 Seungwon Ryu
Seungwon Ryu Joon Seok Park
Joon Seok Park Hye Young Kim
Hye Young Kim Ji Hyung Kim
Ji Hyung Kim- 1Department of Biomedical Sciences, Seoul National University College of Medicine, Seoul, South Korea
- 2Institute of Allergy and Clinical Immunology, Seoul National University Medical Research Center, Seoul, South Korea
- 3Department of Microbiology and Immunobiology, Harvard Medical School, Boston, MA, United States
- 4College of Life Sciences and Biotechnology, Korea University, Seoul, South Korea
Regulation of T cell-mediated immunity in the lungs is critical for prevention of immune-related lung disorders and for host protection from pathogens. While the prevalent view of pulmonary T cell responses is based on peptide recognition by antigen receptors, called T cell receptors (TCR), on the T cell surface in the context of classical major histocompatibility complex (MHC) molecules, novel pathways involving the presentation of lipid antigens by cluster of differentiation 1 (CD1) molecules to lipid-reactive T cells are emerging as key players in pulmonary immune system. Whereas, genetic conservation of group II CD1 (CD1d) in mouse and human genomes facilitated numerous in vivo studies of CD1d-restricted invariant natural killer T (iNKT) cells in lung diseases, the recent development of human CD1-transgenic mice has made it possible to examine the physiological roles of group I CD1 (CD1a-c) molecules in lung immunity. Here, we discuss current understanding of the biology of CD1-reactive T cells with a specific focus on their roles in several pulmonary disorders.
Introduction
The respiratory system comprises the airways and lungs; as such, it is exposed continuously to foreign materials. Consequently, the immune system in the respiratory system experiences both a huge load and a great variety of foreign antigens. This is significant because this exposure can induce immune reactions that damage lung tissue, thereby transiently or permanently impairing vital respiratory functions. The unique exposure profile of the lung has led to the evolution of immune responses that are not observed in lymphoid organs (1). For example, the surface layer of the lung is composed of airway epithelium, which is a specialized tissue that bears cilia. The epithelial cells secrete mucus that traps inhaled foreign materials; the cilia then move the mucus out of the respiratory system. The lung also harbors a considerable number of innate immune cells, including alveolar macrophages, interstitial macrophages, dendritic cells (DCs), and innate lymphoid cells, all of which act as first line defenders and activate the adaptive immune system, namely, B and T cells. T cells play a particularly important role in controlling immune responses associated with various lung diseases.
Classical T cells recognize foreign or self-antigens complexed with major histocompatibility complex (MHC) I and II molecules on the surface of antigen-presenting cells. When these complexes are recognized by T cell receptors (TCRs), the T cells differentiate into effector cells capable of secreting inflammatory cytokines and cytotoxic proteins that orchestrate downstream immune responses. The role of classical T cells in respiratory health and disease has been well studied [Chen and Kolls published a review on these cells (2)]. However, recent evidence suggests that unconventional subsets of T cells that react to lipid antigens presented
on cluster of differentiation 1 (CD1) molecules also play critical roles in pulmonary immune responses. While the percentage of CD1-reactive T cells in the lung is unclear, a previous study reports a significant percentage of these cells among circulating TCRαβ+ cells (7% CD4+ and 0.2% CD4−CD8−) within human PBMC population (3).
While CD1 proteins are expressed in every mammalian species that was examined, humans express five isoforms of CD1, categorized as group 1 molecules (CD1a, CD1b, and CD1c), group 2 molecules (CD1d), and group 3 molecules (CD1e) (4–7). All CD1 molecules, apart from CD1e, display lipid antigens within their hydrophobic antigen-binding groove. The T cells that recognize lipids bound to group 2 CD1 (CD1d) molecules are known as natural killer T (NKT) cells; these are the best studied among the CD1-restricted T cells. This reflects the fact that, of all the CD1 molecules, only the CD1d gene is conserved in mice and humans: mice lack the other four CD1 isoforms. The presence of CD1d in mice greatly facilitated research on NKT cells, which has shown that these cells play pivotal roles in development of asthma, pulmonary infection, fibrosis, and other pulmonary disorders. NKT cells represent 5–10% of all T cells in the lungs of adult mice (8). Of the iNKT cells in the lung, 0.08% of CD45+ cells are present in the lung parenchyma and 0.2% are present in the lung vasculature (9). By contrast, T cells in the lungs that are restricted by the group 1 CD1 molecules (CD1a–c) are less well studied. However, a number of recent studies suggest that these T cells also participate actively in immune responses in barrier organs such as the skin and the lungs.
T cells are particularly promising targets in terms of treatments for various lung diseases (2). To facilitate development of these therapies, a better understanding of lipid-reactive T cells is required. To address this, this review will describe the general features of lipid-reactive T cells and the roles they play in development of various pulmonary disorders, including asthma, chronic obstructive pulmonary disorder (COPD), fibrosis, infection, and cancer.
The CD1 Family of Lipid Antigen-Presenting Molecules
Classical MHC class I and II molecules present peptide antigens to T cells. By contrast, CD1 molecules present hydrophobic antigens such as lipids to T cells (9). Structurally, CD1 molecules resemble MHC class I molecules in that they comprise an α-chain bearing three domains (α-1, α-2, and α-3), which is bound non-covalently to β2-microglobulin (β2m). However, CD1 molecules have some unique characteristics that distinguish them from MHC class I molecules. First, human CD1 genes are located on chromosome 1, whereas human MHC genes are located on chromosome 6 (in mice, CD1 genes are on chromosome 3 and MHC genes are on chromosome 17) (7). Second, CD1 molecules are less polymorphic. Third, MHC I and II molecules have six pockets in their antigen-binding groove (denoted A–F) whereas the binding groove of CD1 molecules harbors at least two antigen-binding pockets, named A′ and F′; however, these pockets are narrower and deeper than the A–F pockets in MHC molecules. In addition, these pockets are enriched in hydrophobic residues, which aid the stable binding of lipids to the CD1 groove. The subfamilies of CD1 molecules differ in terms of the size (volume and shape) and properties of these antigen-binding pockets. As a result, the CD1 molecules as a group can present a variety of hydrophobic antigens to T cells (10). Various foreign- and self- antigens that react with CD1-reactive T cells have been identified. These antigens include lipids, phospholipids, glycolipids, and lipopeptides with a large spectrum of size and polarity (10). In general, the hydrocarbon tails, usually alkyl chains, of lipids are buried in the pocket of CD1 molecules and the polar portions protrude, thereby providing a template for TCR engagement (11). Recent studies suggest that many lipid ligands of CD1 molecules, especially CD1c, do not interact directly with TCR; rather they affect the interaction between the TCR and CD1 molecules, thereby allowing or blocking activation of autoreactive T cells (11, 12).
All CD1 isoforms, except CD1e, present antigen. Mice express only CD1d (13), while other mammals (ranging from alpacas to sloths) harbor different combinations of the five CD1 isoforms [these are summarized in the Table 1 from (7)]. CD1e participates in presentation of lipid antigens only indirectly: it trims and transfers lipid antigens prior to presentation to other CD1 molecules (14, 15). CD1a-c molecules are expressed by professional antigen-presenting cells and thymocytes. In particular, Langerhans cells prominently express CD1a while DCs express CD1b and marginal zone B cells express CD1c. The group 2 CD1 molecule, CD1d, is expressed by both hematopoietic and non-hematopoietic cells in various organs, including skin, liver, and colon (16, 17). These differential expression patterns of CD1 molecules suggest that the individual CD1 isoforms may shape local T cell responses by presenting tissue-specific lipids. Moreover, when blood monocytes and hematopoietic CD34+ progenitor cells are cultured with granulocyte-macrophage colony stimulating factor (GM-CSF) and IL-4, CD1a expression is induced (18–21). This suggests that CD1 expression can be controlled as necessary in vivo.
CD1 molecules load and present lipid antigens in a manner distinct from that of MHC molecules (22, 23). Initially, they are loaded with self-antigen immediately after synthesis in the endoplasmic reticulum (ER); this process is aided by a lipid-transfer protein (LTP) called microsomal triglyceride transfer protein (24, 25). The CD1 molecules then undergo intracellular trafficking (like other proteins synthesized in the ER). Thus, they enter the secretory pathway, which traffics them from the ER to the Golgi apparatus and then finally to the plasma membrane. Sometimes, CD1 molecules exchange their self-antigen for an exogenous lipid antigen at either the plasma membrane or in various endosomal compartments, including early and late endosomes and lysosomes (26). Different CD1 molecules preferentially enter different endosomal compartments due to their disparate sorting motifs. For example, CD1b bears the AP3 sorting motif, which promotes entry into lysosomes (17). In addition, CD1a molecules lack sorting motifs and are therefore recycled to early endocytic compartments such as Birbeck granules, a specialized organelle found in Langerhans cells. Trafficking of CD1a molecules to this compartment is important for lipid antigen presentation to CD1a-restricted T cells (27). These disparate trafficking pathways suggest that different CD1 molecules capture distinct subsets of foreign antigens. In the endosomal compartments, capture of exogenous lipids is mediated by LTPs, including the CD1e molecule. For example, the self-lipid antigens expressed on CD1d molecules are removed and replaced by foreign lipid antigens with the help of a lysosomal LTP called saposin (28, 29). It should be noted that while some CD1 molecules retain the self-antigen that they received at the ER, they can also be loaded with another self-antigen. These CD1: self-antigen complexes can be recognized by autoreactive T cells. The original self-antigen can also function as a chaperone that promotes CD1 stability and trafficking to the plasma membrane, or as a determinant of activation of autoreactive T cell activation (12, 17).
Lipid-Reactive T Cells
Group 1 CD1-Restricted T Cells
Research on group 1 CD1 molecules (CD1a-c) is limited by the fact that mice lack homologs of these proteins. Therefore, most studies have been conducted in vitro using human cells. T cells that recognize CD1a-c are more common in human peripheral blood than T cells that recognize CD1d: ~2%, ~1%, and ~7% of αβTCR+ cells in human peripheral blood recognize CD1a, CD1b, and CD1c, respectively, whereas only ~0.1% of αβTCR+ cells recognize CD1d (3, 30). This indicates the need for further studies on the functions of T cells that are restricted by CD1a-c, even though there are some discrepancies regarding their percentages in different models/individuals.
Difficulties associated with studying CD1a-c-restricted T cells can be overcome by using a humanized CD1 transgenic mouse model (hCD1Tg) (31, 32) or humanized SCID mice that have been engrafted with human thymus, liver, and CD34+ hematopoietic cells (33). Felio et al. used hCD1Tg mice to examine responses of CD1a-c-restricted T cells to Mycobacterium tuberculosis (Mtb) infection. They showed that Mtb-responsive CD1a-c-restricted T cells did not respond quickly to the infection; rather, they became activated later. Moreover, upon second stimulation, they showed boosted responses. Thus, they do not have the innate immune cell-like activities of NKT cells, which are restricted by CD1d and exhibit strong early responses; rather, they more closely resemble classical adaptive lymphocytes (31). This finding was validated by de Lalla et al., who showed that CD1a-c-restricted and self-reactive T cells within adult PBMCs are more likely to be memory T cells than are the same cell populations in umbilical cord blood (3). However, CD1a-c-restricted T cells do not differ from CD1d-restricted T cells in terms of their ability to secrete Th1/Th17 (31, 34) and Th2 (35, 36) cytokines. The percentage and number of CD1d-restricted T cells in the mouse spleen increase with age (37). However, in human blood, age is inversely related with the frequency of CD1d-restricted T cells (38), suggesting that aging-associated accumulation of NKT cells may depend on the species or environment. Classical T cells also exhibit age-related changes in memory phenotype (3).
Although our understanding of development of CD1a-c-restricted T cells remains poor, Li et al. followed the development of HJ1, a human T cell clone restricted by CD1b; they did this by creating a double transgenic mouse that expressed the TCR of the clone and human CD1b (HJ1Tg/hCD1Tg mice) (34). They showed that to develop properly, HJ1 T cells must encounter CD1b-expressing hematopoietic cells in the thymus. In the absence of CD1b-mediated thymic selection, most HJ1 T cells remained at the CD4+CD8+ double-positive (DP) stage and did not undergo positive selection. However, if CD1b-expressing hematopoietic cells were present, HJ1 T cells underwent positive and negative selection. During this process, the cells changed from DP cells to DPdull and then to double-negative cells. These changes were accompanied by expression of activation markers CD69, CD122, and CD44. They also expressed PLZF (promyelocytic leukemia zinc finger, which is encoded by Zbtb16), as did CD1d-restricted invariant NKT (iNKT) cells. These observations suggest that CD1a-c-restricted T cells may develop via the same developmental pathway used by iNKT cells (34). However, further research into the mechanism by which CD1a-c-restricted T cells develop, and into the endogenous lipids involved in this process, is needed.
Group 2 CD1-Restricted T Cells
Group 2 CD1 (CD1d)-restricted T cells are called NKT cells. They are a small subset of CD1-restricted αβ T cells that express both NK cell markers (NK1.1 in mice and CD161 in humans) and TCRs. They respond quickly during the early phase of immune reactions by secreting cytokines. They also play a critical role in bridging the innate and adaptive immune responses (39).
There are two subsets of NKT cells. Type I NKT cells are referred to as invariant NKT (iNKT) cells because they have a semi-invariant TCR that is encoded by the Vα14-Jα18 or Vα24-Jα18 TCRα gene segments in mice and humans, respectively. iNKT cells respond strongly to α-galactosylceramide (α-GalCer), a glycolipid ligand first isolated from a marine sponge, when it is presented on CD1d molecules (40). Several other endogenous ligands, including α-linked glycosylceramides (41) and β-linked glycosylceramides (42) stimulate iNKT cells. However, the ligands that are responsible for the selection of iNKT cells in the thymus remain unclear at present. However, the fact that the β-linked glycosylceramides used for some research turned out to be contaminated with α-linked anomers, which are very potent stimulators of iNKT cells, negates some of the findings (41, 43, 44). In addition to α-linked monoglycosylceramides, isoglobotrihexosylceramide (iGb3), a lysosomal glycosphingolipid, is an endogenous ligand for iNKT cells (45). Mice deficient in hexosaminidase b (Hexb), which converts iGb4 to iGb3, fail to develop iNKT cells, supporting the in vivo significance of iGb3 as an endogenous ligand (45). However, iGb3 synthase-deficient mice exhibit normal development of iNKT cells (46). Still, given that iGb3 stimulates iNKT cells in a TCR-dependent manner, it might be a bona fide endogenous ligand. Several exogenous ligands derived from bacteria and fungi have been identified. Glycosylceramides derived from the cell wall of Sphingomonas, and glycolipids from Streptococcus pneumoniae, activate iNKT cells in a CD1d-dependent manner (47–49). In addition to bacterial products, a fungal glycolipid called asperamide B can be presented on CD1d, thereby stimulating both human and mouse iNKT cells (50). Interestingly, microbial products can activate iNKT cells via indirect mechanisms by inducing endogenous glycolipid ligands (51, 52). TLR pathways facilitate biosynthesis of glycosphingolipids, which are endogenous ligands for iNKT cells. Of note, cytokines take part in the TLR-mediated iNKT cell activation. TLR4 and TLR9 mediate IFN-γ production by iNKT cells, not IL-4 production, in IL-12- and type I IFN dependent manners, respectively (51). Such cytokine contributions require TCR engagement for the optimal iNKT activation. However, it is also shown that cytokines themselves could activate iNKT cells independently to TCR triggers at certain levels (51–53). Moreover, cytokines and antigens are differentially required for cytokine production by iNKT cells, depending on the stimulants (54). TLR-induced, not ligand-driven, IFN-γ production by iNKT cells is impaired in the absence of IL-12. On the other hand, ligand- or TLR- mediated IL-4 production by iNKT cells does not require IL-12. During infections, iNKT cells produce IFN-γ in a manner dependent on IL-12 and IL-18; However, expression of IL-4 is independent of IL-12 but dependent on CD1 ligands and MyD88 (54, 55). Interestingly, IL-4 production in iNKT cells is prominent under Th2 inflammatory settings found in airway inflammation (56).
Despite expressing an invariant TCR, iNKT cells are quite heterogenous. When Watarai et al. examined expression of IL17RB (a receptor for IL-25) and CD4 in response to IL-2, they identified three subpopulations of thymic iNKT cells; namely, a CD4+IL17RB+ population that produces Th2 and Th17 cytokines (IL-13, IL-9, IL-10, IL-17A, and IL-22), a CD4−IL17RB+ population that produces Th17 cytokines (IL-17 and IL-22), and a CD4+IL17RB− subset that produces IFN-γ (57). These findings were validated by the study of Lee et al., who showed that NKT cells can be classified as T-bet-expressing NKT1, GATA-3-expressing NKT2, and RORγt-expressing NKT17 lineages (58). Recently, two studies identified new NKT subsets; namely, Bcl-6-expressing follicular helper NKTFH (59) and Nfil3-expressing NKT10 (60, 61) cells. In terms of thymic development (Figure 1), iNKT cells develop from a common lymphoid progenitor. Interestingly, however, positive selection of iNKT cells is mediated by DP thymocytes (CD4+CD8+), which present the lipid-CD1d complex, rather than by thymic epithelial cells (62). After positive selection, iNKT cells progress from stage 0 to stage 3 (63). Thus, CD24highCD44lowNK1.1− stage 0 NKT precursors first develop into immature stage 1 cells that express CD24low; they also express GATA-3 and therefore produce IL-4 (64). Stage 1 cells then become CD24lowCD44highNK1.1− stage 2 NKT cells, which eventually develop into mature CD24lowCD44highNK1.1+ stage 3 NKT cells that produce IFN-γ (65–67). PLZF is thought to be the key transcription factor that drives NKT cell differentiation (68, 69). While all NKT cells at all stages express Zbtb16 (the gene that encodes PLZF), conditional knockout of Zbtb16 in NKT cells showed that NKT cells cannot develop into immature stage 2 NKT cells without PLZF; mutant mice accumulated stage 1 NKT precursors in the thymus (68). However, considering that NK1.1 is not expressed by all mouse strains, another model of NKT development has been proposed; namely, that the NKT1, NKT2, and NKT17 lineages develop separately from a common NKT precursor rather than by passing sequentially from stage 0 to stage 3 (58, 70).
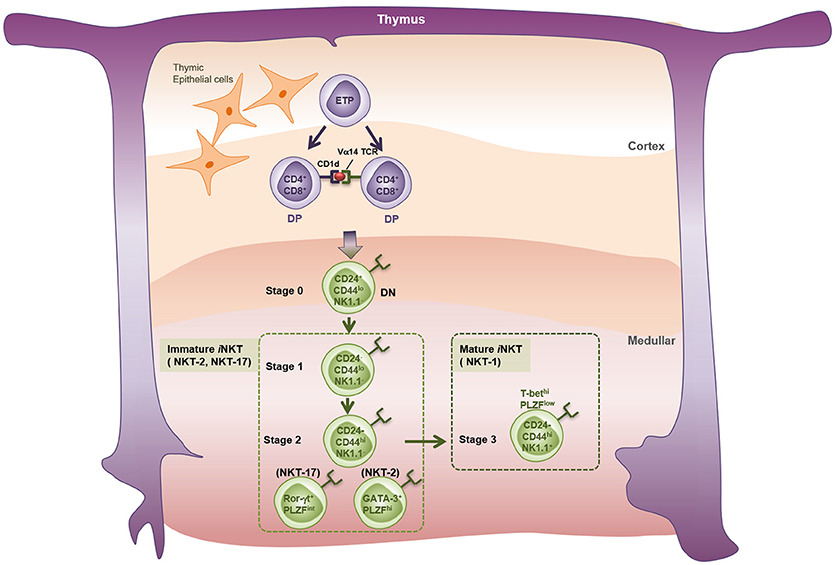
Figure 1. Thymic development of iNKT cells. iNKT cell precursors originate from the common thymic precursors of conventional T cells. Early T-cell precursors (ETP) give rise to CD4 and CD8 double positive (DP) thymocytes expressing TCRβ chain and rearranged TCRα chains in the thymic cortex. Thymocytes bearing Vα14 TCRs undergo the positive selection by CD1d-expressng DP thymocytes, but not by thymic cortical epithelial cells, and enter the lineage of iNKT cells. Endogenous lipid antigens responsible for the development of iNKT cells are not fully described. Classically, the thymic iNKT cells detected by α-GalCer-loaded CD1d-tetramer are divided into four developmental stages based on their surface markers. Stage 0: CD24+CD44−NK1.1−, Stage 1: CD24−CD44−NK1.1−, Stage 2: CD24−CD44+NK1.1−, Stage 3: CD24−CD44+NK1.1+. A new perspective has categorized thymic iNKT cells into three subtypes on the basis of expression patterns of specific transcription factors and cytokines. NKT-1: T-bethiPLZFlo and IFN-γ+, NKT-2: GATA-3+PLZFhi and IL-4+, NKT-17: Rorγt+PLZFinter, and IL-17+.
The second subset of NKT cells, namely, type II NKT cells, express a variable TCR that is restricted by CD1d but is not recognize by α-GalCer (71). Instead, type II NKT cells can be stimulated by sulfatide, β-GalCer, phosphatidylinositol, and phosphatidylglycerol (72). Studies of type II NKT cells are limited by a lack of specific markers for these cells and by a paucity of techniques that permit their analysis (by contrast, iNKT cells can be readily and specifically detected using CD1d/α-GalCer tetramers) (73). However, several experimental strategies have enabled study of the functions of type II NKT cells; these include the use of Jα18-deficient IL-4 reporter mice (74) and 24αβ transgenic mice that express the TCR of a type II NKT cell clone (75). These studies showed that, like iNKT cells, type II NKT cells develop via a linear maturation process marked by changes in expression of CD44 and NK1.1; moreover, like iNKT cells, they differentiate into NKT1, NKT2, and NKT17 cells, as shown by their expression of key transcription factors. While these cells remain relatively poorly characterized, it is possible that type II NKT cells may contribute to lung pathology in humans; indeed, evidence suggests that their close cousins, type I NKT cells, play a role in lung disorders (see below for more details). Moreover, humans harbor more type II NKT cells than type I NKT cells (76). Although the CD1-reactive T cell population in the lung has not been analyzed thoroughly, the spatial distribution of different subsets of iNKT cells in the mouse lung was investigated using a novel approach aimed at distinguishing iNKT cells in lung parenchyma from those in lung vasculature (9). The dominant iNKT subset in lung parenchyma was iNKT17, whereas that in lung vasculature was iNKT1. In both compartments, iNKT2 was the least populating subset. Accumulation of pulmonary iNKT cells relies on the CXCR6-CXCL16 axis (77). By contrast, intratracheal injection of an α-GalCer analog PBS57 promotes extravasation of CD1d-restricted T cells to the lung parenchyma via interaction between CCR4 and CCL17 (8).
Lipid-Reactive T Cells in Pulmonary Disorders
Asthma
Asthma is a well-known allergic disease of the lung that affects over 7% of adults in the United States of America (78). Symptoms include shortness of breath, coughing, wheezing, and airway hyper-responsiveness (AHR). Asthma can be classified broadly into two phenotypes, allergic and non-allergic, based on production of critical immune mediators. Allergic asthma accounts for more than 50% of asthma cases. It is known as a “Th2 disorder” because type 2 cytokines are pivotal for progression of the disease (79). While conventional Th2 cells contribute to development of allergic asthma, many studies suggest that lipid-reactive NKT cells and innate lymphoid cells may also play a role in this pathology (80, 81).
A variety of studies suggested that CD1d-restricted NKT cells could be critical inducers of asthma in murine models. An ovalbumin (OVA)-induced model of asthma first showed that mice lacking iNKT cells (i.e., CD1d- or Vα14-Jα18-deficient mice) do not develop AHR, which is a critical feature of asthma (82–85). Mechanistically, IL-4 and IL-13 which are produced by iNKT cells, are essential for development of OVA-induced AHR (83). Other asthma-related features, including airway eosinophilia, the elevation of both type 2 cytokine levels in bronchoalveolar lavage fluid (BALF), and allergen-specific IgE levels in serum, are also dependent on iNKT cells. Moreover, iNKT cells induce asthma even when CD4+ T cells are absent; when MHC II-deficient mice (which lack conventional CD4+ T cells but have iNKT cells) were challenged with α-GalCer, the mice developed AHR equally as well as the wild-type mice (86). The contribution of iNKT cells to development of asthma has also been tested with respect to real-world allergens (rather than OVA or α-GalCer, which are experimental triggers). iNKT-deficient mice develop less allergic asthma compared to wild-type mice when exposed to ragweed (87), house dust extracts (88), or a fungus (Aspergillus fumigatus) (50). In addition, Pichavant et al. showed that iNKT cells expressing IL-17A are essential for development of non-allergic asthma induced by exposure to ozone, another real-world cause of asthma (89).
However, studies by other groups suggest that iNKT cells are not required for allergic airway inflammation characterized by eosinophilia and type 2 cytokine production (90–93). These studies show that airway inflammation is dependent on MHC class II molecules, not on CD1d molecules (90, 92). Also, β2m-deficient mice, which are unable to express CD1d molecules, develop asthma despite lacking iNKT cells (91, 93, 94). However, Koh et al. suggested that some NKT cells in β2m-deficient mice are restricted to a β2m-independent form of CD1d, and these cells can compensate for deficiency of conventional iNKT cells. Despite this possibility, iNKT cells might become more dispensable than T cells with respect to inducing AHR when mice are challenged repeatedly with antigens such as OVA (90, 92).
It should be noted that Jα18 (or Traj18) knockout mice, which are often used to study the roles of iNKT cells and type II NKT cells, show a significant (>60%) reduction in TCRα chain diversity (95). A recent study attempted to resolve this issue by developing a new type of Traj18 knockout mouse strain lacking iNKT cells but with normal TCRα diversity. When OVA- and cockroach allergen-induced asthma were assessed in these mice, knockout mice showed significantly less severe AHR than wild-type mice (96). Taken together, the results from murine models of asthma are conflicting; however, iNKT cells may contribute to the pathogenesis of asthma under certain conditions.
Akbari et al. ( 97) conducted flow cytometry experiments and found that BALF from patients with moderate to severe asthma contained a very high percentage of iNKT cells (about 60% of all CD3+ cells); BALF from healthy subjects contained very few iNKT cells (97). It should be noted, however, that several other groups found low percentages (<2%) of iNKT cells in BALF from adult asthmatics; these percentages did not differ significantly from those in control subjects detected using CD1d tetramers, an anti-Vα24 antibody, or an anti-Vβ11 antibody (98–101). It was proposed that differences in the percentage of iNKT cells in BALF, sputum, or bronchial biopsies may be due to nonspecific binding of CD1d-tetramers, or to inappropriate gating on iNKT cells (102). Other human studies noted increased percentages of iNKT cells in BALF from childhood asthma patients (0.435% of αβ T cells in asthma vs. 0.116% of αβ T cells in control subjects) (103), and in the lungs of patients with severe asthma compared with patients with well controlled asthma (104). Thus, the link between the percentage of iNKT cells in human lung or bronchial specimens from asthma patients and disease pathogenesis remains still unclear.
A functional study from the United Kingdom, which was based on a human allergen challenge model of asthma, revealed that the percentage of iNKT cells in bronchial biopsies taken 24 h after allergen challenge increased by 15% along with the increase in AHR (105). Moreover, Agea et al. showed that the PBMCs from subjects who were allergic to cypress pollen responded to two specific lipid antigens in the pollen, namely, phosphatidylcholine and phosphatidylethanolamine. These lipid antigens, which are loaded onto CD1 molecules, stimulated T cells within the PBMCs in CD1a- or CD1d-dependent fashion (106). Thus, both CD1a- and CD1d-restricted T cells may participate in allergic reactions in humans.
Although most studies to date have focused on the pathogenic roles of iNKT cells in asthma, iNKT cells also have protective functions under specific preconditions. For example, when young mice are infected with influenza A virus, they are protected against allergen-induced AHR when they become adults (107). CD4−CD8− DN NKT cells showing high expression of CD38 produce IFN-γ and induce CD4 T cell proliferation in a contact dependent manner (108). Glycolipids from Helicobacter pylori activate suppressive NKT cells (107). Notably, Sharma et al. showed that benzo[a]pyrene, a classical environmental air pollutant that causes respiratory disease, suppressed IFN-γ production by CD1a- and CD1d-restricted T cells in vitro by downregulating expression of CD1a and CD1d by human DCs (109). Thus, it appears that certain subsets of NKT cells, such as IFN-γ-producing NKT cells, may suppress AHR.
Because research into the roles of CD1d-restricted iNKT cells in asthma has produced conflicting results, comprehensive studies aimed at characterizing the cause and identifying the contribution made by these cells are needed. Also, we should not overlook the fact that iNKT cells alter the cytokine milieu and affect asthmatic responses under certain conditions. Further studies should determine the role of CD1a-c restricted T cells in the pathogenesis of asthma.
Chronic Obstructive Pulmonary Disease (COPD)
COPD is another chronic pulmonary disease that causes difficulty in breathing; however, it differs from asthma in several aspects. First, it is not an allergic disease. Second, the airway obstruction is largely irreversible. Third, smoking is a well-known risk factor for COPD. Fourth, it is characterized pathologically by either chronic obstructive bronchiolitis or emphysema (110). Although one study shows that the percentage of iNKT cells in induced sputum from COPD patients are not different compared to that in control subjects (101), others suggest that iNKT cells contribute to the pathogenesis of COPD. For example, mice with chronic lung disease caused by Sendai virus infection harbor large numbers of alternatively-activated IL-13-secreting macrophages in the lung. Experiments with CD1d or Jα18 knockout mice revealed that iNKT cells are required to generate virus-induced IL-13-producing macrophages. Thus, iNKT cells facilitate chronic airway inflammation in mice (111). The same authors showed that a similar mechanism may also participate in COPD. Immunofluorescence staining of lung tissues from patients with COPD and from normal controls revealed that the former harbored significantly higher numbers of Vα24+ iNKT cells and IL-13+CD68+ macrophages (111). Wang et al. also showed that patients with COPD harbor higher percentages of CD56+CD3+ NKT cells in the blood than control subjects (although these data should be interpreted carefully due to the possibility of confounders, i.e., smoking, with respect to NKT cells and COPD) (112). In addition, Pichavant et al. found that oxidative stress generated by cigarette smoke activates DCs and airway epithelial cells, which in turn cause iNKT cells to accumulate in the lung and release IL-17 (113). Furthermore, when mice are challenged repeatedly with intranasal α-GalCer, iNKT cells are activated and the mice develop COPD-like symptoms, including mucus hypersecretion and pulmonary emphysema (114). Taken together, these results suggest that iNKT cells might play a pathogenic role in COPD. To date, no study has investigated the role of CD1a-c-restricted T cells in COPD. However, limited number of studies in humans evaluated CD1a expression in DCs from COPD patients (115–117). These studies suggested either no difference or an increase in the number of alveolar langerin-positive and CD1a+ immature DCs in response to chronic cigarette smoking, which contribute to the COPD pathology. The role of CD1a-c-restricted T cells in COPD remains unclear.
Pulmonary Fibrosis
Pulmonary fibrosis is a collection of chronic progressive lung diseases characterized by abnormal wound healing, which in turn generates thickened and stiff lung tissue. The causes of lung fibrosis are numerous; however, most studies on the role of iNKT cells focused on idiopathic pulmonary fibrosis (IPF). IPF is a chronic, progressive fibrotic lung disease that is diagnosed (when no other cause of fibrosis can be found) according to the results of histology or high-resolution CT. With the exception of the conditionally recommended use of pirfenidone and nintedanib (118), no drugs improve the survival of patients with IPF. Therefore, better understanding of the disease is required (119). A common animal model used to study the pathophysiology of IPF is bleomycin-treated mouse; bleomycin is a chemotherapeutic agent that causes lung fibrosis as a side effect. Jα18 knockout and CD1d knockout bleomycin-treated mice exhibited exacerbated symptoms of lung fibrosis (120). Moreover, treatment with α-GalCer ameliorated lung fibrosis in an iNKT cell-dependent manner (121). Of note IFN-γ produced by iNKT cells was responsible for iNKT-mediated protection against fibrosis (120). However, other studies suggest either that iNKT cells are not necessary for lung pathogenesis, or that they play a pathogenic role, when mice are not treated with exogenous ligands (121, 122). Despite this discrepancy with respect to the impact of endogenous ligands or cytokine-driven activation of iNKT cells on disease pathogenesis, exogenous ligand-induced activation of iNKT cells consistently protects against lung fibrosis (120–122). Thus, iNKT cells likely protect against lung fibrosis. In contrast to CD1d-restricted iNKT cells, the function of CD1a-c-restricted T cells has not been studied either human or mouse models of pulmonary fibrosis.
Lung Infection (Pneumonia)
Pneumonia is an inflammatory lung disease caused by viral, bacterial, or fungal infections (123). Unlike the other pulmonary diseases discussed above, many lines of evidence suggest that group 1 CD1-restricted T cells respond to respiratory infections.
Pulmonary infection with Mtb results in an atypical pneumonia called pulmonary tuberculosis (TB). It is a significant public health concern because TB is a highly contagious airborne disease. Moreover, despite development of the M. bovis-derived Bacillus Calmette-Guérin (BCG) vaccine long time ago, it is still difficult to prevent TB in adults. These factors, along with the high prevalence of TB, its disease course, and practical issues regarding long-term treatment regimens, have led to great interest in understanding Mtb infection and in developing new vaccines that will curb TB (124). In 1994, Beckman et al. showed that mycolic acid derived from the Mtb cell wall binds to CD1b (125). Later, Moody et al. found that mannosyl-beta1-phosphodolichol and didehydroxymycobactins, another Mtb lipid and a lipopeptide antigens, activate CD1c- and CD1a- restricted T cells, respectively, as well as peripheral blood cells, from patients with TB (126, 127). The percentages of the group 1 CD1-responding T cells in individuals exposed to Mtb are higher than those in control individuals. Furthermore, they produce IFN-γ responses to lipid antigens derived from Mtb, suggesting protective role against the infection (3, 128). However, group 1 CD1-restrictred T cells produce not only Th1 cytokines, but also Th17 and Th2 cytokines (35, 129, 130). Therefore, further studies should investigate physiological role of Th17 and Th2 cytokine-producing group 1 CD1-restricted T cells in infection. Once the human group 1 CD1 transgenic (hCD1Tg) mice became available, more specific functional studies of CD1-restricted T cells in TB infection have been performed. As mentioned above, Felio et al. showed that, when hCD1Tg mice are infected with Mtb, group 1 CD1-restricted T cells exhibit adaptive cell-type responses, and release IFN-γ at a late time-point after primary exposure to Mtb; however, release is much faster after subsequent exposure (31). Contradictory results were reported by Zhao et al.; When they infected hCD1Tg mice (that were also transgenic for a mycolic acid-specific CD1b-restricted TCR) with TB, they found that CD1b-restricted T cells responded more quickly than Mtb-specific CD4+ T cells to Mtb challenge (131). These disparate reaction patterns may reflect differences in the methods used by Felio et al. and Zhao et al. with respect to antigen challenge; the former used intraperitoneal injection of Mtb (31), whereas the latter used intranasal challenge (131). In any case, these studies show that group 1 CD1-restricted T cells participate in immune responses to Mtb. Moreover, peripheral blood from BCG-immunized subjects contains a high percentage of group 1 CD1-restricted CD8 T cells, which react with live BCG-infected DCs (132). Interestingly, when Parlato et al. compared the gene expression patterns of Mtb antigen-challenged DCs from patients with active and latent TB infection, they found that DCs from patients with active TB lacked CD1a and CD1c expression. By contrast, DCs from patients with latent TB exhibited upregulated expression of CD1a. This suggests that, even though group 1 CD1-restricted T cells may help to protect the host from TB by producing IFN-γ, specific CD1a–c-restricted subsets of these cells may respond to the same infection in different ways (133).
Group 2 CD1-restricted T cells (specifically, iNKT cells) may be involved in immune responses to Mtb. A mouse study showed that injection of an anti-CD1d antibody exacerbated the symptoms of TB, which suggests that iNKT cells may have anti-microbial functions (134). In addition, Sada-Ovalle et al. found that adoptive transfer of iNKT cells reduced the bacterial burden in Mtb-infected mice by producing IFN-γ and killing intracellular bacteria (55). This protective function of iNKT cells suggests that vaccination with common lipid antigens may boost the beneficial anti-TB functions of CD1-restricted T cells (135).
Since human immune responses differ according to the pathogen encountered, it is possible that lipid-reactive T cells respond quite differently to infectious agent other than Mtb. Nevertheless, it is noteworthy that various studies show that iNKT cells also largely play protective roles against other pathogens such as Streptococcus pneumoniae and Saccharopolyspora rectivirgula, the most common causes of community-acquired pneumonia (48, 136) and Farmer's lung (137) respectively. Exceptions include a few cases of non-common pneumonia occurring in adults. For example, the causative agent of atypical pneumonia is Chlamydia trachomatis. Patients become infected with C. trachomatis either after sexual activity or by vertical transmission from an infected mother. Bilenki et al. showed that, when a mouse model of chlamydial pneumonia is injected with α-GalCer and then infected with C. muridarum, the α-GalCer-activated NKT cells increase the chlamydial burden and worsen the symptoms (138).
Contrary to microbial infections in which CD1-reactive T cells are mainly activated through microbial lipid-CD1-dependent mechanisms, viral infections are a good model in which to demonstrate the CD1- or TCR-independent mechanisms underlying CD1-restricted T cell activation. Unfortunately, no study has examined the role of group 1 CD1-restricted T cells in pulmonary viral infection, except during systemic viral infections such as HIV (139), which might cause pulmonary infection. Therefore, we focus here on the biology of iNKT cells in viral infection models.
Influenza virus is a representative respiratory virus that causes seasonal flu and pneumonia with high morbidity and mortality (140). Several studies showed that NKT cells control lung inflammation during influenza A virus (IAV) infection through dampening suppression of the virus-specific T cell responses or controlling the IAV-associated lethal inflammation. During IAV (PR8, H1N1) infection, iNKT cells suppress myeloid-derived suppressor cells (MDSCs), which contribute to lung pathology and inhibit the activity of the virus-specific immune cells (141). The MDSC suppressive activity was inhibited by iNKT cells, which were activated in endogenous lipids:CD1d-dependnet manner. MDSCs derived from Hexβ−/− mice (unable to produce endogenous lipid ligands) failed to activate iNKT cells, thereby sustained their suppressive capability. In another study showing a protective role of iNKT cells in IAV (Scotland, H3N2) infection, however, the underlying mechanism was not mediated by MDSCs, but by a positive effect of iNKT cells on the migration of respiratory dendritic cells to the draining lymph nodes, a phenomenon that favors the expansion of virus-specific CD8+ T cells (142). Also, iNKT cells suppressed the fatal inflammation by lysing infected inflammatory monocytes (the dominant immune cell in IAV-infected lungs) in a CD1d-dependent fashion (143). In contrast to the CD1d-dependent actions of iNKT cells, a cytokine-mediated, CD1d-independent iNKT cell activation was shown to limit pulmonary inflammation and a secondary bacterial infection during IAV infection. RORγt+ iNKT cells (iNKT17) secrete a lung-protective cytokine, IL-22, in response to stimulation by IL-1β and IL-23 secreted by IAV-infected DCs (144, 145). Interestingly, the reduction of the burden of secondary bacterial infection (Streptococcus pneumoniae) after IAV infection was observed when recombinant IL-22 was treated intranasally (146). This new strategy draws an attention considering the severe consequences from the co-infections (147, 148). Although we could speculate that there is no lipid antigen directly from viral genome in cells with virus infection, it should be verified with the in vivo system which enabled to distinguish the origin of effector functions (either TCR-dependent or -independent manner) in iNKT cells. A Nur77gfp reporter system, which expresses GFP in response to TCR stimulation only, was used to show that IFN-γ-producing iNKT cells are induced during infection with murine cytomegalovirus (MCMV) without any change in GFP expression (149). It is now anticipated that the previously reported (i.e., not restricted to infection models) effector functions of CD1-restricted T cells will be re-evaluated using novel approaches to better understand the contribution of the TCR to iNKT cell activation.
Lung Cancer
The role of lipid-reactive T cells (especially α-GalCer-reactive iNKT cells) in tumors began attracting attention when Kobayashi et al. (150) showed that iNKT cells play protective roles in a B16 melanoma mouse model (150). Although CD1-reactive T cells are best known for their ability to produce cytokines, iNKT cells (like NK cells and cytotoxic T cells) are also cytotoxic; thus, they may be potential targets for therapies aimed at inducing anti-tumor immunity (151). The first in vivo iNKT cell-based immunotherapy regimen tested in patients with a variety of cancers, including breast, colon, liver, skin, kidney, prostate, peritoneum, and lung cancer. The study showed that, when iNKT cells were activated by treatment with α-GalCer-loaded DCs, they contributed to the tumor suppressive environment and reduced the size of the tumors (152). It is well recognized that iNKT cells protect against lung cancers, particularly non-small cell lung cancers (NSCLC), by producing IFN-γ and cytotoxic proteins (153–155). The percentage of NKT cells (CD56+ CD3+ T cells) in the blood of NSCLC and SCLC (small cell lung cancer) patients is increased compared to healthy controls (156). In agreement with the beneficial role of iNKT cells in lung cancer, the numbers of iNKT cells were compared following the cancers of differing histologic type and stage. It was found that the number was: (1) lower in adenocarcinoma, which has the worse prognosis than in squamous cell carcinoma (157); (2) lower in stage 1 than in stage IV cancers (156). Another human study showed that the percentage of Vα24 NKT cells in the PBMC of lung cancer patients was 0.01–0.03%; however, patients with recurrent cancers harbored a significantly lower percentage of NKT cells than healthy donors (158). Collectively, the data suggest that more immunosuppressive environments encountered at the later stages of lung cancer reduce the percentage of iNKT cells; this may allow the cancer to escape immune surveillance. In line with this idea, one study shows that injecting in vitro-expanded autologous iNKT cells may be an effective form of iNKT cell-based immunotherapy (159).
Several studies have assessed the role of type II NKT cells in tumor immunity. One such study was conducted by Terabe et al., who showed that when CD1d knockout and Jα18 knockout mice were used as CT26 colon carcinoma metastasis to the lungs, both knockouts developed significantly lower numbers of lung tumor nodules, compared to wild-type mice. However, this effect was more pronounced in CD1d knockout mice (which lack both type I and II CD1d-restricted NKT cells) than in Jα18 knockout mice (which lack only type I CD1d-restricted NKT cells). This suggests that type II NKT may suppress tumor metastasis or growth (160). However, evidence suggests that type I NKT cells promote immunity to CT26 cancer cells while type II NKT cells suppress it. Moreover, an absence of type I NKT cells is associated with stronger type II NKT cell-mediated suppression, whereas type II NKT cells appear to downregulate protective tumor immunity mediated by type I NKT cells. These findings suggest that type I and II NKT cells may counter the effects of each other in a balanced manner (161).
Conclusion and Perspectives
Here, we discussed the role of CD1-restricted T cells in lung diseases, including asthma, COPD, pulmonary fibrosis, infection, and cancer. Studies on group 2 CD1-restricted T cells are prolific. Less is known about group 1 CD1-reactive T cells because mice lack group 1 CD1 genes. Nevertheless, the current state of knowledge suggests that, like conventional T cells, group 1 and 2 CD1-restricted T cells can play either a protective role or a pathogenic role in lung disease (Table 1).
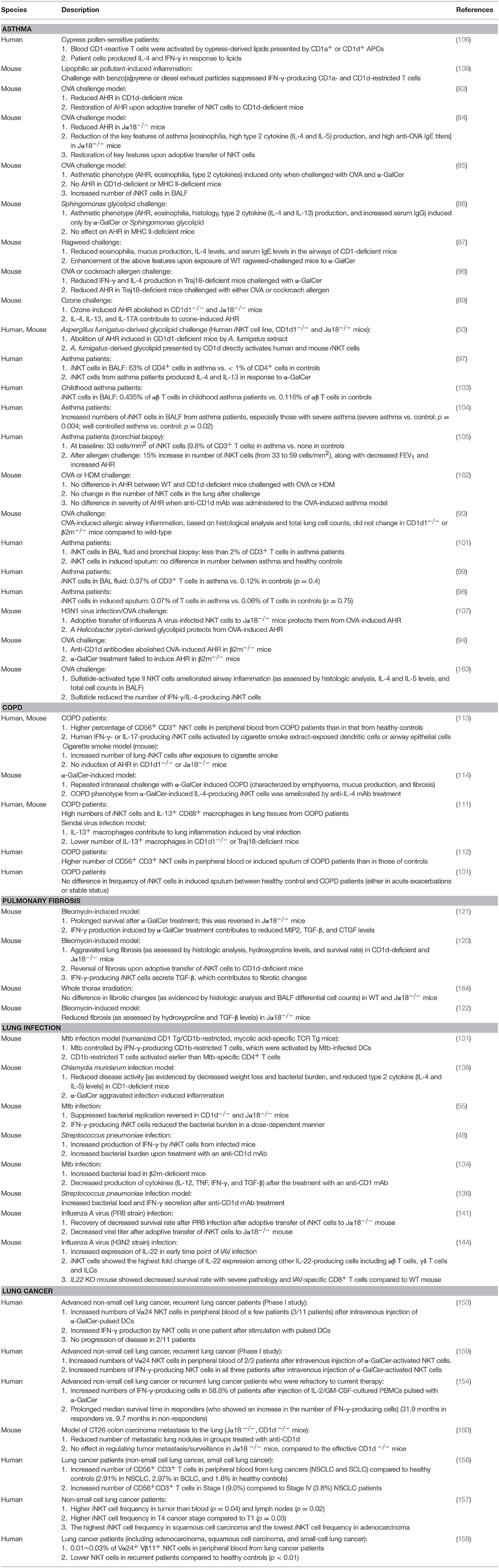
Table 1. Studies of lipid-reactive T cells in pulmonary disorders.
Despite the large body of work on CD1-restricted T-cell biology, many questions remain in this field, as follows. First, our understanding of how group 1 CD1-restricted T cells participate in lung diseases is limited. The recent development of transgenic mouse models expressing human CD1a, CD1b, or CD1c will improve this situation. Indeed, two studies based on these mice showed that they generate group 1 CD1-restricted T cell responses that are consistent with the responses of human T cell lines (129, 130). While these studies mainly showed that CD1-restricted T cells participate in skin inflammation, these group 1 CD1 transgenic mice may be useful for examining the in vivo roles of CD1-restricted T cells in other diseases. Second, no study has confirmed that autoreactive CD1-restricted T cells contribute to development of lung diseases. By contrast, several reports suggest that self-lipid-reactive T cells may participate in the pathology of psoriasis, which is an autoimmune skin disease (129, 165). Autoimmune pulmonary diseases are either primary autoimmune diseases or systemic autoimmune diseases that show pulmonary manifestations: the latter include systemic sclerosis, systemic lupus erythematosus, rheumatoid arthritis, and Sjogren's syndrome (166). Given that self-lipid-reactive T cells are associated with development of psoriatic skin, it is highly likely that there are self-lipid antigens that could promote the lung manifestations observed in systemic autoimmune diseases. In addition, lipid antigens in the lungs still need to be defined. An allergic reaction in the lungs can be induced by house dust mite and ozone, which activate CD1-restricted T cells (35, 89). These substances have the potential to generate a neoantigen for CD1-restricted T cells. For example, house dust mite contains phospholipase A2 (PLA2), which converts self-lipids into neoantigens. Similarly, it is speculated that ozone may chemically modify self-lipids to render antigenic properties against CD1. Notably, many lipid ligands of CD1c do not make direct contact with the TCR to trigger CD1-mediated autoreactivity (12). Further investigation is required to understand the circumstances in which CD1-autoreactive T cells are activated and how they are involved in lung disorders (Figure 2). Finally, the role of NKT cells in asthma should be re-evaluated. There is a large body of work on the role of CD1-restricted T cells in asthma, which is the most studied disease in this field. This work has provided ample evidence that CD1-restricted T cells, especially NKT cells, are involved in the pathology of asthma in mouse models. However, controversy about the role of these cells in asthmatic humans arose in the late 2000s when some studies reported that asthmatics have low percentages of NKT cells (98, 99, 101). It seems quite possible, for example, that NKT cells play an important role in a subset of patients who are allergic to lipids. This possibility is supported by the fact that the triggering antigen in patients with allergic asthma is very heterogeneous; these patients show distinct individual patterns of reactivity to diverse allergens (167). While the most common allergens in patients with asthma do not contain lipid components, it seems possible that at least some patients would be allergic to lipid components that can activate NKT cells.
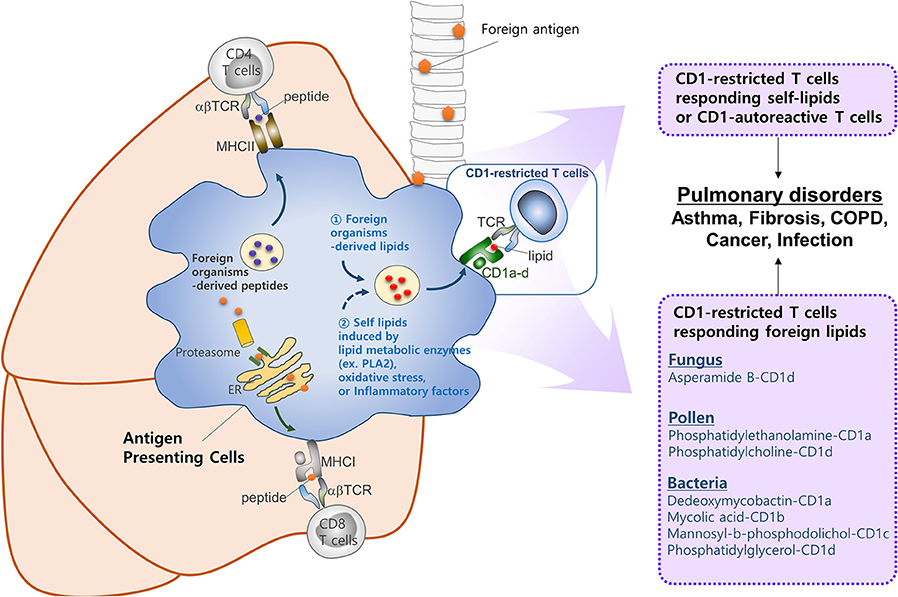
Figure 2. CD1-reactive T cells in pulmonary disorders. T cells play a critical role in pulmonary immune responses to both non-self antigens (such as microbiota and allergens) and self-antigens. In general, T cells are activated upon recognition of peptide antigens presented to the TCR by classical MHCI and MHCII molecules. However, increasing evidence suggests that T cells recognizing lipid antigens presented by CD1 molecules play a significant role in immune responses in the lung. CD1 molecules can display a broad range of lipid antigens derived from foreign organisms (e.g., fungi, pollen, and Mycobacterium tuberculosis). Oxidative stress or inflammatory factors released by host, or lipid metabolic enzymes (e.g., PLA2) derived from foreign organisms, might be responsible for activating CD1-restricted T cells in the lung. However, further studies are required to identify self-lipid antigens, the mechanisms by which such antigens are generated, the circumstances under which CD1-autoreactive T cells are activated, and the roles they play in lung disorders.
This review summarizes current knowledge of CD1-restricted T cells, their specific lipid antigens, and their role in lung disease. Further research is likely to reveal hitherto unsuspected contributions to the pathogenesis of various lung diseases.
Author Contributions
SR, JP, JK, and HK wrote and revised the manuscript.
Funding
HK was supported by a grant from the Korea Healthcare Technology R&D Project, Ministry for Health and Welfare Affairs, Republic of Korea (HI15C1736). JK was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2018R1D1A1B07048813).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
MHC, major histocompatibility complex; TCR, T cell receptor; IFN, interferon; IL, interleukin; CD1, cluster of differentiation 1; COPD, chronic obstructive pulmonary disease; ER, endoplasmic reticulum; PBMC, peripheral blood mononuclear cell; hCD1Tg, human CD1 transgenic; Mtb, Mycobacterium tuberculosis; NKT, natural killer T cells; DP, double-positive; α-GalCer, α-galactosylceramide; iGb3, isoglobotrihexosylceramide; AHR, airway hyper-responsiveness; OVA, ovalbumin; BALF, bronchoalveolar lavage fluid; β2M, β2-microglobulin; MDSC, myeloid-derived suppressor cell; IAV, influenza A virus; IPF, idiopathic pulmonary fibrosis; TB, tuberculosis; BCG, Bacillus Calmette-Guérin; DC, dendritic cell; NSCLC, non-small cell lung cancer; SCLC, small cell lung cancer; ILC, innate lymphoid cell; FEV1, forced expiratory volume in one second; MIP, macrophage inflammatory protein; CTGF, connective tissue growth factor; TNF, tumor necrosis factor.
References
1. Lloyd CM, Marsland BJ. Lung homeostasis: influence of age, microbes, and the immune system. Immunity (2017) 46:549–61. doi: 10.1016/j.immuni.2017.04.005
2. Chen K, Kolls JK. T cell-mediated host immune defenses in the lung. Annu Rev Immunol. (2013) 31:605–33. doi: 10.1146/annurev-immunol-032712-100019
3. de Lalla C, Lepore M, Piccolo FM, Rinaldi A, Scelfo A, Garavaglia C, et al. High-frequency and adaptive-like dynamics of human CD1 self-reactive T cells. Eur J Immunol. (2011) 41:602–10. doi: 10.1002/eji.201041211
4. Martin LH, Calabi F, Milstein C. Isolation of CD1 genes: a family of major histocompatibility complex-related differentiation antigens. Proc Natl Acad Sci USA. (1986) 83:9154–8. doi: 10.1073/pnas.83.23.9154
5. Yu CY, Milstein C. A physical map linking the five CD1 human thymocyte differentiation antigen genes. EMBO J. (1989) 8:3727–32.
6. Adams EJ. Lipid presentation by human CD1 molecules and the diverse T cell populations that respond to them. Curr Opin Immunol. (2014) 26:1–6. doi: 10.1016/j.coi.2013.09.005
7. Reinink P, Van Rhijn I. Mammalian CD1 and MR1 genes. Immunogenetics (2016) 68:515–23. doi: 10.1007/s00251-016-0926-x
8. Scanlon ST, Thomas SY, Ferreira CM, Bai L, Krausz T, Savage PB, et al. Airborne lipid antigens mobilize resident intravascular NKT cells to induce allergic airway inflammation. J Exp Med. (2011) 208:2113–24. doi: 10.1084/jem.20110522
9. Lee YJ, Wang H, Starrett GJ, Phuong V, Jameson SC, Hogquist KA. Tissue-specific distribution of iNKT cells impacts their cytokine response. Immunity (2015) 43:566–78. doi: 10.1016/j.immuni.2015.06.025
10. Moody DB, Zajonc DM, Wilson IA. Anatomy of CD1-lipid antigen complexes. Nat Rev Immunol. (2005) 5:387–99. doi: 10.1038/nri1605
11. Cotton RN, Shahine A, Rossjohn J, Moody DB. Lipids hide or step aside for CD1-autoreactive T cell receptors. Curr Opin Immunol. (2018) 52:93–9. doi: 10.1016/j.coi.2018.04.013
12. Wun KS, Reijneveld JF, Cheng TY, Ladell K, Uldrich AP, Le Nours J, et al. T cell autoreactivity directed toward CD1c itself rather than toward carried self lipids. Nat Immunol. (2018) 19:397–406. doi: 10.1038/s41590-018-0065-7
13. Calabi F, Jarvis JM, Martin L, Milstein C. Two classes of CD1 genes. Eur J Immunol. (1989) 19:285–92. doi: 10.1002/eji.1830190211
14. de la Salle H, Mariotti S, Angenieux C, Gilleron M, Garcia-Alles LF, Malm D, et al. Assistance of microbial glycolipid antigen processing by CD1e. Science (2005) 310:1321–4. doi: 10.1126/science.1115301
15. Facciotti F, Cavallari M, Angenieux C, Garcia-Alles LF, Signorino-Gelo F, Angman L, et al. Fine tuning by human CD1e of lipid-specific immune responses. Proc Natl Acad Sci USA. (2011) 108:14228–33. doi: 10.1073/pnas.1108809108
16. Dougan SK, Kaser A, Blumberg RS. CD1 expression on antigen-presenting cells. Curr Top Microbiol Immunol. (2007) 314:113–41. doi: 10.1007/978-3-540-69511-0_5
17. Van Kaer L, Wu L, Joyce S. Mechanisms and consequences of antigen presentation by CD1. Trends Immunol. (2016) 37:738–54. doi: 10.1016/j.it.2016.08.011
18. Palucka KA, Taquet N, Sanchez-Chapuis F, Gluckman JC. Dendritic cells as the terminal stage of monocyte differentiation. J Immunol. (1998) 160:4587–95.
19. Rougier N, Schmitt D, Vincent C. IL-4 addition during differentiation of CD34 progenitors delays maturation of dendritic cells while promoting their survival. Eur J Cell Biol. (1998) 75:287–93. doi: 10.1016/S0171-9335(98)80124-6
20. Rosenzwajg M, Canque B, Gluckman JC. Human dendritic cell differentiation pathway from CD34+ hematopoietic precursor cells. Blood (1996) 87:535–44.
21. Kasinrerk W, Baumruker T, Majdic O, Knapp W, Stockinger H. CD1 molecule expression on human monocytes induced by granulocyte-macrophage colony-stimulating factor. J Immunol. (1993) 150:579–84.
22. Moody DB, Cotton RN. Four pathways of CD1 antigen presentation to T cells. Curr Opin Immunol. (2017) 46:127–33. doi: 10.1016/j.coi.2017.07.013
23. Barral DC, Brenner MB. CD1 antigen presentation: how it works. Nat Rev Immunol. (2007) 7:929–41. doi: 10.1038/nri2191
24. Dougan SK, Salas A, Rava P, Agyemang A, Kaser A, Morrison J, et al. Microsomal triglyceride transfer protein lipidation and control of CD1d on antigen-presenting cells. J Exp Med. (2005) 202:529–39. doi: 10.1084/jem.20050183
25. Zeissig S, Dougan SK, Barral DC, Junker Y, Chen Z, Kaser A, et al. Primary deficiency of microsomal triglyceride transfer protein in human abetalipoproteinemia is associated with loss of CD1 function. J Clin Invest. (2010) 120:2889–99. doi: 10.1172/JCI42703
26. Sugita M, Grant EP, van Donselaar E, Hsu VW, Rogers RA, Peters PJ, et al. Separate pathways for antigen presentation by CD1 molecules. Immunity (1999) 11:743–52. doi: 10.1016/S1074-7613(00)80148-X
27. Cernadas M, Cavallari M, Watts G, Mori L, De Libero G, Brenner MB. Early recycling compartment trafficking of CD1a is essential for its intersection and presentation of lipid antigens. J Immunol. (2010) 184:1235–41. doi: 10.4049/jimmunol.0804140
28. Leon L, Tatituri RV, Grenha R, Sun Y, Barral DC, Minnaard AJ, et al. Saposins utilize two strategies for lipid transfer and CD1 antigen presentation. Proc Natl Acad Sci USA. (2012) 109:4357–64. doi: 10.1073/pnas.1200764109
29. Salio M, Ghadbane H, Dushek O, Shepherd D, Cypen J, Gileadi U, et al. Saposins modulate human invariant Natural Killer T cells self-reactivity and facilitate lipid exchange with CD1d molecules during antigen presentation. Proc Natl Acad Sci USA. (2013) 110:E4753–61. doi: 10.1073/pnas.1310050110
30. Young MH, Gapin L. Group 1 CD1-restricted T cells take center stage. Eur J Immunol. (2011) 41:592–4. doi: 10.1002/eji.201141408
31. Felio K, Nguyen H, Dascher CC, Choi HJ, Li S, Zimmer MI, et al. CD1-restricted adaptive immune responses to Mycobacteria in human group 1 CD1 transgenic mice. J Exp Med. (2009) 206:2497–509. doi: 10.1084/jem.20090898
32. Kobayashi C, Shiina T, Tokioka A, Hattori Y, Komori T, Kobayashi-Miura M, et al. GM-CSF-independent CD1a expression in epidermal Langerhans cells: evidence from human CD1A genome-transgenic mice. J Invest Dermatol. (2012) 132:241–4. doi: 10.1038/jid.2011.280
33. Lockridge JL, Chen X, Zhou Y, Rajesh D, Roenneburg DA, Hegde S, et al. Analysis of the CD1 antigen presenting system in humanized SCID mice. PLoS ONE (2011) 6:e21701. doi: 10.1371/journal.pone.0021701
34. Li S, Choi HJ, Felio K, Wang CR. Autoreactive CD1b-restricted T cells: a new innate-like T-cell population that contributes to immunity against infection. Blood (2011) 118:3870–8. doi: 10.1182/blood-2011-03-341941
35. Jarrett R, Salio M, Lloyd-Lavery A, Subramaniam S, Bourgeois E, Archer C, et al. Filaggrin inhibits generation of CD1a neolipid antigens by house dust mite-derived phospholipase. Sci Transl Med. (2016) 8:325ra18. doi: 10.1126/scitranslmed.aad6833
36. Subramaniam S, Aslam A, Misbah SA, Salio M, Cerundolo V, Moody DB, et al. Elevated and cross-responsive CD1a-reactive T cells in bee and wasp venom allergic individuals. Eur J Immunol. (2016) 46:242–52. doi: 10.1002/eji.201545869
37. Faunce DE, Palmer JL, Paskowicz KK, Witte PL, Kovacs EJ. CD1d-restricted NKT cells contribute to the age-associated decline of T cell immunity. J Immunol. (2005) 175:3102–9. doi: 10.4049/jimmunol.175.5.3102
38. Jing Y, Gravenstein S, Chaganty NR, Chen N, Lyerly KH, Joyce S, et al. Aging is associated with a rapid decline in frequency, alterations in subset composition, and enhanced Th2 response in CD1d-restricted NKT cells from human peripheral blood. Exp Gerontol. (2007) 42:719–32. doi: 10.1016/j.exger.2007.01.009
39. Taniguchi M, Seino K, Nakayama T. The NKT cell system: bridging innate and acquired immunity. Nat Immunol. (2003) 4:1164–5. doi: 10.1038/ni1203-1164
40. Brossay L, Chioda M, Burdin N, Koezuka Y, Casorati G, Dellabona P, et al. CD1d-mediated recognition of an alpha-galactosylceramide by natural killer T cells is highly conserved through mammalian evolution. J Exp Med. (1998) 188:1521–8. doi: 10.1084/jem.188.8.1521
41. Kain L, Webb B, Anderson BL, Deng S, Holt M, Costanzo A, et al. The identification of the endogenous ligands of natural killer T cells reveals the presence of mammalian alpha-linked glycosylceramides. Immunity (2014) 41:543–54. doi: 10.1016/j.immuni.2014.08.017
42. Brennan PJ, Tatituri RV, Brigl M, Kim EY, Tuli A, Sanderson JP, et al. Invariant natural killer T cells recognize lipid self antigen induced by microbial danger signals. Nat Immunol. (2011) 12:1202–11. doi: 10.1038/ni.2143
43. Brennan PJ, Tatituri RV, Heiss C, Watts GF, Hsu FF, Veerapen N, et al. Activation of iNKT cells by a distinct constituent of the endogenous glucosylceramide fraction. Proc Natl Acad Sci USA. (2014) 111:13433–8. doi: 10.1073/pnas.1415357111
44. Parekh VV, Singh AK, Wilson MT, Olivares-Villagomez D, Bezbradica JS, Inazawa H, et al. Quantitative and qualitative differences in the in vivo response of NKT cells to distinct alpha- and beta-anomeric glycolipids. J Immunol. (2004) 173:3693–706. doi: 10.4049/jimmunol.173.6.3693
45. Zhou D, Mattner J, Cantu C 3rd, Schrantz N, Yin N, Gao Y, et al. Lysosomal glycosphingolipid recognition by NKT cells. Science (2004) 306:1786–9. doi: 10.1126/science.1103440
46. Porubsky S, Speak AO, Luckow B, Cerundolo V, Platt FM, Grone HJ. Normal development and function of invariant natural killer T cells in mice with isoglobotrihexosylceramide (iGb3) deficiency. Proc Natl Acad Sci USA. (2007) 104:5977–82. doi: 10.1073/pnas.0611139104
47. Mattner J, Debord KL, Ismail N, Goff RD, Cantu C 3rd, Zhou D, et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature (2005) 434:525–9. doi: 10.1038/nature03408
48. Kinjo Y, Illarionov P, Vela JL, Pei B, Girardi E, Li X, et al. Invariant natural killer T cells recognize glycolipids from pathogenic Gram-positive bacteria. Nat Immunol. (2011) 12:966–74. doi: 10.1038/ni.2096
49. Kinjo Y, Tupin E, Wu D, Fujio M, Garcia-Navarro R, Benhnia MR, et al. Natural killer T cells recognize diacylglycerol antigens from pathogenic bacteria. Nat Immunol. (2006) 7:978–86. doi: 10.1038/ni1380
50. Albacker LA, Chaudhary V, Chang YJ, Kim HY, Chuang YT, Pichavant M, et al. Invariant natural killer T cells recognize a fungal glycosphingolipid that can induce airway hyperreactivity. Nat Med. (2013) 19:1297–304. doi: 10.1038/nm.3321
51. Paget C, Mallevaey T, Speak AO, Torres D, Fontaine J, Sheehan KC, et al. Activation of invariant NKT cells by toll-like receptor 9-stimulated dendritic cells requires type I interferon and charged glycosphingolipids. Immunity (2007) 27:597–609. doi: 10.1016/j.immuni.2007.08.017
52. Salio M, Speak AO, Shepherd D, Polzella P, Illarionov PA, Veerapen N, et al. Modulation of human natural killer T cell ligands on TLR-mediated antigen-presenting cell activation. Proc Natl Acad Sci USA. (2007) 104:20490–5. doi: 10.1073/pnas.0710145104
53. Nagarajan NA, Kronenberg M. Invariant NKT cells amplify the innate immune response to lipopolysaccharide. J Immunol. (2007) 178:2706–13. doi: 10.4049/jimmunol.178.5.2706
54. Brigl M, Tatituri RV, Watts GF, Bhowruth V, Leadbetter EA, Barton N, et al. Innate and cytokine-driven signals, rather than microbial antigens, dominate in natural killer T cell activation during microbial infection. J Exp Med. (2011) 208:1163–77. doi: 10.1084/jem.20102555
55. Sada-Ovalle I, Chiba A, Gonzales A, Brenner MB, Behar SM. Innate invariant NKT cells recognize Mycobacterium tuberculosis-infected macrophages, produce interferon-gamma, and kill intracellular bacteria. PLoS Pathog. (2008) 4:e1000239. doi: 10.1371/journal.ppat.1000239
56. O'Brien TF, Bao K, Dell'Aringa M, Ang WX, Abraham S, Reinhardt RL. Cytokine expression by invariant natural killer T cells is tightly regulated throughout development and settings of type-2 inflammation. Mucosal Immunol. (2016) 9:597–609. doi: 10.1038/mi.2015.78
57. Watarai H, Sekine-Kondo E, Shigeura T, Motomura Y, Yasuda T, Satoh R, et al. Development and function of invariant natural killer T cells producing TH2- and TH17-cytokines. PLoS Biol. (2012) 10:e1001255. doi: 10.1371/journal.pbio.1001255
58. Lee YJ, Holzapfel KL, Zhu J, Jameson SC, Hogquist KA. Steady-state production of IL-4 modulates immunity in mouse strains and is determined by lineage diversity of iNKT cells. Nat Immunol. (2013) 14:1146–54. doi: 10.1038/ni.2731
59. Chang PP, Barral P, Fitch J, Pratama A, Ma CS, Kallies A, et al. Identification of Bcl-6-dependent follicular helper NKT cells that provide cognate help for B cell responses. Nat Immunol. (2011) 13:35–43. doi: 10.1038/ni.2166
60. Sag D, Krause P, Hedrick CC, Kronenberg M, Wingender G. IL-10-producing NKT10 cells are a distinct regulatory invariant NKT cell subset. J Clin Invest. (2014) 124:3725–40. doi: 10.1172/JCI72308
61. Lynch L, Michelet X, Zhang S, Brennan PJ, Moseman A, Lester C, et al. Regulatory iNKT cells lack expression of the transcription factor PLZF and control the homeostasis of T(reg) cells and macrophages in adipose tissue. Nat Immunol. (2015) 16:85–95. doi: 10.1038/ni.3047
62. Bendelac A. Positive selection of mouse NK1+ T cells by CD1-expressing cortical thymocytes. J Exp Med. (1995) 182:2091–6. doi: 10.1084/jem.182.6.2091
63. Gapin L. Development of invariant natural killer T cells. Curr Opin Immunol. (2016) 39:68–74. doi: 10.1016/j.coi.2016.01.001
64. Kim PJ, Pai SY, Brigl M, Besra GS, Gumperz J, Ho IC. GATA-3 regulates the development and function of invariant NKT cells. J Immunol. (2006) 177:6650–9. doi: 10.4049/jimmunol.177.10.6650
65. Benlagha K, Kyin T, Beavis A, Teyton L, Bendelac A. A thymic precursor to the NK T cell lineage. Science (2002) 296:553–5. doi: 10.1126/science.1069017
66. Pellicci DG, Hammond KJ, Uldrich AP, Baxter AG, Smyth MJ, Godfrey DI. A natural killer T (NKT) cell developmental pathway iInvolving a thymus-dependent NK1.1−CD4+ CD1d-dependent precursor stage. J Exp Med. (2002) 195:835–44. doi: 10.1084/jem.20011544
67. Benlagha K, Wei DG, Veiga J, Teyton L, Bendelac A. Characterization of the early stages of thymic NKT cell development. J Exp Med. (2005) 202:485–92. doi: 10.1084/jem.20050456
68. Savage AK, Constantinides MG, Han J, Picard D, Martin E, Li B, et al. The transcription factor PLZF directs the effector program of the NKT cell lineage. Immunity (2008) 29:391–403. doi: 10.1016/j.immuni.2008.07.011
69. Kovalovsky D, Uche OU, Eladad S, Hobbs RM, Yi W, Alonzo E, et al. The BTB-zinc finger transcriptional regulator PLZF controls the development of invariant natural killer T cell effector functions. Nat Immunol. (2008) 9:1055–64. doi: 10.1038/ni.1641
70. Kwon DI, Lee YJ. Lineage Differentiation program of invariant natural killer T cells. Immune Netw. (2017) 17:365–77. doi: 10.4110/in.2017.17.6.365
71. Jahng A, Maricic I, Aguilera C, Cardell S, Halder RC, Kumar V. Prevention of autoimmunity by targeting a distinct, noninvariant CD1d-reactive T cell population reactive to sulfatide. J Exp Med. (2004) 199:947–57. doi: 10.1084/jem.20031389
72. Dasgupta S, Kumar V. Type II NKT cells: a distinct CD1d-restricted immune regulatory NKT cell subset. Immunogenetics (2016) 68:665–76. doi: 10.1007/s00251-016-0930-1
73. Matsuda JL, Naidenko OV, Gapin L, Nakayama T, Taniguchi M, Wang CR, et al. Tracking the response of natural killer T cells to a glycolipid antigen using CD1d tetramers. J Exp Med. (2000) 192:741–54. doi: 10.1084/jem.192.5.741
74. Zhao J, Weng X, Bagchi S, Wang CR. Polyclonal type II natural killer T cells require PLZF and SAP for their development and contribute to CpG-mediated antitumor response. Proc Natl Acad Sci USA. (2014) 111:2674–9. doi: 10.1073/pnas.1323845111
75. Weng X, Liao CM, Bagchi S, Cardell SL, Stein PL, Wang CR. The adaptor protein SAP regulates type II NKT-cell development, cytokine production, and cytotoxicity against lymphoma. Eur J Immunol. (2014) 44:3646–57. doi: 10.1002/eji.201444848
76. Dhodapkar MV, Kumar V. Type II NKT cells and their emerging role in health and disease. J Immunol. (2017) 198:1015–21. doi: 10.4049/jimmunol.1601399
77. Germanov E, Veinotte L, Cullen R, Chamberlain E, Butcher EC, Johnston B. Critical role for the chemokine receptor CXCR6 in homeostasis and activation of CD1d-restricted NKT cells. J Immunol. (2008) 181:81–91. doi: 10.4049/jimmunol.181.1.81
78. McCracken JL, Veeranki SP, Ameredes BT, Calhoun WJ. Diagnosis and management of asthma in adults: a review. JAMA (2017) 318:279–90. doi: 10.1001/jama.2017.8372
79. Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. (2015) 16:45–56. doi: 10.1038/ni.3049
80. DeKruyff RH, Yu S, Kim HY, Umetsu DT. Innate immunity in the lung regulates the development of asthma. Immunol Rev. (2014) 260:235–48. doi: 10.1111/imr.12187
81. Morita H, Moro K, Koyasu S. Innate lymphoid cells in allergic and nonallergic inflammation. J Allergy Clin Immunol. (2016) 138:1253–64. doi: 10.1016/j.jaci.2016.09.011
82. Cui J, Watanabe N, Kawano T, Yamashita M, Kamata T, Shimizu C, et al. Inhibition of T helper cell type 2 cell differentiation and immunoglobulin E response by ligand-activated Valpha14 natural killer T cells. J Exp Med. (1999) 190:783–92. doi: 10.1084/jem.190.6.783
83. Akbari O, Stock P, Meyer E, Kronenberg M, Sidobre S, Nakayama T, et al. Essential role of NKT cells producing IL-4 and IL-13 in the development of allergen-induced airway hyperreactivity. Nat Med. (2003) 9:582–8. doi: 10.1038/nm851
84. Lisbonne M, Diem S, de Castro Keller A, Lefort J, Araujo LM, Hachem P, et al. Cutting edge: invariant V alpha 14 NKT cells are required for allergen-induced airway inflammation and hyperreactivity in an experimental asthma model. J Immunol. (2003) 171:1637–41. doi: 10.4049/jimmunol.171.4.1637
85. Kim JO, Kim DH, Chang WS, Hong C, Park SH, Kim S, et al. Asthma is induced by intranasal coadministration of allergen and natural killer T-cell ligand in a mouse model. J Allergy Clin Immunol. (2004) 114:1332–8. doi: 10.1016/j.jaci.2004.09.004
86. Meyer EH, Goya S, Akbari O, Berry GJ, Savage PB, Kronenberg M, et al. Glycolipid activation of invariant T cell receptor+ NK T cells is sufficient to induce airway hyperreactivity independent of conventional CD4+ T cells. Proc Natl Acad Sci USA. (2006) 103:2782–7. doi: 10.1073/pnas.0510282103
87. Bilenki L, Yang J, Fan Y, Wang S, Yang X. Natural killer T cells contribute to airway eosinophilic inflammation induced by ragweed through enhanced IL-4 and eotaxin production. Eur J Immunol. (2004) 34:345–54. doi: 10.1002/eji.200324303
88. Wingender G, Rogers P, Batzer G, Lee MS, Bai D, Pei B, et al. Invariant NKT cells are required for airway inflammation induced by environmental antigens. J Exp Med. (2011) 208:1151–62. doi: 10.1084/jem.20102229
89. Pichavant M, Goya S, Meyer EH, Johnston RA, Kim HY, Matangkasombut P, et al. Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17. J Exp Med. (2008) 205:385–93. doi: 10.1084/jem.20071507
90. Das J, Eynott P, Jupp R, Bothwell A, Van Kaer L, Shi Y, et al. Natural killer T cells and CD8+ T cells are dispensable for T cell-dependent allergic airway inflammation. Nat Med. (2006) 12:1345–6. author reply 1347. doi: 10.1038/nm1206-1345
91. Brown DR, Fowell DJ, Corry DB, Wynn TA, Moskowitz NH, Cheever AW, et al. Beta 2-microglobulin-dependent NK1.1+ T cells are not essential for T helper cell 2 immune responses. J Exp Med. (1996) 184:1295–304.
92. Korsgren M, Persson CG, Sundler F, Bjerke T, Hansson T, Chambers BJ, et al. Natural killer cells determine development of allergen-induced eosinophilic airway inflammation in mice. J Exp Med. (1999) 189:553–62. doi: 10.1084/jem.189.3.553
93. Zhang Y, Rogers KH, Lewis DB. Beta 2-microglobulin-dependent T cells are dispensable for allergen-induced T helper 2 responses. J Exp Med. (1996) 184:1507–12. doi: 10.1084/jem.184.4.1507
94. Koh YI, Kim HY, Meyer EH, Pichavant M, Akbari O, Yasumi T, et al. Activation of nonclassical CD1d-restricted NK T cells induces airway hyperreactivity in beta 2-microglobulin-deficient mice. J Immunol. (2008) 181:4560–9. doi: 10.4049/jimmunol.181.7.4560
95. Bedel R, Matsuda JL, Brigl M, White J, Kappler J, Marrack P, et al. Lower TCR repertoire diversity in Traj18-deficient mice. Nat Immunol. (2012) 13:705–6. doi: 10.1038/ni.2347
96. Chandra S, Zhao M, Budelsky A, de Mingo Pulido A, Day J, Fu Z, et al. A new mouse strain for the analysis of invariant NKT cell function. Nat Immunol. (2015) 16:799–800. doi: 10.1038/ni.3203
97. Akbari O, Faul JL, Hoyte EG, Berry GJ, Wahlstrom J, Kronenberg M, et al. CD4+ invariant T-cell-receptor+ natural killer T cells in bronchial asthma. N Engl J Med. (2006) 354:1117–29. doi: 10.1056/NEJMoa053614
98. Brooks CR, Weinkove R, Hermans IF, van Dalen CJ, Douwes J. Invariant natural killer T cells and asthma: immunologic reality or methodologic artifact? J Allergy Clin Immunol. (2010) 126:882–5. doi: 10.1016/j.jaci.2010.06.041
99. Mutalithas K, Croudace J, Guillen C, Siddiqui S, Thickett D, Wardlaw A, et al. Bronchoalveolar lavage invariant natural killer T cells are not increased in asthma. J Allergy Clin Immunol. (2007) 119:1274–6. doi: 10.1016/j.jaci.2007.02.021
100. Thomas SY, Lilly CM, Luster AD. Invariant natural killer T cells in bronchial asthma. N Engl J Med. (2006) 354:2613–6. doi: 10.1056/NEJMc066189
101. Vijayanand P, Seumois G, Pickard C, Powell RM, Angco G, Sammut D, et al. Invariant natural killer T cells in asthma and chronic obstructive pulmonary disease. N Engl J Med. (2007) 356:1410–22. doi: 10.1056/NEJMoa064691
102. Thomas SY, Chyung YH, Luster AD. Natural killer T cells are not the predominant T cell in asthma and likely modulate, not cause, asthma. J Allergy Clin Immunol. (2010) 125:980–4. doi: 10.1016/j.jaci.2010.01.032
103. Pham-Thi N, de Blic J, Le Bourgeois M, Dy M, Scheinmann P, Leite-de-Moraes MC. Enhanced frequency of immunoregulatory invariant natural killer T cells in the airways of children with asthma. J Allergy Clin Immunol. (2006) 117:217–8. doi: 10.1016/j.jaci.2005.09.052
104. Matangkasombut P, Marigowda G, Ervine A, Idris L, Pichavant M, Kim HY, et al. Natural killer T cells in the lungs of patients with asthma. J Allergy Clin Immunol. (2009) 123:1181–5. doi: 10.1016/j.jaci.2009.02.013
105. Reynolds C, Barkans J, Clark P, Kariyawasam H, Altmann D, Kay B, et al. Natural killer T cells in bronchial biopsies from human allergen challenge model of allergic asthma. J Allergy Clin Immunol. (2009) 124:860–2. doi: 10.1016/j.jaci.2009.07.022
106. Agea E, Russano A, Bistoni O, Mannucci R, Nicoletti I, Corazzi L, et al. Human CD1-restricted T cell recognition of lipids from pollens. J Exp Med. (2005) 202:295–308. doi: 10.1084/jem.20050773
107. Chang YJ, Kim HY, Albacker LA, Lee HH, Baumgarth N, Akira S, et al. Influenza infection in suckling mice expands an NKT cell subset that protects against airway hyperreactivity. J Clin Invest. (2011) 121:57–69. doi: 10.1172/JCI44845
108. Chuang YT, Leung K, Chang YJ, DeKruyff RH, Savage PB, Cruse R, et al. A natural killer T-cell subset that protects against airway hyperreactivity. J Allergy Clin Immunol. (2018). doi: 10.1016/j.jaci.2018.03.022. [Epub ahead of print].
109. Sharma M, Zhang X, Zhang S, Niu L, Ho SM, Chen A, et al. Inhibition of endocytic lipid antigen presentation by common lipophilic environmental pollutants. Sci Rep. (2017) 7:2085. doi: 10.1038/s41598-017-02229-7
110. Barnes PJ, Burney PG, Silverman EK, Celli BR, Vestbo J, Wedzicha JA, et al. Chronic obstructive pulmonary disease. Nat Rev Dis Primers (2015) 1:15076. doi: 10.1038/nrdp.2015.76
111. Kim EY, Battaile JT, Patel AC, You Y, Agapov E, Grayson MH, et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med. (2008) 14:633–40. doi: 10.1038/nm1770
112. Wang J, Urbanowicz RA, Tighe PJ, Todd I, Corne JM, Fairclough LC. Differential activation of killer cells in the circulation and the lung: a study of current smoking status and chronic obstructive pulmonary disease (COPD). PLoS ONE (2013) 8:e58556. doi: 10.1371/journal.pone.0058556
113. Pichavant M, Remy G, Bekaert S, Le Rouzic O, Kervoaze G, Vilain E, et al. Oxidative stress-mediated iNKT-cell activation is involved in COPD pathogenesis. Mucosal Immunol. (2014) 7:568–78. doi: 10.1038/mi.2013.75
114. Tsao CC, Tsao PN, Chen YG, Chuang YH. Repeated activation of lung invariant NKT cells results in chronic obstructive pulmonary disease-like symptoms. PLoS ONE (2016) 11:e0147710. doi: 10.1371/journal.pone.0147710
115. Van Pottelberge GR, Bracke KR, Demedts IK, De Rijck K, Reinartz SM, van Drunen CM, et al. Selective accumulation of langerhans-type dendritic cells in small airways of patients with COPD. Respir Res. (2010) 11:35. doi: 10.1186/1465-9921-11-35
116. Verhoeven GT, Hegmans JP, Mulder PG, Bogaard JM, Hoogsteden HC, Prins JB. Effects of fluticasone propionate in COPD patients with bronchial hyperresponsiveness. Thorax (2002) 57:694–700. doi: 10.1136/thorax.57.8.694
117. Soler P, Moreau A, Basset F, Hance AJ. Cigarette smoking-induced changes in the number and differentiated state of pulmonary dendritic cells/Langerhans cells. Am Rev Respir Dis. (1989) 139:1112–7. doi: 10.1164/ajrccm/139.5.1112
118. Raghu G, Rochwerg B, Zhang Y, Garcia CA, Azuma A, Behr J, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. (2015) 192:e3–19. doi: 10.1164/rccm.201506-1063ST
119. Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet (2017) 389:1941–52. doi: 10.1016/S0140-6736(17)30866-8
120. Kim JH, Kim HY, Kim S, Chung JH, Park WS, Chung DH. Natural killer T (NKT) cells attenuate bleomycin-induced pulmonary fibrosis by producing interferon-gamma. Am J Pathol. (2005) 167:1231–41. doi: 10.1016/S0002-9440(10)61211-4
121. Kimura T, Ishii Y, Morishima Y, Shibuya A, Shibuya K, Taniguchi M, et al. Treatment with alpha-galactosylceramide attenuates the development of bleomycin-induced pulmonary fibrosis. J Immunol. (2004) 172:5782–9. doi: 10.4049/jimmunol.172.9.5782
122. Grabarz F, Aguiar CF, Correa-Costa M, Braga TT, Hyane MI, Andrade-Oliveira V, et al. Protective role of NKT cells and macrophage M2-driven phenotype in bleomycin-induced pulmonary fibrosis. Inflammopharmacology (2018) 26:491–504. doi: 10.1007/s10787-017-0383-7
123. Musher DM, Thorner AR. Community-acquired pneumonia. N Engl J Med. (2014) 371:1619–28. doi: 10.1056/NEJMra1312885
124. Pai M, Behr MA, Dowdy D, Dheda K, Divangahi M, Boehme CC, et al. Tuberculosis. Nat Rev Dis Primers (2016) 2:16076. doi: 10.1038/nrdp.2016.76
125. Beckman EM, Porcelli SA, Morita CT, Behar SM, Furlong ST, Brenner MB. Recognition of a lipid antigen by CD1-restricted alpha beta+ T cells. Nature (1994) 372:691–4. doi: 10.1038/372691a0
126. Moody DB, Ulrichs T, Muhlecker W, Young DC, Gurcha SS, Grant E, et al. CD1c-mediated T-cell recognition of isoprenoid glycolipids in Mycobacterium tuberculosis infection. Nature (2000) 404:884–8. doi: 10.1038/35009119
127. Moody DB, Young DC, Cheng TY, Rosat JP, Roura-Mir C, O'Connor PB, et al. T cell activation by lipopeptide antigens. Science (2004) 303:527–31. doi: 10.1126/science.1089353
128. Ulrichs T, Moody DB, Grant E, Kaufmann SH, Porcelli SA. T-cell responses to CD1-presented lipid antigens in humans with Mycobacterium tuberculosis infection. Infect Immun. (2003) 71:3076–87. doi: 10.1128/IAI.71.6.3076-3087.2003
129. Kim JH, Hu Y, Yongqing T, Kim J, Hughes VA, Le Nours J, et al. CD1a on Langerhans cells controls inflammatory skin disease. Nat Immunol. (2016) 17:1159–66. doi: 10.1038/ni.3523
130. Bagchi S, He Y, Zhang H, Cao L, Van Rhijn I, Moody DB, et al. CD1b-autoreactive T cells contribute to hyperlipidemia-induced skin inflammation in mice. J Clin Invest. (2017) 127:2339–52. doi: 10.1172/JCI92217
131. Zhao J, Siddiqui S, Shang S, Bian Y, Bagchi S, He Y, et al. Mycolic acid-specific T cells protect against Mycobacterium tuberculosis infection in a humanized transgenic mouse model. Elife (2015) 4:08525. doi: 10.7554/eLife.08525
132. Kawashima T, Norose Y, Watanabe Y, Enomoto Y, Narazaki H, Watari E, et al. Cutting edge: major CD8 T cell response to live bacillus Calmette-Guerin is mediated by CD1 molecules. J Immunol. (2003) 170:5345–8. doi: 10.4049/jimmunol.170.11.5345
133. Parlato S, Chiacchio T, Salerno D, Petrone L, Castiello L, Romagnoli G, et al. Impaired IFN-alpha-mediated signal in dendritic cells differentiates active from latent tuberculosis. PLoS ONE (2018) 13:e0189477. doi: 10.1371/journal.pone.0189477
134. Szalay G, Zugel U, Ladel CH, Kaufmann SH. Participation of group 2 CD1 molecules in the control of murine tuberculosis. Microbes Infect. (1999) 1:1153–7. doi: 10.1016/S1286-4579(99)00248-8
135. Dascher CC, Hiromatsu K, Xiong X, Morehouse C, Watts G, Liu G, et al. Immunization with a mycobacterial lipid vaccine improves pulmonary pathology in the guinea pig model of tuberculosis. Int Immunol. (2003) 15:915–25. doi: 10.1093/intimm/dxg091
136. Christaki E, Diza E, Giamarellos-Bourboulis EJ, Papadopoulou N, Pistiki A, Droggiti DI, et al. NK and NKT cell depletion alters the outcome of experimental pneumococcal pneumonia: relationship with regulation of interferon-gamma production. J Immunol Res. (2015) 2015:532717. doi: 10.1155/2015/532717
137. Hwang SJ, Kim S, Park WS, Chung DH. IL-4-secreting NKT cells prevent hypersensitivity pneumonitis by suppressing IFN-gamma-producing neutrophils. J Immunol. (2006) 177:5258–68. doi: 10.4049/jimmunol.177.8.5258
138. Bilenki L, Wang S, Yang J, Fan Y, Joyee AG, Yang X. NK T cell activation promotes Chlamydia trachomatis infection in vivo. J Immunol. (2005) 175:3197–206. doi: 10.4049/jimmunol.175.5.3197
139. Kelly H, Mandraju R, Coelho-dos-Reis JG, Tsuji M. Effects of HIV-1-induced CD1c and CD1d modulation and endogenous lipid presentation on CD1c-restricted T-cell activation. BMC Immunol. (2013) 14:4. doi: 10.1186/1471-2172-14-4
140. Taubenberger JK, Morens DM. The pathology of influenza virus infections. Annu Rev Pathol. (2008) 3:499–522. doi: 10.1146/annurev.pathmechdis.3.121806.154316
141. De Santo C, Salio M, Masri SH, Lee LY, Dong T, Speak AO, et al. Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J Clin Invest. (2008) 118:4036–48. doi: 10.1172/JCI36264
142. Paget C, Ivanov S, Fontaine J, Blanc F, Pichavant M, Renneson J, et al. Potential role of invariant NKT cells in the control of pulmonary inflammation and CD8+ T cell response during acute influenza A virus H3N2 pneumonia. J Immunol. (2011) 186:5590–602. doi: 10.4049/jimmunol.1002348
143. Kok WL, Denney L, Benam K, Cole S, Clelland C, McMichael AJ, et al. Pivotal advance: invariant NKT cells reduce accumulation of inflammatory monocytes in the lungs and decrease immune-pathology during severe influenza A virus infection. J Leukoc Biol. (2012) 91:357–68. doi: 10.1189/jlb.0411184
144. Ivanov S, Renneson J, Fontaine J, Barthelemy A, Paget C, Fernandez EM, et al. Interleukin-22 reduces lung inflammation during influenza A virus infection and protects against secondary bacterial infection. J Virol. (2013) 87:6911–24. doi: 10.1128/JVI.02943-12
145. Paget C, Ivanov S, Fontaine J, Renneson J, Blanc F, Pichavant M, et al. Interleukin-22 is produced by invariant natural killer T lymphocytes during influenza A virus infection: potential role in protection against lung epithelial damages. J Biol Chem. (2012) 287:8816–29. doi: 10.1074/jbc.M111.304758
146. Barthelemy A, Sencio V, Soulard D, Deruyter L, Faveeuw C, Le Goffic R, et al. Interleukin-22 immunotherapy during severe influenza enhances lung tissue integrity and reduces secondary bacterial systemic invasion. Infect Immun. (2018) 86:e00706–17. doi: 10.1128/IAI.00706-17
147. Joseph C, Togawa Y, Shindo N. Bacterial and viral infections associated with influenza. Influenza Other Respir Viruses (2013) 7(Suppl 2):105–13. doi: 10.1111/irv.12089
148. Brundage JF. Interactions between influenza and bacterial respiratory pathogens: implications for pandemic preparedness. Lancet Infect Dis. (2006) 6:303–12. doi: 10.1016/S1473-3099(06)70466-2
149. Holzapfel KL, Tyznik AJ, Kronenberg M, Hogquist KA. Antigen-dependent versus -independent activation of invariant NKT cells during infection. J Immunol. (2014) 192:5490–8. doi: 10.4049/jimmunol.1400722
150. Kobayashi E, Motoki K, Uchida T, Fukushima H, Koezuka Y. KRN7000, a novel immunomodulator, and its antitumor activities. Oncol Res. (1995) 7:529–34.
151. Kawano T, Nakayama T, Kamada N, Kaneko Y, Harada M, Ogura N, et al. Antitumor cytotoxicity mediated by ligand-activated human V alpha24 NKT cells. Cancer Res. (1999) 59:5102–5.
152. Nieda M, Okai M, Tazbirkova A, Lin H, Yamaura A, Ide K, et al. Therapeutic activation of Valpha24+Vbeta11+ NKT cells in human subjects results in highly coordinated secondary activation of acquired and innate immunity. Blood (2004) 103:383–9. doi: 10.1182/blood-2003-04-1155
153. Ishikawa A, Motohashi S, Ishikawa E, Fuchida H, Higashino K, Otsuji M, et al. A phase I study of alpha-galactosylceramide (KRN7000)-pulsed dendritic cells in patients with advanced and recurrent non-small cell lung cancer. Clin Cancer Res. (2005) 11:1910–7. doi: 10.1158/1078-0432.CCR-04-1453
154. Motohashi S, Nagato K, Kunii N, Yamamoto H, Yamasaki K, Okita K, et al. A phase I-II study of alpha-galactosylceramide-pulsed IL-2/GM-CSF-cultured peripheral blood mononuclear cells in patients with advanced and recurrent non-small cell lung cancer. J Immunol. (2009) 182:2492-501. doi: 10.4049/jimmunol.0800126
155. Nagato K, Motohashi S, Ishibashi F, Okita K, Yamasaki K, Moriya Y, et al. Accumulation of activated invariant natural killer T cells in the tumor microenvironment after alpha-galactosylceramide-pulsed antigen presenting cells. J Clin Immunol. (2012) 32:1071–81. doi: 10.1007/s10875-012-9697-9
156. Al Omar SY, Marshall E, Middleton D, Christmas SE. Increased numbers but functional defects of CD56+CD3+ cells in lung cancer. Int Immunol. (2012) 24:409–15. doi: 10.1093/intimm/dxr122
157. Pyszniak M, Rybojad P, Pogoda K, Jablonka A, Bojarska-Junak A, Tabarkiewicz J. Percentages of NKT cells in the tissues of patients with non-small cell lung cancer who underwent surgical treatment. Kardiochir Torakochirurgia Pol. (2014) 11:34–9. doi: 10.5114/kitp.2014.41928
158. Konishi J, Yamazaki K, Yokouchi H, Shinagawa N, Iwabuchi K, Nishimura M. The characteristics of human NKT cells in lung cancer–CD1d independent cytotoxicity against lung cancer cells by NKT cells and decreased human NKT cell response in lung cancer patients. Hum Immunol. (2004) 65:1377–88. doi: 10.1016/j.humimm.2004.09.003
159. Motohashi S, Ishikawa A, Ishikawa E, Otsuji M, Iizasa T, Hanaoka H, et al. A phase I study of in vitro expanded natural killer T cells in patients with advanced and recurrent non-small cell lung cancer. Clin Cancer Res. (2006) 12(20 Pt 1):6079–86. doi: 10.1158/1078-0432.CCR-06-0114
160. Terabe M, Swann J, Ambrosino E, Sinha P, Takaku S, Hayakawa Y, et al. A nonclassical non-Valpha14Jalpha18 CD1d-restricted (type II) NKT cell is sufficient for down-regulation of tumor immunosurveillance. J Exp Med. (2005) 202:1627–33. doi: 10.1084/jem.20051381
161. Ambrosino E, Terabe M, Halder RC, Peng J, Takaku S, Miyake S, et al. Cross-regulation between type I and type II NKT cells in regulating tumor immunity: a new immunoregulatory axis. J Immunol. (2007) 179:5126–36. doi: 10.4049/jimmunol.179.8.5126
162. McKnight CG, Morris SC, Perkins C, Zhu Z, Hildeman DA, Bendelac A, et al. NKT cells contribute to basal IL-4 production but are not required to induce experimental asthma. PLoS ONE (2017) 12:e0188221. doi: 10.1371/journal.pone.0188221
163. Zhang G, Nie H, Yang J, Ding X, Huang Y, Yu H, et al. Sulfatide-activated type II NKT cells prevent allergic airway inflammation by inhibiting type I NKT cell function in a mouse model of asthma. Am J Physiol Lung Cell Mol Physiol. (2011) 301:L975–84. doi: 10.1152/ajplung.00114.2011
164. Paun A, Bergeron ME, Haston CK. NKT deficient mice are not spared lung disease after exposure to thoracic radiotherapy. Radiat Res. (2014) 181:369–75. doi: 10.1667/RR13581.1
165. Cheung KL, Jarrett R, Subramaniam S, Salimi M, Gutowska-Owsiak D, Chen YL, et al. Psoriatic T cells recognize neolipid antigens generated by mast cell phospholipase delivered by exosomes and presented by CD1a. J Exp Med. (2016) 213:2399–412. doi: 10.1084/jem.20160258
166. Mathai SC, Danoff SK. Management of interstitial lung disease associated with connective tissue disease. BMJ (2016) 352:h6819. doi: 10.1136/bmj.h6819
Keywords: pulmonary disorders, lipid antigens, CD1 molecules, CD1-restricted T cells, natural killer T cells
Citation: Ryu S, Park JS, Kim HY and Kim JH (2018) Lipid-Reactive T Cells in Immunological Disorders of the Lung. Front. Immunol. 9:2205. doi: 10.3389/fimmu.2018.02205
Received: 03 June 2018; Accepted: 05 September 2018;
Published: 26 September 2018.
Edited by:
Luc Van Kaer, Vanderbilt University, United StatesReviewed by:
François Trottein, Centre National de la Recherche Scientifique (CNRS), FranceMark L. Lang, University of Oklahoma Health Sciences Center, United States
Seddon Y. Thomas, National Institute of Environmental Health Sciences (NIEHS), United States
Copyright © 2018 Ryu, Park, Kim and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hye Young Kim, aHlraW0xMUBzbnUuYWMua3I=
Ji Hyung Kim, amF5X2tpbUBrb3JlYS5hYy5rcg==