
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 25 September 2018
Sec. Microbial Immunology
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02151
This article is part of the Research Topic Secondary Respiratory Infections in the Context of Acute and Chronic Pulmonary Diseases View all 13 articles
 Radha Gopal1†
Radha Gopal1† Benjamin Lee2†Kevin J. McHugh1Helen E. Rich1Krishnaveni Ramanan1Sivanarayana Mandalapu1Michelle E. Clay1Philip J. Seger1
Benjamin Lee2†Kevin J. McHugh1Helen E. Rich1Krishnaveni Ramanan1Sivanarayana Mandalapu1Michelle E. Clay1Philip J. Seger1 Richard I. Enelow3Michelle L. Manni1
Richard I. Enelow3Michelle L. Manni1 Keven M. Robinson4
Keven M. Robinson4 Javier Rangel-Moreno5
Javier Rangel-Moreno5 John F. Alcorn1*
John F. Alcorn1*Influenza is a common respiratory virus that infects between 5 and 20% of the US population and results in 30,000 deaths annually. A primary cause of influenza-associated death is secondary bacterial pneumonia. We have previously shown that influenza induces type I interferon (IFN)-mediated inhibition of Type 17 immune responses, resulting in exacerbation of bacterial burden during influenza and Staphylococcus aureus super-infection. In this study, we investigated the role of STAT2 signaling during influenza and influenza-bacterial super-infection in mice. Influenza-infected STAT2−/− mice had increased morbidity, viral burden, and inflammation when compared to wild-type mice. Despite an exaggerated inflammatory response to influenza infection, we found increased bacterial control and survival in STAT2 deficient mice during influenza-MRSA super-infection compared to controls. Further, we found that increased bacterial clearance during influenza-MRSA super-infection is not due to rescue of Type 17 immunity. Absence of STAT2 was associated with increased accumulation of M1, M2 and M1/M2 co-expressing macrophages during influenza-bacterial super-infection. Neutralization of IFNγ (M1) and/or Arginase 1 (M2) impaired bacterial clearance in Stat2−/− mice during super-infection, demonstrating that pulmonary macrophages expressing a mixed M1/M2 phenotype promote bacterial control during influenza-bacterial super-infection. Together, these results suggest that the STAT2 signaling is involved in suppressing macrophage activation and bacterial control during influenza-bacterial super-infection. Further, these studies reveal novel mechanistic insight into the roles of macrophage subpopulations in pulmonary host defense.
Influenza A virus infection causes significant mortality and morbidity worldwide. It is estimated that influenza infection causes 3 million hospitalizations and 250,000 deaths, globally each year (1). The innate immune system senses invading influenza viruses through a variety of pattern recognition receptors, inducing the production of inflammatory cytokines and chemokines, including type I (IFNα/β) and type III interferons (IFNλ) (2–4). Type I IFNs (IFNα/β) bind to the heterodimer of IFNaR1 and IFNaR2 receptors, associated with Jak1 and Tyk2 kinases, respectively, to produce signaling effects during influenza infection (5). Signaling through IFNaR1/2 leads to phosphorylation of STAT1 and STAT2, activating downstream complex formation with IRF9, and transcription of IFN stimulated genes (ISGs), thereby controlling viral burden. Type II IFN (IFNγ) activates only STAT1 signaling through IFNγR1 and IFNγR2 and plays a role in cell-mediated immunity (6, 7). Type III IFNs signal through IFNLR and IL10R2 to induce ISGs similar to those induced by type I IFN (7). Type I, type II and type III IFNs have been shown to have important roles during influenza infection (7–9). In the absence of type I or type II IFN during influenza infection, increased granulocytic or lymphocytic inflammation, respectively, has been reported (7). Absence of STAT1 signaling specifically resulted in an increased granulocytic and Th2-skewed response (7), and both STAT1 and STAT2 were crucial for viral control and survival in mice (9).
Secondary bacterial pneumonia during influenza infection is a common cause of influenza-associated hospitalization and mortality during both seasonal and pandemic outbreaks (10, 11). During the 1918 influenza pandemic, Streptococcus pneumoniae was the most common bacteria isolated from influenza-bacteria super-infected patients (10). However, recent reports have shown that Staphylococcus aureus is now the most frequent super-infecting bacteria (10, 12). We have shown that during influenza-bacterial super-infection, influenza-induced type I IFN inhibited S. aureus-induced Type 17 immunity and associated antimicrobial peptide (AMP) production (13, 14). Further, we and others have shown that mice lacking the type I IFN receptor cleared S. aureus and S. pneumoniae better than wild-type (WT) mice during super-infection (13, 15). While influenza infection alters host defense to S. aureus, the converse is also likely true; S. aureus increases influenza burden in the lung, possibly by affecting STAT1-STAT2 dimerization during super-infection (13, 16). These data suggest that influenza-induced STAT1 and STAT2 signaling is critical to mediating susceptibility to secondary bacterial pneumonia. We have recently shown that STAT1 is involved in increasing bacterial burden through suppression of the Type 17 immune response during influenza-bacterial super-infection (17). However, little is known regarding the specific role of STAT2 in super-infection.
Since type I and type III IFN signaling relies on STAT2, while type II IFN signals solely through STAT1, examination of STAT2 deficiency enables a more targeted evaluation of type I and type III IFN-mediated immune responses. In this study, we investigated the role of STAT2 signaling during influenza infection and influenza-bacterial super-infection by infecting WT and Stat2−/− mice with influenza A/PR/8/34 followed by MRSA (USA300) challenge, and evaluating subsequent survival, morbidity, viral and bacterial burden, and inflammatory responses. We then elucidated differential host responses in Stat2−/− mice using RNA expression, flow cytometry, immunohistochemistry and in vitro macrophage culture. Further, we investigated the role of hematopoietic and non-hematopoietic STAT2 signaling during influenza-bacterial super-infection. These studies are the first to define the role of STAT2 signaling in influenza, bacterial super-infection and identify a novel macrophage-dependent mechanism of susceptibility to secondary bacterial pneumonia.
WT C57BL/6 (6 to 8-week-old) mice were purchased from Taconic Farms (Germantown, NY). Stat2−/− mice on C57BL/6 background were a kind gift from Dr. Christian Schindler, Columbia University, NY (18), and colonies were subsequently maintained under specific pathogen-free conditions. In vivo studies were performed on age matched adult male mice, unless otherwise indicated. All experiments were approved by the University of Pittsburgh IACUC (19).
Influenza A/PR/8/34 (influenza H1N1) was propagated in chicken eggs as described (20). Mice were infected with 100 plaque-forming units (PFU) of influenza in 40 μl of sterile PBS, unless otherwise noted. MRSA, USA 300, was provided by Dr. Alice Prince, Columbia University, NY. MRSA stocks were grown overnight in casein hydrolysate yeast extract-containing modified broth medium at 37°C and diluted to an inoculum of 5 × 107 CFU in 50 μl of sterile PBS. MRSA dosing was calculated using OD660 measurement of overnight cultures and application of an extinction coefficient. For survival experiments, 2 × 108 CFU of MRSA were delivered. All infections were performed on isoflurane-anesthetized mice via oropharyngeal aspiration. For super-infection experiments, mice were challenged with influenza or vehicle and then infected with MRSA or vehicle on day 6 after influenza infection (13, 21, 22). Mouse tissues were collected 24 h after MRSA or vehicle challenge. To neutralize IFNγ, mice were treated with 300 μg anti-IFNγ (XMG1.2) antibody in 200 μl sterile PBS (BioXCell, West Lebanon, NH) or rat IgG isotype control via intraperitoneal (IP) injection on days 4 and 6 post-influenza infection. To neutralize arginase, mice were treated with 100 μg N-−hydroxy-nor-L-arginine (nor-NOHA) via IP injection on days −1, 0, 3, and 6 post-influenza.
Lungs were perfused with 1 ml of sterile PBS and cell differential counts were performed on cytospin smears from bronchoalveolar lavage (BAL) fluid stained with Protocol Hema 3 staining (Fisher Scientific, Kalamazoo, MI). The cranial lobe of the right lung was homogenized in sterile PBS by mechanical grinding, for quantification of bacterial burden by plating serial dilutions and for cytokine production measurement by Lincoplex (Millipore, Billerica, MA), Bio-plex (Bio-Rad, Hercules, CA), or ELISA (R&D Systems, Minneapolis, MN). The middle and caudal lobes of the right lung were snap-frozen and homogenized in liquid nitrogen for RNA isolation using the Absolutely RNA Miniprep Kit (Agilent Technologies, Santa Clara, CA). Gene expression was analyzed by RT-PCR utilizing commercially available Taqman primer and probe sets (Applied Biosystems, Foster City, CA). Fold changes in mRNA expression were calculated using the ΔΔCT method, and were normalized to the endogenous housekeeping gene hypoxanthine-guanine phosphoribosyltransferase (HPRT). The left lobe of the lung was pressure inflated and fixed in 10% neutral-buffered formalin for histology, or collected in DMEM media for flow cytometry. Histology was scored by a sample blinded pathologist.
Mouse lungs were aseptically dissected into small sections, digested for 30 min at 37°C in 1 mg/mL collagenase media, and passed through 70 μm filters (23). Single cell suspensions were stained with anti-CD3 (145-2C11), CD4 (RM4-5), CD11b (M1/70), Ly6C (HK1.4), CD80(16-10A1), and macrophage galactose lectin (MGL) (LOM-14). After the singlets were gated from total cells, macrophages were gated for CD11b+Ly6C+ cells. Further, these cells were gated for CD80 and MGL to determine macrophage phenotype as M1 (CD80+) and/or M2 (MGL+). The frequency of CD11b+Ly6C+, CD11b+Ly6C+ CD80+, CD11b+ Ly6C+ CD80+ MGL+, and CD11b+ Ly6C+ MGL+ cells were calculated from the frequency of total cells. For intracellular staining, cells were stimulated for 4 h with 50 ng/mL of phorbol myristate acetate (PMA) and 750 ng/mL ionomycin (Sigma-Aldrich, St Louis, MO) with Golgi plug (BD Pharmingen, San Diego, CA) added 1 h into stimulation. After stimulation, cells were surface stained, permeabilized with cytofix-cytoperm solution (BD Pharmingen, San Diego, CA), and stained with antibodies specific for IL-17 (TC11-18H10), IL-22 (Poly5164) for 30 min at 4°C. The percentage of IL-17+ and IL-22+ cells were determined from gating on CD3+CD4+ T cells. Cells were collected in a Becton Dickinson FACS Aria flow cytometer with FACS Diva software (BD, Franklin Lakes, NJ). Flow cytometric analysis was performed using FlowJo (Tree Star, Ashland, OR).
Left lung lobes were perfused and stored in 10% neutral buffered formalin. Paraffin-embedded sections were stained with hematoxylin and eosin (H&E) and pulmonary inflammation was evaluated by microscopy. For immunofluorescent staining, formalin-fixed lung sections were incubated at 60°C, and quickly immersed in xylene to remove paraffin. Sections were then hydrated in 96% alcohol and PBS. Antigen retrieval was performed by incubating slides in boiling Dako Target Retrieval Solution (S1699, DAKO Cytomation), followed by blocking with 5% (v/v) normal donkey (017-000-121, Jackson ImmunoResearch Laboratories) serum and Fc block (BD Pharmingen, San Diego, CA). Sections were stained with antibodies against inducible NO synthase (iNOS) (goat anti-mouse iNOS, clone M-19; Santa Cruz Biotechnology Inc.), F4/80 (clone Cl:A3-1, MCA497GA, Bio-Rad), and arginase-1 (Arg1) (rabbit anti–arginase I, clone H-52; Santa Cruz Biotechnology Inc.). Primary antibodies were detected with Alexa Fluor 568-donkey anti-goat Ig G (H+L) cross adsorbed (A-11057; Invitrogen) to detect iNOS, donkey anti-rabbit Ig G (H+L) antibody conjugated to FITC (711-095-152, Jackson Immuno Research Laboratories) to visualize Arg1. Slides were incubated with biotin-F(ab')2 donkey anti-rat (712-006-153 Jackson Immuno Research Laboratories), followed by Cy5-Streptavidin (405209, Biolegend) to reveal the location of F4/80+ macrophages. Vectashield anti-fade mounting medium with DAPI (H-1200, Vector Laboratories) was used to counterstain tissues and to detect nuclei. Images were obtained with a Zeiss Axioplan 2 microscope and recorded with a Zeiss AxioCam digital camera. iNOS, F4/80, and Arg1 positive cells were enumerated in three random 200x fields per lung sample, and the average number of iNOS+, Arg1+ or double positive macrophages was calculated. Samples were analyzed in a blinded fashion.
Arginase-1 activity was quantified in lung BAL using the QuantiChrom Arginase Assay Kit as per the manufacturer's instructions (BioAssay Systems). In BAL samples, total protein was measured by using BCA Protein Assay kit (Pierce Biotechnology, Rockford, IL). Protein concentration was adjusted to 100 μg/ml per sample. Arg-1 activity is expressed as U/l of sample.
Nitrite was quantified in BAL samples by using Griess reagent as per the manufacturer's instructions (Promega Corporation).
RNA was isolated from C57BL/6 and Stat2−/− mouse lungs using the Agilent RNA miniprep kit. RNA integrity was determined using an Agilent 2100 bio analyzer. mRNA was purified by using Sera-Mag Oligo(dT) Beads, fragmented with magnesium-catalyzed hydrolysis, and reverse transcribed into cDNA using random primers (Superscript II; Invitrogen). Then, cDNA underwent end repair with T4 DNA polymerase and Klenow DNA polymerase, followed by the addition of “A” bases to the 3′ end and ligation to adaptor oligonucleotides. Products from the ligation were run on a 2% agarose gel. A gel slice consisting of the 200 bp region (±25 bp) was excised and used as a template for PCR amplification. The final PCR product was purified, denatured with 2 N NaOH, and diluted to 10–12 pM prior to cluster amplification on a single-read flow cell v4, as outlined in the Single-Read Cluster Generation Kit v4 (Illumina). The flow cell was sequenced on an Illumina Genome Analyzer II. The data were analyzed as previously described (24). Full sequencing data has been uploaded to the National Center for Biotechnology Information Gene Expression Omnibus, GSE119029.
Bone marrow cells were isolated from the femurs and tibias of mice, and grown for 7 days in complete DMEM media supplemented with 20 ng/mL GM-CSF (PeproTech, NJ), as previously described (25). On day 7, non-adherent cells (BMDCs) were collected, and adherent cells (BMDMs) were recovered using cell scraper (Genemate, Kaysville, UT) or by gentle mechanical scraping.
Bone marrow derived cells were plated to 1 × 106 cells/mL and cells in 1 mL of media in 24 well tissue culture-treated plates, rested overnight at 37°C in 5% CO2, then treated with IFN-β (10 units/mL) or IFN-γ (10 ng/mL) (R&D Systems, MN). Twenty-four hours later, cell culture supernatants were harvested and cells were lysed in RLT buffer and frozen at −80°C for RNA extraction using the RNAeasy mini kit (QIAGEN, Germantown, MD. Gene expression was analyzed as described above.
BMDMs were incubated with fluorescein isothiocyanate or Alexa Fluor-647 (Molecular Probes) —labeled MRSA (10 MOI) for 30 min. After washing, flow cytometry was performed to determine bacterial uptake. To determine bacterial killing by macrophages, BMDMs were incubated with MRSA for 1 h. Cells were washed with PBS and the remaining macrophages were incubated with gentamycin (300 μl/ml) for 15 min to kill extracellular bacteria. Cells were washed and incubated for an additional hour to determine killing of intracellular bacteria. Cells were washed and lysed with 0.5 ml of 0.02% Triton X-100 in PBS, and plated to determine the percentage of intracellular bacterial killing.
To generate BMC mice, C57BL/6 (Thy1.1), C57BL/6 (Thy1.2), and Stat2−/− (Thy 1.2) mice were fed with Sulfa-Trimm diet containing 1.2% sulfamethoxazole and 0.2% trimethoprim for 2 weeks before irradiation (26, 27). Mice were sub-lethally irradiated twice with two doses of 600 rad delivered 4 h apart. Mice were reconstituted with 1 × 107 bone marrow cells from either C57BL/6 (Thy1.1), C57BL/6 (Thy1.2), or Stat2−/− (Thy 1.2) mice as previously described (26). Mice were allowed to reconstitute for 9 weeks.
Data were analyzed using GraphPad Prism software. Experiments were repeated 2 to 5 times. All data are presented as mean ± SEM, unless otherwise noted. Significance was determined by unpaired Student's t test or one-way ANOVA followed by post-hoc Bonferroni or Tukey's test for multiple comparisons. Mortality data was analyzed by Log-rank (Mantel-Cox) test.
To determine the role of STAT2 signaling during influenza infection, WT and Stat2−/− mice were infected with influenza. By day 6 of influenza infection, influenza-challenged Stat2−/− mice showed significantly more weight loss than WT mice and increased lung viral burden (Figures 1A,B). In males, infection was sub-lethal in both Stat2−/− and WT mice, but Stat2−/− mice showed markedly delayed recovery, requiring 35 days to return to baseline weight, versus 14 days for WT mice (Figure 1C). In females, Stat2−/− mice showed increased mortality compared to WT during influenza infection (Figure 1D). Next, we determined the cellular inflammatory response to influenza infection in WT and Stat2−/− mice. There was a significantly greater number of polymorphonuclear cells (PMN, neutrophils) observed in bronchoalveolar lavage (BAL) from Stat2−/− mice when compared to WT (Figure 1E). Interestingly, Stat2−/− mice had significantly fewer numbers of macrophages and lymphocytes in BAL compared to WT mice (Figure 1E).
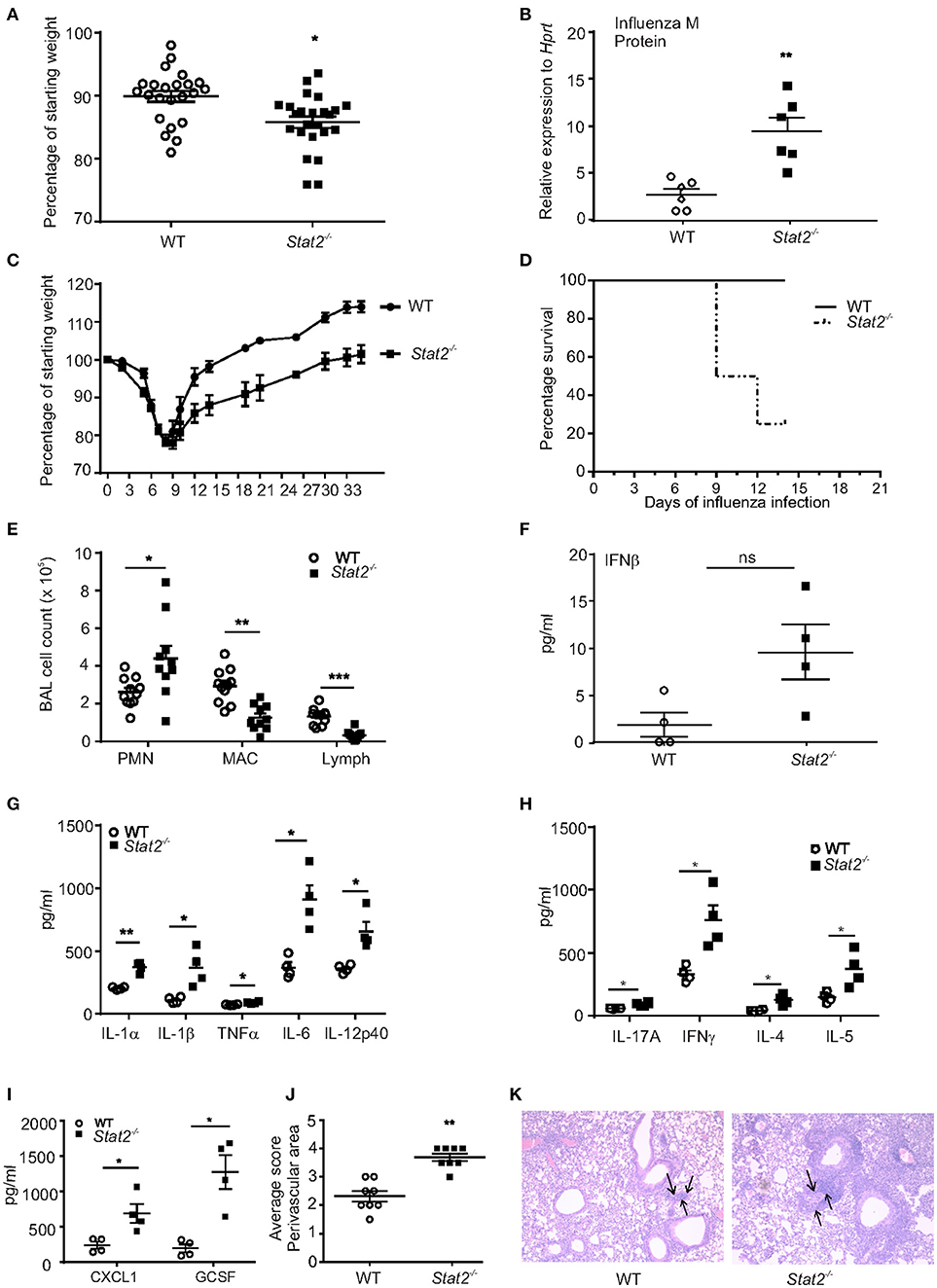
Figure 1. Stat2−/− mice have increased susceptibility to influenza infection. (A) WT, Stat2−/− male 6–8 weeks mice were infected with 100 PFU of influenza, percentage weight loss on day 6 of influenza infection, N = 22–24 per group. (B) Viral burden was measured by influenza M protein expression 6 days following influenza infection in whole lung, N = 6 per group. (C) Weight loss during 35 days following influenza infection, N = 8 per group. (D) Percentage survival following influenza infection in females, N = 8 per group. (E) BAL samples collected on day 6 following influenza infection from mice lungs, cytospin the cells and differential counts were made N = 10–12 per group. Right upper lung lobes were homogenized in PBS, (F) IFNβ levels were measured in BAL samples by using ELISA, N = 4 per group. (G) IL-1α, IL-1β, TNFα, IL-6, and IL-12p40, (H) IL-17A, IFNγ, IL-4, and IL-5 (I) CXCL1, and GCSF levels were measured by Luminex assay, N = 4 per group. Representative data shown from 3 or more experiments are shown (J) On day 6 post infection, lungs were fixed in 10% formalin, embedded in paraffin, stained with H&E, lung perivascular areas (arrows) were scored. (K) Representative histology pictures are shown. Original magnification for H&E sections × 100. Data are represented as mean±SEM, two tailed Student's t-test, *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant.
Next, we determined whether type I IFN levels were altered during influenza infection. We found a trend toward increased levels of IFNβ in Stat2−/− mice when compared to WT mice during primary influenza infection (Figure 1F). Influenza infection induced a variety of inflammatory cytokines such as IL-1β, IL-6, TNFα, IL-8, CCL2, CCL5, and CXCL10 from epithelial and immune cells (2). We found an increase in the levels of the inflammatory cytokines IL-1α, IL-1β, TNFα, IL-6, IL-12p40, IL-17, and IFNγ, and increased levels of the Type 2 cytokines IL-4 and IL-5, in influenza-infected Stat2−/− mice when compared to WT mice (Figures 1G,H).
It has been shown that IL-17 signaling induces the chemokines CXCL1, CXCL2, CXCL5, and CXCL8 to mediate granulopoiesis and increased neutrophil recruitment to mucosal sites (28, 29). Consistent with the increase in PMNs, we observed an increase in CXCL1 and the granulopoietic factor, G-CSF in Stat2−/− when compared to WT mice (Figure 1I). Further, scoring of pulmonary inflammation indicated an increase in perivascular infiltration of cells in the lungs of Stat2−/− mice when compared to WT mice (Figures 1J,K). Together, these data demonstrate that influenza-infected Stat2−/− mice have more severe influenza infection, which is associated with an increased inflammatory response, compared to WT mice.
We have previously shown that influenza-associated IFNβ attenuated host bacterial defense due to suppression of Type 17 immunity (13, 14). Also, we recently identified that Type 17 immunity is rescued in the absence of STAT1 signaling during influenza-bacterial super-infection (17). In this study, we examined the effects of MRSA challenge on day 6 of influenza infection in WT and Stat2−/− mice. One day following MRSA challenge, super-infected WT mice had significantly increased lung bacterial burden compared to mice infected with MRSA alone, demonstrating impaired bacterial clearance from the lung caused by preceding influenza infection (Figure 2A). However, Stat2−/− mice were rescued from this clearance defect (Figure 2A). To assess survival in the context of super-infection, adult female mice were subjected to influenza/MRSA super-infection using a dose of MRSA previously determined to be lethal to WT mice. Given the increased severity of influenza infection seen in females, mice were infected with a lower dose of influenza to minimize the confounding effects of lethal influenza infection. Under these conditions, Stat2−/− mice showed significantly delayed mortality (Figure 2B). Next, we found that BAL fluid from both Stat2−/− and WT mice had significantly greater numbers of neutrophils compared to macrophages or lymphocytes (Figure 2C). However, we observed no significant differences in neutrophils, macrophages, or lymphocytes between super-infected WT and Stat2−/− mice (Figure 2C). Further, we found no differences in the levels of cytokines IL-1α, IL-1β, IL-6, TNFα, IL-12p40, IL-17A, IL-10, CXCL1, and GCSF between WT and Stat2−/− mice (Figures 2D–F). However, the levels of IFNγ, IL-4 and IL-5 were elevated in Stat2−/− mice during super-infection (Figure 2E). In agreement with similar cellularity and local production of pro-inflammatory cytokines, histological scoring showed no differences in perivascular inflammation (Figures 2G,H). These findings demonstrate that Stat2−/− mice are rescued from impaired bacterial clearance in the lung during super-infection, which translated into a detectable survival advantage, but this was independent of any noticeable differences in lung inflammation.
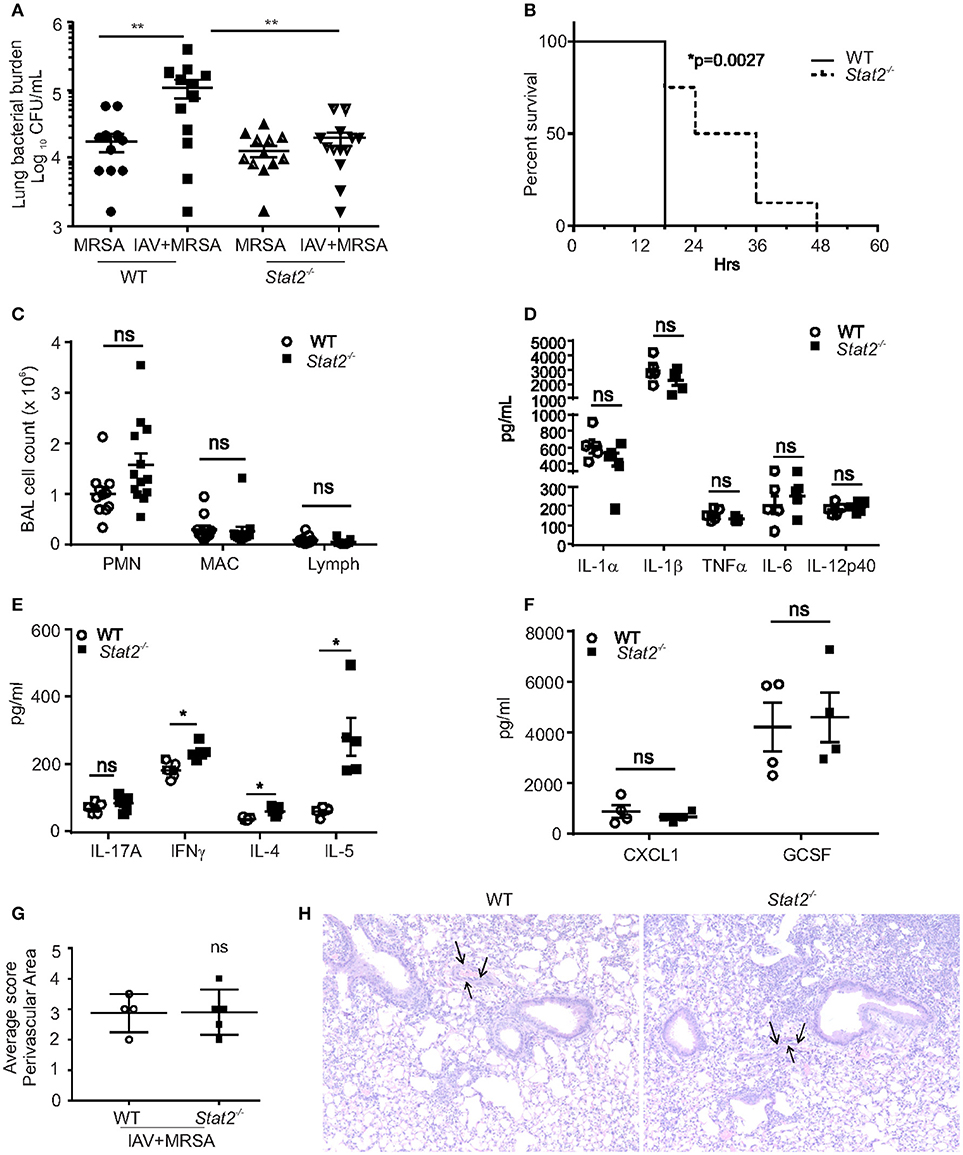
Figure 2. Stat2−/− are rescued from impaired bacterial clearance from the lung following influenza infection. (A) WT or Stat2−/− mice were infected with 100 PFU of influenza for 6 days then challenged with 5 × 107 cfu of MRSA for one additional day. Right upper lung lobes were homogenized in PBS and bacterial burden was determined. N = 12–13 per group. (B) Adult female mice were infected with 66 PFU influenza A PR/8/H1N1 followed by challenge with 2 × 108 CFU MRSA and the survival was determined, N = 7–8 per group. (C) BAL samples collected from co-infected B6 and Stat2−/− mice, cytospin the cells and differential counts were made, N = 11–13 per group. Right Upper lung lobes were homogenized in PBS, and (D) IL-1α, IL-1β, TNFα, IL-6, IL-12p40, (E) IL-17A, IFNγ, IL-4, IL-5 (F) CXCL1, and GCSF levels were measured by luminex assay, N = 4 per group. Representative data shown from three or more experiments. Lungs were fixed in 10% formalin, embedded in paraffin, (G) perivascular areas (arrows) were scored in formalin fixed lungs by staining with H&E (H) representative figures are shown, N = 4 per group. Original magnification for H&E sections × 100. Data are represented as mean±SEM. Data analyzed using 2-tailed Student's t-test or One way ANOVA followed by Bonferroni test for multiple comparisons, *p < 0.05, **p < 0.01, ns, not significant.
We next addressed whether the increased bacterial clearance observed in Stat2−/− mice was due to altered Type 17 immunity. Interestingly, numbers of IL-17 and IL-22 positive CD4 cells, and the levels of Type 17 cytokines (IL-17, IL-22, and IL-23) were not altered in Stat2−/− mice compared to WT mice (Figures S1A–E). In further support of this finding, no differences in expression levels of the Th17 transcription factors Rorc and Rora, and Th17 immune mediated antimicrobial peptide (AMP) such as Lcn2 and Reg3b production in the lungs were detected between Stat2−/− and WT mice (data not shown). These data suggest that protection in Stat2−/− mice is not mediated by rescue of Type 17 immunity.
Next, to determine the pathways associated with this increased bacterial clearance in Stat2−/− mice, we performed RNA sequencing analyses. We found increased expression of Type 1 (IFNγ) and Type 2 (IL-4 and IL-13) cytokines in Stat2−/− mice compared to WT (Figures 2E, 3A,B). In response to IFNγ stimulation, macrophages increase their production of nitric oxide and reactive oxygen intermediates to become M1 type macrophages, which exhibit proinflammatory and antibacterial activity (30, 31) Accordingly, we found increased RNA abundance of genes associated with the M1 phenotype including Nos2, Tnfa, Tnfsf13b, Batf2, Ido1, and chemokines such as Cxcl9, Ccl5, and Ccl8 in the lungs of Stat2−/− mice compared to WT during super-infection (Figure 3A).
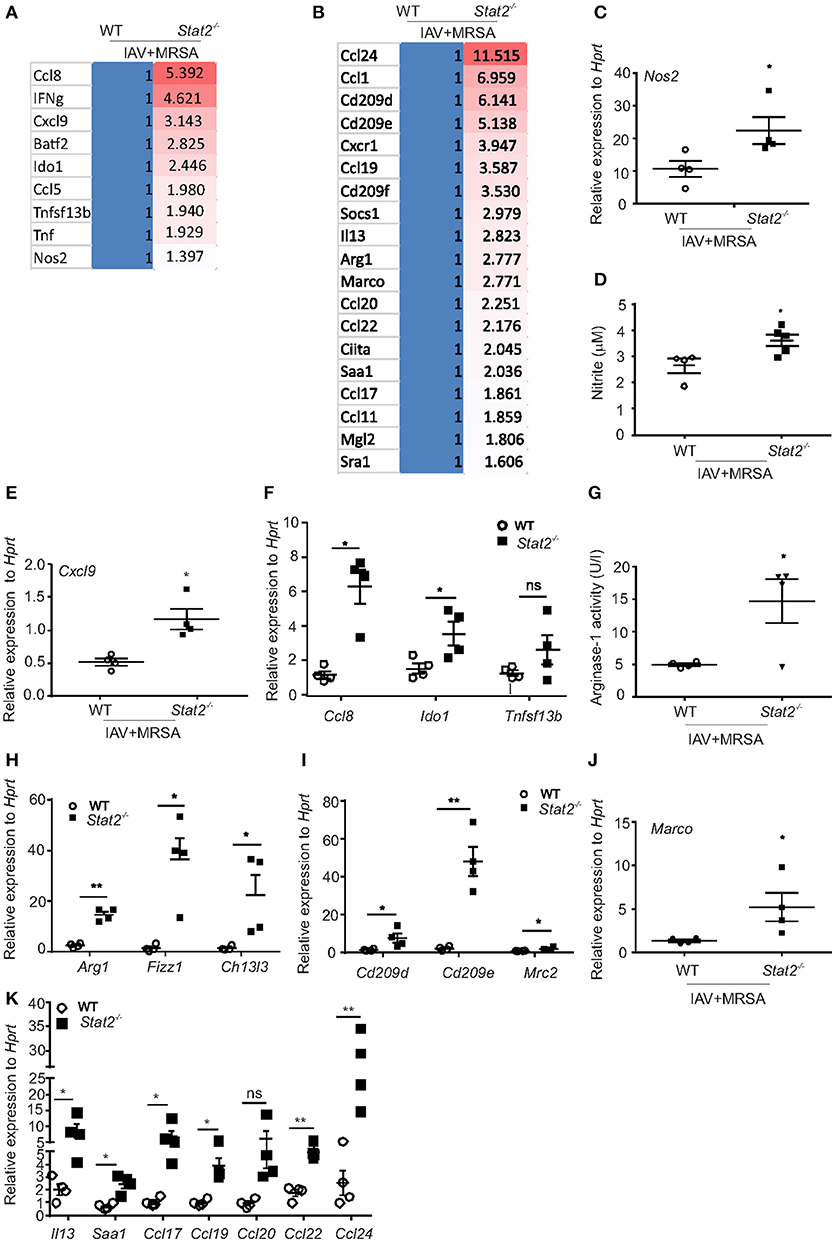
Figure 3. M1 and M2 macrophages are associated with increased bacterial clearance in Stat2−/− during influenza-MRSA super-infection. WT or Stat2−/− mice were infected with 100 PFU of influenza for 6 days then challenged with 5 × 107 cfu of MRSA for one additional day. Gene expression analyses were measured in lung by RNAseq analysis. Heat-map representing RNA abundance associated with M1 (A) and M2 (B) macrophages from WT and Stat2−/− mice, N = 4 per group. (C) Nos2, (D) Nitrite, (E) Cxcl9, (F) Ccl8, Ido1, Tnfsf13b were analyzed by RT-PCR, N = 4 per group. (G) Arginase-1 activity was determined from BAL samples from both WT and Stat2−/− mice infected with influenza and MRSA super-infection, N = 4 per group. (H) Arg1, Fizz1, and Chi3l3, (I) Cd209d, Cd209e and Mrc2 (J) Marco mRNA expression levels were determined by RT-PCR, N = 3–4 per group. (K) Il13 and Saa1, Ccl17, Ccl19, Ccl20, Ccl22, and Ccl24 mRNA expression levels were determined by RT-PCR, N = 4 per group. Representative data shown from three or more experiments. Data are represented as mean±SEM. Data analyzed using 2-tailed Student's t-test, *p < 0.05, **p < 0.01, ns, not significant.
In response to IL-4, IL-13, and glucocorticoids, macrophages express markers of the M2 phenotype, which is characterized by induction of Arg1, chitinase 3-like 3 (Ym), found in inflammatory zone-1 (Fizz1), resistin-like molecule (Relmα/Retnla), chemokines such as CCL17, CCL20, and CCL22, scavenger receptors (CD36, Macrophage receptor with collagenous structure or MARCO), and c-type lectin receptors or CLRs (CD209, Macrophage Galactose Lectin or MGL) (30, 32). Accordingly, we found increased RNA abundance of Arg1, Socs1, Sra1, Saa1, Ciita, Marco, Mgl2, Cd209d, Cd209e, Cd209f, Ccl1, Ccl11, Ccl17, Ccl19, Ccl20, Ccl22, and Ccl24 genes in Stat2−/− mice when compared to WT mice during super-infection (Figure 3B).
Next, we confirmed the observed RNA abundance of the classical M1 and M2 macrophage markers. We found increased gene expression of Nos2 and Nitrite in Stat2−/− mice compared to WT with super-infection (Figures 3C,D). IFNγ-inducible chemokines CXCL9, CXCL10, and CXCL11 are ligands for the receptor CXCR3, which is essential in the attraction of effector lymphocytes to inflammatory sites (33, 34). Accordingly, we found increased mRNA expression of Cxcl9 in influenza/MRSA infected Stat2−/− mice compared to WT mice (Figure 3E). Further, we found increased expression of M1 macrophage markers Ccl8, Ido1, and Tnfsf13b in Stat2−/− mice during super-infection when compared to WT mice (Figure 3F). These data confirm the increased expression of genes associated with M1 macrophages in Stat2−/− mice during influenza-bacterial super-infection.
Next, we found increased mRNA expression and activity levels of Arg1, mRNA expression of Fizz, Chi3l3, Cd209d, Cd209e, Mrc2 (Cd206), Marco, Il13, Saa1 (Serum amyloid A1 protein), Ccl17, Ccl19, Ccl20, Ccl22, and Ccl24 in Stat2−/− mice compared to WT mice during super-infection (Figures 3G–K). Together, these data show that in the absence of STAT2 signaling, both the M1 and M2 macrophage phenotypes are enhanced during super-infection.
IFNγ signals through the IFNGR1/ IFNGR2 complex and activates the transcription factor STAT1, thereby inducing ISGs. We found increased levels of IFNγ (Figure 2E) and IFNγ-induced ISGs (Figure 3E) in Stat2−/− mice during influenza-bacterial super-infection. Next, we determined whether STAT1 is altered due to increased levels of IFNγ in Stat2−/− mice during influenza-bacterial super-infection. As expected, we found increased STAT1 expression in Stat2−/− mice when compared to WT mice during influenza-bacterial super-infection (Figure S2A).
IL-4 activates the STAT6 signaling pathway through activation of transcription factors PPARγ and PPARδ and drives M2 polarization (31, 35). Here, we tested whether STAT6 signaling is altered due to increased IL-4 levels in Stat2−/− mice during influenza-bacterial super-infection. However, we found no differences in STAT6 expression levels in between WT and Stat2−/− mice during influenza-bacterial super-infection (Figure S2B). Further, we measured the RNA expression of Pparg from WT and Stat2−/− mice during influenza-MRSA super-infection. Interestingly, we found that Pparg expression was suppressed in Stat2−/− mice during influenza-bacterial super-infection (Figure S2C). Together, these data suggest that in the absence of STAT2, there is alteration of other associated signaling pathways during super-infection.
Next, we determined the frequency of M1 and M2 macrophages by using flow cytometry. CD80 and MGL (macrophage galactose lectin) have been shown to be markers for M1 and M2 macrophages, respectively (36, 37). We found a significant increase in the percentage of CD11b+Ly6C+ cells in influenza/MRSA infected Stat2−/− lungs when compared to WT mice by flow cytometry (Figure 4A). Further, we found increased frequency of M1 (CD80+) and M1/M2 (CD80+MGL+) co-expressing macrophages in Stat2−/− mice when compared to WT during influenza-bacterial super-infection (Figures 4B,C). Interestingly, we found no differences in M2 (MGL+) macrophages in between WT and Stat2−/− mice in super-infection (Figure 4D). Further, we found higher frequency of M1/M2 co-expressing macrophages when compared to M1 or M2 populations (Figures 4B–E).
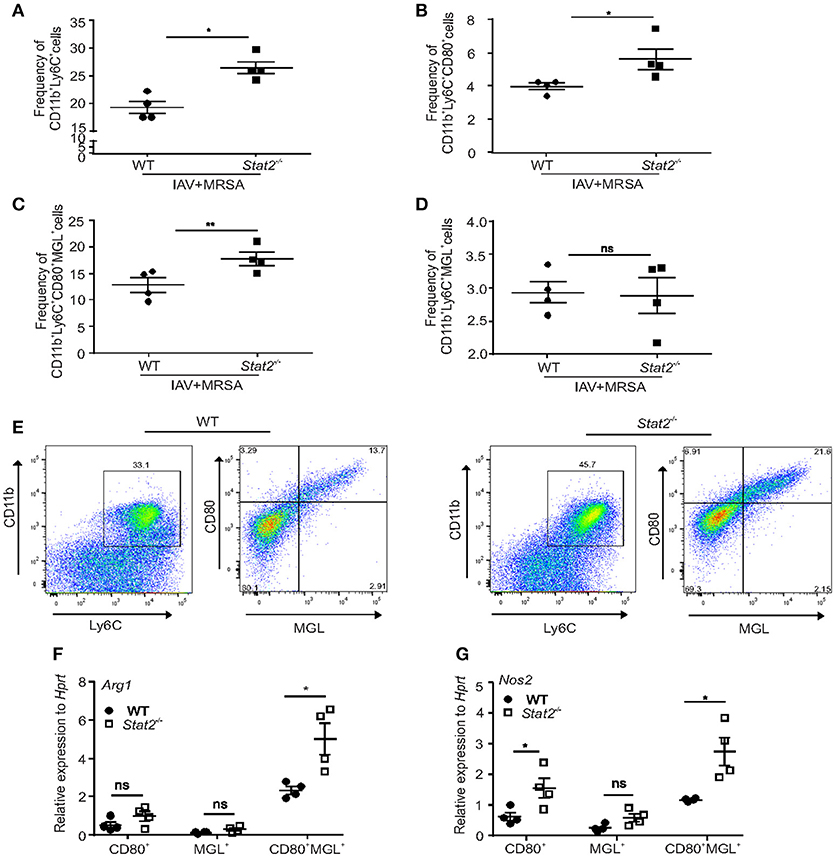
Figure 4. Increased frequency of M1/M2 macrophages in Stat2−/− during influenza-MRSA super-infection. WT or Stat2−/− mice were infected with 100 PFU of influenza for 6 days then challenged with 5 × 107 cfu of MRSA for one additional day. (A) Frequency of CD11b+Ly6C+ cells, (B) CD11b+Ly6C+CD80+ cells, (C) CD11b+Ly6C+MGL+ cells (D) CD11b+Ly6C+CD80+MGL+ cells from lungs were determined from influenza and MRSA super infection by flow cytometry, N = 4 per group. Double positive CD11b+Ly6C+ cells were gated for CD80 and MGL to identify the CD11b+Ly6C+CD80+, CD11b+Ly6C+MGL+, CD11b+Ly6C+CD80+MGL+ cells. (E) The representative figures were shown. CD80+, MGL+, CD80+MGL+ cells were sorted from the lung using FACS and Arg1 (F) NOS2 (G) RNA expression was analyzed using RT-PCR. N = 4 per group. Data analyzed using 2-tailed Student's t-test, *p < 0.05, **p < 0.01, ns, not significant.
To then determine the specific macrophages involved in production of Arg1 and Nos2, we sorted M1, M2, and M1/M2 cells by using specific antibodies, and determined the RNA expression levels of Nos2 and Arg1 (Figures 4F,G). As expected, we found increased expression of Nos2 in M1 and M1/M2 macrophages. Next, we found no differences in induction of Arg1 in M1 and M2 macrophages. However, we found increased expression of Arg1 in dual M1/M2 macrophages. These data suggest that these M1/M2 macrophages induce both Arg1 and Nos2 in influenza-bacterial super-infection. To further characterize specific Arg1+ and iNOS+ macrophage localization in the lung, we used an IHC technique using iNOS and Arg1 antibodies.
To determine whether the increase in M1 and M2 macrophages is influenza or MRSA dependent, we infected WT or Stat2−/− mice with influenza or MRSA or super-infection, and determined the number of iNOS+F4/80+ and Arg1+F4/80+ macrophages in the lung by IHC. We found increased numbers of iNOS+F4/80+ and Arg1+F4/80+ cells during influenza infection and super-infection in Stat2−/− mice compared to WT mice (Figure 5A). In WT mice, we observed a higher number of iNOS+F4/80+ cells during MRSA and super-infection compared to influenza infection alone. In contrast, we observed a decrease in the number of Arg1+F4/80+ cells in MRSA and super-infection compared to WT mice infected with influenza alone (Figure 5B). Further, no differences were observed between WT and Stat2−/− mice in the number of iNOS+F4/80+ and Arg1+F4/80+ cells during MRSA infection alone (Figures 5A,B). Next, we found increased gene expression of Nos2 and Arg1 in Stat2−/− mice when compared to WT mice during influenza, but not MRSA infection (Figures 5C,D). These data indicate that influenza infection, not MRSA infection, is likely the cause of the increased number of Arg1+ macrophages seen in Stat2−/− mice during super-infection.
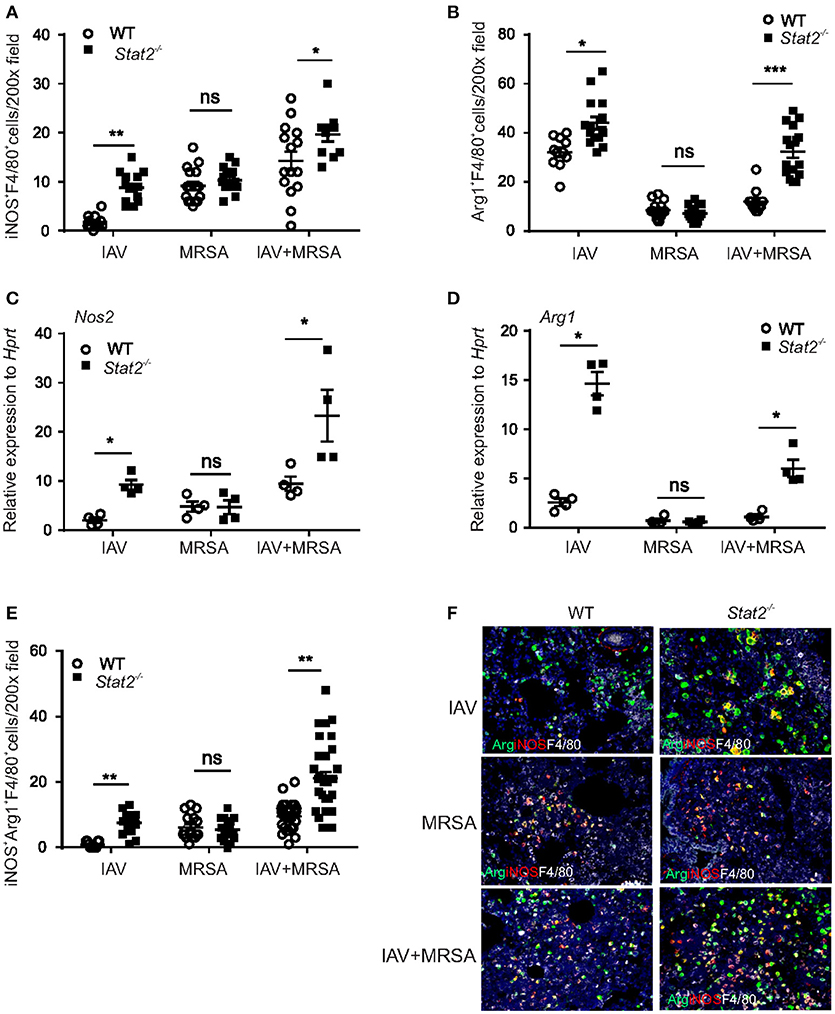
Figure 5. Increased M1 and M2 macrophage expression in Stat2−/− is dependent on influenza but not MRSA infection. WT or Stat2−/− mice were infected with 100 PFU of influenza for 6 days or 5 × 107 cfu of MRSA for one day or super-infection as described in methods. (A) iNOS+F4/80+, N = 11–16 (B) Arg1+F4/80+ producing cells were determined from lungs by IHC, N = 11–16. (C) Nos2, (D) Arg1 gene expression was analyzed from lungs by RT-PCR, N = 4 per group. (E) iNOS+Arg1+F4/80+ producing cells were determined from lung by immunohistochemistry, N = 11–32 per group. (F) Representative figures, × 200 magnification fields are shown. Data are represented as mean±SEM. Data analyzed using two-tailed Student's t-test, *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant.
The M1 and M2 macrophage phenotypes are thought to antagonize each other to establish inflammatory or anti-inflammatory responses (38). M1 macrophages induce L-hydroxy-arginine and citrulline through NOS2 induction, which is a potent inhibitor of arginase (39), to promote inflammation. Conversely, M2 macrophage induction of arginase antagonizes M1 markers to promote repair and resolution of infection (39). As we seen in flow cytometry data, we found an increased number of “dual function” iNOS+Arg1+F4/80+ cells during and super-infection in Stat2−/− mice compared to WT mice (Figures 5E,F). However, no significant differences were observed in the number of dual function iNOS+Arg1+F4/80+ cells between WT and Stat2−/− mice during MRSA infection (Figures 5E,F). Together, these data show that in the absence of STAT2 signaling, the increase in Type 1 and Type 2 cytokines during influenza infection creates a pulmonary environment that supports both M1 and M2 macrophage differentiation after super-infection. Unexpectedly and contrary to the idea that there is antagonism between M1 and M2 macrophages, we found increased accumulation of M1, M2 and M1/M2 co-expressing cells in the lungs of Stat2−/− mice after super-infection.
We next examined whether M1 and/or M2 macrophages are required for the improved bacterial clearance in Stat2−/− mice during super infection. We treated both influenza-infected WT and Stat2−/− mice with anti-IFNγ, followed by super-infection with MRSA, and measured subsequent bacterial burden. We found that blocking IFNγ decreased bacterial clearance in Stat2−/− mice (Figure 6A). IFNγ neutralization showed a trend toward decreased bacterial clearance in WT mice as well. Next, we found fewer iNOS+F4/80+ cells in anti-IFNγ-treated WT and Stat2−/− mice compared to mice treated with isotype controls (Figure 6B). Further, we found a decreased number of dual function iNOS+Arg1+F4/80+ in mice treated with anti-IFNγ antibody (Figures 6C,D). However, consistent with the role of IFNγ as an inducer of the M1 phenotype, we found no significant differences in the number of total Arg1+F4/80+ cells with IFNγ neutralization (Figure S3A).
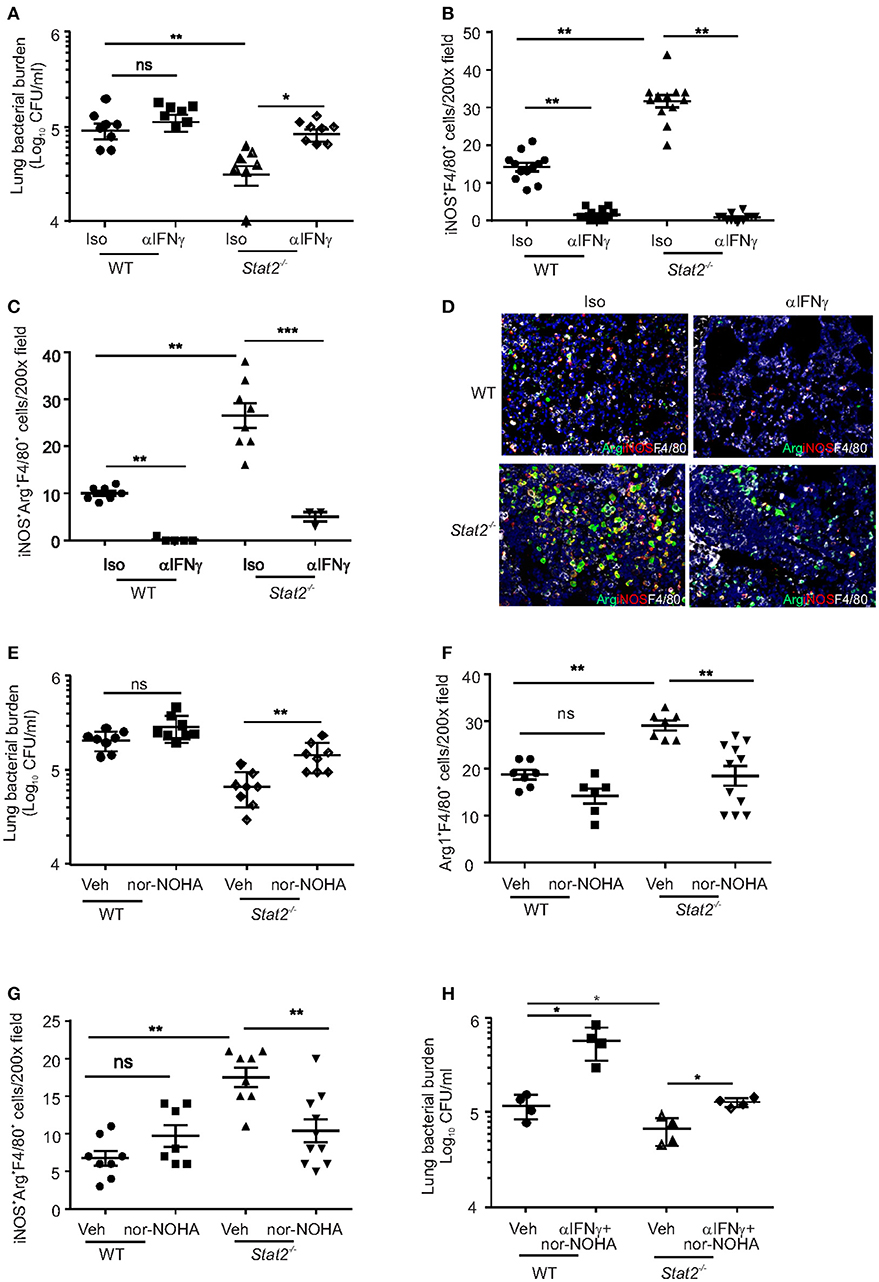
Figure 6. M1/M2 co-expressing macrophages are required for increased bacterial clearance in Stat2−/− during influenza-MRSA super-infection. WT or Stat2−/− mice were infected with 100 PFU of influenza for 6 days then challenged with 5 × 107 cfu of MRSA for one additional day. Mice were treated with 300 μg of anti-IFNγ antibodies or 300 μg of rat IgG isotype controls as described in methods. (A) Bacterial burden was measured, N = 7–8 per group, (B) iNOS+F4/80+ producing cells, N = 12 per group (C) iNOS+Arg1+F4/80+ producing cells, N = 3–7 per group (D) were determined by IHC, representative figures x200 magnification fields are shown. WT or Stat2−/− mice were infected with 100 PFU of influenza for 6 days then challenged with 5 × 107 cfu of MRSA for one additional day. Mice were treated with Vehicle (DMSO) or N-−hydroxy-nor-L-arginine (nor-NOHA) as described in methods (E) Bacterial burden, N = 8 per group, (F) Arg+F4/80+ cells, N = 6–10 per group (G) iNOS+Arg+F4/80+ producing cells were determined, N = 7–10 per group. Representative pictures from two experiments are shown. (G) WT or Stat2−/− mice were infected with 100 PFU of influenza for 6 days then challenged with 5 × 107 cfu of MRSA for one additional day. Mice were treated with 300 μg of anti-IFNγ antibodies and N-−hydroxy-nor-L-arginine (nor-NOHA) or vehicle as described in methods (H) Bacterial burden was measured, N = 4 per group, Data are represented as mean±SEM. Data analyzed using 2-tailed Student's t-test or One way ANOVA followed by Bonferroni test for multiple comparisons, *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant.
Next, we determined whether M2 macrophage activation was required to control MRSA during super-infection in Stat2−/− super-infected mice. Arg1 was neutralized using nor-NOHA in both WT and Stat2−/− mice subjected to influenza/MRSA super-infection. Inhibition of Arg1 during super-infection increased bacterial burden in Stat2−/−, but not WT mice (Figure 6E). Further, we observed a decrease in the number of Arg1+F4/80+ macrophages and dual function iNOS+Arg1+F4/80+ macrophages in Stat2−/− mice treated with nor-NOHA when compared to Stat2−/− mice treated with vehicle (Figures 6F,G). Interestingly, we found increased numbers of iNOS+F4/80+ macrophages in both WT and Stat2−/− mice upon nor-NOHA treatment during influenza-bacterial infection (Figure S3B). Despite the presence of M1 macrophages during Arg1/M2 blockade and M2 macrophages during IFNγ/M1 blockade, bacterial control was suppressed in Stat2−/− mice during super-infection. These data suggest that neither M1 nor M2 macrophages alone are as proficient at MRSA clearance. We then inhibited both IFNγ and Arg1 simultaneously during super-infection in WT and Stat2−/− mice (Figure 6H). Inhibition of both M1 and M2 pathways increased bacterial burden in both WT and Stat2−/− mice. These data together suggest that activation of M1/M2 co-expressing macrophages is required for increased bacterial clearance during super-infection in Stat2−/− mice.
We then examined bacterial uptake and killing by macrophages from WT and Stat2−/− mice. BMDMs isolated from WT and Stat2−/− mice were incubated with fluorescent-labeled MRSA for 30 min. Bacterial uptake percentage was determined by the number of FITC+ BMDMs by flow cytometry. It has been shown that the FITC dye can be quenched in response to pH changes in endosomes (40, 41). Therefore, we compared phagocytosis by using FITC or Alexa Fluor-647-labeled bacteria. We found increased bacterial uptake in Stat2−/− mice compared to WT mice in both FITC or Alexa Fluor-647-labeled MRSA (Figures 7A,B). These data show that macrophages from Stat2−/− mice can bind and take up more bacteria than WT mice. Further, under basal conditions we found increased mRNA expression of Mrc2 and Cd209e in Stat2−/− BMDMs compared to WT BMDMs (Figures 7C,D). However, no significant differences in the expression levels of Arg1, Chi3l3, and Marco were observed (Figures 7E–G). These results suggest that increased expression of the M2 phenotype-associated receptors MRC2 and CD209 is likely to be a mechanism involved in increased binding ability in BMDMs from Stat2−/− mice.
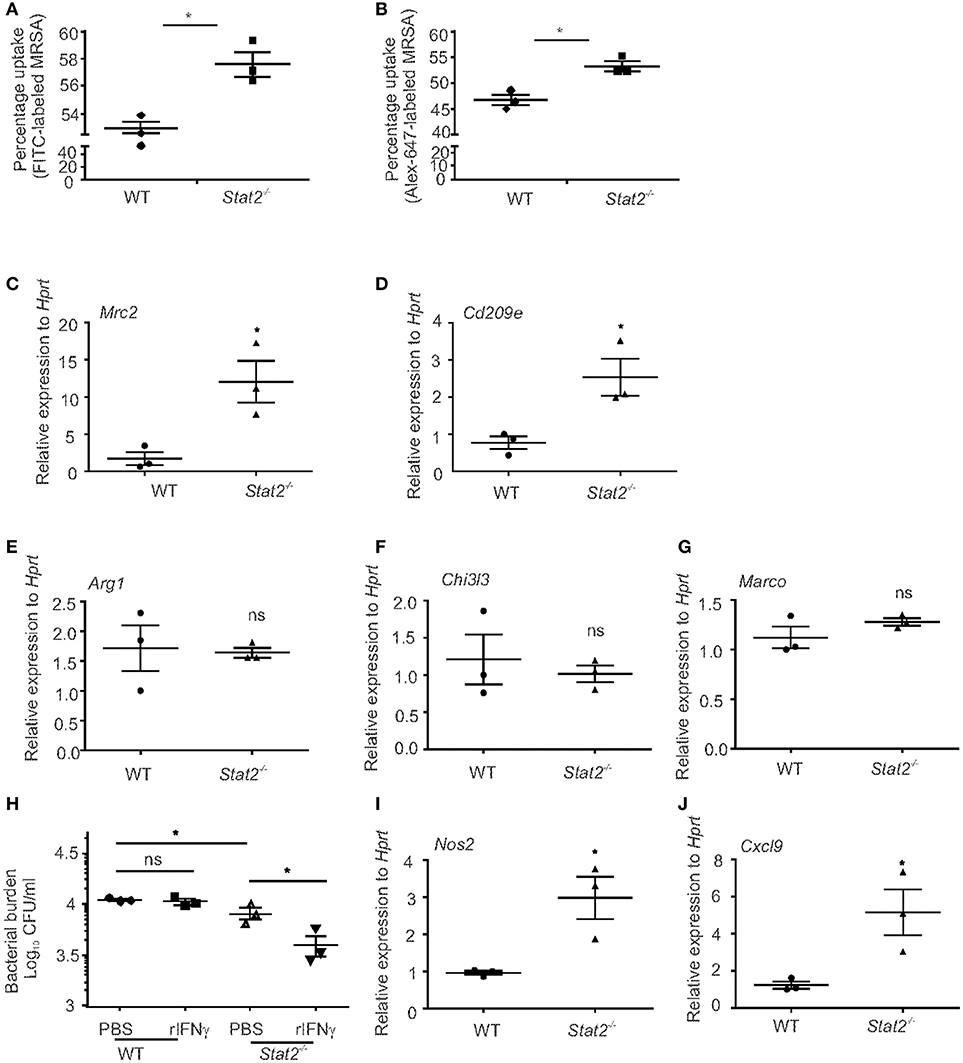
Figure 7. Increased bacterial uptake and killing efficiency in BMDMs from Stat2−/− mice BMDMs were generated and infected with FITC or Alex-647-labeled MRSA (10 MOI) for 30 min, washed, fixed with 1% formaldehyde and the number of FITC+ (A) or Alex-647+ (B) BMDMs were determined by Flow cytometry. (C) Mrc2 and (D) Cd209e, (E) Arg1, (F) Chi3l3, (G) Marco gene expression levels were measured by RT-PCR. (H) BMDMs were generated, treated with recombinant IFNγ (10 μg/ml) for 24 h, and infected with MRSA (10 MOI) and bacterial killing was determined. (I) Nos2 and (J) Cxcl9 gene expression levels were measured from naïve BMDMs, N = triplicates per treatment. Representative pictures from two experiments are shown. Data are represented as mean±SEM. Data analyzed using 2-tailed Student's t-test, *p < 0.05, ns, not significant.
To test the ability of these BMDMs to kill MRSA, we performed a bacterial killing assay using BMDMs treated with mouse IFNγ. We found that Stat2−/− BMDMs have increased killing ability at baseline and following IFNγ treatment when compared to WT BMDMs (Figure 7H). However, in BMDMs from WT mice, IFNγ treatment had no effect on bacterial killing (Figure 7H). Next, we found that macrophages from Stat2−/− mice had increased expression of Nos2 and Cxcl9 compared to WT mice (Figures 7I,J). These data suggest that macrophages from Stat2−/− mice have an enhanced ability to kill bacteria due to increased activation and nitric oxide production, and have the potential to attract CXCR3+ effector lymphocytes that can amplify M1 activation. These results together show that STAT2 modulates the expression of CLRs and iNOS production by macrophages, and thus negatively impacts their protective functions during super-infection.
To further confirm that increased bacterial control in Stat2−/− mice during influenza-bacterial super infection is due to M1/M2 co-expressing macrophage activation, we generated bone marrow chimeric (BMC) mice. We transferred hematopoietic cells from WT or Stat2−/− mice to irradiated WT or STAT2 mice and vice versa. We used WT C57BL/6 mice with Thy1.1 or Thy 1.2 markers and Stat2−/− mice bred on a Thy1.2 background. After 9 weeks of bone marrow reconstitution, we infected mice with both influenza and MRSA. Consistent with our previous data, we found higher lung bacterial burden in WT BMC mice (WT host/WT BM) than Stat2−/− mice (Stat2−/− host/Stat2−/− BM) during influenza-bacterial super-infection (Figure 8A). As expected, we found decreased bacterial burden in hematopoietic Stat2−/− BMC mice (WT host/Stat2−/− BM) compared to WT BMC mice (WT host/ WT BM) during super-infection (Figure 8A). Interestingly, we also found decreased bacterial burden in non-hematopoietic Stat2−/− BMC (Stat2−/− host/WT BM) compared to WT BMC mice (WT host/ WT BM) during super-infection (Figure 8A).
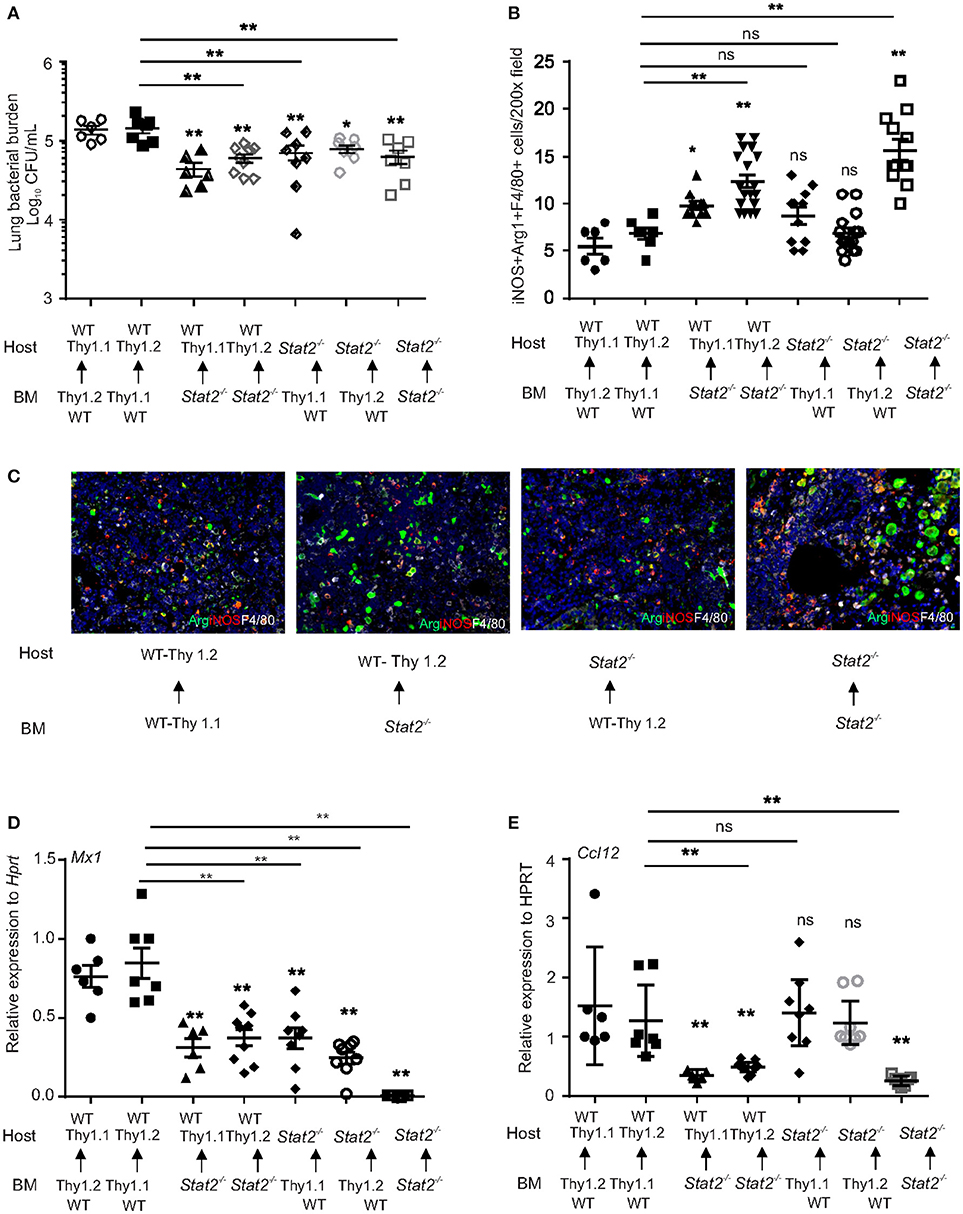
Figure 8. Increased bacterial control in cells from hematopoietic or non-hematopoietic compartments of Stat2−/− mice. WT BMC (Thy1.1 host, Thy 1.2 BM or Thy 1.2 host, Thy 1.1 BM), Stat2−/− BMC (Stat2−/− host, Stat2−/− BM), Hematopoietic Stat2−/− BMC mice (Thy 1.1 or Thy 1.2 host, Stat2−/− BM), non- hematopoietic Stat2−/− BMC (Stat2−/− host, Thy 1.1 or Thy 1.2 BM) were generated as described in methods. These mice were infected with 100 PFU of influenza for 6 days then challenged with 5 × 107 cfu of MRSA for one additional day. (A) Mice were sacrificed and right upper lung lobes were homogenized in PBS and bacterial burden was measured. (B) iNOS+Arg1+F4/80+ producing cells were determined from lung by immunohistochemistry, N = 6–19 per group. (C) Representative figures, × 200 magnification fields are shown. (D) Mx1, (E) Ccl12 mRNA expression was measured, N = 6–9 per group. Data are represented as mean±SEM. Data analyzed using One way ANOVA followed by Bonferroni test for multiple comparisons, *p < 0.05, **p < 0.01, ns, not significant.
Next, we found increased expression of iNOS+Arg1+ macrophages in hematopoietic Stat2−/− BMC (WT host/Stat2−/− BM) and Stat2−/− BMC mice (Stat2−/− host/Stat2−/− BM), suggesting that hematopoietic cells are involved in bacterial control in Stat2−/− mice during influenza-MRSA super-infection (Figures 8B,C). Also, no significant differences were observed in the expression of iNOS+Arg1+ macrophages between WT BMC mice (WT host/WT BM) and non-hematopoietic Stat2−/− BMC mice (Stat2−/− host/WT BM). These data suggest that iNOS+Arg1+ macrophages mediate bacterial control in hematopoietic Stat2−/− BMC (WT host/ Stat2−/− BM) and Stat2−/− BMC mice (Stat2−/− host/Stat2−/− BM) during influenza-MRSA super-infection. However, we found increased bacterial clearance in non-hematopoietic Stat2−/− BMC mice (Stat2−/− host/WT BM) during influenza-MRSA super-infection.
Type I and type II interferons induce interferon stimulated genes (ISGs) to establish anti-viral response responses (42–44). Based on RNAseq data and RT-PCR analysis, we identified that expression of type I IFN-induced genes such as Mx1, Lhx2, and CCL12 was suppressed in Stat2−/− mice during influenza-MRSA super-infection (Figures S4A–D). However, the type II IFN-induced chemokine CXCL9 was not suppressed during influenza-MRSA super-infection (Figure S4E). Further, Mx1 and Ccl12 gene expression was suppressed in bone marrow dendritic cells (BMDCs) from Stat2−/− mice stimulated with IFNβ compared to BMDCs from WT mice (Figures S4F,G). These data confirm that type I IFN, but not type II IFN responses are significantly attenuated in the lungs of Stat2−/− mice during influenza infection.
To determine if ISG expression in Stat2−/− BMC mice was suppressed, we measured the expression of Mx1 and Ccl12. We found decreased expression of Mx1 in Stat2−/− BMC (Stat2−/− host/Stat2−/− BM), hematopoietic Stat2−/− BMC (WT host/ Stat2−/− BM), and non-hematopoietic Stat2−/− BMC mice (Stat2−/− host/WT BM) compared to WT BMC mice (WT host/WT BM) (Figure 8D). The expression of Mx1 was further suppressed in Stat2−/− BMC mice (Stat2−/− host/Stat2−/− BM) when compared to hematopoietic Stat2−/− BMC (WT host/ Stat2−/− BM) and non-hematopoietic Stat2−/− BMC mice (Stat2−/− host/WT BM) (Figure 8D). These data suggest that both hematopoietic and non-hematopoietic cells are involved in the expression of Mx1 in response to influenza-bacterial super-infection.
Next, we determined the expression of Ccl12 in the BMC mice. We found decreased expression of Ccl12 in Stat2−/− BMC (Stat2−/− host/Stat2−/− BM) and hematopoietic Stat2−/− BMC mice (WT host/ Stat2−/− BM) as compared to WT BMC (WT host/WT BM) (Figure 8E). However, the expression of Ccl12 was not suppressed in Stat2−/− non-hematopoietic BMC mice (Stat2−/− host/WT BM) (Figure 8E). These data suggest that STAT2 signaling in hematopoietic cells is crucial in induction of CCL12 during influenza-bacterial super-infection. Further, these data suggest that the absence of stromal cell STAT2 resulted in decreased ISG expression and improved bacterial control in Stat2−/− non-hematopoietic BMC mice (Stat2−/− host/WT BM) during influenza-bacterial super-infection.
Next, we determined the influenza viral burden in the BMC mice. We found higher viral burden in Stat2−/− BMC (Stat2−/− host/Stat2−/− BM) and non-hematopoietic Stat2−/− BMC mice (Stat2−/− host/WT BM) when compared to WT BMC (WT host/WT BM) and hematopoietic Stat2−/− BMC (WT host/ Stat2−/− BM) (Figure S5). These data suggest that non-hematopoietic cells are involved in STAT2-mediated viral control during influenza-MRSA super-infection.
During influenza infection, epithelial cells, macrophages and dendritic cells all induce type I and type III IFNs, proinflammatory cytokines and chemokines (2). Type I IFN signaling mediates lung pathology and infiltration of granulocytes during influenza infection (7). Absence of STAT1 or STAT2 signaling compromises viral control and survival in mice (9). In the current study, we have demonstrated that in the absence of STAT2 signaling the type I IFN response is impaired. This was associated with increased viral burden, inflammation, and pathology in the lungs during influenza infection. These results show that STAT2-dependent signaling is crucial in controlling influenza burden and inflammatory immune responses during primary influenza infection. Consistent with these findings, elevated levels of type I IFNs during influenza infection correlates with disease severity in outbred mice (45).
Influenza-associated secondary bacterial pneumonia is a serious complication of influenza infection (10, 11, 46). Reducing morbidity and mortality requires insight into the immune mechanisms that alter susceptibility to secondary bacterial infection. In this study, we show that STAT2 deficiency improves survival and rescues the impairment of bacterial clearance from the lung otherwise observed during influenza-bacterial super-infection. Further, we have identified increased accumulation of M1, M2 and M1/M2 co-expressing macrophages by influenza-MRSA super-infection in the setting of STAT2 deficiency as a novel mechanism that mediates this protection.
We have previously demonstrated that influenza-induced type I IFN-mediated suppression of Type 17 responses to both Gram (+) (S. aureus) and Gram (−) bacteria (Pseudomonas aeruginosa, Escherichia coli) during influenza-bacterial super-infection (13, 14). In these studies, loss of Type 17 immune responses was associated with exacerbation of secondary bacterial challenge during influenza infection. Further, we have also shown increased bacterial control in Ifnar−/− mice during super-infection, suggesting that influenza-induced type I IFN is a critical mediator of antibacterial immune suppression (13). Also, we have recently shown increased Type 17 immunity in STAT1−/− mice during super-infection (17). Therefore, we hypothesized that STAT2 deficiency would rescue suppression of Type 17 responses during influenza infection. Surprisingly, we found that the increased bacterial clearance we observed in the absence of STAT2 was not due to rescue of the Type 17 immune response. These data suggest that Type 17 immunity is predominantly regulated by STAT1 and not STAT2 during super-infection. This finding prompted further investigation into the mechanism of protection in Stat2−/− mice.
Since Type 17 responses were not associated with the observed phenotype, we next explored other possible mechanisms involved in bacterial control. In our study, we found no differences in neutrophils, macrophages and lymphocyte numbers in BAL of WT and Stat2−/− mice during influenza-bacterial super-infection. A possible role for neutrophils in bacterial killing exists, as shown by a trend toward increased neutrophil numbers in BAL from Stat2−/− mice. However, based on RNAseq data analysis, we found increased RNA abundance of M1 and M2 macrophage markers, and further characterized the RNA expression, frequency and immunolocalization of these cells. We then determined the role of these macrophages in bacterial control in Stat2−/− mice during influenza-bacterial super-infection.
M1 macrophages are known to be involved in pathogen defense and inflammation, whereas M2 macrophages are thought to have a suppressive or regulatory role during inflammation (30, 47). In the current study, we found the majority of CD11b+Ly6C+ macrophages are CD80+/MGL+ (M1/M2) positive. Also, we found increased frequency of M1 (CD80+), but not M2 (MGL+) single positive cells in Stat2−/− mice during influenza-bacterial super-infection. Further, these M1/M2 cells had increased expression levels of both Arg1 and Nos2 in Stat2−/− mice during influenza-bacterial super-infection. We then confirmed these flow cytometry findings by IHC and observed an increase in Arg1+iNOS+F4/80+ cells in Stat2−/− mice. These data confirm the ability of STAT2 to regulate macrophage phenotype during pneumonia, and identify an M1/M2 dual phenotype macrophage population in the context of influenza-associated secondary bacterial infection.
Further, in accordance with increased levels of IFNγ and ISGs, we found increased Stat1 expression levels in Stat2−/− mice when compared to WT mice. This suggests that in the absence of STAT2 signaling, other pathways are activated as a compensatory effect. However, Stat6 expression is not altered in the absence of STAT2 in super-infection. IFNγ is known to suppress expression of the nuclear receptor PPARγ (48, 49). We also found suppression of Pparg expression in Stat2−/− mice during influenza-bacterial super-infection. Alterations in IFNγ and PPARγ signaling may be a possible mechanism by which M1/M2 macrophage populations are altered in Stat2−/− mice in super-infection.
In Stat2−/− mice, iNOS+ macrophages were elevated during super-infection. This contrasted with WT mice in which iNOS+ macrophages were only induced in the setting of bacterial challenge. In contrast, Arg1+ macrophages were decreased during both MRSA and super-infection in WT mice. However, in Stat2−/− mice the Arg1+ cells were increased in influenza-bacterial super-infection, but not MRSA infection, suggesting that the M2 induction during super-infection is driven by influenza infection. Chen et al. (15) correlated an increased number of M2 macrophages during influenza infection with suppression of the protective immune response to bacterial super-infection (50). In contrast, one study has shown that S. aureus priming increased M2 macrophages and anti-inflammatory responses to influenza challenge (51). In the current study, we found an increased number of Arg1+ macrophages associated with increased bacterial control in Stat2−/− mice during super-infection, and that WT mice suffered from impaired bacterial clearance despite the presence of Arg1+ at a level similar to that seen during MRSA infection alone. Also, we found that Arg1- and iNOS-expressing dual function macrophages were significantly higher in Stat2−/− mice. The presence of M1/M2 co-expressing macrophages has recently been described in response to Toxoplasma gondii infection (52), but to our knowledge is a novel finding in pulmonary host defense.
Promotion of bacterial killing by IFNγ is well established for intracellular pathogens such as Mycobacterium tuberculosis and Toxoplasma gondii. However, information regarding its role in bacterial killing of S. aureus is limited. Studies have shown that influenza-induced IFNγ inhibits pneumococcal control during super-infection (53). In this study, we found a trend in increased bacterial burden in IFNγ neutralization in WT mice during super-infection. However, no differences were observed in WT BMDMs in bacterial killing in response to IFNγ treatment. Observed discrepancies in the role of IFNγ during super-infection might be due to differences in the dose and the strain of bacteria. We found that improved bacterial control seen during influenza-bacterial super-infection in Stat2−/− mice was lost upon neutralization of IFNγ. This was associated with a loss of iNOS+ and iNOS+Arg1+ dual-function cells, suggesting that increased IFNγ in Stat2−/− mice during super-infection mediates the protective phenotype by driving induction of M1-polarized macrophages. However, it is known that IFNγ increases neutrophil nitrite production and increases phagocytosis (54–56). It is also possible that other reactive oxygen species are involved in increased IFNγ-mediated bacterial clearance in Stat2−/− mice during super-infection. Similarly, we found that neutralization of Arg1 decreased bacterial clearance in Stat2−/− mice during super-infection in association with an attenuated number of iNOS+Arg1+ dual function macrophages. Ultimately, bacterial control in Stat2−/− mice during super-infection was compromised when the number of iNOS+Arg1+ dual function macrophages was diminished. Further, we confirmed this by decreased bacterial clearance upon both Arg1 and IFNγ neutralization in both WT and Stat2−/− mice.
Further, BMC studies showed increased bacterial control in both Stat2−/− BMC and Stat2−/− hematopoietic BMC mice during influenza-bacterial infection, confirming the role of macrophage STAT2 in suppressing bacterial control during influenza-bacterial super-infection. However, bacterial control in non-hematopoietic Stat2−/− mice is likely due to altered macrophage phenotype, as influenza-induced ISGs are reduced in these mice. Further, increased influenza viral burden in non-hematopoietic Stat2−/− mice indicated a role for stromal STAT2 signaling in inducing ISGs. Collectively, these data indicate a cell intrinsic role for STAT2 signaling in macrophages and an accessory role for stromal STAT2 signaling via regulation of ISG expression.
In addition to quantitative differences in critical macrophage phenotypes, we have demonstrated that macrophages from Stat2−/− mice showed important qualitative differences compared to WT cells. We demonstrated that BMDM from Stat2−/− mice had improved MRSA uptake at baseline. This increased efficiency was unaffected by any infection challenge. Increased uptake was associated with expression of multiple C-type lectin receptors (CLRs), which are primarily expressed in monocytes in the lung, and are involved in phagocytosis of a variety of pulmonary pathogens (57, 58). However, these receptors favor the entry of influenza by acting as a receptor for viral attachment (59, 60). In this study, we found increased expression of CLRs and increased phagocytic ability in BMDMs from Stat2−/− mice. These results suggest that, even though these receptors favor influenza infection, they may help control secondary bacterial infection in Stat2−/− mice by improving bacterial uptake. In addition to increased bacterial uptake, BMDM from Stat2−/− mice displayed increased bacterial killing compared to WT mice upon IFNγ treatment. Naive BMDMs from Stat2−/− mice also displayed increased Nos2 and Cxcl9 expression compared to cells from WT mice, suggesting that Stat2−/− macrophages have inherently increased expression of Nos2 and enhanced bacterial killing ability compared to WT macrophages.
In summary, we have shown that STAT2 signaling decreases influenza viral burden and inflammatory immune responses during influenza infection, at the cost of inhibiting bacterial control during subsequent bacterial challenge by suppressing a distinct M1/M2 dual function macrophage population during influenza-bacterial super-infection. Together our data show a novel role of influenza induced type I IFN-mediated STAT2 signaling in inhibiting bacterial control through suppression of macrophage activity during influenza and influenza/MRSA super-infection. STAT2 and dual function M1/M2 macrophage activation may be a potential target for the treatment or prevention of influenza-bacterial super-infection.
All mouse experiments were approved by the University of Pittsburgh IACUC, Protocol #: 17071194; PHS Assurance Number: D16-00118.
RG, BL, JR-M, and JA designed the experiments. RG, BL, KM, HR, KR, SM, MC, PS, RE, MM, KMR, and JR-M performed the experiments. RG, BL, JR-M, and JA performed the analyses. RG, BL, HR, MM, KMR, JR-M, and JA drafted and edited the manuscript.
This work was supported by NIH R01HL107380 (JA) and T32 HD071834 (BL).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2018.02151/full#supplementary-material
FIGURE S1. No differences in Th17 response between WT and Stat2−/− mice following MRSA, and influenza-MRSA super-infection. WT or Stat2−/− mice were infected with 100 PFU of influenza A/PR/8/H1N1 or PBS by oropharyngeal aspiration for 6 days then challenged with 5 × 107 cfu of MRSA or PBS by oropharyngeal aspiration for one additional day. Frequency of (A) IL-17+ or (B) IL-22+ cells were determined by flow cytometry. The percentage of IL-17+ and IL-22+ cells were calculated from the percentage of CD3+CD4+ T cells. WT or Stat2−/− mice were infected with influenza-MRSA as described above. Lung lobes were homogenized with PBS, and the levels of (C) IL-23, (D) IL-17 were measured by Luminex analysis, and (E) IL-22 levels measured by ELISA. Data are represented as mean ± SEM. Data analyzed using two-tailed Student's t-test, ns, not significant.
FIGURE S2. Stat2−/− mice have altered other STAT1 and STA6 signaling pathways during influenza-bacterial super-infection. WT or Stat2−/− mice were infected with either 100 PFU of influenza A PR/8/H1N1 or influenza for 6 days and then challenged with 5 × 107 cfu of MRSA by oropharyngeal aspiration for one additional day, (A) Stat1, (B) Stat6, (C) Pparg gene expression relative to Hprt was analyzed by RT-PCR. N = 4 per group. Data are represented as mean ± SEM. Data analyzed using two-tailed Student's t-test, *p ≤ 0.05, ns, not significant.
FIGURE S3. M1 and M2 macrophages are altered in Stat2−/− mice in response to Arg1 and IFNγ neutralization during influenza-MRSA super-infection. WT or Stat2−/− mice were infected with 100 PFU of influenza for 6 days then challenged with 5 × 107 cfu of MRSA for one additional day. Mice were treated with 300 μg of anti-IFNγ antibodies or 300 μg of rat IgG isotype controls as described in methods. (A) Arg1+F4/80+ producing cells, were determined by IHC, N = 4–13 per group. WT or Stat2−/− mice were infected with 100 PFU of influenza for 6 days then challenged with 5 × 107 cfu of MRSA for one additional day. Mice were treated with Vehicle (DMSO) or N-hydroxy-nor-L-arginine (nor-NOHA) as described in methods. (B) iNOS+F4/80+ cells, N = 6–9 per group. Data are represented as mean ± SEM. Data analyzed using two-tailed Student's t-test, *p ≤ 0.05, **p ≤ 0.01, ns, not significant.
FIGURE S4. Stat2−/− mice have deficient type I IFN responses with preserved type II IFN responses. (A) WT and Stat2−/− male 6–8 weeks mice were infected with 100 PFU of influenza A/PR/8/H1N1 by oropharyngeal aspiration for 6 days then challenged with 5 × 107 cfu of MRSA by oropharyngeal aspiration for one additional day. Overall gene expression was measured in lung by RNAseq analysis, Heat-map representing the fold induction of Interferon stimulated genes (ISGs) from WT and Stat2−/− mice following influenza-MRSA super-infection, N = 4 per group. WT or Stat2−/− mice were infected with either 100 PFU of influenza A PR/8/H1N1 or influenza for 6 days and then challenged with 5 × 107 cfu of MRSA by oropharyngeal aspiration for one additional day, (B) Mx1, (C) Lhx2, and (D) Ccl12, (E) Cxcl9 gene expression relative to Hprt was analyzed by RT-PCR. N = 3–4 per group. Bone marrow-derived dendritic cells (1 × 106) were cultured in vitro and stimulated in vitro with IFNβ (6.5 units/mL) or IFNγ (20 ng/mL) for 24 h, (F) Mx1, (G) Ccl12 gene expression in cell RNA was analyzed by qRT-PCR. The cells were treated in triplicates. Each data point from panel F and G were the number of replicates per treatment. Data are represented as mean ± SEM. Data analyzed using two-tailed Student's t-test, *p ≤ 0.05, **p ≤ 0.01, ns, not significant.
FIGURE S5. Increased influenza burden in cells from non-hematopoietic compartments of Stat2−/− mice. WT BMC (Thy1.1 host, Thy 1.2 BM or Thy 1.2 host, Thy 1.1 BM), Stat2−/− BMC (Stat2−/− host, Stat2−/− BM), Hematopoietic Stat2−/− BMC mice (Thy 1.1 or Thy 1.2 host, Stat2−/− BM), non-hematopoietic Stat2−/− BMC (Stat2−/− host, Thy 1.1 or Thy 1.2 BM) were generated as described in methods. These mice were infected with 100 PFU of influenza for 6 days then challenged with 5 × 107 cfu of MRSA for one additional day. Mice were sacrificed and RNA expression of Influenza M protein was measured from the lungs by PCR, N = 6–9 per group. Data are represented as mean ± SEM. Data analyzed using One way ANOVA followed by Bonferroni test for multiple comparisons, **p ≤ 0.01, ns, not significant.
1. Simonsen L, Clarke MJ, Williamson GD, Stroup DF, Arden NH, Schonberger LB. The impact of influenza epidemics on mortality: introducing a severity index. Am J Public Health (1997) 87:1944–50.
2. Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol. (2014) 14:315–28. doi: 10.1038/nri3665
3. Wu S, Metcalf JP, Wu W. Innate immune response to influenza virus. Curr Opin Infect Dis. (2011) 24:235–40. doi: 10.1097/QCO.0b013e328344c0e3
4. Pulendran B, Maddur MS. Innate immune sensing and response to influenza. Curr Top Microbiol Immunol. (2015) 386:23–71. doi: 10.1007/82_2014_405
5. Durbin RK, Kotenko SV, Durbin JE. Interferon induction and function at the mucosal surface. Immunol Rev. (2013) 255:25–39. doi: 10.1111/imr.12101
6. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. (2005) 5:375–86. doi: 10.1038/nri1604
7. Durbin JE, Fernandez-Sesma A, Lee CK, Rao TD, Frey AB, Moran TM, et al. Type I IFN modulates innate and specific antiviral immunity. J Immunol. (2000) 164:4220–8. doi: 10.4049/jimmunol.164.8.4220
8. Garcia-Sastre A, Durbin RK, Zheng H, Palese P, Gertner R, Levy DE, et al. The role of interferon in influenza virus tissue tropism. J Virol. (1998) 72:8550–8.
9. Jewell NA, Cline T, Mertz SE, Smirnov SV, Flano E, Schindler C, et al. Lambda interferon is the predominant interferon induced by influenza A virus infection in vivo. J Virol. (2010) 84:11515–22. doi: 10.1128/JVI.01703-09
10. Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis. (2008) 198:962–70. doi: 10.1086/591708
11. Hers JF, Masurel N, Mulder J. Bacteriology and histopathology of the respiratory tract and lungs in fatal Asian influenza. Lancet. (1958) 2:1141–3.
12. Rothberg MB, Haessler SD. Complications of seasonal and pandemic influenza. Crit Care Med. (2010) 38(4 Suppl.):e91–7. doi: 10.1097/CCM.0b013e3181c92eeb
13. Kudva A, Scheller EV, Robinson KM, Crowe CR, Choi SM, Slight SR, et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J Immunol. (2011) 186:1666–74. doi: 10.4049/jimmunol.1002194
14. Lee B, Robinson KM, McHugh KJ, Scheller EV, Mandalapu S, Chen C, et al. Influenza-induced type I interferon enhances susceptibility to gram-negative and gram-positive bacterial pneumonia in mice. Am J Physiol Lung Cell Mol Physiol. (2015) 309:L158–67. doi: 10.1152/ajplung.00338.2014
15. Shahrara S, Pickens SR, Mandelin AM II, Karpus WJ, Huang Q, Kolls JK, et al. IL-17-mediated monocyte migration occurs partially through CC chemokine ligand 2/monocyte chemoattractant protein-1 induction. J Immunol. (2010) 184:4479–87. doi: 10.4049/jimmunol.0901942
16. Warnking K, Klemm C, Loffler B, Niemann S, van Kruchten A, Peters G, et al. Super-infection with Staphylococcus aureus inhibits influenza virus-induced type I IFN signalling through impaired STAT1-STAT2 dimerization. Cell Microbiol. (2015) 17:303–17. doi: 10.1111/cmi.12375
17. Lee B, Gopal R, Manni ML, McHugh KJ, Mandalapu S, Robinson KM, et al. STAT1 Is required for suppression of Type 17 immunity during influenza and bacterial superinfection. ImmunoHorizons (2017) 1:81–91. doi: 10.4049/immunohorizons.1700030
18. Park C, Li S, Cha E, Schindler C. Immune response in Stat2 knockout mice. Immunity. (2000) 13:795–804. doi: 10.1016/S1074-7613(00)00077-7
19. Curzer HJ, Perry G, Wallace MC, Perry D. The three Rs of animal research: what they mean for the institutional animal care and use committee and why. Sci Eng Ethics (2016) 22:549–65. doi: 10.1007/s11948-015-9659-8
20. Braciale TJ. Immunologic recognition of influenza virus-infected cells. II. Expression of influenza A matrix protein on the infected cell surface and its role in recognition by cross-reactive cytotoxic T cells. J Exp Med. (1977) 146:673–89.
21. Robinson KM, McHugh KJ, Mandalapu S, Clay ME, Lee B, Scheller EV, et al. Influenza A virus exacerbates Staphylococcus aureus pneumonia in mice by attenuating antimicrobial peptide production. J Infect Dis. (2014) 209:865–75. doi: 10.1093/infdis/jit527
22. Robinson KM, Choi SM, McHugh KJ, Mandalapu S, Enelow RI, Kolls JK, et al. Influenza A exacerbates Staphylococcus aureus pneumonia by attenuating IL-1beta production in mice. J Immunol. (2013) 191:5153–9. doi: 10.4049/jimmunol.1301237
23. Gopal R, Khader SA. Vaccines against tuberculosis: moving forward with new concepts. Expert Rev Vaccines (2013) 12:829–31. doi: 10.1586/14760584.2013.814836
24. Eddens T, Elsegeiny W, Nelson MP, Horne W, Campfield BT, Steele C, et al. Eosinophils contribute to early clearance of pneumocystis murina infection. J Immunol. (2015) 195:185–93. doi: 10.4049/jimmunol.1403162
25. Crowe CR, Chen K, Pociask DA, Alcorn JF, Krivich C, Enelow RI, et al. Critical role of IL-17RA in immunopathology of influenza infection. J Immunol. (2009) 183:5301–10. doi: 10.4049/jimmunol.0900995
26. Gopal R, Monin L, Slight S, Uche U, Blanchard E, Fallert Junecko BA, et al. Unexpected role for IL-17 in protective immunity against hypervirulent Mycobacterium tuberculosis HN878 infection. PLoS Pathog. (2014) 10:e1004099. doi: 10.1371/journal.ppat.1004099
27. Slight SR, Rangel-Moreno J, Gopal R, Lin Y, Fallert Junecko BA, Mehra S, et al. CXCR5(+) T helper cells mediate protective immunity against tuberculosis. J Clin Invest. (2013) 123:712–26. doi: 10.1172/JCI65728
28. Kolls JK, Khader SA. The role of Th17 cytokines in primary mucosal immunity. Cytokine Growth Factor Rev. (2010) 21:443–8. doi: 10.1016/j.cytogfr.2010.11.002
29. Witowski J, Pawlaczyk K, Breborowicz A, Scheuren A, Kuzlan-Pawlaczyk M, Wisniewska J, et al. IL-17 stimulates intraperitoneal neutrophil infiltration through the release of GRO alpha chemokine from mesothelial cells. J Immunol. (2000) 165:5814–21. doi: 10.4049/jimmunol.165.10.5814
30. Gordon S. Alternative activation of macrophages. Nat Rev Immunol. (2003) 3:23–35. doi: 10.1038/nri978
31. Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. (2006) 177:7303–11. doi: 10.4049/jimmunol.177.10.7303
32. Ho VW, Sly LM. Derivation and characterization of murine alternatively activated (M2) macrophages. Methods Mol Biol. (2009) 531173–85. doi: 10.1007/978-1-59745-396-7_12
33. Crawford MA, Burdick MD, Glomski IJ, Boyer AE, Barr JR, Mehrad B, et al. Interferon-inducible CXC chemokines directly contribute to host defense against inhalational anthrax in a murine model of infection. PLoS Pathog. (2010) 6:e1001199. doi: 10.1371/journal.ppat.1001199
34. Colvin RA, Campanella GS, Sun J, Luster AD. Intracellular domains of CXCR3 that mediate CXCL9, CXCL10, and CXCL11 function. J Biol Chem. (2004) 279:30219–27. doi: 10.1074/jbc.M403595200
35. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. (2014) 6:13. doi: 10.12703/P6-13
36. Raes G, Brys L, Dahal BK, Brandt J, Grooten J, Brombacher F, et al. Macrophage galactose-type C-type lectins as novel markers for alternatively activated macrophages elicited by parasitic infections and allergic airway inflammation. J Leukoc Biol. (2005) 77:321–7. doi: 10.1189/jlb.0304212
37. Li W, Katz BP, Spinola SM. Haemophilus ducreyi-induced interleukin-10 promotes a mixed M1 and M2 activation program in human macrophages. Infect Immun. (2012) 80:4426–34. doi: 10.1128/IAI.00912-12
38. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. (2011) 11:723–37. doi: 10.1038/nri3073
39. Hesse M, Modolell M, La Flamme AC, Schito M, Fuentes JM, Cheever AW, et al. Differential regulation of nitric oxide synthase-2 and arginase-1 by type 1/type 2 cytokines in vivo: granulomatous pathology is shaped by the pattern of L-arginine metabolism. J Immunol. (2001) 167:6533–44. doi: 10.4049/jimmunol.167.11.6533
40. Murphy RF. Analysis and isolation of endocytic vesicles by flow cytometry and sorting: demonstration of three kinetically distinct compartments involved in fluid-phase endocytosis. Proc Natl Acad Sci USA. (1985) 82:8523–6. doi: 10.1073/pnas.82.24.8523
41. Sokolovska A, Becker CE, Stuart LM. Measurement of phagocytosis, phagosome acidification, and intracellular killing of Staphylococcus aureus. Curr Protoc Immunol. (2012) Chapter 14:Unit14.30. doi: 10.1002/0471142735.im1430s99
42. Liu SY, Sanchez DJ, Aliyari R, Lu S, Cheng G. Systematic identification of type I and type II interferon-induced antiviral factors. Proc Natl Acad Sci USA. (2012) 109:4239–44. doi: 10.1073/pnas.1114981109
43. Schoggins JW, Rice CM. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol. (2011) 1:519–25. doi: 10.1016/j.coviro.2011.10.008
44. Dussurget O, Bierne H, Cossart P. The bacterial pathogen Listeria monocytogenes and the interferon family: type I, type II and type III interferons. Front Cell Infect Microbiol. (2014) 4:50. doi: 10.3389/fcimb.2014.00050
45. McHugh KJ, Mandalapu S, Kolls JK, Ross TM, Alcorn JF.A novel outbred mouse model of 2009 pandemic influenza and bacterial co-infection severity.PLoS ONE (2013) 8:e82865. doi: 10.1371/journal.pone.0082865
46. Rynda-Apple A, Robinson KM, Alcorn JF. Influenza and bacterial superinfection: illuminating the immunologic mechanisms of disease. Infect Immun. (2015) 83:3764–70. doi: 10.1128/IAI.00298-15
47. Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. (2009) 27:451–83. doi: 10.1146/annurev.immunol.021908.132532
48. Waite KJ, Floyd ZE, Arbour-Reily P, Stephens JM. Interferon-gamma-induced regulation of peroxisome proliferator-activated receptor gamma and STATs in adipocytes. J Biol Chem. (2001) 276:7062–8. doi: 10.1074/jbc.M007894200
49. Qiao Y, Kang K, Giannopoulou E, Fang C, Ivashkiv LB. IFN-gamma induces histone 3 lysine 27 trimethylation in a small subset of promoters to stably silence gene expression in human macrophages. Cell Rep. (2016) 16:3121–9. doi: 10.1016/j.celrep.2016.08.051
50. Chen WH, Toapanta FR, Shirey KA, Zhang L, Giannelou A, Page C, et al. Potential role for alternatively activated macrophages in the secondary bacterial infection during recovery from influenza. Immunol Lett. (2012) 141:227–34. doi: 10.1016/j.imlet.2011.10.009
51. Wang J, Li F, Sun R, Gao X, Wei H, Li LJ, et al. Bacterial colonization dampens influenza-mediated acute lung injury via induction of M2 alveolar macrophages. Nat Commun. (2013) 42106. doi: 10.1038/ncomms3106
52. Patil V, Zhao Y, Shah S, Fox BA, Rommereim LM, Bzik DJ, et al. Co-existence of classical and alternative activation programs in macrophages responding to Toxoplasma gondii. Int J Parasitol. (2014) 44:161–4. doi: 10.1016/j.ijpara.2013.08.003
53. Sun K, Metzger DW. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat Med. (2008) 14:558–64. doi: 10.1038/nm1765
54. Ellis TN, Beaman BL. Interferon-gamma activation of polymorphonuclear neutrophil function. Immunology (2004) 112:2–12. doi: 10.1111/j.1365-2567.2004.01849.x
55. Bovolenta C, Gasperini S, McDonald PP, Cassatella MA. High affinity receptor for IgG (Fc gamma RI/CD64) gene and STAT protein binding to the IFN-gamma response region (GRR) are regulated differentially in human neutrophils and monocytes by IL-10. J Immunol. (1998) 160:911–9.
56. Hansen BD, Finbloom DS. Characterization of the interaction between recombinant human interferon-gamma and its receptor on human polymorphonuclear leukocytes. J Leukoc Biol. (1990) 47:64–9. doi: 10.1002/jlb.47.1.64
57. Cambi A, Netea MG, Mora-Montes HM, Gow NA, Hato SV, Lowman DW, et al. Dendritic cell interaction with Candida albicans critically depends on N-linked mannan. J Biol Chem. (2008) 283:20590–9. doi: 10.1074/jbc.M709334200
58. Tailleux L, Pham-Thi N, Bergeron-Lafaurie A, Herrmann JL, Charles P, Schwartz O, et al. DC-SIGN induction in alveolar macrophages defines privileged target host cells for mycobacteria in patients with tuberculosis. PLoS Med. (2005) 2:e381. doi: 10.1371/journal.pmed.0020381
59. Ng WC, Londrigan SL, Nasr N, Cunningham AL, Turville S, Brooks AG, et al. The C-type lectin langerin functions as a receptor for attachment and infectious entry of influenza a virus. J Virol. (2015) 90:206–21. doi: 10.1128/JVI.01447-15
Keywords: influenza, Staphylococcus aureus, super-infection, STAT2, macrophages, lung, pneumonia
Citation: Gopal R, Lee B, McHugh KJ, Rich HE, Ramanan K, Mandalapu S, Clay ME, Seger PJ, Enelow RI, Manni ML, Robinson KM, Rangel-Moreno J and Alcorn JF (2018) STAT2 Signaling Regulates Macrophage Phenotype During Influenza and Bacterial Super-Infection. Front. Immunol. 9:2151. doi: 10.3389/fimmu.2018.02151
Received: 12 April 2018; Accepted: 31 August 2018;
Published: 25 September 2018.
Edited by:
Laurel L. Lenz, University of Colorado, United StatesReviewed by:
Dane Parker, Columbia University, United StatesCopyright © 2018 Gopal, Lee, McHugh, Rich, Ramanan, Mandalapu, Clay, Seger, Enelow, Manni, Robinson, Rangel-Moreno and Alcorn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John F. Alcorn, am9obi5hbGNvcm5AY2hwLmVkdQ==
† These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.