 Hiroaki Takatori1,2*
Hiroaki Takatori1,2* Takashi Ito
Takashi Ito Ayako Matsuki
Ayako Matsuki Hiroshi Nakajima
Hiroshi Nakajima
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 04 September 2018
Sec. Cytokines and Soluble Mediators in Immunity
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.02004
This article is part of the Research Topic Interleukin-33 Biology in Tissue Development, Homeostasis and Disease View all 20 articles
Interleukin-33 (IL-33) plays multiple roles in tissue homeostasis, prevention of parasitic infection, and induction of allergic inflammation. Especially, IL-33-ST2 (IL-1RL1) axis has been regarded as the villain in allergic diseases such as asthma and atopic dermatitis and in autoimmune diseases such as rheumatoid arthritis. Indeed, a number of studies have indicated that IL-33 produced by endothelial cells and epithelial cells plays a critical role in the activation and expansion of group 2 innate lymphoid cells (ILC2s) which cause allergic inflammation by producing large amounts of IL-5 and IL-13. However, mechanisms that antagonize IL-33-ST2-mediated allergic responses remain largely unknown. Recently, several groups including our group have demonstrated cellular and molecular mechanisms that could suppress excessive activation of ILC2s by the IL-33-ST2 axis. In this review, we summarize recent progress in the regulatory mechanisms of IL-33-ST2-mediated allergic responses. Selective targeting of the IL-33-ST2 axis would be a promising strategy in the treatment of allergic diseases.
Interleukin-33 (IL-33) is a member of the IL-1 cytokine family and mainly expressed in non-hematopoietic cells such as endothelial cells, epithelial cells, fibroblast-like cells, and myofibroblasts during homeostasis and in response to inflammation (1–5). IL-33 functions on target cells by binding to a heterodimeric receptor composed of suppression of tumorigenicity 2 (ST2, also known as IL-1RL1) and co-receptor, IL-1 receptor accessory protein (IL-1RAcP) (6, 7). Triggering of ST2/IL-1RAcP by IL-33 activates intracellular signaling pathways, which is initiated by homotypic protein-protein interactions with the adaptor molecule MyD88 and the further recruitment of IRAKs and TRAF6, leading to the expression of several inflammatory mediators through the activation of nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) pathways (8, 9). IL-33 activates a variety of tissue-resident immune cells that express ST2, including group 2 innate lymphoid cells (ILC2s), mast cells, Th2 cells, regulatory T cells (Tregs), natural killer (NK) cells, eosinophils, basophils, dendritic cells, and alternatively activated macrophages (10–12).
Regarding in vivo relevance of IL-33-ST2 axis, it has been shown that bronchial epithelium is an important reservoir of IL-33 in the lung and that IL-33 expression is elevated in the airways of bronchial asthma along with disease severity (13). It has also been shown that increased IL-33 expression is associated with enhanced reticular basement membrane thickness in endobronchial tissues in children with severe therapy-resistant asthma (14). In addition, it has been reported that airway remodeling is absent in ST2−/− mice in house dust mite (HDM)-induced neonatal asthma models (14). Moreover, IL-33 expression is not affected by steroid treatment in neonatal HDM-treated mice and in endobronchial tissues from children with severe therapy-resistant asthma (14). These findings suggest that IL-33-ST2 axis causes refractoriness in the allergic asthma.
Recently, numerous studies analyzing patient samples and murine models have revealed that ILC2s produce large amounts of IL-5 and IL-13 in response to IL-33 and IL-25 and that the activation of ILC2s by IL-33-ST2-mediated signaling contributes to anti-helminth responses and to the development of various allergic diseases such as asthma, atopic dermatitis, allergic rhinitis, and chronic rhinosinusitis (10–12, 15, 16). Furthermore, IL-33-ST2-mediated signaling seems to be involved in the development and exacerbation of non-allergic inflammatory conditions including arthritis, chronic obstructive pulmonary disease (COPD) with cigarette smoke, experimental autoimmune encephalomyelitis (EAE), periodontitis, and retina inflammation in murine models (12), although it remains unclear whether IL-33-ST2-ILC2s axis is involved in the pathogenesis of corresponding diseases in humans. Because IL-33-ST2-mediated signaling seems critical for the induction of excessive inflammatory responses in many conditions, it would be worthy of further investigation elucidating regulatory mechanisms against IL-33-ST2-mediated signaling to develop therapeutic strategies for these inflammatory diseases. In this review, we summarize recent progress in the regulatory mechanisms for IL-33-ST2-mediated allergic inflammation. Comprehensive reviews of the biology of IL-33 and its involvement in non-allergic conditions including autoimmune diseases and cancer have recently be described elsewhere (5, 9, 12).
Bioactivity of IL-33-ST2 pathways is regulated by several distinct mechanisms (Figure 1). It has been shown that many of the single nucleotide polymorphisms (SNPs) in the human IL-33 gene associated with asthma are located in the promoter and intron 1, both of which are important for gene expression (17). Imai et al. have shown that keratinocyte-specific over-expression of IL-33 results in the spontaneous development of an atopic dermatitis (AD)-like skin disease, along with the activation of ST2+ ILC2s (18). In addition, transgenic mice expressing IL-33 by a keratin 14 promoter spontaneously developed keratoconjunctivitis (19). These findings suggest that the expression level of IL-33 is critical for the induction of allergic inflammation and thus the suppression of IL-33 expression seems to be a good therapeutic strategy for the treatment of allergic diseases. In this regard, we have recently reported that IL-22, which belongs to the IL-10 family of cytokines, induces the expression of Reg3γ from lung epithelial cells in mice and that Reg3γ suppresses HDM-induced IL-33 expression and accumulation of ILC2s in the lung (20) (Figure 1). These results indicate that the IL-22-Reg3γ pathway is one of endogenous mechanisms that inhibit IL-33 expression. TGF-β is also shown to inhibit IL-33 expression in lung epithelial cells (21).
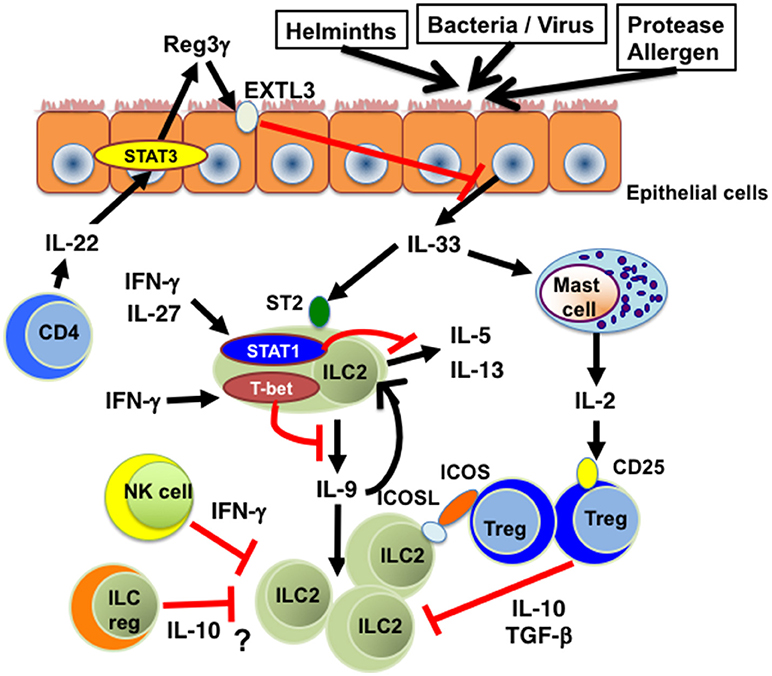
Figure 1. The inhibitory effects of cytokines on IL-33-ST2-ILC2 axis. Th1 cell-related cytokines such as IFN-γ and IL-27 inhibit IL-33-induced cytokine production and proliferation of ILC2s in a STAT1-dependent manner. T-bet expressed in ILC2s suppresses IL-9 production and subsequent expansion of ILC2s. IL-22 produced by CD4+ T cells suppresses IL-33 production from lung epithelial cells through Reg3γ induction. IL-2 produced by IL-33-stimulated mast cells promotes the expansion of Tregs, which suppress ILC2-mediated inflammation through the production of IL-10 and TGF-β. ICOS-ICOSL-mediated cell contact is also involved in Treg-mediated ILC2 suppression. In addition, IFN-γ produced by NK cells inhibits cytokine production and function of ILC2s. Moreover, IL-10-producing ILCregs could be a suppresser of IL-33-ST2-ILC2-mediated inflammation. EXTL3, exostatin-like 3; ICOS, inducible T-cell costimulator; ICOSL, inducible T-cell costimulator ligand; ILC2, group 2 innate lymphoid cells; ILCreg, regulatory innate lymphoid cell. NK cell, natural killer cell; Reg3γ, regenerating islet-derived protein 3 gamma; STAT1, signal transducer and activator or transcription 1; STAT3, signal transducer and activator or transcription 3; ST2, suppression of tumorigenicity 2; T-bet, T-box transcription factor expressed in T-cells; Treg, regulatory T cell.
Recent studies indicate that microRNAs (miRNAs) also play pivotal roles in the regulation of the IL-33-ST2 axis. miRNAs are short noncoding RNAs that regulate the expression of various cytokines and pathogen recognition receptors at post-transcriptional levels by binding to the 3′-untranslated region (UTR) (22, 23). Regarding the regulation of IL-33 expression, Tang et al. have reported that miR-200b and miR-200c are significantly reduced in asthmatic patients and the induction of miR-200b and miR-200c decreases IL-33 expression in lung epithelial cells by binding 3′UTR of IL-33 mRNA (24). miR-487b has also been shown to suppress IL-33 expression in bone marrow-derived macrophages (25) and lung epithelial cells (26). On the other hand, RNA binding protein Mex-3B upregulates IL-33 expression by inhibiting miR-487b-3p-mediated repression of IL-33 (26). Additionally, it has been shown that miR-155−/− mice exhibit impaired IL-33 levels in lung in response to allergen challenge, suggesting the involvement of miR-155 in the induction of IL-33 expression (27). These findings indicate that multiple miRNAs are involved in the regulation of IL-33 expression during allergic responses.
The bioactivity of IL-33 is also regulated by posttranslational modification of IL-33 (5) (Figure 2). IL-33 is known to accumulate in the nucleus of producing cells by binding to histones and chromatin (1, 3, 28, 29). The N-terminal domain of IL-33 contains a chromatin-binding motif and a predicted nuclear localization sequence (28, 29). Importantly, the deletion of N-terminal domain containing a chromatin-binding motif has been shown to lead to early lethality in mice by constitutive secretion of IL-33 in serum, which causes severe multi-organ inflammation with infiltration of various immune cells including eosinophils, monocytes, neutrophils, and macrophages (30). These findings indicate that sequestration of IL-33 in the nucleus of IL-33-producing (or necrotic) cells is important to avoid the constitutive release of IL-33 and subsequent systemic inflammation. Interestingly, Osbourn et al. have recently shown that a product secreted by the parasite Heligmosomoides polygyrus (H. polygyrus Alarmin Release Inhibitor; HpARI) binds to IL-33 and also to nuclear DNA via its N-terminal complement control protein module pair (CCP1/2), tethering active IL-33 within necrotic cells, preventing its release, and inhibiting initiation of allergic responses (31). These findings suggest that H. polygyrus may escape from IL-33-mediated anti-parasitic responses via the production of HpARI.
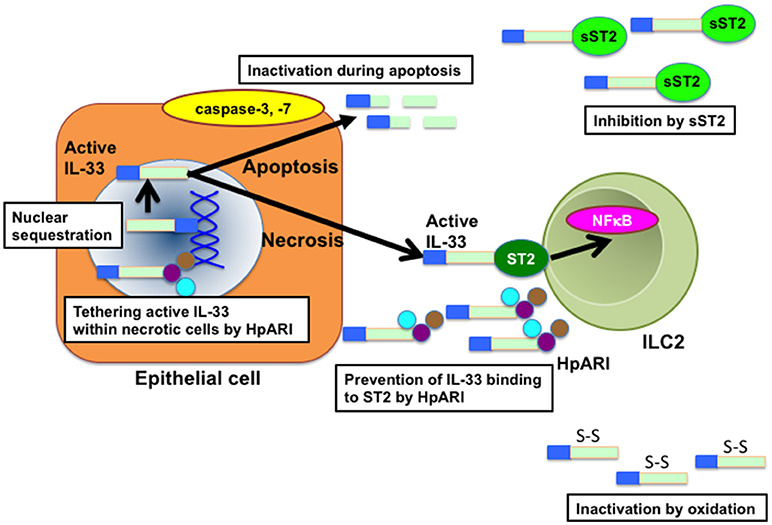
Figure 2. Molecular mechanisms that affect IL-33 bioactivity. Nuclear localization of IL-33 by binding to histone and chromatin is essential for the suppression of IL-33 bioactivity. At cell necrosis, active IL-33 is released from the nuclei of producing cells. Released active IL-33 is abrogated by several mechanisms such as inhibition by decoy receptor sST2 and inactivation by the conformational change. In addition, active IL-33 is cleaved and inactivated by caspases such as caspase-3 and caspase-7 during apoptosis of producing cells. HpARI secreted by H. polygylus inhibits the release of IL-33 by binding directly to active IL-33 and nuclear DNA, tethering active IL-33 within necrotic cells. HpARI also prevents binding of active IL-33 to ST2. HpARI, Heligmosomooides polygyrus alarmin release inhibitor; NFκB, nuclear factor-kappa B. sST2, soluble suppression of tumorigenicity 2.
It has also been reported that processing of IL-33 by proteases is crucial to regulate the bioactivity of IL-33 (Figure 2). IL-33 is cleaved by caspase-3 and caspase-7 during apoptosis, which generate two inactive IL-33 products (32–34). In addition, caspase-1 has been shown to cleave and inactivate IL-33 (32, 35, 36). Consistently, the administration of IL-33 greatly enhanced HDM-induced eosinophilic inflammation in caspase-1-deficient mice as compared with that in wild-type (WT) mice (36). Interestingly, cleaved chitin by acidic mammalian chitinase (AMCase) promotes the inactivation of IL-33 by inducing the activation of caspase-1 and subsequent activation of caspase-7, whereas uncleaved chitin promotes the release of IL-33 (35–37). Moreover, Cayrol et al. have recently shown that full-length IL-33 is cleaved by proteases derived from various environmental allergens such as fungi, HDM, cockroaches, and pollens and that the cleaved short mature forms of IL-33 are potent inducers of allergic airway inflammation (38). These findings suggest that the bioactivity of IL-33 is intricately regulated by endogenous and exogenous proteases.
The bioactivity of IL-33 is also controlled by the regulation of IL-33 binding to the receptor (Figure 2). IL-33 is shown to be rapidly inactivated in the extracellular lung environment by the oxidation of four critical cysteine residues and the formation of two disulfide bridges in the IL-1-like cytokine domain, resulting in an extensive conformational change that leads to the disruption of ST2 binding site (39). On the other hand, soluble spliced variant of ST2 (sST2), which directly binds to IL-33 and inhibits IL-33 bioactivity as a decoy receptor (40–42) is highly produced by mast cells and Th2 cells in asthmatic patients (40).
IL-33-ST2 signaling is also inhibited by SIGIRR (single immunoglobulin IL-1R-related molecule) which forms a complex with ST2 upon IL-33 stimulation (43). FBXL19, an orphan member of the Skp1-Cullin-F-box family of E3 ubiquitin ligases, selectively binds to ST2 to induce polyubiquitination and subsequent degradation of ST2 (44). Interestingly, IL-33 itself diminishes ST2 expression in mouse lung epithelial cells (MLE12 cells) in an FBXL19-dependent manner as one of negative feedback mechanisms (44).
Recent studies have shown that IL-33-ST2-ILC2s axis is inhibited by several distinct mechanisms (Figure 1). It has been reported that pro-inflammatory cytokines such as type 1 interferons and Th1 cell-inducing cytokines such as IFN-γ and IL-27 inhibit IL-33-ST2-ILC2s axis in vivo as well as in vitro. Type 1 interferons including IFN-β suppress the proliferation and cytokine production of IL-33-activated murine and human ILC2s in a manner dependent on interferon-stimulated gene factor 3 (ISGF3) (45). Both IFN-γ and IL-27 have also been shown to inhibit cytokine production by ILC2s in IL-33-injected mice in a STAT1-dependent manner (45–48). In addition, STAT1-deficient mice have been shown to exhibit significantly increased IL-33 expression as compared with WT mice during respiratory syncytial virus (RSV) infection (49).
Both IFN-γ and IL-27 induce the expression of T-bet, a master regulator of Th1 cells (50), in CD4+ T cells in a STAT1-dependent manner (51, 52). In this regard, we found that T-bet is expressed in lung ILC2s upon IFN-γ stimulation and that T-bet inhibits IL-33-induced accumulation of ILC2s in the lung and subsequent eosinophilic airway inflammation (16). Importantly, IL-9 production is significantly enhanced in T-bet−/− ILC2s and the neutralization of IL-9 markedly attenuates IL-33-induced eosinophilic airway inflammation in T-bet−/− mice (16). These results indicate that T-bet expressed in ILC2s plays an inhibitory role in IL-33-ST2-induced accumulation of lung ILC2s by inhibiting IL-9 production, consistent with a previous report showing that IL-9 promotes cell survival of ILC2s in an autocrine mechanism (53).
Regarding the other extracellular factors involved in the regulation of IL-33 bioactivity, it has been shown that prostaglandin D2 (PGD2) in combination with IL-33 stimulates IL-13 secretion and ST2 expression in human ILC2s through CRTH2 (chemoattractant receptor-homologous molecule expressed on Th2 cells) (54, 55). On the other hand, lipoxin A4 (LXA4), which is a natural pro-resolving ligand for ALX/FPR2 receptors, decreases cytokine production by ILC2s in response to PGD2 and IL-33 (56). PGE2 and PGI2 have also been shown to exhibit a profound inhibitory effect on IL-33-mediated ILC2 expansion and cytokine production in mice and humans (57–59). Mechanistic studies revealed that PGE2- EP4 (E-prostanoid 4)-cyclic AMP signaling leads to the suppression of GATA3 and ST2 expression in ILC2s (59). These reports suggest that lipid mediators also seem to be critical factors for modulation of IL-33-ST2-ILC2 axis.
In addition, Laffont et al. have reported that androgen receptor-mediated signaling decreases the numbers of ILC2 progenitors in bone marrow and suppresses IL-33-induced ILC2-mediated airway inflammation (60). Moreover, upon IL-33 administration, the number of ILC2s is significantly increased in gonadectomized male mice as compared to sham-operated male mice (61). Consistently, testosterone attenuates alternaria extract-induced cytokine expression of ILC2s and eosinophils infiltration by decreasing the numbers of lung ILC2s (61). These results indicate that the imbalance in sexual hormones could be involved in the regulation of IL-33-mediated inflammatory diseases.
Recently, Moriyama et al. have reported that murine ILC2s express high levels of β2-adrenergic receptor (β2AR) (62) and the treatment with β2AR agonist (salmeterol) inhibits IL-33-induced IL-5 and IL-13 production of ILC2s, suggesting that the IL-33-ST2-ILC2 axis could be regulated by nervous systems including β2AR signaling (63). Moreover, Thio et al. have shown that butyrate inhibits IL-33-induced GATA3 expression and cytokine production in ILC2s (64). On the other hand, Suzuki et al. have shown that IL-33 enhances the expression of Spred1 (sprouty-related Ena/VASP homology 1 domain-containing protein) in ILC2s, which negatively regulates ILC2 expansion and cytokine production by suppressing the Ras-ERK pathway (65).
Several cell types have been shown to regulate IL-33-ST2-ILC2 axis (Figure 1) in allergy. Roy et al. have shown that chymase derived from mast cells degrades IL-33 and that IL-33-induced TNF-α response is enhanced in chymase-deficient mice compared with WT mice (66). Moreover, it has been shown that IL-2 produced by IL-33-stimulated mast cells promotes the expansion of IL-10-producing CD4+ CD25+ Foxp3+ Tregs, thereby suppressing the development of IL-33-induced airway eosinophilia (67). These findings suggest that mast cells suppress IL-33-ST2-mediated inflammation through at least two distinct mechanisms. On the other hand, Rigas et al. have shown that iTregs attenuate IL-33-induced airway hyperreactivity by inhibiting IL-5 and IL-13 production from ILC2s and that the interaction of inducible T-cell costimulator ligand (ICOSL) on ILC2s with ICOS on Tregs is required for Treg-mediated suppression of ILC2 function (68). IFN-γ-producing NK cells also inhibit the cytokine expression and function of ILC2s (69).
More recently, Wang et al. have demonstrated that a regulatory subpopulation of ILCs (called ILCregs), which produce IL-10 and TGF-β, exists in murine and human intestine (70) and that ILCregs suppress intestinal inflammation driven by ILC1s and ILC3s via the secretion of IL-10 (70). Although it is reported that ILCregs isolated from the intestine do not suppress IL-33-induced IL-5 and IL-13 production from ILC2s (60), it remains unclear whether ILCregs are involved in the down-regulation of ILC2-mediated allergic responses in vivo.
Regarding the IL-33-ST2 axis in human allergic diseases, genome-wide association studies have clarified that a variety of SNPs in IL33 gene and IL1RL1 (ST2) gene are associated with asthma susceptibility (17). Additionally, Savenije et al. have reported that SNPs of IL1RAP and TRAF6, a downstream molecule of the IL-33-ST2 pathway, are associated with a phenotype of intermediate-onset wheeze, which is closely associated with sensitization (71), suggesting that polymorphisms of genes involved in IL-33-ST2 axis are associated with onset of asthma in childhood. Consistently, it has been shown that serum sST2 levels correlate well with the severity of asthma exacerbation (72). Moreover, a rare loss of function mutation in IL-33 gene has been shown to cause reduced numbers of eosinophils in blood and to protect against asthma (73). These finding suggest that the IL-33-ST2 axis seems to be a rational target in the treatment of allergic diseases.
Based on these findings, biologics including three anti-IL-33 monoclonal antibodies (ANB020; AnaptysBio, AMG282; Genentech, and REGN3500; Regeneron Pharmaceuticals) and an anti-ST2 monoclonal antibody (GSK3772847; GlaxoSmithKline) have already been under development and clinical trials have commenced (http://www.clinicaltrials.gov/). A phase 2 study of ANB020 for peanut allergy have been completed, and subjects are currently being recruited in phase 2 trials for severe eosinophilic asthma and atopic dermatitis. Phase 1 trials of AMG282 for mild atopic asthma and chronic rhinosinusitis with nasal polyps have also been completed. In addition, REGN3500 is being examined for asthma in a phase 1 trial. ST2 -targeting therapies with GSK3772847 for asthma are also currently in clinical trials. Because it is currently difficult to delete ILC2s in vivo, the neutralization of IL-33-ST2 pathway is a practical approach to regulate IL-33-ST2-ILC2 axis for the treatment of refractory allergic diseases.
In the past two decades, IL-33 has been regarded as an important initiator of immune responses. Additionally, recent studies have provided strong evidence that IL-33-ST2-ILC2s axis is one of central pathways in the pathogenesis of allergic inflammation. Accordingly, biologics targeting IL-33-ST2-ILC2 axis have already been under development. In addition, recent studies have shown that IL-33-ST2-ILC2s axis is negatively regulated by both cell intrinsic and extrinsic mechanisms. Given that IL-33-ST2-ILC2s axis seems to be deeply involved in the initiation phase of allergic diseases, these studies on the inhibitory mechanisms have yielded critical insights into the pathogenic mechanism underlying the onset of allergic diseases. Although further studies are needed to address how IL-33-ST2-ILC2s axis is negatively regulated in pathological situations in more detail, selective targeting of IL-33-ST2-ILC2s axis would be a promising strategy for the treatment of allergic diseases.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported in part by Grants-in-Aids for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, the Japanese Government, LGS Program (Leading Graduate School at Chiba University), MEXT, and Institute for Global Prominent Research, Chiba University, Japan.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, et al. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci USA. (2007) 104:282–7. doi: 10.1073/pnas.0606854104
2. Küchler AM, Pollheimer J, Balogh J, Sponheim J, Manley L, Sorensen DR, et al. Nuclear interleukin-33 is generally expressed in resting endothelium but rapidly lost upon angiogenic or proinflammatory activation. Am J Pathol. (2008) 173:1229–42. doi: 10.2353/ajpath.2008.080014
3. Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin'? PLoS ONE (2008) 3:e3331. doi: 10.1371/journal.pone.0003331
4. Pichery M, Mirey E, Mercier P, Lefrancais E, Dujardin A, Ortega N, et al. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel IL-33-LacZ gene trap reporter strain. J Immunol. (2012) 188:3488–95. doi: 10.4049/jimmunol.1101977
5. Cayrol C, Girard JP. Interleukin-33 (IL-33): a nuclear cytokine from the IL-1 family. Immunol Rev. (2018) 281:154–68. doi: 10.1111/imr.12619
6. Lingel A, Weiss TM, Niebuhr M, Pan B, Appleton BA, Wiesmann C, et al. Structure of IL-33 and its interaction with the ST2 and IL-1RAcP receptors–insight into heterotrimeric IL-1 signaling complexes. Structure (2009) 17:1398–410. doi: 10.1016/j.str.2009.08.009
7. Liu X, Hammel M, He Y, Tainer JA, Jeng US, Zhang L, et al. Structural insights into the interaction of IL-33 with its receptors. Proc Natl Acad Sci USA. (2013) 110:14918–23. doi: 10.1073/pnas.1308651110
8. Martin MU. Special aspects of interleukin-33 and the IL-33 receptor complex. Semin Immunol. (2013) 25:449–57. doi: 10.1016/j.smim.2013.10.006
9. Braun H, Afonina IS, Mueller C, Beyaert R. Dichotomous function of IL-33 in health and disease: from biology to clinical implications. Biochem Pharmacol. (2018) 148:238–52. doi: 10.1016/j.bcp.2018.01.010
10. Cayrol C, Girard JP. IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol. (2014) 31:31–7. doi: 10.1016/j.coi.2014.09.004
11. Molofsky AB, Savage AK, Locksley RM. Interleukin-33 in tissue homeostasis, injury, and inflammation. Immunity (2015) 42:1005–19. doi: 10.1016/j.immuni.2015.06.006
12. Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. (2016) 16:676–89. doi: 10.1016/j.ejphar.2015.04.047
13. Préfontaine D, Nadigel J, Chouiali F, Audusseau S, Semlali A, Chakir J, et al. Increased IL-33 expression by epithelial cells in bronchial asthma. J Allergy Clin Immunol. (2010) 125:752–4. doi: 10.1016/j.jaci.2009.12.935
14. Saglani S, Lui S, Ullmann N, Campbell GA, Sherburn RT, Mathie SA, et al. IL-33 promotes airway remodeling in pediatric patients with severe steroid-resistant asthma. J Allergy Clin Immunol. (2013) 132:676–85.e13. doi: 10.1016/j.jaci.2013.04.012
15. Kim BS, Wojno ED, Artis D. Innate lymphoid cells and allergic inflammation. Curr Opin Immunol. (2013) 25:738–44. doi: 10.1016/j.coi.2013.07.013
16. Matsuki A, Takatori H, Makita S, Yokota M, Tamachi T, Suto A, et al. T-bet inhibits innate lymphoid cell-mediated eosinophilic airway inflammation by suppressing IL-9 production. J Allergy Clin Immunol. (2017) 139:1355–67.e6. doi: 10.1016/j.jaci.2016.08.022
17. Grotenboer NS, Ketelaar ME, Koppelman GH, Nawijn MC. Decoding asthma: translating genetic variation in IL33 and IL1RL1 into disease pathophysiology. J Allergy Clin Immunol. (2013) 131:856–65. doi: 10.1016/j.jaci.2012.11.028
18. Imai Y, Yasuda K, Sakaguchi Y, Haneda T, Mizutani H, Yoshimoto T, et al. Skin-specific expression of IL-33 activates group 2 innate lymphoid cells and elicits atopic dermatitis-like inflammation in mice. Proc Natl Acad Sci USA. (2013) 110:13921–6. doi: 10.1073/pnas.1307321110
19. Imai Y, Hosotani Y, Ishikawa H, Yasuda K, Nagai M, Jitsukawa O, et al. Expression of IL-33 in ocular surface epithelium induces atopic keratoconjunctivitis with activation of group 2 innate lymphoid cells in mice. Sci Rep. (2017) 7:10053. doi: 10.1038/s41598-017-10227-y
20. Ito T, Hirose K, Saku A, Kono K, Takatori H, Tamachi T, et al. IL-22 induces Reg3γ and inhibits allergic inflammation in house dust mite-induced asthma models. J Exp Med. (2017) 214:3037–50. doi: 10.1084/jem.20162108
21. Richards CD, Izakelian L, Dubey A, Zhang G, Wong S, Kwofie K, et al. Regulation of IL-33 by oncostatin M in mouse lung epithelial cells. Mediators Inflamm. (2016) 2016:9858374. doi: 10.1155/2016/9858374
22. Foster PS, Plank M, Collison A, Tay HL, Kaiko GE, Li J, et al. The emerging role of microRNAs in regulating immune and inflammatory responses in the lung. Immunol Rev. (2013) 253:198–215. doi: 10.1111/imr.12058
23. Fleshner M, Crane CR. Exosomes, DAMPs and miRNA: Features of stress physiology and immune homeostasis. Trends Immunol. (2017) 38:768–76. doi: 10.1016/j.it.2017.08.002
24. Tang X, Wu F, Fan J, Jin Y, Wang J, Yang G. Posttranscriptional regulation of Interleukin-33 expression by microRNA-200 in bronchial asthma. Mol Ther. (2018) 26:1808–17. doi: 10.1016/j.ymthe.2018.04.016
25. Xiang Y, Eyers F, Herbert C, Tay HL, Foster PS, Yang M. MicroRNA-487b is a negative regulator of macrophage activation by targeting IL-33 production. J Immunol. (2016) 196:3421–8. doi: 10.4049/jimmunol.1502081
26. Yamazumi Y, Sasaki O, Imamura M, Oda T, Ohno Y, Shiozaki-Sato Y, et al. The RNA binding protein Mex-3B is required for IL-33 induction in the development of allergic airway inflammation. Cell Rep. (2016) 16:2456–71. doi: 10.1016/j.celrep.2016.07.062
27. Johansson K, Malmhäll C, Ramos-Ramírez P, Rådinger M. MicroRNA-155 is a critical regulator of type 2 innate lymphoid cells and IL-33 signaling in experimental models of allergic airway inflammation. J Allergy Clin Immunol. (2017) 139:1007–16.e9. doi: 10.1016/j.jaci.2016.06.035
28. Baekkevold ES, Roussigné M, Yamanaka T, Johansen FE, Jahnsen FL, Amalric F, et al. Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Am J Pathol. (2003) 163:69–79. doi: 10.1016/S0002-9440(10)63631-0
29. Roussel L, Erard M, Cayrol C, Girard JP. Molecular mimicry between IL-33 and KSHV for attachment to chromatin through the H2A-H2B acidic pocket. EMBO Rep. (2008) 9:1006–12. doi: 10.1038/embor.2008.145
30. Bessa J, Meyer CA, de Vera Mudry MC, Schlicht S, Smith SH, Iglesias A, et al. Altered subcellular localization of IL-33 leads to non-resolving lethal inflammation. J Autoimmun. (2014) 55:33–41. doi: 10.1016/j.jaut.2014.02.012
31. Osbourn M, Soares DC, Vacca F, Cohen ES, Scott IC, Gregory WF, et al. HpARI protein secreted by a helminth parasite suppresses Interleukin-33. Immunity (2017) 47:739–51.e5. doi: 10.1016/j.immuni.2017.09.015
32. Cayrol C, Girard JP. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci USA. (2009) 106:9021–6. doi: 10.1073/pnas.0812690106
33. Lüthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity (2009) 31:84–98. doi: 10.1016/j.immuni.2009.05.007
34. Ali S, Nguyen DQ, Falk W, Martin MU. Caspase 3 inactivates biologically active full length interleukin-33 as a classical cytokine but does not prohibit nuclear translocation. Biochem Biophys Res Commun. (2010) 391:1512–6. doi: 10.1016/j.bbrc.2009.12.107
35. Kim LK, Morita R, Kobayashi Y, Eisenbarth SC, Lee CG, Elias J, et al. AMCase is a crucial regulator of type 2 immune responses to inhaled house dust mites. Proc Natl Acad Sci USA. (2015) 112:E2891–9. doi: 10.1073/pnas.1507393112
36. Madouri F, Guillou N, Fauconnier L, Marchiol T, Rouxel N, Chenuet P, et al. Caspase-1 activation by NLRP3 inflammasome dampens IL-33-dependent house dust mite-induced allergic lung inflammation. J Mol Cell Biol. (2015) 7:351–65. doi: 10.1093/jmcb/mjv012
37. Lamkanfi M, Kanneganti TD, Van Damme P, Vanden Berghe T, Vanoverberghe I, Vandekerckhove J, et al. Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol Cell Proteomics (2008) 7:2350–63. doi: 10.1074/mcp.M800132-MCP200
38. Cayrol C, Duval A, Schmitt P, Roga S, Camus M, Stella A, et al. Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat Immunol. (2018) 19:375–85. doi: 10.1038/s41590-018-0067-5
39. Cohen ES, Scott IC, Majithiya JB, Rapley L, Kemp BP, England E, et al. Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat Commun. (2015) 6:8327. doi: 10.1038/ncomms9327
40. Hayakawa H, Hayakawa M, Kume A, Tominaga S. Soluble ST2 blocks interleukin-33 signaling in allergic airway inflammation. J Biol Chem. (2007) 282:26369–80. doi: 10.1074/jbc.M704916200
41. Bandara G, Beaven MA, Olivera A, Gilfillan AM, Metcalfe DD. Activated mast cells synthesize and release soluble ST2-a decoy receptor for IL-33. Eur J Immunol. (2015) 45:3034–44. doi: 10.1002/eji.201545501
42. Hayakawa H, Hayakawa M, Tominaga SI. Soluble ST2 suppresses the effect of interleukin-33 on lung type 2 innate lymphoid cells. Biochem Biophys Rep. (2016) 5:401–7. doi: 10.1016/j.bbrep.2016.02.002
43. Bulek K, Swaidani S, Qin J, Lu Y, Gulen MF, Herjan T, et al. The essential role of single Ig IL-1 receptor-related molecule/Toll IL-1R8 in regulation of Th2 immune response. J Immunol. (2009) 182:2601–9. doi: 10.4049/jimmunol.0802729
44. Zhao J, Wei J, Mialki RK, Mallampalli DF, Chen BB, Coon T, et al. F-box protein FBXL19-mediated ubiquitination and degradation of the receptor for IL-33 limits pulmonary inflammation. Nat Immunol. (2012) 13:651–8. doi: 10.1038/ni.2341
45. Duerr CU, McCarthy CD, Mindt BC, Rubio M, Meli AP, Pothlichet J, et al. Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat Immunol. (2016) 17:65–75. doi: 10.1038/ni.3308
46. Kudo F, Ikutani M, Seki Y, Otsubo T, Kawamura YI, Dohi T, et al. Interferon-γ constrains cytokine production of group 2 innate lymphoid cells. Immunology (2016) 147:21–9. doi: 10.1111/imm.12537
47. Mchedlidze T, Kindermann M, Neves AT, Voehringer D, Neurath MF, Wirtz S. IL-27 suppresses type 2 immune responses in vivo via direct effects on group 2 innate lymphoid cells. Mucosal Immunol. (2016) 9:1384–94. doi: 10.1038/mi.2016.20
48. Moro K, Kabata H, Tanabe M, Koga S, Takeno N, Mochizuki M, et al. Interferon and IL-27 antagonize the function of group 2 innate lymphoid cells and type 2 innate immune responses. Nat Immunol. (2016) 17:76–86. doi: 10.1038/ni.3309
49. Stier MT, Goleniewska K, Cephus JY, Newcomb DC, Sherrill TP, Boyd KL, et al. STAT1 represses cytokine-producing group 2 and group 3 innate lymphoid cells during viral infection. J Immunol. (2017) 199:510–9. doi: 10.4049/jimmunol.1601984
50. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell (2000) 100:655–69. doi: 10.1016/S0092-8674(00)80702-3
51. Lighvani AA, Frucht DM, Jankovic D, Yamane H, Aliberti J, Hissong BD, et al. T-bet is rapidly induced by interferon-gamma in lymphoid and myeloid cells. Proc Natl Acad Sci USA. (2001) 98:15137–42. doi: 10.1073/pnas.261570598
52. Takeda A, Hamano S, Yamanaka A, Hanada T, Ishibashi T, Mak TW, et al. Cutting edge: role of IL-27/WSX-1 signaling for induction of T-bet through activation of STAT1 during initial Th1 commitment. J Immunol. (2003) 170:4886–90. doi: 10.4049/jimmunol.170.10.4886
53. Turner JE, Morrison PJ, Wilhelm C, Wilson M, Ahlfors H, Renauld JC, et al. IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J Exp Med. (2013) 210:2951–65. doi: 10.1084/jem.20130071
54. Chang JE, Doherty TA, Baum R, Broide D. Prostaglandin D2 regulates human type 2 innate lymphoid cell chemotaxis. J Allergy Clin Immunol. (2014) 133:899–901.e3. doi: 10.1016/j.jaci.2013.09.020
55. Xue L, Salimi M, Panse I, Mjösberg JM, McKenzie AN, Spits H, et al. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J Allergy Clin Immunol. (2014) 133:1184–94. doi: 10.1016/j.jaci.2013.10.056
56. Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S, et al. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci Transl Med. (2013) 5:174ra26. doi: 10.1126/scitranslmed.3004812
57. Zhou W, Toki S, Zhang J, Goleniewksa K, Newcomb DC, Cephus JY, et al. Prostaglandin I2 signaling and inhibition of group 2 innate lymphoid cell responses. Am J Respir Crit Care Med. (2016) 193:31–42. doi: 10.1164/rccm.201410-1793OC
58. Maric J, Ravindran A, Mazzurana L, Björklund Å, Van Acker A, Rao A, et al. Prostaglandin E2 suppresses human group 2 innate lymphoid cell function. J Allergy Clin Immunol. (2018) 141:1761–73.e6. doi: 10.1016/j.jaci.2017.09.050
59. Zhou Y, Wang W, Zhao C, Wang Y, Wu H, Sun X, et al. Prostaglandin E2 inhibits group 2 innate lymphoid cell activation and allergic airway inflammation through E-prostanoid 4-cyclic adenosine monophosphate signaling. Front Immunol. (2018) 9:501. doi: 10.3389/fimmu.2018.00501
60. Laffont S, Blanquart E, Savignac M, Cénac C, Laverny G, Metzger D, et al. Androgen signaling negatively controls group 2 innate lymphoid cells. J Exp Med. (2017) 214:1581–92. doi: 10.1084/jem.20161807
61. Cephus JY, Stier MT, Fuseini H, Yung JA, Toki S, Bloodworth MH, et al. Testosterone attenuates group 2 innate lymphoid cell-mediated airway inflammation. Cell Rep. (2017) 21:2487–99. doi: 10.1016/j.celrep.2017.10.110
62. Moriyama S, Brestoff JR, Flamar AL, Moeller JB, Klose CSN, Rankin LC, et al. β2-adrenergic receptor-mediated negative regulation of group 2 innate lymphoid cell responses. Science (2018) 359:1056–61. doi: 10.1126/science.aan4829
63. Veiga-Fernandes H, Artis D. Neuronal-immune system cross-talk in homeostasis. Science (2018) 359:1465–6. doi: 10.1126/science.aap9598
64. Thio CL, Chi PY, Lai AC, Chang YJ. Regulation of type 2 innate lymphoid cell-dependent airway hyperreactivity by butyrate. J Allergy Clin Immunol. (2018). doi: 10.1016/j.jaci.2018.02.032. [Epub ahead of print].
65. Suzuki M, Morita R, Hirata Y, Shichita T, Yoshimura A. Spred1, a suppressor of the Ras-ERK pathway, negatively regulates expansion and function of group 2 innate lymphoid cells. J Immunol. (2015) 195:1273–81. doi: 10.4049/jimmunol.1500531
66. Roy A, Ganesh G, Sippola H, Bolin S, Sawesi O, Dagälv A, et al. Mast cell chymase degrades the alarmins heat shock protein 70, biglycan, HMGB1, and interleukin-33 (IL-33) and limits danger-induced inflammation. J Biol Chem. (2014) 289:237–50. doi: 10.1074/jbc.M112.435156
67. Morita H, Arae K, Unno H, Miyauchi K, Toyama S, Nambu A, et al. An Interleukin-33-mast cell-Interleukin-2 axis suppresses papain-induced allergic inflammation by promoting regulatory T cell numbers. Immunity (2015) 43:175–86. doi: 10.1016/j.immuni.2015.06.021
68. Rigas D, Lewis G, Aron JL, Wang B, Banie H, Sankaranarayanan I, et al. Type 2 innate lymphoid cell suppression by regulatory T cells attenuates airway hyperreactivity and requires inducible T-cell costimulator-inducible T-cell costimulator ligand interaction. J Allergy Clin Immunol. (2017) 139:1468–77.e2. doi: 10.1016/j.jaci.2016.08.034
69. Bi J, Cui L, Yu G, Yang X, Chen Y, Wan X. NK cells alleviate lung inflammation by negatively regulating group 2 innate lymphoid cells. J Immunol. (2017) 198:3336–44. doi: 10.4049/jimmunol.1601830
70. Wang S, Xia P, Chen Y, Qu Y, Xiong Z, Ye B, et al. Regulatory innate lymphoid cells control innate intestinal inflammation. Cell (2017) 171:201–16.e18. doi: 10.1016/j.cell.2017.07.027
71. Savenije OE, Mahachie John JM, Granell R, Kerkhof M, Dijk FN, de Jongste JC, et al. Association of IL33-IL-1 receptor-like 1 (IL1RL1) pathway polymorphisms with wheezing phenotypes and asthma in childhood. J Allergy Clin Immunol. (2014) 134:170–7. doi: 10.1016/j.jaci.2013.12.1080
72. Oshikawa K, Kuroiwa K, Tago K, Iwahana H, Yanagisawa K, Ohno S, et al. Elevated soluble ST2 protein levels in sera of patients with asthma with an acute exacerbation. Am J Respir Crit Care Med. (2001) 164:277–81. doi: 10.1164/ajrccm.164.2.2008120
Keywords: IL-33, ST2, innate lymphoid cells (ILCs), regulatory T cells (Tregs), epithelial cells
Citation: Takatori H, Makita S, Ito T, Matsuki A and Nakajima H (2018) Regulatory Mechanisms of IL-33-ST2-Mediated Allergic Inflammation. Front. Immunol. 9:2004. doi: 10.3389/fimmu.2018.02004
Received: 28 May 2018; Accepted: 14 August 2018;
Published: 04 September 2018.
Edited by:
Hui-Rong Jiang, University of Strathclyde, United KingdomReviewed by:
Fons Van De Loo, Radboud University Medical Center, NetherlandsCopyright © 2018 Takatori, Makita, Ito, Matsuki and Nakajima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hiroaki Takatori, dGFrYXRvcmloQGZhY3VsdHkuY2hpYmEtdS5qcA==
Hiroshi Nakajima, bmFrYWppbWhAZmFjdWx0eS5jaGliYS11Lmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.