 Shigeharu Ueki
Shigeharu Ueki Akira Hebisawa2
Akira Hebisawa2 Koichiro Asano
Koichiro Asano Josiane S. Neves
Josiane S. Neves
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 11 October 2018
Sec. Molecular Innate Immunity
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02346
This article is part of the Research Topic Severe Eosinophilic Disorders: Mechanisms and Clinical Management View all 14 articles
Allergic bronchopulmonary aspergillosis (ABPA) is characterized by an early allergic response and late-phase lung injury in response to repeated exposure to Aspergillus antigens, as a consequence of persistent fungal colonization of the airways. Here, we summarize the clinical and pathological features of ABPA, focusing on thick mucus plugging, a key observation in ABPA. Recent findings have indicated that luminal eosinophils undergo cytolytic extracellular trap cell death (ETosis) and release filamentous chromatin fibers (extracellular traps, ETs) by direct interaction with Aspergillus fumigatus. Production of ETs is considered to be an innate immune response against non-phagocytable pathogens using a “trap and kill” mechanism, although eosinophil ETs do not promote A. fumigatus damage or killing. Compared with neutrophils, eosinophil ETs are composed of stable and condensed chromatin fibers and thus might contribute to the higher viscosity of eosinophilic mucus. The major fate of massively accumulated eosinophils in the airways is ETosis, which potentially induces the release of toxic granule proteins and damage-associated molecular patterns, epithelial damage, and further decreases mucus clearance. This new perspective on ABPA as a luminal hypereosinophilic disease with ETosis/ETs could provide a better understanding of airway mucus plugging and contribute to future therapeutic strategies for this challenging disease.
Allergic bronchopulmonary aspergillosis (ABPA) is a disease entity first proposed by Hinson and colleagues as bronchopulmonary aspergillosis in 1952 (1). ABPA develops mainly in adolescent and adult patients with asthma or cystic fibrosis. It has been estimated that 2.5% (0.7–3.5%) of adult patients with asthma suffer from ABPA (2). Clinically, it is characterized by peripheral blood eosinophilia, increased levels of serum IgE, an immediate skin reaction and/or specific IgE/IgG antibodies to Aspergillus fumigatus due to type I and III hypersensitivity reactions, and radiographic findings including pulmonary opacities, central bronchiectasis, and mucus plugs (3–5). Systemic corticosteroids and/or anti-fungal drugs are effective, although approximately half of ABPA patients experience relapse (6, 7).
Besides blood eosinophilia, a massive accumulation of eosinophils and clustering of these immune cells in the bronchial lumen, resulting in bronchial impaction, are hallmarks of ABPA. Clinically, this has been defined by different terms depending on the context, such as “allergic mucin,” “high attenuation mucus,” or “allergic mucus plugs.” Considerable evidence has indicated the close association between the sputum eosinophil count and/or the eosinophil granule protein concentration and asthma severity (8, 9), although much less attention has been paid to luminal eosinophils (and their lytic components) in ABPA. This may be simply because of the lack of an explicit relationship and/or the difficulties in measuring protein concentrations due to the inspissated bronchial secretions. In this review, we discuss the clinical features of ABPA, focusing on new insights into the fate of eosinophils and their “cell debris” in the airways.
Germination and saprophytic growth of fungi in the mucus are interesting and unique features of ABPA. Aspergillus fumigatus is the major causative fungus of ABPA, but other Aspergillus spp., such as A. flavus, A. niger, and A. oryzae, can also cause ABPA, although less frequently. Schizophyllum commune, a filamentous basidiomycete commonly found on the rotten wood of trees, can cause similar pathology and a condition known as allergic bronchopulmonary mycosis (ABPM) (7, 10–12). Because fungal hyphae are immunologically active expressing and releasing various proteases and pathogen-associated molecular patterns (13–15), repeated exposures to airborne fragments of fungal hyphae, either dead or alive, can cause asthma and hypersensitivity pneumonitis. For the development of ABPM, by contrast, inhalation of viable fungi as conidia, not their hyphae fragments, and their germination in the lower airways is essential. A. fumigatus has advantages for the development of ABPA/ABPM over other fungi because of the small size of its conidia (3–6 μm) and thermophilicity (16). The conidia of S. commune are also small (3–4 μm), and prefer a relatively high temperature (30–35°C) to germinate (17). A. fumigatus conidia are also known for their high dispersibility due to their remarkable hydrophobicity (18).
Unlike fungal infections, germinated hyphae cannot penetrate the lung tissues in the presence of a normal immune system and bronchial structure. Therefore, A. fumigatus remains in the mucus plugs of the bronchi. Compared with other fungi, A. fumigatus has another advantage in this process in that it induces the Muc5ac gene, one of the mucin genes, and mucus production in bronchial epithelial cells (19). Induction of Muc5ac gene expression is dependent on the high serine protease activity of A. fumigatus, which activates membrane-bound TNFα-converting enzyme and TGF-α, stimulating epidermal growth factor receptors. Compared with A. fumigatus, other fungi, such as Penicillium and Alternaria, and other Aspergillus spp., such as A. flavus and A. niger have lower serine protease activity (19, 20).
In the first description of ABPA, Hinson and colleagues advocated that the features of ABPA included (i) wheezing and blood eosinophilia, (ii) repetitive infiltrations visible on chest X-ray, and (iii) “allergic” (eosinophilic) mucus plugs with fungal hyphae (AMwF) (1). In 1967, Scadding (21) reported the presence of central bronchiectasis by bronchography, with the initial mucus-filled bronchi being less apparent but bronchial ectasis remaining. The diagnostic criteria for ABPA in the pre-computerized tomography era, proposed by Rosenberg and Patterson (22), included pulmonary opacities and central bronchiectasis.
Currently, the radiographic features of ABPA include bronchiectasis, mucoid impaction, pulmonary opacities, mosaic attenuation, centrilobular nodules, tree-in-bud opacities, and pleuropulmonary fibrosis (3). Pulmonary opacities, which are usually transient, are frequently observed during the course of the disease, with 89% of ABPA cases with bronchiectasis demonstrating pulmonary opacities/ground grass attenuation in a nationwide survey in Japan (7); however, this is not classed as a specific feature of this disease. Central bronchiectasis with peripheral tapering of bronchi has been considered as a relatively specific finding for ABPA, but bronchiectasis can extend to the peripheral bronchi in 33–63% of cases (23, 24). Mucus plugs in the bronchi are common in ABPA, and may present as high attenuation mucus (HAM) with a CT density higher than the values of paraspinal skeletal muscle (25) or 70 Hounsfield units (26). Magnetic resonance imaging of HAM showed hypodense lesions in T1- and T2-weighed images, suggesting that the mucus is desiccated or inspissated (27). HAM can be observed in more than half of the cases with mucus plugs due to ABPA (7, 23), and is more specific for this disease. The presence of HAM also correlates with a higher number of eosinophils in the peripheral blood and greater susceptibility to disease relapse (6, 28).
Regarding the pathological features of ABPA, Katzenstein and colleagues emphasized the presence of AMwF, bronchocentric granulomatosis with tissue eosinophilia (BrCG-eo), eosinophilic bronchiolitis, and eosinophilic pneumonia (29). In 1988, Bosken and coworkers indicated, based on the investigation of surgically resected specimens, that mucoid impaction of bronchi with allergic mucin or BrCG-eo, together with fungal hyphae detected in the lesion, were sufficient for the diagnosis of ABPA/ABPM (30). However, the diagnostic criteria proposed by Rosenberg and Patterson (22) excluded pathological findings; instead, clinical, radiological, and laboratory findings, especially allergy tests, were included.
AMwF was detected in the biopsy samples by bronchoscopy, expectorated airway secretions, and the resected lungs. AMwF has been described by pathologists as follows (31, 32). “AMwF could be found in any bronchi of any lobes, including lobar and segmental bronchi and 3rd−5th bifurcated small bronchi. AMwF was typically white-yellowish, dark-yellowish, or yellow-greenish in color, and sticky with elastic soft or elastic hard in consistency. The shape ranged from branching and clubbing to lumpy. The H&E stained section of the AMwF demonstrated concentric multiple layers consisting of eosinophils, Charcot-Leyden crystals, and fibrin (32, 33). The eosinophils might be viable, necrobiotic (with pyknotic nuclei and nuclear dusts) or necrotic. The latter two types formed inspissated eosinophil aggregates with indistinct cell margins and occasionally exhibited many fissures likely due to dehydration inside of AMwF, demonstrating ‘fir-tree structure’ (33). Fungi were usually found in the clusters of eosinophils as fragmented hyphae or branching septate hyphae, and occasionally in the mucus surrounding the eosinophil clusters. While fungi floats in the air as unicellular spores, they reside in the AMwF as multicellular hyphae, suggesting the proliferation of fungi.”
Central bronchiectasis and peripheral lung lesions in ABPM were suggested as secondary pathological changes after the formation of AMwF. BrCG-eo, eosinophilic bronchiolitis, and eosinophilic pneumonia were found to be in the airways peripheral to the AMwF plugs, previously described as follows (32). “The investigation of the resected lungs of ABPM patients revealed the airway walls plugged by AMwF were invaded with eosinophils and small round cells including plasma cells and were ulcerated, resulting in eosinophilic infiltration into the allergic mucin. The intense inflammation in the bronchial walls destructed and dissipated airway structures i.e., elastic fibers and smooth muscles, and even bronchial cartilages. Central bronchiectasis could be caused by the vulnerability of diseased bronchial walls and by expanding AMwF. In the exudate from BrCG-eo and eosinophilic bronchiolitis, inspissated eosinophil aggregates, fir-tree structures and fungi were detected, similar to the AMwF. However, the inspissated eosinophil aggregates and fir-tree structures were fragmented and the number of fungi was smaller than that in the central lesions of AMwF (30–33). These findings suggested that the peripheral lung lesions might be formed by the bronchogenic spread of AMwF to the peripheral lung.” Thus, careful morphological analysis has indicated the AMwF is an initial and crucial step in the development of ABPA.
Eosinophils are bone marrow-derived, short-lived, non-dividing granulocytes that have been implicated as integral components of allergic airway inflammation. Eosinophils contain ~200 granules per cell (34), which are preformed stores of the specific granule proteins (35). The highly cationic granule proteins possess cytotoxicity by disrupting the integrity of lipid bilayers, exerting neurotoxic properties, RNase activities, and participating in the generation of reactive oxidants and radical species (36, 37). Since eosinophil degranulation does not occur in the circulation (38), it is critical to understand the actual mode of degranulation and cell fate in the airways. Our recent studies indicated eosinophil cell-death mediated degranulation, i.e., eosinophil ETosis, which might play an important role in the generation of mucus plugs (39, 40).
In 2004, Brinkmman and colleagues provided the first evidence that neutrophils release filamentous chromatin structures, i.e., neutrophil extracellular traps (NETs) (41). A subsequent study revealed that the novel cell death process mediates the release of NETs and was designated NETosis (42). NETosis has unique features that are different from apoptosis and necrosis. Unlike apoptosis, nuclear DNA fragmentation is spared during the process of NETosis. Histone citrullination induces chromatin decondensation and granules are intracellularly disrupted before plasma membrane disintegration, thereby NETs are associated with various antimicrobial molecules (including histones, elastase, myeloperoxidase, cathepsin, and lactoferrin). Once NETs are released, they immobilize and potentially kill pathogens (41, 43, 44). Based on this, roles for these structures in preventing microbial spread and in creating an antimicrobial environment have been postulated.
Since chromatin traps are cytolytically released by other leukocytes, such as macrophages (45) and mast cells (46, 47), “ETosis” has been proposed as a similar cell death pathway (48). Extracellular trap (ET) formation is now considered to be a common mechanism of the innate immune system in vertebrates (49). A recent report showed that even invertebrate cells are capable of ETosis, indicating an ancient and evolutionary conserved mechanism (50). Eosinophil ETosis (EETosis) was first reported as a mechanism of “cytolysis” (also called lytic degranulation, necrosis, primary lysis, and eosinophil lysis) that has been observed (but often overlooked) in eosinophilic inflammatory diseases (35). This finding indicates that eosinophil cytolysis does not represent a process of passive/accidental necrosis, rather eosinophils are actively selecting their death program at the inflammatory site. The process of EETosis is akin to that of neutrophils but notably differs in that intact granules are released extracellularly (35, 51–53).
It is well-established that the process of leukocyte activation and ET release plays a role in health and disease, including innate immunity, autoimmunity, metabolic disorders, malignancies, and coagulation (44, 46, 54–57). Excess production of ETs could be pathogenic (49); for instance, large amounts of NETs contribute to the thickness of airway fluids from patients with cystic fibrosis (58). Inhalation of recombinant human DNase improved lung function in cystic fibrosis patients, indicating the pathogenicity of NETs (58–60). Similar to NETs, EETs provide a sticky scaffold for secretions, a fact confirmed by the decrease in viscosity following disruption of EET polymers with DNase (61). Recently, we have shown abundant EETs associated with clusters of cell-free extracellular granules in the bronchial mucus plugs of ABPA patients (39, 40). We also observed a drastic decrease in EETs in the bronchoscopically-obtained secretions from an ABPA patient after corticosteroid treatment, which correlated with clinical improvements (40).
It is noteworthy that NETs and EETs have different attributes in terms of stability and structure. The protein content of ETs plays an important role in its mechanical properties (62). Compared with neutrophils, eosinophils contain far less proteases, and thus eosinophil chromatin is spared from endogenous protease digestion (61, 63). Neutrophil elastase promotes the chromatin decondensation of NETs by proteolytic processing of histones (64). In vitro, NETs deteriorated within 24 h, whereas EETs were stable for at least 7 days (61). Electron microscopy showed that released NETs consisted of 5–10-nm smooth stretches (composed of stacked cylindrical nucleosomes) and 25–50-nm globular domains (41, 65), although EETs consisted mostly of chromatin fibers with diameters of 25–35 nm in conjunction with larger fibers (35). In terms of innate immune responses, staunch fibers might offer an advantage by immobilizing and hampering the progression of large parasites (66), and may also pathologically contribute to the highly viscous nature of eosinophil-dominant airway secretions, as clinically observed in ABPA, but also in other diseases, such as eosinophil chronic rhinosinusitis (51, 61), eosinophilic otitis media (63, 67), and severe asthma (40, 68).
There is evidence in vitro that human eosinophils rapidly activate in response to various stimuli by releasing mitochondrial DNA via a non-cytolytic mechanism (69, 70). The mitochondrial DNA would be present in mucus plugs since EETosis liberates total cellular contents through plasma membrane disintegration. However, mitochondrial DNA, lacking the histone and nucleosome structure, is only 16 kbp in size, constituting <1% of total cellular DNA (e.g., genomic DNA has 3.2 billion bp, which equates to 2 m in length/cell) (71). In addition, human eosinophils contain a small number of mitochondria (~30/cell) (72). Therefore, nuclear-derived chromatin fibers are a major component contributing to viscosity (35), as evidenced by positive staining of airway EETs with specific antibodies against histones/citrullinated histones (39, 61).
The immunopathological mechanisms that underlie the molecular interactions between A. fumigatus and eosinophils are also of interest (see review (73)). Our in vitro studies have shown that ETs are released by lytic human eosinophils in direct response to A. fumigatus (39). When human eosinophils were co-cultured with A. fumigatus, EETs were released in a time and ratio (fungus: cell) dependent manner (39).
Unlike necrosis, ETosis is an active form of cell death and is frequently accompanied by the production of reactive oxygen species (ROS) (35, 41, 43, 52, 69, 70, 74–76); however, we observed that human eosinophils release EETs in vitro in response to A. fumigatus in a ROS-independent manner (39). Regarding Aspergillus specifically, neutrophils are described to respond to different morphotypes of this fungus by releasing NETs in a ROS-dependent process (77–79). However, the ROS-independent release of NETs was observed when stimulated by bacteria (80), parasites (81), fungi (Candida and Paracoccidioides brasiliensis conidia) (82, 83), and other stimuli (84–86). The participation of ROS in the process of ETosis might be essentially dependent on the stimuli, time point, or microorganisms examined.
Mechanistically, A. fumigatus induced EETosis in vitro is a process dependent on the Syk tyrosine kinase pathway (known to mediate cell signaling via different classes of receptors involved in fungus recognition) and adhesion molecule CD11b β-integrin (which recognizes β-glucan, a molecular pattern commonly found in the cell wall of fungi) (39). Consistent with these findings, CD11b has been described to play an important role in eosinophil interactions with the fungus Alternaria alternata (87). Previous studies with eosinophils and A. alternata suggest a critical role for the CD11b I domain in eosinophil activation and degranulation, but not for the lectin domain, which recognizes the β-glucan (87). In neutrophils, both the C-type lectin receptor dectin-1 and the β-integrin CD11b/CD18 (CR3) have been implicated in the recognition of β-glucans on the cell surface of A. fumigatus (79, 88, 89). However, we did not detect the expression of dectin-1 protein in human blood eosinophils (39).
Airway eosinophils display enhanced responses to a variety of ligands and become further activated to degranulate (90, 91). Human eosinophils undergo ETosis in response to immobilized IgG or IgA (used as in vitro defined models of immunoglobulin-coated pathogens including parasites), calcium ionophore, IL-5/GM-CSF with platelet activating factor, or phorbol myristate acetate to liberate ETs (35, 68, 92). In addition to the direct response to A. fumigatus, it is possible that locally produced immunoglobulins, cytokines, and other mediators could activate eosinophils to induce EETosis. Indeed, a recent study indicated that IgG-type autoantibodies were present in the airways of patients with severe eosinophilic asthma, potentially induced by the release of EETs (68). It is also noteworthy that biological molecules associated with the fungal cell wall, such as chitin and β-glucan from Aspergillus species, induce eosinophilic Th2 inflammation in mouse lungs (93–95).
EETs exhibited intimate contact with A. fumigatus conidia and with cell-free extracellular granules, indicating that EETs provide an adhesive surface for organelle and microorganism entrapment (39). The highly hydrophobic surface of A. fumigatus might be easily entrapped by EETs, since EETs ensnare microorganisms mainly via hydrophobic interactions (39, 61). It has been reported that NETs may efficiently kill microbial pathogens (96–99); however, several fungi may resist the antimicrobial effect of NETs (79, 83, 100, 101).
Interestingly, our study revealed that human EETs did not promote the damage or killing of A. fumigatus (39). Eosinophils exhibit killing activity against different pathogens both in vitro and in experimental models (reviewed in 102). In vitro, Yoon and colleagues demonstrated that human eosinophils release their cytotoxic granule proteins in response to A. alternata and kill the fungus in a contact-dependent manner (87). Mouse bone marrow-derived eosinophils exhibited killing activity against A. fumigatus in vitro, which did not require cell contact (103). By contrast, using a Th2-dominant murine model of chronic invasive aspergilosis, eosinophil-deficient mice showed decreased morbidity and improved clearance of A. fumigatus (94). Intriguingly, studies based on mouse experimental models revealed that Th2-type immunity to chronic fungi exposition is generally accompanied by detrimental allergic inflammation, including tissue eosinophilia, goblet cell hyperplasia, and airway remodeling, but no signs of hyphae or invasive fungal growth were reported at any time after conidia challenge (104). Thus, the development of an animal model showing fungal colonization of the airways similar to that observed in ABPA pathology is still lacking. It remains unclear whether these discrepancies are due to the differences between mouse and human eosinophils (105) and/or experimental settings. Nevertheless, A. fumigatus resistance to EET killing activity might be an important feature of ABPA pathogenesis, possibly explaining the previously suggested “innate immunological defect” in this disease (3).
The rheological properties of mucus and the mucociliary transport system function as a self-cleaning mechanism for the respiratory tract (106). However, difficult-to-remove eosinophilic airway secretions are associated with disease severity (107, 108). As described above, the lower protease content and stable chromatin traps of eosinophils contribute to airway mucus viscosity. Most recently, Dunican and coworkers indicated that oxidants generated by eosinophil granule protein oxidize cysteine thiol groups to stiffen airway mucus (109).
We propose that EETosis contributes significantly to the fate of luminal eosinophils and plays a critical role in the pathogenesis of ABPA (Figure 1). Under Th2 conditions, blood eosinophils actively accumulate in the airway tissue. Eosinophils are usually eliminated by migration into the airway lumen followed by mucociliary clearance (110). Unlike apoptosis that produces fragmented DNA, luminal eosinophils undergo ETosis by direct interaction with fungi and/or local stimuli to release a sticky chromatin structure. EETs contribute to the increased viscosity of mucus rather than the direct elimination of fungi. Eosinophil cytolysis also releases intact granules and a wide range of nuclear and cytosolic damage-associated molecular patterns (92). Free granules may act as a reservoir of toxic granule proteins and secretion-competent extracellular organelles (35, 111). Perpetuating epithelial damage may inhibit the effectiveness of ciliary beating, thereby decreasing mucus transport and also resulting in the release of alamins and further eosinophilic inflammation. Thus, EETosis might promote a perpetuating cycle of thickening secretions (51). However, this new perspective requires further evaluation.
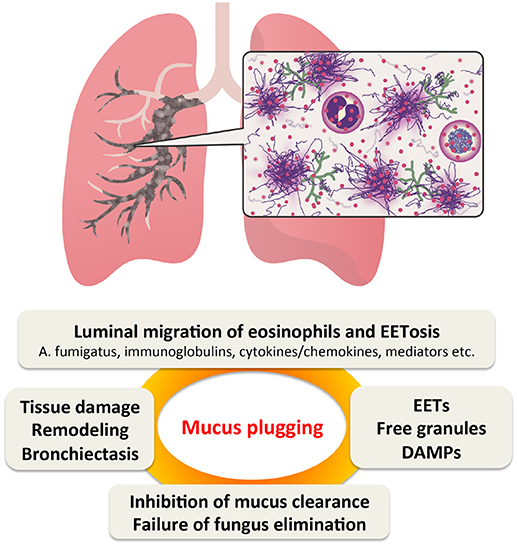
Figure 1. Proposed perpetuating cycle of mucus plugging caused by eosinophil ETosis (EETosis). In Th2-type airway inflammation, eosinophils are the major effector cells. They are supplied from the blood and massively accumulate in the bronchial lumen, where they are highly activated by fungi and other stimulants. EETosis-mediated cytolysis releases their total cellular contents including eosinophil extracellular traps (EETs), which contributes to the higher viscosity of eosinophilic mucus. Persistent fungal colonization of the airways and inhibition of “cytotoxic” mucus clearance further contributes to tissue damage, resulting in bronchiectasis. DMAPs, damage-associated molecular patterns.
As reviewed by Persson and Uller (112), cytolysis has often been overlooked by researchers, although several clinical studies illustrated that cytolysis accounted for a major proportion of bronchial eosinophils in severe and lethal asthma (112). Because apoptosis is a rare event among tissue-migrated eosinophils, ETosis-mediated cytolysis and luminal entry are likely to be the major fates of these cells. Indeed, Charcot–Leyden crystals, often observed in mucus plugs, were generated in association with the EETosis process (53). Considering the clinical features of ABPA, it is conceivable that increased eosinophil turnover, rather than prolonged lifespan, might be a major cause of luminal eosinophilia. It should be recognized that EETosis-mediated cytolysis and EETs are potentially important therapeutic targets in ABPA, as well as in other eosinophilic inflammatory diseases.
SU wrote and finalize the manuscript. AH and MK contributed pathological part; KA contributed clinical part. JN wrote cell biology part.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are grateful to Noriko Tan, Satomi Misawa, and Kaori Kato (Akita University) for their outstanding assistance. This study was funded in part by a Research Grant on Allergic Disease and Immunology from the Japan Agency for Medical Research and Development under Grant Number JP18ek0410026, Japanese Society of Laboratory Medicine Found for the Promotion of Scientific Research, and JSPS KAKENHI 15KK0329, 16K08926 (SU). JN thanks the Brazilian agencies Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ–grant number E-26/203.312/2017) e Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq–grant number 309216/2017-6) for the financial support. We also thank Kate Fox, DPhil, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
ABPA, allergic bronchopulmonary aspergillosis; ABPM, allergic bronchopulmonary mycosis; AMwF, allergic mucus plugs with fungal hyphae; BrCG-eo, bronchocentric granulomatosis with tissue eosinophilia; ET, extracellular trap; EET, eosinophil extracellular trap; ETosis, extracellular trap cell death; EETosis, eosinophil extracellular trap cell death; HAM, high attenuation mucus; NET, neutrophil extracellular trap; NETosis, neutrophil extracellular trap cell death; ROS, reactive oxygen species
1. Hinson KF, Moon AJ, Plummer NS. Broncho-pulmonary aspergillosis; a review and a report of eight new cases. Thorax (1952) 7:317–33. doi: 10.1136/thx.7.4.317
2. Denning DW, Pleuvry A, Cole DC. Global burden of allergic bronchopulmonary aspergillosis with asthma and its complication chronic pulmonary aspergillosis in adults. Med Mycol. (2013) 51:361–70. doi: 10.3109/13693786.2012.738312
3. Agarwal R, Chakrabarti A, Shah A, Gupta D, Meis JF, Guleria R, et al. Allergic bronchopulmonary aspergillosis: review of literature and proposal of new diagnostic and classification criteria. Clin Exp Allergy (2013) 43:850–73. doi: 10.1111/cea.12141
4. Knutsen AP, Bush RK, Demain JG, Denning DW, Dixit A, Fairs A, et al. Fungi and allergic lower respiratory tract diseases. J Allergy Clin Immunol. (2012) 129:280–91; quiz 292–3. doi: 10.1016/j.jaci.2011.12.970
5. Kosmidis C, Denning DW. The clinical spectrum of pulmonary aspergillosis. Thorax (2015) 70:270–7. doi: 10.1136/thoraxjnl-2014-206291
6. Agarwal R, Khan A, Gupta D, Aggarwal AN, Saxena AK, Chakrabarti A. An alternate method of classifying allergic bronchopulmonary aspergillosis based on high-attenuation mucus. PLoS ONE (2010) 5:e15346. doi: 10.1371/journal.pone.0015346
7. Oguma T, Taniguchi M, Shimoda T, Kamei K, Matsuse H, Hebisawa A, et al. Allergic bronchopulmonary aspergillosis in Japan: a nationwide survey. Allergol Int. (2018) 67:79–84. doi: 10.1016/j.alit.2017.04.011
8. Mukherjee M, Nair P. Blood or sputum eosinophils to guide asthma therapy? Lancet Respir Med. (2015) 3:824–5. doi: 10.1016/S2213-2600(15)00419-1
9. Persson C, Uller L. Resolution of leucocyte-mediated mucosal diseases. A novel in vivo paradigm for drug development. Br J Pharmacol. (2012) 165:2100–9. doi: 10.1111/j.1476-5381.2011.01772.x
10. Kamei K, Unno H, Nagao K, Kuriyama T, Nishimura K, Miyaji M. Allergic bronchopulmonary mycosis caused by the basidiomycetous fungus Schizophyllum commune. Clin Infect Dis. (1994) 18:305–9. doi: 10.1093/clinids/18.3.305
11. Ishiguro T, Takayanagi N, Kagiyama N, Shimizu Y, Yanagisawa T, Sugita Y. Clinical characteristics of biopsy-proven allergic bronchopulmonary mycosis: variety in causative fungi and laboratory findings. Internal Med. (2014) 53:1407–11. doi: 10.2169/internalmedicine.53.2230
12. Chowdhary A, Agarwal K, Kathuria S, Gaur SN, Randhawa HS, Meis JF. Allergic bronchopulmonary mycosis due to fungi other than Aspergillus: a global overview. Crit Rev Microbiol. (2014) 40:30–48. doi: 10.3109/1040841X.2012.754401
13. Romani L. Immunity to fungal infections. Nat Rev Immunol. (2011) 11:275–88. doi: 10.1038/nri2939
14. Fukutomi Y, Taniguchi M. Sensitization to fungal allergens: resolved and unresolved issues. Allergol Int. (2015) 64:321–31. doi: 10.1016/j.alit.2015.05.007
15. Portnoy JM, Williams PB, Barnes CS. Innate immune responses to fungal allergens. Curr Allergy Asthma Rep. (2016) 16:62. doi: 10.1007/s11882-016-0643-4
16. Araujo R, Rodrigues AG. Variability of germinative potential among pathogenic species of Aspergillus. J Clin Microbiol. (2004) 42:4335–7. doi: 10.1128/JCM.42.9.4335-4337.2004
17. Imtiaj A, Jayasinghe C, Lee GW, Kim HY, Shim MJ, Rho HS, et al. Physicochemical requirement for the vegetative growth of Schizophyllum commune collected from different ecological origins. Mycobiology (2008) 36:34–9. doi: 10.4489/MYCO.2008.36.1.034
18. Kwon-Chung KJ, Sugui JA. Aspergillus fumigatus–what makes the species a ubiquitous human fungal pathogen? PLoS Pathog. (2013) 9:e1003743. doi: 10.1371/journal.ppat.1003743
19. Oguma T, Asano K, Tomomatsu K, Kodama M, Fukunaga K, Shiomi T, et al. Induction of mucin and MUC5AC expression by the protease activity of Aspergillus fumigatus in airway epithelial cells. J Immunol. (2011) 187:999–1005. doi: 10.4049/jimmunol.1002257
20. Alp S, Arikan S. Investigation of extracellular elastase, acid proteinase and phospholipase activities as putative virulence factors in clinical isolates of Aspergillus species. J Basic Microbiol. (2008) 48:331–7. doi: 10.1002/jobm.200700349
22. Rosenberg M, Patterson R, Mintzer R, Cooper BJ, Roberts M, Harris KE. Clinical and immunologic criteria for the diagnosis of allergic bronchopulmonary aspergillosis. Ann Intern Med. (1977) 86:405–14. doi: 10.7326/0003-4819-86-4-405
23. Reiff DB, Wells AU, Carr DH, Cole PJ, Hansell DM. CT findings in bronchiectasis: limited value in distinguishing between idiopathic and specific types. AJR Am J Roentgenol. (1995) 165:261–7. doi: 10.2214/ajr.165.2.7618537
24. Agarwal R, Khan A, Garg M, Aggarwal AN, Gupta D. Chest radiographic and computed tomographic manifestations in allergic bronchopulmonary aspergillosis. World J Radiol. (2012) 4:141–50. doi: 10.4329/wjr.v4.i4.141
25. Agarwal R, Gupta D, Aggarwal AN, Saxena AK, Chakrabarti A, Jindal SK. Clinical significance of hyperattenuating mucoid impaction in allergic bronchopulmonary aspergillosis: an analysis of 155 patients. Chest (2007) 132:1183–90. doi: 10.1378/chest.07-0808
26. Phuyal S, Garg MK, Agarwal R, Gupta P, Chakrabarti A, Sandhu MS, Khandelwal N. High-attenuation mucus impaction in patients with allergic bronchopulmonary aspergillosis: objective criteria on high-resolution computed tomography and correlation with serologic parameters. Curr Probl Diagn Radiol. (2016) 45:168–73. doi: 10.1067/j.cpradiol.2015.07.006
27. Garg MK, Gupta P, Agarwal R, Sodhi KS, Khandelwal N. MRI: a new paradigm in imaging evaluation of allergic bronchopulmonary aspergillosis? Chest (2015) 147:e58–9. doi: 10.1378/chest.14-2347
28. Agarwal R, Khan A, Aggarwal AN, Varma N, Garg M, Saikia B, et al. Clinical relevance of peripheral blood eosinophil count in allergic bronchopulmonary aspergillosis. J Infect Public Health (2011) 4:235–43. doi: 10.1016/j.jiph.2011.08.006
29. Katzenstein AL, Liebow AA, Friedman PJ. Bronchocentric granulomatosis, mucoid impaction, and hypersensitivity reactions to fungi. Am Rev Respir Dis. (1975) 111:497–537.
30. Bosken CH, Myers JL, Greenberger PA, Katzenstein AL. Pathologic features of allergic bronchopulmonary aspergillosis. Am J Surg Pathol. (1988) 12:216–22. doi: 10.1097/00000478-198803000-00007
31. Aubry MC, Fraser R. The role of bronchial biopsy and washing in the diagnosis of allergic bronchopulmonary aspergillosis. Mod Pathol. (1998) 11:607–11.
32. Hebisawa A, Tamura A, Kurashima A, Oobayashi C, Kawamata M, Maeda M, et al. Pathologic reconsideration on allergic bronchopulmonary aspergillosis and mycosis. Nihon Kokyuki Gakkai Zasshi (1998) 36:330–7.
33. Jelihovsky T. The structure of bronchial plugs in mucoid impaction, bronchocentric granulomatosis and asthma. Histopathology (1983) 7:153–67. doi: 10.1111/j.1365-2559.1983.tb02232.x
34. Moqbel R LP, Adamko DJ, Odemuyiwa SO. Biology of Eosinophils, Philadelphia, PA: Elsevier Science (2009).
35. Ueki S, Melo RC, Ghiran I, Spencer LA, Dvorak AM, Weller PF. Eosinophil extracellular DNA trap cell death mediates lytic release of free secretion-competent eosinophil granules in humans. Blood (2013) 121:2074–83. doi: 10.1182/blood-2012-05-432088
36. Kita H. Eosinophils: multifunctional and distinctive properties. Int Arch Allergy Immunol. (2013) 161(Suppl. 2):3–9. doi: 10.1159/000350662
37. Weller PF, Spencer LA. Functions of tissue-resident eosinophils. Nat Rev Immunol. (2017) 17:746–60. doi: 10.1038/nri.2017.95
38. Malm-Erjefalt M, Greiff L, Ankerst J, Andersson M, Wallengren J, Cardell LO, et al. Circulating eosinophils in asthma, allergic rhinitis, and atopic dermatitis lack morphological signs of degranulation. Clin Exp Allergy (2005) 35:1334–40. doi: 10.1111/j.1365-2222.2005.02335.x
39. Muniz VS, Silva JC, Braga YAV, Melo RCN, Ueki S, Takeda M, et al. Eosinophils release extracellular DNA traps in response to Aspergillus fumigatus. J Allergy Clin Immunol. (2018) 141:571–585 e7. doi: 10.1016/j.jaci.2017.07.048
40. Omokawa A, Ueki S, Kikuchi Y, Takeda M, Asano M, Sato K, et al. Mucus plugging in allergic bronchopulmonary aspergillosis: implication of the eosinophil DNA traps. Allergol Int. (2018) 67:280–2. doi: 10.1016/j.alit.2017.08.002
41. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
42. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. (2007) 176:231–41. doi: 10.1083/jcb.200606027
43. Urban CF, Reichard U, Brinkmann V, Zychlinsky A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol. (2006) 8:668–76. doi: 10.1111/j.1462-5822.2005.00659.x
44. Delgado-Rizo V, Martinez-Guzman MA, Iniguez-Gutierrez L, Garcia-Orozco A, Alvarado-Navarro A, Fafutis-Morris M. Neutrophil extracellular traps and its implications in inflammation: an overview. Front Immunol. (2017) 8:81. doi: 10.3389/fimmu.2017.00081
45. Chow OA, von Kockritz-Blickwede M, Bright AT, Hensler ME, Zinkernagel AS, Cogen AL, et al. Statins enhance formation of phagocyte extracellular traps. Cell Host Microbe (2010) 8:445–54. doi: 10.1016/j.chom.2010.10.005
46. Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, et al. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol. (2011) 187:490–500. doi: 10.4049/jimmunol.1100123
47. von Kockritz-Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby-Teglund A, Rohde M, et al. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood (2008) 111:3070–80. doi: 10.1182/blood-2007-07-104018
48. Wartha F, Henriques-Normark B. ETosis: a novel cell death pathway. Sci Signal (2008) 1:pe25. doi: 10.1126/stke.121pe25
49. Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol. (2007) 5:577–82. doi: 10.1038/nrmicro1710
50. Robb CT, Dyrynda EA, Gray RD, Rossi AG, Smith VJ. Invertebrate extracellular phagocyte traps show that chromatin is an ancient defence weapon. Nat Commun. (2014) 5:4627. doi: 10.1038/ncomms5627
51. Ueki S, Tokunaga T, Fujieda S, Honda K, Hirokawa M, Spencer LA, et al. Eosinophil ETosis and DNA traps: a new look at Eosinophilic inflammation. Curr Allergy Asthma Rep. (2016) 16:54. doi: 10.1007/s11882-016-0634-5
52. Muniz VS, Baptista-Dos-Reis R, Neves JS. Functional extracellular eosinophil granules: a bomb caught in a trap. Int Arch Allergy Immunol. (2013) 162:276–82. doi: 10.1159/000354934
53. Ueki S, Tokunaga T, Melo RCN, Saito H, Honda K, Fukuchi M, et al. Charcot-Leyden crystal formation is closely associated with eosinophil extracellular trap cell death. Blood (2018). doi: 10.1182/blood-2018-04-842260. [Epub ahead of print].
54. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra20. doi: 10.1126/scitranslmed.3001201
55. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. (2013) 5:178ra40. doi: 10.1126/scitranslmed.3005580
56. Wang Y, Xiao Y, Zhong L, Ye D, Zhang J, Tu Y, et al. Increased neutrophil elastase and proteinase 3 and augmented NETosis are closely associated with beta-cell autoimmunity in patients with type 1 diabetes. Diabetes (2014) 63:4239–48. doi: 10.2337/db14-0480
57. Sollberger G, Tilley DO, Zychlinsky A. Neutrophil extracellular traps: the biology of chromatin externalization. Dev Cell (2018) 44:542–53. doi: 10.1016/j.devcel.2018.01.019
58. Manzenreiter R, Kienberger F, Marcos V, Schilcher K, Krautgartner WD, Obermayer A, et al. Ultrastructural characterization of cystic fibrosis sputum using atomic force and scanning electron microscopy. J Cyst Fibros. (2012) 11:84–92. doi: 10.1016/j.jcf.2011.09.008
59. Yang CL, Chilvers M, Montgomery M, Nolan SJ. Dornase alfa for cystic fibrosis. Paediatr Respirat Rev. (2016) 4:CD001127. doi: 10.1002/14651858.CD001127
60. George PM, Banya W, Pareek N, Bilton D, Cullinan P, Hodson ME, et al. Improved survival at low lung function in cystic fibrosis: cohort study from 1990 to 2007. BMJ (2011) 342:d1008. doi: 10.1136/bmj.d1008
61. Ueki S, Konno Y, Takeda M, Moritoki Y, Hirokawa M, Matsuwaki Y, et al. Eosinophil extracellular trap cell death-derived DNA traps: their presence in secretions and functional attributes. J Allergy Clin Immunol. (2016) 137:258–67. doi: 10.1016/j.jaci.2015.04.041
62. Pires RH, Felix SB, Delcea M. The architecture of neutrophil extracellular traps investigated by atomic force microscopy. Nanoscale (2016) 8:14193–202. doi: 10.1039/C6NR03416K
63. Ueki S, Ohta N, Takeda M, Konno Y, Hirokawa M. Eosinophilic otitis media: the aftermath of Eosinophil extracellular trap cell death. Curr Allergy Asthma Rep. (2017) 17:33. doi: 10.1007/s11882-017-0702-5
64. Papayannopoulos V, Staab D, Zychlinsky A. Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving DNase therapy. PLoS ONE (2011) 6:e28526. doi: 10.1371/journal.pone.0028526
65. Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. (2009) 5:e1000639. doi: 10.1371/journal.ppat.1000639
66. Munoz-Caro T, Rubio RM, Silva LM, Magdowski G, Gartner U, McNeilly TN, et al. Leucocyte-derived extracellular trap formation significantly contributes to Haemonchus contortus larval entrapment. Parasit Vectors (2015) 8:607. doi: 10.1186/s13071-015-1219-1
67. Ohta N, Ueki S, Konno Y, Hirokawa M, Kubota T, Tomioka-Matsutani S, et al. ETosis-derived DNA trap production in middle ear effusion is a common feature of eosinophilic otitis media. Allergol Int. (2018) 67:414–6. doi: 10.1016/j.alit.2017.11.007
68. Mukherjee M, Bulir DC, Radford K, Kjarsgaard M, Huang CM, Jacobsen EA, et al. Sputum autoantibodies in patients with severe eosinophilic asthma. J Allergy Clin Immunol. (2018) 141:1269–79. doi: 10.1016/j.jaci.2017.06.033
69. Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med. (2008) 14:949–53. doi: 10.1038/nm.1855
70. Morshed M, Yousefi S, Stockle C, Simon HU, Simon D. Thymic stromal lymphopoietin stimulates the formation of eosinophil extracellular traps. Allergy (2012) 67:1127–37. doi: 10.1111/j.1398-9995.2012.02868.x
71. Persson C, Ueki S. Lytic eosinophils produce extracellular DNA traps as well as free eosinophil granules. J Allergy Clin Immunol. (2018) 141:1164. doi: 10.1016/j.jaci.2017.05.047
72. Peachman KK, Lyles DS, Bass DA. Mitochondria in eosinophils: functional role in apoptosis but not respiration. Proc Natl Acad Sci USA. (2001) 98:1717–22. doi: 10.1073/pnas.98.4.1717
73. Figueiredo RT, Neves JS. Eosinophils in fungal diseases: an overview. J Leukoc Biol. (2018) 104:49–60. doi: 10.1002/JLB.4MR1117-473R
74. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. (2009) 16:1438–44. doi: 10.1038/cdd.2009.96
75. Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. (2011) 21:290–304. doi: 10.1038/cr.2010.150
76. Douda DN, Khan MA, Grasemann H, Palaniyar N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci USA. (2015) 112:2817–22. doi: 10.1073/pnas.1414055112
77. Bruns S, Kniemeyer O, Hasenberg M, Aimanianda V, Nietzsche S, Thywissen A, et al. Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin rod. PLoS Pathog. (2010) 6:e1000873. doi: 10.1371/journal.ppat.1000873
78. Rohm MA, Grimm MJ, D'Auria AC, Almyroudis NG, Segal BH, Urban CF. NADPH oxidase promotes neutrophil extracellular trap formation in pulmonary aspergillosis. Infect Immun. (2014) 82:1766–77. doi: 10.1128/IAI.00096-14
79. Gazendam RP, van Hamme JL, Tool AT, Hoogenboezem M, van den Berg JM, Prins JM, et al. Human neutrophils use different mechanisms to kill Aspergillus fumigatus conidia and hyphae: evidence from phagocyte defects. J Immunol. (2016) 196:1272–83. doi: 10.4049/jimmunol.1501811
80. Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. (2010) 185:7413–25. doi: 10.4049/jimmunol.1000675
81. Gabriel C, McMaster WR, Girard D, Descoteaux A. Leishmania donovani promastigotes evade the antimicrobial activity of neutrophil extracellular traps. J Immunol. (2010) 185:4319–27. doi: 10.4049/jimmunol.1000893
82. Byrd AS, O'Brien XM, Johnson CM, Lavigne LM, Reichner JS. An extracellular matrix-based mechanism of rapid neutrophil extracellular trap formation in response to Candida albicans. J Immunol. (2013) 190:4136–48. doi: 10.4049/jimmunol.1202671
83. Mejia SP, Cano LE, Lopez JA, Hernandez O, Gonzalez A. Human neutrophils produce extracellular traps against Paracoccidioides brasiliensis. Microbiology (2015) 161:1008–17. doi: 10.1099/mic.0.000059
84. Arai Y, Nishinaka Y, Arai T, Morita M, Mizugishi K, Adachi S, et al. Uric acid induces NADPH oxidase-independent neutrophil extracellular trap formation. Biochem Biophys Res Commun. (2014) 443:556–61. doi: 10.1016/j.bbrc.2013.12.007
85. Parker H, Dragunow M, Hampton MB, Kettle AJ, Winterbourn CC. Requirements for NADPH oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J Leukoc Biol. (2012) 92:841–9. doi: 10.1189/jlb.1211601
86. Hosseinzadeh A, Thompson PR, Segal BH, Urban CF. Nicotine induces neutrophil extracellular traps. J Leukoc Biol. (2016) 100:1105–12. doi: 10.1189/jlb.3AB0815-379RR
87. Yoon J, Ponikau JU, Lawrence CB, Kita H. Innate antifungal immunity of human eosinophils mediated by a beta 2 integrin, CD11b. J Immunol. (2008) 181:2907–15. doi: 10.4049/jimmunol.181.4.2907
88. Steele C, Rapaka RR, Metz A, Pop SM, Williams DL, Gordon S, et al. The beta-glucan receptor dectin-1 recognizes specific morphologies of Aspergillus fumigatus. PLoS Pathog. (2005) 1:e42. doi: 10.1371/journal.ppat.0010042
89. van Bruggen R, Drewniak A, Jansen M, van Houdt M, Roos D, Chapel H, et al. Complement receptor 3, not Dectin-1, is the major receptor on human neutrophils for beta-glucan-bearing particles. Mol Immunol. (2009) 47:575–81. doi: 10.1016/j.molimm.2009.09.018
90. Bates ME, Sedgwick JB, Zhu Y, Liu LY, Heuser RG, Jarjour NN, et al. Human airway eosinophils respond to chemoattractants with greater eosinophil-derived neurotoxin release, adherence to fibronectin, and activation of the Ras-ERK pathway when compared with blood eosinophils. J Immunol. (2010) 184:7125–33. doi: 10.4049/jimmunol.0900634
91. Clark K, Simson L, Newcombe N, Koskinen AM, Mattes J, Lee NA, et al. Eosinophil degranulation in the allergic lung of mice primarily occurs in the airway lumen. J Leukoc Biol. (2004) 75:1001–9. doi: 10.1189/jlb.0803391
92. Yamauchi Y, Ueki S, Konno Y, Ito W, Takeda M, Nakamura Y, et al. The effect of hepatocyte growth factor on secretory functions in human eosinophils. Cytokine (2016) 88:45–50. doi: 10.1016/j.cyto.2016.08.013
93. Van Dyken SJ, Garcia D, Porter P, Huang X, Quinlan PJ, Blanc PD, et al. Fungal chitin from asthma-associated home environments induces eosinophilic lung infiltration. J Immunol. (2011) 187:2261–7. doi: 10.4049/jimmunol.1100972
94. O'Dea EM, Amarsaikhan N, Li H, Downey J, Steele E, Van Dyken SJ, et al. Eosinophils are recruited in response to chitin exposure and enhance Th2-mediated immune pathology in Aspergillus fumigatus infection. Infect Immun. (2014) 82:3199–205. doi: 10.1128/IAI.01990-14
95. Van Dyken SJ, Mohapatra A, Nussbaum JC, Molofsky AB, Thornton EE, Ziegler SF, et al. Chitin activates parallel immune modules that direct distinct inflammatory responses via innate lymphoid type 2 and gammadelta T cells. Immunity (2014) 40:414–24. doi: 10.1016/j.immuni.2014.02.003
96. Short KR, von Kockritz-Blickwede M, Langereis JD, Chew KY, Job ER, Armitage CW, et al. Antibodies mediate formation of neutrophil extracellular traps in the middle ear and facilitate secondary pneumococcal otitis media. Infect Immun. (2014) 82:364–70. doi: 10.1128/IAI.01104-13
97. Narayana Moorthy A, Narasaraju T, Rai P, Perumalsamy R, Tan KB, Wang S, et al. In vivo and in vitro studies on the roles of neutrophil extracellular traps during secondary pneumococcal pneumonia after primary pulmonary influenza infection. Front Immunol. (2013) 4:56. doi: 10.3389/fimmu.2013.00056
98. de Buhr N, Neumann A, Jerjomiceva N, von Kockritz-Blickwede M, Baums CG. Streptococcus suis DNase SsnA contributes to degradation of neutrophil extracellular traps (NETs) and evasion of NET-mediated antimicrobial activity. Microbiology (2014) 160:385–95. doi: 10.1099/mic.0.072199-0
99. Berends ET, Horswill AR, Haste NM, Monestier M, Nizet V, von Kockritz-Blickwede M. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J Innate Immun. (2010) 2:576–86. doi: 10.1159/000319909
100. McCormick A, Heesemann L, Wagener J, Marcos V, Hartl D, Loeffler J, et al. NETs formed by human neutrophils inhibit growth of the pathogenic mold Aspergillus fumigatus. Microbes Infect. (2010) 12:928–36. doi: 10.1016/j.micinf.2010.06.009
101. Springer DJ, Ren P, Raina R, Dong Y, Behr MJ, McEwen BF, et al. Extracellular fibrils of pathogenic yeast Cryptococcus gattii are important for ecological niche, murine virulence and human neutrophil interactions. PLoS ONE (2010) 5:e10978. doi: 10.1371/journal.pone.0010978
102. Ravin KA, Loy M. The eosinophil in infection. Clin Rev Allergy Immunol. (2016) 50:214–27. doi: 10.1007/s12016-015-8525-4
103. Lilly LM, Scopel M, Nelson MP, Burg AR, Dunaway CW, Steele C. Eosinophil deficiency compromises lung defense against Aspergillus fumigatus. Infect Immun. (2014) 82:1315–25. doi: 10.1128/IAI.01172-13
104. Hogaboam CM, Blease K, Mehrad B, Steinhauser ML, Standiford TJ, Kunkel SL, et al. Chronic airway hyperreactivity, goblet cell hyperplasia, and peribronchial fibrosis during allergic airway disease induced by Aspergillus fumigatus. Am J Pathol. (2000) 156:723–32. doi: 10.1016/S0002-9440(10)64775-X
105. Kita H. Eosinophils: multifaceted biological properties and roles in health and disease. Immunol Rev. (2011) 242:161–77. doi: 10.1111/j.1600-065X.2011.01026.x
106. King M. Experimental models for studying mucociliary clearance. Eur Respir J. (1998) 11:222–8. doi: 10.1183/09031936.98.11010222
107. Vlaminck S, Vauterin T, Hellings PW, Jorissen M, Acke F, Van Cauwenberge P, et al. The importance of local eosinophilia in the surgical outcome of chronic rhinosinusitis: a 3-year prospective observational study. Am J Rhinol Allergy (2014) 28:260–4. doi: 10.2500/ajra.2014.28.4024
108. Healy DY, Leid JG, Sanderson AR, Hunsaker DH. Biofilms with fungi in chronic rhinosinusitis. Otolaryngol Head Neck Surg. (2008) 138:641–7. doi: 10.1016/j.otohns.2008.02.002
109. Dunican EM, Elicker BM, Gierada DS, Nagle SK, Schiebler ML, Newell JD, et al. Mucus plugs in patients with asthma linked to eosinophilia and airflow obstruction. J Clin Invest. (2018) 128:997–1009. doi: 10.1172/JCI95693
110. Uller L, Persson CG, Kallstrom L, Erjefalt JS. Lung tissue eosinophils may be cleared through luminal entry rather than apoptosis: effects of steroid treatment. Am J Respir Crit Care Med. (2001) 164:1948–56. doi: 10.1164/ajrccm.164.10.2011135
111. Neves JS, Perez SA, Spencer LA, Melo RC, Reynolds L, Ghiran I, et al. Eosinophil granules function extracellularly as receptor-mediated secretory organelles. Proc Natl Acad Sci USA. (2008) 105:18478–83. doi: 10.1073/pnas.0804547105
Keywords: eosinophil, extracellular traps, extracellular trap cell death, allergic bronchopulmonary aspergillosis, inflammation, mucus plugs, ETosis, NETosis
Citation: Ueki S, Hebisawa A, Kitani M, Asano K and Neves JS (2018) Allergic Bronchopulmonary Aspergillosis–A Luminal Hypereosinophilic Disease With Extracellular Trap Cell Death. Front. Immunol. 9:2346. doi: 10.3389/fimmu.2018.02346
Received: 14 June 2018; Accepted: 21 September 2018;
Published: 11 October 2018.
Edited by:
Uday Kishore, Brunel University London, United KingdomReviewed by:
Lakshna Mahajan, University of Delhi, IndiaCopyright © 2018 Ueki, Hebisawa, Kitani, Asano and Neves. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shigeharu Ueki, c2hpZ2VoYXJ1LnVla2lAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.