
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 26 September 2018
Sec. Viral Immunology
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02180
This article is part of the Research Topic Emerging Viruses: Host Immunity and Novel Therapeutic Interventions View all 17 articles
 Mathilde Laureti1,2†
Mathilde Laureti1,2† Divya Narayanan1,2†Julio Rodriguez-Andres1,2†
Divya Narayanan1,2†Julio Rodriguez-Andres1,2† John K. Fazakerley1,2
John K. Fazakerley1,2 Lukasz Kedzierski1,2*
Lukasz Kedzierski1,2*Flaviviruses are emerging and re-emerging arthropod-borne pathogens responsible for significant mortality and morbidity worldwide. The genus comprises more than seventy small, positive-sense, single-stranded RNA viruses, which are responsible for a spectrum of human and animal diseases ranging in symptoms from mild, influenza-like infection to fatal encephalitis and haemorrhagic fever. Despite genomic and structural similarities across the genus, infections by different flaviviruses result in disparate clinical presentations. This review focusses on two haemorrhagic flaviviruses, dengue virus and yellow fever virus, and two neurotropic flaviviruses, Japanese encephalitis virus and Zika virus. We review current knowledge on host-pathogen interactions, virus entry strategies and tropism.
The Flavivirus genus consists of more than 70 small, positive-sense, single-stranded RNA viruses transmitted by arthropods, in particular mosquitos and ticks. These include globally important human pathogens such as West Nile virus (WNV), Japanese encephalitis virus (JEV), dengue virus (DENV), Murray Valley encephalitis virus (MVE), tick-borne encephalitis virus (TBEV), Yellow Fever virus (YFV), and Zika virus (ZIKV). These viruses are responsible for some of the most severe arbovirus infections affecting humans, pose a serious threat to global health and have the potential to cause severe outbreaks. These are exemplified by the global distribution of DENV (1), the recent ZIKV outbreak in South America (2), YFV outbreaks in Africa (3), and Brazil (4) and the spread of WNV across North America (5). Flavivirus infections range from asymptomatic, through mild fever and arthralgia to life threatening haemorrhagic or encephalitic diseases (6). Flaviviruses are also able to persist in patients and can be responsible for long-term morbidities (7). There are no antiviral treatments for flavivirus infection currently in clinical use, and despite licensed vaccines against several of the viruses including YFV, JEV, TBEV, or DENV, outbreaks still occur highlighting challenges in implementing effective vaccination programs (8).
The flaviviral genome of ~11 kb contains a single open reading frame flanked by untranslated regions, and encodes 3 structural proteins (C, M, and E) and 7 non-structural proteins (NS). The mature virion features a surface densely covered with E glycoproteins and M proteins and a core consisting of capsid (C) protein and the RNA genome (9, 10). The entry into the target cell is dependent on E protein contact with its cognate receptor. E protein initially binds to attachment factors such as glycosaminoglycans. This effectively increases viral density on the cell surface, leading to high affinity receptor binding (11). The E protein ectodomain consists of three domains (E-DI, E-DII, E-DIII) of which E-DIII is thought to interact with attachment factors and receptors (12). E-DIII domain's importance is highlighted by the fact that a vast majority of potent, neutralizing antibodies has been mapped to this region. Nevertheless, anti-DI and DII antibodies, although less potent, show broader cross-reactivity and form a major pool of anti-E protein specific immonoglobulins (13). Receptor binding is followed by clathrin-mediated endocytosis (14), which is considered to be a major mechanism of flavivirus cell entry, although there are exceptions described below. This leads to formation of endosomes and low pH dependent changes in the E glycoprotein with subsequent membrane fusion and release of nucleocapsid into the cytosol (15). In vitro, flaviviruses are able to infect a plethora of cell lines originating from rodents, non-human primates, humans and mosquitos. However in vivo, fewer cell types seem able to support flavivirus replication (16). A wide range of cell surface receptors has been implicated in flavivirus entry into different cells types (11). Amongst the entry receptors postulated to be involved in flavivirus entry, the best characterized to date include αvβ3 integrins (17, 18), C-type lectin receptors (CLR) (19–23), phosphatidylserine receptors TIM (T-cell immunoglobulin and mucin domain) and TYRO3, AXL and MER (TAM) (24). Recent studies indicate that flaviviruses can produce a range of structurally different virions. This structural heterogeneity may expand tissue tropism and ability to infect different cell types both in invertebrate and vertebrate hosts (25).
Flaviviruses are deposited into the skin epidermis by a mosquito bite where they encounter cells permissive to infection such as keratinocytes and skin dendritic cells (Langerhans cells) (26). Dendritic cells in particular appear to be a common initial target for flaviviruses. When infected, dendritic cells migrate to lymphoid organs where viral replication takes place allowing for flavivirus dissemination into circulation and internal organs (12). Viruses such DENV (27), JEV (28), ZIKV (29) have been shown to infect skin dendritic cells, and although there are no reports on YFV infecting Langerhans cells, it can nevertheless infect other types of dendritic cells (30). This interaction is mediated by DC-SIGN for JEV (28) and ZIKV (29), but appears to be DC-SIGN independent in case of DENV (31) and YFV (30).
Many flaviviruses are neuroinvasive and neurovirulent and cause central nervous system (CNS) damage (32). Neuroinvasive infections are observed with JEV, TBEV, and WNV (33, 34), and occasionally with haemorrhagic viruses including DENV (35). There is a paucity of knowledge regarding factors involved in CNS cell entry. While CLRs and TIMs are expressed by cells of the CNS (36–38), they are not expressed by neurons (39–41). However, members of the TAM family of receptors are expressed by different neuronal subtypes (42), though they are dispensable for ZIKV infection as ZIKV was able to infect and replicate in TAM receptor knockout mice (43).
As natural vectors, mosquitos and ticks are highly permissive to flavivirus infection. The virus can replicate in a range of arthropod tissues and cells (44, 45). Given that flaviviruses have only one glycoprotein, it seems likely that the mechanism of entry into vertebrate and invertebrate cells is evolutionarily conserved. A number of the cellular receptors implicated in flavivirus entry into mosquito cell lines overlaps with those identified for mammalian cells (46). Some flaviviruses are more selective regarding their arthropod host than others. For example, DENV is spread mainly by Aedes spp. mosquitos (6), WNV by Culex spp. (47), whereas JEV is transmitted by Aedes, Anopheles and Culex spp. (48). There appears to be a more restricted receptor repertoire used by flaviviruses for insect cell entry compared to mammalian cell entry. The range of clinical manifestations of flaviviral infection in the mammalian host suggests that these viruses may use a wide range of receptors. Mammalian tissues in general offer much greater range of receptors compared to invertebrates.
Identification of flavivirus entry receptors, particularly those involved in CNS infection, could lead to identification of novel therapeutic targets. For this review we will focus on four major flaviviruses of humans—DENV, JEV, ZIKV, and YFV, and discuss the differences and similarities in their mechanisms of entry into arthropod and mammalian cells.
DENV is one of the most common mosquito-borne viruses, mainly transmitted by Aedes aegypti mosquitoes, and occasionally by Ae. albopictus. Symptoms of DENV infection range from fever and muscle and joint pain (Dengue fever) to potentially life threatening haemorrhagic fever or shock syndrome. While DENV was endemic in <10 countries in the 1970's, it is presently a threat in over 128 countries and is responsible for almost 400 million human infections every year. About 24% of infections manifest in severe clinical symptoms. There is currently no treatment for DENV serotypes. There are four virus serotypes, and recovery from one serotype provides lifelong homologous immunity (49).
DENV is an icosahedral particle of 50 nm with a positive, single-stranded RNA genome of 10–11 kb (50). As in other flaviviruses, E protein is involved in receptor binding and fusion (51) and has the ability to bind to a wide range of cellular receptors to initiate DENV entry. The E-DIII domain has a role in cellular recognition (52) and has been suggested as a target for the development of a DENV vaccine (53).
Over the years, several cell membrane receptors involved in DENV entry have been identified. These include carbohydrate molecules (54–56), lectins (57, 58), and claudin-1 cell receptors (59). Carbohydrate molecules such as glycosaminoglycans (GAGs), sulphated polysaccharides, and glycosphingolipids (GSL) are widely expressed cell surface co-receptors for DENV entry and are believed to enhance viral entry efficiency. The highly sulphated form of GAGs, the heparan sulfates (HS) and heparan sulfates proteoglycans (HSPG), are essential for cellular adhesion to extracellular matrix and binding of polypeptide growth factors involved in intracellular signaling (56). It has been suggested that DENV first contacts HSPG, and that this weak interaction facilitates binding of virus to other receptors, which then results in virus internalization (55). Several studies have shown that pre-treatment with heparin can reduce or block DENV-2 infection (60, 61). However, the efficiency of inhibition of viral entry was dependent on numerous factors, such as the virus strains and the target cell (61). GSLs, a member of the same family of carbohydrate molecules as HS, are ubiquitous cellular components of eukaryotic plasma membranes that can also facilitate entry and binding of virus (54). However, GSLs are not required for DENV entry as the virus was able to enter GSL-deficient cells (62).
Cellular C-type lectin receptors (CLRs) are part of the host immune response to fungal, bacterial and viral infections (57). CLRs in mammalian cells include DC-SIGN/L-SIGN, mannose receptors (MR), and CLEC5A. DC-SIGN receptors are widely known because of their association with HIV. These receptors are also involved in DENV binding and internalization into dendritic cells (63). MR has been found to be the primary DENV cell receptor in macrophages. The CLEC5A receptor cooperates with DC-SIGN or MR to increase DENV binding and stability (58).
Other studies have suggested claudin-1 as a putative cell receptor for DENV entry through a direct interaction with the viral prM protein. Claudins are vital components of tight junction complexes and are essential for normal permeability of the epithelia (59, 64). DENV-2 entry was significantly reduced in claudin-1 deficient cells (59). Also, it has been demonstrated that caudin-1 is upregulated early in infection in order to facilitate entry and downregulated in late stage of infection (64).
Protein binding assays and mass spectrometry analysis have identified several additional potential flavivirus cellular receptors (65–67). Among them, the tubulin and tubulin-like proteins in C6/36 Ae. albopictus cell line (65). Heat shock proteins (HSPs) of ~70 kDa and 80 kDa were also identified as cellular receptors for all four DENV serotypes in C6/36 cell line (66, 68). HSPs are chaperone proteins involved in the regulation of folding and unfolding of cellular, and upon infection, viral proteins (69). The 70 kDa protein, also known as heat shock cognate protein (HSC70) or HSPA8, acts as a chaperone protein during DENV entry (70, 71). Modulation of HSC70 expression was observed during DENV-2 infection, with an increase on the cell membrane during infection, suggesting that DENV-2 utilizes HSC70 for entry into mosquito cells (67). In addition to its role in viral entry, HSP70 is involved in virion biogenesis and RNA replication (71). It appears that all four DENV serotypes are dependent on this chaperone protein family, which makes HSP70 proteins an interesting target for the design of a tetravalent DENV therapy or vaccine (71).
HSP90, another heat shock protein, can also act as chaperone. This protein interacts with six DENV proteins (69). While the involvement of HSP70 and HSP90 in DENV binding to host cells has been reported (70–72), these proteins are not involved in internalization of virus into the host cell (73).
As mentioned before, TIM/TAM family receptors have been implicated in flavivirus entry. DENV express on its surface phosphatidylserine (PS) and phosphatidylethanolamine (PE) molecules. Both PS and PE are known to directly interact with TIM/TAM receptors and DENV is able to enhance its entry by exploiting these interactions (74).
After binding to cellular receptors, internalization of viral particles occurs. For DENV, internalization occurs via pH-dependent endocytosis. Several endocytosis pathways are currently known, but clathrin-mediated endocytosis is the main pathway for DENV (75). The DENV use of clathrin-mediated endocytosis was demonstrated in C6/36 mosquito cells by biochemical inhibition of cell receptors (76), and in several human cell lines through siRNA silencing of genes associated with clathrin-mediated receptors (75, 77, 78). While this inhibition and specific gene silencing resulted in a decrease in viral load, a complete inhibition was not achieved, suggesting the existence of alternative entry pathways in mosquito and mammalian cells.
In addition to the exploitation of clathrin-mediated endocytosis, the host immune system can also promote viral entry (79). This phenomenon, known as antibody-dependent enhancement (ADE), was first described in 1964 by Hawkes (80) for WNV and JEV, and observed for DENV more than a decade later (81). Antibody-virus complexes are internalized by phagocytosis via Fc gamma receptors (FcγR) into macrophages, monocytes and dendritic cells (82) (Figure 1), thus facilitating virus entry (83). It has recently been shown that ADE increases membrane fusion activity, promoting DENV entry (79). Moreover, prM antibodies have the capacity to convert non-infectious, immature DENV particles into infectious particles and enhance their infectivity to levels comparable to wild-type virus (50). ADE has been linked to the observation that one flavivirus infection can enhance another (84). However, a recent study showed that ADE is dependent on the level of neutralizing antibodies, particularly IgG and IgM (85); only patients with a low level of neutralizing antibodies showed enhancement of DENV infection (85). Antibodies, even at low concentration, against the EDIII domain were able to block viral entry of the four DENV serotypes without inducing antibody-dependent enhancement (86). However, high IgG titres were observed in patients with ADE following DENV infection, in particular IgG1 levels were the highest in patients with dengue fever or shock syndrome (87). ADE has been recently identified as consequence of sensitisation with Dengvaxia quadrivalent vaccine, leading to severe vaccine-enhanced disease resulting in hospitalization (88).
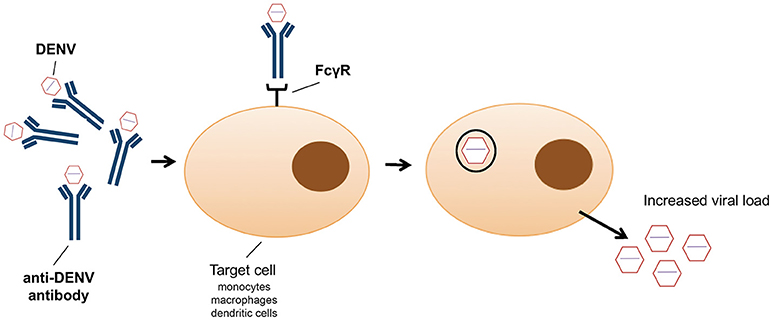
Figure 1. Schematic representation of antibody dependent enhancement (ADE) entry into monocytes, macrophages and dendritic cells as employed by dengue virus.
Globally, Japanese encephalitis is the most clinically important arboviral encephalitis, with an estimated annual prevalence of up to 50,000 cases (89). As is the case with most arboviral encephalitic infections, humans are dead-end hosts unable to develop a sufficiently high viremia to transmit to feeding mosquitos. The majority of JEV infections are asymptomatic. Approximately a third of clinical cases are fatal and half of survivors have neurological or neuropsychiatric sequelae with symptoms resembling Parkinsonian movement disorders, poliomyelitis-like paralysis or impaired cognition (90). Disease is most common in children up to 14 years of age. JEV has been expanding its endemic areas in Asia (91) and poses an unpredictable and emerging global threat.
In humans, JEV has been found in different anatomical compartments and a variety of cell types is able to support its replication. These include endothelial cells, granulocytes, dendritic cells, macrophages and cells of the CNS including astrocytes, neurons and microglia (92). The virus spreads from dermal tissues (93) to lymphoid organs (94) and during the acute stage of infection can be found in blood (95). Although highly neuroinvasive, the mechanism of JEV entry into the CNS is unclear. Transport along the olfactory nerve and across the blood brain barrier have been implicated in JEV invasion of the CNS (96, 97). Studies in rodent models indicate that the blood brain barrier is disrupted following neuroinvasion (98), and might be a consequence of invasion rather than an entry route. Once in the brain, JEV can infect pericytes (99), astrocytes (100) and microglia (101), and has a predilection for developing neurons and neuronal progenitors (102, 103). As described below, a number of receptors mediate entry into different cells types. The distinctive neuronal tropism suggests the existence of JEV-specific receptors in the CNS, but their nature remains elusive (104).
In vitro studies on mouse neuroblastoma cells indicate heat shock protein (HSP) 70 as a putative entry receptor present on neuronal cells (105). This has not been corroborated by in vivo experiments, but in human hepatoma Huh7 cells, HSP70 is required for entry (106). Recently, a member of the HSP70 family, glucose-regulated protein (GRP) 78, has been implicated in JEV entry into Neuro2a and BHK-21 cells (107, 108). In addition to HSP70 and GRP78, HSP90β also interacts with E protein and may be used by JEV to enter mammalian cells (109). Another member of the HSP70 family is heat shock cognate (HSC) protein 70. HSC70 has been suggested to be a receptor for entry into mosquito cells (110). HSC70 isoform D is essential for clathrin-dependent endocytosis of JEV into C6/36 cells (111). Clathrin dependence seems to be critical for JEV entry into mammalian cells with the exception of neuronal cells (112), where JEV internalization into rodent neuroblastoma cell lines has been shown to be clathrin-independent (113, 114) and independent of HSP70 family proteins. JEV can enter human neuronal cells by caveolin-mediated endocytosis (115), a process that is receptor-independent (116). Interestingly, JEV has been shown to utilize the dopaminergic signal transduction pathway to increase neuronal susceptibility to infection (117). Infection of human dopaminergic neuroblastoma cells in vitro leads to increased levels of secreted dopamine and activation of the phospholipase C cascade. The latter enhances formation of structures known as focal adhesions on the cell surface and increases JEV binding and entry. One of the main components of focal adhesions is αvβ3 integrin that recruits vimentin to the cell surface (118), and is involved in JEV binding and infection of BHK-21 cells (18). Vimentin is a putative JEV receptor (119, 120). Thus, by signaling through dopamine D2 receptors and activating the phospholipase C cascade, JEV induces recruitment of surface molecules that enhance and propagate infection in adjacent cells. Enhanced infection of dopaminergic neurons also explains why JEV is predominantly found in brain areas rich in these cells including the thalamus and the midbrain (121, 122). Whereas JEV infection of neurons may be most directly relevant for disease, other cells types are also likely to have an important role in the disease process. Microglial cells may be a viral reservoir due to long-term, high level of virus production in these cells (123). CD4 has been identified as a major receptor for JEV entry into microglia (124). Presumably, CD4 can be used by JEV to enter other CD4 positive cells such as T cells, macrophages or dendritic cells. Published data are scarce, however, JEV productive infection of splenic macrophages and T cells has been reported in a mouse model of infection (125). T lymphocytes have also been reported as a reservoir of latent JEV in asymptomatic children following recovery from acute infection (126). The involvement of CD4 in microglial cell entry has not been reported for any other flavivirus, however CD4 is the main receptor for retroviral entry and is primarily localized in lipid rafts (127). As mentioned above, HSP70 in lipid rafts is involved in JEV entry into human cells and in general lipid rafts play a critical role in JEV entry (128, 129). Moreover, lipid rafts, as well as clathrin-coated pits and caveolae, contain sphingolipids such as sphingomyelin (SM) that is involved in JEV attachment and entry (130). Studies in SM synthase 1 deficient mice infected with JEV showed a reduction in disease, indicating a role for SM in JEV infection models (130).
Despite advances in identification of new receptors associated with JEV entry and its clear tropism for neuronal cells in the CNS, the identity of a specific neuronal receptor remains elusive. Notably, JEV has the ability to infect cells in the absence of above mentioned putative receptors although at a reduced rate (104). This suggests that the entry process involves multi protein interactions with high degree of redundancy and a single, specific entry receptor might not exist. Alternatively, the inability to identify such a receptor highlights the limitations of in vitro systems commonly used to investigate virus-cell interactions.
ZIKV is a mosquito-borne emerging pathogen that poses significant public health concerns due to recent rapidly expanding outbreaks. ZIKV was relatively unknown until 2007, when an outbreak occurred in Yap Island (Micronesia) (131). The virus was first isolated in the Zika Forest in Uganda from a rhesus monkey in 1947. In 1948 a second isolate from Ae. africanus mosquitoes was obtained from the same forest (132). Prior to the recent serious outbreak in French Polynesia, New Caledonia, the Cook Islands and Easter Island in 2013 and 2014 (133), Zika has not been reported to cause significant disease. Data from French Polynesia during the ZIKV epidemic documented the occurrence of Guillain-Barre syndrome and other neurological complications (134). The pathogenesis of ZIKV infection is poorly understood and involves a multifaceted interaction between viral and host factors. ZIKV has shown a significant tropism to the CNS and causes neurodegeneration, particularly of neural progenitor cells (135–137). ZIKV is also the only flavivirus known to have teratogenic effects in humans, including microcephaly, intracranial calcification and fetal death (138). As a result, the World Health Organization announced in 2016 that the ZIKV outbreak was a health emergency of international concern (139). Like other flaviviruses, ZIKV likely enters host cells through endocytosis instigated by an interaction of E glycoprotein with cell surface receptors. Identification of the entry receptor(s) for ZIKV is essential to understanding viral tropism and pathogenesis, and could lead to the development of novel therapeutics to treat the infection.
The first barrier for the virus to enter the host cell is the skin epidermis. ZIKV is transmitted by Aedes spp. mosquitoes, which deposit virus in the epidermis and dermis during the blood meal. Both dermal fibroblast and epidermal keratinocytes are permissive to ZIKV infection as are skin dendritic cells. Several entry receptors including the innate immune receptor DC-SIGN, transmembrane protein TIM-1 and TAM receptors (TYRO3, AXL, MER), have been shown to facilitate entry and enhance ZIKV infection (29). RNA silencing of TIM-1 and AXL in subsets of human skin cells showed a significant reduction in ZIKV titre in AXL knockdown, and in double AXL and TIM-1 knockdown, indicating that AXL is a major receptor for ZIKV entry at least in human skin cells. However, a recent study (43) investigating different infection routes of ZIKV, including subcutaneous, transplacental, vaginal, and intracranial infections in wild-type and TAM receptor null mice, showed no difference in viral titres. TAM receptors, at least in mice, are therefore not essential for ZIKV infection. Interestingly, WNV infection of neurons can be enhanced in mice lacking AXL and MER. This increase in infectivity was associated with changes in blood brain barrier permeability (140), suggesting that AXL and MER do not serve exclusively as receptors and might have other roles in WNV infection of the brain.
To reach the fetal brain, ZIKV must first be transported to the fetal circulation, and cross the placental barrier. The placental barrier is composed of placental barrier cells, trophoblasts and fetal endothelial cells, which separate the fetal blood in capillaries from maternal blood. ZIKV has been reported in the amniotic fluid of fetuses in Brazil (141). This observation strengthened the association of ZIKV with microcephaly in neonates. Moreover, it has been shown that microcephaly caused by maternal viral infection in mice could result from direct viral infection of the fetus via the trans-placental route as well as from a placental inflammatory response that affects fetal development (142). ZIKV can efficiently infect fetal endothelial cells, whereas WNV and DENV do not, highlighting ZIKV unique tropism among flaviviruses (143). These differences between flaviviruses are due to ZIKV ability to efficiently use AXL receptor to enter fetal endothelial cells (143).
TIM-1 was also observed to have an important role in placental entry of ZIKV (144). ZIKV was able to infect different human primary placental cell types and explants from chorionic villi. AXL, TYRO3, and TIM-1 were present in the primary placental cells and are found at the uterine-placental interface. Particularly high expression of TIM-1 has been observed in cells where maternal blood perfused placenta including basal decidua and neighboring chorionic villi. Expression of AXL and TYRO3 varied with explant donor, gestational age and cell type. Specific pharmacological inhibition of TIM-1 by duramycin (145) could inhibit ZIKV infection at the uterine-placental interface, indicating that TIM-1 is a putative receptor for ZIKV placental cell entry. However, the role of AXL, variation in the expression of AXL and TYRO3 in pregnant women and whether TIM-1 is the sole receptor for ZIKV infection of the placenta need further study.
Numerous studies on ZIKV infection in mice having defective interferon signaling, including IFNα/β knockout mice (146, 147), double knockout of IFNα/β and IFNγ (148), and triple knockout of IRF-3,-5,-7 (136, 149) showed viremia, microcephaly and death in young mice and viremia with recovery in adult mice. However, cell death and reduced proliferation was observed for adult neural stem cells (136) suggesting possible long term effects in adult brain followin ZIKV infection. ZIKV also infects other cell types, especially in the eye. ZIKV-inoculated mice develop ocular defects including conjunctivitis, pan uveitis, and infection of the optic nerve, cornea, iris, and ganglion and bipolar cells in the retina (150). AXL is expressed at high levels in retinal progenitor cells (151) suggesting a possible role in ZIKV infection of ocular cells. However, the ocular abnormalities were shown to be independent of AXL or MER, given that AXL−/−, MER−/−, and AXL−/− MER−/− double knockout mice sustained levels of infection similar to those of control animals. Nevertheless, AXL might have a role in ZIKV infection of glial cells via Gas6 mediated activation of AXL kinase (152).
In vitro and in vivo systems to study ZIKV infection of neural cells have been developed. ZIKV neuro-infection models using cultured neural precursor cells (NPCs), cortical organoids, mouse brains, and human fetal brain materials have been studied (153). Microcephaly in these models is associated with inflammation, reduced proliferation of NPCs and neuronal cell death. ZIKV-BR can infect mouse fetuses and infection of pregnant mice also causes disease in embryos with intrauterine growth restriction, including signs of microcephaly (154). This study also demonstrated that ZIKV-BR infects human cortical progenitor cells, increasing the rate of cell death. ZIKV was found to directly infect human NPCs with high efficiency providing a plausible explanation for the observed developmental phenotypes and associated teratogenicity in the neonatal brains (137). Based on previous studies (29, 151, 152), AXL is a strong candidate receptor for the entry of ZIKV into cells of the developing brain. The potential role of AXL to facilitate ZIKV infection of the neonatal brain was explored by determining, at a single cell level, RNA expression profiles in the developing human cerebral cortex (151). The study revealed a higher expression of AXL in the radial cells and neural stem cells of the developing brain throughout neurogenesis and in capillaries and astrocytes. However, loss of AXL expression following CRISPR/Cas9 gene editing, did not affect ZIKV infectivity into hNPCs or cerebral organoids (155).
As is the case with JEV, the identity of the receptors involved in ZIKV receptor mediated endocytosis remains to be elucidated. There might be tissue specificity in receptor mediated viral entry, with variation in receptor repertoire in the skin, placenta, neurons, and other cell types. Alternatively, given ZIKV unique ability to cross the placenta and infect developing neurons in the fetal brain, there might be some as yet unidentified receptors facilitating this process.
YFV is the prototype and namesake virus of the Flavivirus genus; flavi means yellow in Latin. When infecting humans YFV replicates in liver, heart, kidneys, and lungs causing a broad spectrum of clinical symptoms. These vary from asymptomatic infection to renal and hepatic failures with severe haemorrhagic disease (156). A live attenuated YFV vaccine 17D was created over 70 years ago and has been used safely in over 500 million people. The parent strain of 17D is the virulent Asibi strain (157) isolated in Africa in 1927. 17D was passaged more than 230 times in mouse and chicken embryonic tissue. The adaptation of 17D to grow in tissue culture resulted in loss of viscerotropism, neurotropism, and mosquito tropism (156), making it an ideal candidate for a vaccine. The genome of both strains has been sequenced (158). The extensive passage history gave rise to 68 nucleotide mutations and 32 amino acid substitutions. Most of the genetic differences occur in the envelope (E) protein gene (157). Interestingly, the molecular determinants and mechanisms of this attenuation remain largely unknown. It has been suggested that the differences in the E protein and its involvement in cell entry are determinants of the difference in pathogenicity between the 17D and Asibi strains (159, 160). Mutations in the E gene have been suggested to allow the 17D strain to bind and enter hosts cells more efficiently.
YFV shares genome organization and entry by clarithin-mediated endocytosis (CME) with most of the other flaviviruses. During YFV infection, the E protein binds to an unknown entry receptor that traffics the virion to endosomes. Similarly to other flaviviruses, increase in acidification of the endosome results in conformational changes in the E protein, membrane fusion and nucleocapsid release into the cytoplasm (156). The vaccine strain uses a clathrin- and caveolin-independent, but dynamin-2-dependent, pathway for infection (160). Dynamin-2 is a GTPase involved in cleaving off endocytic vesicles from the plasma membrane (161). The entry pathway of the 17D strain was further characterized as Rac1, Pak1, and cortactin independent (160). Clarithin-independent entry has been reported to mediate the internalization of a variety of viruses, such as rotavirus, human rhinovirus, influenza, and interestingly, JEV vaccine strain in neuronal cells (114, 162–164). Cells infected with 17D have been found to produce more viral RNA and INF-β, IL-29, ISG56, CCL5, and CXCL10 mRNA than those infected with the parental Asibi strain. In addition, 17D infected cells secrete INF-β, whereas cells infected with the Asibi strain do not. Virus entry through a clathrin-independent pathway allows for more efficient virion delivery into endosomes or protection from degradation, relative to entry via the classical clathrin-mediated route. This former entry route has been suggested to allow for a higher amount of viral RNA released into the cytoplasm (160). Viral RNA in the cytoplasm is detected by RIG-I, MDA5 and TLR7 (165), triggering strong innate immune responses. The Asibi strain on other hand, replicates at lower levels and inhibits the innate immune system. This difference in entry mechanism has been suggested to account for the differences in cytokine response between the two YFV strains, though further mutations in other proteins, such as NS2A could also be involved.
Glycoprotein E is responsible for receptor-mediated attachment of flaviviruses to the host cell and membrane fusion. Although E protein of different flaviviruses share approximately 40% sequence identity (e.g., DENV and TBEV), their overall structural features are almost identical and this is assumed to apply to all flaviviruses (166). Cell entry is facilitated by a conserved peptide of 16 amino acids, located in E-DII region of the envelope glycoprotein (167). This conservation, coupled with highly organized conformational changes upon exposure to low pH (168), suggests evolutionary constraints allowing flaviviruses to enter both mammalian and arthropod cells. Yet, flavivirus receptors show diversity and significant cell type specificity. It is not unusual that a single molecule can bind to variety of targets as exemplified by immunoglobulins. However, their diversity and specificity are governed by V(D)J recombination, while the flaviviral glycoprotein E is conserved. The flavivirus infection is a consequence of multiple complex interactions between the virus and the target cell. It is clear that the flavivirus can exploit different endocytic routes that can be either clathrin or caveolae dependent or independent. The neurotropism of specific flaviviruses raises the question, is there a single specific neuronal receptor? What is the identity of this receptor and is the same receptor being used by all encephalitic flaviviruses? Another unresolved question is whether all flaviviruses share the same features of infection in the developing brain, or whether viruses such as microcephaly-causing ZIKV, exhibit a different infection pattern. It is also relevant to note that the expression of entry receptors (e.g., CLRs or TAM) does not account for flavivirus tropism and cellular models lacking those receptors are still permissive to infection. Identifying the relevant entry receptors is essential to deciphering the mechanisms of pathogenesis, tropism and viral biology. A better understanding of those processes will uncover new strategies for designing therapeutics and vaccines against flaviviruses.
ML, DN, JR-A, and LK wrote sections of the manuscript, JF and LK edited and critically evaluated the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Messina JP, Brady OJ, Scott TW, Zou C, Pigott DM, Duda KA, et al. Global spread of dengue virus types: mapping the 70 year history. Trends Microbiol. (2014) 2:138–46. doi: 10.1016/j.tim.2013.12.011
2. Weaver SC, Costa F, Garcia-Blanco MA, Ko AI, Ribeiro GS, Saade G, et al. Zika virus: History, emergence, biology, and prospects for control. Antiviral Res. (2016) 30:69–80. doi: 10.1016/j.antiviral.2016.03.010
3. Kraemer MUG, Faria NR, Reiner RC Jr Golding N, Nikolay B, Stasse S, et al. Spread of yellow fever virus outbreak in angola and the democratic republic of the Congo 2015-16: a modelling study. Lancet Infect Dis. (2017) 7:330–8. doi: 10.1016/S1473-3099(16)30513-8
4. Hamer DH, Angelo K, Caumes E, van Genderen PJJ, Florescu SA, Popescu CP, et al. Fatal Yellow Fever in Travelers to Brazil, 2018. MMWR Morb Mortal Wkly Rep. (2018) 67:340–1. doi: 10.15585/mmwr.mm6711e1
5. Hayes EB, Gubler DJ. West Nile virus: epidemiology and clinical features of an emerging epidemic in the United States. Annu Rev Med. (2006) 7:181–94. doi: 10.1146/annurev.med.57.121304.131418
6. Gould EA, Solomon T. Pathogenic flaviviruses. Lancet (2008) 71:500–9. doi: 10.1016/S0140-6736(08)60238-X
7. Mlera L, Melik W, Bloom M E. The role of viral persistence in flavivirus biology. Pathog Dis. (2014) 1:137–63. doi: 10.1111/2049-632X.12178
8. Collins MH, Metz S W. Progress and works in progress: update on flavivirus vaccine development. Clin Ther. (2017) 9:1519–36. doi: 10.1016/j.clinthera.2017.07.001
9. Sirohi D, Chen Z, Sun L, Klose T, Pierson TC, Rossmann MG, et al. The 3.8 A resolution cryo-EM structure of Zika virus. Science (2016) 52:467–70. doi: 10.1126/science.aaf5316
10. Zhang X, Ge P, Yu X, Brannan JM, Bi G, Zhang Q, et al. Cryo-EM structure of the mature dengue virus at 3.5-A resolution. Nat Struct Mol Biol. (2013) 20:105–10. doi: 10.1038/nsmb.2463
11. Perera-Lecoin M, Meertens L, Carnec X, Amara A. Flavivirus entry receptors: an update. Viruses (2013) 6:69–88. doi: 10.3390/v6010069
12. Pierson TC, Kielian M. Flaviviruses: braking the entering. Curr Opin Virol. (2013) 3:3–12. doi: 10.1016/j.coviro.2012.12.001
13. Sun H, Chen Q, Lai H. Development of antibody therapeutics against flaviviruses. Int J Mol Sci. (2017) 19:E54. doi: 10.3390/ijms19010054
14. Smit JM, Moesker B, Rodenhuis-Zybert I, Wilschut J. Flavivirus cell entry and membrane fusion. Viruses (2011) 3:160–71. doi: 10.3390/v3020160
15. Stiasny K, Fritz R, Pangerl K, Heinz F X. Molecular mechanisms of flavivirus membrane fusion. Amino Acids (2011) 1:1159–63. doi: 10.1007/s00726-009-0370-4
16. Noisakran S, Onlamoon N, Songprakhon P, Hsiao H M, Chokephaibulkit K, Perng G C. Cells in dengue virus infection in vivo. Adv Virol. (2010) 2010:164878. doi: 10.1155/2010/164878
17. Chu JJ, Ng M L. Interaction of West Nile virus with alpha v beta 3 integrin mediates virus entry into cells. J Biol Chem. (2004) 79:54533–41. doi: 10.1074/jbc.M410208200
18. Fan W, Qian P, Wang D, Zhi X, Wei Y, Chen H, et al. Integrin alphavbeta3 promotes infection by Japanese encephalitis virus. Res Vet Sci. (2017) 11:67–74. doi: 10.1016/j.rvsc.2016.12.007
19. Davis CW, Nguyen HY, Hanna SL, Sanchez MD, Doms RW, Pierson T C. West Nile virus discriminates between DC-SIGN and DC-SIGNR for cellular attachment and infection. J Virol. (2006) 80:1290–301. doi: 10.1128/JVI.80.3.1290-1301.2006
20. Davis CW, Mattei LM, Nguyen HY, Ansarah-Sobrinho C, Doms RW, Pierson T C. The location of asparagine-linked glycans on West Nile virions controls their interactions with CD209 (dendritic cell-specific ICAM-3 grabbing nonintegrin). J Biol Chem. (2006) 81:37183–94. doi: 10.1074/jbc.M605429200
21. Miller JL, de Wet BJ, Martinez-Pomares L, Radcliffe CM, Dwek RA, Rudd PM, et al. The mannose receptor mediates dengue virus infection of macrophages. PLoS Pathog (2008) 4:e17. doi: 10.1371/journal.ppat.0040017
22. Chen ST, Liu RS, Wu MF, Lin YL, Chen SY, Tan DT, et al. CLEC5A regulates Japanese encephalitis virus-induced neuroinflammation and lethality. PLoS Pathog (2012) 8:e1002655. doi: 10.1371/journal.ppat.1002655
23. Cheng G, Cox J, Wang P, Krishnan MN, Dai J, Qian F, et al. AC-type lectin collaborates with a CD45 phosphatase homolog to facilitate West Nile virus infection of mosquitoes. Cell (2010) 42:714–25. doi: 10.1016/j.cell.2010.07.038
24. Meertens L, Carnec X, Lecoin MP, Ramdasi R, Guivel-Benhassine F, Lew E, et al. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe (2012) 2:544–57. doi: 10.1016/j.chom.2012.08.009
25. Rey FA, Stiasny K, Heinz F X. Flavivirus structural heterogeneity: implications for cell entry. Curr Opin Virol. (2017) 4:132–9. doi: 10.1016/j.coviro.2017.06.009
26. Briant L, Despres P, Choumet V, Misse D. Role of skin immune cells on the host susceptibility to mosquito-borne viruses. Virology (2014) 4:64–465, 26–32. doi: 10.1016/j.virol.2014.06.023
27. Wu SJ, Grouard-Vogel G, Sun W, Mascola JR, Brachtel E, Putvatana R, et al. Human skin Langerhans cells are targets of dengue virus infection. Nat Med. (2000) 6:816–20. doi: 10.1038/77553
28. Wang P, Hu K, Luo S, Zhang M, Deng X, Li C, et al. DC-SIGN as an attachment factor mediates Japanese encephalitis virus infection of human dendritic cells via interaction with a single high-mannose residue of viral E glycoprotein. Virology (2016) 88:108–19. doi: 10.1016/j.virol.2015.11.006
29. Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, et al. Biology of Zika Virus Infection in Human Skin Cells. J Virol. (2015) 9:8880–96. doi: 10.1128/JVI.00354-15
30. Barba-Spaeth G, Longman RS, Albert ML, Rice C M. Live attenuated yellow fever 17D infects human DCs and allows for presentation of endogenous and recombinant T cell epitopes. J Exp Med. (2005) 02:1179–84. doi: 10.1084/jem.20051352
31. Lozach PY, Burleigh L, Staropoli I, Navarro-Sanchez E, Harriague J, Virelizier JL, et al. Dendritic cell-specific intercellular adhesion molecule 3-grabbing non-integrin (DC-SIGN)-mediated enhancement of dengue virus infection is independent of DC-SIGN internalization signals. J Biol Chem. (2005) 80:23698–708. doi: 10.1074/jbc.M504337200
32. Neal J W. Flaviviruses are neurotropic, but how do they invade the CNS? J Infect. (2014) 9:203–15. doi: 10.1016/j.jinf.2014.05.010
34. Kramer LD, Li J, Shi P Y. West Nile virus. Lancet Neurol. (2007) 6:171–81. doi: 10.1016/S1474-4422(07)70030-3
35. Domingues RB, Kuster GW, Onuki-Castro FL, Souza VA, Levi JE, Pannuti C S. Involvement of the central nervous system in patients with dengue virus infection. J Neurol Sci. (2008) 2:67:36–40. doi: 10.1016/j.jns.2007.09.040
36. Linehan SA, Martinez-Pomares L, Stahl PD, Gordon S. Mannose receptor and its putative ligands in normal murine lymphoid and nonlymphoid organs: In situ expression of mannose receptor by selected macrophages, endothelial cells, perivascular microglia, and mesangial cells, but not dendritic cells. J Exp Med. (1999) 89:1961–72.
37. Burudi EM, Riese S, Stahl PD A. Regnier-Vigouroux: Identification and functional characterization of the mannose receptor in astrocytes. Glia (1999) 5:44–55.
38. Mukhtar M, Harley S, Chen P, BouHamdan M, Patel C, Acheampong E, et al. Primary isolated human brain microvascular endothelial cells express diverse HIV/SIV-associated chemokine coreceptors and DC-SIGN and L-SIGN. Virology (2002) 297:78–88. doi: 10.1006/viro.2002.1376
39. Galea I, Palin K, Newman TA, Van Rooijen N, Perry VH, Boche D. Mannose receptor expression specifically reveals perivascular macrophages in normal, injured, and diseased mouse brain. Glia (2005) 9:375–84. doi: 10.1002/glia.20124
40. Prodinger C, Bunse J, Kruger M, Schiefenhovel F, Brandt C, Laman JD, et al. CD11c-expressing cells reside in the juxtavascular parenchyma and extend processes into the glia limitans of the mouse nervous system. Acta Neuropathol. (2011) 21:445–58. doi: 10.1007/s00401-010-0774-y
41. Todorich B, Zhang X, Slagle-Webb B, Seaman WE, Connor J R. Tim-2 is the receptor for H-ferritin on oligodendrocytes. J Neurochem. (2008) 107:1495–505. doi: 10.1111/j.1471-4159.2008.05678.x
42. Prieto AL, Weber JL, Lai C. Expression of the receptor protein-tyrosine kinases Tyro-3, Axl, and mer in the developing rat central nervous system. J Comp Neurol. (2000) 25:295–314. doi: 10.1002/1096-9861(20000918)425:2<295::AID-CNE11>3.0.CO;2-G
43. Hastings AK, Yockey LJ, Jagger BW, Hwang J, Uraki R, Gaitsch HF, et al. TAM receptors are not required for zika virus infection in Mice. Cell Rep. (2017) 9:558–68. doi: 10.1016/j.celrep.2017.03.058
44. Girard YA, Klingler KA, Higgs S. West Nile virus dissemination and tissue tropisms in orally infected Culex pipiens quinquefasciatus. Vector Borne Zoonotic Dis (2004) 4:109–22. doi: 10.1089/1530366041210729
45. Salazar MI, Richardson JH, Sanchez-Vargas I, Olson KE, Beaty B J. Dengue virus type 2: replication and tropisms in orally infected Aedes aegypti mosquitoes. BMC Microbiol. (2007) 7:9. doi: 10.1186/1471-2180-7-9
46. Vega-Almeida TO, Salas-Benito M M. A. De Nova-Ocampo, Del Angel RM, Salas-Benito J. S. Surface proteins of C6/36 cells involved in dengue virus 4 binding and entry. Arch Virol. (2013) 58:1189–207. doi: 10.1007/s00705-012-1596-0
47. Lima-Camara TN. Emerging arboviruses and public health challenges in Brazil. Rev Saude Publica (2016) 50:S0034-89102016000100602. doi: 10.1590/S1518-8787.2016050006791
48. Sucharit S, Surathin K, Shrestha S R. Vectors of Japanese encephalitis virus (JEV): species complexes of the vectors. Southeast Asian J Trop Med Public Health (1989) 20:611–21.
50. Rodenhuis-Zybert IA, Wilschut J, Smit J M. Dengue virus life cycle: Viral and host factors modulating infectivity. Cell Mol Life Sci. (2010) 67:2773–86. doi: 10.1007/s00018-010-0357-z
51. Watterson D, Kobe B, Young P R. Residues in domain III of the dengue virus envelope glycoprotein involved in cell-surface glycosaminoglycan binding. J Gen Virol. (2012) 3:72–82. doi: 10.1099/vir.0.037317-0
52. Tripathi NK, Shrivastava A, Biswal KC, Rao PV L. Recombinant dengue virus type 3 envelope domain III protein from Escherichia coli. Biotechnol J. (2011) 6:604–8. doi: 10.1002/biot.201000399
53. Chiang CY, Pan CH, Chen MY, Hsieh CH, Tsai JP, Liu HH, et al. Immunogenicity of a novel tetravalent vaccine formulation with four recombinant lipidated dengue envelope protein domain IIIs in mice. Sci Rep. (2016) 6:30648. doi: 10.1038/srep30648
54. Aoki C, Hidari KIPJ, Itonori S, Yamada A, Takahashi N, Kasama T, et al. Identification and characterization of carbohydrate molecules in mammalian cells recognized by dengue virus type 2. J Biochem. (2006) 139:607–14. doi: 10.1093/jb/mvj067
55. Hilgard P, Stockert R. Heparan sulfate proteoglycans initiate dengue virus infection of hepatocytes. Hepatology (2000) 2:1069–77. doi: 10.1053/jhep.2000.18713
56. Kato D, Era S, Watanabe I, Arihara M, Sugiura N, Kimata K, et al. Antiviral activity of chondroitin sulphate E targeting dengue virus envelope protein. Antiviral Res. (2010) 8:236–43. doi: 10.1016/j.antiviral.2010.09.002
57. Hoving JC, Wilson GJ, Brown G D. Signalling C-type lectin receptors, microbial recognition and immunity. Cell Microbiol. (2014) 6:185–94. doi: 10.1111/cmi.12249
58. Lo YL, Liou GG, Lyu JH, Hsiao M, Hsu TL, Wong CH. Dengue virus infection is through a cooperative interaction between a mannose receptor and CLEC5A on macrophage as a multivalent hetero-complex. PLoS ONE (2016) 1:e0166474. doi: 10.1371/journal.pone.0166474
59. Che P, Tang H, Li Q. The interaction between claudin-1 and dengue viral prM/M protein for its entry. Virology (2013) 46:303–13. doi: 10.1016/J.VIROL.2013.08.009
60. Dalrymple N, Mackow E R. Productive dengue virus infection of human endothelial cells is directed by heparan sulfate-containing proteoglycan receptors. J Virol. (2011) 5:9478–85. doi: 10.1128/JVI.05008-11
61. Germi R, Crance JM, Garin D, Guimet J, Lortat-Jacob H, Ruigrok RW, et al. Heparan sulfate-mediated binding of infectious dengue virus type 2 and yellow fever virus. Virology (2002) 92:162–8. doi: 10.1006/viro.2001.1232
62. Wang K, Wang J, Sun T, Bian G, Pan W, Feng T, et al. Glycosphingolipid GM3 is Indispensable for Dengue Virus Genome Replication. Int J Biol Sci. (2016) 2:872–83. doi: 10.7150/ijbs.15641
63. Liu P, Ridilla M, Patel P, Betts L, Gallichotte E, Shahidi L, et al. Beyond attachment: Roles of DC-SIGN in dengue virus infection. Traffic (2017) 8:218–31. doi: 10.1111/tra.12469
64. Gao F, Duan X, Lu X, Liu Y, Zheng L, Ding Z, et al. Novel binding between pre-membrane protein and claudin-1 is required for efficient dengue virus entry. Biochem Biophys Res Commun. (2010) 91:952–7. doi: 10.1016/J.BBRC.2009.11.172
65. >H.-Chee Y, AbuBakar S. Identification of a 48kDa tubulin or tubulin-like C6/36 mosquito cells protein that binds dengue virus 2 using mass spectrometry. Biochem Biophys Res Commun. (2004) 20:11–17. doi: 10.1016/j.bbrc.2004.05.124
66. Mercado-Curiel RF, Esquinca-Avilés HA, Tovar R, Díaz-Badillo A, Camacho-Nuez M M. d. L. Muñoz: The four serotypes of dengue recognize the same putative receptors in Aedes aegypti midgut and Ae. albopictus cells. BMC Microbiol. (2006) 6:85. doi: 10.1186/1471-2180-6-85
67. Paingankar MS, Gokhale MD, Deobagkar D N. Dengue-2-virus-interacting polypeptides involved in mosquito cell infection. Arch Virol. (2010) 55:1453–61. doi: 10.1007/s00705-010-0728-7
68. Cao-Lormeau VM. Dengue viruses binding proteins from Aedes aegypti and Aedes polynesiensis salivary glands. Virol J. (2009) 6:35. doi: 10.1186/1743-422X-6-35
69. Srisutthisamphan K, Jirakanwisal K, Ramphan S, Tongluan N, Kuadkitkan A, Smith D R. Hsp90 interacts with multiple dengue virus 2 proteins. Sci Rep. (2018) 8:4308. doi: 10.1038/s41598-018-22639-5
70. Salas-Benito J, Reyes-Del Valle J, Salas-Benito M, Ceballos-Olvera I, Mosso C, del Angel RM. Evidence that the 45-kD Glycoprotein, part of a putative dengue virus receptor complex in the mosquito cell line C6/36, is a heat-shock-related protein. Am J Trop Med Hygiene (2007) 7:283–90. doi: 10.4269/ajtmh.2007.77.283
71. Taguwa S, Maringer K, Li X, Bernal-Rubio D, Rauch JN, Gestwicki JE, et al. Defining Hsp70 subnetworks in dengue virus replication reveals key vulnerability in flavivirus infection HHS public access. Cell (2015) 9:1108–23. doi: 10.1016/j.cell.2015.10.046
72. Reyes-Del Valle J, Chávez-Salinas S, Medina F, Del Angel RM. Heat shock protein 90 and heat shock protein 70 are components of dengue virus receptor complex in human cells. J Virol. (2005) 9:4557–67. doi: 10.1128/JVI.79.8.4557-4567.2005
73. Cabrera-Hernandez A, Thepparit C, Suksanpaisan L, Smith DR. Dengue virus entry into liver (HepG2) cells is independent of hsp90 and hsp70. J Med Virol. (2007) 9:386–92. doi: 10.1002/jmv.20786
74. Dejarnac O, Hafirassou M L, Chazal M, Versapuech M, Gaillard J, Perera-Lecoin M, et al. TIM-1 ubiquitination mediates dengue virus Entry. Cell Rep. (2018) 3:1779–93. doi: 10.1016/j.celrep.2018.04.013
75. Alhoot MA, Wang SM, Sekaran SD. Inhibition of dengue virus entry and multiplication into monocytes using RNA interference. PLoS Negl Trop Dis. (2011) 5:e1410. doi: 10.1371/journal.pntd.0001410
76. Acosta EG, Castilla V, Damonte EB. Infectious dengue-1 virus entry into mosquito C6/36 cells. Virus Res. (2011) 60:173–9. doi: 10.1016/J.VIRUSRES.2011.06.008
77. Alhoot MA, Wang SM, Sekaran SD. RNA Interference mediated inhibition of dengue virus multiplication and entry in HepG2 Cells. PLoS ONE (2012) 7:e34060. doi: 10.1371/journal.pone.0034060
78. Acosta EG, Castilla V, Damonte EB. Alternative infectious entry pathways for dengue virus serotypes into mammalian cells. Cell Microbiol. (2009) 1:1533–49. doi: 10.1111/j.1462-5822.2009.01345.x
79. Flipse J, Diosa-Toro MA, Hoornweg TE, van de Pol DPI, Urcuqui-Inchima S, Smit JM. Antibody-dependent enhancement of dengue virus infection in primary human macrophages; balancing higher fusion against antiviral responses. Sci Rep. (2016) 6:29201. doi: 10.1038/srep29201
80. Hawkes RA. Enhancement of the infectivity of arboviruses by specific antisera produced in domestic fowls. Aust J Exp Biol Med Sci. (1964) 42:465–82.
81. Halstead SB, O'Rourke EJ. Dengue viruses and mononuclear phagocytes. I. Infection enhancement by non-neutralizing antibody. J Exp Med. (1977) 46:201–17.
82. Boonnak K, Dambach K M, Donofrio G C, Tassaneetrithep B, Marovich MA. Cell type specificity and host genetic polymorphisms influence antibody-dependent enhancement of dengue virus infection. J Virol. (2011) 5:1671–83. doi: 10.1128/JVI.00220-10
83. Gollins SW, Porterfield JS. Flavivirus infection enhancement in macrophages: radioactive and biological studies on the effect of antibody on viral fate. J Gen Virol. (1984) 65(Pt 8):1261–72.
84. Priyamvada L, Quicke KM, Hudson WH, Onlamoon N, Sewatanon J, Edupuganti S, et al. Human antibody responses after dengue virus infection are highly cross-reactive to Zika virus. Proc Natl Acad Sci USA. (2016) 13:7852–7. doi: 10.1073/pnas.1607931113
85. Ly MHP, Moi ML, Vu TBH, Tun MMN, Saunders T, Nguyen CN, et al. Dengue virus infection-enhancement activity in neutralizing antibodies of healthy adults before dengue season as determined by using FcγR-expressing cells. BMC Infect Dis. (2018) 8:31. doi: 10.1186/s12879-017-2894-7
86. Ramasamy V, Arora U, Shukla R, Poddar A, Shanmugam RK, White LJ, et al. A tetravalent virus-like particle vaccine designed to display domain III of dengue envelope proteins induces multi-serotype neutralizing antibodies in mice and macaques which confer protection against antibody dependent enhancement in AG129 mice. PLoS Negl Trop Dis. (2018) 2:e0006191. doi: 10.1371/journal.pntd.0006191
87. Thein S, Aaskov J, Myint TT, Shwe TN, Saw TT, Zaw A. Changes in levels of anti-dengue virus IgG subclasses in patients with disease of varying severity. J Med Virol. (1993) 106:102–6.
88. Halstead SB. Dengvaxia sensitizes seronegatives to vaccine enhanced disease regardless of age. Vaccine (2017) 5:6355–8. doi: 10.1016/j.vaccine.2017.09.089
90. Mackenzie JS, Gubler DJ, Petersen LR. Emerging flaviviruses: the spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. Nat Med. (2004) 10(12 Suppl):S98–109. doi: 10.1038/nm1144
91. Misra UK, Kalita J. Overview: Japanese encephalitis. Prog Neurobiol. (2010) 1:108–20. doi: 10.1016/j.pneurobio.2010.01.008
92. Lannes N, Summerfield A, Filgueira L. Regulation of inflammation in Japanese encephalitis. J Neuroinflammation (2017) 4:158. doi: 10.1186/s12974-017-0931-5
93. Agrawal T, Sharvani V, Nair D, Medigeshi GR. Japanese encephalitis virus disrupts cell-cell junctions and affects the epithelial permeability barrier functions. PLoS ONE (2013) 8:e69465. doi: 10.1371/journal.pone.0069465
94. Nagata N, Iwata-Yoshikawa N, Hayasaka D, Sato Y, Kojima A, Kariwa H, et al. The pathogenesis of 3 neurotropic flaviviruses in a mouse model depends on the route of neuroinvasion after viremia. J Neuropathol Exp Neurol. (2015) 4:250–60. doi: 10.1097/NEN.0000000000000166
95. Sapkal GN, Wairagkar NS, Ayachit VM, Bondre VP, Gore MM. Detection and isolation of Japanese encephalitis virus from blood clots collected during the acute phase of infection. Am J Trop Med Hyg. (2007) 7:1139–45. doi: 10.4269/ajtmh.2007.77.1139
96. Myint KS, Raengsakulrach B, Young G D, Gettayacamin M, Ferguson L M, Innis BL, et al. Production of lethal infection that resembles fatal human disease by intranasal inoculation of macaques with Japanese encephalitis virus. Am J Trop Med Hyg (1999) 60:338–42.
97. Liou ML, Hsu CY. Japanese encephalitis virus is transported across the cerebral blood vessels by endocytosis in mouse brain. Cell Tissue Res. (1998) 93:389–94
98. Li F, Wang Y, Yu L, Cao S, Wang K, Yuan J, et al. Viral infection of the central nervous system and neuroinflammation precede blood-brain barrier disruption during japanese encephalitis virus infection. J Virol. (2015) 9:5602–14. doi: 10.1128/JVI.00143-15
99. Chen CJ, Ou YC, Li JR, Chang CY, Pan HC, Lai CY, et al. Infection of pericytes in vitro by Japanese encephalitis virus disrupts the integrity of the endothelial barrier. J Virol. (2014) 8:1150–61. doi: 10.1128/JVI.02738-13
100. Chang CY, Li JR, Chen WY, Ou YC, Lai CY, Hu YH, et al. Disruption of in vitro endothelial barrier integrity by Japanese encephalitis virus-Infected astrocytes. Glia (2015) 63:1915–32. doi: 10.1002/glia.22857
101. Myint KS, Kipar A, Jarman RG, Gibbons RV, Perng GC, Flanagan B, et al. Neuropathogenesis of Japanese encephalitis in a primate model. PLoS Negl Trop Dis. (2014) 8:e2980. doi: 10.1371/journal.pntd.0002980
102. Kimura-Kuroda J, Ichikawa M, Ogata A, Nagashima K, Yasui K. Specific tropism of Japanese encephalitis virus for developing neurons in primary rat brain culture. Arch Virol. (1993) 130:477–84.
103. Das S, Basu A. Japanese encephalitis virus infects neural progenitor cells and decreases their proliferation. J Neurochem. (2008) 6:1624–36. doi: 10.1111/j.1471-4159.2008.05511.x
104. Nain M, Abdin MZ, Kalia M, Vrati S. Japanese encephalitis virus invasion of cell: allies and alleys. Rev Med Virol. (2016) 6:129–41. doi: 10.1002/rmv.1868
105. Das S, Laxminarayana SV, Chandra N, Ravi V, Desai A. Heat shock protein 70 on Neuro2a cells is a putative receptor for Japanese encephalitis virus. Virology (2009) 85:47–57. doi: 10.1016/j.virol.2008.10.025
106. Zhu YZ, Cao MM, Wang WB, Wang W, Ren H, Zhao P, et al. Association of heat-shock protein 70 with lipid rafts is required for Japanese encephalitis virus infection in Huh7 cells. J Gen Virol. (2012) 93(Pt. 1):61–71. doi: 10.1099/vir.0.034637-0
107. Nain M, Mukherjee S, Karmakar SP, Paton AW, Paton JC, Abdin MZ, et al. GRP78 is an important host factor for Japanese Encephalitis virus entry and replication in mammalian cells. J Virol. (2017) 91:e02274–16. doi: 10.1128/JVI.02274-16
108. Wu YP, Chang CM, Hung CY, Tsai MC, Schuyler SC, Wang RY. Japanese encephalitis virus co-opts the ER-stress response protein GRP78 for viral infectivity. Virol J. (2011) 8:128. doi: 10.1186/1743-422X-8-128
109. Hung CY, Tsai MC, Wu YP, Wang RY. Identification of heat-shock protein 90 beta in Japanese encephalitis virus-induced secretion proteins. J Gen Virol. (2011) 92(Pt. 12):2803–9. doi: 10.1099/vir.0.033993-0
110. Ren J, Ding T, Zhang W, Song J, Ma W. Does Japanese encephalitis virus share the same cellular receptor with other mosquito-borne flaviviruses on the C6/36 mosquito cells? Virol J. (2007) 4:83. doi: 10.1186/1743-422X-4-83
111. Chuang CK, Yang TH, Chen TH, Yang CF, Chen WJ. Heat shock cognate protein 70 isoform D is required for clathrin-dependent endocytosis of Japanese encephalitis virus in C6/36 cells. J Gen Virol. (2015) 96(Pt. 4):793–803. doi: 10.1099/jgv.0.000015
112. Liu CC, Zhang YN, Li ZY, Hou JX, Zhou J, Kan L, et al. Rab5 and Rab11 are required for clathrin-dependent endocytosis of Japanese Encephalitis virus in BHK-21 Cells. J Virol. (2017) 91:e01113–17. doi: 10.1128/JVI.01113-17
113. Zhu YZ, Xu QQ, Wu DG, Ren H, Zhao P, Lao WG, et al. Japanese encephalitis virus enters rat neuroblastoma cells via a pH-dependent, dynamin and caveola-mediated endocytosis pathway. J Virol. (2012) 6:13407–22. doi: 10.1128/JVI.00903-12
114. Kalia M, Khasa R, Sharma M, Nain M, Vrati S. Japanese encephalitis virus infects neuronal cells through a clathrin-independent endocytic mechanism. J Virol. (2013) 7:148–62. doi: 10.1128/JVI.01399-12
115. Xu Q, Cao M, Song H, Chen S, Qian X, Zhao P, et al. Caveolin-1-mediated Japanese encephalitis virus entry requires a two-step regulation of actin reorganization. Future Microbiol. (2016) 1:1227–48. doi: 10.2217/fmb-2016-0002
116. Liu P, Rudick M, Anderson RG. Multiple functions of caveolin-1. J Biol Chem. (2002) 77:41295–8. doi: 10.1074/jbc.R200020200
117. Simanjuntak Y, Liang JJ, Lee YL, Lin YL. Japanese Encephalitis virus exploits Dopamine D2 receptor-phospholipase C to target dopaminergic human neuronal cells. Front Microbiol. (2017) 8:651. doi: 10.3389/fmicb.2017.00651
118. Bhattacharya R, Gonzalez AM, Debiase PJ, Trejo HE, Goldman RD, Flitney FW, et al. Recruitment of vimentin to the cell surface by beta3 integrin and plectin mediates adhesion strength. J Cell Sci. (2009) 122(Pt. 9):1390–400. doi: 10.1242/jcs.043042
119. Das S, Ravi V, Desai A. Japanese encephalitis virus interacts with vimentin to facilitate its entry into porcine kidney cell line. Virus Res. (2011) 160:404–8. doi: 10.1016/j.virusres.2011.06.001
120. Liang JJ, Yu CY, Liao CL, Lin YL. Vimentin binding is critical for infection by the virulent strain of Japanese encephalitis virus. Cell Microbiol. (2011) 3:1358–70. doi: 10.1111/j.1462-5822.2011.01624.x
121. Kalita J, Misra UK. Comparison of CT scan and MRI findings in the diagnosis of Japanese encephalitis. J Neurol Sci. (2000) 74:3–8. doi: 10.1016/S0022-510X(99)00318-4
122. Kumar S, Kalita J, Saxena V, Khan MY, Khanna VK, Sharma S, et al. Some observations on the tropism of Japanese encephalitis virus in rat brain. Brain Res. (2009) 268:135–41. doi: 10.1016/j.brainres.2009.02.051
123. Thongtan T, Cheepsunthorn P, Chaiworakul V, Rattanarungsan C, Wikan N, Smith DR. Highly permissive infection of microglial cells by Japanese encephalitis virus: a possible role as a viral reservoir. Microbes Infect. (2010) 2:37–45. doi: 10.1016/j.micinf.2009.09.013
124. Thongtan T, Wikan N, Wintachai P, Rattanarungsan C, Srisomsap C, Cheepsunthorn P, et al. Characterization of putative Japanese encephalitis virus receptor molecules on microglial cells. J Med Virol. (2012) 4:615–23. doi: 10.1002/jmv.23248
125. Mathur A, Bharadwaj M, Kulshreshtha R, Rawat S, Jain A, Chaturvedi UC. Immunopathological study of spleen during Japanese encephalitis virus infection in mice. Br J Exp Pathol. (1988) 9:423–32.
126. Sharma S, Mathur A, Prakash V, Kulshreshtha R, Kumar R, Chaturvedi UC. Japanese encephalitis virus latency in peripheral blood lymphocytes and recurrence of infection in children. Clin Exp Immunol. (1991) 5:85–9.
127. Popik W, Alce TM, Au WC. Human immunodeficiency virus type 1 uses lipid raft-colocalized CD4 and chemokine receptors for productive entry into CD4(+) T cells. J Virol. (2002) 6:4709–22. doi: 10.1128/JVI.76.10.4709-4722.2002
128. Lee C J, Lin HR, Liao CL, Lin YL. Cholesterol effectively blocks entry of flavivirus. J Virol. (2008) 2:6470–80. doi: 10.1128/JVI.00117-08
129. Das S, Chakraborty S, Basu A. Critical role of lipid rafts in virus entry and activation of phosphoinositide 3' kinase/Akt signaling during early stages of Japanese encephalitis virus infection in neural stem/progenitor cells. J Neurochem. (2010) 15:537–49. doi: 10.1111/j.1471-4159.2010.06951.x
130. Taniguchi M, Tasaki T, Ninomiya H, Ueda Y, Kuremoto KI, Mitsutake S, et al. Sphingomyelin generated by sphingomyelin synthase 1 is involved in attachment and infection with Japanese encephalitis virus. Sci Rep. (2016) 6:37829. doi: 10.1038/srep37829
131. Duffy MR, Chen TH, Hancock WT, Powers AM, Kool JL, Lanciotti RS, et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N Engl J Med. (2009) 60:2536–43. doi: 10.1056/NEJMoa0805715
132. Dick GW, Kitchen SF, Haddow AJ. Zika virus. Isolations I, and serological specificity. Trans R Soc Trop Med Hyg. (1952) 6:509–20.
133. Petersen LR, Jamieson DJ, Powers AM, Honein MA. Zika Virus. N Engl J Med. (2016) 74:1552–63. doi: 10.1056/NEJMra1602113
134. Cao-Lormeau VM, Roche C, Teissier A, Robin E, Berry AL, Mallet HP, et al. Zika Virus, French Polynesia, South Pacific 2013. Emerg Infect Dis. (2014) 20:1085–6. doi: 10.3201/eid2006.140138
135. Dang J, Tiwari SK, Lichinchi G, Qin Y, Patil VS, Eroshkin AM, et al. Zika virus depletes neural progenitors in human cerebral organoids through activation of the innate immune receptor TLR3. Cell Stem Cell (2016) 9:258–65. doi: 10.1016/j.stem.2016.04.014
136. Li H, Saucedo-Cuevas L, Regla-Nava JA, Chai G, Sheets N, Tang W, et al. Zika virus infects neural progenitors in the adult mouse brain and alters proliferation. Cell Stem Cell (2016) 9:593–8. doi: 10.1016/j.stem.2016.08.005
137. Tang H, Hammack C, Ogden SC, Wen Z, Qian X, Li Y, et al. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell (2016) 8:587–90. doi: 10.1016/j.stem.2016.02.016
138. Mlakar J, Korva M, Tul N, Popovic M, Poljsak-Prijatelj M, Mraz J, et al. Zika virus associated with microcephaly. N Engl J Med. (2016) 74:951–8. doi: 10.1056/NEJMoa1600651
139. Gulland A. Zika virus is a global public health emergency, declares WHO. BMJ (2016) 52:i657. doi: 10.1136/bmj.i657
140. Miner JJ, Daniels BP, Shrestha B, Proenca-Modena JL, Lew ED, Lazear HM, et al. The TAM receptor Mertk protects against neuroinvasive viral infection by maintaining blood-brain barrier integrity. Nat Med. (2015) 1:1464–72. doi: 10.1038/nm.3974
141. Calvet G, Aguiar RS, Melo ASO, Sampaio SA, de Filippis I, Fabri A, et al. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: a case study. Lancet Infect Dis. (2016) 6:653–60. doi: 10.1016/s1473-3099(16)00095-5
142. Adibi JJ, Marques ET A, Cartus A, Beigi RH. Teratogenic effects of the Zika virus and the role of the placenta. Lancet (2016) 87:1587–90. doi: 10.1016/S0140-6736(16)00650-4
143. Richard AS, Shim BS, Kwon YC, Zhang R, Otsuka Y, Schmitt K, et al. AXL-dependent infection of human fetal endothelial cells distinguishes Zika virus from other pathogenic flaviviruses. Proc Natl Acad Sci USA. (2017) 14:2024–9. doi: 10.1073/pnas.1620558114
144. Tabata T, Petitt M, Puerta-Guardo H, Michlmayr D, Wang C, Fang-Hoover J, et al. Zika virus targets different primary human placental cells suggesting two routes for vertical transmission. Cell Host Microb. (2016) 20:155–66. doi: 10.1016/j.chom.2016.07.002
145. Richard AS, Zhang A, Park SJ, Farzan M, Zong M, Choe H. Virion-associated phosphatidylethanolamine promotes TIM1-mediated infection by Ebola, dengue, and West Nile viruses. Proc Natl Acad Sci USA. (2015) 112:14682–7. doi: 10.1073/pnas.1508095112
146. Dowall SD, Graham VA, Rayner E, Atkinson B, Hall G, Watson RJ, et al. A susceptible mouse model for Zika virus infection. PLoS Negl Trop Dis. (2016) 10:e0004658. doi: 10.1371/journal.pntd.0004658
147. Rossi SL, Tesh RB, Azar SR, Muruato AE, Hanley KA, Auguste AJ, et al. Characterization of a novel murine model to study Zika virus. Am J Trop Med Hyg. (2016) 4:1362–9. doi: 10.4269/ajtmh.16-0111
148. Aliota MT, Caine EA, Walker EC, Larkin KE, Camacho E, Osorio JE. Characterization of Lethal Zika virus infection in AG129 mice. PLoS Negl Trop Dis. (2016) 10:e0004682. doi: 10.1371/journal.pntd.0004682
149. Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, et al. A mouse model of Zika virus pathogenesis. Cell Host Microb. (2016) 9:720–30. doi: 10.1016/j.chom.2016.03.010
150. Miner JJ, Sene A, Richner JM, Smith AM, Santeford A, Ban N, et al. Zika virus infection in mice causes panuveitis with shedding of virus in tears. Cell Rep. (2016) 6:3208–18. doi: 10.1016/j.celrep.2016.08.079
151. Nowakowski TJ, Pollen AA, Di Lullo E, Sandoval-Espinosa C, Bershteyn M, Kriegstein AR. Expression analysis highlights AXL as a Candidate Zika virus entry receptor in neural stem cells. Cell Stem Cell (2016) 8:591–6. doi: 10.1016/j.stem.2016.03.012
152. Meertens L, Labeau A, Dejarnac O, Cipriani S, Sinigaglia L, Bonnet-Madin L, et al. Axl Mediates ZIKA virus entry in human glial cells and modulates innate immune responses. Cell Rep. (2017) 8:324–33. doi: 10.1016/j.celrep.2016.12.045
153. Li H, Saucedo-Cuevas L, Shresta S, Gleeson JG. The neurobiology of Zika virus. Neuron (2016) 2:949–58. doi: 10.1016/j.neuron.2016.11.031
154. Cugola FR, Fernandes IR, Russo FB, Freitas BC, Dias JL, Guimaraes KP, et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature (2016) 34:267–71. doi: 10.1038/nature18296
155. Wells MF, Salick MR, Wiskow O, Ho DJ, Worringer KA, Ihry RJ, et al. Genetic ablation of AXL does not protect human neural progenitor cells and cerebral organoids from Zika Virus infection. Cell Stem Cell (2016) 9:703–08. doi: 10.1016/j.stem.2016.11.011
156. Monath TP, Barrett ADT. Pathogenesis and pathophysiology of yellow fever. Adv Virus Res. (2003) 60:343–95. doi: 10.1016/S0065-3527(03)60009-6
157. Hahn CS, Dalrymple JM, Strauss JH, Rice CM. Comparison of the virulent Asibi strain of yellow fever virus with the 17D vaccine strain derived from it. Proc Natl Acad Sci USA. (1987) 4:2019–23.
158. Rice C, Lenches E, Eddy Shin S, Sheets R, Strauss J. Nucleotide sequence of yellow fever virus: implications for flavivirus gene expression and evolution. Science (1985) 29:726–33. doi: 10.1126/science.4023707
159. Lee E, Lobigs ME. Protein Domain III determinants of yellow fever virus 17D Vaccine strain enhance binding to Glycosaminoglycans, impede virus spread, and attenuate virulence. J Virol. (2008) 2:6024–33. doi: 10.1128/jvi.02509-07
160. Fernandez-Garcia MD, Meertens L, Chazal M, Hafirassou ML, Dejarnac O, Zamborlini A, et al. Vaccine and wild-type strains of yellow fever virus engage distinct entry mechanisms and differentially stimulate antiviral immune responses. mBio (2016) 7:e01956-1. doi: 10.1128/mBio.01956-15
161. Mayor S, Pagano RE. Pathways of clathrin-independent endocytosis. Nat Rev Mol Cell Biol. (2007) 8:603. doi: 10.1038/nrm2216
162. Sánchez-San Martín C, López T, Arias CF, López S. Characterization of rotavirus cell entry. J Virol. (2004) 8:2310–8. doi: 10.1128/jvi.78.5.2310-2318.2004
163. Khan AG, Pickl-Herk A, Gajdzik L, Marlovits TC, Fuchs R, Blaas D. Human Rhinovirus 14 enters Rhabdomyosarcoma cells expressing ICAM-1 by a Clathrin-, Caveolin-, and Flotillin-independent pathway. J Virol. (2010) 4:3984–92. doi: 10.1128/jvi.01693-09
164. Sieczkarski SB, Whittaker GR. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J Virol. (2002) 6:10455–64. doi: 10.1128/jvi.76.20.10455-10464.2002
165. Mandl JN, Akondy R, Lawson B, Kozyr N, Staprans SI, Ahmed R, et al. Distinctive TLR7 signaling, type I IFN production, and attenuated innate and adaptive immune responses to yellow fever virus in a primate reservoir host. J Immunol. (2011) 86:6406–16. doi: 10.4049/jimmunol.1001191
166. Stiasny K, Heinz FX. Flavivirus membrane fusion. J Gen Virol. (2006) 87(Pt. 10):2755–66. doi: 10.1099/vir.0.82210-0
167. Seligman SJ. Constancy and diversity in the flavivirus fusion peptide. Virol J. (2008) 5:27. doi: 10.1186/1743-422X-5-27
Keywords: flaviviruses, Japanese encephalitis virus, Zika virus (ZIKV), dengue virus, yellow fever virus, entry receptor
Citation: Laureti M, Narayanan D, Rodriguez-Andres J, Fazakerley JK and Kedzierski L (2018) Flavivirus Receptors: Diversity, Identity, and Cell Entry. Front. Immunol. 9:2180. doi: 10.3389/fimmu.2018.02180
Received: 12 May 2018; Accepted: 04 September 2018;
Published: 26 September 2018.
Edited by:
Alan Chen-Yu Hsu, University of Newcastle, AustraliaReviewed by:
Alec Jay Hirsch, Oregon Health & Science University, United StatesCopyright © 2018 Laureti, Narayanan, Rodriguez-Andres, Fazakerley and Kedzierski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lukasz Kedzierski, bHVrYXN6a0B1bmltZWxiLmVkdS5hdQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.