5 is also associated with RA, juvenile idiopathic arthritis and primary biliary cirrhosis. However, the function of ATG5 in patients with SSc requires further investigation (17).
The TGF-β superfamily consists of TGF-βs, bone morphogenetic proteins (BMPs) and activin. These proteins activate TGF-β signaling by binding to their membrane-anchored serine/threonine kinase receptors TGF-βRI or TGF-βRII. The canonical TGF-β signaling pathway is regulated by Smad proteins. Smad proteins are divided into three types. R-Smads are receptor-regulated Smads, including TGF-β/activin-specific Samd2 and Smad3, BMP-specific Smad1, Smad5 and Smad8. I-Smads are inhibitory Smads and include Smad6 and Smad7. Co-Smad is the common Smad and is represented by Smad4. The Smad proteins regulate the transcription of various genes. Many transcription factors and co-factors are also involved in this process; transcription factors include Mixer, FoxH1, E2F, and Runx-related proteins, co-activators include p300 and CBP, and co-repressors include c-Ski and SnoN (18) (Figure 1A).
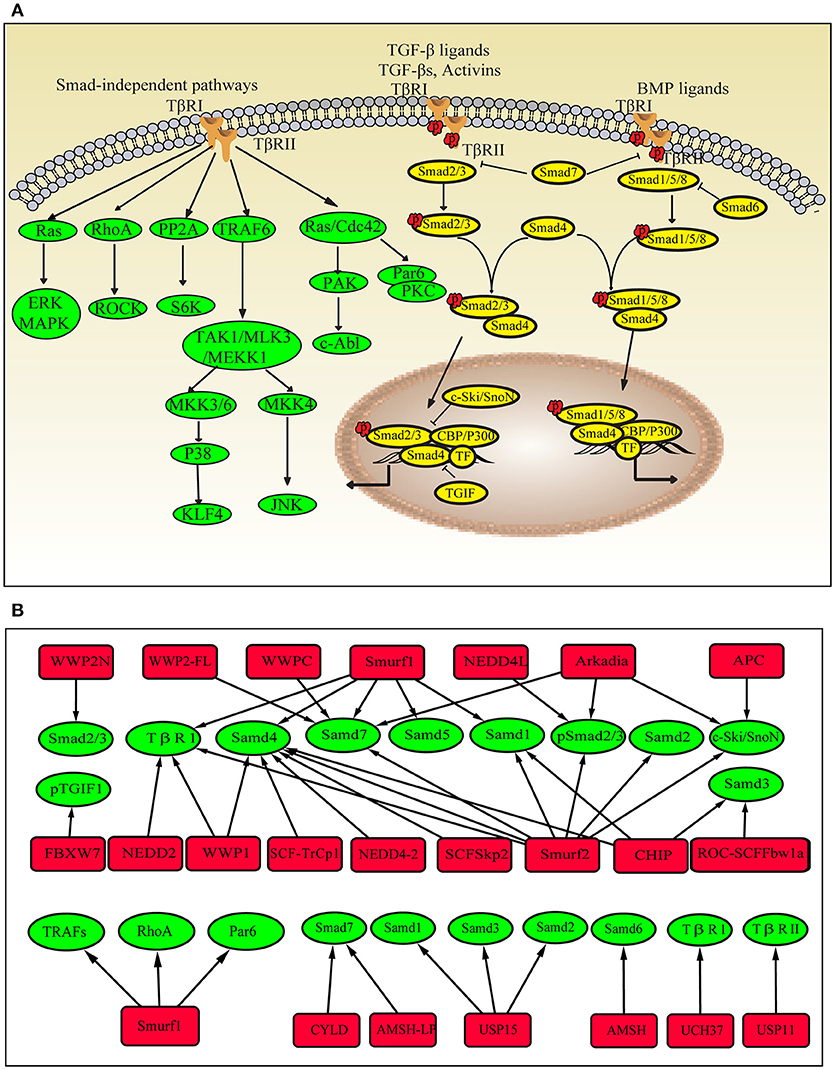
Figure 1. Ubiquitin modifications in the TGF-β pathway. (A) Schematic showing the Smad-dependent and Smad-independent TGF-β signaling pathways. (B) Ubiquitinating enzymes, DUBs and target proteins in the TGF-β pathway. Red square nodes represent enzymes, green circles represent target proteins.
TGF-β is commonly viewed as playing a critical role in the fibrosis process. Dysregulation of the TGF-β signaling pathway is involved in the pathogenesis of SSc (19). High levels of TGF-β and its regulated genes have been detected in skin biopsies and were positively correlated with the severity of SSc (20). The TGF-β neutralizing antibody fresolimumab exerts anti-fibrotic effects on patients with SSc, but its use is also accompanied by a high incidence of keratoacanthomas, which limits its use in long-term treatment. Therefore, the development of new drugs that target downstream mediators of TGF-β signaling is important (21, 22).
Many E3 ligases are involved in TGF-β/SMAD signaling, including Smurfs (E6-accessory protein C-terminus, HECT), WWP family (HECT type), NEDD4L (HECT type), Arkadia (RING-H2 finger domain), CHIP (C-terminus of HSC70-interacting protein), β-TrCP (Skp1-Cullin-F-box (SCF)-type ubiquitin ligase), and Fbxw7 (SCF type). TGF-β/SMAD signaling regulates the transcription of various genes, including negative regulators, such as I-Smads and Smurfs. When TGF-β signaling is activated, I-Smads and Smurfs interact in the nucleus and translocate to the cytoplasm (23, 24). Smad7 recruits WWP1 and NEDD4L to the active TGF-β receptor complexes and induces the degradation of the complexes (25, 26). Smurf1 ubiquitinates Smad1 and Smad5 (27, 28), and Smurf2 ubiquitinates Smad1 and Smad2 under steady-state conditions (29, 30). Arkadia and NEDD4L ubiquitinate phospho-Smad2/3 (31, 32), whereas CHIP regulates the abundance of Smad1 and Smad3 (33, 34). Smurfs, WWP1, NEDD4-2, CHIP, and β-TrCP conjugate polyubiquitin chains onto Smad4 and mediate its degradation (35). As mentioned above, E3 ligases mainly function as inhibitors of TGF-β signaling. However, they have also been shown to enhance TGF-β signaling. Arkadia degrades the negative regulators of TGF-β signaling, such as Smad7, c-Ski, and SnoN (36). Smad7 is also ubiquitinated by WWP-C and WWP2-FL (37). Phosphorylated TGIF1 (TGF-β-induced factor 1) is a transcriptional repressor of TGF-β signaling that is degraded by the ubiquitin ligase complex containing Fbxw7 (38).
Ubiquitination also plays important roles in Smad-independent TGF-β pathways. TGF-β induces the ubiquitination and degradation of KLF4 (Krüppel-like factor 4), which is important for TGF-β-mediated regulation of transcription (39). The TGF-β/RhoA pathway is required for the progression of the epithelial-mesenchymal transition (EMT). Smurf1 targets RhoA for degradation (40). In TGF-β-induced anti-inflammatory signaling, Smurf1 ubiquitinates TRAFs (TNF receptor-associated factors), and Smurf2 interacts with TRAF2 and ubiquitinates TNF receptor 2, thereby inhibiting downstream signaling (41). In fibroblasts from patients with SSc, Smurf2 is upregulated after stimulation with TGF-β (42), and the Smad7-Smurf-mediated inhibitory effect is impaired (43). Ubiquitination also promotes Smad2/3 signaling and increases collagen I accumulation by stabilizing Ha-Ras, which is independent of TGF-β activation. All of these mechanisms eventually contribute to collagen overproduction. The dysregulation of E3 ubiquitin ligases involved in TGF-β signaling has also been observed in patients with other fibrotic diseases and in animal models. The levels of MDM2 (RING-type) and FIEL1 (HECT-Type E3) are increased in lung tissues from patients with idiopathic pulmonary fibrosis (IPF) (44, 45). Smurf2 is upregulated in the fibroblasts present in hypertrophic scars (46). Smurf1, Smurf2, Arkadia and Hrd1 (Synoviolin, RING-type) levels are increased in animals with unilateral ureteral obstruction-induced renal fibrosis (47–49). Smurf2 and Synoviolin are upregulated in a liver fibrosis model (50–52). NEDD4 and Pellino1 (RING-type) expression are increased in keloid fibroblasts (53) and cardiac fibroblasts, respectively (54). These E3 ubiquitin ligases not only directly mediate the degradation of components of the TGF-β signaling pathway but also participate in inducing the transition of epithelial cells to mesenchymal cells, enhancing fibroblast proliferation and invasiveness, and increasing TGF-β production.
DUBs have also been implicated in TGF-β signaling. UCH37 and USP11 were shown to deubiquitinate TβRI or TβRII, which is important for early steps in the TGF-β signaling pathway (55–57). High levels of USP11 have been detected in lung tissues from patients with IPF and bleomycin-induced mice, whereas inhibition of USP11 expression attenuates TGF-β signaling (57). CYLD deubiquitinates Smad7 and inhibits TGF-β signaling (58). In mice with liver fibrosis, CYLD ameliorates hepatocellular damage and liver fibrogenesis (59). USP15 deubiquitinates mono-ubiquitinated R-Smads and is required for proper TGF-β signaling. Other DUBs, such as AMSH (the associated molecule with the Src homology 3 domain of the signal-transducing adaptor molecule) and AMSH-like protein cleave K63-linked ubiquitin chains, which are associated with I-Smads and inhibit their functions (60–62) (Figure 1B).
The canonical WNT pathway is closely related to the regulation of β-catenin and its potential to modulate transcription. When WNT signaling is inactivated, β-catenin is phosphorylated (pβ-catenin) in the cytoplasm by a multiprotein complex (Axin/APC complex) composed of Axin, APC, CK1 and GSKβ. The pβ-catenin protein is immediately degraded by the UPS and is rarely detected in normal cells. Upon stimulation, WNT binds to the receptor Frizzled (Fz) and the coreceptors LRP5/6, recruits the cytoplasmic effector protein Disheveled (Dvl) and inhibits the Axin/APC complex. Consequently, the high levels of cytosolic β-catenin are translocated to the nucleus and regulate the transcription of target genes via β-catenin/TCF complexes. Based on accumulating evidence from recent studies, ubiquitin modification also plays important roles in regulating the WNT pathway (63).
WNT signaling plays pivotal roles in developmental processes and tissue homeostasis. The canonical WNT/β-catenin pathway elicits fibrotic responses both directly and through TGF-β (2). High levels of activated β-catenin and its regulated gene AXIN2 have been in skin and lung tissues from patients with SSc, as well as in animal models of fibrosis (64, 65). SSc autoantibodies and oxidative DNA damage mediate Wnt inhibitor factor 1 (WIF-1) silencing and promote WNT activation and subsequent fibrosis. Strategies that restore the expression of WIF-1 prevent collagen accumulation in vivo. Microarray studies have also revealed the activation of WNT/β-catenin pathways in the skin tissues from patients with SSc. According to the results from clinical trials, treatments targeting the WNT/β-catenin pathway (tankyrase and porcupine inhibitors) are effective, well-tolerated and safe for long-term application (66). C-82, which targets the β-catenin/CBP interaction, is now in phase I/II clinical trials for SSc therapy. Therefore, the molecular mechanisms regulating the WNT/β-catenin pathway must be completely identified (67, 68).
The cell-surface receptor Fz is ubiquitinated by the transmembrane E3 ligases ZNRF3 and RNF43 and is deubiquitinated by UBPY/Ub-specific protease 8 (USP8) for recycling to the plasma membrane (69, 70). LRP6 is retained in the endoplasmic reticulum due to palmitoylation and monoubiquitylation, suggesting that an E3 ligase and DUBs participate in this process; however, the types of ubiquitin chains remain unknown (71). Dvl proteins (Dvl1, Dvl2 and Dvl3) play key roles in both canonical and noncanonical WNT signaling. Multiple E3 ligases that ubiquitinate Dvl negatively regulate WNT signaling (72). The DUBs CYLD and USP14 remove the K63-linked polyubiquitin chain from Dvl (73, 74). Axin is degraded by the E3 ligase RNF146 and Smurf1 and Smurf2, which interact with LRP5/6 (75–77). HectD1 ubiquitinates APC, promotes its interaction with Axin and negatively regulates WNT signaling (78), whereas USP15 protects the APC from ubiquitin-mediated degradation (79). β-TrCP assembles K48-linked polyubiquitin chains onto β-catenin and mainly regulates the nuclear pool of β-catenin (80). The ubiquitin ligase Jade-1 also mediates β-catenin ubiquitination and is responsible for degrading cytoplasmic β-catenin (81). Unlike β-TrCP and Jade-1, Rad6B (an E2 ubiquitin-conjugating enzyme) and EDD (an E3 ubiquitin ligase) ubiquitinate β-catenin and increase its activity (82, 83) (Figure 2).
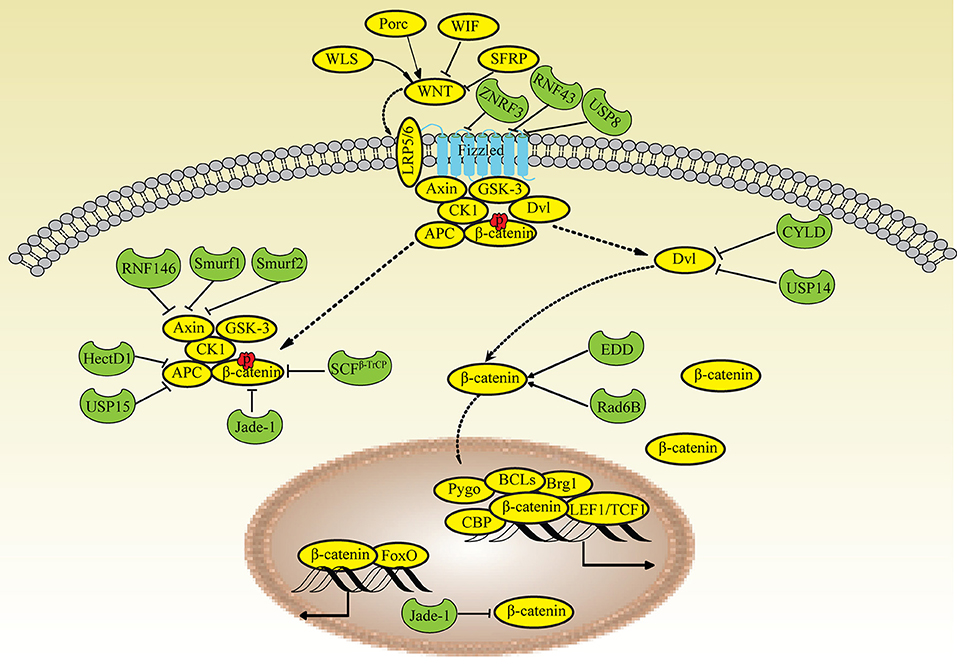
Figure 2. Ubiquitin modifications in the WNT/β-catenin pathway. When WNT signaling is inactivated, β-catenin is phosphorylated (pβ-catenin) in the cytoplasm by the Axin/APC complex and degraded by Jade-1 and β-TrCP. Upon stimulation, WNT binds to the receptor Frizzled and the coreceptors LRP5/6, recruits Dvl and inhibits the Axin/APC complex, leading to the translocation of high levels of cytosolic β-catenin into the nucleus. Fz is ubiquitinated by ZNRF3 and RNF43 and deubiquitinated by USP8. Dvl is deubiquitinated by CYLD and USP14. Axin is degraded by RNF146, Smurf2, and Smurf1. APC is ubiquitinated by HectD1 and deubiquitinated by USP15. Rad6B and EDD ubiquitinate β-catenin; Jade-1 also ubiquitinates β-catenin in the nucleus.
STAT3 belongs to the transcription factor family that transduces cellular signals from a number of soluble growth factors and cytokines, including PDGF, epidermal growth factor (EGF) and IL-6 family cytokines. STAT3 plays critical roles in several biological processes, including cell proliferation, differentiation and migration. Upon stimulation, cytoplasmic STAT3 is phosphorylated, dimerizes, and then translocates to the nucleus to regulate the transcription of target genes. Recently, STAT3 was shown to integrate multiple profibrotic signals and was identified as a key checkpoint in fibroblast activation. STAT3 is considered a potential target for SSc treatment (4, 84). The STAT3 dimerization inhibitor S3I-201 exerts strong anti-fibrotic effects on animal models of SSc. STAT3 is ubiquitinated with lysine-63-linked ubiquitin chains by TRAF6 (tumor necrosis factor receptor-associated factor 6) (85), which exhibits E3 ubiquitin ligase activity. STAT3 is also ubiquitinated and degraded by the E3 ligase COP1 (86).
Bortezomib is a reversible 20S proteasome inhibitor and the first drug approved to treat multiple myeloma. In the UPS, the proteasome is the final step in protein degradation and a valuable target for developing potential drugs. Bortezomib derivatives, such as carfilzomib and marizomib, are in various phases of clinical trials as potential treatments for several malignancies (87). In fibroblasts from patients with SSc, proteasome inhibitors replenish human dermal fibroblasts, degrade the extracellular matrix and exert anti-fibrotic effects (88). The proteasome inhibitor MG-132, synthetic lactacystin, and bortezomib decrease the expression of type I collagen and tissue inhibitor of metalloproteinase-1 and increase the production of metalloproteinase-1 in a dose-dependent manner (89). However, proteasome inhibitors have been reported to induce resistance and side effects, and a combination of several UPS inhibitors targeting different components may overcome this challenge (6).
Studies of E1 enzyme inhibitors are rare due to their lack of specificity as therapies. Several inhibitors of E2 enzymes are in preclinical research stages, but their role in SSc has been poorly investigated. The specificity of the UPS depends mainly on E3 ubiquitin ligases. The FIEL1 inhibitor BC-1485 ameliorates lung fibrosis in a mouse model (44). The Synoviolin inhibitor LS-102 reduces endoplasmic reticulum stress-induced collagen secretion from lung epithelial cells, suggesting that it might be a potential treatment for IPF (90). In patients with SSc, the TGF-β and WNT/β-catenin signaling pathways and STAT3 are mainly regulated by E3 ligases at multiple levels, which presents the potential for specific substrates for drug target design. Erioflorin, which is isolated from Eriophyllum lanatum, has been shown to block β-TrCP and affect WNT/β-catenin signaling (91). Specific inhibitors or antagonists of other E3 ligases, such as Smurf1, Smurf2 and NEDD4, have not yet been discovered.
DUBs deubiquitinate and rescue substrates from proteasomal degradation, and thus are regarded other potential targets for drug development. WP1130 has been shown to suppress the activity of several DUBs, including UCH37 and USP14, which regulate both TGF-β and WNT/β-catenin signaling. The combination of WP1130 and bortezomib exerts pro-apoptosis and anti-proliferation effects on tumor cells (92). b-AP15 is a small-molecule inhibitor of USP14 and UCHL5. b-AP15 blocks USP14 in a reversible manner and regulates WNT/β-catenin signaling (93). UCHL5 levels are elevated in lung tissues from patients with IPF (80). b-AP15 also reduces the levels of the fibronectin, type I collagen, and SMAD2/3 proteins in lung tissues from mice with fibrosis (94). The USP11 inhibitor mitoxantrone attenuates TGF-β signaling in lung fibroblasts and has been indicated as a potential antifibrotic drug for subjects with fibrosis.
In animal models and clinical trials, the UPS has been validated as a valuable molecular target for the treatment of cancer, asthma and arthritis, as >1,000 proteins have been identified in the UPS. These proteins represent substantial opportunities and challenges for researchers to investigate the molecular mechanisms underlying the addition of ubiquitin chains and the main components. The identification and validation of these components will expand the pool of targets for drug discovery for fibrosis (95).
In this review, we highlight the mechanisms regulating ubiquitination in patients with SSc and explore potential anti-fibrosis drugs. Effective therapies for many fibrotic manifestations in patients with SSc are currently unavailable. Considering the central role of TGF-β signaling, WNT/β-catenin signaling and STAT3 in SSc, the use of UPS inhibitors to selectively disrupt the formation of receptor or co-receptor complexes or block intracellular signaling may yield advances in the development of urgently needed treatments. These drugs are very powerful and might also induce severe side effects because of their unselective action that would limit their widespread use. In the near future, the elucidation of new, potent and highly specific drugs targeting specific UPS components is required. Therefore, investigations of the enzymology of ubiquitination will be of paramount importance in the next few years. Moreover, more studies are needed of enzymes involved in ubiquitination that represent promising drug targets to ameliorate fibrosis in patients with SSc.
YL wrote the first draft. WC, QD, and XZ revised the manuscript. HZ revised the final version and inserted additional information.
This study was funded by grants from the Hunan Provincial Natural Science Foundation (2018JJ3823) and the National Natural Science Foundation of China (81771765, 81373206, and 81671622).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Denton CP, Khanna D. Systemic sclerosis. Lancet (2017) 390:1685–99. doi: 10.1016/S0140-6736(17)30933-9
2. Allanore Y, Simms R, Distler O, Trojanowska M, Pope J, Denton CP, et al. Systemic sclerosis. Nat Rev Dis Primers (2015) 1:15002. doi: 10.1038/nrdp.2015.2
3. Ho YY, Lagares D, Tager AM, Kapoor M. Fibrosis–a lethal component of systemic sclerosis. Nat Rev Rheumatol. (2014) 10:390–402. doi: 10.1038/nrrheum.2014.53
4. McHugh J. Systemic sclerosis: STAT3–a key integrator of profibrotic signalling. Nat Rev Rheumatol. (2017) 13:693. doi: 10.1038/nrrheum.2017.190
5. Yau R, Rape M. The increasing complexity of the ubiquitin code. Nat Cell Biol. (2016) 18:579–86. doi: 10.1038/ncb3358
6. Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med. (2014) 20:1242–53. doi: 10.1038/nm.3739
7. Ebner P, Versteeg GA, Ikeda F. Ubiquitin enzymes in the regulation of immune responses. Crit Rev Biochem Mol Biol. (2017) 52:425–60. doi: 10.1080/10409238.2017.1325829
8. Guo Y, Zhao M, Lu Q. Transcription factor RFX1 is ubiquitinated by E3 ligase STUB1 in systemic lupus erythematosus. Clin Immunol. (2016) 169:1–7. doi: 10.1016/j.clim.2016.06.003
9. Fakhfakh Karray E, Bendhifallah I, Zakraoui L, Hamzaoui K. Association of small ubiquitin-like modifier 4 gene polymorphisms with rheumatoid arthritis in a Tunisian population. Clin Exp Rheumatol. (2011) 29:751.
10. Fukasawa H, Fujigaki Y, Yamamoto T, Hishida A, Kitagawa M. Protein degradation by the ubiquitin-proteasome pathway and organ fibrosis. Curr Med Chem. (2012) 19:893–900. doi: 10.2174/092986712799034941
11. Fujimoto M, Sato S, Ihn H, Kikuchi K, Tamaki T, Tamaki K, et al. Antiubiquitin antibody in localised and systemic scleroderma. Ann Rheum Dis. (1996) 55:399–402. doi: 10.1136/ard.55.6.399
12. Gao L, Emond MJ, Louie T, Cheadle C, Berger AE, Rafaels N, et al. Identification of rare variants in ATP8B4 as a risk factor for systemic sclerosis by whole-exome sequencing. Arthritis Rheumatol. (2016) 68:191–200. doi: 10.1002/art.39449
13. Zuo X, Zhang L, Luo H, Li Y, Zhu H. Systematic approach to understanding the pathogenesis of systemic sclerosis. Clin Genet. (2017) 92:365–71. doi: 10.1111/cge.12946
14. Dieude P, Guedj M, Wipff J, Ruiz B, Riemekasten G, Matucci-Cerinic M, et al. Association of the TNFAIP3 rs5029939 variant with systemic sclerosis in the European Caucasian population. Ann Rheum Dis. (2010) 69:1958–64. doi: 10.1136/ard.2009.127928
15. Allanore Y, Saad M, Dieude P, Avouac J, Distler JH, Amouyel P, et al. Genome-wide scan identifies TNIP1, PSORS1C1, and RHOB as novel risk loci for systemic sclerosis. PLoS Genet. (2011) 7:e1002091. doi: 10.1371/journal.pgen.1002091
16. Keller KE, Yang YF, Sun YY, Sykes R, Acott TS, Wirtz MK. Ankyrin repeat and suppressor of cytokine signaling box containing protein-10 is associated with ubiquitin-mediated degradation pathways in trabecular meshwork cells. Mol Vis. (2013) 19:1639–1655.
17. Mayes MD, Bossini-Castillo L, Gorlova O, Martin JE, Zhou X, Chen WV, et al. Immunochip analysis identifies multiple susceptibility loci for systemic sclerosis. Am J Hum Genet. (2014) 94:47–61. doi: 10.1016/j.ajhg.2013.12.002
18. Biernacka A, Dobaczewski M, Frangogiannis NG. TGF-beta signaling in fibrosis. Growth Factors (2011) 29:196–202. doi: 10.3109/08977194.2011.595714
19. Varga J, Pasche B. Transforming growth factor beta as a therapeutic target in systemic sclerosis. Nat Rev Rheumatol. (2009) 5:200–6. doi: 10.1038/nrrheum.2009.26
20. Farina G, Lafyatis D, Lemaire R, Lafyatis R. A four-gene biomarker predicts skin disease in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheum. (2010) 62:580–8. doi: 10.1002/art.27220
21. Rice LM, Padilla CM, McLaughlin SR, Mathes A, Ziemek J, Goummih S, et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J Clin Invest. (2015) 125:2795–807. doi: 10.1172/JCI77958
22. Lacouture ME, Morris JC, Lawrence DP, Tan AR, Olencki TE, Shapiro GI, et al. Cutaneous keratoacanthomas/squamous cell carcinomas associated with neutralization of transforming growth factor beta by the monoclonal antibody fresolimumab (GC1008). Cancer Immunol Immunother. (2015) 64:437–46. doi: 10.1007/s00262-015-1653-0
23. Ebisawa T, Fukuchi M, Murakami G, Chiba T, Tanaka K, Imamura T, et al. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J Biol Chem. (2001) 276:12477–80. doi: 10.1074/jbc.C100008200
24. Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell (2000) 6:1365–75. doi: 10.1016/S1097-2765(00)00134-9
25. Komuro A, Imamura T, Saitoh M, Yoshida Y, Yamori T, Miyazono K, et al. Negative regulation of transforming growth factor-beta (TGF-beta) signaling by WW domain-containing protein 1 (WWP1). Oncogene (2004) 23:6914–23. doi: 10.1038/sj.onc.1207885
26. Kuratomi G, Komuro A, Goto K, Shinozaki M, Miyazawa K, Miyazono K, et al. NEDD4-2 (neural precursor cell expressed, developmentally down-regulated 4-2) negatively regulates TGF-beta (transforming growth factor-beta) signalling by inducing ubiquitin-mediated degradation of Smad2 and TGF-beta type I receptor. Biochem J. (2005) 386(Pt 3):461–70. doi: 10.1042/BJ20040738
27. Zhu H, Kavsak P, Abdollah S, Wrana JL, Thomsen GH. A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature (1999) 400:687–93. doi: 10.1038/23293
28. Murakami G, Watabe T, Takaoka K, Miyazono K, Imamura T. Cooperative inhibition of bone morphogenetic protein signaling by Smurf1 and inhibitory Smads. Mol Biol Cell (2003) 14:2809–17. doi: 10.1091/mbc.e02-07-0441
29. Lin X, Liang M, Feng XH. Smurf2 is a ubiquitin E3 ligase mediating proteasome-dependent degradation of Smad2 in transforming growth factor-beta signaling. J Biol Chem. (2000) 275:36818–22. doi: 10.1074/jbc.C000580200
30. Zhang Y, Chang C, Gehling DJ, Hemmati-Brivanlou A, Derynck R. Regulation of Smad degradation and activity by Smurf2, an E3 ubiquitin ligase. Proc Natl Acad Sci USA. (2001) 98:974–9. doi: 10.1073/pnas.98.3.974
31. Gao S, Alarcon C, Sapkota G, Rahman S, Chen PY, Goerner N, et al. Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-beta signaling. Mol Cell (2009) 36:457–68. doi: 10.1016/j.molcel.2009.09.043
32. Mavrakis KJ, Andrew RL, Lee KL, Petropoulou C, Dixon JE, Navaratnam N, et al. Arkadia enhances Nodal/TGF-beta signaling by coupling phospho-Smad2/3 activity and turnover. PLoS Biol. (2007) 5:e67. doi: 10.1371/journal.pbio.0050067
33. Xin H, Xu X, Li L, Ning H, Rong Y, Shang Y, et al. CHIP controls the sensitivity of transforming growth factor-beta signaling by modulating the basal level of Smad3 through ubiquitin-mediated degradation. J Biol Chem. (2005) 280:20842–50. doi: 10.1074/jbc.M412275200
34. Wang L, Liu YT, Hao R, Chen L, Chang Z, Wang HR, et al. Molecular mechanism of the negative regulation of Smad1/5 protein by carboxyl terminus of Hsc70-interacting protein (CHIP). J Biol Chem. (2011) 286:15883–94. doi: 10.1074/jbc.M110.201814
35. Moren A, Imamura T, Miyazono K, Heldin CH, Moustakas A. Degradation of the tumor suppressor Smad4 by WW and HECT domain ubiquitin ligases. J Biol Chem. (2005) 280:22115–23. doi: 10.1074/jbc.M414027200
36. Nagano Y, Mavrakis KJ, Lee KL, Fujii T, Koinuma D, Sase H, et al. Arkadia induces degradation of SnoN and c-Ski to enhance transforming growth factor-beta signaling. J Biol Chem. (2007) 282:20492–501. doi: 10.1074/jbc.M701294200
37. Soond SM, Chantry A. Selective targeting of activating and inhibitory smads by distinct WWP2 ubiquitin ligase isoforms differentially modulates TGFbeta signalling and EMT. Oncogene (2011) 30:2451–62. doi: 10.1038/onc.2010.617
38. Bengoechea-Alonso MT, Ericsson J. Tumor suppressor Fbxw7 regulates TGFbeta signaling by targeting TGIF1 for degradation. Oncogene (2010) 29:5322–8. doi: 10.1038/onc.2010.278
39. Hu D, Wan Y. Regulation of Kruppel-like factor 4 by the anaphase promoting complex pathway is involved in TGF-beta signaling. J Biol Chem. (2011) 286:6890–901. doi: 10.1074/jbc.M110.179952
40. Andrews PS, Schneider S, Yang E, Michaels M, Chen H, Tang J, et al. Identification of substrates of SMURF1 ubiquitin ligase activity utilizing protein microarrays. Assay Drug Dev Technol. (2010) 8:471–87. doi: 10.1089/adt.2009.0264
41. Carpentier I, Coornaert B, Beyaert R. Smurf2 is a TRAF2 binding protein that triggers TNF-R2 ubiquitination and TNF-R2-induced JNK activation. Biochem Biophys Res Commun. (2008) 374:752–7. doi: 10.1016/j.bbrc.2008.07.103
42. Zuscik MJ, Rosier RN, Schwarz EM. Altered negative regulation of transforming growth factor beta signaling in scleroderma: potential involvement of SMURF2 in disease. Arthritis Rheum. (2003) 48:1779–80. doi: 10.1002/art.11158
43. Asano Y, Ihn H, Yamane K, Kubo M, Tamaki K. Impaired Smad7-Smurf-mediated negative regulation of TGF-beta signaling in scleroderma fibroblasts. J Clin Invest. (2004) 113:253–64. doi: 10.1172/JCI16269
44. Lear T, McKelvey AC, Rajbhandari S, Dunn SR, Coon TA, Connelly W, et al. Ubiquitin E3 ligase FIEL1 regulates fibrotic lung injury through SUMO-E3 ligase PIAS4. J Exp Med. (2016) 213:1029–46. doi: 10.1084/jem.20151229
45. Kusko RL, Brothers JF II, Tedrow J, Pandit K, Huleihel L, Perdomo C, et al. Integrated genomics reveals convergent transcriptomic networks underlying chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2016) 194:948–60. doi: 10.1164/rccm.201510-2026OC
46. Zhang Z, Finnerty CC, He J, Herndon DN. Smad ubiquitination regulatory factor 2 expression is enhanced in hypertrophic scar fibroblasts from burned children. Burns (2012) 38:236–46. doi: 10.1016/j.burns.2011.08.012
47. Li L, Shen Y, Ding Y, Liu Y, Su D, Liang X. Hrd1 participates in the regulation of collagen I synthesis in renal fibrosis. Mol Cell Biochem. (2014) 386:35–44. doi: 10.1007/s11010-013-1843-z
48. Fukasawa H, Yamamoto T, Togawa A, Ohashi N, Fujigaki Y, Oda T, et al. Down-regulation of Smad7 expression by ubiquitin-dependent degradation contributes to renal fibrosis in obstructive nephropathy in mice. Proc Natl Acad Sci USA. (2004) 101:8687–92. doi: 10.1073/pnas.0400035101
49. Liu FY, Li XZ, Peng YM, Liu H, Liu YH. Arkadia-Smad7-mediated positive regulation of TGF-beta signaling in a rat model of tubulointerstitial fibrosis. Am J Nephrol. (2007) 27:176–83. doi: 10.1159/000100518
50. Hasegawa D, Fujii R, Yagishita N, Matsumoto N, Aratani S, Izumi T, et al. E3 ubiquitin ligase synoviolin is involved in liver fibrogenesis. PLoS ONE (2010) 5:e13590. doi: 10.1371/journal.pone.0013590
51. Cai Y, Shen XZ, Zhou CH, Wang JY. Abnormal expression of Smurf2 during the process of rat liver fibrosis. Chin J Dig Dis. (2006) 7:237–45. doi: 10.1111/j.1443-9573.2006.00275.x
52. Cai Y, Zhou CH, Fu D, Shen XZ. Overexpression of Smad ubiquitin regulatory factor 2 suppresses transforming growth factor-beta mediated liver fibrosis. J Dig Dis. (2012) 13:327–34. doi: 10.1111/j.1751-2980.2012.00592.x
53. Chung S, Nakashima M, Zembutsu H, Nakamura Y. Possible involvement of NEDD4 in keloid formation; its critical role in fibroblast proliferation and collagen production. Proc Jpn Acad Ser B Phys Biol Sci. (2011) 87:563–73. doi: 10.2183/pjab.87.563
54. Song J, Zhu Y, Li J, Liu J, Gao Y, Ha T, et al. Pellino1-mediated TGF-beta1 synthesis contributes to mechanical stress induced cardiac fibroblast activation. J Mol Cell Cardiol. (2015) 79:145–56. doi: 10.1016/j.yjmcc.2014.11.006
55. Wicks SJ, Haros K, Maillard M, Song L, Cohen RE, Dijke PT, et al. The deubiquitinating enzyme UCH37 interacts with Smads and regulates TGF-beta signalling. Oncogene (2005) 24:8080–4. doi: 10.1038/sj.onc.1208944
56. Cutts AJ, Soond SM, Powell S, Chantry A. Early phase TGFbeta receptor signalling dynamics stabilised by the deubiquitinase UCH37 promotes cell migratory responses. Int J Biochem Cell Biol. (2011) 43:604–12. doi: 10.1016/j.biocel.2010.12.018
57. Jacko AM, Nan L, Li S, Tan J, Zhao J, Kass DJ, et al. De-ubiquitinating enzyme, USP11, promotes transforming growth factor β-1 signaling through stabilization of transforming growth factor β receptor II. Cell Death Dis. (2016) 7:e2474. doi: 10.1038/cddis.2016.371
58. Zhao Y, Thornton AM, Kinney MC, Ma CA, Spinner JJ, Fuss IJ, et al. The deubiquitinase CYLD targets Smad7 protein to regulate transforming growth factor beta (TGF-beta) signaling and the development of regulatory T cells. J Biol Chem. (2011) 286:40520–30. doi: 10.1074/jbc.M111.292961
59. Pannem RR, Dorn C, Hellerbrand C, Massoumi R. Cylindromatosis gene CYLD regulates hepatocyte growth factor expression in hepatic stellate cells through interaction with histone deacetylase 7. Hepatology (2014) 60:1066–81. doi: 10.1002/hep.27209
60. Inui M, Manfrin A, Mamidi A, Martello G, Morsut L, Soligo S, et al. USP15 is a deubiquitylating enzyme for receptor-activated SMADs. Nat Cell Biol. (2011) 13:1368–75. doi: 10.1038/ncb2346
61. Itoh F, Asao H, Sugamura K, Heldin CH, ten Dijke P, Itoh S. Promoting bone morphogenetic protein signaling through negative regulation of inhibitory Smads. EMBO J. (2001) 20:4132–42. doi: 10.1093/emboj/20.15.4132
62. Ibarrola N, Kratchmarova I, Nakajima D, Schiemann WP, Moustakas A, Pandey A, et al. Cloning of a novel signaling molecule, AMSH-2, that potentiates transforming growth factor beta signaling. BMC Cell Biol. (2004) 5:2. doi: 10.1186/1471-2121-5-2
63. Nusse R, Clevers H. Wnt/beta-catenin signaling, disease, and emerging therapeutic modalities. Cell (2017) 169:985–99. doi: 10.1016/j.cell.2017.05.016
64. Lam AP, Herazo-Maya JD, Sennello JA, Flozak AS, Russell S, Mutlu GM, et al. Wnt coreceptor Lrp5 is a driver of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2014) 190:185–95. doi: 10.1164/rccm.201401-0079OC
65. Wei J, Fang F, Lam AP, Sargent JL, Hamburg E, Hinchcliff ME, et al. Wnt/beta-catenin signaling is hyperactivated in systemic sclerosis and induces Smad-dependent fibrotic responses in mesenchymal cells. Arthritis Rheum. (2012) 64:2734–45. doi: 10.1002/art.34424
66. Beyer C, Reichert H, Akan H, Mallano T, Schramm A, Dees C, et al. Blockade of canonical Wnt signalling ameliorates experimental dermal fibrosis. Ann Rheum Dis. (2013) 72:1255–8. doi: 10.1136/annrheumdis-2012-202544
67. Distler JH, Feghali-Bostwick C, Soare A, Asano Y, Distler O, Abraham DJ. Review: frontiers of antifibrotic therapy in systemic sclerosis. Arthritis Rheumatol. (2017) 69:257–67. doi: 10.1002/art.39865
68. Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov. (2014) 13:513–32. doi: 10.1038/nrd4233
69. Koo BK, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M, et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature (2012) 488:665–9. doi: 10.1038/nature11308
70. Hao HX, Xie Y, Zhang Y, Charlat O, Oster E, Avello M, et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature (2012) 485:195–200. doi: 10.1038/nature11019
71. Abrami L, Kunz B, Iacovache I, van der Goot FG. Palmitoylation and ubiquitination regulate exit of the Wnt signaling protein LRP6 from the endoplasmic reticulum. Proc Natl Acad Sci USA. (2008) 105:5384–9. doi: 10.1073/pnas.0710389105
72. Gao C, Xiao G, Hu J. Regulation of Wnt/beta-catenin signaling by posttranslational modifications. Cell Biosci. (2014) 4:13. doi: 10.1186/2045-3701-4-13
73. Tauriello DV, Haegebarth A, Kuper I, Edelmann MJ, Henraat M, Canninga-van Dijk MR, et al. Loss of the tumor suppressor CYLD enhances Wnt/beta-catenin signaling through K63-linked ubiquitination of Dvl. Mol Cell. (2010) 37:607–19. doi: 10.1016/j.molcel.2010.01.035
74. Jung H, Kim BG, Han WH, Lee JH, Cho JY, Park WS, et al. Deubiquitination of dishevelled by Usp14 is required for Wnt signaling. Oncogenesis (2013) 2:e64. doi: 10.1038/oncsis.2013.28
75. Zhang Y, Liu S, Mickanin C, Feng Y, Charlat O, Michaud GA, et al. RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat Cell Biol. (2011) 13:623–9. doi: 10.1038/ncb2222
76. Kim S, Jho EH. The protein stability of Axin, a negative regulator of Wnt signaling, is regulated by Smad ubiquitination regulatory factor 2 (Smurf2). J Biol Chem. (2010) 285:36420–26. doi: 10.1074/jbc.M110.137471
77. Fei C, Li Z, Li C, Chen Y, Chen Z, He X, et al. Smurf1-mediated Lys29-linked nonproteolytic polyubiquitination of axin negatively regulates Wnt/beta-catenin signaling. Mol Cell Biol. (2013) 33:4095–105. doi: 10.1128/MCB.00418-13
78. Tran H, Bustos D, Yeh R, Rubinfeld B, Lam C, Shriver S, et al. HectD1 E3 ligase modifies adenomatous polyposis coli (APC) with polyubiquitin to promote the APC-axin interaction. J Biol Chem. (2013) 288:3753–67. doi: 10.1074/jbc.M112.415240
79. Huang X, Langelotz C, Hetfeld-Pechoc BK, Schwenk W, Dubiel W. The COP9 signalosome mediates beta-catenin degradation by deneddylation and blocks adenomatous polyposis coli destruction via USP15. J Mol Biol. (2009) 391:691–702. doi: 10.1016/j.jmb.2009.06.066
80. Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, Pavletich NP. Structure of a beta-TrCP1-Skp1-beta-catenin complex: destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Mol Cell. (2003) 11:1445–56. doi: 10.1016/S1097-2765(03)00234-X
81. Chitalia VC, Foy RL, Bachschmid MM, Zeng L, Panchenko MV, Zhou MI, et al. Jade-1 inhibits Wnt signalling by ubiquitylating beta-catenin and mediates Wnt pathway inhibition by pVHL. Nat Cell Biol. (2008) 10:1208–16. doi: 10.1038/ncb1781
82. Shekhar MP, Gerard B, Pauley RJ, Williams BO, Tait L. Rad6B is a positive regulator of beta-catenin stabilization. Cancer Res. (2008) 68:1741–50. doi: 10.1158/0008-5472.CAN-07-2111
83. Hay-Koren A, Caspi M, Zilberberg A, Rosin-Arbesfeld R. The EDD E3 ubiquitin ligase ubiquitinates and up-regulates beta-catenin. Mol Biol Cell (2011) 22:399–411. doi: 10.1091/mbc.e10-05-0440
84. Chakraborty D, Sumova B, Mallano T, Chen CW, Distler A, Bergmann C, et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat Commun. (2017) 8:1130. doi: 10.1038/s41467-017-01236-6
85. Wei J, Yuan Y, Jin C, Chen H, Leng L, He F, et al. The ubiquitin ligase TRAF6 negatively regulates the JAK-STAT signaling pathway by binding to STAT3 and mediating its ubiquitination. PLoS ONE (2012) 7:e49567. doi: 10.1371/journal.pone.0049567
86. Dallavalle C, Albino D, Civenni G, Merulla J, Ostano P, Mello-Grand M, et al. MicroRNA-424 impairs ubiquitination to activate STAT3 and promote prostate tumor progression. J Clin Invest. (2016) 126:4585–602. doi: 10.1172/JCI86505
87. Ao N, Chen Q, Liu G. The small molecules targeting ubiquitin-proteasome system for cancer therapy. Comb Chem High Throughput Screen. (2017) 20:403–13. doi: 10.2174/1386207320666170710124746
88. Koca SS, Ozgen M, Dagli F, Tuzcu M, Ozercan IH, Sahin K, et al. Proteasome inhibition prevents development of experimental dermal fibrosis. Inflammation (2012) 35:810–7. doi: 10.1007/s10753-011-9380-y
89. Fineschi S, Reith W, Guerne PA, Dayer JM, Chizzolini C. Proteasome blockade exerts an antifibrotic activity by coordinately down-regulating type I collagen and tissue inhibitor of metalloproteinase-1 and up-regulating metalloproteinase-1 production in human dermal fibroblasts. FASEB J. (2006) 20:562–4. doi: 10.1096/fj.05-4870fje
90. Nakajima F, Aratani S, Fujita H, Yagishita N, Ichinose S, Makita K, et al. Synoviolin inhibitor LS-102 reduces endoplasmic reticulum stress-induced collagen secretion in an in vitro model of stress-related interstitial pneumonia. Int J Mol Med. (2015) 35:110–6. doi: 10.3892/ijmm.2014.1984
91. Blees JS, Bokesch HR, Rubsamen D, Schulz K, Milke L, Bajer MM, et al. Erioflorin stabilizes the tumor suppressor Pdcd4 by inhibiting its interaction with the E3-ligase beta-TrCP1. PLoS ONE (2012) 7:e46567. doi: 10.1371/journal.pone.0046567
92. Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, Donato NJ. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res. (2010) 70:9265–76. doi: 10.1158/0008-5472.CAN-10-1530
93. Tian Z, D'Arcy P, Wang X, Ray A, Tai YT, Hu Y, et al. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood (2014) 123:706–16. doi: 10.1182/blood-2013-05-500033
94. Nan L, Jacko AM, Tan J, Wang D, Zhao J, Kass DJ, et al. Ubiquitin carboxyl-terminal hydrolase-L5 promotes TGFbeta-1 signaling by de-ubiquitinating and stabilizing Smad2/Smad3 in pulmonary fibrosis. Sci Rep. (2016) 6:33116. doi: 10.1038/srep33116
Keywords: SSc, ubiquitination, TGF-β, WNT/β-catenin, STAT3
Citation: Long Y, Chen W, Du Q, Zuo X and Zhu H (2018) Ubiquitination in Scleroderma Fibrosis and Its Treatment. Front. Immunol. 9:2383. doi: 10.3389/fimmu.2018.02383
Received: 19 June 2018; Accepted: 25 September 2018;
Published: 17 October 2018.
Edited by:
Raffaele De Palma, Università degli Studi della Campania “Luigi Vanvitelli” Caserta, ItalyReviewed by:
Gianluca Moroncini, Università Politecnica delle Marche, ItalyCopyright © 2018 Long, Chen, Du, Zuo and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Honglin Zhu, aG9uZ2xpbnpodUBjc3UuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.