
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 01 October 2018
Sec. Primary Immunodeficiencies
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02191
This article is part of the Research Topic Etiopathogenesis of Systemic Sclerosis: An Update View all 11 articles
 Sokratis A. Apostolidis1†Giuseppina Stifano2†Tracy Tabib1Lisa M. Rice2Christina M. Morse1Bashar Kahaleh3Robert Lafyatis1*
Sokratis A. Apostolidis1†Giuseppina Stifano2†Tracy Tabib1Lisa M. Rice2Christina M. Morse1Bashar Kahaleh3Robert Lafyatis1*Objective: The mechanisms that lead to endothelial cell (EC) injury and propagate the vasculopathy in Systemic Sclerosis (SSc) are not well understood. Using single cell RNA sequencing (scRNA-seq), our goal was to identify EC markers and signature pathways associated with vascular injury in SSc skin.
Methods: We implemented single cell sorting and subsequent RNA sequencing of cells isolated from SSc and healthy control skin. We used t-distributed stochastic neighbor embedding (t-SNE) to identify the various cell types. We performed pathway analysis using Gene Set Enrichment Analysis (GSEA) and Ingenuity Pathway Analysis (IPA). Finally, we independently verified distinct markers using immunohistochemistry on skin biopsies and qPCR in primary ECs from SSc and healthy skin.
Results: By combining the t-SNE analysis with the expression of known EC markers, we positively identified ECs among the sorted cells. Subsequently, we examined the differential expression profile between the ECs from healthy and SSc skin. Using GSEA and IPA analysis, we demonstrated that the SSc endothelial cell expression profile is enriched in processes associated with extracellular matrix generation, negative regulation of angiogenesis and epithelial-to-mesenchymal transition. Two of the top differentially expressed genes, HSPG2 and APLNR, were independently verified using immunohistochemistry staining and real-time qPCR analysis.
Conclusion: ScRNA-seq, differential gene expression and pathway analysis revealed that ECs from SSc patients show a discrete pattern of gene expression associated with vascular injury and activation, extracellular matrix generation and negative regulation of angiogenesis. HSPG2 and APLNR were identified as two of the top markers of EC injury in SSc.
Vascular injury is a hallmark event in the pathogenesis of Systemic Sclerosis (SSc) (1). Endothelial dysfunction happens early in the course of the disease and drives some of the most prominent clinical manifestations of SSc, including Raynaud's phenomenon (RP), telangiectasias, gastric antral vascular ectasias (GAVE), pulmonary arterial hypertension (PAH), and SSc renal crisis (SRC) (1, 2). Loss of nailfold capillaries, nailfold microhemorrhages, and “giant” capillaries are a very useful tool for physicians to diagnose SSc and underlines the importance of microvascular damage in the disease progression (3). Histopathologic examination of the affected vessels reveals extensive intimal hyperplasia, adventitial fibrosis and vascular smooth muscle hypertrophy that lead to luminal narrowing and ultimately occlusion and thrombosis (4, 5). The result is progressive tissue hypoxia, recurrent cycles of ischemia—reperfusion injury and inflammatory changes. In the majority of SSc patients, vascular changes precede the onset of fibrosis suggesting that endothelial injury is central in the pathogenesis of the disease (6–8) and can involve organs in which fibrosis is not traditionally seen such as the kidneys.
The exact mechanisms that lead to endothelial cell injury and propagate the vasculopathy in SSc are not well understood. The presence of tissue hypoxia should promote compensatory angiogenesis. However, this process is defective in SSc patients who exhibit impaired neovascularization and loss of capillaries and arterioles leading to painful digital ulcerations, PAH and SRC (1, 9, 10). A complex network of interactions between endothelial cells, pericytes, myofibroblasts, and the extracellular matrix (ECM) has been implicated in the pathogenesis of SSc (8). It is currently unclear what drives the activation of fibroblasts and the increased ECM deposition responsible for the fibrotic changes seen in SSc. The endothelial cell injury has been proposed to play a prominent role through the production of activating cytokines by SSc endothelial cells (2, 11), disruption of vascular permeability and extravasation of growth factors (1), induction of hypoxia (2), and possibly by contributing to the pool of myofibroblasts through endothelial-to-mesenchymal transition (12).
Altered gene expression, alternative splicing and epigenetic mechanisms have been shown to contribute to the aberrant endothelial function (13–15). Prior gene expression profiling studies (16–19) and proteome-side analyses (13) have shed light onto the molecular pathways affected in SSc patients. However, these studies do not address the discrete contributions of the implicated cell subsets or individual cells and they do not account for cellular heterogeneity and differential cell composition of the target tissues. Thus, interpretation of their results is limited.
In this report, we implemented single cell sorting and subsequent RNA sequencing of cells isolated from SSc and healthy control (HC) skin. We present evidence that scRNA-seq provides a robust platform for cellular identification that allows for gene expression analysis at the single cell level and accounts for cellular heterogeneity. We focus on skin endothelial cells and define the differential in situ gene expression profile in SSc patients. Using pathway analysis software, we highlight the implicated molecular pathways. Finally, we verify independently on skin biopsies using immunohistochemistry and on primary endothelial cells using qPCR that APLNR and HSPG2 represent markers highly expressed in endothelial cells from SSc skin and can potentially be used as surrogates of endothelial dysfunction in SSc patients.
The Boston University Medical Center Institutional Review Board (Boston, MA, USA) reviewed and approved the conduct of this study. Informed consent was obtained from patients with diffuse cutaneous SSc [according to diagnostic (20) and subtype (21) criteria] and healthy subjects. Skin biopsies were obtained from the dorsal mid forearm and immediately collected in PBS for single cell isolation. The modified Rodnan skin score (MRSS) was determined for each patient on the day of the biopsy (22).
For the qPCR studies with primary endothelial cells, human microvascular endothelial cells (MVECs) were isolated as described previously (23) from skin biopsies of four diffuse cutaneous SSc patients and four age and sex-matched healthy controls. Informed consent was obtained in compliance with the Institutional Review Board of Human Studies of University of Toledo. All patients fulfilled the American College of Rheumatology criteria for the diagnosis of SSc; they were not on immunosuppressive or steroid therapy and none had digital ulcers or PAH.
Skin digestion was performed using the whole skin dissociation kit for human (130-101-540, Macs Miltenyi Biotec). Enzymatic digestion was completed in 2 h, followed by mechanical dissociation using gentleMacs Dissociator running the gentleMACS program h_skin_01.
Live cells were stained using NucBlue Live Cell Stain ReadyProbes reagent (Hoechst33342), and sorted using fluorescence-activated cell sorting (FACS) with a Beckman Coulter MoFlo Legacy, excited with multi line UV and detected with 450/20 band pass filter. Cells were deposited with cyclone in TCL buffer (Qiagen) on a 96-well plate, and stored at −80°C until RNA-seq processing.
RNA-seq was performed using the SmartSeq2 protocol. The SmartSeq2 libraries were prepared according to the SmartSeq2 protocol (24) with some modifications (25). The Smart-Seq2 data was processed at the Broad Institute using a standard computational pipeline. Libraries were barcoded by cell. They were sequenced using Illumina NextSeq platform. Data was deconvoluted by barcode and aligned using Tophat version 2.0.10 (26). Transcripts were quantified using the Cufflinks suite version 2.2.1 (27). Cuffnorm files were analyzed using the R environment for statistical computing (version 3.2.1). Using R, we performed t-distributed stochastic neighbor embedding (t-SNE) analysis, k-means clustering and hierarchical clustering. The following packages were used in R: tsne, rtsne, heatmap.2, rorc, gplots, ggplot2, hmisc, reshape, stringr, mixtools, reshape2, vioplot, seurat. The following parameters were used for t-SNE plots: perplexity 30, max iterations at default of 1000, initial dimensions at 10 and theta 0.0. Pathway analysis was performed using the Gene Set Enrichment Analysis software (GSEA) developed by the Broad Institute (28). Our dataset was compared against the following reference genesets: extracellular matrix, KEGG ECM receptor interactions, hallmark epithelial mesenchymal transition, positive regulation of angiogenesis, negative regulation of angiogenesis. Data was also analyzed with the Ingenuity Pathway Analysis (IPA, QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis).
Microvascular endothelial cells (MVECs) were isolated from the biopsy samples and purified using CD31 magnetic beads as previously described (23) and cultured in Clonetics Endothelial Cell Basal Medium-2 (EBM-2) supplemented with EGM-2-MV growth factors (EGM-2) at 37°C in 5% CO2. Normal control dermal MVECs were similarly derived from healthy adult donors who were matched with the SSc patients for age, sex and race.
Immunohistochemistry for HSPG2 was performed using an anti-HSPG2 antibody (anti-Perlecan, mouse IgG1 Antibody, clone 5D7-2E4, Millipore Sigma), using a staining protocol as previously described (29). Four healthy control skin biopsies and six scleroderma skin biopsies were stained with anti-HSPG2 antibody. Quantitative real-time PCR was performed using primary endothelial cells (MVECs) for APLNR and ACTB control, using the ddCT method as previously described (30), with the following TaqMan probes on a 7300 Real-Time PCR system (Applied Biosystems):
APLNR: Hs00270873_s1, FAM-MGB, Cat. #4453320 (ThermoFisher Scientific, Applied Biosystems).
ACTB: Hs01060665_g1, FAM-MGB, Cat. #4448892 (ThermoFisher Scientific, Applied Biosystems).
Immunofluorescent single and dual antibody staining using tyramide signal amplification (Tyramide SuperBoost Kits with Alexa Fluor Tyramides; ThermoFisher Scientific, Waltham, MA) were performed on formalin fixed paraffin embedded SSc skin tissues. Tissue sections (5 μm thick) were depariffinized and rehydrated followed by heat induced antigen retrieval in citrate buffer pH6.0 (Vector Labs, Burlingame, CA) for 10 min then allowed to cool for 10 min. Blocking was achieved by using 3% H2O2 followed by 10% goat serum (ThermoFisher Scientific, Waltham, MA) for 1 h each. Dual antibody staining was performed using combinations of mouse monoclonal anti-HSPG Perlecan antibody (1:100; Millipore, Temecula, CA); anti-human Von Willebrand Factor mouse monoclonal antibody (1:100; Dako, Santa Clara, CA); anti-human Von Willebrand Factor rabbit polyclonal antibody (1:50); Sigma Aldrich, St. Louis,MO) and Apelin receptor rabbit monoclonal antibody(clone: 5H5L9, 1:500, ThermoFisher Scientific, Waltham, MA). All primary antibodies were incubated overnight at 4°C. Enzymatic development was performed using appropriate goat anti-rabbit or goat anti-mouse Poly-HRP conjugated secondary antibodies (ThermoFisher Scientific, Waltham, MA) for 1 h followed by Alexa Fluor 488 or 594 labeled tyramide solution and completed with reaction stop solution (ThermoFisher Scientific, Waltham, MA) Dual antibody stained samples underwent the same staining process; after the first antibody development was complete tissue again underwent an additional heat induced antigen retrieval, blocking, incubation with compatible primary antibody, poly-HRP secondary antibody and development with spectrally compatible tryamide Alexa Fluor. Slides underwent nuclear staining with Hoeschst stain (1:2000; ThermoFisher Scientific, Waltham, MA). All wash steps consisted of PBS washes 3 times 10 min each. Finally, slides were cover slipped, using ProLong Diamnond Antifade Mountant (ThermoFisher Scientific, Waltham, MA). Images were taken using an Olympus FLUOVIEW FV1000 (Olympus, Waltham, MA) confocal laser-scanning microscope.
In order to discover the altered regulation of gene expression in diffuse cutaneous systemic sclerosis (dcSSc), we analyzed skin biopsies from one dcSSc patient and one age and sex-matched healthy control. Supplementary Figure 1 shows H&E staining of the skin of the dSSc patient with notable fibrosis and inflammatory infiltration. The skin biopsies were digested and single cell suspensions were used to FACS sort single cells in individual wells, which were subsequently used for cDNA library creation and down-stream RNA sequencing. We successfully sequenced 88 cells from the healthy skin biopsy and 96 cells from the SSc skin biopsy.
For our initial goal to visualize and ultimately define the various cell subsets in the dataset, we used t-distributed stochastic neighbor embedding (t-SNE), a method of unsupervised learning for dimensionality reduction. 2D projection of the t-SNE effectively reduced the dimensionality of the data, revealing clustering patterns that represent distinct cellular populations (Supplementary Figure 2). Next, we performed K-means clustering analysis of the t-SNE output. First, we calculated the recommended number of k-means centers to be 10 using the “elbow criterion” (Supplementary Figure 3). Subsequently, we overlaid the k-means clustering on the t-SNE projection to visualize individual clusters (Figure 1A).
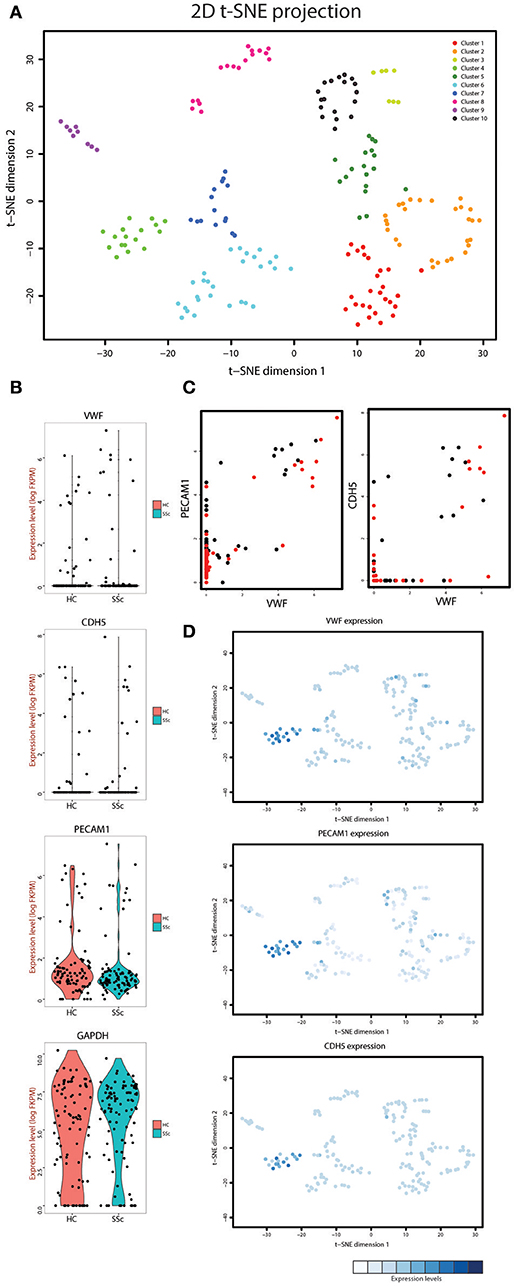
Figure 1. (A) t-SNE analysis of the cells isolated from the systemic sclerosis (SSc) and healthy control (HC) skin. Overlaid is k-means clustering with a defined number of 10 clusters with every cluster represented by a different color. (B) Violin plots showing the expression levels and density of expression for the VWF, CDH5, PECAM1, and GAPDH genes in the cells derived from HC and SSc skin. (C) Co-expression plots of VWF with PECAM1 (left) and CDH5 (right) in both HC (black dots) and SSc (red dots) skin cells. (D) Overlay of the t-SNE analysis with the expression level of VWF, PECAM1, and CDH5 for each cell. The expression levels for each gene were normalized within the respective graph.
In order to define the cluster that represents the endothelial cell population in our dataset we employed known endothelial cell markers. Von Willebrand factor (gene name VWF), platelet endothelial cell adhesion molecule (gene name PECAM1) and vascular endothelial cadherin (gene name CDH5) were expressed in a distinct group of cells in our dataset (Figure 1B). Furthermore, plotting VWF against PECAM1 and CDH5 demonstrated that the cells expressing high levels of VWF were also the cells that expressed increased levels of PECAM1 and CDH5 (Figure 1C). Next, we overlaid the expression of these genes on the t-SNE projection plot (Figure 1D) and were able to identify cluster 4 as the endothelial cell cluster (see Figure 1 for cluster numbering). This cluster contained 9 cells from healthy control skin and 8 cells from SSc skin. We used the gene expression profiles of these cells for the rest of the analysis.
After defining the endothelial cells in our dataset, we identified differentially expressed genes between HC and SSc ECs of the skin. We focused on the two-fold up-regulated or two-fold down-regulated genes in SSc compared to HC endothelial cells (Supplementary Figure 4 and Supplementary Table 1). Differential expression was performed using excel. Pair-wise comparison was done with t-test and FDR applied to account for multiple tests. Only genes that that had >2.0 or < −2.0 FC in a statistically significant manner were included. Figure 2A shows the genes upregulated by at least two-fold in a statistically significant manner (top bin) that contain already established markers of endothelial injury and activation, such as the Apelin receptor APLNR (31–36), as well as previously identified markers of vascular dysfunction in SSc, such as THBS1 (16, 37–39) and VWF (13, 40–44). The top bin also includes components of the extracellular matrix, including the heparan sulfate proteoglycan 2 (gene name HSPG2) that was previously shown to be implicated in fibrotic processes (45–47) including SSc-associated fibrosis (48), wound healing (49, 50) and TGF-β signaling (51, 52). Violin plots for the expression of VWF, THBS1, APLNR, and HSPG2 demonstrate that these genes are upregulated in endothelial cells from SSc skin compared to HC skin (Figure 2B).
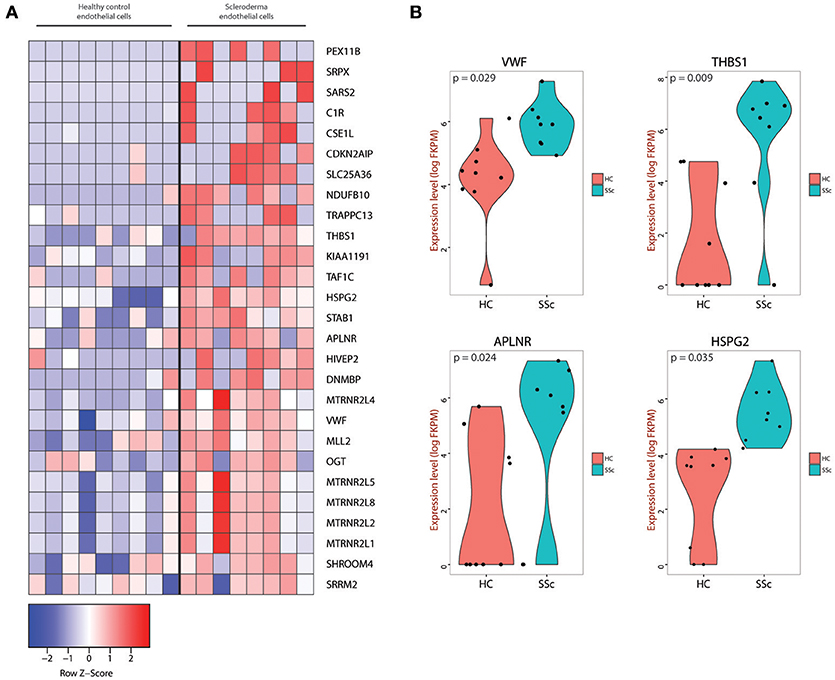
Figure 2. (A) Heatmap of genes that were at least two-fold upregulated in SSc endothelial cells compared to endothelial cells isolated from healthy skin. (B) Violin plots for the VWF, THBS1, APLNR, and HSPG2 genes showing the expression level and density of expression for the endothelial cells from HC and SSc skin. The corresponding p-values are also depicted for each gene.
In order to understand the pathways enriched in our dataset and to find gene signatures that are positively or negatively regulated in SSc endothelial cells compared to their healthy counterparts, we used Gene Set Enrichment Analysis (GSEA) (28) and Ingenuity Pathway Analysis (IPA, Qiagen). GSEA provides a software platform to analyze gene expression data through pairwise comparison of the dataset of interest with known biological processes and pathways. ECM alterations are a prominent feature in SSc pathogenesis. Intimal thickening and adventitial fibrosis are commonly seen in biopsies of SSc skin and are the pathologic representations of defective angiogenesis, skin fibrosis and vascular wall remodeling. Previous functional studies have shown that endothelial cells play a central role by promoting the fibrotic intimal lesions through interaction with vascular smooth muscle cells and pericytes (1, 53) and possibly by TGF-β mediated endothelial-to-mesenchymal transition (1, 54, 55). Using genesets that represent the ECM production and ECM receptor interactions, we found that the SSc endothelial cell gene expression profile correlated weakly with ECM (p = 0.38, Figure 3A, ECM) but more strongly in the complex interactions associated with ECM formation and receptor interactions (p = 0.0001, Figure 3A, ECM receptor interaction). Using a signature geneset for epithelial-to-mesenchymal transition (EMT), we show that SSc endothelial cells showed a trend toward enrichment in EMT-associated genes (p = 0.284, Figure 3B). Finally, in order to better understand the effect of SSc in angiogenesis, we employed two different GSEA sets: one corresponding to negative and one to positive regulation of angiogenesis. SSc endothelial cells demonstrated enrichment of genes associated with negative regulation of angiogenesis (p = 0.018, Figure 3C); conversely, genes associated with the positive regulation of angiogenesis showed a trend toward downregulation in SSc. (p = 0.2703, Figure 3D). Thus, GSEA demonstrates that the SSc endothelial cell expression profile is enriched in processes associated with ECM generation, weakly associated with EMT, and detects angiogenesis to be negatively regulated in SSc endothelial cells.
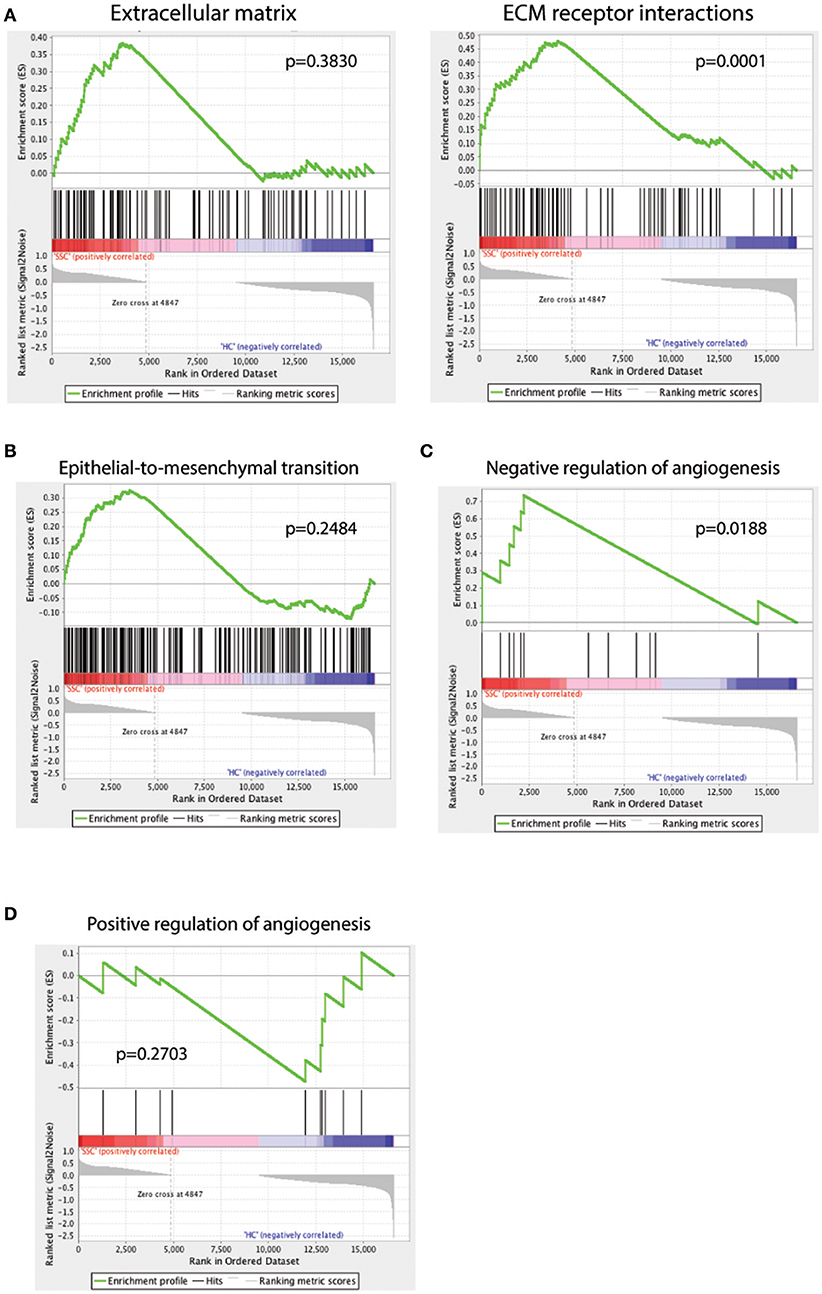
Figure 3. GSEA analysis of the scRNAseq dataset against the GSEA geneset for extracellular matrix (A, left) and extracellular matrix (ECM) receptor interactions (A, right), epithelial-to-mesenchymal transition (B), negative regulation of angiogenesis (C) and positive regulation of angiogenesis (D). A positive enrichment score on the y-axis indicates positive correlation with the SSc endothelial cell group and a negative enrichment score indicates a negative correlation.
In order to find the top biologic processes, signatures, and pathways associated with our dataset, we used Ingenuity Pathway Analysis (Qiagen). Examining the canonical pathways enriched in our database, we found the SSc endothelial cells show enrichment in pathways associated with inhibition of angiogenesis, acute phase response, complement activation and matrix metalloproteinases (Figure 4).
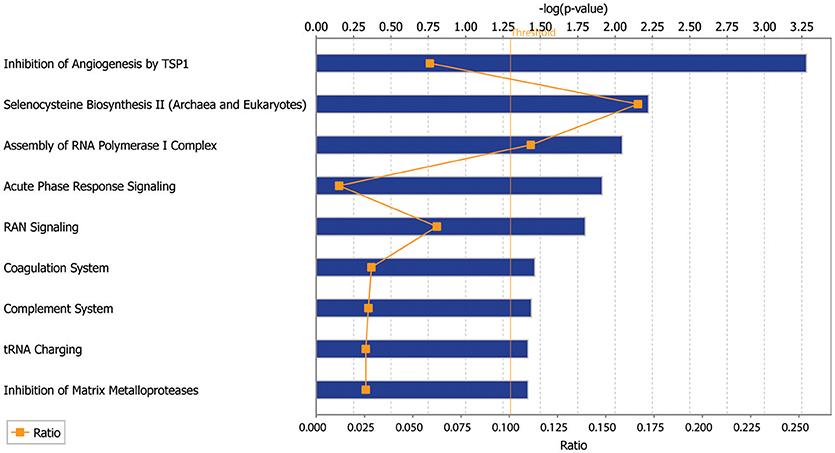
Figure 4. Ingenuity Pathway Analysis (IPA) of the scRNAseq database showing the top canonical pathways enriched in the endothelial cells derived from SSc skin compared to those from healthy control skin.
Complementing our scRNA-seq experiments, we independently verified our findings using distinct experimental protocols. First, we stained skin biopsies of SSc skin and HC skin with the extracellular matrix protein HSPG2, one of the differentially expressed genes in the scRNA-seq experiments (Figure 2). HSPG2 staining was more robust in SSc skin, especially in the perivascular areas (Figure 5A). HSPG2 has been shown to be regulated in a TGF-β dependent manner (51, 52). Both APLNR and HSPG2 co-stained by immunoflorescence with VWF, confirming that they are expressed by endothelial cells in SSc skin (Figure 5D). In order to explore the effect of TGF-β signaling blockade on HSPG2 in SSc patients, we used our previously generated microarray data from our clinical trial of a TGF-β blocking antibody, fresolimumab (for patient characteristics, study details, outcomes and microarray data, refer to (16), Clinicaltrials.gov NCT01284322, GEO database accession number GSE55036). In this trial, SSc patients were treated for 7 and 24 weeks with fresolimumab (Figure 5B) and HSPG2 expression was reduced by the end of 24 weeks in a statistically significant manner (ANOVA, p value 0.027), indicating that HSPG2 expression is inhibited by TGF-β blockade, coincident with the decrease in disease activity and skin inflammation seen with fresolimumab.
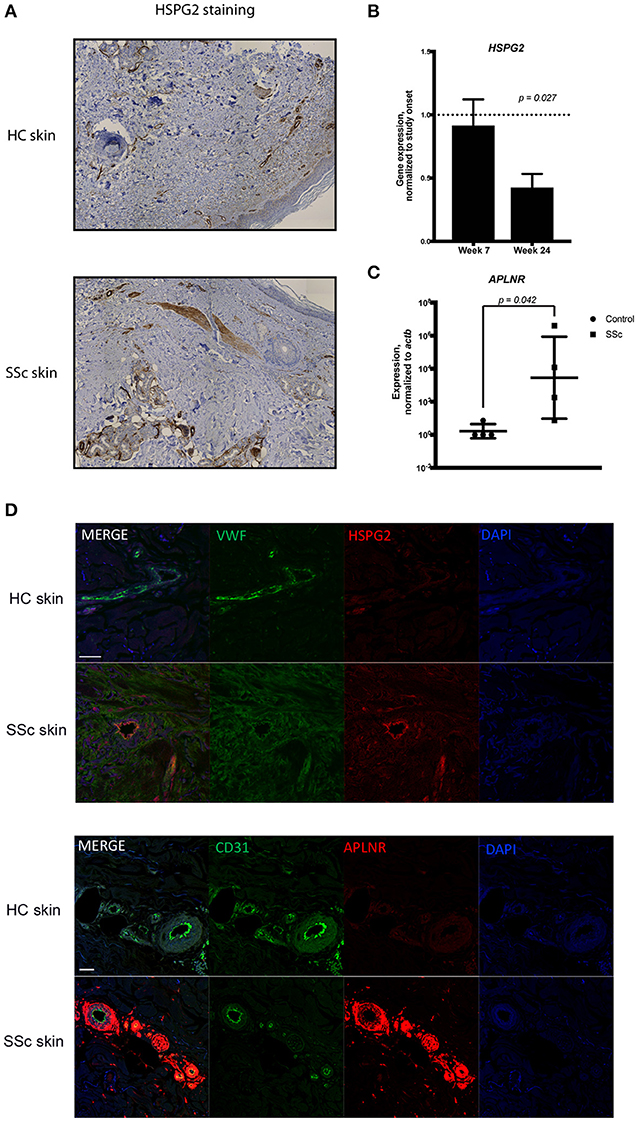
Figure 5. (A) Immunohistochemistry of skin biopsies from healthy and SSc skin showing the expression of HSPG2. (B) The microarray data from the Fresolimumab clinical trial were analyzed to show the expression of HSPG2 gene within the study subjects during treatment with fresolimumab at week 7 and week 24. Expression levels are normalized to the onset of study at the time of enrollment. Data were analyzed using ANOVA and shown is the p-value. (C) qPCR analysis of the expression of APLNR in MVECs isolated from healthy and scleroderma skin. Data are normalized to ACTB. t-test analysis was performed and the p-value is shown. (D) Dual antibody immunofluorescent (IF) staining of formalin fixed paraffin embedded scleroderma and control skin biopsies sectioned in 5 μm sections. Upper panels show dual IF staining of HSPG2 (green) and Von Willebrand factor (red) co-localizing on blood vessels. Lower panels show dual IF staining of Von Willebrand factor (green) and APLNR (red) co-localizing on blood vessels in scleroderma skin with DAPI nuclear stain. Scale bar = 50mm.
APLNR was another gene found to be increased in the SSc endothelial cells using scRNA-seq (Figure 2). To independently verify this result, we studied APLNR gene expression in microvascular endothelial cells (MVECs) isolated from the skin of four distinct SSc patients and four healthy controls using qPCR. APLNR was more highly expressed by SSc primary endothelial cells compared to HC endothelial cells (Figure 5C).
Vascular injury is a central event in the pathophysiology of SSc. However, our knowledge regarding the pathogenesis and pathways involved in the generation of endothelial cell injury is limited. Here, we provide a comprehensive analysis of scRNA-seq data generated from cells derived from SSc and healthy skin. We show that scRNA-seq provides a robust platform for identifying specific cell subtypes and more importantly differential gene expression profiles and signatures in the cell subpopulations of interest at the single cell level.
Using scRNA-seq, we identified distinct markers that can provide insight in the development of endothelial cell injury, including markers that have been previously linked with the pathogenesis of SSc, such as Thrombospondin 1 and Von Willebrand Factor (13, 16, 37–44). Additional genes identified by our analysis, notably APLNR and HSPG2, have not been previously linked to SSc pathogenesis. These genes are of particular interest as they have been associated with vascular activation and dysfunction as well as fibrosis in different settings (31–36, 45–50, 56).
Apelin and Elabela are both ligands of the Apelin receptor (APLNR/APJ), a G-protein coupled receptor found on endothelial cells (57). These ligands play central roles in cardiovascular development and angiogenesis (31, 35, 56, 58). The Apelin/Elabela-APLNR signaling is critical in regulating vascular maturation and APLNR –/– mice are embryonically lethal secondary to cardiovascular defects (33). APLNR expression is induced in conditions of hypoxia (32, 36). APLNR signaling has not been investigated in SSc other than data suggesting that elevated Apelin levels are associated with more severe vascular disease (59). The Apelin/Elabela-APLNR signaling has been implicated in pulmonary arterial hypertension, one of the major complications of SSc (60, 61). In this setting, increased Apelin activity appears beneficial and receptor agonists are being considered as therapies. In this context, increased APLNR expression might represent a positive adaptive response to hypoxia, fibrosis and/or other vascular injury in the SSc skin microenvironment. On the other hand, APLNR has been associated with pathological retinal angiogenesis and telangectasias (34, 62) and, thus, it might contribute to the dysregulated angiogenesis seen in SSc.
The HSPG2 gene codes for the extracellular matrix protein perlecan, which represents a main component of the blood vessel basement membrane (63). HSPG2 has been implicated in several fibrotic processes, including liver fibrosis and desmoplastic tumors (45–47). Interestingly, a C-terminal fragment of HSPG2 was found to be a main fibrogenic mediator produced by apoptotic SSc endothelial cells (48). Specifically, the C-terminal HSPG2 fragment produced by apoptotic SSc endothelial cells induced PI3K-dependent resistance to apoptosis in fibroblasts and activated myofibroblast differentiation (48). HSPG2-deficient mice exhibit delayed wound healing (49), impaired angiogenesis (49, 52) and decreased TGF-β production in the mouse skin (50). Reciprocally, TGF-β signaling induces HSPG2 promoter activity (51). Analysis of the fresolimumab clinical trial microarray data suggests that HSPG2 gene expression in SSc skin is regulated by TGF-β. Thus, TGFβ up-regulation of HSPG2 may mediate a profibrotic response to vascular injury in SSc skin.
Our pathway analysis on the single-cell platform demonstrates that the pathways of extracellular matrix generation, inhibition of angiogenesis, epithelial to mesenchymal transition and matrix metalloproteinase production are highly activated and centrally involved in the pathophysiology of endothelial cell injury in SSc patients. A recent study has highlighted the potential importance of endothelial-mesenchymal transition in SSc (64). Our pathway analysis, showing a trend toward upregulated epithelial-mesenchymal transition, supports this notion.
A limitation of the current study is that only one patient and one control biopsy were analyzed by scRNA-seq. However, ancillary studies examining RNA expression in cells and skin immunohistochemistry support the generalizability of the scRNA-seq findings. We anticipate with emerging technologies that more cells and more robust signatures might be found.
Vascular injury occurs early in the disease course of SSc. Raynaud's phenomenon and other vascular malformations can precede the development of overt disease for years to decades. Understanding the endothelial cell injury pathways has the potential of allowing early identification of patients and more accurate prognostication. In addition, our data point to several upregulated endothelial cell genes in SSc skin and implicate these genes in distinct pathways in SSc pathogenesis. Our results provide the framework for the development of biomarkers representing vascular injury and therapeutic targets to be further explored.
This study was carried out in accordance with the recommendations of IRB committee of Boston University Medical Center. All subjects gave written informed consent in accordance with the Declaration of Helsinki.
SA, GS, TT, LR, and CM designed and performed experiments, analyzed the data, and prepared the manuscript. BK analyzed the data and prepared the manuscript. RL designed experiments, analyzed the data and prepared the manuscript.
Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases under award number 2P50 AR060780 of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2018.02191/full#supplementary-material
Supplementary Figure 1. H&E staining of the skin from the dcSSc patients used for the scRNA-seq showing the extensive fibrosis (arrows) and inflammatory infiltration (arrowheads).
Supplementary Figure 2. Two-dimensional projection of the t-SNE analysis of the cells isolated from the systemic sclerosis (SSc) and healthy control (HC) skin. Cells were pooled together to facilitate cell subset identification.
Supplementary Figure 3. The “elbow” criterion used to determine the optimal number of clusters to be used for the k-means clustering used in conjunction with the t-SNE analysis.
Supplementary Figure 4. Heatmap with hierarchical clustering of genes that were at least two-fold up-regulated or down-regulated in SSc endothelial cells compared to endothelial cells isolated from healthy skin.
Supplementary Table 1. Genes regulated < 2-fold in SSc compared to healthy skin endothelial cells.
1. Matucci-Cerinic M, Kahaleh B, Wigley FM. Review: evidence that systemic sclerosis is a vascular disease. Arthritis Rheum. (2013) 65:1953–62. doi: 10.1002/art.37988
2. Altorok N, Wang Y, Kahaleh B. Endothelial dysfunction in systemic sclerosis. Curr Opin Rheumatol. (2014) 26:615–20. doi: 10.1097/BOR.0000000000000112
3. Cutolo M, Sulli A, Pizzorni C, Accardo S. Nailfold videocapillaroscopy assessment of microvascular damage in systemic sclerosis. J Rheumatol. (2000) 27:155–60.
4. D'Angelo WA, Fries JF, Masi AT, Shulman LE. Pathologic observations in systemic sclerosis (scleroderma). A study of fifty-eight autopsy cases and fifty-eight matched controls. Am J Med. (1969) 46:428–40. doi: 10.1016/0002-9343(69)90044-8
5. Norton WL, Nardo JM. Vascular disease in progressive systemic sclerosis (scleroderma). Ann Intern Med. (1970) 73:317–24.
6. Cipriani P, Marrelli A, Liakouli V, Di Benedetto P, Giacomelli R. Cellular players in angiogenesis during the course of systemic sclerosis. Autoimmun Rev. (2011) 10:641–6. doi: 10.1016/j.autrev.2011.04.016
7. Kahaleh B. Vascular disease in scleroderma: mechanisms of vascular injury. Rheum Dis Clin North Am. (2008) 34:57–71. doi: 10.1016/j.rdc.2007.12.004
8. Wigley FM. Vascular disease in scleroderma. Clin Rev Allergy Immunol. (2009) 36:150–75. doi: 10.1007/s12016-008-8106-x
9. Kuwana M, Okazaki Y. Brief report: impaired in vivo neovascularization capacity of endothelial progenitor cells in patients with systemic sclerosis. Arthritis Rheumatol. (2014) 66:1300–5. doi: 10.1002/art.38326
10. Manetti M, Guiducci S, Ibba-Manneschi L, Matucci-Cerinic M. Impaired angiogenesis in systemic sclerosis: the emerging role of the antiangiogenic VEGF(165)b splice variant. Trends Cardiovasc Med. (2011) 21:204–10. doi: 10.1016/j.tcm.2012.05.011
11. Serrati S, Chilla A, Laurenzana A, Margheri F, Giannoni E, Magnelli L, et al. Systemic sclerosis endothelial cells recruit and activate dermal fibroblasts by induction of a connective tissue growth factor (CCN2)/transforming growth factor beta-dependent mesenchymal-to-mesenchymal transition. Arthritis Rheum. (2013) 65:258–69. doi: 10.1002/art.37705
12. Hashimoto N, Phan SH, Imaizumi K, Matsuo M, Nakashima H, Kawabe T, et al. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. (2010) 43:161–72. doi: 10.1165/rcmb.2009-0031OC
13. van Bon L, Affandi AJ, Broen J, Christmann RB, Marijnissen RJ, Stawski L, et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N Engl J Med. (2014) 370:433–43. doi: 10.1056/NEJMoa1114576
14. Manetti M, Guiducci S, Romano E, Ceccarelli C, Bellando-Randone S, Conforti ML, et al. Overexpression of VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, leads to insufficient angiogenesis in patients with systemic sclerosis. Circ Res. (2011) 109:e14–26. doi: 10.1161/CIRCRESAHA.111.242057
15. Noda S, Asano Y, Takahashi T, Akamata K, Aozasa N, Taniguchi T, et al. Decreased cathepsin V expression due to Fli1 deficiency contributes to the development of dermal fibrosis and proliferative vasculopathy in systemic sclerosis. Rheumatology (2013) 52:790–9. doi: 10.1093/rheumatology/kes379
16. Rice LM, Padilla CM, McLaughlin SR, Mathes A, Ziemek J, Goummih S, et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J Clin Invest. (2015) 125:2795–807. doi: 10.1172/JCI77958
17. Whitfield ML, Finlay DR, Murray JI, Troyanskaya OG, Chi JT, Pergamenschikov A, et al. Systemic and cell type-specific gene expression patterns in scleroderma skin. Proc Natl Acad Sci USA. (2003) 100:12319–24. doi: 10.1073/pnas.1635114100
18. Gardner H, Shearstone JR, Bandaru R, Crowell T, Lynes M, Trojanowska M, et al. Gene profiling of scleroderma skin reveals robust signatures of disease that are imperfectly reflected in the transcript profiles of explanted fibroblasts. Arthritis Rheum. (2006) 54:1961–73. doi: 10.1002/art.21894
19. Milano A, Pendergrass SA, Sargent JL, George LK, McCalmont TH, Connolly MK, et al. Molecular subsets in the gene expression signatures of scleroderma skin. PLoS ONE (2008) 3:e2696. doi: 10.1371/annotation/05bed72c-c6f6-4685-a732-02c78e5f66c2
20. Masi AT, Rodnan G, Medsger T Jr, Altman R, D'Angelo W, Fries J. Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Preliminary criteria for the classification of systemic sclerosis (scleroderma). Arthritis Rheum. (1980) 23:581–90.
21. LeRoy EC, Black C, Fleischmajer R, Jablonska S, Krieg T, Medsger TA Jr, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. (1988) 15:202–5.
22. Furst DE, Clements PJ, Steen VD, Medsger T Jr, Masi AT, D'Angelo WA, et al. The modified Rodnan skin score is an accurate reflection of skin biopsy thickness in systemic sclerosis. J Rheumatol. (1998) 25:84–8.
23. Normand J, Karasek MA. A method for the isolation and serial propagation of keratinocytes, endothelial cells, and fibroblasts from a single punch biopsy of human skin. In Vitro Cell Dev Biol Anim. (1995) 31:447–55. doi: 10.1007/BF02634257
24. Picelli S, Faridani OR, Björklund ÅK, Winberg G, Sagasser S, Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. (2014) 9:171–81. doi: 10.1038/nprot.2014.006
25. Trombetta JJ, Gennert D, Lu D, Satija R, Shalek AK, Regev A. Preparation of single-cell RNA-seq libraries for next generation sequencing. Curr Protoc Mol Biol. (2014) suppl 107:4.22.1–4.22.17. doi: 10.1002/0471142727.mb0422s107
26. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. (2013) 14:R36. doi: 10.1186/gb-2013-14-4-r36
27. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. (2012) 7:562–78. doi: 10.1038/nprot.2012.016
28. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. (2005) 102:15545–50. doi: 10.1073/pnas.0506580102
29. Nazari B, Rice LM, Stifano G, Barron AM, Wang YM, Korndorf T, et al. Altered dermal fibroblasts in systemic sclerosis display podoplanin and CD90. Am J Pathol. (2016) 186:2650–64. doi: 10.1016/j.ajpath.2016.06.020
30. Farina G, Lafyatis D, Lemaire R, Lafyatis R. A four-gene biomarker predicts skin disease in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheum. (2010) 62:580–8. doi: 10.1002/art.27220
31. Cox CM, D'Agostino SL, Miller MK, Heimark RL, Krieg PA. Apelin, the ligand for the endothelial G-protein-coupled receptor, APJ, is a potent angiogenic factor required for normal vascular development of the frog embryo. Dev Biol. (2006) 296:177–89. doi: 10.1016/j.ydbio.2006.04.452
32. Eyries M, Siegfried G, Ciumas M, Montagne K, Agrapart M, Lebrin F, et al. Hypoxia-induced apelin expression regulates endothelial cell proliferation and regenerative angiogenesis. Circ Res. (2008) 103:432–40. doi: 10.1161/CIRCRESAHA.108.179333
33. Kang Y, Kim J, Anderson JP, Wu J, Gleim SR, Kundu RK, et al. Apelin-APJ signaling is a critical regulator of endothelial MEF2 activation in cardiovascular development. Circ Res. (2013) 113:22–31. doi: 10.1161/CIRCRESAHA.113.301324
34. Kasai A, Ishimaru Y, Higashino K, Kobayashi K, Yamamuro A, Yoshioka Y, et al. Inhibition of apelin expression switches endothelial cells from proliferative to mature state in pathological retinal angiogenesis. Angiogenesis (2013) 16:723–34. doi: 10.1007/s10456-013-9349-6
35. Kasai A, Shintani N, Oda M, Kakuda M, Hashimoto H, Matsuda T, et al. Apelin is a novel angiogenic factor in retinal endothelial cells. Biochem Biophys Res Commun. (2004) 325:395–400. doi: 10.1016/j.bbrc.2004.10.042
36. Zhang J, Liu Q, Hu X, Fang Z, Huang F, Tang L, et al. Apelin/APJ signaling promotes hypoxia-induced proliferation of endothelial progenitor cells via phosphoinositide-3 kinase/Akt signaling. Mol Med Rep. (2015) 12:3829–34. doi: 10.3892/mmr.2015.3866
37. Chen Y, Leask A, Abraham DJ, Kennedy L, Shi-Wen X, Denton CP, et al. Thrombospondin 1 is a key mediator of transforming growth factor beta-mediated cell contractility in systemic sclerosis via a mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK)-dependent mechanism. Fibrogenesis Tissue Repair (2011) 4:9. doi: 10.1186/1755-1536-4-9
38. Macko RF, Gelber AC, Young BA, Lowitt MH, White B, Wigley FM, et al. Increased circulating concentrations of the counteradhesive proteins SPARC and thrombospondin-1 in systemic sclerosis (scleroderma). Relationship to platelet and endothelial cell activation. J Rheumatol. (2002) 29:2565–70.
39. Mimura Y, Ihn H, Jinnin M, Asano Y, Yamane K, Tamaki K. Constitutive thrombospondin-1 overexpression contributes to autocrine transforming growth factor-beta signaling in cultured scleroderma fibroblasts. Am J Pathol. (2005) 166:1451–63. doi: 10.1016/S0002-9440(10)62362-0
40. Scheja A, Akesson A, Geborek P, Wildt M, Wollheim CB, Wollheim FA, et al. Von Willebrand factor propeptide as a marker of disease activity in systemic sclerosis (scleroderma). Arthritis Res. (2001) 3:178–82. doi: 10.1186/ar295
41. Blann AD, Sheeran TP, Emery P. von Willebrand factor: increased levels are related to poor prognosis in systemic sclerosis and not to tissue autoantibodies. Br J Biomed Sci. (1997) 54:5–9.
42. Herrick AL, Illingworth K, Blann A, Hay CR, Hollis S, Jayson MI. Von Willebrand factor, thrombomodulin, thromboxane, beta-thromboglobulin and markers of fibrinolysis in primary Raynaud's phenomenon and systemic sclerosis. Ann Rheum Dis. (1996) 55:122–7. doi: 10.1136/ard.55.2.122
43. Kahaleh MB, Osborn I, LeRoy EC. Increased factor VIII/von Willebrand factor antigen and von Willebrand factor activity in scleroderma and in Raynaud's phenomenon. Ann Intern Med. (1981) 94(4 pt 1):482–4. doi: 10.7326/0003-4819-94-4-482
44. Pendergrass SA, Hayes E, Farina G, Lemaire R, Farber HW, Whitfield ML, et al. Limited systemic sclerosis patients with pulmonary arterial hypertension show biomarkers of inflammation and vascular injury. PLoS ONE (2010) 5:e12106. doi: 10.1371/journal.pone.0012106
45. Gallai M, Kovalszky I, Knittel T, Neubauer K, Armbrust T, Ramadori G. Expression of extracellular matrix proteoglycans perlecan and decorin in carbon-tetrachloride-injured rat liver and in isolated liver cells. Am J Pathol. (1996) 148:1463–71.
46. Warren CR, Grindel BJ, Francis L, Carson DD, Farach-Carson MC. Transcriptional activation by NFkappaB increases perlecan/HSPG2 expression in the desmoplastic prostate tumor microenvironment. J Cell Biochem. (2014) 115:1322–33. doi: 10.1002/jcb.24788
47. Baiocchini A, Montaldo C, Conigliaro A, Grimaldi A, Correani V, Mura F, et al. Extracellular matrix molecular remodeling in human liver fibrosis evolution. PLoS ONE (2016);11:e0151736. doi: 10.1371/journal.pone.0151736
48. Laplante P, Raymond MA, Gagnon G, Vigneault N, Sasseville AM, Langelier Y, et al. Novel fibrogenic pathways are activated in response to endothelial apoptosis: implications in the pathophysiology of systemic sclerosis. J Immunol. (2005) 174:5740–9. doi: 10.4049/jimmunol.174.9.5740
49. Zhou Z, Wang J, Cao R, Morita H, Soininen R, Chan KM, et al. Impaired angiogenesis, delayed wound healing and retarded tumor growth in perlecan heparan sulfate-deficient mice. Cancer Res. (2004) 64:4699–702. doi: 10.1158/0008-5472.CAN-04-0810
50. Shu C, Smith SM, Melrose J. The heparan sulphate deficient Hspg2 exon 3 null mouse displays reduced deposition of TGF-beta1 in skin compared to C57BL/6 wild type mice. J Mol Histol. (2016) 47:365–74. doi: 10.1007/s10735-016-9677-0
51. Iozzo RV, Pillarisetti J, Sharma B, Murdoch AD, Danielson KG, Uitto J, et al. Structural and functional characterization of the human perlecan gene promoter. Transcriptional activation by transforming growth factor-beta via a nuclear factor 1-binding element. J Biol Chem. (1997) 272:5219–28. doi: 10.1074/jbc.272.8.5219
52. Sharma B, Handler M, Eichstetter I, Whitelock JM, Nugent MA, Iozzo RV. Antisense targeting of perlecan blocks tumor growth and angiogenesis in vivo. J Clin Invest. (1998) 102:1599–608. doi: 10.1172/JCI3793
53. Rajkumar VS, Sundberg C, Abraham DJ, Rubin K, Black CM. Activation of microvascular pericytes in autoimmune Raynaud's phenomenon and systemic sclerosis. Arthritis Rheum. (1999) 42:930–41. doi: 10.1002/1529-0131(199905)42:5 < 930::AID-ANR11>3.0.CO;2-1
54. Zvaifler NJ. Relevance of the stroma and epithelial-mesenchymal transition (EMT) for the rheumatic diseases. Arthritis Res Ther. (2006) 8:210. doi: 10.1186/ar1963
55. Li Z, Jimenez SA. Protein kinase Cdelta and c-Abl kinase are required for transforming growth factor beta induction of endothelial-mesenchymal transition in vitro. Arthritis Rheum. (2011) 63:2473–83. doi: 10.1002/art.30317
56. Kidoya H, Takakura N. Biology of the apelin-APJ axis in vascular formation. J Biochem. (2012) 152:125–31. doi: 10.1093/jb/mvs071
57. Ho L, van Dijk M, Chye STJ, Messerschmidt DM, Chng SC, Ong S, et al. ELABELA deficiency promotes preeclampsia and cardiovascular malformations in mice. Science (2017) 357:707–13. doi: 10.1126/science.aam6607
58. Chng SC, Ho L, Tian J, Reversade B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Dev Cell (2013) 27:672–80. doi: 10.1016/j.devcel.2013.11.002
59. Aozasa N, Asano Y, Akamata K, Noda S, Masui Y, Yamada D, et al. Serum apelin levels: clinical association with vascular involvements in patients with systemic sclerosis. J Eur Acad Dermatol Venereol. (2013) 27:37–42. doi: 10.1111/j.1468-3083.2011.04354.x
60. Yang P, Read C, Kuc RE, Buonincontri G, Southwood M, Torella R, et al. Elabela/toddler is an endogenous agonist of the apelin APJ receptor in the adult cardiovascular system, and exogenous administration of the peptide compensates for the downregulation of its expression in pulmonary arterial hypertension. Circulation (2017) 135:1160–73. doi: 10.1161/CIRCULATIONAHA.116.023218
61. Andersen CU, Hilberg O, Mellemkjaer S, Nielsen-Kudsk JE, Simonsen U. Apelin and pulmonary hypertension. Pulm Circ. (2011) 1:334–46. doi: 10.4103/2045-8932.87299
62. McKenzie JA, Fruttiger M, Abraham S, Lange CA, Stone J, Gandhi P, et al. Apelin is required for non-neovascular remodeling in the retina. Am J Pathol. (2012) 180:399–409. doi: 10.1016/j.ajpath.2011.09.035
63. Noonan DM, Fulle A, Valente P, Cai S, Horigan E, Sasaki M, et al. The complete sequence of perlecan, a basement membrane heparan sulfate proteoglycan, reveals extensive similarity with laminin A chain, low density lipoprotein-receptor, and the neural cell adhesion molecule. J Biol Chem. (1991) 266:22939–47.
Keywords: ScRNA-seq, HSPG2, APLNR, systemic sclerosis, endothelial dysfunction
Citation: Apostolidis SA, Stifano G, Tabib T, Rice LM, Morse CM, Kahaleh B and Lafyatis R (2018) Single Cell RNA Sequencing Identifies HSPG2 and APLNR as Markers of Endothelial Cell Injury in Systemic Sclerosis Skin. Front. Immunol. 9:2191. doi: 10.3389/fimmu.2018.02191
Received: 01 February 2018; Accepted: 04 September 2018;
Published: 01 October 2018.
Edited by:
Megan Anne Cooper, Washington University in St. Louis, United StatesReviewed by:
Mirko Manetti, Università degli Studi di Firenze, ItalyCopyright © 2018 Apostolidis, Stifano, Tabib, Rice, Morse, Kahaleh and Lafyatis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert Lafyatis, rlafyatis@gmail.com
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.