 Ifrah Sohail
Ifrah Sohail Santhosh Mukundan
Santhosh Mukundan Susan Zelewski
Susan Zelewski M. Nadeem Khan
M. Nadeem Khan- Biomedical Sciences, University of North Dakota, Grand Forks, ND, United States
Streptococcus pneumoniae (Spn) is a colonizer of the human nasopharynx (NP), causing a variety of infections in humans including otitis media, pneumonia, sepsis, and meningitis. The NP is an immune permissive site which allows for the persistence of commensal bacteria. Acute or chronic respiratory airway inflammation constitutes a significant risk factor for the manifestation of Spn infections. The inflammatory conditions caused by an upper respiratory viral infection or respiratory conditions such as allergic asthma and chronic obstructive pulmonary disorders (COPDs) are implicated in the dysregulation of airway inflammation and tissue damage, which compromise the respiratory barrier integrity. These immune events promote bacterial outgrowth leading to Spn dissemination and invasion into the bloodstream. Therefore, suppression of inflammation and restoration of respiratory barrier integrity could contain Spn infections manifesting in the backdrop of an inflammatory disease condition. The gained knowledge could be harnessed in the design of novel therapeutic interventions to circumvent Spn bacterial infections.
Spn Carriage, Diseases, and Vaccines
Spn diseases constitute a major global health problem (1). It is estimated that about one million US adults contract Spn pneumonia each year, which accounts for 5–7% of annual mortality (2), and the mortality rate for invasive Spn diseases (IPDs; sepsis and meningitis) is even higher (3). The vulnerable human populations who are at the highest risk of developing Spn infections include infants, the elderly, and immunocompromised patients (4, 5); thus, these populations have a much higher incidence of IPDs (6). Currently available pneumococcal conjugate vaccines (PCVs) target the bacterial capsule antigens and have contributed to a global reduction in the Spn disease burden (7). However, the replacement of capsular serotypes has occurred (8, 9), thus requiring an expansion of the valency of current PCV formulations. Therefore, despite continued vaccination programs, Spn infections continue to occur and account for significant clinical and economic burden.
Spn establishes asymptomatic carriage in the NP of about 20–40% of healthy adults, with even higher frequency in infant and elderly populations (10). Since carriage is a prerequisite for Spn infections and diseases (11, 12), at-risk populations are more frequently and persistently colonized by Spn (13, 14). The transition of carriage into disease depends on multiple risk factors such as age, inflammatory conditions, geographical area, socio-economic conditions, genetics, and immune system. Additionally, carriage is associated with bacterial dissemination, which leads to the widespread acquisition of Spn bacteria in the community (14). However, carriage serves as a double-edged sword: while it constitutes an indispensable state for Spn infections, carriage also activates innate and adaptive immunity in the NP, leading to the generation of a Spn antigen-specific protective immune response against colonizing Spn serotypes (15–18). In mouse models, Spn carriage has been shown to trigger a mild inflammatory event resulting in the activation of Toll-Like Receptor-2 (TLR2) and inflammasome (16, 19). Additionally, Spn carriage develops antigen-specific antibody and T-cell responses in mouse and human experimental carriage models (20).
Currently available Spn conjugate vaccines protect against invasive diseases by preventing the acquisition of Spn bacterial carriage of the vaccine serotypes (21). PCVs induce anti-capsular opsonic antibodies which lead to the elimination of Spn carriage of vaccine serotypes in the NP (12, 22). However, the elimination of vaccine serotypes has led to the emergence of non-vaccine colonizing and disease-causing replacement serotypes (9, 23). Protein-based Spn vaccines are envisioned to protect against Spn infections in a serotype-independent manner (24, 25). However, protein-based Spn vaccines may or may not eliminate carriage from the NP. Some protein-based Spn vaccines aim to maintain low-density stable carriage instead of eliminating it (22–24). It is expected that the maintenance of low-density carriage will limit bacterial virulence evolution, and, consequently, could contain the emergence of new capsule serotypes (24). However, advanced infection models and a substantial amount of data are required to develop this concept further and understand the effect of carriage persistence in the at-risk populations. It should be noted that carriage persistence and its beneficial aspects may differ from one Spn serotype to another, since Spn has a vast serotype repertoire with diverse colonization kinetics in humans and mouse models, and some invasive serotypes are less effective colonizers (26, 27). Therefore, further studies are required to establish the duration of stable asymptomatic carriage specific to each serotype. Given the nature of Spn as a bridge between a commensal and a pathogen, these factors are essential to consider for the development of advanced vaccination or therapeutic strategies that favor the retention of low-density Spn carriage in the NP. The answers to these questions necessitate the development of advanced animal infection models that mimic the natural Spn infections.
Respiratory Inflammation and Spn Infections
Respiratory conditions involving dysregulated and damaging airway inflammation constitute a significant risk factor for the commencement of Spn bacterial infections (28–34). While baseline inflammation is required for the gradual clearance of carriage in the NP, hyper and dysregulated inflammation is implicated in epithelial inflammation and tissue damage, which compromises airway barrier integrity and promotes Spn outgrowth and dissemination to sterile tissues (34–36). Therefore, a carefully orchestrated inflammatory response is required for the resolution of airway bacterial infections (37, 38), and defects in the regulation of inflammation that arise from respiratory conditions become a contributor to tissue pathology, thus leading to an increased permissiveness for Spn infections (Figure 1).
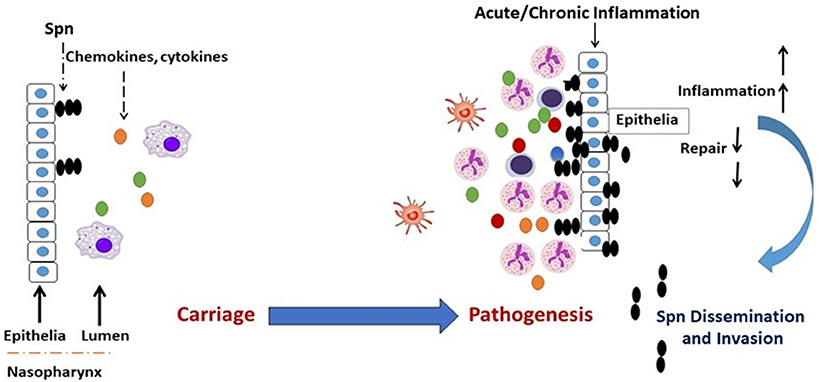
Figure 1. Transition of Spn commensal colonization to disease. Acute or chronic airway inflammation caused by influenza virus or chronic conditions such as COPD or allergic asthma promote Spn bacterial outgrowth and compromises respiratory tissue barrier function, which leads to the invasion of Spn bacteria into the bloodstream establishing invasive infection. Colored dots (green, yellow, red, blue) represent chemokines and cytokines.
Influenza Virus-Induced Airway Inflammation and Spn Infections
Airway co-infection with a respiratory virus constitutes the most significant risk factor for Spn infections (36, 39). Clinically, over 50% of young children (<2 years of age) are colonized by Spn and other otopathogens in the NP, and there is a significant correlation between the development of bacterial acute otitis media (AOM) and pneumonia with viral upper respiratory co-infections (40–42). A prior influenza viral infection in the NP/lung promotes secondary Spn bacterial infections. Similarly, a concurrent viral infection in Spn-colonized hosts results in the transition of commensal low-density Spn carriage into a Spn infection. Traditionally, a majority of murine influenza-Spn co-infection models introduced Spn in influenza-infected hosts, allowing Spn to utilize influenza-mediated airway changes to manifest the disease (43–46). This model system could be used to study immune mechanisms implicated in the development of post-influenza secondary Spn infections. However, since a significant proportion of humans is colonized by Spn and other otopathogens in the NP, the introduction of influenza in Spn-colonized mice also mimics the natural Spn infections (47). The latter infection condition is more appropriate to study the transition from commensal colonizer to disease-causing Spn during an influenza viral co-infection (Figure 1). Therefore, both of the aforementioned experimental models should be used to investigate the role of influenza virus in the manifestation of Spn infections.
Airway co-infection is characterized by complex interactions between co-infecting pathogens and the host, leading to the disruption of physical barriers, dysregulation of immune responses, and delays in a return to homeostasis (48, 49). Respiratory viruses such as influenza, RSV, parainfluenza, and adenovirus promote Spn diseases in the setting of a co-infection (40, 50–54). Influenza virus replication in alveolar epithelia promotes the recruitment of CCR2hi inflammatory monocytes (IMs) and upregulates receptors for bacterial adherence (55–57). IMs act as a double-edged sword; they are required for viral resolution but are implicated in first-line pulmonary epithelial and tissue damage, leading to viral spread (56, 58). CCR2-deficient mice (CCR2−/−) lacking monocytes and monocyte-derived cells significantly limit lung tissue damage (59). Additionally, CCR2−/− co-infected (influenza-Spn) mice show reduced epithelial damage and are able to control Spn invasion (55). Alveolar macrophages (AMs) are resident phagocytic cells that provide the first line of defense against bacterial infections in the lungs (60). AMs are phagocytic, anti-inflammatory cells which maintain lung sterility and control pulmonary inflammation by preventing an influx of neutrophils during the early infection stage (61). An influenza infection promotes the apoptosis of AMs, leading to an increased permissiveness for Spn bacterial infections (43). The restoration of AMs in influenza-infected lungs confers resistance to Spn disease (62). Neutrophils are potent phagocytic cells with known anti-bacterial functions (63), and they massively accumulate in influenza and influenza-Spn co-infected mice. Prior reports suggest an impairment of neutrophil phagocytosis and intracellular reactive oxygen production in influenza-infected mice (64, 65), and neutrophil depletion was shown to be associated with an increased severity for Spn lung infection (64). However, dysregulated neutrophil recruitment is also associated with damage and pathology in infected tissues (66). While neutrophils have been shown to have a protective function in single Spn infections (67), their role in influenza-Spn co-infections remains unclear.
The levels of lung TNF-α elevate during a Spn-influenza co-infection, and its depletion exacerbates Spn infection (55). However, TNF-α responses are severely impaired during the early phase of co-infections (55), leading to a failure to control bacterial outgrowth and Spn infection. IL-6 limits influenza-induced inflammation and lung pathology as IL-6-deficient mice exhibited higher fibroblast accumulation, a lower extracellular matrix (ECM) turnover, and higher mortality (68). The critical role of IL-17 receptor (IL-17RA) signaling has been shown in acute immunopathology of influenza-infected lungs, as IL-17RA knockout mice had reduced tissue damage, reduced neutrophil numbers, and increased survival (69). Mucosal pre-exposure to Th17-inducing adjuvants have been shown to exacerbate pathology after an influenza infection (70), further establishing the role of IL-17 responses in influenza acute pathology. Therefore, an influenza infection promotes damaging inflammation in the airway, leading to permissiveness for Spn bacterial infections. A better understanding of the immune mediators implicated in influenza-mediated tissue pathology could lead to the development of therapeutic interventions to contain influenza pathology and subsequent Spn infections.
Chronic Inflammation and Spn Infections
Spn causes significant morbidity and mortality worldwide in patients with chronic respiratory diseases, and vaccination is the most significant tool to contain Spn diseases among allergy and COPD patients (71). Several recent studies have shown a clear association between acute exacerbation and the isolation of bacterial species such as Spn, Moraxella catarrhalis, and Haemophilus influenzae, among the most frequently associated bacterial organisms (72). Consequently, Centers for Disease Control and Prevention (CDC) recommends the use of influenza and Spn vaccines (PCVs) in people with COPD and asthma conditions (71).
Allergy, Asthma Airway Pathology, and Spn Infections
Asthma is a complex chronic inflammatory disease characterized by airway hyperresponsiveness, reversible airflow obstruction, and airway inflammation (73). The most common trigger for asthma is the continuous exposure to allergens, of which fungal agents are important factors (74, 75), and there is evidence for the presence of fungal sensitization in patients with asthma (76). Fungal asthma promotes a Th2-type response that mediates the production of cytokines such as IL-4, IL-5, IL-13, and IL-33 (77, 78), which leads to the recruitment of complex multi-factorial leukocyte eosinophils (79, 80). In murine models, Th2 cells play an important role in eosinophilic inflammation in fungal allergic asthma. Epithelial cell-derived cytokines such as thymic stromal lymphoprotein (TSLP), IL-25, and IL-33 promote eosinophilia by inducing IL-5 production (81). Additionally, dysregulation of the IL-17F/IL-17RC axis has been shown to predispose allergic inflammation in murine models of fungal allergic asthma (82). Besides murine models of fungal allergic asthma, mouse models using the house dust mite (HDM) also feature similarities to human allergic asthma, including eosinophilic lung inflammation and cytokines primarily associated with Th2-type inflammation (83). These animal models are used for evaluating the efficacy of anti-asthma drugs and exacerbations of bacterial/viral infections.
Allergic asthma consists of diverse immune phenotypes exhibiting differential lung pathology, remodeling of the respiratory tract, and mucociliary bronchial clearance (75, 84, 85). These confounding inflammatory immune mechanisms may promote or resist microbial infections in allergic human and animal infection models. Eosinophils are generally considered as major effector cells of asthma, and the eosinophilic response to viral infection has predominantly been shown to have a negative effect on human health (86). However, several studies have shown eosinophils may promote viral clearance and antiviral host defense. A recent study has shown that pulmonary eosinophilia linked with fungal allergic respiratory inflammation promotes antiviral host defenses against the influenza virus by promoting CD8+ T cell immunity (87). However, other studies have shown that viral infections exacerbate asthma and thereby promote bacterial infections (88). Since, influenza infections are an important determining factor for the development and exacerbation of Spn infections and a diverse spectrum of influenza strains cause infections in humans, animal models of influenza-Spn co-infections utilizing different influenza strains are required to study the role of asthma inflammation in influenza-dependent Spn diseases. The relationship between allergic airway inflammation and Spn pneumonia is not well understood. Recent studies report that asthma is associated with an increased risk of invasive Spn diseases (89, 90), and higher rates of Spn NP colonization, sinusitis, and otitis media have also been reported among individuals with asthma (91, 92). Additionally, in vivo model systems demonstrated that allergic airway inflammation was associated with a trend toward increased extrapulmonary Spn infections, highlighting the role of allergic airway inflammation in the development of invasive Spn diseases. Further studies that mimic diverse features of asthma phenotypes are needed to elucidate the precise mechanisms of the inflammatory response to Spn infections and the effectiveness of Spn vaccination in patients with asthma.
COPD Airway Pathology and Spn Infections
COPD is a heterogeneous entity that includes a variety of obstructive diseases that differ considerably on their mechanisms of action and responses to treatment. COPD is characterized by smoke-induced hypersecretion of mucus and emphysema in the pulmonary gas exchanging units (93). COPD and asthma are two distinct diseases with significantly different mechanisms of chronic inflammatory reactions (94). In COPD, the inflammation-associated changes are demonstrated predominantly in small airways and lung parenchyma, and result in tissue destruction with progressive, irreversible airflow restriction. The main changes in asthma are found in larger airways and may cause their intermittent and usually reversible obstruction (95, 96). Airway and alveolar epithelial cells play a central role in COPD inflammation (93). Epithelial cells are the primary source of proteolytic enzymes and chemoattractants responsible for the recruitment of immune effector cells implicated in the amplification and persistence of airway inflammation (93), and nitric oxide has been shown to be the most representative biomarker in exhaled air, originated from respiratory epithelium (97). Dendritic cells and macrophages are key innate immune effector cells in COPD inflammation (98). Activated macrophages and dendritic cells release inflammatory cytokines implicated in COPD pathology, such as TNF-α, IL-6, IL-8, IL-1β, and TGF-β (93, 98). Recent studies also implicate the pathogenic role of IL-17, since IL-17RA-deficient mice were protected from airway inflammation and fibrosis in COPD-like models (99). Additionally, lymphocytes and neutrophil recruitment in the inflamed airway/lungs further promote airway damage (61). The persistent inflammation in COPD allows for pathological airway and vascular remodeling, which involves the deposition of an extracellular matrix in sub-epithelial layers and hypertrophy of smooth muscles, leading to the thickening of airway walls and the narrowing of bronchial lumen. Therefore, COPD-mediated pulmonary pathology compromises tissue barrier integrity, which leads to an increased permissiveness for bacterial infections (100).
Spn plays an important role in causing acute exacerbations in patients with chronic respiratory diseases (101). A number of Spn serotypes have shown to be associated with colonization and exacerbations in COPD patients (72, 101–103). Currently available Spn vaccines could cover almost 50% of the Spn serotypes in COPD patients (72). However, due to the evolution of non-vaccine Spn serotypes and the limitation of currently available Spn vaccines, understanding the role of COPD inflammation in the development of Spn diseases is central to designing an intervention to prevent Spn colonization and diseases. Pulmonary inflammation, mucociliary dysfunction, and airway remodeling are central components of COPD-mediated immune pathology, implicated in the permissiveness of airway infections (104). The tracheobronchial mucilliary movement is central to maintaining the sterility of the lower respiratory tract by transporting bacteria trapped in mucus toward the pharynx. COPD causes mucociliary dysfunction, thus results in the promotion of persistent airway bacterial colonization (105, 106). Therefore, anti-inflammatory therapies intended to improve mucociliary clearance have the potential to modulate a pulmonary defense against microbial pathogens.
Conclusion
Despite currently available vaccines, Spn continues to colonize human populations, and Spn carriage thus represents a significant medical problem. Airway tissue damage and hyper-inflammation is one of the most significant risk factors for the transition of Spn carriage into a disease. An acute or chronic inflammatory condition caused by airway viral infection or conditions such as COPD and asthma are major risk factors for the manifestation of Spn diseases, and CDC recommends the use of seasonal influenza and Spn vaccines in these human populations. A carefully orchestrated inflammatory response is required for the resolution of airway bacterial infections, and defects in the regulation of inflammation that arise from respiratory conditions become a contributor to tissue pathology, thus leading to an increased permissiveness for Spn bacterial infections. Traditionally, a majority of murine infection models employed in the investigating of Spn diseases typically used Spn single infection conditions. These mice models used infection inocula in low or high volumes intended to develop colonization or a pneumonia infection. However, a significant shift has occurred with the development of mouse infection models mimicking the natural infections of Spn bacteria. Influenza-Spn co-infection models are increasingly being used to study the role of viral-induced acute airway inflammation in Spn infections. Similarly, mouse models of chronic inflammation such as allergy or COPD provide a platform to study Spn infections in conditions similar to natural bacterial infections. Therefore, the utilization of risk factors in murine infection models will lead to a better understanding of the dynamics of microbial interplay and the inflammatory mechanisms implicated in Spn infections. The knowledge gained could be harnessed in the design of novel therapeutic interventions to circumvent Spn bacterial infections in at-risk human populations.
Author Contributions
MK conceived the idea, drafted the outline of the article, and supervised through all stages of its preparation. IS collected the review literature and participated in the writing of first draft. SG, SM and SZ participated in the writing of first draft.
Funding
The study was supported by NIH Center of Biomedical Research Excellence (CoBRE), Department of Biomedical Sciences, University of North Dakota.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Richmond P. Vaccines for other neonatal infections: pneumococcal disease: a major global health problem of young children. Expert Rev Vaccin. (2004) 3:379–82. doi: 10.1586/14760584.3.4.379
2. Drijkoningen JJ, and Rohde GG. (2014). Pneumococcal infection in adults: burden of disease. Clin Microbiol Infect. 20 (Suppl. 5):45–51. doi: 10.1111/1469-0691.12461
3. Moore JX, Donnelly JP, Griffin R, Howard G, Safford MM, Wang HE. Defining sepsis mortality clusters in the United States. Crit Care Med. (2016) 44:1380–7. doi: 10.1097/CCM.0000000000001665
4. Vanderkooi OG, Church DL, MacDonald J, Zucol F, Kellner JD. Community-based outbreaks in vulnerable populations of invasive infections caused by Streptococcus pneumoniae serotypes 5 and 8 in Calgary, Canada. PLoS ONE 6:e28547. doi: 10.1371/journal.pone.0028547
5. Steenhoff AP, Wood SM, Rutstein RM, Wahl A, McGowan KL, Shah SS. Invasive pneumococcal disease among human immunodeficiency virus-infected children, 1989-2006. Pediatr Infect Dis J. (2008) 27:886–91. doi: 10.1097/INF.0b013e3181734f8f
6. Bhagwanjee S, Ugarte S. Sepsis in vulnerable populations. Glob Heart (2014) 9:281–8. doi: 10.1016/j.gheart.2014.08.008
7. Berical AC, Harris D, Dela Cruz CS, Possick JD. Pneumococcal vaccination strategies. An update and perspective. Ann Am Thorac Soc. (2016) 13:933–44. doi: 10.1513/AnnalsATS.201511-778FR
8. Croucher NJ, Finkelstein JA, Pelton SI, Mitchell PK, Lee GM, Parkhill J, et al. Population genomics of post-vaccine changes in pneumococcal epidemiology. Nat Genet. (2013) 45:656–63. doi: 10.1038/ng.2625
9. Croucher NJ, Harris SR, Fraser C, Quail MA, Burton J, van der Linden M, et al. Rapid pneumococcal evolution in response to clinical interventions. Science (2011) 331:430–4. doi: 10.1126/science.1198545
10. Lijek RS, Weiser JN. Co-infection subverts mucosal immunity in the upper respiratory tract. Curr Opin Immunol. (2012) 24:417–23. doi: 10.1016/j.coi.2012.05.005
11. Bogaert D, De Groot R, Hermans PW. Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect Dis. (2004) 4:144–54. doi: 10.1016/S1473-3099(04)00938-7
12. Simell B, Auranen K, Kayhty H, Goldblatt D, Dagan R, O'Brien KL, et al. The fundamental link between pneumococcal carriage and disease. Expert Rev Vaccin. (2012) 11:841–55. doi: 10.1586/erv.12.53
13. Weinberger DM, Grant LR, Weatherholtz RC, Warren JL, O'Brien KL, Hammitt LL. Relating pneumococcal carriage among children to disease rates among adults before and after the introduction of conjugate vaccines. Am J Epidemiol. (2016) 183:1055–62. doi: 10.1093/aje/kwv283
14. Sa-Leao R, Nunes S, Brito-Avo A, Alves CR, Carrico JA, Saldanha J, et al. High rates of transmission of and colonization by Streptococcus pneumoniae and Haemophilus influenzae within a day care center revealed in a longitudinal study. J Clin Microbiol. (2008) 46:225–34. doi: 10.1128/JCM.01551-07
15. Richards L, Ferreira DM, Miyaji EN, Andrew PW, Kadioglu A. The immunising effect of pneumococcal nasopharyngeal colonisation; protection against future colonisation and fatal invasive disease. Immunobiology (2010) 215:251–63. doi: 10.1016/j.imbio.2009.12.004
16. McNeela EA, Burke A, Neill DR, Baxter C, Fernandes VE, Ferreira D, et al. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog. 6:e1001191. doi: 10.1371/journal.ppat.1001191
17. Kadioglu A, Coward W, Colston MJ, Hewitt CR, Andrew PW. CD4-T-lymphocyte interactions with pneumolysin and pneumococci suggest a crucial protective role in the host response to pneumococcal infection. Infect Immun. (2004) 72:2689–97. doi: 10.1128/IAI.72.5.2689-2697.2004
18. Khan MN, Pichichero ME. The host immune dynamics of pneumococcal colonization: implications for novel vaccine development. Hum Vaccin Immunother. (2014) 10:3688–99. doi: 10.4161/21645515.2014.979631
19. Richard AL, Siegel SJ, Erikson J, Weiser JN. TLR2 signaling decreases transmission of Streptococcus pneumoniae by limiting bacterial shedding in an infant mouse Influenza A co-infection model. PLoS Pathog. 10:e1004339. doi: 10.1371/journal.ppat.1004339
20. Gritzfeld JF, Wright AD, Collins AM, Pennington SH, Wright AK, Kadioglu A, et al. Experimental human pneumococcal carriage. J Vis Exp. (2013) 72:1–5. doi: 10.3791/50115
21. Jochems SP, Weiser JN, Malley R, Ferreira DM. The immunological mechanisms that control pneumococcal carriage. PLoS Pathog. (2017) 13:e1006665. doi: 10.1371/journal.ppat.1006665
22. Song JY, Moseley MA, Burton RL, Nahm MH. Pneumococcal vaccine and opsonic pneumococcal antibody. J Infect Chemother. (2013) 19:412–25. doi: 10.1007/s10156-013-0601-1
23. Pichichero ME, Casey JR. Emergence of a multiresistant serotype 19A pneumococcal strain not included in the 7-valent conjugate vaccine as an otopathogen in children. JAMA (2007) 298:1772–8. doi: 10.1001/jama.298.15.1772
24. Pichichero ME, Khan MN, Xu Q. Next generation protein based Streptococcus pneumoniae vaccines. Hum Vaccin Immunother. (2016) 12:194–205. doi: 10.1080/21645515.2015.1052198
25. Pichichero ME. Pneumococcal whole-cell and protein-based vaccines: changing the paradigm. Expert Rev Vaccin. (2017) 16:1181–90. doi: 10.1080/14760584.2017.1393335
26. Khan MN, Coleman JR, Vernatter J, Varshney AK, Dufaud C, Pirofski LA. An ahemolytic pneumolysin of Streptococcus pneumoniae manipulates human innate and CD4+ T-cell responses and reduces resistance to colonization in mice in a serotype-independent manner. J Infect Dis. (2014) 210:1658–69. doi: 10.1093/infdis/jiu321
27. Tigoi CC, Gatakaa H, Karani A, Mugo D, Kungu S, Wanjiru E, et al. Rates of acquisition of pneumococcal colonization and transmission probabilities, by serotype, among newborn infants in Kilifi District, Kenya. Clin Infect Dis. (2012) 55:180–8. doi: 10.1093/cid/cis371
28. Feemster KA, Li Y, Localio AR, Shults J, Edelstein P, Lautenbach E, et al. Risk of invasive pneumococcal disease varies by neighbourhood characteristics: implications for prevention policies. Epidemiol Infect. (2013) 141:1679–89. doi: 10.1017/S095026881200235X
29. Kerr AR, Irvine JJ, Search JJ, Gingles NA, Kadioglu A, Andrew PW, et al. Role of inflammatory mediators in resistance and susceptibility to pneumococcal infection. Infect Immun. (2002) 70:1547–57. doi: 10.1128/IAI.70.3.1547-1557.2002
30. Adler H, Ferreira DM, Gordon SB, Rylance J. Pneumococcal capsular polysaccharide immunity in the elderly. Clin Vaccin Immunol. (2017) 24:e00004–17. doi: 10.1128/CVI.00004-17
31. Mosser JF, Grant LR, Millar EV, Weatherholtz RC, Jackson DM, Beall B, et al. Nasopharyngeal carriage and transmission of Streptococcus pneumoniae in American Indian households after a decade of pneumococcal conjugate vaccine use. PLoS ONE 9:e79578. doi: 10.1371/journal.pone.0079578
32. Abdullahi O, Nyiro J, Lewa P, Slack M, Scott JA. The descriptive epidemiology of Streptococcus pneumoniae and Haemophilus influenzae nasopharyngeal carriage in children and adults in Kilifi district, Kenya. Pediatr Infect Dis J. (2008) 27:59–64. doi: 10.1097/INF.0b013e31814da70c
33. Esposito S, Mari D, Bergamaschini L, Orenti A, Terranova L, Ruggiero L, et al. Pneumococcal colonization in older adults. Immun Ageing (2016) 13:2. doi: 10.1186/s12979-016-0057-0
34. Short KR, Reading PC, Wang N, Diavatopoulos DA, Wijburg OL. Increased nasopharyngeal bacterial titers and local inflammation facilitate transmission of Streptococcus pneumoniae. MBio (2012) 3:e00255-12. doi: 10.1128/mBio.00255-12
35. Nakamura S, Davis KM, Weiser JN. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J Clin Invest. (2011) 121:3657–65. doi: 10.1172/JCI57762
36. Cauley LS, Vella AT. Why is coinfection with influenza virus and bacteria so difficult to control? Discov Med. (2015) 19:33–40.
37. Puchta A, Verschoor CP, Thurn T, and Bowdish DM. Characterization of inflammatory responses during intranasal colonization with Streptococcus pneumoniae. J Vis Exp. (2014):e50490. doi: 10.3791/50490
38. Ferreira DM, Neill DR, Bangert M, Gritzfeld JF, Green N, Wright AK, et al. Controlled human infection and rechallenge with Streptococcus pneumoniae reveals the protective efficacy of carriage in healthy adults. Am J Respir Crit Care Med. (2013) 187:855–64. doi: 10.1164/rccm.201212-2277OC
39. Wolter N, Tempia S, Cohen C, Madhi SA, Venter M, Moyes J, et al. High nasopharyngeal pneumococcal density, increased by viral coinfection, is associated with invasive pneumococcal pneumonia. J Infect Dis. (2014) 210:1649–57. doi: 10.1093/infdis/jiu326
40. Pettigrew MM, Gent JF, Pyles RB, Miller AL, Nokso-Koivisto J, Chonmaitree T. Viral-bacterial interactions and risk of acute otitis media complicating upper respiratory tract infection. J Clin Microbiol. (2011) 49:3750–5. doi: 10.1128/JCM.01186-11
41. Marom T, Nokso-Koivisto J, Chonmaitree T. Viral-bacterial interactions in acute otitis media. Curr Allergy Asthma Rep. (2012) 12:551–8. doi: 10.1007/s11882-012-0303-2
42. Xu Q, Wischmeyer J, Gonzalez E, Pichichero ME. Nasopharyngeal polymicrobial colonization during health, viral upper respiratory infection and upper respiratory bacterial infection. J Infect. (2017) 75:26–34. doi: 10.1016/j.jinf.2017.04.003
43. Ghoneim HE, Thomas PG, McCullers JA. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. J Immunol. (2013) 191:1250–9. doi: 10.4049/jimmunol.1300014
44. Robinson KM, Lee B, Scheller EV, Mandalapu S, Enelow RI, Kolls JK, et al. The role of IL-27 in susceptibility to post-influenza Staphylococcus aureus pneumonia. Respir Res. (2015) 16:10. doi: 10.1186/s12931-015-0168-8
45. Smith AM, Adler FR, Ribeiro RM, Gutenkunst RN, McAuley JL, McCullers JA, et al. Kinetics of coinfection with influenza A virus and Streptococcus pneumoniae. PLoS Pathog 9:e1003238. doi: 10.1371/journal.ppat.1003238
46. Sharma-Chawla N, Sender V, Kershaw O, Gruber AD, Volckmar J, Henriques-Normark B, et al. Influenza A virus infection predisposes hosts to secondary infection with different Streptococcus pneumoniae serotypes with similar outcome but serotype-specific manifestation. Infect Immun. (2016) 84:3445–57. doi: 10.1128/IAI.00422-16
47. Khan MN, Xu Q, Pichichero ME. Protection against Streptococcus pneumoniae invasive pathogenesis by a protein-based vaccine is achieved by suppression of nasopharyngeal bacterial density during influenza A virus coinfection. Infect Immun. (2017) 85:e00530–16. doi: 10.1128/IAI.00530-16
48. Damjanovic D, Divangahi M, Kugathasan K, Small CL, Zganiacz A, Brown EG, et al. Negative regulation of lung inflammation and immunopathology by TNF-alpha during acute influenza infection. Am J Pathol. (2011) 179:2963–76. doi: 10.1016/j.ajpath.2011.09.003
49. Robinson KM, Kolls JK, Alcorn JF. The immunology of influenza virus-associated bacterial pneumonia. Curr Opin Immunol. (2015) 34:59–67. doi: 10.1016/j.coi.2015.02.002
50. McCullers JA. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat Rev Microbiol. (2014) 12:252–62. doi: 10.1038/nrmicro3231
51. Avadhanula V, Rodriguez CA, Devincenzo JP, Wang Y, Webby RJ, Ulett GC, et al. Respiratory viruses augment the adhesion of bacterial pathogens to respiratory epithelium in a viral species- and cell type-dependent manner. J Virol. (2006) 80:1629–36. doi: 10.1128/JVI.80.4.1629-1636.2006
52. Verhoeven D, Xu Q, Pichichero ME. Differential impact of respiratory syncytial virus and parainfluenza virus on the frequency of acute otitis media is explained by lower adaptive and innate immune responses in otitis-prone children. Clin Infect Dis. (2014) 59:376–83. doi: 10.1093/cid/ciu303
53. Hament JM, Kimpen JL, Fleer A, Wolfs TF. Respiratory viral infection predisposing for bacterial disease: a concise review. FEMS Immunol Med Microbiol. (1999) 26:189–95. doi: 10.1111/j.1574-695X.1999.tb01389.x
54. Murrah KA, Turner RL, Pang B, Perez AC, Reimche JL, King LB, et al. Replication of type 5 adenovirus promotes middle ear infection by Streptococcus pneumoniae in the chinchilla model of otitis media. Pathog Dis. (2015) 73:1–8. doi: 10.1111/2049-632X.12216
55. Ellis GT, Davidson S, Crotta S, Branzk N, Papayannopoulos V, Wack A. TRAIL+ monocytes and monocyte-related cells cause lung damage and thereby increase susceptibility to influenza-Streptococcus pneumoniae coinfection. EMBO Rep. (2015) 16:1203–18. doi: 10.15252/embr.201540473
56. Lin SJ, Lo M, Kuo RL, Shih SR, Ojcius DM, Lu J, et al. The pathological effects of CCR2+ inflammatory monocytes are amplified by an IFNAR1-triggered chemokine feedback loop in highly pathogenic influenza infection. J Biomed Sci. (2014) 21:99. doi: 10.1186/s12929-014-0099-6
57. McCullers JA. Insights into the interaction between influenza virus and pneumococcus. Clin Microbiol Rev. (2006) 19:571–82. doi: 10.1128/CMR.00058-05
58. Monton C, Torres A. Lung inflammatory response in pneumonia. Monaldi Arch Chest Dis. (1998) 53:56–63.
59. Lin KL, Sweeney S, Kang BD, Ramsburg E, Gunn MD. CCR2-antagonist prophylaxis reduces pulmonary immune pathology and markedly improves survival during influenza infection. J Immunol. (2011) 186:508–15. doi: 10.4049/jimmunol.1001002
60. Goldstein E, Lippert W, Warshauer D. Pulmonary alveolar macrophage. Defender against bacterial infection of the lung. J Clin Invest. (1974) 54:519–28. doi: 10.1172/JCI107788
61. Moldoveanu B, Otmishi P, Jani P, Walker J, Sarmiento X, Guardiola J, et al. Inflammatory mechanisms in the lung. J Inflamm Res. (2009) 2:1–11.
62. Bansal S, Yajjala VK, Bauer C, Sun K. IL-1 signaling prevents alveolar macrophage depletion during influenza and Streptococcus pneumoniae coinfection. J Immunol. (2018) 200:1425–33. doi: 10.4049/jimmunol.1700210
63. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. (2006) 6:173–82. doi: 10.1038/nri1785
64. McNamee LA, Harmsen AG. Both influenza-induced neutrophil dysfunction and neutrophil-independent mechanisms contribute to increased susceptibility to a secondary Streptococcus pneumoniae infection. Infect Immun. (2006) 74:6707–21. doi: 10.1128/IAI.00789-06
65. Engelich G, White M, Hartshorn KL. Neutrophil survival is markedly reduced by incubation with influenza virus and Streptococcus pneumoniae: role of respiratory burst. J Leukoc Biol. (2001) 69:50–6.
66. Kruger P, Saffarzadeh M, Weber AN, Rieber N, Radsak M, von Bernuth H, et al. Neutrophils: between host defence, immune modulation, and tissue injury. PLoS Pathog. 11:e1004651. doi: 10.1371/journal.ppat.1004651
67. Wilson R, Cohen JM, Jose RJ, de Vogel C, Baxendale H, Brown JS. Protection against Streptococcus pneumoniae lung infection after nasopharyngeal colonization requires both humoral and cellular immune responses. Mucosal Immunol. (2015) 8:627–39. doi: 10.1038/mi.2014.95
68. Yang ML, Wang CT, Yang SJ, Leu CH, Chen SH, Wu CL, et al. IL-6 ameliorates acute lung injury in influenza virus infection. Sci Rep. (2017) 7:43829. doi: 10.1038/srep43829
69. Crowe CR, Chen K, Pociask DA, Alcorn JF, Krivich C, Enelow RI, et al. Critical role of IL-17RA in immunopathology of influenza infection. J Immunol. (2009) 183:5301–10. doi: 10.4049/jimmunol.0900995
70. Gopal R, Rangel-Moreno J, Fallert Junecko BA, Mallon DJ, Chen K, Pociask DA, et al. Mucosal pre-exposure to Th17-inducing adjuvants exacerbates pathology after influenza infection. Am J Pathol. (2014) 184:55–63. doi: 10.1016/j.ajpath.2013.09.012
71. Mirsaeidi M, Ebrahimi G, Allen MB, Aliberti S. Pneumococcal vaccine and patients with pulmonary diseases. Am J Med. (2014) 127;886 e1–8. doi: 10.1016/j.amjmed.2014.05.010
72. Mantero M, Aliberti S, Azzari C, Moriondo M, Nieddu F, Blasi F, et al. Role of Streptococcus pneumoniae infection in chronic obstructive pulmonary disease patients in Italy. Ther Adv Respir Dis. (2017) 11:403–7. doi: 10.1177/1753465817728479
73. Kudo M, Ishigatsubo Y, Aoki I. Pathology of asthma. Front Microbiol. (2013) 4:263. doi: 10.3389/fmicb.2013.00263
74. Agarwal R, Gupta D. Severe asthma and fungi: current evidence. Med Mycol. (2011) 49(Suppl. 1):S150–7. doi: 10.3109/13693786.2010.504752
75. Ghosh S, Hoselton SA, Schuh JM. Allergic Inflammation in Aspergillus fumigatus-induced fungal asthma. Curr Allergy Asthma Rep. (2015) 15:59. doi: 10.1007/s11882-015-0561-x
76. Denning DW, O'Driscoll BR, Hogaboam CM, Bowyer P, Niven RM. The link between fungi and severe asthma: a summary of the evidence. Eur Respir J. (2006) 27:615–26. doi: 10.1183/09031936.06.00074705
77. Allard JB, Rinaldi L, Wargo MJ, Allen G, Akira S, Uematsu S, et al. Th2 allergic immune response to inhaled fungal antigens is modulated by TLR-4-independent bacterial products. Eur J Immunol. (2009) 39:776–88. doi: 10.1002/eji.200838932
78. Sjoberg LC, Nilsson AZ, Lei Y, Gregory JA, Adner M, Nilsson GP. Interleukin 33 exacerbates antigen driven airway hyperresponsiveness, inflammation and remodeling in a mouse model of asthma. Sci Rep. (2017) 7:4219. doi: 10.1038/s41598-017-03674-0
79. Possa SS, Leick EA, Prado CM, Martins MA, Tiberio IF. Eosinophilic inflammation in allergic asthma. Front Pharmacol. (2013) 4:46. doi: 10.3389/fphar.2013.00046
80. Ghosh S, Hoselton SA, Dorsam GP, Schuh JM. Eosinophils in fungus-associated allergic pulmonary disease. Front Pharmacol. (2013) 4:8. doi: 10.3389/fphar.2013.00008
81. Kouzaki H, Matsumoto K, Kato T, Tojima I, Shimizu S, Shimizu T. Epithelial cell-derived cytokines contribute to the pathophysiology of eosinophilic chronic rhinosinusitis. J Interferon Cytokine Res. (2016) 36:169–79. doi: 10.1089/jir.2015.0058
82. De Luca A, Pariano M, Cellini B, Costantini C, Villella VR, Jose SS, et al. The IL-17F/IL-17RC axis promotes respiratory allergy in the proximal airways. Cell Rep. (2017) 20:1667–80. doi: 10.1016/j.celrep.2017.07.063
83. Woo LN, Guo WY, Wang X, Young A, Salehi S, Hin A, et al. A 4-week model of House Dust Mite (HDM) induced allergic airways inflammation with airway remodeling. Sci Rep. (2018) 8:6925. doi: 10.1038/s41598-018-24574-x
84. Sethi S. Bacterial infection and the pathogenesis of COPD. Chest (2000) 117:286S−91S. doi: 10.1378/chest.117.5_suppl_1.286S
85. Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med. (2010) 363:2233–47. doi: 10.1056/NEJMra0910061
86. Jacobsen EA, Helmers RA, Lee JJ, Lee NA. The expanding role(s) of eosinophils in health and disease. Blood (2012) 120:3882–90. doi: 10.1182/blood-2012-06-330845
87. Samarasinghe AE, Melo RC, Duan S, LeMessurier KS, Liedmann S, Surman SL, et al. Eosinophils promote antiviral immunity in mice infected with influenza A virus. J Immunol. (2017) 198:3214–26. doi: 10.4049/jimmunol.1600787
88. Gern JE. Virus/allergen interaction in asthma exacerbation. Ann Am Thorac Soc. (2015) 12(Suppl. 2):S137–43. doi: 10.1513/AnnalsATS.201503-153AW
89. Talbot TR, Hartert TV, Mitchel E, Halasa NB, Arbogast PG, Poehling KA, et al. Asthma as a risk factor for invasive pneumococcal disease. N Engl J Med. (2005) 352:2082–90. doi: 10.1056/NEJMoa044113
90. Kwak BO, Choung JT, Park YM. The association between asthma and invasive pneumococcal disease: a nationwide study in Korea. J Korean Med Sci. (2015) 30:60–5. doi: 10.3346/jkms.2015.30.1.60
91. Esposito S, Terranova L, Patria MF, Marseglia GL, Miraglia del Giudice M, Bodini A, et al. Streptococcus pneumoniae colonisation in children and adolescents with asthma: impact of the heptavalent pneumococcal conjugate vaccine and evaluation of potential effect of thirteen-valent pneumococcal conjugate vaccine. BMC Infect Dis. (2016) 16, 12. doi: 10.1186/s12879-016-1335-3
92. MacIntyre EA, Heinrich J. Otitis media in infancy and the development of asthma and atopic disease. Curr Allergy Asthma Rep. (2012) 12:547–50. doi: 10.1007/s11882-012-0308-x
93. Angelis N, Porpodis K, Zarogoulidis P, Spyratos D, Kioumis I, Papaiwannou A, et al. Airway inflammation in chronic obstructive pulmonary disease. J Thorac Dis. (2014) 6(Suppl. 1):S167–72. doi: 10.3978/j.issn.2072-1439.2014.03.07
94. Cukic V, Lovre V, Dragisic D, Ustamujic A. Asthma and Chronic Obstructive Pulmonary Disease (COPD)-differences and similarities. Mater Sociomed. (2012) 24:100–5. doi: 10.5455/msm.2012.24.100-105
95. Broide DH. Immunologic and inflammatory mechanisms that drive asthma progression to remodeling. J Allergy Clin Immunol. (2008) 121:560–70; quiz 571-2. doi: 10.1016/j.jaci.2008.01.031
96. Sethi S, Mahler DA, Marcus P, Owen CA, Yawn B, Rennard S. Inflammation in COPD: implications for management. Am J Med. (2012) 125:1162–70. doi: 10.1016/j.amjmed.2012.06.024
97. Hatziagorou E, Tsanakas J. Assessment of airway inflammation with exhaled NO measurement. Hippokratia (2007) 11:51–62.
98. Nurwidya F, Damayanti T, Yunus F. The role of innate and adaptive immune cells in the immunopathogenesis of chronic obstructive pulmonary disease. Tuberc Respir Dis. (2016) 79:5–13. doi: 10.4046/trd.2016.79.1.5
99. Yanagisawa H, Hashimoto M, Minagawa S, Takasaka N, Ma R, Moermans C, et al. Role of IL-17A in murine models of COPD airway disease. Am J Physiol Lung Cell Mol Physiol. (2017) 312:L122–30. doi: 10.1152/ajplung.00301.2016
100. Shukla SD, Fairbairn RL, Gell DA, Latham RD, Sohal SS, Walters EH, et al. An antagonist of the platelet-activating factor receptor inhibits adherence of both nontypeable Haemophilus influenzae and Streptococcus pneumoniae to cultured human bronchial epithelial cells exposed to cigarette smoke. Int J Chron Obstruct Pulmon Dis. (2016) 11:1647–55. doi: 10.2147/COPD.S108698
101. Domenech A, Ardanuy C, Balsalobre L, Marti S, Calatayud L, De la Campa AG, et al. (2012). Pneumococci can persistently colonize adult patients with chronic respiratory disease. J Clin Microbiol. 50, 4047–53. doi: 10.1128/JCM.02056-12
102. Domenech A, Ardanuy C, Calatayud L, Santos S, Tubau F, Grau I, et al. Serotypes and genotypes of Streptococcus pneumoniae causing pneumonia and acute exacerbations in patients with chronic obstructive pulmonary disease. J Antimicrob Chemother. (2011) 66:487–93. doi: 10.1093/jac/dkq480
103. Bogaert D, van der Valk P, Ramdin R, Sluijter M, Monninkhof E, Hendrix R, et al. Host-pathogen interaction during pneumococcal infection in patients with chronic obstructive pulmonary disease. Infect Immun. (2004) 72:818–23. doi: 10.1128/IAI.72.2.818-823.2004
104. Hirota N, Martin JG. Mechanisms of airway remodeling. Chest (2013) 144:1026–32. doi: 10.1378/chest.12-3073
105. Leung JM, Tiew PY, Mac Aogain M, Budden KF, Yong VF, Thomas SS, et al. The role of acute and chronic respiratory colonization and infections in the pathogenesis of COPD. Respirology (2017) 22:634–50. doi: 10.1111/resp.13032
Keywords: carriage, inflammation, pneumococcus, pathogenesis, co-infection
Citation: Sohail I, Ghosh S, Mukundan S, Zelewski S and Khan MN (2018) Role of Inflammatory Risk Factors in the Pathogenesis of Streptococcus pneumoniae. Front. Immunol. 9:2275. doi: 10.3389/fimmu.2018.02275
Received: 03 February 2018; Accepted: 12 September 2018;
Published: 02 October 2018.
Edited by:
Donata Medaglini, Università degli Studi di Siena, ItalyReviewed by:
Rosalinda Sorrentino, Università degli Studi di Salerno, ItalyRita Carsetti, Bambino Gesù Ospedale Pediatrico (IRCCS), Italy
Copyright © 2018 Sohail, Ghosh, Mukundan, Zelewski and Khan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Nadeem Khan, bmFkZWVtLmtoYW5AbWVkLnVuZC5lZHU=