 Redmond P. Smyth
Redmond P. Smyth Matteo Negroni
Matteo Negroni Andrew M. Lever4,5
Andrew M. Lever4,5 Julia C. Kenyon
Julia C. Kenyon
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 19 September 2018
Sec. Viral Immunology
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02097
This article is part of the Research TopicThe Past and the Future of Human Immunity under Viral Evolutionary PressureView all 15 articles
The central dogma of molecular biology describes the flow of genetic information from DNA to protein via an RNA intermediate. For many years, RNA has been considered simply as a messenger relaying information between DNA and proteins. Recent advances in next generation sequencing technology, bioinformatics, and non-coding RNA biology have highlighted the many important roles of RNA in virtually every biological process. Our understanding of RNA biology has been further enriched by a number of significant advances in probing RNA structures. It is now appreciated that many cellular and viral biological processes are highly dependent on specific RNA structures and/or sequences, and such reliance will undoubtedly impact on the evolution of both hosts and viruses. As a contribution to this special issue on host immunity and virus evolution, it is timely to consider how RNA sequences and structures could directly influence the co-evolution between hosts and viruses. In this manuscript, we begin by stating some of the basic principles of RNA structures, followed by describing some of the critical RNA structures in both viruses and hosts. More importantly, we highlight a number of available new tools to predict and to evaluate novel RNA structures, pointing out some of the limitations readers should be aware of in their own analyses.
Mutation rates of viral genomes are extremely high when compared with those of eukaryotic cells; RNA virus polymerases typically possess error rates of 10−4 to 10−6 per base (1). Such rapid mutation is a strategy by which they can evade host adaptive immune responses (2). Antiviral defenses of the innate immune system, which is less genetically flexible than the adaptive response, enable a very broad range of recognition from which it is difficult for viruses to escape, even given their high error rate. To counter the innate immune system, viruses have developed strategies to block its activation. This arms race prompts the immune system to develop counter measures to recognize and eliminate the virus, whilst viruses that survive and transmit successfully are those that have evolved to escape it. When considering the evolutionary pressures on both virus and host, research has often focused on the protein sequences needed by each. A recent explosion in our understanding of the functions of RNA, however, leads us to consider instead the role of RNA itself in driving the evolution of viruses and of human immunity.
RNA is a truly multifunctional molecule. It directs ribosomes (themselves RNA based enzymes) to produce proteins, but also regulates cellular activity by interacting directly with proteins or nucleic acids and by catalyzing biochemical reactions (ribozymes). Indeed, RNA is now implicated in almost every cellular process, including immune defense. It is also recognized as being key to viral infection processes. RNA multifunctionality comes from its ability to fold into complex three-dimensional structures that can often switch conformation to effect different functions such as binding other RNA molecules or proteins. The initial fold of an RNA molecule depends primarily on its sequence and is established by Watson-Crick pairing of complementary bases into stem-loop structures that then orientate themselves relative to one another [for a general review of RNA structure see (3)]. This three-dimensional positioning can be stabilized by non-canonical interactions or structures such as pseudoknots, which occur where the nucleotides of a loop region base pair intramolecularly with complementary nucleotides. An example is shown in Figure 1A, within the complex structural element known as an IRES (internal ribosome entry site). As for proteins, single nucleotide mutations can alter the three-dimensional structure of the RNA, with corresponding deleterious or positive effects on its function; RNA structures are hence substrates for, and drivers of, viral evolution. For example, random mutation may confer a new beneficial function on a given structure that is then selectively favored by evolution.
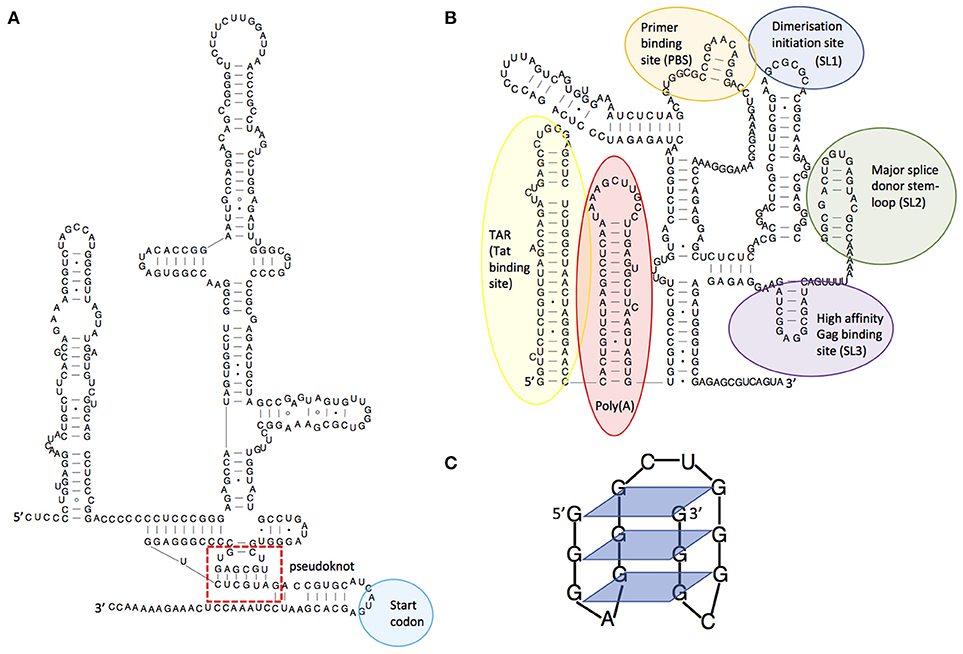
Figure 1. Examples of RNA structures. (A) Structural diagram of the hepatitis C virus IRES structure, showing its pseudoknot. Adapted from Malygin et al.(4), (B) Structural model of the HIV-1 5'leader RNA, highlighting some of the important RNA structures it contains. Adapted from Kenyon et al. (5), (C) An RNA guanine quadruplex structure, showing the 4-way bonds between guanines in each plane. Adapted from Frees et al. (6).
Viruses are known to be extremely thrifty with their genomes to maximize replication speed, using strategies such as overlapping reading frames and polycistronic mRNAs. Similarly, they often contain functional RNA structures within both coding and non-coding regions. For example, the first 500 nucleotides of the HIV-1 genome is densely packed with structured domains that control key steps of the replication cycle including transcription, translation, export, packaging, and reverse transcription (7–9) and structural switches that aid their regulation (5, 10–12). Functionality encoded within an RNA structure is often a requisite for successful initiation or completion of viral replication. Many viruses, particularly those of the picornaviridae, initiate translation from IRES elements (13). RNA structures facilitating frameshifting enable viruses to encode multiple proteins from a single RNA (14). Additionally, viral RNA structures may directly or indirectly impact cellular immunity. In HIV-1, the transactivation response element (TAR, Figure 1B) regulates transcription of the genomic RNA and gene expression (15) thereby playing a central role in determining the level of virus detected by the immune system; at its most extreme leading to complete evasion of the immune response through latency (16). Frameshift structures or splicing regulators qualitatively and quantitatively manage the amounts of proteins produced by viruses, and hence those that are seen by the immune system (17). For example, some strains of Influenza A virus encode a second open reading frame (ORF) in segment three which is accessed by a low frequency +1 ribosome frameshifting event (18). This ORF produces PA-X, a protein which modulates inflammatory, apoptotic, and T-lymphocyte signaling pathways (18). Viruses with larger genomes can even produce their own microRNAs (19) or long non-coding RNAs (lncRNAs) that control cellular functions (20, 21). Dengue virus expresses a subgenomic RNA that has been shown to inhibit interferon expression by binding to TRIM25 (22). This subgenomic RNA is produced when a cellular 5′-to-3′ exoribonuclease stalls at a stable pseudoknot RNA structure in the 3′UTR; small substitutions within this structure modulate viral fitness and pathogenicity through their effects on the immune system. The dengue 3'UTR also folds differently in humans and insects (23) leading to production of different immunomodulatory non-coding RNAs in each host type (22, 23). This is one mechanism by which the same viral genome can both effectively replicate in human and insect cells and counteract these two divergent immune systems.
The immune system senses viral RNA using different mechanisms, including the recognition of viral single-stranded RNA by Toll-like receptors (24). The importance of RNA structures in viral replication is so fundamental that they are directly recognized by the innate immune response. Not only has the innate immune system evolved to recognize double-stranded RNA (dsRNA) within viral genomes or genomic replication intermediates, often via MDA5, it has also evolved to recognize the double-stranded parts of conserved RNA elements like IRESs (13) or 3' stem-loops, via RIG-I [reviewed in (25)]. The importance of this in maintaining broad antiviral defense is reflected in the fact that such RNA structures are also formed by DNA viruses in their protein-coding RNAs. The cellular double-stranded RNA recognition system not only leads to production of interferon, but was recently shown to upregulate NKG2D ligand, thus alerting NK cells to the presence of virus and enabling destruction of the infected cell (26). Viruses have evolved strategies to mask recognition of their RNAs, such as 5' cap-snatching (27), but their need to maintain certain critical RNA structures means that they struggle to avoid recognition entirely.
The presence of essential conserved viral RNA structures can constrain the ability of viruses to evolve and to evade the immune response. Some RNA viruses have optimized their genome structure and organization to facilitate viral evolution during co-infection of the same cell with different viral strains. One widespread strategy is genome segmentation leading to reassortment (seen in rotaviruses and influenza viruses) (28). Another common strategy is template switching during replication leading to recombination and the formation of genome chimeras (seen in retroviruses) (29). Reassortment and recombination are non-random processes that are known to depend on RNA sequence and structure, but the underlying mechanisms are often poorly understood. Both processes may be facilitated by inter-molecular interactions (30, 31), and evolution can be both promoted or inhibited by intra-molecular RNA structure.
In HIV-1, each virion contains two copies of the RNA genome, which in the case of co-infection of the same cell can originate from two different proviruses. These are non-covalently joined as a dimer via stem-loop 1 (SL1, Figure 1B). During viral replication, reverse transcriptase (RT) switches template, thereby adding template switching generated errors to its inherently low fidelity, producing the genetic diversity that allows HIV-1 to rapidly escape the immune system and antiretroviral therapy (2). Sequence incompatibility preventing formation of heterodimers of genomic RNA at SL1 has been shown to be a major restriction to inter-subtype recombination, with only a 3-nt sequence difference being sufficient to disrupt this (32). When considering distantly-related strains of HIV with compatible SL1 sequences, the main factor governing recombination is the degree of local sequence similarity (33, 34) However, in more closely-related viral sequences, RNA structures strongly influence recombination locally (35–37). This has been shown to be the case for well-defined RNA structures within env such as the C2 hairpin and the Rev responsive element (RRE) (38). It has been suggested that this evolved to favor the stepwise folding of proteins during translation, but it has also been shown to favor the occurrence of recombination in these same regions (37). As a consequence, recombination may shuffle whole domains of proteins thus generating structural variants that escape immune recognition, particularly for quaternary epitopes generated by the juxtaposition of different protein domains. RNA structures and sequences have also been shown to influence the fidelity of reverse transcription such that stable secondary structures enhance the number and type of mutations incorporated (39, 40). The regions of env encoding the external parts of the viral surface glycoprotein gp120 are under strong positive selection by the humoral immune system. Perhaps counterintuitively they present a lower degree of RNA structure; however, this can be accounted for by such rapid viral mutation that the RNA is unable to conserve base-pairing. As studies have shown, poorly structured RNA regions are reverse transcribed with higher fidelity, which paradoxically limits the rate of introduction of mutations in these highly variable regions of the genome (39).
RNA viruses with segmented genomes can undergo reassortment, leading to the exchange of entire gene segments, potentially giving rise to new viral strains to which humans have no previous immunity. In influenza A, reassortment has been historically associated with the emergence of pandemic strains, including the most recent H1N1 2009 pandemic which contained influenza gene segments from human, avian and swine lineages (41, 42). It is thought that packaging sequences within each gene segment direct their selective incorporation into virus particles (43, 44) and recent work suggests a mechanism where packaging signals mediate RNA-RNA interactions that would guide their incorporation. It therefore follows that any preferential interactions or incompatibilities between vRNP segments would then regulate genetic reassortment and influenza evolution (45). It is possible that improved understanding of this process would help to better predict the emergence of pandemic influenza.
In addition to genome diversification through recombination and reassortment, RNA structures influence immune escape by modulating the viral proteome. The use of frameshifting or alternative start codons is often controlled by viral RNA structures, resulting in the translation of viral peptides or proteins in a different reading frame. The resulting peptides are often antigenic and may act to dilute the presentation of conventional viral peptides on the surface of the infected cell (46). In some viruses there is an evolutionary constraint to maintain translation-impeding RNA structures in genes that encode good T cell epitopes, thus maintaining their translation at levels too low to trigger T cell recognition and killing. The EBNA-1 RNA from Epstein-Barr virus for example regulates its own translation in cis (47) and the evolutionary pressure on this gene comes from the need to maintain G-quadruplex RNA structures (Figure 1C) that act as “steric blocks” to ribosomes. When these structures are destabilized, cells infected with the resulting mutant virus are more readily seen by T cells than those infected with wild-type virus (48).
Viruses also appear to be under pressure to maintain unstructured regions in their genome: there is a bias in HIV-1 toward the use of A's in the retroviral genome; this biases codon usage and ultimately even the amino acid composition of the viral proteins (49). Adenosines are vastly overrepresented in the single-stranded regions of RNA and underrepresented in double-stranded regions; their only binding partner, U, can also pair with G, which may explain the single-stranded nature of A-rich regions. Artificial introduction of extensive synonymous A to G mutations in pol led to increased stability of the dimeric genome inside the virion, and reduced reverse transcription as a result (50). The signature distribution of Adenosine frequency and its relation to local RNA structure was thought to be maintained by the influence of RNA secondary structures on reverse transcription. Changing the A ratio in local areas by only including codons found in natural isolates of HIV-1, did not affect replication efficiency in vitro (51) however it is possible that in vivo viruses use this strategy of maintaining parts of their genome as single-stranded in order to avoid innate immune recognition by RIG-I or MDA5. Many viruses also need to maintain a low number of CpG dinucleotides in their genome in order to avoid recognition by the zinc-finger antiviral protein (ZAP) (52). When HIV-1 codon usage was humanized, affecting the native RNA structure, a reduced IFN-α/β response was observed (53), suggesting that the maintenance of specific structures in the viral genome comes at the cost of greater recognition by the innate immune response. Despite this, the viral genome apparently undergoes positive selection for the maintenance of many specific RNA structures: when synonymous mutations were introduced extensively into the viral genome a decrease in infectivity was observed that could be attributed to an expected alteration of splicing pattern and/or modification of RNA structures (50, 54).
In terms of the evolution of the human genome, the degree of variation at the MHC class I locus is positively correlated with local pathogen richness, for which viruses are postulated to play an important role. This is particularly evident for HLA B (55). The importance of RNA structure in influencing the generation of T cell epitopes, either through translational enhancing, blocking, or frameshifting mechanisms, means that RNA structures within viruses must have influenced the evolution of the HLA locus. For example, macaques that have the correct MHC-I allele to present an antigenic cryptic peptide derived from the env ORF are better able to control simian immunodeficiency virus (SIV) infection (56) which would be expected to be a driver for the maintenance of this MHC allele within the population. As mentioned above, the mechanisms controlling the generation of cryptic translational products are often RNA structure-dependent.
Viral RNA structures have also influenced the evolution of the innate immune system; as previously discussed, hallmarks of viral RNAs are targeted by conserved RNA-binding proteins such as RIG-I. More specific antiviral proteins have also evolved, however. APOBEC3 proteins target retroviral genomes and are incorporated into viral particles (57). These host cell-derived cytidine deaminases bind to the viral RNA and mutate it during the reverse transcription process, leading to non-functional virus. It has been reported that regions of the genome under strong purifying selection present an underrepresentation of APOBEC3 target sequences, a signature of a strong pressure for limiting the occurrence of mutations in certain regions of the genome (39). Retroviruses have also developed direct strategies to counteract these proteins, often by encoding proteins that bind to them directly. Interestingly, APOBEC3s have recently been shown to bind to the same motifs in the viral RNA genome as the viral NC structural protein involved in genome packaging. This suggests a competitive relationship may have developed between host cell and viral factors, for binding to the same viral RNA structures (58).
RNA functionality is best understood through its structure, but RNA structure determination is extremely challenging. Although it is formed based on simple base pairing rules, for RNA molecules of biologically relevant sizes there are an astronomical number of possible structural permutations, meaning that RNA structure cannot be predicted easily from base pairing rules alone. Biophysical methods such as crystallography (59) and NMR (60) are each able to determine RNA structure at atomic resolution but both have difficulty with large RNA substrates. This is evidenced by the paucity of atomic resolution RNA structures, compared to their protein equivalents. For example, the RCSB databank holds structural data for over 100,000 proteins, but contains only around 1000 RNA structures. This difficulty arises because RNA molecules tend to adopt long flexible shapes with weak tertiary interactions that are prone to misfolding (61). Furthermore, the negatively charged phosphate groups on the surface of an RNA molecule can impose technical challenges as they hinder crystal packing. Newer techniques are emerging to address this gap including small-angle X-ray scattering (62), single molecule FRET (63, 64), and atomic force microscopy (65).
RNA secondary structure is currently most commonly resolved using a combination of (i) phylogenetic approaches (ii), structure prediction algorithms, and (iii) experimental methods with chemical/enzymatic probes. Whilst these methodologies cannot determine RNA structure at atomic resolution, they are nevertheless able to generate models that provide useful biological insights (8, 66, 67). Indeed, RNA structure determination is currently undergoing a revolution thanks to advances in next generation sequencing technology that have transformed traditional biochemical assays into powerful tools that can characterize thousands of RNA structures in single experiments (68–71). The most widely used methods take advantage of chemical probes, such as dimethyl sulfate (DMS) and selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) reagents, that differentially react with single stranded vs double stranded RNA. Knowledge of whether a nucleotide is likely to be base paired or not can significantly improve the accuracy of RNA structure predictions from thermodynamic folding algorithms when included as an energetic consideration in the modeling programme, known as a pseudo free energy parameter (72). For example, if chemical probes show a nucleotide is single-stranded, the modeling algorithm adds an energetic penalty to structures that include it in a double-stranded region. The programme then displays the most energetically favorable structures, that fit all of the data best. Several chemical probes can penetrate cells and virions, which is important for understanding RNA function in vivo, such as the binding sites of regulatory proteins (9, 73). Further characterization of RNA structure-function relationships can be obtained using specialized approaches, such as mutational interference mapping experiment (MIME) (74) and cross-linking SHAPE (XL-SHAPE), where protein binding sites are mapped using UV cross-linking in parallel with SHAPE probing (75), and by CLIP (crosslinking-immunoprecipitation sequencing) related methodologies (76). More recently, RNA proximity ligation has emerged as a new class of RNA structural probing technique for the direct detection of long-range base pairing or inter-molecular interactions (77–84). These types of interactions are commonly found in viral genomes/regulatory RNAs and are difficult to identify with other methodologies. As the immune system is known to be regulated by non-coding RNAs (85), the ability to detect direct interactions between viral and cellular RNAs will be particularly important for future understanding of virus-host interactions.
JM conceived the review. RS, MN, AL, JM, and JK wrote the manuscript.
This work was supported by the Helmholtz Association (VH-NG-1347 to RS), Sidaction (AI25-1-02335 to MN), The Biomedical Research Centre UK and Clinical Academic Reserve UK (to AL), and the Medical Research Council UK (MR/N022939/1 to AL and JK). JM is recipient of funding from Australian National Health and Medical Research Council project grant App1121697.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Sanjuan R, Domingo-Calap P. Mechanisms of viral mutation. Cell Mol Life Sci. (2016) 73:4433–48. doi: 10.1007/s00018-016-2299-6
2. Smyth RP, Davenport MP, Mak J. The origin of genetic diversity in HIV-1. Virus Res. (2012) 169:415–29. doi: 10.1016/j.virusres.2012.06.015
3. Batey RT, Rambo RP, Doudna JA. Tertiary motifs in RNA structure and folding. Angew Chem Int Ed Engl. (1999) 38:2326–43.
4. Malygin AA, Kossinova OA, Shatsky IN, Karpova GG. HCV IRES interacts with the 18S rRNA to activate the 40S ribosome for subsequent steps of translation initiation. Nucleic Acids Res. (2013) 41:8706–14. doi: 10.1093/nar/gkt632
5. Kenyon JC, Prestwood LJ, Le Grice SF, Lever AM. In-gel probing of individual RNA conformers within a mixed population reveals a dimerization structural switch in the HIV-1 leader. Nucleic Acids Res. (2013) 41:e174. doi: 10.1093/nar/gkt690
6. Frees S, Menendez C, Crum M, Bagga PS. QGRS-Conserve: a computational method for discovering evolutionarily conserved G-quadruplex motifs. Hum Genomics (2014) 8:8. doi: 10.1186/1479-7364-8-8
7. Paillart JC, Dettenhofer M, Yu XF, Ehresmann C, Ehresmann B, Marquet R. First snapshots of the HIV-1 RNA structure in infected cells and in virions. J Biol Chem. (2004) 279:48397–403. doi: 10.1074/jbc.M408294200
8. Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW Jr, Swanstrom R, et al. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature (2009) 460:711–6. doi: 10.1038/nature08237
9. Wilkinson KA, Gorelick RJ, Vasa SM, Guex N, Rein A, Mathews DH, et al. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol. (2008) 6:e96. doi: 10.1371/journal.pbio.0060096
10. Keane SC, Van V, Frank HM, Sciandra CA, McCowin S, Santos J, et al. NMR detection of intermolecular interaction sites in the dimeric 5'-leader of the HIV-1 genome. Proc Natl Acad Sci USA. (2016) 113:13033–8. doi: 10.1073/pnas.1614785113
11. Mailler E, Bernacchi S, Marquet R, Paillart JC, Vivet-Boudou V, Smyth RP. The Life-cycle of the HIV-1 gag-RNA complex. Viruses (2016) 8:E248. doi: 10.3390/v8090248
12. Lu K, Heng X, Garyu L, Monti S, Garcia EL, Kharytonchyk S, et al. NMR detection of structures in the HIV-1 5'-leader RNA that regulate genome packaging. Science (2011) 334:242–5. doi: 10.1126/science.1210460
13. Fernandez N, Buddrus L, Pineiro D, Martinez-Salas E. Evolutionary conserved motifs constrain the RNA structure organization of picornavirus IRES. FEBS Lett. (2013) 587:1353–8. doi: 10.1016/j.febslet.2013.03.005
14. Houck-Loomis B, Durney MA, Salguero C, Shankar N, Nagle JM, Goff SP, et al. An equilibrium-dependent retroviral mRNA switch regulates translational recoding. Nature (2011) 480:561–4. doi: 10.1038/nature10657
15. Kao SY, Calman AF, Luciw PA, Peterlin BM. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product Nature (1987) 330:489–93.
16. Ruelas DS, Greene WC. An integrated overview of HIV-1 latency. Cell (2013) 155:519–29. doi: 10.1016/j.cell.2013.09.044
17. Napthine S, Ling R, Finch LK, Jones JD, Bell S, Brierley I, et al. Protein-directed ribosomal frameshifting temporally regulates gene expression. Nat Commun. (2017) 8:15582. doi: 10.1038/ncomms15582
18. Hayashi T, MacDonald LA, Takimoto T. Influenza A virus protein PA-X contributes to viral growth and suppression of the host antiviral and immune responses. J Virol. (2015) 89:6442–52. doi: 10.1128/JVI.00319-15
19. Pfeffer S, Zavolan M, Grasser FA, Chien M, Russo JJ, Ju J, et al. Identification of virus-encoded microRNAs. Science (2004) 304:734–6. doi: 10.1126/science.1096781
20. Reeves MB, Davies AA, McSharry BP, Wilkinson GW, Sinclair JH. Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science (2007) 316:1345–8. doi: 10.1126/science.1142984
21. Zhang X, Ma X, Jing S, Zhang H, Zhang Y. Non-coding RNAs and retroviruses. Retrovirology (2018) 15:20. doi: 10.1186/s12977-018-0403-8
22. Manokaran G, Finol E, Wang C, Gunaratne J, Bahl J, Ong EZ, et al. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science (2015) 350:217–21. doi: 10.1126/science.aab3369
23. Kieft JS, Rabe JL, Chapman EG. New hypotheses derived from the structure of a flaviviral Xrn1-resistant RNA: conservation, folding, and host adaptation. RNA Biol. (2015) 12:1169–77. doi: 10.1080/15476286.2015.1094599
24. Jensen S, Thomsen AR. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol. (2012) 86:2900–10. doi: 10.1128/JVI.05738-11
25. Kell AM, Gale M. RIG-I in RNA virus recognition. Virology (2015) 479–80:110–21. doi: 10.1016/j.virol.2015.02.017
26. Esteso G, Guerra S, Vales-Gomez M, Reyburn HT. Innate immune recognition of double-stranded RNA triggers increased expression of NKG2D ligands after virus infection. J Biol Chem. (2017) 292:20472–80. doi: 10.1074/jbc.M117.818393
27. Dias A, Bouvier D, Crepin T, McCarthy AA, Hart DJ, Baudin F, et al. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature (2009) 458:914–8. doi: 10.1038/nature07745
28. McDonald SM, Nelson MI, Turner PE, Patton JT. Reassortment in segmented RNA viruses: mechanisms and outcomes. Nat Rev Microbiol. (2016) 14:448–60. doi: 10.1038/nrmicro.2016.46
29. Levy DN, Aldrovandi GM, Kutsch O, Shaw GM. Dynamics of HIV-1 recombination in its natural target cells. Proc Natl Acad Sci USA. (2004) 101:4204–9. doi: 10.1073/pnas.0306764101
30. Gavazzi C, Yver M, Isel C, Smyth RP, Rosa-Calatrava M, Lina B, et al. A functional sequence-specific interaction between influenza A virus genomic RNA segments. Proc Natl Acad Sci USA. (2013) 110:16604–9. doi: 10.1073/pnas.1314419110
31. Paillart JC, Shehu-Xhilaga M, Marquet R, Mak J. Dimerization of retroviral RNA genomes: an inseparable pair. Nat Rev Microbiol. (2004) 2:461–72. doi: 10.1038/nrmicro903
32. Chin MP, Rhodes TD, Chen J, Fu W, Hu WS. Identification of a major restriction in HIV-1 intersubtype recombination. Proc Natl Acad Sci USA. (2005) 102:9002–7. doi: 10.1073/pnas.0502522102
33. Baird HA, Gao Y, Galetto R, Lalonde M, Anthony RM, Giacomoni V, et al. Influence of sequence identity and unique breakpoints on the frequency of intersubtype HIV-1 recombination. Retrovirology (2006) 3:91. doi: 10.1186/1742-4690-3-91
34. Magiorkinis G, Paraskevis D, Vandamme A-M, Magiorkinis E, Sypsa V, Hatzakis A. In vivo characteristics of human immunodeficiency virus type 1 intersubtype recombination: determination of hot spots and correlation with sequence similarity. J Gen Virol. (2003) 84:2715–22. doi: 10.1099/vir.0.19180-0
35. Baird HA, Galetto R, Gao Y, Simon-Loriere E, Abreha M, Archer J, et al. Sequence determinants of breakpoint location during HIV-1 intersubtype recombination. Nucleic Acids Res. (2006) 34:5203–16. doi: 10.1093/nar/gkl669
36. Smyth RP, Schlub TE, Grimm AJ, Waugh C, Ellenberg P, Chopra A, et al. Identifying recombination hot spots in the HIV-1 genome. J Virol. (2014) 88:2891–902. doi: 10.1128/JVI.03014-13
37. Simon-Loriere E, Martin DP, Weeks KM, Negroni M. RNA structures facilitate recombination-mediated gene swapping in HIV-1. J Virol. (2010) 84:12675–82. doi: 10.1128/JVI.01302-10
38. Simon-Loriere E, Galetto R, Hamoudi M, Archer J, Lefeuvre P, Martin DP, et al. Molecular mechanisms of recombination restriction in the envelope gene of the human immunodeficiency virus. PLoS Pathog. (2009) 5:e1000418. doi: 10.1371/journal.ppat.1000418
39. Geller R, Domingo-Calap P, Cuevas JM, Rossolillo P, Negroni M, Sanjuan R. The external domains of the HIV-1 envelope are a mutational cold spot. Nat Commun. (2015) 6:8571. doi: 10.1038/ncomms9571
40. Harrison GP, Mayo MS, Hunter E, Lever AM. Pausing of reverse transcriptase on retroviral RNA templates is influenced by secondary structures both 5' and 3' of the catalytic site. Nucleic Acids Res. (1998) 26:3433–42.
41. Cohen J, Enserink M. Swine flu. After delays WHO agrees: the 2009 pandemic has begun. Science (2009) 324:1496–7. doi: 10.1126/science.324_1496
42. Smith GJ, Vijaykrishna D, Bahl J, Lycett SJ, Worobey M, Pybus OG, et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature (2009) 459:1122–5. doi: 10.1038/nature08182
43. Gerber M, Isel C, Moules V, Marquet R. Selective packaging of the influenza A genome and consequences for genetic reassortment. Trends Microbiol. (2014) 22:446–55. doi: 10.1016/j.tim.2014.04.001
44. Hutchinson EC, von Kirchbach JC, Gog JR, Digard P. Genome packaging in influenza A virus. J Gen Virol. (2010) 91:313–28. doi: 10.1099/vir.0.017608-0
45. Essere B, Yver M, Gavazzi C, Terrier O, Isel C, Fournier E, et al. Critical role of segment-specific packaging signals in genetic reassortment of influenza A viruses. Proc Natl Acad Sci USA. (2013) 110:E3840–8. doi: 10.1073/pnas.1308649110
46. Maness NJ, Wilson NA, Reed JS, Piaskowski SM, Sacha JB, Walsh AD, et al. Robust, vaccine-induced CD8(+) T lymphocyte response against an out-of-frame epitope. J Immunol. (2010) 184:67–72. doi: 10.4049/jimmunol.0903118
47. Blake N. Immune evasion by gammaherpesvirus genome maintenance proteins. J Gen Virol. (2010) 91:829–46. doi: 10.1099/vir.0.018242-0
48. Murat P, Zhong J, Lekieffre L, Cowieson NP, Clancy JL, Preiss T, et al. G-quadruplexes regulate Epstein-Barr virus-encoded nuclear antigen 1 mRNA translation. Nat Chem Biol. (2014) 10:358–64. doi: 10.1038/nchembio.1479
49. van Hemert FJ, van der Kuyl AC, Berkhout B. The A-nucleotide preference of HIV-1 in the context of its structured RNA genome. RNA Biol. (2013) 10:211–5. doi: 10.4161/rna.22896
50. Keating CP, Hill MK, Hawkes DJ, Smyth RP, Isel C, Le SY, et al. The A-rich RNA sequences of HIV-1 pol are important for the synthesis of viral cDNA. Nucleic Acids Res. (2009) 37:945–56. doi: 10.1093/nar/gkn1015
51. Klaver B, van der Velden Y, van Hemert F, van der Kuyl AC, Berkhout B. HIV-1 tolerates changes in A-count in a small segment of the pol gene. Retrovirology (2017) 14:43. doi: 10.1186/s12977-017-0367-0
52. Takata MA, Goncalves-Carneiro D, Zang TM, Soll SJ, York A, Blanco-Melo D, et al. CG dinucleotide suppression enables antiviral defence targeting non-self RNA. Nature (2017) 550:124–7. doi: 10.1038/nature24039
53. Vabret N, Bailly-Bechet M, Najburg V, Muller-Trutwin M, Verrier B, Tangy F. The biased nucleotide composition of HIV-1 triggers type I interferon response and correlates with subtype D increased pathogenicity. PLoS ONE (2012) 7:e33502. doi: 10.1371/journal.pone.0033502
54. Takata MA, Soll SJ, Emery A, Blanco-Melo D, Swanstrom R, Bieniasz PD. Global synonymous mutagenesis identifies cis-acting RNA elements that regulate HIV-1 splicing and replication. PLoS Pathog. (2018) 14:e1006824. doi: 10.1371/journal.ppat.1006824
55. Prugnolle F, Manica A, Charpentier M, Guegan JF, Guernier V, Balloux F. Pathogen-driven selection and worldwide HLA class I diversity. Curr Biol. (2005) 15:1022–7. doi: 10.1016/j.cub.2005.04.050
56. Yant LJ, Friedrich TC, Johnson RC, May GE, Maness NJ, Enz AM, et al. The high-frequency major histocompatibility complex class I allele Mamu-B*17 is associated with control of simian immunodeficiency virus SIVmac239 replication. J Virol. (2006) 80:5074–7. doi: 10.1128/JVI.80.10.5074-5077.2006
57. Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature (2002) 418:646–50. doi: 10.1038/nature00939
58. York A, Kutluay SB, Errando M, Bieniasz PD. The RNA binding specificity of human APOBEC3 proteins resembles that of HIV-1 nucleocapsid. PLoS Pathog. (2016) 12:e1005833. doi: 10.1371/journal.ppat.1005833
60. Scott LG, Hennig M. RNA structure determination by NMR. Methods Mol Biol. (2008) 452:29–61. doi: 10.1007/978-1-60327-159-2_2
61. Woodson SA. Taming free energy landscapes with RNA chaperones. RNA Biol. (2010) 7:677–86. doi: 10.4161/rna.7.6.13615
62. Bhandari YR, Jiang W, Stahlberg EA, Stagno JR, Wang YX. Modeling RNA topological structures using small angle X-ray scattering. Methods (2016) 103:18–24. doi: 10.1016/j.ymeth.2016.04.015
63. Stephenson JD, Kenyon JC, Symmons MF, Lever AM. Characterizing 3D RNA structure by single molecule FRET. Methods (2016) 103:57–67. doi: 10.1016/j.ymeth.2016.02.004
64. Stephenson JD, Li H, Kenyon JC, Symmons M, Klenerman D, Lever AM. Three-dimensional RNA structure of the major HIV-1 packaging signal region. Structure (2013) 21:951–62. doi: 10.1016/j.str.2013.04.008
65. Schon P. Imaging and force probing RNA by atomic force microscopy. Methods (2016) 103:25–33. doi: 10.1016/j.ymeth.2016.05.016
66. Rausch JW, Sztuba-Solinska J. Le Grice SFJ Probing the structures of viral RNA regulatory elements with SHAPE and related methodologies. Front Microbiol. (2017) 8:2634. doi: 10.3389/fmicb.2017.02634
67. Jayaraman D, Kenyon JC. New windows into retroviral RNA structures. Retrovirology (2018) 15:11. doi: 10.1186/s12977-018-0393-6
68. Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature (2014) 505:701–5. doi: 10.1038/nature12894
69. Ding Y, Tang Y, Kwok CK, Zhang Y, Bevilacqua PC, Assmann SM. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature (2014) 505:696–700. doi: 10.1038/nature12756
70. Underwood JG, Uzilov AV, Katzman S, Onodera CS, Mainzer JE, Mathews DH, et al. FragSeq: transcriptome-wide RNA structure probing using high-throughput sequencing. Nat Methods (2010) 7:995–1001. doi: 10.1038/nmeth.1529
71. Wan Y, Qu K, Ouyang Z, Kertesz M, Li J, Tibshirani R, et al. Genome-wide measurement of RNA folding energies. Mol Cell. (2012) 48:169–81. doi: 10.1016/j.molcel.2012.08.008
72. Deigan KE, Li TW, Mathews DH, Weeks KM. Accurate SHAPE-directed RNA structure determination. Proc Natl Acad Sci USA. (2009) 106:97–102. doi: 10.1073/pnas.0806929106
73. Watters KE, Abbott TR, Lucks JB. Simultaneous characterization of cellular RNA structure and function with in-cell SHAPE-Seq. Nucleic Acids Res. (2016) 44:e12. doi: 10.1093/nar/gkv879
74. Smyth RP, Despons L, Huili G, Bernacchi S, Hijnen M, Mak J, et al. Mutational interference mapping experiment (MIME) for studying RNA structure and function. Nat Methods (2015) 12:866–72. doi: 10.1038/nmeth.3490
75. Kenyon JC, Prestwood LJ, Lever AM. A novel combined RNA-protein interaction analysis distinguishes HIV-1 Gag protein binding sites from structural change in the viral RNA leader. Sci Rep. (2015) 5:14369. doi: 10.1038/srep14369
76. Bieniasz PD, Kutluay SB. CLIP-related methodologies and their application to retrovirology. Retrovirology (2018) 15:35. doi: 10.1186/s12977-018-0417-2
77. Weidmann CA, Mustoe AM, Weeks KM. Direct duplex detection: an emerging tool in the RNA structure analysis toolbox. Trends Biochem Sci. (2016) 41:734–736. doi: 10.1016/j.tibs.2016.07.001
78. Helwak A, Kudla G, Dudnakova T, Tollervey D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell (2013) 153:654–65. doi: 10.1016/j.cell.2013.03.043
79. Kudla G, Granneman S, Hahn D, Beggs JD, Tollervey D. Cross-linking, ligation, and sequencing of hybrids reveals RNA-RNA interactions in yeast. Proc Natl Acad Sci USA. (2011) 108:10010–5. doi: 10.1073/pnas.1017386108
80. Ramani V, Qiu R, Shendure J. High-throughput determination of RNA structure by proximity ligation. Nat Biotechnol. (2015) 33:980–4. doi: 10.1038/nbt.3289
81. Lu Z, Zhang QC, Lee B, Flynn RA, Smith MA, Robinson JT, et al. RNA duplex map in living cells reveals higher-order transcriptome structure. Cell (2016) 165:1267–79. doi: 10.1016/j.cell.2016.04.028
82. Sharma E, Sterne-Weiler T, O'Hanlon D, Blencowe BJ. Global mapping of human RNA-RNA interactions. Mol Cell. (2016) 62:618–26. doi: 10.1016/j.molcel.2016.04.030
83. Aw JG, Shen Y, Wilm A, Sun M, Lim XN, Boon KL, et al. In vivo mapping of eukaryotic RNA interactomes reveals principles of higher-order organization and regulation. Mol Cell. (2016) 62:603–17. doi: 10.1016/j.molcel.2016.04.028
84. Nguyen TC, Cao X, Yu P, Xiao S, Lu J, Biase FH, et al. Mapping RNA-RNA interactome and RNA structure in vivo by MARIO. Nat Commun. (2016) 7:12023. doi: 10.1038/ncomms12023
Keywords: RNA structure, viral evolution, secondary structure, immune evasion, viral RNA
Citation: Smyth RP, Negroni M, Lever AM, Mak J and Kenyon JC (2018) RNA Structure—A Neglected Puppet Master for the Evolution of Virus and Host Immunity. Front. Immunol. 9:2097. doi: 10.3389/fimmu.2018.02097
Received: 10 June 2018; Accepted: 24 August 2018;
Published: 19 September 2018.
Edited by:
Gkikas Magiorkinis, National and Kapodistrian University of Athens, GreeceReviewed by:
Subrata H. Mishra, Johns Hopkins University, United StatesCopyright © 2018 Smyth, Negroni, Lever, Mak and Kenyon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matteo Negroni, bS5uZWdyb25pQGlibWMtY25ycy51bmlzdHJhLmZy
Julia C. Kenyon, amNrMzNAY2FtLmFjLnVr
† These authors have contributed equally to this work and are co-first authors
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.