 Kazuyuki Nakagome
Kazuyuki Nakagome Makoto Nagata
Makoto Nagata- 1Department of Respiratory Medicine, Saitama Medical University, Saitama, Japan
- 2Allergy Center, Saitama Medical University, Saitama, Japan
Eosinophils are involved in the development of asthma exacerbation. Recent studies have suggested that sputum and blood eosinophil counts are important factors for predicting asthma exacerbation. In severe eosinophilic asthma, anti-interleukin (IL)-5 monoclonal antibody decreases blood eosinophil count and asthma exacerbation frequency. However, even in the absence of IL-5, eosinophilic airway inflammation can be sufficiently maintained by the T helper (Th) 2 network, which comprises a cascade of vascular cell adhesion molecule-1/CC chemokines/eosinophil growth factors, including granulocyte-macrophage colony-stimulating factor (GM-CSF). Periostin, an extracellular matrix protein and a biomarker of the Th2 immune response in asthma, directly activates eosinophils in vitro. A major cause of asthma exacerbation is viral infection, especially rhinovirus (RV) infection. The expression of intercellular adhesion molecule (ICAM)-1, a cellular receptor for the majority of RVs, on epithelial cells is increased after RV infection, and adhesion of eosinophils to ICAM-1 can upregulate the functions of eosinophils. The expressions of cysteinyl leukotrienes (cysLTs) and CXCL10 are upregulated in virus-induced asthma. CysLTs can directly provoke eosinophilic infiltration in vivo and activate eosinophils in vitro. Furthermore, eosinophils express the CXC chemokine receptor 3, and CXCL10 activates eosinophils in vitro. Both eosinophils and neutrophils contribute to the development of severe asthma or asthma exacerbation. IL-8, which is an important chemoattractant for neutrophils, is upregulated in some cases of severe asthma. Lipopolysaccharide (LPS), which induces IL-8 from epithelial cells, is also increased in the lower airways of corticosteroid-resistant asthma. IL-8 or LPS-stimulated neutrophils increase the transbasement membrane migration of eosinophils, even in the absence of chemoattractants for eosinophils. Therefore, eosinophils are likely to contribute to the development of asthma exacerbation through several mechanisms, including activation by Th2 cytokines, such as IL-5 or GM-CSF or by virus infection-related proteins, such as CXCL10, and interaction with other cells, such as neutrophils.
Introduction
Bronchial asthma is a chronic disease with airway hyperresponsiveness (AHR), reversible airflow limitation, and airway inflammation (1, 2). Asthma is recognized as a heterogeneous disease that has different phenotypes with distinct clinical characteristics, or different endotypes with distinct functional or pathophysiological mechanisms including eosinophilic asthma or non-eosinophilic asthma (3, 4). Recent studies have suggested that eosinophils play important roles in the development of asthma exacerbation (5–7). Therefore, suppressing eosinophilic inflammation and distinguishing eosinophilic from non-eosinophilic asthma may be useful for the treatment or prevention of asthma exacerbation. In the present review, the involvement and possible role of eosinophils in asthma exacerbation is discussed.
Ethics Statement
Our studies in this review were approved by the Institutional Review Board of Saitama Medical University Hospital, and written informed consent was obtained from the patients.
Role of Eosinophilic Inflammation in Asthma
Eosinophils, which tend to accumulate at sites of allergic inflammation, contribute to the development of bronchial asthma. They release a number of mediators, including specific granule proteins, such as major basic protein (MBP), radical oxygen species, cytokines, such as granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin (IL)-8, and lipid mediators, such as cysteinyl leukotrienes (cysLTs) (8, 9). However, previous studies investigating the effectiveness of anti-IL-5 monoclonal antibody (mAb) treatment for asthmatics have suggested that eosinophils may only play a small role (10, 11). For example, it has been reported that anti-IL-5 mAb reduces the sputum or blood eosinophil count, but has no effect on histamine-induced AHR or allergen-induced late asthmatic response (10), which suggests that eosinophils do not play a role in the development of AHR or allergen-induced airflow obstruction.
By contrast, the role of eosinophils in the development of airway remodeling has been established at a relatively early phase (12). Eosinophil-deficient mice are reportedly protected from peribronchiolar collagen deposition (13). Eosinophils produce transforming growth factor (TGF)-β (14), which may contribute to airway fibrosis. Additionally, eosinophils can produce cysLTs (15) and be a major cellular source of cysLTs in the airways of individuals with seasonal allergic asthma or aspirin-exacerbated respiratory disease (16, 17), which also contribute to airway remodeling. Anti-IL-5 mAb suppresses airway remodeling (reduction of tenascin, lumican, and procollagen III) as well as airway eosinophils expressing mRNA for TGF-β1 and concentrations of TGF-β1 in the bronchoalveolar lavage (BAL) fluid of asthmatics (18).
As for the role of eosinophils in asthma exacerbation, recent studies have reported that sputum and blood eosinophil counts are important factors for predicting asthma exacerbation (5, 6, 7). Treatment for normalizing sputum eosinophil counts can help prevent asthma exacerbation (5), and blood eosinophil counts are associated with exacerbation frequency (6, 7). Furthermore, in severe asthmatic patients with sustained blood or sputum eosinophilia, anti-IL-5 treatment decreases both blood eosinophil counts and asthma exacerbation frequency (19–21). Eosinophil-derived granule products, such as MBP mediate epithelial cell damage, thereby, inducing AHR (22, 23). In accordance with these findings, anti-IL-5 mAb is now prescribed for patients with severe eosinophil-dominant asthma in the clinical setting.
However, even with anti-IL-5 mAb, eosinophils can still accumulate and activate in the airways of asthmatics. One study reported that anti-IL-5 mAb may be insufficient to inhibit eosinophil activation in the airway (24). To accumulate in asthmatic airways, circulating eosinophils needs to adhere to vascular endothelial cells and then migrate over cells, which are regulated primarily by cytokines or chemokines induced by a number of cells, such as T helper (Th) 2 cells (Figure 1) (25). The crucial step for selective eosinophil recruitment is likely the adhesion of eosinophils with endothelial cells through the α4 integrin/vascular cell adhesion molecule (VCAM)-1 (25–27). The expression of VCAM-1 in endothelial cells is upregulated by IL-4 and IL-13, after which, blood eosinophils adhere spontaneously to VCAM-1 (28, 29). The interaction of eosinophils with VCAM-1 induces eosinophil superoxide anion (O) generation and degranulation, and therefore may be the first step of eosinophil activation (28–30).
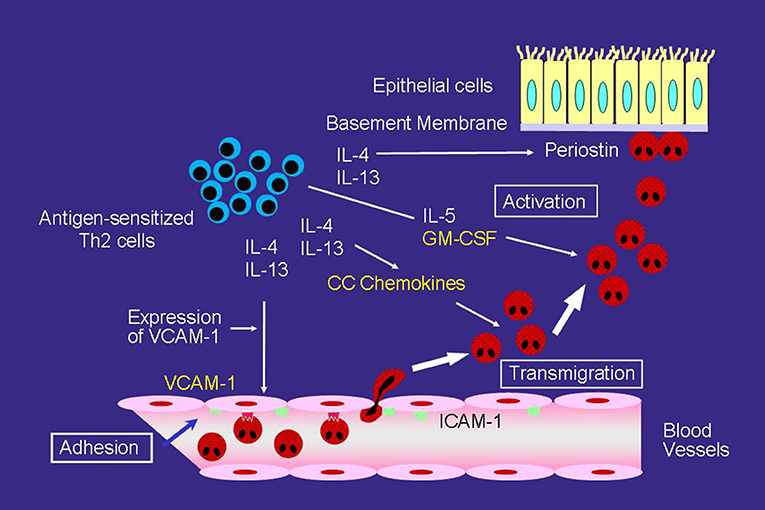
Figure 1. Mechanisms of eosinophilic airway inflammation in bronchial asthma. IL-5 plays an important role in the development of eosinophilic airway inflammation. However, in the absence of IL-5, the Th2 network, which includes a cascade of VCAM-1/CC chemokines/GM-CSF, can maintain eosinophilic infiltration and activation. Yellow text indicates a cascade of VCAM-1/CC chemokines/GM-CSF.
After adhering to the endothelial cells, CC chemokines, such as eotaxin and regulated upon activation, normal T-cell expressed and secreted (RANTES) effectively induce eosinophil transmigration over endothelial cells expressing VCAM-1 (Figure 1) (25, 31). A number of studies have reported an increase in CC chemokines in the airways of asthmatic patients. The airway expression of eotaxin and its receptor, CCR3, are elevated in atopic asthmatics compared with normal controls (32). In patients with acute eosinophilic pneumonia, monocyte chemotactic protein (MCP)-4, which is also a CCR3 ligand, is involved in the development of eosinophil transendothelial migration (33); however, the role of MCP-4 in asthma has yet to be fully clarified.
GM-CSF play an important role in eosinophil activation after migration process, even without IL-5 (Figure 1). GM-CSF induces eosinophil O generation and the release of specific granule proteins in vitro when incubated with VCAM-1 or intercellular cell adhesion molecule (ICAM)-1 (29). Furthermore, GM-CSF, but not IL-5, activates eosinophils of airways after segmental allergen challenge (34, 35). These findings suggest that in the absence of IL-5, the Th2 network, which includes a cascade of VCAM-1/CC chemokines/GM-CSF, is likely the primary pathway for maintaining eosinophilic infiltration and activation in asthma (25).
Moreover, cysLTs may be involved in the eosinophil accumulation in the airways of asthma. Inhalation of LTE4 stimulates the accumulation of eosinophils in asthmatic airways (36). LTD4 upregulates the β2 integrin expression of human eosinophils and increases eosinophil adhesiveness to ICAM-1 in vitro (37). Furthermore, β2 integrin-enhanced adhesion increases the effector functions of eosinophils. Therefore, cysLT-induced β2 integrin activation may be a key process in regard to cell activation in asthmatics (25, 29). In addition, LTD4 induces eosinophil transendothelial migration, O generation, and the release of specific granule proteins primarily through β2 integrin and the cysLT1 receptor (38). Furthermore, leukotriene receptor antagonist (LTRA) suppresses eosinophil airway inflammation in vivo (39–41). These findings suggest that cysLTs, along with the Th2 network, contribute to the development and maintenance of airway eosinophilic inflammation in asthma.
Periostin is an extracellular matrix protein that is highly expressed in the airways of asthmatics in response to Th2 cytokines, such as IL-13 (42), and is a biomarker of Th2-mediated immune responses in bronchial asthma (43, 44). Periostin also functions as a matricellular protein (42) that binds to cellular receptors and activates cells, including eosinophils. Periostin directly induces eosinophil adhesion, O generation, and degranulation through the αMβ2 integrin in vitro (45).
Interactions of Viral Infection and Eosinophils in the Development of Asthma Exacerbation
Viral infection, especially rhinovirus (RV) infection, is a major cause of asthma exacerbation. In some community-based studies, viral infections have been identified in 80–85% of cases involving asthma exacerbation, and RV was found to be involved in about 65% of the patients in whom the causative virus was identified (46, 47). RVs have tremendous diversity (48). In addition to about 100 classical serotypes of the RV species A (RV-A) and B, over 60 types of RV-C were recently discovered by molecular techniques (48). Recent clinical data suggests that RV-C (49, 50) or RV-C and RV-A (51, 52) can induce more severe illness or asthma exacerbations, compared with other RVs, such as RV-B.
The numbers of not only neutrophils, but also eosinophils, increase in asthmatic airways during or after a viral infection (53–55). Experimental RV infection induces increased recruitment of eosinophils into the airway after segmental allergen challenge in allergic rhinitis patients, but not in non-allergic volunteers (53). Viral infection increases the eosinophil count in the airway epithelium of patients with allergic asthma (55), and high levels of eosinophilic cationic protein are observed in the sputum of asthmatic patients with viral infection (54). Therefore, eosinophils are indeed recruited to and activated in asthmatic airways during or after a viral infection.
Recent studies have suggested that the presence of eosinophil inflammation may be a risk factor for virus-related asthma exacerbation (56, 57). High fractional exhaled nitric oxide and sputum eosinophils are associated with an increased risk of future virus-induced exacerbations (57). Epithelial cells are damaged by eosinophil-derived granule products, such as MBP (23), and this increases the susceptibility to RV infection (Figure 2) (58). Furthermore, eosinophils can suppress the RV-induced expression of interferons (IFNs), anti-viral cytokines, including IFN-λ from epithelial cells, likely through the production of TGF-β, resulting in an increased quantity of RV (Figure 2) (56). Therefore, reducing the eosinophil count could be a reasonable strategy for suppressing virus-induced asthma exacerbation.
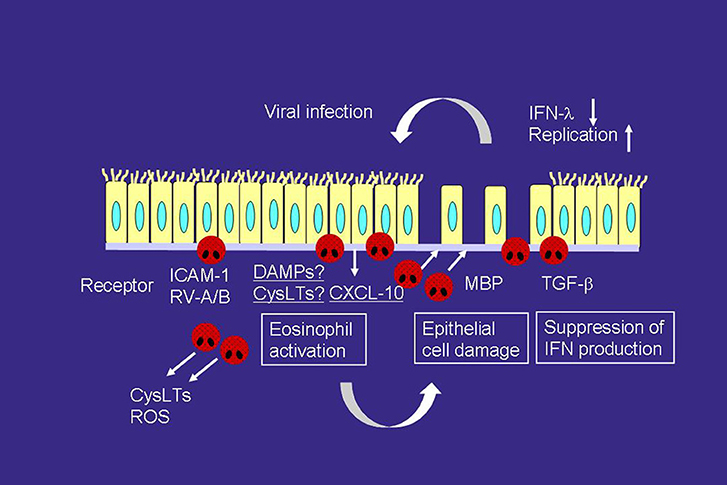
Figure 2. Interactions of viral infection and eosinophils in the development of asthma exacerbation. RV infection releases a variety of mediators, including CXCL-10 and cysLTs, which can directly activate eosinophils and induce asthma exacerbation, from airway epithelial cells. RV infection increases the expression of ICAM-1, which also activates eosinophils. On the other hand, activated eosinophils release MBP, which induces epithelial cell damage. Eosinophils also produce TGF-β, which can suppress the expression of IFN-λ, anti-viral cytokines, on epithelial cells. Therefore, eosinophilic airway inflammation can increase susceptibility to RV infection. ROS, reactive oxygen species; TGF, transforming growth factor.
ICAM-1 is a cellular receptor for the majority of RV-A (major) and all of RV-B (59), and RV infection increases ICAM-1 expression on epithelial cells (60). ICAM-1 is also an adhesion molecule, and adhesion of eosinophils to ICAM-1 can activate the functions of eosinophils (28, 29). Therefore, eosinophil adhesion to epithelial cells via ICAM-1 may activate eosinophils during RV-induced asthma exacerbation (Figure 2). Cadherin-related family member 3 (CDHR3) is a recently found receptor for RV-C (61). In this context, a coding single nucleotide polymorphism (SNP) in CDHR3 has been shown to be related to the severe exacerbation of childhood asthma (62). Moreover, this SNP has been reported to increase the expression of the CDHR3 protein on the cell surface (61, 62), resulting in increased RV-C binding and progeny yields (61). Because the cadherin family members are involved in cell adhesion, eosinophil adhesion to CDHR3 may activate eosinophil functions in a manner similar to that as ICAM-1.
CXCL10 may also play a role in the virus-induced asthma exacerbation (Figure 2). RV infection produces CXCL10 from bronchial epithelial cells in vitro and in vivo (63). Specifically, concentrations of serum CXCL10 are elevated in virus or RV-induced asthma; correlations are reported between higher levels of CXCL10 and disease severity, including airflow limitation (63). CXCL10 induces eosinophil adhesion, O generation, eosinophil-derived neurotoxin release, and cytokine production through CXCR3, expressed on eosinophils, in vitro (64). As for other CXCR3 ligands, CXCL9 is involved in severe asthma (65) and produced from epithelial cells by RV infection (66). CXCL9 induces eosinophil adhesion, O generation, and eosinophil-derived neurotoxin release in vitro (64), whereas it inhibits eosinophil chemotaxis through a CCR3-dependent mechanism (67, 68).
CysLTs are upregulated in the asthmatic airways during virus or RV-induced exacerbation (69, 70). Respiratory syncytial virus (RSV) induces LTC4 synthase expression on bronchial epithelial cells (71). Therefore, cysLTs are likely to be involved in virus-induced eosinophilic inflammation (Figure 2), and LTRA may be useful for virus-induced asthma treatment. The LTRA montelukast suppresses the respiratory symptoms of RSV bronchiolitis (72) as well as the frequency of virus-induced asthma exacerbation (73). Moreover, montelukast inhibits eosinophil adhesion induced by CXCL10 and ICAM-1 in vitro (74), both are virus-infection-related proteins.
Innate immune responses play roles in the development of eosinophilic airway inflammation; this process involves type 2 innate lymphoid cells (ILC2) as well as epithelial cell-related cytokines including IL-33, IL-25, and thymic stromal lymphopoietin, (75, 76). The ILC2 stimulated by these cytokines produce IL-5 and IL-13 and induce eosinophilic inflammation. In fact, ILC2 are upregulated in severe asthmatics (77). Recent studies have suggested that innate immune responses contribute to virus-induced asthma exacerbation. For example, IL-33-dependent type 2 inflammation plays an important role in RV-induced asthma exacerbation in vivo (78).
During viral infections, damage-associated molecular pattern molecules (DAMPs) can be released by stressed or damaged cells, and function as endogenous danger signals (79). Damaged epithelial cells are capable of inducing eosinophilic migration, specific granule protein release, and cytokine production, likely via the release of DAMPs (80). Uric acid (UA) or adenosine triphosphate (ATP), an important DAMP, activates eosinophil functions in vitro (81, 82); however, the role of DAMPs in the development of asthma exacerbation remains unclear.
Interactions of Neutrophils and Eosinophils in the Development of Severe Asthma or Asthma Exacerbation
Both neutrophilic and eosinophilic inflammation may play roles in severe asthma [(83–85)]. Neutrophilic inflammation has been shown to be involved in the pathogenesis of asthma exacerbation (86), which occurs frequently in severe asthma. The European Network for Understanding Mechanisms of Severe Asthma (ENFUMOSA) study suggested that compared with patients with mild-to-moderate asthma, those with severe asthma have both a greater sputum neutrophil count and an increased release of eosinophil-derived mediators (84). IL-8 plays an important role in the accumulation of neutrophils in inflammation sites, and IL-8 expression is upregulated in the airways of severe asthmatic patients (87, 88). In addition, we reported that neutrophils that had migrated to IL-8 induce the transbasement membrane migration of eosinophils in vitro, even without eosinophil chemoattractants (89); this neutrophil-induced eosinophil migration is suppressed by LTB4 antagonist or platelet-activating factor (PAF) antagonist. LTB4 and PAF are potent chemotactic factors for eosinophils (90, 91); therefore, IL-8-stimulated neutrophils can lead to eosinophil accumulation in asthmatic airways through LTB4 or PAF (92).
Lipopolysaccharide (LPS) may play a role in inducing IL-8 or neutrophilic inflammation in the airway of severe asthmatics. In the BAL fluid of asthmatic children, LPS levels correlate with airway neutrophils or IL-8 (93). Furthermore, the BAL fluid LPS and genes associated with LPS signaling activation are higher in corticosteroid-resistant asthma (94). Furthermore, a positive correlation is observed between IL-8 mRNA expression in BAL cells and the amount of LPS in BAL fluid (94). Several studies have suggested that Gram-negative bacteria or house dust plays a role in the LPS upregulation in the airways of severe asthmatics. We previously reported that LPS-stimulated neutrophils induce the transbasement membrane migration of eosinophils in vitro (95).
IL-17 is another candidate for the upregulation of IL-8 expression (96). Sputum IL-17 concentration correlates with the clinical severity of asthma (97), and the airway expression of IL-17 is increased in severe asthmatics only (98). Furthermore, a correlation between the number of bronchial cells that produce IL-17 and the number of bronchial neutrophils and frequency of asthma exacerbation has been reported (86). In addition, we reported that the dopamine D1-like receptor antagonist attenuates the Th17-mediated immune response and neutrophilic airway inflammation in mice (99) this could be reasonable strategy for controlling neutrophilic airway inflammation in severe asthma or asthma exacerbation.
The role of neutrophil extracellular traps (NETs) in the pathogenesis of asthma exacerbation has recently been highlighted. In a mouse model, RV infection triggered a double-stranded DNA (dsDNA) release that was associated with the formation of NETs; this is known as NETosis (100). Furthermore, in humans, a significant correlation is identified between the release of host dsDNA after RV infection and the exacerbation of type-2 allergic inflammation (100).
Role of Mast Cells and Prostaglandin (PG) D2 in Eosinophilic Inflammation of Asthma
Mast cells also play roles in the development of severe asthma. Mast cell numbers and PGD2 concentrations are increased in the lower airway of patients with severe asthma (101, 102). Mast cells are major cellular sources of PGD2, and D prostanoid (DP) and chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2) are receptors for PGD2 (103). Recently, the role of CRTH2 in the pathogenesis of asthma has been highlighted. CRTH2 is expressed on Th2 cells, ILC2, eosinophils, and basophils (103). PGD2 induces chemotaxis in eosinophils through CRTH2 (104), and CRTH2 antagonist suppresses eosinophil chemotaxis and respiratory burst (105). CRTH2 antagonists are already being developed (103, 106), and clinical trial data suggest that CRTH2 antagonists may target eosinophilic asthma (103, 107).
Conclusion
Eosinophils are likely to contribute to the development of asthma exacerbation. This process can involve cytokines, such as IL-5 or GM-CSF, chemokines, such as CCR3 ligands, matricellular proteins, a danger signal, and other cells, such as neutrophils or mast cells.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by a grant from the Ministry of Education, Culture, Sports, Science and Technology (21790783 and 15K09228).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bochner BS, Undem BJ, Lichtenstein LM. Immunological aspects of allergic asthma. Annu Rev Immunol. (1994) 12:295–335. doi: 10.1146/annurev.iy.12.040194.001455
2. Barnes PJ. Pathophysiology of allergic inflammation. Immunol Rev. (2011) 242:31–50. doi: 10.1111/j.1600-065X.2011.01020.x
3. Lötvall J, Akdis CA, Bacharier LB, Bjermer L, Casale TB, Custovic A, et al. Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. J Allergy Clin Immunol. (2011) 127:355–60. doi: 10.1016/j.jaci.2010.11.037
4. Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. (2012) 18:716–25. doi: 10.1038/nm.2678
5. Green RH, Brightling CE, McKenna S, Hargadon B, Parker D, Bradding P, et al. Asthma exacerbations and sputum eosinophil counts: a randomised controlled trial. Lancet (2002) 360:1715–21. doi: 10.1016/S0140-6736(02)11679-5
6. Price DB, Rigazio A, Campbell JD, Bleecker ER, Corrigan CJ, Thomas M, et al. Blood eosinophil count and prospective annual asthma disease burden: a UK cohort study. Lancet Respir Med. (2015) 3, 849–58. doi: 10.1016/S2213-2600(15)00367-7
7. Denlinger LC, Phillips BR, Ramratnam S, Ross K, Bhakta NR, Cardet JC, et al. Inflammatory and comorbid features of patients with severe asthma and frequent exacerbations. Am J Respir Crit Care Med. (2017) 195:302–13. doi: 10.1164/rccm.201602-0419OC
8. Weller PF. Human eosinophils. J Allergy Clin Immunol. (1997) 100:283–7. doi: 10.1016/S0091-6749(97)70237-9
9. Gleich GJ. Mechanisms of eosinophil-associated inflammation. J Allergy Clin Immunol. (2000) 105:651–63. doi: 10.1067/mai.2000.105712
10. Leckie MJ, ten Brinke A, Khan J, Diamant Z, O'Connor BJ, Walls CM, et al. (2000). Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet 356, 2144–2148. doi: 10.1016/S0140-6736(00)03496-6
11. Flood-Page P, Swenson C, Faiferman I, Matthews J, Williams M, Brannick L, et al. A study to evaluate safety and efficacy of mepolizumab in patients with moderate persistent asthma. Am J Respir Crit Care Med. (2007) 176:1062–71. doi: 10.1164/rccm.200701-085OC
12. Kay AB, Phipps S, Robinson DS. A role for eosinophils in airway remodelling in asthma. Trends Immunol. (2004) 25:477–82. doi: 10.1016/j.it.2004.07.006
13. Humbles AA, Lloyd CM, McMillan SJ, Friend DS, Xanthou G, McKenna EE, et al. A critical role for eosinophils in allergic airways remodeling. Science (2004) 305:1776–9. doi: 10.1126/science.1100283
14. Wong DT, Elovic A, Matossian K, Nagura N, McBride J, Chou MY, et al. Eosinophils from patients with blood eosinophilia express transforming growth factor β1. Blood (1991) 78:2702–7.
15. Owen WF Jr, Soberman RJ, Yoshimoto T, Sheffer AL, Lewis RA, Austen KF. Synthesis and release of leukotriene C4 by human eosinophils. J Immunol. (1987) 138:532–8.
16. Cowburn AS, Sladek K, Soja J, Adamek L, Nizankowska E, Szczeklik A, et al. Overexpression of leukotriene C4 synthase in bronchial biopsies from patients with aspirin-intolerant asthma. J Clin Invest. (1998) 101:834–46. doi: 10.1172/JCI620
17. Seymour ML, Rak S, Aberg D, Riise GC, Penrose JF, Kanaoka Y, et al. Leukotriene and prostanoid pathway enzymes in bronchial biopsies of seasonal allergic asthmatics. Am J Respir Crit Care Med. (2001) 164:2051–6. doi: 10.1164/ajrccm.164.11.2008137
18. Flood-Page P, Menzies-Gow A, Phipps S, Ying S, Wangoo A, Ludwig MS, et al. Anti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J Clin Invest. (2003) 112, 1029–1036. doi: 10.1172/JCI17974
19. Nair P, Pizzichini MM, Kjarsgaard M, Inman MD, Efthimiadis A, Pizzichini E, et al. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med. (2009) 360:985–93. doi: 10.1056/NEJMoa0805435
20. Haldar P, Brightling CE, Hargadon B, Gupta S, Monteiro W, Sousa A, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. (2009) 360:973–84. doi: 10.1056/NEJMoa0808991
21. Pavord ID, Korn S, Howarth P, Bleecker ER, Buhl R, Keene ON, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet (2012) 380:651–9. doi: 10.1016/S0140-6736(12)60988-X
22. Gleich GJ, Flavahan NA, Fujisawa T, Vanhoutte PM. The eosinophil as a mediator of damage to respiratory epithelium: a model for bronchial hyperreactivity. J Allergy Clin Immunol. (1988) 81:776–81. doi: 10.1016/0091-6749(88)90931-1
23. Wardlaw AJ, Dunnette S, Gleich GJ, Collins JV, Kay AB. Eosinophils and mast cells in bronchoalveolar lavage in subjects with mild asthma. Relation. bronchial hyperreactivity. Am Rev Respir Dis. (1988) 137:62–9. doi: 10.1164/ajrccm/137.1.62
24. Kelly EA, Esnault S, Liu LY, Evans MD, Johansson MW, Mathur S, et al. Mepolizumab attenuates airway eosinophil numbers, but not their functional phenotype, in asthma. Am J Respir Crit Care Med. (2017) 196:1385–95. doi: 10.1164/rccm.201611-2234OC
25. Nakagome K, Nagata M. Pathogenesis of airway inflammation in bronchial asthma. Auris Nasus Larynx (2011) 38:555–63. doi: 10.1016/j.anl.2011.01.011
26. Schleimer RP, Sterbinsky SA, Kaiser J, Bickel CA, Klunk DA, Tomioka K, et al. (1992). IL-4 induces adherence of human eosinophils and basophils but not neutrophils to endothelium. Association with expression of VCAM-1. J Immunol. 148:1086–92.
27. Bochner BS. Cellular adhesion and its antagonism. J Allergy Clin Immunol. (1997) 100:581–5. doi: 10.1016/S0091-6749(97)70158-1
28. Nagata M, Sedgwick JB, Bates ME, Kita H, Busse WW. Eosinophil adhesion to vascular cell adhesion molecule-1 activates superoxide anion generation. J Immunol. (1995) 155:2194–202.
29. Nagata M, Sedgwick JB, Kita H, Busse WW. Granulocyte macrophage colony-stimulating factor augments ICAM-1 and VCAM-1 activation of eosinophil function. Am J Respir Cell Mol Biol. (1998) 19:158–66. doi: 10.1165/ajrcmb.19.1.3001
30. Nagata M, Sedgwick JB, Vrtis R, Busse WW. Endothelial cells upregulate eosinophil superoxide generation via VCAM-1 expression. Clin Exp Allergy (1999) 29:550–61. doi: 10.1046/j.1365-2222.1999.00506.x
31. Nagata M, Yamamoto H, Tabe K, Sakamoto Y. Eosinophil transmigration across VCAM-1-expressing endothelial cells is upregulated by antigen-stimulated mononuclear cells. Int Arch Allergy Immunol. (2001) 125:7–11. doi: 10.1159/000053844
32. Ying S, Robinson DS, Meng Q, Rottman J, Kennedy R, Ringler DJ, et al. Enhanced expression of eotaxin and CCR3 mRNA and protein in atopic asthma. Association with airway hyperresponsiveness and predominant co-localization of eotaxin mRNA to bronchial epithelial and endothelial cells Eur J Immunol. (1997) 27:3507–16. doi: 10.1002/eji.1830271252
33. Nakagome K, Shoda H, Shirai T, Nishihara F, Soma T, Uchida Y, et al. Eosinophil transendothelial migration induced by the bronchoalveolar lavage fluid of acute eosinophilic pneumonia. Respirology (2017) 22:913–21. doi: 10.1111/resp.12982
34. Liu LY, Sedgwick JB, Bates ME, Vrtis RF, Gern JE, Kita H, et al. (2002). Decreased expression of membrane IL-5 receptor alpha on human eosinophils: I. Loss of membrane IL-5 receptor alpha on airway eosinophils and increased soluble IL-5 receptor alpha in the airway after allergen challenge. J Immunol. 169:6452–8. doi: 10.4049/jimmunol.169.11.6452
35. Liu LY, Sedgwick JB, Bates ME, Vrtis RF, Gern JE, Kita H, et al. (2002). Decreased expression of membrane IL-5 receptor alpha on human eosinophils: II. IL-5 down-modulates its receptor via a proteinase-mediated process. J Immunol. 169:6459–66. doi: 10.4049/jimmunol.169.11.6459
36. Laitinen LA, Laitinen A, Haahtela T, Vilkka V, Spur BW, Lee TH. Leukotriene E4 and granulocytes infiltration into asthmatic airways. Lancet (1993) 341:989–90. doi: 10.1016/0140-6736(93)91073-U
37. Nagata M, Saito K, Tsuchiya T, Sakamoto Y. Leukotriene D4 upregulates eosinophil adhesion via the cysteinyl leukotriene 1 receptor. J Allergy Clin Immunol. (2002) 109:676–80. doi: 10.1067/mai.2002.122841
38. Saito K, Nagata M, Kikuchi I, Sakamoto Y. Leukotriene D4 and eosinophil transendothelial migration, superoxide generation, and degranulation via β2 integrin. Ann Allergy Asthma Immunol. (2004) 93:594–600. doi: 10.1016/S1081-1206(10)61269-0
39. Pizzichini E, Leff JA, Reiss TF, Hendeles L, Boulet LP, Wei LX, et al. Montelukast reduces airway eosinophilic inflammation in asthma: a randomized, controlled trial. Eur Respir J. (1999) 14:12–8. doi: 10.1034/j.1399-3003.1999.14a04.x
40. Obase Y, Shimoda T, Tomari SY, Mitsuta K, Kawano T, Matsuse H, et al. Effects of pranlukast on chemical mediators in induced sputum on provocation tests in atopic and aspirin-intolerant asthmatic patients. Chest (2002) 121:143–50. doi: 10.1378/chest.121.1.143
41. Bjermer L, Bisgaard H, Bousquet J, Fabbri LM, Greening AP, Haahtela T, et al. Montelukast and fluticasone compared with salmeterol and fluticasone in protecting against asthma exacerbation in adults: one year, double blind, randomised, comparative trial. BMJ (2003) 327:891. doi: 10.1136/bmj.327.7420.891
42. Izuhara K, Arima K, Ohta S, Suzuki S, Inamitsu M, Yamamoto K. Periostin in allergic inflammation. Allergol Int. (2014) 63:143–51. doi: 10.2332/allergolint.13-RAI-0663
43. Jia G, Erickson RW, Choy DF, Mosesova S, Wu LC, Solberg OD, et al. Bronchoscopic exploratory research study of biomarkers in corticosteroid-refractory asthma (BOBCAT) study group. Periostin is a systemic biomarker of eosinophilic airway inflammation in asthmatic patients. J Allergy Clin Immunol. (2012) 130:647–54. doi: 10.1016/j.jaci.2012.06.025
44. Kanemitsu Y, Matsumoto H, Izuhara K, Tohda Y, Kita H, Horiguchi T, et al. Increased periostin associates with greater airflow limitation in patients receiving inhaled corticosteroids. J Allergy Clin Immunol. (2013) 132:305–12. doi: 10.1016/j.jaci.2013.04.050
45. Noguchi T, Nakagome K, Kobayashi T, Uchida Y, Soma T, Nakamoto H, et al. Periostin upregulates the effector functions of eosinophils. J Allergy Clin Immunol. (2016) 138:1449–52. doi: 10.1016/j.jaci.2016.05.020
46. Johnson SL, Pattemore PK, Sanderson G, Smith S, Lampe F, Josephs L, et al. Community study of role of viral infections in exacerbations of asthma in 9–11 year old children. BMJ (1995) 310:1225–9. doi: 10.1136/bmj.310.6989.1225
47. Friedlander SL, Busse WW. The role of rhinovirus in asthma exacerbations. J Allergy Clin Immunol. (2005) 116:267–73. doi: 10.1016/j.jaci.2005.06.003
48. Palmenberg AC, Spiro D, Kuzmickas R, Wang S, Djikeng A, Rathe JA, et al. Sequencing and analyses of all known human rhinovirus genomes reveal structure and evolution. Science (2009) 324:55–9. doi: 10.1126/science.1165557
49. Miller EK, Khuri-Bulos N, Williams JV, Shehabi AA, Faouri S, Al Jundi I, et al. Human rhinovirus C associated with wheezing in hospitalised children in the Middle East. J Clin Virol. (2009) 46:85–9. doi: 10.1016/j.jcv.2009.06.007
50. Bizzintino J, Lee WM, Laing IA, Vang F, Pappas T, Zhang G, et al. Association between human rhinovirus C and severity of acute asthma in children. Eur Respir J. (2011) 37:1037–42. doi: 10.1183/09031936.00092410
51. Lee WM, Lemanske RF Jr, Evans MD, Vang F, Pappas T, Gangnon R, et al. Human rhinovirus species and season of infection determine illness severity. Am J Respir Crit Care Med. (2012) 186:886–91. doi: 10.1164/rccm.201202-0330OC
52. Nakagome K, Bochkov YA, Ashraf S, Brockman-Schneider RA, Evans MD, Pasic TR, et al. Effects of rhinovirus species on viral replication and cytokine production. J Allergy Clin Immunol. (2014) 134:332–41. doi: 10.1016/j.jaci.2014.01.029
53. Calhoun WJ, Dick EC, Schwartz LB, Busse WW. A common cold virus, rhinovirus 16, potentiates airway inflammation after segmental antigen bronchoprovocation in allergic subjects. J Clin Invest. (1994) 94:2200–8. doi: 10.1172/JCI117581
54. Fraenkel DJ, Bardin PG, Sanderson G, Lampe F, Johnston SL, Holgate ST. Lower airways inflammation during rhinovirus colds in normal and in asthmatic subjects. Am J Respir Crit Care Med. (1995) 151:879–86.
55. Pizzichini MM, Pizzichini E, Efthimiadis A, Chauhan AJ, Johnston SL, Hussack P, et al. Asthma and natural colds. Inflammatory indices induced sputum; a feasibility study. Am J Respir Crit Care Med. (1998) 158:1178–84. doi: 10.1164/ajrccm.158.4.9712082
56. Mathur SK, Fichtinger PS, Kelly JT, Lee WM, Gern JE, Jarjour NN. Interaction between allergy and innate immunity: model for eosinophil regulation of epithelial cell interferon expression. Ann Allergy Asthma Immunol. (2013) 111:25–31. doi: 10.1016/j.anai.2013.05.010
57. Bjerregaard A, Laing IA, Backer V, Sverrild A, Khoo SK, Chidlow G, et al. High fractional exhaled nitric oxide and sputum eosinophils are associated with an increased risk of future virus-induced exacerbations: a prospective cohort study. Clin Exp Allergy (2017) 47:1007–13. doi: 10.1111/cea.12935
58. Jakiela B, Brockman-Schneider R, Amineva S, Lee WM, Gern JE. Basal cells of differentiated bronchial epithelium are more susceptible to rhinovirus infection. Am J Respir Cell Mol Biol. (2008) 38:517–23. doi: 10.1165/rcmb.2007-0050OC
59. Greve JM, Davis G, Meyer AM, Forte CP, Yost SC, Marlor CW, et al. The major human rhinovirus receptor is ICAM-1. Cell (1989) 56:839–47. doi: 10.1016/0092-8674(89)90688-0
60. Grünberg K, Sharon RF, Hiltermann TJ, Brahim JJ, Dick EC, Sterk PJ, et al. Experimental rhinovirus 16 infection increases intercellular adhesion molecule-1 expression in bronchial epithelium of asthmatics regardless of inhaled steroid treatment. Clin Exp Allergy (2000) 30:1015–23. doi: 10.1046/j.1365-2222.2000.00854.x
61. Bochkov YA, Watters K, Ashraf S, Griggs TF, Devries MK, Jackson DJ, et al. Cadherin-related family member 3, a childhood asthma susceptibility gene product, mediates rhinovirus C binding and replication. Proc Natl Acad Sci USA. (2015) 112:5485–90. doi: 10.1073/pnas.1421178112
62. Bønnelykke K, Sleiman P, Nielsen K, Kreiner-Møller E, Mercader JM, Belgrave D, et al. A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat Genet. (2014) 46:51–5. doi: 10.1038/ng.2830
63. Wark PA, Bucchieri F, Johnston SL, Gibson PG, Hamilton L, Mimica J, et al. IFN-γ-induced protein 10 is novel biomarker of rhinovirus induced asthma exacerbation. J Allergy Clin Immunol. (2007) 120:586–93. doi: 10.1016/j.jaci.2007.04.046
64. Takaku Y, Nakagome K, Kobayashi T, Hagiwara K, Kanazawa M, Nagata M. IFN-γ-inducible protein of 10 kDa upregulates the effector functions of eosinophils through β2 integrin and CXCR3. Respir Res. (2011) 12:138. doi: 10.1186/1465-9921-12-138
65. Xie M, Mustovich AT, Jiang Y, Trudeau JB, Ray A, Ray P, et al. IL-27 and type 2 immunity in asthmatic patients: association with severity, CXCL9, and signal transducer and activator of transcription signaling. J Allergy Clin Immunol. (2015) 135:386–94. doi: 10.1016/j.jaci.2014.08.023
66. Tan KS, Ong HH, Yan Y, Liu J, Li C, Ong YK, et al. In vitro model of fully differentiated human nasal epithelial cells infected with rhinovirus reveals epithelium-initiated immune responses. J Infect Dis. (2018) 217:906–15. doi: 10.1093/infdis/jix640
67. Fulkerson PC, Zimmermann N, Brandt EB, Muntel EE, Doepker MP, Kavanaugh JL, et al. Negative regulation of eosinophil recruitment to the lung by the chemokine monokine induced by IFN-gamma (Mig, CXCL9). Proc Natl Acad Sci USA. (2004) 101:1987–92. doi: 10.1073/pnas.0308544100
68. Fulkerson PC, Zhu H, Williams DA, Zimmermann N, Rothenberg ME. CXCL9 inhibits eosinophil responses by a CCR3- and Rac2-dependent mechanism. Blood (2005) 106:436–43. doi: 10.1182/blood-2005-02-0489
69. van Schaik SM, Tristram DA, Nagpal IS, Hintz KM, Welliver RC III, Welliver RC. Increased production of IFN-γ and cysteinyl leukotrienes in virus-induced wheezing. J. Allergy Clin Immunol. (1999) 103:630–6. doi: 10.1016/S0091-6749(99)70235-6
70. Gentile DA, Fireman P, Skoner DP. Elevations of local leukotriene C4 levels during viral upper respiratory tract infections. Ann Allergy Asthma Immunol. (2003) 91:270–4. doi: 10.1016/S1081-1206(10)63529-6
71. Sun LH, Chen AH, Yang ZF, Chen JJ, Guan WD, Wu JL, et al. Respiratory syncytial virus induces leukotriene C4 synthase expression in bronchial epithelial cells. Respirology (2018) 18:40–6. doi: 10.1111/resp.12188
72. Bisgaard H, Study Group on Montelukast and Respiratory Syncytial Virus. A randomized trial of montelukast in respiratory syncytial virus postbronchiolitis. Am J Respir Crit Care Med. (2003) 167:379–83. doi: 10.1164/rccm.200207-747OC
73. Bisgaard H, Zielen S, Garcia-Garcia ML, Johnston SL, Gilles L, Menten J, et al. Montelukast reduces asthma exacerbations in 2- to 5-year-old children with intermittent asthma. Am J Respir Crit Care Med. (2005) 171:315–22. doi: 10.1164/rccm.200407-894OC
74. Noguchi T, Nakagome K, Kobayashi T, Ueda Y, Uchida Y, Soma T, et al. Effect of LTRA on IP-10-induced eosinophil adhesion to ICAM-1. Allergol Int. (2016) 65:S62–4. doi: 10.1016/j.alit.2016.04.015
75. Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N, et al. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol. (2008) 20:791–800. doi: 10.1093/intimm/dxn037
76. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature (2010) 464:1367–70. doi: 10.1038/nature08900
77. Smith SG, Chen R, Kjarsgaard M, Huang C, Oliveria JP, O'Byrne PM, et al. Increased numbers of activated group 2 innate lymphoid cells in the airways of patients with severe asthma and persistent airway eosinophilia. J Allergy Clin Immunol. (2016) 137:75–86. doi: 10.1016/j.jaci.2015.05.037
78. Jackson DJ, Makrinioti H, Rana BM, Shamji BW, Trujillo-Torralbo MB, Footitt J, et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am J Respir Crit Care Med. (2014) 190:1373–82. doi: 10.1164/rccm.201406-1039OC
79. Medzhitov R. Origin and physiological roles of inflammation. Nature (2008) 454:428–35. doi: 10.1038/nature07201
80. Stenfeldt AL, Wenneras C. Danger signals derived from stressed and necrotic epithelial cells activate human eosinophils. Immunology (2004) 112:605–14. doi: 10.1111/j.1365-2567.2004.01906.x
81. Kobayashi T, Kouzaki H, Kita H. Human eosinophils recognize endogenous danger signal crystalline uric acid and produce proinflammatory cytokines mediated by autocrine ATP. J Immunol. (2010) 184:6350–8. doi: 10.4049/jimmunol.0902673
82. Kobayashi T, Soma T, Noguchi T, Nakagome K, Nakamoto H, Kita H, et al. ATP drives eosinophil effector responses through P2 purinergic receptors. Allergol Int. (2015) 64:S30–6. doi: 10.1016/j.alit.2015.04.009
83. Wenzel SE, Schwartz LB, Langmack EL, Halliday JL, Trudeau JB, Gibbs RL, et al. Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am J Respir Crit Care Med. (1999) 160:1001–8. doi: 10.1164/ajrccm.160.3.9812110
84. The ENFUMOSA and Study Group. The ENFUMOSA cross-sectional European multicentre study of the clinical phenotype of chronic severe asthma. European network for understanding mechanisms of severe asthma. Eur Respir J. (2003) 22:470–7. doi: 10.1183/09031936.03.00261903
85. Kikuchi S, Nagata M, Kikuchi I, Hagiwara K, Kanazawa M. Association between neutrophilic and eosinophilic inflammation in patients with severe persistent asthma. Int Arch Allergy Immunol. (2005) 137:7–11. doi: 10.1159/000085425
86. Ricciardolo FLM, Sorbello V, Folino A, Gallo F, Massaglia GM, Favatà G, et al. Identification of IL-17F/frequent exacerbator endotype in asthma. J Allergy Clin Immunol. (2017) 140:395–406. doi: 10.1016/j.jaci.2016.10.034
87. Shannon J, Ernst P, Yamauchi Y, Olivenstein R, Lemiere C, Foley S, et al. Differences in airway cytokine profile in severe asthma compared to moderate asthma. Chest (2008) 133:420–6. doi: 10.1378/chest.07-1881
88. Kikuchi S, Kikuchi I, Takaku Y, Kobayashi T, Hagiwara K, Kanazawa M, et al. Neutrophilic inflammation and CXC chemokines in patients with refractory asthma. Int Arch Allergy Immunol. (2009) 149:87–93. doi: 10.1159/000211379
89. Kikuchi I, Kikuchi S, Kobayashi T, Hagiwara K, Sakamoto Y, Kanazawa M, et al. Eosinophil trans-basement membrane migration induced by interleukin-8 and neutrophils. Am J Respir Cell Mol Biol. (2006) 34:760–5. doi: 10.1165/rcmb.2005-0303OC
90. Coëffier E, Joseph D, Vargaftig BB. LTB4, a potent chemotactic factor for purified guinea-pig eosinophils: interference of PAF-acether antagonists. Int J Immunopharmacol. (1991) 13:273–80. doi: 10.1016/0192-0561(91)90108-J
92. Nakagome K, Matsushita S, Nagata M. Neutrophilic inflammation in severe asthma. Int Arch Allergy Immunol. (2012) 158:96–102. doi: 10.1159/000337801
93. Hauk PJ, Krawiec M, Murphy J, Boguniewicz J, Schiltz A, Goleva E, et al. Neutrophilic airway inflammation and association with bacterial lipopolysaccharide in children with asthma and wheezing. Pediatr Pulmonol. (2008) 43:916–23. doi: 10.1002/ppul.20880
94. Goleva E, Hauk PJ, Hall CF, Liu AH, Riches DW, Martin RJ, et al. Corticosteroid-resistant asthma is associated with classical antimicrobial activation of airway macrophages. J Allergy Clin Immunol. (2008) 122:550–9. doi: 10.1016/j.jaci.2008.07.007
95. Nishihara F, Nakagome K, Kobayashi T, Noguchi T, Araki R, Uchida Y, et al. Trans-basement membrane migration of eosinophils induced by LPS-stimulated neutrophils from human peripheral blood in vitro. ERJ Open Res. (2015) 1:00003-2015. doi: 10.1183/23120541.00003-2015
96. Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. (2007) 25:821–52. doi: 10.1146/annurev.immunol.25.022106.141557
97. Barczyk A, Pierzchala W, Sozanska E. Interleukin-17 in sputum correlates with airway hyperresponsiveness to methacholine. Respir Med. (2003) 97:726–33. doi: 10.1053/rmed.2003.1507
98. Al-Ramli W, Préfontaine D, Chouiali F, Martin JG, Olivenstein R, Lemière C, et al. TH17-associated cytokines (IL-17A and IL-17F) in severe asthma. J Allergy Clin Immunol. (2009) 123:1185–7. doi: 10.1016/j.jaci.2009.02.024
99. Nakagome K, Imamura M, Okada H, Kawahata K, Inoue T, Hashimoto K, et al. Dopamine D1-like receptor antagonist attenuates Th17-mediated immune response and ovalbumin antigen-induced neutrophilic airway inflammation. J Immunol. (2011) 186:5975–82. doi: 10.4049/jimmunol.1001274
100. Toussaint M, Jackson DJ, Swieboda D, Guedán A, Tsourouktsoglou TD, Ching YM, et al. Host DNA released by NETosis promotes rhinovirus-induced type-2 allergic asthma exacerbation. Nat Med. (2017) 23:681–91. doi: 10.1038/nm.4332
101. Balzar S, Fajt ML, Comhair SA, Erzurum SC, Bleecker E, Busse WW, et al. Mast cell phenotype, location, and activation in severe asthma. Data from the Severe Asthma Research Program. Am J Respir Crit Care Med. (2011) 183:299–309. doi: 10.1164/rccm.201002-0295OC
102. Fajt ML, Gelhaus SL, Freeman B, Uvalle CE, Trudeau JB, Holguin F, et al. Prostaglandin D2 pathway upregulation: relation to asthma severity, control, and TH2 inflammation. J Allergy Clin Immunol. (2013) 131:1504–12. doi: 10.1016/j.jaci.2013.01.035
103. Singh D, Ravi A, Southworth T. CRTH2 antagonists in asthma: current perspectives. Clin Pharmacol. (2017) 9:165–173. doi: 10.2147/CPAA.S119295
104. Hirai H, Tanaka K, Yoshie O, Ogawa K, Kenmotsu K, Takamori Y, et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med. (2001) 193:255–61. doi: 10.1084/jem.193.2.255
105. Royer JF, Schratl P, Lorenz S, Kostenis E, Ulven T, Schuligoi R, et al. A novel antagonist of CRTH2 blocks eosinophil release from bone marrow, chemotaxis and respiratory burst. Allergy (2007) 62:1401–9. doi: 10.1111/j.1398-9995.2007.01452.x
106. Barnes N, Pavord I, Chuchalin A, Bell J, Hunter M, Lewis T, et al. A randomized, double-blind, placebo-controlled study of the CRTH2 antagonist OC000459 in moderate persistent asthma. Clin Exp Allergy (2012) 42:38–48. doi: 10.1111/j.1365-2222.2011.03813.x
107. Gonem S, Berair R, Singapuri A, Hartley R, Laurencin MFM, Bacher G, et al. Fevipiprant, a prostaglandin D2 receptor 2 antagonist, in patients with persistent eosinophilic asthma: a single-centre, randomised, double-blind, parallel-group, placebo-controlled trial. Lancet Respir Med. (2016) 4:699–707. doi: 10.1016/S2213-2600(16)30179-5
Keywords: bronchial asthma, eosinophils, neutrophils, periostin, rhinovirus
Citation: Nakagome K and Nagata M (2018) Involvement and Possible Role of Eosinophils in Asthma Exacerbation. Front. Immunol. 9:2220. doi: 10.3389/fimmu.2018.02220
Received: 30 May 2018; Accepted: 07 September 2018;
Published: 28 September 2018.
Edited by:
Shigeharu Ueki, Akita University, JapanReviewed by:
Bruno L. Diaz, Universidade Federal do Rio de Janeiro, BrazilCalman Prussin, Knopp Biosciences LLC, United States
Copyright © 2018 Nakagome and Nagata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazuyuki Nakagome, bmFrYWdvbWVrLXRreUB1bWluLmFjLmpw