
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 04 September 2018
Sec. Inflammation
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02006
This article is part of the Research TopicRole of Lipids in the Dynamics of Allergic Airway InflammationView all 10 articles
 Nincy Debeuf1,2
Nincy Debeuf1,2 Bart N. Lambrecht1,2,3*
Bart N. Lambrecht1,2,3*Asthma is a common lung disease affecting 300 million people worldwide. Allergic asthma is recognized as a prototypical Th2 disorder, orchestrated by an aberrant adaptive CD4+ T helper (Th2/Th17) cell immune response against airborne allergens, that leads to eosinophilic inflammation, reversible bronchoconstriction, and mucus overproduction. Other forms of asthma are controlled by an eosinophil-rich innate ILC2 response driven by epithelial damage, whereas in some patients with more neutrophilia, the disease is driven by Th17 cells. Dendritic cells (DCs) and macrophages are crucial regulators of type 2 immunity in asthma. Numerous lipid mediators including the eicosanoids prostaglandins and leukotrienes influence key functions of these cells, leading to either pro- or anti-inflammatory effects on disease outcome. In this review, we will discuss how eicosanoids affect the functions of DCs and macrophages in the asthmatic lung and how this leads to aberrant T cell differentiation that causes disease.
Eicosanoids are an important class of biologically active molecules, comprising prostanoids, leukotrienes (LTs) and lipoxins that have important pro- and anti-inflammatory effects in asthma. Under a variety of non-specific activation stimuli, such as pro-inflammatory mediators and other stress, the precursor molecule arachidonic acid (AA) is released from membrane phospholipids by cytosolic phospholipase A2. AA can be enzymatically converted either to prostanoids [prostaglandin (PG) and thromboxane] by COX enzymes or to LT and lipoxins by lipoxygenases (LOXs) (Figure 1).
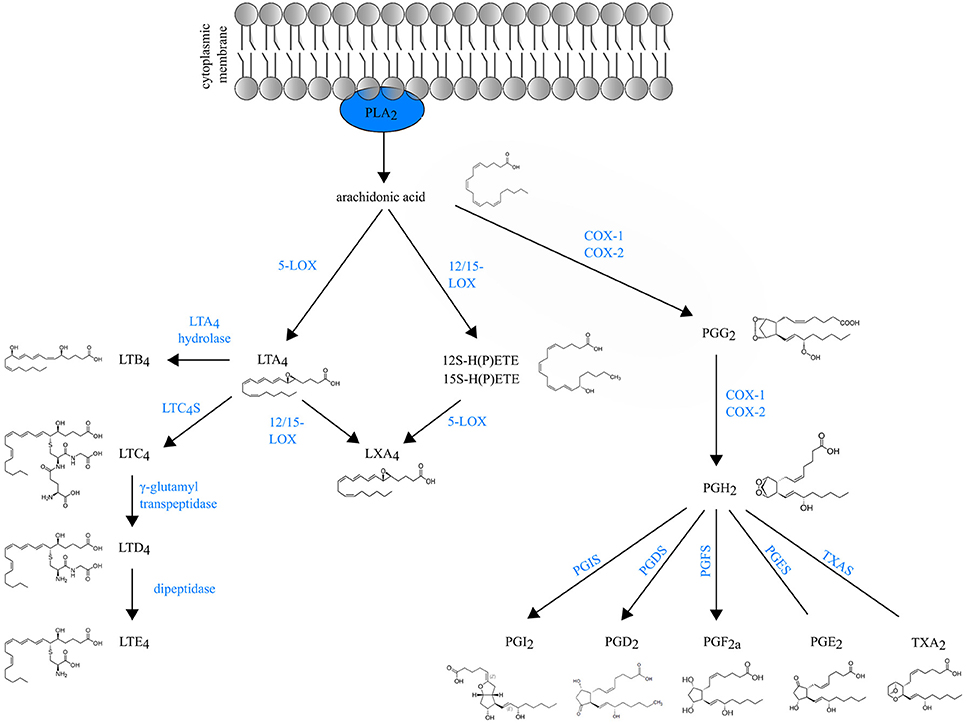
Figure 1. Schematic overview of eicosanoid biosynthesis. Arachidonic acid that is released from membrane phospholipids by cytosolic phospholipase A2 (PLA2), can be enzymatically converted either to prostaglandins (PG) and thromboxane (TXA2) by COX enzymes or to LT and lipoxins (LXA4) by lipoxygenases (LOXs).
Prostanoids The COX isozymes (constitutive COX-1 and inducible COX-2) catalyze the formation of PGG2, which is then reduced to the intermediate PGH2 through peroxidase activity. Various cell-specific PG synthases convert PGH2 to biologically active products, such as PGE2, PGI2, PGD2 and PGF2a and thromboxane (TXA2) (1). The differential expression and the distribution of these enzymes within cells present at sites of inflammation will determine the profile of prostanoid production. For instance, mast cells predominantly generate PGD2 through their expression of hematopoietic PGD synthase (hPGDS). Through microsomal PGE2 synthase (mPGES-1), PGE2 is produced by virtually all lung cell types, but the most abundant sources are epithelial cells, fibroblasts, and macrophages (1). Prostanoids act in both paracrine and autocrine fashion through G protein-coupled receptors (GPCRs) on the surface of target cells. Interestingly, the distribution of prostanoid receptors on immune cells differs from the distribution of prostanoid-specific synthases. Prostanoid synthases are mainly expressed on innate immune cells, whereas prostanoid receptors are expressed on both innate and adaptive immune system leukocytes (2). So, during inflammation, activated innate immune cells will produce prostanoids that act on lymphocytes in a paracrine manner and also modulate their own function in an autocrine way (3).
Leukotrienes are generated by LOX enzymes. The different LOX enzymes are named based on their positional specificity of AA oxygenation. For instance, 12-LOX oxygenates AA at carbon 12, resulting in 12-hydro(peroxy)eicosatetraenoic acid [12-H(P)ETE] (4). Since the human leukocyte-type 12-LOX is very similar to reticulocyte-type 15-LOX, these enzymes are often referred to in the literature as 12/15-LOXs (5). Furthermore, mice do not express 15-LOX and only express the leukocyte-derived 12-LOX. Because murine 12-LOX is also able to generate 15-H(P)ETE, the enzyme is often designated as 12/15 LOX as well (6).
5-lipoxygenase (5-LOX) generates the leukotriene LTA4, an unstable intermediate, which is converted to the chemoattractant LTB4 or to nonchemotactic LTC4 by the cytosolic LTA4 hydrolase enzyme or leukotriene C4 synthase (LTC4S) respectively. LTC4 is exported to the extracellular space and is further converted to the unstable LTD4 and subsequently to the stable end-metabolite LTE4 (7). LTC4, LTD4 and LTE4 belong to the so-called cysteinyl leukotrienes, due to the presence of the amino acid cysteine in their structure. There are at least three different cysteinyl leukotriene receptors (CysLTR1, CysLTR2, and CysLTR3). LTE4 preferably binds CysLTR3 (8), whereas LTC4 binds CysLTR2 and LTD4 binds both CysLTR1 and CysLTR2 (9, 10).
Leukotrienes are predominantly produced by leukocytes, hence their name. However, the specific profile of LTs produced depends on the cell type. Neutrophils produce exclusively LTB4, whereas mast cells, basophils and eosinophils mainly produce cysLTs. Macrophages and DCs synthesize both LTB4 and cysLTs (11).
Lipoxins (LXA4 and LXB4) are short-lived eicosanoids that are derived from arachidonic acid through sequential activity of 5-LOX and 12/15-LOX. 15-LOX is a key enzyme for lipoxin generation in the human lung and is expressed by many cells during inflammation, including macrophages, eosinophils and bronchial epithelial cells (12–14).
Asthma is a chronic inflammatory disease of the airways, characterized by reversible bronchoconstriction, airway remodeling and mucus production. Most childhood-onset asthma and half of the adult-onset asthma cases are allergic, identified by a positive skin prick test or the detection of serum IgE antibodies against common antigens, such as plant and tree pollen, animal dander, house dust mites (HDM) and fungal spores. Virtually all cell types relevant to Th2 pathology such as Th2 cells, ILC2s, mast cells, basophils, epithelial cells, smooth muscle cells and fibroblasts generate LT and/or PG mediators, and/or express receptors for those eicosanoids (Figure 2). Among prostanoids, PGD2 released from mast cells, has long been implicated in allergic diseases (15). PGD2 is known to have chemotactic effects on eosinophils, basophils, Th2 lymphocytes and ILC2s acting via the DP2/CRTh2 receptor (16, 17) and in this way contributes to airway hyperresponsiveness, IgE and cytokine secretion (18–20). PGD2 levels and the number of CRTH2+ cells are increased in bronchoalveolar lavage (BAL) fluids from severe asthmatics compared to those with milder disease (21). Several CRTH2 antagonists have shown encouraging results in clinical trials for asthma, further supporting for the role of PGD2 in allergic diseases and its potential as a therapeutic target (22).
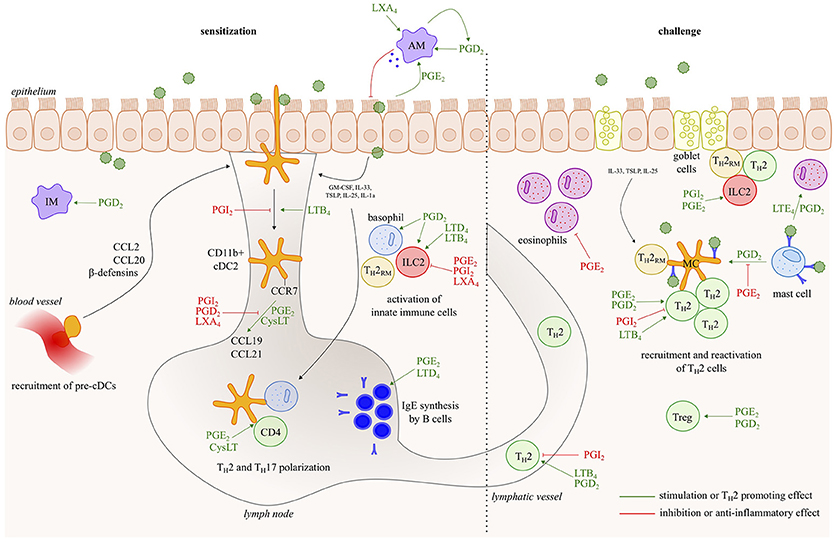
Figure 2. Eicosanoids have multiple effects in allergic asthma. In response to allergens and/or instructive cytokines by epithelial cells and innate immune cells, dendritic cells activate Th2 immunity in the draining lymph nodes, leading to IgE responses and to Th2 effector cells that control eosinophilic airway inflammation, goblet cell metaplasia and bronchial hyperreactivity upon return to the lung. Virtually all cell types relevant to Th2 pathology such as Th2 cells, ILC2s, mast cells, basophils and epithelial cells, generate leukotriene/prostaglandin mediators, and/or express receptors for those eicosanoids. The figure represents a schematic overview of eicosanoid functions described in this review.
Although cyclooxygenase and its products, PGs, have been traditionally linked to all four cardinal signs of inflammation (redness, swelling, heat, pain), prostanoids may also have an inhibitory role on inflammatory cells. This discrepancy can be explained by the fact that typical inflammation hallmarks are the result of actions on microvasculature, hypothalamus and nerves, rather than on immune cells. In mouse models of asthma, PGs have pleiotropic effects. PGI2 can abolish asthma development by inhibition of DC activation and Th2 cell migration (23–25), whereas PGE2 can reduce mast cell secretory responses (26–29) and chemotaxis of eosinophils (30). Furthermore, both PGI2 and PGE2 can inhibit cytokine release of both Th1 and Th2 CD4 T cells and macrophages (31, 32). Treg differentiation and function is also promoted by PGE2 (33, 34).
Prostanoids are also able to inhibit airway remodeling and mucus secretion in asthma models in vivo (35–37). It has been shown that PGE2 induces fibroblast apoptosis (38), abolishes myofibroblast differentiation (39) and inhibits proliferation of airway smooth muscle cells (40).
In asthma patients, inhalation of exogenous PGE2 or its analogs results in bronchodilatation and protection against early- and late-phase bronchoconstriction induced by various asthma triggers (41–43). Despite the benefits of inhaled PGE2, it has also been reported that prostanoids can induce irritancy of the upper airway resulting in a reflex cough. However, this can be overcome by treatment with a receptor-selective agonist, as cough is exclusively mediated via the EP3 receptor (44).
In contrary to the bronchodilatory properties of the prostaglandin PGE2, CysLTs are an important cause of allergen-induced bronchoconstriction (45). Indeed, treatment with Cysteinyl LT receptor 1 antagonists (LTRAs) attenuates allergen-induced increases in airway hyperresponsiveness (46, 47). Furthermore LTRAs partially attenuate allergen-induced airway eosinophilia (47, 48), demonstrating a more extensive role for LTs in asthma. Indeed, CysLTs that are also released from mast cells, particularly LTE4, can cause eosinophil chemotaxis in allergic asthmatics (49). Interestingly, CysLT levels are also increased in BAL fluid (50) and in urine after allergen challenge (51). Currently, LTRAs (such as montelukast) are clinically available. Although these drugs are superior to placebo at decreasing asthmatic symptoms and exacerbations, LTRAs are not recommended as first line therapy for asthma. The reason for this is that they are generally inferior to inhaled corticosteroids in anti-inflammatory and clinical effects1. Furthermore, about one third of the asthma patients does not respond to LTRAs (52).
Another type of leukotriene, LTB4, through its actions on the BLT1 receptor, is an activator and chemoattractant for different cell types such as T cells (53, 54) and DCs (55). OVA-induced allergic inflammation was completely abolished in BLT1 deficient mice, demonstrating the importance of BLT1 and its ligand LTB4 in the development of allergic airway inflammation (56).
Lipoxins have a pro-resolution role in allergic airway inflammation. In severe asthmatics, blood LXA4 levels and leukocyte LXA4 generation are reduced compared to those with milder disease (57–59). In a mouse model of asthma, administration of a stable analog of LXA4 resulted in a diminished airway hyperresponsiveness and pulmonary inflammation (60, 61). Similar results were obtained with resolvins and protectins. Those mediators are also generated by LOX enzymes, but are derived from omega-3 polyunsaturated fatty acids instead of the substrate arachidonic acid (62–64).
Finally, absence of all eicosanoids impairs the induction of a Th2 response and reduces airway inflammation. This has been shown with mice lacking group V secretory phospholipase A2 (sPLA2), which is the enzyme that releases AA from membrane lipids and catalyzes the first step of eicosanoid generation. Deletion of sPLA2 attenuates cell migration and airway hyperresponsiveness, whereas sPLA2 overexpression is associated with severe asthma (65–68). An impaired antigen capture activity and maturation of DCs is responsible for the inhibition of asthma development in sPLA2−/− mice (69).
In allergic asthma, airway DCs take up allergens across the epithelial barrier and subsequently activate Th2 immunity in the draining lymph nodes, leading to IgE responses and to Th2 effector cells that control eosinophilic airway inflammation, goblet cell metaplasia and bronchial hyperreactivity upon return to the lung (70, 71). The central role for DCs in the development of allergic asthma has been demonstrated in numerous studies. Adoptive transfer of GM-CSF-cultured bone marrow-derived DCs (BMDCs) or splenic DCs that were pulsed with ovalbumin (OVA) antigen in vitro can sensitize mice, leading to a Th2 response and eosinophilic inflammation upon challenges with OVA aerosol (72, 73). Likewise, DCs originating from the lungs of allergen-exposed mice are also able to induce sensitization when transferred to naive recipients (74, 75). This holds also true for chronic asthma models as repeated DC injection into the lung induces irreversible airway remodeling, characterized by subepithelial collagen deposition and increased peribronchial airway smooth muscle volume (76).
In addition to these studies demonstrating that DCs are sufficient for induction of Th2 immunity in the lung, DCs are also required for inducing a Th2 response to allergens, even in very young mice before weaning (77). Depletion of lung DCs in CD11c-DTR transgenic mice during the first exposure to the inhaled HDM allergen impeded the development of lung eosinophilia and Th2 cytokine production (74). Likewise, DCs are also required for optimal Th2 immunity against other allergens, such as papain and helminths (78, 79).
Beside the crucial role of DCs in inducing Th2 immunity in naïve animals, DCs have also a non-redundant role during the secondary immune response (76, 80). During the challenge phase, DCs are closely located to antigen-specific T cells around the airways and large blood vessels (81). Here, they might secrete chemokines to attract effector T cells or they might restimulate resident memory T cells by providing costimulatory molecules (75, 82).
Murine lungs in steady state contain three major subsets of DCs with specific phenotype and functions; pDCs, IRF8-dependent XCR1+ CD103+ cDC1s and IRF4-dependent CD11b+ SIRPα+ cDC2s. However, during inflammation monocyte-derived DCs (MCs) emerge, coming from monocytes that migrate to the local tissue and upregulate the expression of CD11c and MHC-II (75, 83). They can be distinguished from CD11b+ cDCs by the expression of the Fc receptors CD64 and MAR1 (75). Various studies have shown that CD11b+ cDC2s are the responsible DC subtype for Th2/17 induction upon allergen challenge (75, 84–86). MCs rather play a role during the effector of the immune response, by interacting with effector Th2 cells that migrate back to the lung or with resident-memory T cells (87). In contrast to CD11b+ cDC2s, CD103+ cDC1s play a redundant role in the HDM-driven asthma model (75). There is even literature suggesting that cDC1s induce a tolerogenic response to inhaled allergens (88–90). An immunoregulatory role has also been described for pDCs. Indeed, it has been shown that pDCs in the lung are essential to induce inhalation tolerance to harmless antigens like OVA (91, 92). Furthermore, depletion of pDCs during sensitization or challenge to OVA or HDM allergen might exacerbate inflammation, as immunoregulatory regulatory T cells fail to function properly in the absence of pDCs (91, 93, 94).
Although DCs express PRRs and can sense the environment directly, the epithelium has been shown to be equally important in activating DCs in response to allergens (95). As this is beyond the scope of this review, we refer to Hammad and Lambrecht for a recent review describing the role of epithelial cytokines in the activation of DCs during allergic inflammation (96). In brief, DCs get activated by epithelial cytokines like IL-33, GM-CSF, IL-1a, IL-25, and thymic stromal lymphopoietin (TSLP). The same cytokines also activate ILC2s, basophils and Th2 effector cells to become cytokine producing cells and contribute to the initiation of a Th2 response (70, 71, 97). The release of epithelial cytokines is elicited by environmental stimuli of asthma, such as HDM, viruses, diesel particles and cigarette smoke. On the other hand, protective environments, such as farm dust or lipopolysaccharide exposure, have the potential to suppress this cytokine release and DC activation (98).
The control of DC migration is pivotal for the initiation of cellular immune responses. Upon activation by inflammatory stimuli, DCs upregulate the chemokine receptor CCR7 and home to lymphoid organs, where the CCR7 ligands CCL19 and CCL21 are expressed. This migratory capacity of DCs requires environmental instruction by PGE2. PGE2 has no effect on the expression of CCR7 on DCs, but couples CCR7 expression to signal transduction pathways such as activation of cAMP-dependent protein kinase A (PKA) and Rho Kinase (99). These signals allow the DCs to start migration, among other by inducing a rapid disassembly of podosomes (100). Surprisingly, PGE2 was only required at early time points of maturation to enable DC chemotaxis, whereas PGE2 addition has no effect during terminal maturation. Mouse DCs exclusively rely on EP4 receptor triggering for migration, whereas human MCs require a signal mediated by EP2 or EP4 either alone or in combination (101, 102).
In contrary to PGE2, PGD2, and PGI2 inhibit the maturation and migration of DCs. In the skin, Angeli and colleagues showed that parasite-derived PGD2 inhibits the migration of epidermal Langerhans cells to the skin draining lymph nodes and affects the subsequent cutaneous inflammatory reaction (103). Similarly, intratracheal instillation of FITC-OVA together with PGD2 inhibits the migration of FITC+ lung DC to draining LNs. Activation of the DP1 receptor was responsible for this inhibition (104). DP1 activation also lowers the expression of costimulatory molecules on DCs and enhances the induction of Foxp3+ Treg cells, resulting in an abolished asthma phenotype (34). Inhalation of iloprost, a stable PGI2 analog, also suppressed the cardinal features of asthma by interfering with the function of lung myeloid DC. Furthermore, iloprost-treated DCs no longer induced Th2 differentiation from naive T cells or boosted effector cytokine production in primed Th2 cells, showing that the effect of iloprost was DC intrinsic (23).
CysLT enhance the migration of DCs. Indeed, DCs lacking the LTC4 transporter multidrug resistance-associated protein 1 (MRP1) failed to migrate to the lymph nodes, whereas exogenous LTC4 or LTD4 could restore this migration. However, these CysLTs only promoted optimal chemotaxis to the chemokine CCL19, but not to other related chemokines (105). On the other hand, lipoxins were able to inhibit DC migration (106).
Upon DC-T cell encounter, DCs produce cytokines that drive Th differentiation. The secretion pattern of these cytokines, and thus the Th1/Th2 balance can be modulated by a variety of biologically active mediators synthesized by innate and adaptive immune cells. Eicosanoids such as PGE2 exert a great impact on this regulation. For instance, the ratio of PGE2 and IL-12, both produced by APCs, may control the balance between Th1 and Th2 immunity (107). Basically, it has been shown that PGE2 is a potent inhibitor of IL-12 production (108) and in this way favors a Th2 response (109–112). PGE2 also inhibits the secretion of TNF-α from murine DCs (113, 114). The inhibitory role of PGE2 on DC cytokine secretion can also be indirect by inducing IL-10 secretion (108, 113, 115). Due to its inhibitory effect on IL-12, PGE2 also indirectly inhibits IFN-γ secretion by T cells and NK cells (110, 116).
Beside the Th2 inducing role for PGE2, it has also been reported that PGE2-treated DCs can induce Th1 and Th17 responses. Adding PGE2 together with TNF-α to human BMDCs stimulates IL-12 production by DCs, favoring a Th1 response (117–121). PGE2 also stimulates IL-23 production by cultured BMDCs and promotes in this way Th17 differentiation (122, 123).
Prostanoids can also directly modify production of Th cytokines from polarized T cells. PGE2 can favor Th2 immunity by inhibiting IL-2 and IFN-γ production by Th1 cells, but not the production of IL-4 by Th2 cells (124, 125). However, in a mouse model of asthma, PGE2 has also been shown to inhibit Th2 responses via direct effects on the EP2 receptor on T cells (126). Furthermore, PGE2 regulates Th17 cell differentiation and cytokine secretion directly through EP2/EP4 receptor signaling on T cells (127). Via DP1, PGD2 can block the expression of the Th1 cytokine IFN-γ. Furthermore, Th2 cytokine secretion is increased through CRTH2 signaling (128). On the other hand, PGI2 can directly inhibit production of Th2 cytokines from Th2 polarized mouse splenic CD4+ cells (32, 129), thus directly exhibiting lower levels of Th2 response.
Less is known about the role of leukotrienes on T cell polarization. Machida et al. reported that in vitro treatment with LTRAs modifies the cytokine profile of DCs (130). By in vivo administration of LTRAs, Okunishi and colleagues showed that LTs promote DC antigen presentation and both Th1 and Th2 polarizing cytokine secretion (131).
Furthermore, using and CysLTR1−/− mice, it has been demonstrated that leukotrienes are crucial for the initiation of a Th2 response upon HDM-dependent Dectin-2 activation on DCs (132). Through CysLTR1, LTD4 can induce IL-4 secretion by ILC2s, contributing to Th2 polarization as well (133). CysLTR1 can also be up-regulated in activated CD4+ T cells themselves and can mediate their chemotaxis to LTD4, but whether cysLTs exert a direct effect on cytokine production by CD4+ T cells remains unclear (134). This is different from the leukotriene LTB4, which increases cytokine production by T cells (135), but does not affect antigen presentation and cytokine production by DCs (55). Strikingly, mice deficient in CysLTR2 or adoptive transfer of DCs lacking CysLTR2 developed markedly enhanced Th2 immunity to HDM. In fact, CysLTR2 negatively regulates cell surface expression and receptor signaling of DCs (136). Thus, the biologic activity of CysLTs can be tightly regulated by competition between the different expressed CysLT receptors.
12/15-LOX enzymes, required for lipoxin synthesis, are also involved in the modulation of Th2 cytokine secretion. In response to IL-13, DCs secrete the lectin Ym1/2 that might interact with 12/15-LOX in or at the surface of T cells. 12/15-LOX generates 12-HETE that has been shown to reduce Th2 cytokine secretion both in vivo and in vitro. Furthermore, 12-HETE attenuated airway eosinophilia in an OVA-induced allergic asthma model. However, DC-secreted Ym1/2 was able to decrease the expression of 12-HETE, suggesting that the asthma-promoting effects of Ym1/2 might be explained by inhibiting 12/15-LOX on T cells (137).
The most described function of B cells is their production of antigen-specific immunoglobulins. However, in addition to antibody production, activated B cells also play a role as accessory antigen-presenting cells. Although they are not as potent as DCs in priming naïve T cells, they are abundantly present in T-cell inductive sites, express costimulatory molecules and produce cytokines that activate DCs and naïve T cells (138–141). Their antigen-presenting and Th2-promoting effects have also been demonstrated in murine asthma models, with a particular role during secondary challenge and when the antigen dose is limiting (142).
Numerous studies have shown that eicosanoids are required for both the development and function of B lymphocytes (143–147). PGE2 is necessary for IgE production both in vitro and in vivo, by affecting IgE class switching (145, 148–151). Furthermore, PGE2 has been demonstrated to regulate B cell proliferation (152). Interestingly, PGE2 is also able to lower MHCII expression on B cells (148), but whether this affects antigen presentation is still unclear. Leukotrienes, in particular LTD4, can enhance immunoglobulin production as well (153). In contrast, lipoxins have the opposite effect as 12/15-LOX deficiency protects mice from allergic airway inflammation by increasing secretory IgA levels (147).
Strikingly, ILC2s have also been shown to present antigen (154, 155). In response to the parasitic worm Nippostrongylus brasiliensis, MHC class II expression on ILC2s was required for the induction of an efficient Th2 response. ILC2s express the costimulatory receptors CD80 and CD86, acquire and process antigen and interact with antigen-specific T cells. During this interaction, T cell-derived IL-2 promotes ILC2 proliferation and IL-13 production (155) and this can be affected by eicosanoids. PGD2 and CysLTs stimulate Th2 cytokine production from ILC2s (133, 156, 157), whereas other lipid mediators have suppressive roles on ILC2 function. The pro-resolving mediator LXA4 could inhibit ILC2 activation (156) and both PGE2 and PGI2 were able to attenuate ILC2 proliferation, Th2 cytokine generation and resulting type 2 immune response (158, 159).
Lung macrophages can be divided into alveolar macrophages (AMs) and interstitial macrophages (IMs). AMs are most abundantly present and are situated in the alveolar lumen, while IMs are located inside the lung interstitium. During inflammation, a third population emerges, as monocyte-derived macrophages infiltrate the alveolar and interstitial areas. Macrophages express different eicosanoid receptors, such as the receptors for PGE2 and PGD2. Furthermore, macrophages produce both prostaglandins and leukotrienes themselves, allowing autocrine regulation (160).
AMs are sessile, long-lived, and self-renewing cells that derive from fetal monocytes under the influence of GM-CSF (161–164). Several studies have clearly demonstrated that resident AMs induce a tolerogenic response to inhaled antigens (164–171). Use of liposomal clodronate to deplete resident AMs in an OVA or HDM-induced asthma model, favored a Th2 response and subsequently resulted in increased BAL eosinophilia and inflammatory cytokine levels (167, 168). One possible mechanism for this inhibitory role of macrophages is the secretion of SOCS1 and SOCS3 in exosomes and microparticles. The uptake of these particles by alveolar epithelial cells inhibits their activation in a JAK/STAT-dependent way (169). PGE2 is a major epithelium-derived factor mediating SOCS secretion (170) and in this way inhibiting the development of allergic lung inflammation (171). Indeed, in a HDM-dependent asthma model, adoptive transfer of PGE2-treated macrophages led to a reduction in eosinophilia in the allergic lung (171).
Pulmonary inflammation was also reduced if macrophages lacked group V sPLA2, which is the enzyme releasing AA from membrane lipids and is required for both PG and LT synthesis (172). Those macrophages generated less PGE2, resulting in a diminished transglutaminase activity of M2 macrophages (173). Furthermore, by activating the EP4 receptor on macrophages, PGE2 inhibits TNF-α and IL-12 cytokine secretion (31). The ability of lung macrophages to prevent Th2 induction in response to inhaled allergens has also been demonstrated in rats. The replacement of AM of sensitized animals by AM from naive animals completely abolished Th2 polarization by inhibition of DC allergen capture and migration to the lymph nodes (174).
Although PGE2 suppresses type 2 inflammation in most settings, a recent study demonstrated that PGE2 also has pro-inflammatory effects in murine macrophages. Mice lacking microsomal PGE2 synthase 1 (mPGE1) had an attenuated asthma phenotype compared to wild-type controls in response to repetitive inhalation challenges with an extract from the allergenic mold Alternaria alternata, which could be explained by a diminished IL-33 production by murine macrophages (175).
The pro-inflammatory prostanoid PGD2 binds both the DP1 and DP2 receptor on lung macrophages. DP signaling enhances migration and TNF-α secretion of both alveolar and interstitial macrophages. Furthermore, PGD2 also induces KC secretion from macrophages, resulting in neutrophil recruitment in the lung and this neutrophilia could be abolished by macrophage depletion (176). Interestingly, PGD2 synthesis by macrophages is also involved in the enhancement of airway inflammation by virus infections. Respiratory infections with RNA viruses, such as rhinovirus or respiratory syncytial virus (RSV), are associated with asthmatic exacerbations (177). To study the mechanism behind this association, Shiraishi and colleagues administered poly I:C, a synthetic dsRNA, intratracheally in OVA-sensitized rats. Those rats developed an exacerbated asthma phenotype and had elevated PGD2 synthesis in the lung, particularly in AMs. CRTH2-deficient animals did not exhibit a dsRNA-induced increase in eosinophil accumulation, demonstrating the necessary role for PGD2 in dsRNA-induced enhancement of airway inflammation (178).
RSV infection of mice deficient in 5-LOX, an enzyme required for lipoxin synthesis, resulted in stronger lung pathology compared to wildtype mice, due to a lack of alternatively activated macrophages (179, 180). Treatment with LXA4 partially restored this, supporting a pro-resolution role for lipoxins in viral respiratory tract infections (180).
Just as described for DCs, AMs produce leukotrienes in response to HDM-driven Dectin-2 activation. Both an inhibitor of LT production and Dectin-2 blockade could prevent the development of bronchial hyperreactivity and airway inflammation, demonstrating the required role for Dectin-2 dependent leukotriene production in the initiation of allergic airway inflammation (181).
Allergic asthma is a chronic lung disease, driven by a prototypical Th2 response against airborne allergens. Dendritic cells (CD11b + cDC2s) are indispensable and sufficient for the development of allergic asthma, whereas macrophages have merely a tolerogenic role. Eicosanoids, leukotrienes and prostaglandins, influence key functions of these cells. However, given the diverse spectrum of eicosanoids and given the cell-type dependent expression profile of eicosanoid receptors, it is not surprising that the effects of PG/LT can be very distinct depending on the inflammatory context. A particular eicosanoid can have a pro-inflammatory effect on a certain cell type, whereas it can act anti-inflammatory on another. Furthermore, one particular cell type will be exposed to both pro-inflammatory and anti-inflammatory eicosanoids and the balance between those will determine the cellular outcome. Leukotriene receptor antagonists are already in clinical use for the treatment of asthma. In addition, given the multiple roles of prostaglandins in the pathogenesis of asthma, PG a-/antagonists may also have a promising therapeutic effect.
ND wrote the first draft of the manuscript. BL edited the manuscript. Both authors contributed to manuscript revision, read and approved the submitted version.
ND is supported by a Fonds Wetenschappelijk Onderzoek Vlaanderen (FWO) grant (11Y8417N). BL is supported by an European Research Council (ERC)-Consolidator grant (EU789384) and Excellence of Science (EOS) research grant (project 30565447).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. ^National Institutes of Health; National Heart, Lung, and Blood. National Asthma Education and Prevention Program Institute, Expert panel report 3: guidelines for the diagnosis and management of asthma. No. 07–4051 Available from: http://www.nhlbi.nih.gov/guidelines/asthma/asthgdln.htm 2007.
1. Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science (2001) 294:1871–5. doi: 10.1126/science.294.5548.1871
2. Tilley SL, Coffman TM, Koller BH. Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. J Clin Invest. (2001) 108:15–23. doi: 10.1172/JCI200113416
3. Harizi H, Gualde N. The impact of eicosanoids on the crosstalk between innate and adaptive immunity: the key roles of dendritic cells. Tissue Antigens (2005) 65:507–14. doi: 10.1111/j.1399-0039.2005.00394.x
4. Brash AR. Lipoxygenases: occurrence, functions, catalysis, and acquisition of substrate. J Biol Chem. (1999) 274:23679–82. doi: 10.1074/jbc.274.34.23679
5. Kühn H, Walther M, Kuban RJ. Mammalian arachidonate 15-lipoxygenases structure, function, and biological implications. Prostaglandins Other Lipid Mediat. (2002) 68–9:263–290. doi: 10.1016/S0090-6980(02)00035-7
6. Chen XS, Kurre U, Jenkins NA, Copeland NG, Funk CD. cDNA cloning, expression, mutagenesis of C-terminal isoleucine, genomic structure, and chromosomal localizations of murine 12-lipoxygenases. J Biol Chem. (1994) 269:13979–87.
7. Samuchiwal SK, Boyce JA. Role of lipid mediators and control of lymphocyte responses in type 2 immunopathology. J Allergy Clin Immunol. (2018) 141:1182−90. doi: 10.1016/j.jaci.2018.02.006
8. Kanaoka Y, Maekawa A, Austen KF. Identification of GPR99 protein as a potential third cysteinyl leukotriene receptor with a preference for leukotriene E4 ligand. J Biol Chem. (2013) 288:10967–72. doi: 10.1074/jbc.C113.453704
9. Nothacker HP, Wang Z, Zhu Y, Reinscheid RK, Lin SH, Civelli O. Molecular cloning and characterization of a second human cysteinyl leukotriene receptor: discovery of a subtype selective agonist. Mol Pharmacol. (2000) 58:1601–8. doi: 10.1124/mol.58.6.1601
10. Lynch KR, O'Neill GP, Liu Q, Im DS, Sawyer N, Metters KM, et al. Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature (1999) 399:789–93. doi: 10.1038/21658
11. Peters-Golden M, Henderson WR Jr. Leukotrienes. N Engl J Med. (2007) 357:1841–54. doi: 10.1056/NEJMra071371
12. Hunter JA, Finkbeiner WE, Nadel JA, Goetzl EJ, Holtzman MJ. Predominant generation of 15-lipoxygenase metabolites of arachidonic acid by epithelial cells from human trachea. Proc Natl Acad Sci USA. (1985) 82:4633–7. doi: 10.1073/pnas.82.14.4633
13. Levy BD, Romano M, Chapman HA, Reilly JJ, Drazen J, Serhan CN. Human alveolar macrophages have 15-lipoxygenase and generate 15(S)-hydroxy-5,8,11-cis-13-trans-eicosatetraenoic acid and lipoxins. J Clin Invest. (1993) 92:1572–9. doi: 10.1172/JCI116738
14. Serhan CN, Hirsch U, Palmblad J, Samuelsson B. Formation of lipoxin A by granulocytes from eosinophilic donors. FEBS Lett. (1987) 217:242–6. doi: 10.1016/0014-5793(87)80671-3
15. Hardy CC, Robinson C, Tattersfield AE, Holgate ST. The bronchoconstrictor effect of inhaled prostaglandin D2 in normal and asthmatic men. N Engl J Med. (1984) 311:209–13. doi: 10.1056/NEJM198407263110401
16. Hirai H, Tanaka K, Yoshie O, Ogawa K, Kenmotsu K, Takamori Y, et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med. (2001) 193:255–61. doi: 10.1084/jem.193.2.255
17. Nagata K, Hirai H, Tanaka K, Ogawa K, Aso T, Sugamura K, et al. CRTH2, an orphan receptor of T-helper-2-cells, is expressed on basophils and eosinophils and responds to mast cell-derived factor(s). FEBS Lett. (1999) 459:195–9. doi: 10.1016/S0014-5793(99)01251-X
18. Pettipher R, Whittaker M. Update on the development of antagonists of chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2). From lead optimization to clinical proof-of-concept in asthma and allergic rhinitis. J Med Chem. (2012) 55:2915–31. doi: 10.1021/jm2013997
19. Xue L, Salimi M, Panse I, Mjösberg JM, McKenzie ANJ, Spits H, et al. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J Allergy Clin Immunol. (2014) 133:1184–94. doi: 10.1016/j.jaci.2013.10.056
20. Chang JE, Doherty TA, Baum R, Broide D. Prostaglandin D2 regulates human type 2 innate lymphoid cell chemotaxis. J Allergy Clin Immunol. (2014) 133:899–901.e3. doi: 10.1016/j.jaci.2013.09.020
21. Fajt ML, Gelhaus SL, Freeman B, Uvalle CE, Trudeau JB, Holguin F, et al. Prostaglandin D2 pathway upregulation: relation to asthma severity, control, and TH2 inflammation. J Allergy Clin Immunol. (2013) 131:1504–12. doi: 10.1016/j.jaci.2013.01.035
22. George L, Brightling CE. Eosinophilic airway inflammation: role in asthma and chronic obstructive pulmonary disease. Ther Adv Chronic Dis. (2016) 7:34–51. doi: 10.1177/2040622315609251
23. Idzko M, Hammad H, van Nimwegen M, Kool M, Vos N, Hoogsteden HC, et al. Inhaled iloprost suppresses the cardinal features of asthma via inhibition of airway dendritic cell function. J Clin Invest. (2007) 117:464–72. doi: 10.1172/JCI28949
24. Nagao K, Tanaka H, Komai M, Masuda T, Narumiya S, Nagai H. Role of prostaglandin I2 in airway remodeling induced by repeated allergen challenge in mice. Am J Respir Cell Mol Biol. (2003) 29:314–20. doi: 10.1165/rcmb.2003-0035OC
25. Jaffar Z, Ferrini ME, Buford MC, FitzGerald GA, Roberts K. Prostaglandin I2-IP signaling blocks allergic pulmonary inflammation by preventing recruitment of CD4+ Th2 cells into the airways in a mouse model of asthma. J Immunol. (2007) 179:6193–203. doi: 10.4049/jimmunol.179.9.6193
26. Herrerias A, Torres R, Serra M, Marco A, Roca-Ferrer J, Picado C, et al. Subcutaneous prostaglandin E(2) restrains airway mast cell activity in vivo and reduces lung eosinophilia and Th(2) cytokine overproduction in house dust mite-sensitive mice. Int Arch Allergy Immunol. (2009) 149:323–32. doi: 10.1159/000205578
27. Torres R, Pérez M, Marco A, Picado C, de Mora F. A Cyclooxygenase-2 selective inhibitor worsens respiratory function and enhances mast cell activity in ovalbumin-sensitized mice. Arch Bronconeumol. (English Edition) (2009) 45:162–7. doi: 10.1016/j.arbres.2008.04.007
28. Torres R, Herrerias A, Serra-Pagès M, Marco A, Plaza J, Costa-Farré C, et al. Locally administered prostaglandin E2 prevents aeroallergen-induced airway sensitization in mice through immunomodulatory mechanisms. Pharmacol Res. (2013) 70:50–9. doi: 10.1016/j.phrs.2012.12.008
29. Kay LJ, Yeo WW, Peachell PT. Prostaglandin E 2activates EP 2receptors to inhibit human lung mast cell degranulation. Br J Pharmacol. (2006) 147:707–13. doi: 10.1038/sj.bjp.0706664
30. Sturm EM, Schratl P, Schuligoi R, Konya V, Sturm GJ, Lippe IT, et al. Prostaglandin E2 inhibits eosinophil trafficking through E-prostanoid 2 receptors. J Immunol. (2008) 181:7273–83. doi: 10.4049/jimmunol.181.10.7273
31. Nataraj C, Thomas DW, Tilley SL, Nguyen MT, Mannon R, Koller BH, et al. Receptors for prostaglandin E(2) that regulate cellular immune responses in the mouse. J Clin Invest. (2001) 108:1229–35. doi: 10.1172/JCI200113640
32. Zhou W, Blackwell TS, Goleniewska K, O'Neal JF, FitzGerald GA, Lucitt M, et al. Prostaglandin I 2analogs inhibit Th1 and Th2 effector cytokine production by CD4 T cells. J Leukoc Biol. (2006) 81:809–17. doi: 10.1189/jlb.0606375
33. Baratelli F, Lin Y, Zhu L, Yang SC, Heuze-Vourc'h N, Zeng G, et al. Prostaglandin E2 induces FOXP3 Gene expression and T regulatory cell function in human CD4+ T cells. J Immunol. (2005) 175:1483–90. doi: 10.4049/jimmunol.175.3.1483
34. Hammad H, Kool M, Soullié T, Narumiya S, Trottein F, Hoogsteden HC, et al. Activation of the D prostanoid 1 receptor suppresses asthma by modulation of lung dendritic cell function and induction of regulatory T cells. J Exp Med. (2007) 204:357–67. doi: 10.1084/jem.20061196
35. Yamabayashi C, Koya T, Kagamu H, Kawakami H, Kimura Y, Furukawa T, et al. A novel prostacyclin agonist protects against airway hyperresponsiveness and remodeling in mice. Am J Respir Cell Mol Biol. (2012) 47:170–7. doi: 10.1165/rcmb.2011-0350OC
36. Hattori R, Shimizu S, Majima Y, Shimizu T. Prostaglandin E2 receptor EP2, EP3, and EP4 agonists inhibit antigen-induced mucus hypersecretion in the nasal epithelium of sensitized rats. Ann Otol Rhinol Laryngol. (2009) 118:536–41. doi: 10.1177/000348940911800714
37. Lundequist A, Nallamshetty SN, Xing W, Feng C, Laidlaw TM, Uematsu S, et al. Prostaglandin E(2) exerts homeostatic regulation of pulmonary vascular remodeling in allergic airway inflammation. J Immunol. (2010) 184:433–41. doi: 10.4049/jimmunol.0902835
38. Huang SK, White ES, Wettlaufer SH, Grifka H, Hogaboam CM, Thannickal VJ, et al. Prostaglandin E 2induces fibroblast apoptosis by modulating multiple survival pathways. FASEB J. (2009) 23:4317–26. doi: 10.1096/fj.08-128801
39. Kolodsick JE, Peters-Golden M, Larios J, Toews GB, Thannickal VJ, Moore BB. Prostaglandin E 2inhibits fibroblast to myofibroblast transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am J Respir Cell Mol Biol. (2003) 29:537–44. doi: 10.1165/rcmb.2002-0243OC
40. Mori A, Ito S, Morioka M, Aso H, Kondo M, Sokabe M, et al. Effects of specific prostanoid EP receptor agonists on cell proliferation and intracellular Ca2+ concentrations in human airway smooth muscle cells. Eur J Pharmacol. (2011) 659:72–8. doi: 10.1016/j.ejphar.2011.03.001
41. Pavord ID, Wisniewski A, Mathur R, Wahedna I, Knox AJ, Tattersfield AE. Effect of inhaled prostaglandin E2 on bronchial reactivity to sodium metabisulphite and methacholine in patients with asthma. Thorax (1991) 46:633–7. doi: 10.1136/thx.46.9.633
42. Szczeklik A, Mastalerz L, Nizankowska E, Cmiel A. Protective and bronchodilator effects of prostaglandin E and salbutamol in aspirin-induced asthma. Am J Respir Crit Care Med. (1996) 153:567–71. doi: 10.1164/ajrccm.153.2.8564099
43. Sestini P, Armetti L, Gambaro G, Pieroni MG, Refini RM, Sala A, et al. Inhaled PGE2 prevents aspirin-induced bronchoconstriction and urinary LTE4 excretion in aspirin-sensitive asthma. Am J Respir Crit Care Med. (1996) 153:572–5. doi: 10.1164/ajrccm.153.2.8564100
44. Maher SA, Birrell MA, Belvisi MG. Prostaglandin E2 mediates cough via the EP3 receptor: implications for future disease therapy. Am J Respir Crit Care Med. (2009) 180:923–8. doi: 10.1164/rccm.200903-0388OC
45. Adelroth E, Morris MM, Hargreave FE, O'Byrne PM. Airway responsiveness to leukotrienes C4 and D4 and to methacholine in patients with asthma and normal controls. N Engl J Med. (1986) 315:480–4. doi: 10.1056/NEJM198608213150803
46. Hamilton A, Faiferman I, Stober P, Watson RM, O'Byrne PM. Pranlukast, a cysteinyl leukotriene receptor antagonist, attenuates allergen-induced early- and late-phase bronchoconstriction and airway hyperresponsiveness in asthmatic subjects. J Allergy Clin Immunol. (1998) 102:177–83. doi: 10.1016/S0091-6749(98)70083-1
47. Parameswaran K, Watson R, Gauvreau GM, Sehmi R, O'Byrne PM. The effect of pranlukast on allergen-induced bone marrow Eosinophilopoiesis in subjects with asthma. Am J Respir Crit Care Med. (2004) 169:915–20. doi: 10.1164/rccm.200312-1645OC
48. Leigh R, Vethanayagam D, Yoshida M, Watson RM, Rerecich T, Inman MD, et al. Effects of montelukast and budesonide on airway responses and airway inflammation in asthma. Am J Respir Crit Care Med. (2002) 166:1212–7. doi: 10.1164/rccm.200206-509OC
49. Gauvreau GM, Parameswaran KN, Watson RM, O'Byrne PM. Inhaled leukotriene E 4, but not leukotriene D 4, increased airway inflammatory cells in subjects with atopic asthma. Am J Respir Crit Care Med. (2001) 164:1495–500. doi: 10.1164/ajrccm.164.8.2102033
50. Wenzel SE, Larsen GL, Johnston K, Voelkel NF, Westcott JY. Elevated levels of leukotriene C 4in bronchoalveolar lavage fluid from atopic asthmatics after endobronchial allergen challenge. Am Rev Respir Dis. (1990) 142:112–9. doi: 10.1164/ajrccm/142.1.112
51. Manning PJ, Rokach J, Malo JL, Ethier D, Cartier A, Girard Y, et al. Urinary leukotriene E4 levels during early and late asthmatic responses. J Allergy Clin Immunol. (1990) 86:211–20. doi: 10.1016/S0091-6749(05)80068-5
52. Barnes N, Thomas M, Price D, Tate H. The national montelukast survey. J Allergy Clin Immunol. (2005) 115:47–54. doi: 10.1016/j.jaci.2004.10.011
53. Tager AM, Bromley SK, Medoff BD, Islam SA, Bercury SD, Friedrich EB, et al. Leukotriene B4 receptor BLT1 mediates early effector T cell recruitment. Nat Immunol. (2003) 4:982–90. doi: 10.1038/ni970
54. Goodarzi K, Goodarzi M, Tager AM, Luster AD, Andrian von UH. Leukotriene B4 and BLT1 control cytotoxic effector T cell recruitment to inflamed tissues. Nat Immunol. (2003) 4:965–973. doi: 10.1038/ni972
55. Miyahara N, Ohnishi H, Matsuda H, Miyahara S, Takeda K, Koya T, et al. Leukotriene B4 receptor 1 expression on dendritic cells is required for the development of Th2 responses and allergen-induced airway hyperresponsiveness. J Immunol. (2008) 181:1170–8. doi: 10.4049/jimmunol.181.2.1170
56. Terawaki K, Yokomizo T, Nagase T, Toda A, Taniguchi M, Hashizume K, et al. Absence of leukotriene B4 receptor 1 confers resistance to airway hyperresponsiveness and Th2-type immune responses. J Immunol. (2005) 175:4217–25. doi: 10.4049/jimmunol.175.7.4217
57. Planagumà A, Kazani S, Marigowda G, Haworth O, Mariani TJ, Israel E, et al. Airway lipoxin A4 generation and lipoxin A4 receptor expression are decreased in severe asthma. Am J Respir Crit Care Med. (2008) 178:574–82. doi: 10.1164/rccm.200801-061OC
58. Levy BD, Bonnans C, Silverman ES, Palmer LJ, Marigowda G, Israel E. Severe asthma research program, national heart, lung, and blood institute. Diminished lipoxin biosynthesis in severe asthma. Am J Respir Crit Care Med. (2005) 172:824–30. doi: 10.1164/rccm.200410-1413OC
59. Çelik GE, Erkekol FO, Mιsιrlιgil Z, Melli M. Lipoxin A4 levels in asthma: relation with disease severity and aspirin sensitivity. Clin Exp Allergy (2007) 37:1494–501. doi: 10.1111/j.1365-2222.2007.02806.x
60. Levy BD, De Sanctis GT, Devchand PR, Kim E, Ackerman K, Schmidt BA, et al. Multi-pronged inhibition of airway hyper-responsiveness and inflammation by lipoxin A(4). Nat Med. (2002) 8:1018–23. doi: 10.1038/nm748
61. Levy BD, Lukacs NW, Berlin AA, Schmidt B, Guilford WJ, Serhan CN, et al. Lipoxin A4 stable analogs reduce allergic airway responses viamechanisms distinct from CysLT1 receptor antagonism. FASEB J. (2007) 21:3877–84. doi: 10.1096/fj.07-8653com
62. Haworth O, Cernadas M, Yang R, Serhan CN, Levy BD. Resolvin E1 regulates interleukin 23, interferon-γ and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat Immunol. (2008) 9:873–9. doi: 10.1038/ni.1627
63. Levy BD, Kohli P, Gotlinger K, Haworth O, Hong S, Kazani S, et al. Protectin D1 is generated in asthma and dampens airway inflammation and hyperresponsiveness. J Immunol. (2007) 178:496–502. doi: 10.4049/jimmunol.178.1.496
64. Rogerio AP, Haworth O, Croze R, Oh SF, Uddin M, Carlo T, et al. Resolvin D1 and aspirin-triggered resolvin D1 promote resolution of allergic airways responses. J Immunol. (2012) 189:1983–91. doi: 10.4049/jimmunol.1101665
65. Henderson WR, Chi EY, Bollinger JG, Tien Y-T, Ye X, Castelli L, et al. Importance of group X-secreted phospholipase A2 in allergen-induced airway inflammation and remodeling in a mouse asthma model. J Exp Med. (2007) 204:865–77. doi: 10.1084/jem.20070029
66. Muñoz NM, Meliton AY, Arm JP, Bonventre JV, Cho W, Leff AR. Deletion of secretory group V phospholipase A2 attenuates cell migration and airway hyperresponsiveness in immunosensitized mice. J Immunol. (2007) 179:4800–7. doi: 10.4049/jimmunol.179.7.4800
67. Hallstrand TS, Lai Y, Ni Z, Oslund RC, Henderson WR Jr, Gelb MH, et al. Relationship between levels of secreted phospholipase A2 groups IIA and X in the airways and asthma severity. Clin Exp Allergy (2011) 41:801–10. doi: 10.1111/j.1365-2222.2010.03676.x
68. Hallstrand TS, Chi EY, Singer AG, Gelb MH, Henderson WR. Secreted phospholipase A2 group X overexpression in asthma and bronchial hyperresponsiveness. Am J Respir Crit Care Med. (2007) 176:1072–8. doi: 10.1164/rccm.200707-1088OC
69. Giannattasio G, Fujioka D, Xing W, Katz HR, Boyce JA, Balestrieri B. Group V secretory phospholipase A2 reveals its role in house dust mite-induced allergic pulmonary inflammation by regulation of dendritic cell function. J Immunol. (2010) 185:4430–8. doi: 10.4049/jimmunol.1001384
70. Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat Med. (2009) 15:410–6. doi: 10.1038/nm.1946
71. Lambrecht BN, Hammad H. Dendritic cell and epithelial cell interactions at the origin of murine asthma. Ann Am Thorac Soc. (2014) 11 (Suppl. 5):S236–43. doi: 10.1513/AnnalsATS.201405-218AW
72. Lambrecht BN, De Veerman M, Coyle AJ, Gutierrez-Ramos JC, Thielemans K, Pauwels RA. Myeloid dendritic cells induce Th2 responses to inhaled antigen, leading to eosinophilic airway inflammation. J Clin Invest. (2000) 106:551–9. doi: 10.1172/JCI8107
73. Raymond M, Van VQ, Wakahara K, Rubio M, Sarfati M. Lung dendritic cells induce T(H)17 cells that produce T(H)2 cytokines, express GATA-3, and promote airway inflammation. J Allergy Clin Immunol. (2011) 128:192–201.e6. doi: 10.1016/j.jaci.2011.04.029
74. Hammad H, Plantinga M, Deswarte K, Pouliot P, Willart MAM, Kool M, et al. Inflammatory dendritic cells–not basophils–are necessary and sufficient for induction of Th2 immunity to inhaled house dust mite allergen. J Exp Med. (2010) 207:2097–111. doi: 10.1084/jem.20101563
75. Plantinga M, Guilliams M, Vanheerswynghels M, Deswarte K, Branco-Madeira F, Toussaint W, et al. Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity (2013) 38:322–35. doi: 10.1016/j.immuni.2012.10.016
76. van Rijt LS, Vos N, Willart M, Muskens F, Tak PP, van der Horst C, et al. Persistent activation of dendritic cells after resolution of allergic airway inflammation breaks tolerance to inhaled allergens in mice. Am J Respir Crit Care Med. (2011) 184:303–11. doi: 10.1164/rccm.201101-0019OC
77. de Kleer IM, Kool M, de Bruijn MJW, Willart M, van Moorleghem J, Schuijs MJ, et al. Perinatal activation of the interleukin-33 pathway promotes type 2 immunity in the developing lung. Immunity (2016) 45:1285–98. doi: 10.1016/j.immuni.2016.10.031
78. Phythian-Adams AT, Cook PC, Lundie RJ, Jones LH, Smith KA, Barr TA, et al. CD11c depletion severely disrupts Th2 induction and development in vivo. J Exp Med. (2010) 207:2089–96. doi: 10.1084/jem.20100734
79. Tang H, Cao W, Kasturi SP, Ravindran R, Nakaya HI, Kundu K, et al. The T helper type 2 response to cysteine proteases requires dendritic cell-basophil cooperation via ROS-mediated signaling. Nat Immunol. (2010) 11:608–17. doi: 10.1038/ni.1883
80. van Rijt LS, Jung S, KleinJan A, Vos N, Willart M, Duez C, et al. In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J Exp Med. (2005) 201:981–91. doi: 10.1084/jem.20042311
81. Thornton EE, Looney MR, Bose O, Sen D, Sheppard D, Locksley R, et al. Spatiotemporally separated antigen uptake by alveolar dendritic cells and airway presentation to T cells in the lung. J Exp Med. (2012) 209:1183–99. doi: 10.1084/jem.20112667
82. Medoff BD, Seung E, Hong S, Thomas SY, Sandall BP, Duffield JS, et al. CD11b+ myeloid cells are the key mediators of Th2 cell homing into the airway in allergic inflammation. J Immunol. (2009) 182:623–35. doi: 10.4049/jimmunol.182.1.623
83. Guilliams M, Lambrecht BN, Hammad H. Division of labor between lung dendritic cells and macrophages in the defense against pulmonary infections. Mucosal Immunol. (2013) 6:464–73. doi: 10.1038/mi.2013.14
84. Williams JW, Tjota MY, Clay BS, Vander Lugt B, Bandukwala HS, Hrusch CL, et al. Transcription factor IRF4 drives dendritic cells to promote Th2 differentiation. Nat Commun. (2013) 4:2990. doi: 10.1038/ncomms3990
85. Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity (2013) 38:970–83. doi: 10.1016/j.immuni.2013.04.011
86. Norimoto A, Hirose K, Iwata A, Tamachi T, Yokota M, Takahashi K, et al. Dectin-2 promotes house dust mite-induced T helper type 2 and type 17 cell differentiation and allergic airway inflammation in mice. Am J Respir Cell Mol Biol. (2014) 51:201–9. doi: 10.1165/rcmb.2013-0522OC
87. Nakano H, Burgents JE, Nakano K, Whitehead GS, Cheong C, Bortner CD, et al. Migratory properties of pulmonary dendritic cells are determined by their developmental lineage. Mucosal Immunol. (2013) 6:678–91. doi: 10.1038/mi.2012.106
88. Everts B, Tussiwand R, Dreesen L, Fairfax KC, Huang SC-C, Smith AM, et al. Migratory CD103+ dendritic cells suppress helminth-driven type 2 immunity through constitutive expression of IL-12. J Exp Med. (2016) 213:35–51. doi: 10.1084/jem.20150235
89. Semmrich M, Plantinga M, Svensson-Frej M, Uronen-Hansson H, Gustafsson T, Mowat AM, et al. Directed antigen targeting in vivo identifies a role for CD103+ dendritic cells in both tolerogenic and immunogenic T-cell responses. Mucosal Immunol. (2012) 5:150–60. doi: 10.1038/mi.2011.61
90. Khare A, Krishnamoorthy N, Oriss TB, Fei M, Ray P, Ray A. Cutting edge: inhaled antigen upregulates retinaldehyde dehydrogenase in lung CD103+ but not plasmacytoid dendritic cells to induce Foxp3 de novo in CD4+ T cells and promote airway tolerance. J Immunol. (2013) 191:25–9. doi: 10.4049/jimmunol.1300193
91. de Heer HJ, Hammad H, Soullié T, Hijdra D, Vos N, Willart MAM, et al. Essential role of lung plasmacytoid dendritic cells in preventing asthmatic reactions to harmless inhaled antigen. J Exp Med. (2004) 200:89–98. doi: 10.1084/jem.20040035
92. Oriss TB, Ostroukhova M, Seguin-Devaux C, Dixon-McCarthy B, Stolz DB, Watkins SC, et al. Dynamics of dendritic cell phenotype and interactions with CD4+ T cells in airway inflammation and tolerance. J Immunol. (2005) 174:854–63. doi: 10.4049/jimmunol.174.2.854
93. Kool M, van Nimwegen M, Willart MAM, Muskens F, Boon L, Smit JJ, et al. An anti-inflammatory role for plasmacytoid dendritic cells in allergic airway inflammation. J Immunol. (2009) 183:1074–82. doi: 10.4049/jimmunol.0900471
94. Lombardi V, Speak AO, Kerzerho J, Szely N, Akbari O. CD8α+β− and CD8α+β+ plasmacytoid dendritic cells induce Foxp3+ regulatory T cells and prevent the induction of airway hyper-reactivity. Mucosal Immunol. (2012) 5:432–43. doi: 10.1038/mi.2012.20
95. Lambrecht BN, Hammad H. The airway epithelium in asthma. Nat Med. (2012) 18:684–92. doi: 10.1038/nm.2737
96. Hammad H, Lambrecht BN. Barrier epithelial cells and the control of type 2 immunity. Immunity (2015) 43:29–40. doi: 10.1016/j.immuni.2015.07.007
97. Willart MAM, Deswarte K, Pouliot P, Braun H, Beyaert R, Lambrecht BN, et al. Interleukin-1α controls allergic sensitization to inhaled house dust mite via the epithelial release of GM-CSF and IL-33. J Exp Med. (2012) 209:1505–17. doi: 10.1084/jem.20112691
98. Schuijs MJ, Willart MA, Vergote K, Gras D, Deswarte K, Ege MJ, et al. Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells. Science (2015) 349:1106–10. doi: 10.1126/science.aac6623
99. Scandella E, Men Y, Legler DF, Gillessen S, Prikler L, Ludewig B, et al. CCL19/CCL21-triggered signal transduction and migration of dendritic cells requires prostaglandin E2. Blood (2004) 103:1595–601. doi: 10.1182/blood-2003-05-1643
100. van Helden SFG, Krooshoop DJEB, Broers KCM, Raymakers RAP, Figdor CG, van Leeuwen FN. A critical role for prostaglandin E2 in podosome dissolution and induction of high-speed migration during dendritic cell maturation. J Immunol. (2006) 177:1567–74. doi: 10.4049/jimmunol.177.3.1567
101. Legler DF, Krause P, Scandella E, Singer E, Groettrup M. Prostaglandin E2 is generally required for human dendritic cell migration and exerts its effect via EP2 and EP4 receptors. J Immunol. (2006) 176:966–73. doi: 10.4049/jimmunol.176.2.966
102. Harizi H, Grosset C, Gualde N. Prostaglandin E2 modulates dendritic cell function via EP2 and EP4 receptor subtypes. J Leukoc Biol. (2003) 73:756–63. doi: 10.1189/jlb.1002483
103. Angeli V, Faveeuw C, Roye O, Fontaine J, Teissier E, Capron A, et al. Role of the parasite-derived prostaglandin D2 in the inhibition of epidermal Langerhans cell migration during schistosomiasis infection. J Exp Med. (2001) 193:1135–47. doi: 10.1084/jem.193.10.1135
104. Hammad H, de Heer HJ, Soullié T, Hoogsteden HC, Trottein F, Lambrecht BN. Prostaglandin D2 inhibits airway dendritic cell migration and function in steady state conditions by selective activation of the D prostanoid receptor 1. J Immunol. (2003) 171:3936–40. doi: 10.4049/jimmunol.171.8.3936
105. Robbiani DF, Finch RA, Jäger D, Muller WA, Sartorelli AC, Randolph GJ. The leukotriene C4 transporter MRP1 regulates CCL19 (MIP-3β, ELC)–dependent mobilization of dendritic cells to lymph nodes. Cell (2000) 103:757–68. doi: 10.1016/S0092-8674(00)00179-3
106. Aliberti J, Hieny S, Reis e Sousa C, Serhan CN, Sher A. Lipoxin-mediated inhibition of IL-12 production by DCs: a mechanism for regulation of microbial immunity. Nat Immunol. (2002) 3:76–82. doi: 10.1038/ni745
107. Hilkens C, Snijders A, Vermeulen H, Meide PVD, Wierenga E, Kapsenberg M. Accessory cell-derived interleukin-12 and prostaglandin E 2determine the level of interferon- γ produced by activated human CD4 +T cells. Ann N Y Acad Sci. (1996) 795:349–50. doi: 10.1111/j.1749-6632.1996.tb52689.x
108. van der Pouw Kraan TC. Prostaglandin-E2 is a potent inhibitor of human interleukin 12 production. J Exp Med. (1995) 181:775–9. doi: 10.1084/jem.181.2.775
109. Kalinski P, Hilkens CM, Snijders A, Snijdewint FG, Kapsenberg ML. Dendritic cells, obtained from peripheral blood precursors in the presence of PGE2, promote Th2 responses. Adv Exp Med Biol. (1997) 417:363–7.
110. Kalinski P, Hilkens CM, Snijders A, Snijdewint FG, Kapsenberg ML. IL-12-deficient dendritic cells, generated in the presence of prostaglandin E2, promote type 2 cytokine production in maturing human naive T helper cells. J Immunol. (1997) 159:28–35.
111. Kalinski P, Schuitemaker JH, Hilkens CM, Kapsenberg ML. Prostaglandin E2 induces the final maturation of IL-12-deficient CD1a+CD83+ dendritic cells: the levels of IL-12 are determined during the final dendritic cell maturation and are resistant to further modulation. J Immunol. (1998) 161:2804–9.
112. Vieira PL, de Jong EC, Wierenga EA, Kapsenberg ML, Kalinski P. Development of Th1-inducing capacity in myeloid dendritic cells requires environmental instruction. J Immunol. (2000) 164:4507–12. doi: 10.4049/jimmunol.164.9.4507
113. Hedi H, Norbert G. Inhibition of IL-6, TNF-α, and cyclooxygenase-2 protein expression by prostaglandin E2-induced IL-10 in bone marrow-derived dendritic cells. Cell Immunol. (2004) 228:99–109. doi: 10.1016/j.cellimm.2004.04.003
114. Vassiliou E, Jing H, Ganea D. Prostaglandin E2 inhibits TNF production in murine bone marrow-derived dendritic cells. Cell Immunol. (2003) 223:120–32. doi: 10.1016/S0008-8749(03)00158-8
115. Harizi H, Juzan M, Pitard V, Moreau J-F, Gualde N. Cyclooxygenase-2-issued prostaglandin e(2) enhances the production of endogenous IL-10, which down-regulates dendritic cell functions. J Immunol. (2002) 168:2255–63. doi: 10.4049/jimmunol.168.5.2255
116. Walker W, Rotondo D. Prostaglandin E2 is a potent regulator of interleukin-12- and interleukin-18-induced natural killer cell interferon-gamma synthesis. Immunology (2004) 111:298–305. doi: 10.1111/j.1365-2567.2004.01810.x
117. Rieser C, Papesh C, Herold M, Böck G, Ramoner R, Klocker H, et al. Differential deactivation of human dendritic cells by endotoxin desensitization: role of tumor necrosis factor-alpha and prostaglandin E2. Blood (1998) 91:3112–7.
118. Rieser C, Böck G, Klocker H, Bartsch G, Thurnher M. Prostaglandin E2 and tumor necrosis factor alpha cooperate to activate human dendritic cells: synergistic activation of interleukin 12 production. J Exp Med. (1997) 186:1603–8.
119. Steinbrink K, Paragnik L, Jonuleit H, Tüting T, Knop J, Enk AH. Induction of dendritic cell maturation and modulation of dendritic cell-induced immune responses by prostaglandins. Arch Dermatol Res. (2000) 292:437–45. doi: 10.1007/s004030002920437.403
120. Lee YG, Jeong JJ, Nyenhuis S, Berdyshev E, Chung S, Ranjan R, et al. Recruited alveolar macrophages, in response to airway epithelial–derived monocyte chemoattractant protein 1/CCL2, regulate airway inflammation and remodeling in allergic asthma. Am J Respir Cell Mol Biol. (2015) 52:772–84. doi: 10.1165/rcmb.2014-0255OC
121. Rubio MT, Means TK, Chakraverty R, Shaffer J, Fudaba Y, Chittenden M, et al. Maturation of human monocyte-derived dendritic cells (MoDCs) in the presence of prostaglandin E2 optimizes CD4 and CD8 T cell-mediated responses to protein antigens: role of PGE2 in chemokine and cytokine expression by MoDCs. Int Immunol. (2005) 17:1561–72. doi: 10.1093/intimm/dxh335
122. Khayrullina T, Yen J-H, Jing H, Ganea D. In vitro differentiation of dendritic cells in the presence of prostaglandin E2 alters the IL-12/IL-23 balance and promotes differentiation of Th17 cells. J Immunol. (2008) 181:721–35. doi: 10.4049/jimmunol.181.1.721
123. Sheibanie AF, Tadmori I, Jing H, Vassiliou E, Ganea D. Prostaglandin E2 induces IL-23 production in bone marrow-derived dendritic cells. FASEB J. (2004) 18:1318–20. doi: 10.1096/fj.03-1367fje
124. Betz M, Fox BS. Prostaglandin E2 inhibits production of Th1 lymphokines but not of Th2 lymphokines. J Immunol. (1991) 146:108–13.
125. Snijdewint FG, Kalinski P, Wierenga EA, Bos JD, Kapsenberg ML. Prostaglandin E2 differentially modulates cytokine secretion profiles of human T helper lymphocytes. J Immunol. (1993) 150:5321–9.
126. Zasłona Z, Okunishi K, Bourdonnay E, Domingo-Gonzalez R, Moore BB, Lukacs NW, et al. Prostaglandin E2 suppresses allergic sensitization and lung inflammation by targeting the E prostanoid 2 receptor on T cells. J Allergy Clin Immunol. (2014) 133:379–87. doi: 10.1016/j.jaci.2013.07.037
127. Boniface K, Bak-Jensen KS, Li Y, Blumenschein WM, McGeachy MJ, McClanahan TK, et al. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. J Exp Med. (2009) 206:535–48. doi: 10.1084/jem.20082293
128. Tanaka K, Hirai H, Takano S, Nakamura M, Nagata K. Effects of prostaglandin D2 on helper T cell functions. Biochem Biophy Res Commun. (2004) 316:1009–14. doi: 10.1016/j.bbrc.2004.02.151
129. Jaffar Z, Wan K-S, Roberts K. A key role for prostaglandin I2 in limiting lung mucosal Th2, but not Th1, responses to inhaled allergen. J Immunol. (2002) 169:5997–6004. doi: 10.4049/jimmunol.169.10.5997
130. Machida I, Matsuse H, Kondo Y, Kawano T, Saeki S, Tomari S, et al. Cysteinyl leukotrienes regulate dendritic cell functions in a murine model of asthma. J Immunol. (2004) 172:1833–8. doi: 10.4049/jimmunol.172.3.1833
131. Okunishi K, Dohi M, Nakagome K, Tanaka R, Yamamoto K. A novel role of cysteinyl leukotrienes to promote dendritic cell activation in the antigen-induced immune responses in the lung. J Immunol. (2004) 173:6393–402. doi: 10.4049/jimmunol.173.10.6393
132. Barrett NA, Rahman OM, Fernandez JM, Parsons MW, Xing W, Austen KF, et al. Dectin-2 mediates Th2 immunity through the generation of cysteinyl leukotrienes. J Exp Med. (2011) 208:593–604. doi: 10.1084/jem.20100793
133. Doherty TA, Khorram N, Lund S, Mehta AK, Croft M, Broide DH. Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. J Allergy Clin Immunol. (2013) 132:205–13. doi: 10.1016/j.jaci.2013.03.048
134. Prinz I, Gregoire C, Mollenkopf H, Aguado E, Wang Y, Malissen M, et al. The type 1 cysteinyl leukotriene receptor triggers calcium influx and chemotaxis in mouse alpha beta- and gamma delta effector T cells. J Immunol. (2005) 175:713–9. doi: 10.4049/jimmunol.175.2.713
135. Yamaoka KA, Kolb J-P. Leukotriene B4 induces interleukin 5 generation from human T lymphocytes. Eur J Immunol. (1993) 23:2392–8. doi: 10.1002/eji.1830231003
136. Barrett NA, Fernandez JM, Maekawa A, Xing W, Li L, Parsons MW, et al. Cysteinyl leukotriene 2 receptor on dendritic cells negatively regulates ligand-dependent allergic pulmonary inflammation. J Immunol. (2012) 189:4556–65. doi: 10.4049/jimmunol.1201865
137. Cai Y, Kumar RK, Zhou J, Foster PS, Webb DC. Ym1/2 promotes Th2 cytokine expression by inhibiting 12/15(S)-lipoxygenase: identification of a novel pathway for regulating allergic inflammation. J Immunol. (2009) 182:5393–9. doi: 10.4049/jimmunol.0803874
138. Linton P-J, Bautista B, Biederman E, Bradley ES, Harbertson J, Kondrack RM, et al. Costimulation via OX40L expressed by B cells is sufficient to determine the extent of primary CD4 cell expansion and Th2 cytokine secretion in vivo. J Exp Med. (2003) 197:875–83. doi: 10.1084/jem.20021290
139. Rodríguez-Pinto D, Moreno J. B cells can prime naive CD4 +T cells in vivo in the absence of other professional antigen-presenting cells in a CD154-CD40-dependent manner. Eur J Immunol. (2005) 35:1097–105. doi: 10.1002/eji.200425732
140. Liu Q, Liu Z, Rozo CT, Hamed HA, Alem F, Urban JF, et al. The role of B cells in the development of CD4 effector T cells during a polarized Th2 immune response. J Immunol. (2007) 179:3821–30. doi: 10.4049/jimmunol.179.6.3821
141. Dwyer DF, Woodruff MC, Carroll MC, Austen KF, Gurish MF. B cells regulate CD4+ T cell responses to papain following B cell receptor-independent papain uptake. J Immunol. (2014) 193:529–39. doi: 10.4049/jimmunol.1303247
142. Dullaers M, Schuijs MJ, Willart M, Fierens K, van Moorleghem J, Hammad H, et al. House dust mite-driven asthma and allergen-specific T cells depend on B cells when the amount of inhaled allergen is limiting. J Allergy Clin Immunol. (2017) 140:76–88.e7. doi: 10.1016/j.jaci.2016.09.020
143. Yang Q, Shi M, Shen Y, Cao Y, Zuo S, Zuo C, et al. COX-1-derived thromboxane A2 plays an essential role in early B-cell development via regulation of JAK/STAT5 signaling in mouse. Blood (2014) 124:1610–21. doi: 10.1182/blood-2014-03-559658
144. Blaho VA, Buczynski MW, Dennis EA, Brown CR. Cyclooxygenase-1 orchestrates germinal center formation and antibody class-switch via regulation of IL-17. J Immunol. (2009) 183:5644–53. doi: 10.4049/jimmunol.0901499
145. Fedyk ER, Phipps RP. Prostaglandin E2 receptors of the EP2 and EP4 subtypes regulate activation and differentiation of mouse B lymphocytes to IgE-secreting cells. Proc Natl Acad Sci USA. (1996) 93:10978–83. doi: 10.1073/pnas.93.20.10978
146. Gao Y, Nish SA, Jiang R, Hou L, Licona-Limón P, Weinstein JS, et al. Control of T helper 2 responses by transcription factor IRF4-dependent dendritic cells. Immunity (2013) 39:722–32. doi: 10.1016/j.immuni.2013.08.028
147. Hajek AR, Lindley AR, Favoreto S, Carter R, Schleimer RP, Kuperman DA. 12/15-Lipoxygenase deficiency protects mice from allergic airways inflammation and increases secretory IgA levels. J Allergy Clin Immunol. (2008) 122:633–9.e3. doi: 10.1016/j.jaci.2008.06.021
148. Roper RL, Phipps RP. Prostaglandin E2 and cAMP inhibit B lymphocyte activation and simultaneously promote IgE and IgG1 synthesis. J Immunol. (1992) 149:2984–91.
149. Roper RL, Brown DM, Phipps RP. Prostaglandin E2 promotes B lymphocyte Ig isotype switching to IgE. J Immunol. (1995) 154:162–70.
150. Roper RL, Conrad DH, Brown DM, Warner GL, Phipps RP. Prostaglandin E2 promotes IL-4-induced IgE and IgG1 synthesis. J Immunol. (1990) 145:2644–51.
151. Gao Y, Zhao C, Wang W, Jin R, Li Q, Ge Q, et al. Prostaglandins E2 signal mediated by receptor subtype EP2 promotes IgE production in vivo and contributes to asthma development. Sci Rep. (2016) 6:20505. doi: 10.1038/srep20505
152. Murn J, Alibert O, Wu N, Tendil S, Gidrol X. Prostaglandin E2 regulates B cell proliferation through a candidate tumor suppressor, Ptger4. J Exp Med. (2008) 205:3091–103. doi: 10.1084/jem.20081163
153. Lamoureux J, Stankova J, Rolapleszczynski M. Leukotriene D4 enhances immunoglobulin production in CD40-activated human B lymphocytes. J Allergy Clin Immunol. (2006) 117:924–30. doi: 10.1016/j.jaci.2005.12.1329
154. Mirchandani AS, Besnard A-G, Yip E, Scott C, Bain CC, Cerovic V, et al. Type 2 innate lymphoid cells drive CD4+ Th2 cell responses. J Immunol. (2014) 192:2442–8. doi: 10.4049/jimmunol.1300974
155. Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity (2014) 41:283–95. doi: 10.1016/j.immuni.2014.06.016
156. Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S, et al. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci Transl Med. (2013) 5:174ra26. doi: 10.1126/scitranslmed.3004812
157. Salimi M, Stöger L, Liu W, Go S, Pavord I, Klenerman P, et al. Cysteinyl leukotriene E4 activates human group 2 innate lymphoid cells and enhances the effect of prostaglandin D2 and epithelial cytokines. J Allergy Clin Immunol. (2017) 140:1090–100.e11. doi: 10.1016/j.jaci.2016.12.958
158. Maric J, Ravindran A, Mazzurana L, Björklund ÅK, Van Acker A, Rao A, et al. Prostaglandin E 2 suppresses human group 2 innate lymphoid cell function. J Allergy Clin Immunol. (2017) 141:1761–73.e6. doi: 10.1016/j.jaci.2017.09.050
159. Zhou W, Toki S, Zhang J, Goleniewksa K, Newcomb DC, Cephus JY, et al. Prostaglandin I2 signaling and inhibition of group 2 innate lymphoid cell responses. Am J Respir Crit Care Med. (2016) 193:31–42. doi: 10.1164/rccm.201410-1793OC
160. Lukic A, Larssen P, Fauland A, Samuelsson B, Wheelock CE, Gabrielsson S, et al. GM-CSF- and M-CSF-primed macrophages present similar resolving but distinct inflammatory lipid mediator signatures. FASEB J. (2017) 31:4370–81. doi: 10.1096/fj.201700319R
161. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. (2013) 210:1977–92. doi: 10.1084/jem.20131199
162. van de Laar L, Saelens W, De Prijck S, Martens L, Scott CL, Van Isterdael G, et al. Yolk sac macrophages, fetal liver, and adult monocytes can colonize an empty niche and develop into functional tissue-resident macrophages. Immunity (2016) 44:755–68. doi: 10.1016/j.immuni.2016.02.017
163. Westphalen K, Gusarova GA, Islam MN, Subramanian M, Cohen TS, Prince AS, et al. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature (2014) 506:503–6. doi: 10.1038/nature12902
164. Bhattacharya J, Westphalen K. Macrophage-epithelial interactions in pulmonary alveoli. Semin Immunopathol. (2016) 38:461–9. doi: 10.1007/s00281-016-0569-x
165. Upham JW, Strickland DH, Bilyk N, Robinson BW, Holt PG. Alveolar macrophages from humans and rodents selectively inhibit T-cell proliferation but permit T-cell activation and cytokine secretion. Immunology (1995) 84:142–7.
166. Tang C, Ward C, Reid D, Bish R, O'Byrne PM, Walters EH. Normally suppressing CD40 coregulatory signals delivered by airway macrophages to TH2 lymphocytes are defective in patients with atopic asthma. J Allergy Clin Immunol. (2001) 107:863–70. doi: 10.1067/mai.2001.114987
167. Tang C, Inman MD, van Rooijen N, Yang P, Shen H, Matsumoto K, et al. Th type 1-stimulating activity of lung macrophages inhibits Th2-mediated allergic airway inflammation by an IFN-gamma-dependent mechanism. J Immunol. (2001) 166:1471–81. doi: 10.4049/jimmunol.166.3.1471
168. Zasłona Z, Przybranowski S, Wilke C, Van Rooijen N, Teitz-Tennenbaum S, Osterholzer JJ, et al. Resident alveolar macrophages suppress, whereas recruited monocytes promote, allergic lung inflammation in murine models of asthma. J Immunol. (2014) 193:4245–53. doi: 10.4049/jimmunol.1400580
169. Bourdonnay E, Zasłona Z, Penke LRK, Speth JM, Schneider DJ, Przybranowski S, et al. Transcellular delivery of vesicular SOCS proteins from macrophages to epithelial cells blunts inflammatory signaling. J Exp Med. (2015) 212:729–42. doi: 10.1084/jem.20141675
170. Speth JM, Bourdonnay E, Penke LRK, Mancuso P, Moore BB, Weinberg JB, et al. Alveolar epithelial cell-derived prostaglandin E2 serves as a request signal for macrophage secretion of suppressor of cytokine signaling 3 during innate inflammation. J Immunol. (2016) 196:5112–20. doi: 10.4049/jimmunol.1502153
171. Draijer C, Boorsma CE, Reker-Smit C, Post E, Poelstra K, Melgert BN. PGE2-treated macrophages inhibit development of allergic lung inflammation in mice. J Leukoc Biol. (2016) 100:95–102. doi: 10.1189/jlb.3MAB1115-505R
172. Ohta S, Imamura M, Xing W, Boyce JA, Balestrieri B. Group V secretory phospholipase A2 is involved in macrophage activation and is sufficient for macrophage effector functions in allergic pulmonary inflammation. J Immunol. (2013) 190:5927–38. doi: 10.4049/jimmunol.1203202
173. Yamaguchi M, Zacharia J, Laidlaw TM, Balestrieri B. PLA2G5 regulates transglutaminase activity of human IL-4-activated M2 macrophages through PGE2 generation. J Leukoc Biol. (2016) 100:131–41. doi: 10.1189/jlb.3A0815-372R
174. Lauzon-Joset J-F, Marsolais D, Langlois A, Bissonnette EY. Dysregulation of alveolar macrophages unleashes dendritic cell-mediated mechanisms of allergic airway inflammation. Mucosal Immunol. (2014) 7:155–64. doi: 10.1038/mi.2013.34
175. Samuchiwal SK, Balestrieri B, Raff H, Boyce JA. Endogenous prostaglandin E 2amplifies IL-33 production by macrophages through an E prostanoid (EP) 2/EP 4-cAMP-EPAC-dependent pathway. J Biol Chem. (2017) 292:8195–206. doi: 10.1074/jbc.M116.769422
176. Jandl K, Stacher E, Bálint Z, Sturm EM, Maric J, Peinhaupt M, et al. Activated prostaglandin D2 receptors on macrophages enhance neutrophil recruitment into the lung. J Allergy Clin Immunol. (2016) 137:833–43. doi: 10.1016/j.jaci.2015.11.012
177. Johnston SL, Pattemore PK, Sanderson G, Smith S, Lampe F, Josephs L, et al. Community study of role of viral infections in exacerbations of asthma in 9-11 year old children. BMJ (1995) 310:1225–9. doi: 10.1136/bmj.310.6989.1225
178. Shiraishi Y, Asano K, Niimi K, Fukunaga K, Wakaki M, Kagyo J, et al. Cyclooxygenase-2/prostaglandin D2/CRTH2 pathway mediates double-stranded RNA-induced enhancement of allergic airway inflammation. J Immunol. (2008) 180:541–9. doi: 10.4049/jimmunol.180.1.541
179. Shirey KA, Pletneva LM, Puche AC, Keegan AD, Prince GA, Blanco JCG, et al. Control of RSV-induced lung injury by alternatively activated macrophages is IL-4Rα-, TLR4- and IFN-β-dependent. Mucosal Immunol. (2010) 3:291–300. doi: 10.1038/mi.2010.6
180. Shirey KA, Lai W, Pletneva LM, Karp CL, Divanovic S, Blanco JCG, et al. Role of the lipoxygenase pathway in RSV-induced alternatively activated macrophages leading to resolution of lung pathology. Mucosal Immunol. (2013) 7:549–57. doi: 10.1038/mi.2013.71
Keywords: eicosanoids, prostaglandins, leukotrienes, asthma, dendritic cells, macrophages
Citation: Debeuf N and Lambrecht BN (2018) Eicosanoid Control Over Antigen Presenting Cells in Asthma. Front. Immunol. 9:2006. doi: 10.3389/fimmu.2018.02006
Received: 02 July 2018; Accepted: 14 August 2018;
Published: 04 September 2018.
Edited by:
Otto Holst, Forschungszentrum Borstel (LG), GermanyReviewed by:
Subhabrata Moitra, Instituto Salud Global Barcelona (ISGlobal), SpainCopyright © 2018 Debeuf and Lambrecht. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bart N. Lambrecht, YmFydC5sYW1icmVjaHRAdWdlbnQuYmU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.