 Elidiana De Bona1
Elidiana De Bona1 Kárita Cláudia Freitas Lidani1
Kárita Cláudia Freitas Lidani1 Lorena Bavia1
Lorena Bavia1 Zahra Omidian2Luiza Helena Gremski3
Zahra Omidian2Luiza Helena Gremski3 Thaisa Lucas Sandri1,4*†Iara J. de Messias Reason1†
Thaisa Lucas Sandri1,4*†Iara J. de Messias Reason1†
- 1Laboratory of Molecular Immunopathology, Department of Clinical Pathology, Federal University of Paraná, Curitiba, Brazil
- 2Department of Pathology, Division of Immunology, School of Medicine, Johns Hopkins University, Baltimore, MD, United States
- 3Department of Cell Biology, Federal University of Paraná, Curitiba, Brazil
- 4Institute of Tropical Medicine, University of Tübingen, Tübingen, Germany
Chagas disease (CD), a neglected tropical disease caused by the protozoan Trypanosoma cruzi, affects around six million individuals in Latin America. Currently, CD occurs worldwide, becoming a significant public health concern due to its silent aspect and high morbimortality rate. T. cruzi presents different escape strategies which allow its evasion from the host immune system, enabling its persistence and the establishment of chronic infection which leads to the development of chronic Chagas cardiomyopathy (CCC). The potent immune stimuli generated by T. cruzi persistence may result in tissue damage and inflammatory response. In addition, molecular mimicry between parasites molecules and host proteins may result in cross-reaction with self-molecules and consequently in autoimmune features including autoantibodies and autoreactive cells. Although controversial, there is evidence demonstrating a role for autoimmunity in the clinical progression of CCC. Nevertheless, the exact mechanism underlying the generation of an autoimmune response in human CD progression is unknown. In this review, we summarize the recent findings and hypotheses related to the autoimmune mechanisms involved in the development and progression of CCC.
Introduction
Chagas disease (CD) is a neglected tropical disease caused by the protozoan parasite Trypanosoma cruzi, which affects around six million individuals in Latin America (1). CD is increasing as a health threat in countries of Europe, the United States, Canada, Japan, and Australia, where blood transfusion, organ transplantation, and vertical transmission seem to be the main transmission routes (1). According to the World Health Organization, the estimated incidence of CD in the Americas is 30,000, followed by 14,000 deaths and 8,000 infected newborn per year (2).
Chagas disease is a life-threatening and persistent illness, having both acute and chronic phases (3). During the acute phase, which develops within a short time (4–8 weeks) following the infection, the parasite burden is controlled by the acute inflammatory response (4, 5). In order to establish a life-long infection, it is known that T. cruzi evades host immune response and, with this, some patients will remain asymptomatic and with low levels of intracellular parasites (6). The long-term proliferation and persistence of these parasites in the tissue leads to the establishment of the chronic phase of CD (7). Nearly 30–40% of chronically infected patients evolve from asymptomatic condition to symptomatic forms, including cardiac, digestive, or cardiodigestive (Figure 1A) (8).
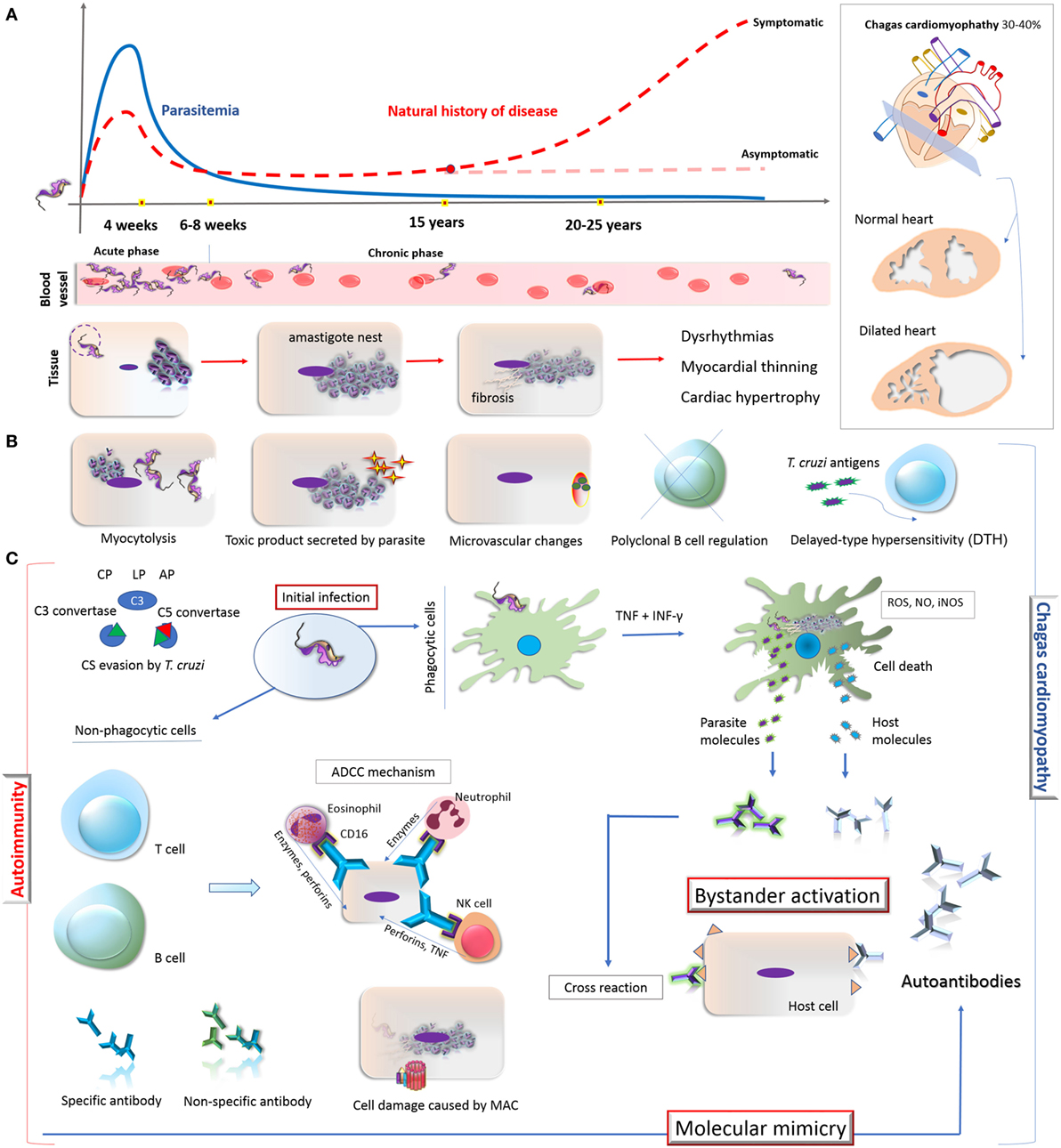
Figure 1. Overview on the natural history of CD, development of cardiomyopathy and its autoimmunity pathophysiological mechanisms. (A) Natural history of CD: the acute phase of Trypanosoma cruzi infection is oligosymptomatic and characterized by high parasitemia, which starts to decrease after 4 weeks. During the chronic phase (6–8 weeks), the parasitemia remains low and some patients (30–40%) might develop Chagas-related symptoms, especially cardiomyopathy. The parasite invades and differentiates in cardiomyocytes, leading to a fibrosis condition and consequently dysrhythmia, myocardial thinning, and cardiac hypertrophy. (B) Direct mechanisms associated with the cardiomyocyte damage: myocytolysis (cell lysis after amastigote differentiate into trypomastigote); toxic molecules produced by the parasite; microvascular changes induced by the parasite (cardiac hypoperfusion); disruption of immune regulation mechanisms in B cell (represented by X); constant presence of T. cruzi antigens triggers T cell-mediated damage and DTH process; autoimmunity (represented by the antibodies in the right). (C) Autoimmunity pathways in chronic CD: T. cruzi presents different escape strategies which enable its evasion from CS activation, allowing its entry in phagocytes, persistence, and the establishment of chronic infection which lead to the development of CCC. The potent immune stimuli generated by T. cruzi persistence (here represented by TNF, IFN-γ, ROS, NO, iNOS production by phagocytic cell) may result in tissue damage and inflammatory response through bystander activation and molecular mimicry. Bystander activation is caused by the exposure of both host and parasite intracellular proteins resulting in potent immune stimuli due to the release of self-antigens that induces the production of autoantibodies. Molecular mimicry occurs when there are structural similarities between T. cruzi-specific molecule and host-molecule, triggering T-cell activation. Specific antibodies from B cells can participate in ADCC mechanism on target cells. Neutrophil, eosinophil, and NK cell interact with these antibodies via CD16 (Fc receptor) and release lytic molecules like enzymes, perforins, or TNF on the target cells, independent of the CS. Moreover, CS activation and constant evasion strategies from T. cruzi could damage the host tissues through MAC formation. Abbreviations: CD, Chagas disease; DTH, delayed-type hypersensitivity; CCC, chronic Chagas cardiomyopathy; CP, classical pathway; LP, lectin pathway; AP, alternative pathway; CS, complement system; TNF, tumor necrosis factor; IFN-γ, interferon; ROS, reactive oxygen species; NO, nitric oxide; iNOS, inducible nitric oxide synthase; ADCC, antibody-dependent cell-mediated cytotoxicity; MAC, membrane attack complex; NK, natural killer cell.
There is a large variability in the outcome of T. cruzi infection, which is possibly due to different pathogenic mechanisms. However, the real contribution of the immunogenetic pattern of the human host, parasite diversity, and persistence, among others that could determine the clinical progression from asymptomatic to symptomatic CD forms remain enigmatic (9–14). In these circumstances, the parasite evasion of both humoral and cellular immune responses may lead to the success of T. cruzi infection and development of chronic CD (6, 9, 15–19).
Despite the contribution of the parasite persistence and the host genetics to the clinical progression of CD, it is known that immune reactivity against cardiac antigens (e.g., cardiac myosin) can occur during the infection in some patients (20, 21), where parasite-induced damage may lead to molecular mimicry between parasite/host proteins epitopes, thereby generating a potent immune stimuli (21). This may exceed the threshold of immune activation acceptable for self-tolerance, resulting in cross-reaction with self-molecules and, eventually, host tissue damage (6, 8, 21–25). Although the development of autoantibodies in CD has been demonstrated in several studies (17, 19, 22, 24, 26–34), its role in the clinical development of the disease has not been clarified. This review aims to address some of the possible mechanisms of autoimmunity involved in CD.
From Infection to Immune Responses Evasion: What is the Consequence of Parasite Persistence?
Trypanosoma cruzi can be transmitted by vectors (bugs from Triatominae subfamily) as well as blood transfusions, organ transplantation, ingestion of food contaminated with the parasite, vertical transmission, among others (35). Through a process called adhesion and recognition, the parasite forms a stable bond with cell surface molecules that serve as adhesion anchors to the cell for invasion (16, 36). In vectorial transmission, the invasion of host cells occurs by metacyclic trypomastigotes, the infective stage of the parasite. After cell invasion, the trypomastigotes differentiate into amastigotes and replicate in the cytosol, where they differentiate into trypomastigotes and, with the rupture of the cell, these reach the bloodstream, spreading the infection to other tissues. Infective T. cruzi metacyclic trypomastigotes have the ability to invade any mammalian cell (7, 37, 38).
Both humoral and cellular immune responses are essential for parasite control (39). In this context, the host response uses several strategies to eliminate the parasite including complement activation (15, 40, 41), opsonization (42), production of specific antibodies (43–45), and antibody-dependent cellular cytotoxicity (46, 47). The complement is part of the innate immunity acting in the first line of host defense against pathogens (48). It comprises more than 35 proteins and can be activated by three pathways: lectin, classical, and alternative (15). As soon as the trypomastigotes reach the host bloodstream, lectin pathway (LP) and alternative pathway (AP) are activated since both pathways do not depend on specific antibody responses (49). Collectins and ficolins recognize and bind to glycosylated and acetylated molecules on the surface of T. cruzi trypomastigotes activating the LP and the AP is spontaneously activated by hydrolysis of C3 (12, 14, 15). As the infection progresses, the host can mount a specific antibody response against T. cruzi that will lead to interaction with C1 complex composed of one molecule of C1q and two molecules each of C1r and C1s—activating the classical pathway (CP) (49). Once activated, proteases from both LP and CP cleave C2 and C4, generating a C3 convertase (C4b2a) which cleaves the central complement component C3 in C3a and C3b. This last fragment binds to C3 convertase forming C5 convertase, which cleaves C5 in C5a and C5b. The fragment C5b binds to C6, C7, C8 and 12–18 copies of C9 and, as a final product of complement activation, the membrane attack complex (MAC) is formed on the target cell (such as epimastigotes), promoting its lysis (50).
Trypanosoma cruzi utilizes its surface proteins (such as T. cruzi calreticulin, trypomastigote decay-accelerating factor, T. cruzi complement regulatory protein—Gp160, T. cruzi complement C2 receptor inhibitor trispanning, and T. cruzi complement regulatory gp58/068) to circumvent complement-mediated lysis and opsonization (15, 51, 52). These proteins disturb the attachment of initial molecules from complement pathways, thereby inhibiting the C3 convertase formation, which is a crucial step in the activation of all three pathways and generation of complement-mediated effects (49, 53).
Metacyclic trypomastigote forms of T. cruzi not only involve the expression of regulatory molecules on parasite’s surface but they also induce membrane-derived vesicles (microvesicles) from host cells, which affects the formation and activation of C3 convertase (C4b2a), resulting in the inhibition of complement activation, increased parasite survival, and eukaryotic cell invasion (54). Moreover, microvesicles derived from both host cells and T. cruzi can fuse, thereby increasing host cell invasion and parasite dissemination (55). Both mechanisms interfere also in the activation of complement (49, 54). Incomplete parasite clearance may lead to immune reactivity that could elicit tissue damage leading to the exposure of neoepitopes and stimulus for autoantibody production (56), triggering mechanisms involved in autoimmunity in chronic CD (8). These mechanisms are demonstrated in Figure 1.
Autoimmunity in CD
The pathogenesis of symptomatic chronic CD is not yet completely understood. Some hypotheses are based on the direct response of the immune system against infected tissues (8, 57, 58). According to these hypotheses, an efficient immune response could result in the substantial reduction in the number of parasites with less tissue damage and lack of clinical manifestations in asymptomatic patients (58). On the other hand, an inefficient immune response would favor parasite persistence in the tissues with consequent injury and fibrosis (59, 60). In addition, the autoimmunity hypothesis suggests that cardiac damage, triggered by parasite persistence, would lead to an exacerbation of the immune response and disruption in self-tolerance, resulting in immune reaction against self-molecules (58). In this case, the autoimmune response would possibly be the reason for the late damage observed in chronic CD (8). In fact, the first evidence of autoimmunity in CD was presented by Cossio and collaborators in 1974 (61), who reported antibodies in sera of chagasic patients that reacted with endocardium, interstitium, and heart blood vessels but were absent in healthy individuals and in patients with non-chagasic cardiomyopathy. The involvement of autoimmunity in the pathogenesis of chronic Chagas cardiomyopathy (CCC) has been extensively studied (18, 24, 25, 62–64), although questions concerning its exact role remain unanswered (58). It is worth mentioning that, in addition to autoimmunity, other mechanisms may contribute to the development of CCC, such as myocytolysis, secretion of toxic molecules by the parasite, microvascular changes induced by the parasite (cardiac hypoperfusion), disruption of immune regulation mechanisms in B cell and T cell-mediated delayed-type hypersensitivity (8, 57, 58, 65, 66) (Figure 1B). In general terms, the autoimmunity in CD has been considered one of the key mechanisms to explain the tissue damage observed in the chronic phase, even in the absence of the parasite in the affected tissues (Figure 1C).
How Does Autoimmunity Lead to Autoantibody Development?
Two main mechanisms support the autoimmunity hypothesis in CCC: bystander activation and molecular mimicry (Figure 1C). The first involves the exposure of intracellular proteins after parasite-induced damage, resulting in the release of self-antigens in an inflammatory environment. In addition, the constant presence of parasite antigens can trigger responses mediated by CD4+ and/or CD8+ T cells, which may be responsible for injuring infected or neighboring tissue cells (59, 67–69). This potent immune stimulus may overcome the threshold of self-tolerance and trigger the production of autoantibodies targeted to multiple antigens (58). In case of molecular mimicry, sequence similarities between foreign and self-peptides result in the cross-activation of autoreactive T or B cells to the host peptides (62, 63, 67). Cumulative evidence of cross-reactivity between T. cruzi and human antigens as well as of autoantibodies affecting structures and functions of the heart muscle have been reported (18, 25, 58).
Several mechanisms involved in the pathogenesis of CD suggest that the autoimmune aggression in the muscle fiber is due to antigenic mimicry to T. cruzi and host molecules. The similarity of antigenic epitopes of the parasite and host tissue leads to cross-reaction and production of autoreactive antibodies. In fact, molecular mimicry is considered the most significant mechanism of autoimmunity in CCC, being a key pathogenic event in disease manifestation. The demonstration that CD4+ T cells from mice with chronic Chagas myocarditis were able to transfer the cardiac damage to healthy mice corroborates this hypothesis (21). In addition, passive transfer of serum and/or antibodies from chagasic patients presenting complex cardiac arrhythmias were able to induce disturbances in the electrogenesis and conduction of adult rabbit hearts, confirming the pathogenicity of CD autoantibodies (70, 71). Moreover, T. cruzi antigens that mimic human host antigens evidence the connection between parasite persistence and autoimmunity (18). The cross-reactivity between host molecules and T. cruzi antigens is listed in Table 1.

Table 1. Cross-reactivity and human autoantibodies described in Chagas disease.
In the course of CD, the most probable routes to the development of autoantibodies include (i) the exposure of intracellular proteins leading to bystander activation mechanism, (ii) molecular mimicry, and (iii) the polyclonal B cell activation (67) (Figure 1). Actually, antibodies against self-antigens such as actin, myosin, myoglobin, DNA, tubulin (85), desmin, and myosin from cardiac muscle (86) were found in animal models of T. cruzi infection. In patients with CD, autoantibodies targeting β1-adrenergic receptors (76) and muscarinic acetylcholine receptors (M2) were found associated with the development of cardiomyopathy (33, 78). Moreover, an association of anti-muscarinic receptors antibodies with ventricular electrical instability and sudden death in patients with CCC has been reported (87, 88). In addition, specific antibodies against cardiac myosin concomitantly with a robust autoreactive T-cell reaction increasing the production of different autoantibodies have been described (27, 89–91). Some of these autoantibodies are listed in Table 1.
The cell injury seen in CCC may be associated with antibody-dependent cell-mediated cytotoxicity (ADCC) since it has been shown that infected mice neutrophil, eosinophil, and natural killer cells interact via Fc receptor with antibodies, releasing lytic molecules such as perforins and tumor necrosis factor, leading to cytotoxic effect of target cells (92–94).
Furthermore, complement activation and formation of MAC on host cell surface may be involved in the cell injury process present in CCC. During the chronic phase of CD, which is known to be associated with ongoing inflammation, complement becomes activated, resulting in the assembly of MAC on endothelial and cardiomyocyte cells and causing tissue damage (95). This event could explain, in part, the active myocarditis and the fibrosis observed in myocardial lesions seen in some patients with chronic CD. The formation of MAC on cardiomyocytes of chagasic patients suggests that cell damage would favor exposure of intracellular molecules and the development of autoantibodies contributing to the autoimmunity process (96) (Figure 1C). Although the presence of autoantibodies might represent a factor involved in cell damage in the chronic phase of CD, the real effect of autoimmunity in the clinical development of the disease is still unknown.
Thus, understanding the autoimmunity hypothesis in CCC development may guide new strategies for the treatment of chronic CD. Nevertheless, one may consider that therapies modulating the immune response are complex and may be a double-edged sword causing side effects since the abrogation of molecules from the immune system in experimental T. cruzi infection have shown to increase parasitemia (97).
Final Considerations
The autoimmune hypothesis in the pathogenesis of CD is a topic of controversial debate, and several studies have demonstrated the involvement of more than one plausible mechanism that could contribute to the tissue damage observed in the chronic phase of the disease. Thus, the presence of the parasite within the tissues could stimulate in a continuous way both humoral and cellular responses activating multiple pathways, such as molecular mimicry and autoantibodies formation, bystander activation, ADCC, and complement activation, contributing to tissue damage and progression to symptomatic forms, including chagasic cardiomyopathy. All the information gathered in this review contributes to highlight points of possible interventions for future development of strategies regarding neutralization, blocking or immunoadsorption of autoantibodies, as well as complement inhibition. Thus, a better understanding of the host immune response during T. cruzi infection and CD progression is a key element to the development of effective vaccines and immunotherapy.
Author Contributions
EB, KCFL, ZO, LB, LHG, TLS, and IJMR participated in the design and writing of the manuscript. TLS and IJMR participated in the design, coordination, and manuscript writing. KCFL developed the figure graphic design.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any personal, professional, commercial, or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by research grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundação Araucária (CP 01/2016 Programa Pesquisa para o Sistema Único de Saúde: Gestão Compartilhada em Saúde—PPSUS Edição 2015 Fundação Araucária-PR/SESA-PR/CNPq/MS-Decit). We acknowledge support by Deutsche Forschungsgemeinschaft and Open Access Publishing Fund of University of Tübingen.
References
1. WHO. Chagas Disease (American Trypanosomiasis) Fact sheet No 340. World Heal Organ (2015). Available from: http://www.who.int/mediacentre/factsheets/fs340/en/ (Accessed: June 25, 2017).
2. PAHO. PAHO WHO General Information – Chagas Disease. (2017). Available from: www.paho.org/hq/index.php?option=com_content&view=article&id=13566&Itemid=40721
3. Pérez-Molina JA, Molina I. Chagas disease. Lancet (2018) 391:82–94. doi:10.1016/S0140-6736(17)31612-4
4. Gutierrez FRS, Guedes PMM, Gazzinelli RT, Silva JS. The role of parasite persistence in pathogenesis of Chagas heart disease. Parasite Immunol (2009) 31:673–85. doi:10.1111/j.1365-3024.2009.01108.x
5. Ramirez JD, Guhl F, Umezawa ES, Morillo CA, Rosas F, Marin-Neto JA, et al. Evaluation of adult chronic Chagas’ heart disease diagnosis by molecular and serological methods. J Clin Microbiol (2009) 47:3945–51. doi:10.1128/JCM.01601-09
6. Cardoso MS, Reis-Cunha JL, Bartholomeu DC. Evasion of the immune response by Trypanosoma cruzi during acute infection. Front Immunol (2016) 6:659. doi:10.3389/fimmu.2015.00659
7. Rassi A, Rassi A, Marin-Neto JA. Chagas disease. Lancet (2010) 375:1388–402. doi:10.1016/S0140-6736(10)60061-X
8. Muñoz-Saravia SG, Haberland A, Wallukat G, Schimke I. Chronic Chagas’ heart disease: a disease on its way to becoming a worldwide health problem: epidemiology, etiopathology, treatment, pathogenesis and laboratory medicine. Heart Fail Rev (2012) 17:45–64. doi:10.1007/s10741-010-9211-5
9. Macêdo V. Indeterminate form of Chagas disease. Mem Inst Oswaldo Cruz (1999) 94:311–6. doi:10.1590/S0074-02761999000700059
10. Lidani KCF, Beltrame MH, Luz PR, Sandri TL, Nisihara RM, De Messias-Reaso IJ. Is pentraxin 3 a cardiovascular marker in patients with chronic Chagas disease? Int J Cardiol (2015) 190:233–5. doi:10.1016/j.ijcard.2015.04.106
11. Sandri TL, Lidani KCF, Andrade FA, Meyer CG, Kremsner PG, de Messias-Reason IJ, et al. Human complement receptor type 1 (CR1) protein levels and genetic variants in chronic Chagas disease. Sci Rep (2018) 8:526. doi:10.1038/s41598-017-18937-z
12. Luz PR, Boldt ABW, Grisbach C, Kun JFJ, Velavan TP, Messias-Reason IJT. Association of L-ficolin levels and FCN2 genotypes with chronic Chagas disease. PLoS One (2013) 8:e60237. doi:10.1371/journal.pone.0060237
13. Boldt ABW, Luz PR, Messias-Reason IJT. MASP2 haplotypes are associated with high risk of cardiomyopathy in chronic Chagas disease. Clin Immunol (2011) 140:63–70. doi:10.1016/j.clim.2011.03.008
14. Luz PR, Miyazaki MI, Chiminacio Neto N, Padeski MC, Barros ACM, Boldt ABW, et al. Genetically determined MBL deficiency is associated with protection against chronic cardiomyopathy in Chagas disease. PLoS Negl Trop Dis (2016) 10:e0004257. doi:10.1371/journal.pntd.0004257
15. Lidani KCF, Bavia L, Ambrosio AR, de Messias-Reason IJ. The complement system: a prey of Trypanosoma cruzi. Front Microbiol (2017) 8:607. doi:10.3389/fmicb.2017.00607
16. Epting CL, Coates BM, Engman DM. Molecular mechanisms of host cell invasion by Trypanosoma cruzi. Exp Parasitol (2010) 126:283–91. doi:10.1016/j.exppara.2010.06.023
17. Goin JC, Borda E, Perez Leiros C, Storino R, Sterin-Borda L. Identification of antibodies with muscarinic cholinergic activity in human Chagas’ disease: pathological implications. J Auton Nerv Syst (1994) 47:45–52. doi:10.1016/0165-1838(94)90064-7
18. Cunha-Neto E, Bilate AM, Hyland KV, Fonseca SG, Kalil J, Engman DM. Induction of cardiac autoimmunity in Chagas heart disease: a case for molecular mimicry. Autoimmunity (2006) 39:41–54. doi:10.1080/08916930500485002
19. Borda E, Pascual J, Cossio P, De La Vega M, Arana R, Sterin-Borda L. A circulating IgG in Chagas’ disease which binds to beta-adrenoceptors of myocardium and modulates their activity. Clin Exp Immunol (1984) 57:679–86.
20. Cunha E, Duranti M, Grubert A, Zingalest B, De Messiast I, Stolf N, et al. Autoimmunity in Chagas disease cardiopathy: biological relevance of a cardiac myosin-specific epitope crossreactive to an immunodominant Trypanosoma cruzi antigen. Immunology (1995) 92:3541–5.
21. Cunha-Neto E, Coelho V, Guilherme L, Fiorelli A, Stolf N, Kalil J. Autoimmunity in Chagas’ disease: identification of cardiac myosin-B13 Trypanosoma cruzi protein crossreactive T cell clones in heart lesions of a chronic Chagas’ cardiomyopathy patient. J Clin Invest (1996) 98:1709–12. doi:10.1172/JCI118969
22. Cunha-Neto E, Kalil J. Autoimmunity in Chagas’ heart disease. Sao Paulo Med J (1995) 113:757–66. doi:10.1590/S1516-31801995000200005
23. Davies JM. Molecular mimicry: can epitope mimicry induce autoimmune disease? Immunol Cell Biol (1997) 75:113–26. doi:10.1038/icb.1997.16
24. Verçosa AFA, Lorena VMB, Carvalho CL, Melo MFAD, Cavalcanti MGA, Silva ED, et al. Chagas’ disease: IgG isotypes against cytoplasmic (CRA) and flagellar (FRA) recombinant repetitive antigens of Trypanosoma cruzi in chronic chagasic patients. J Clin Lab Anal (2007) 21:271–6. doi:10.1002/jcla.20186
25. Nussinovitch U, Shoenfeld Y. Anti-troponin autoantibodies and the cardiovascular system. Heart (2010) 96:1518–24. doi:10.1136/hrt.2010.195255
26. Dos Santos RR, de Oliveira JCR, Rossi MA. Antibodies to neurons in chronic Chagas’ disease. Trans R Soc Trop Med Hyg (1976) 70:167. doi:10.1016/0035-9203(76)90195-4
27. Pontes-De-Carvalho L, Santana CC, Soares MB, Oliveira GG, Cunha-Neto E, Ribeiro-Dos-Santos R. Experimental chronic Chagas’ disease myocarditis is an autoimmune disease preventable by induction of immunological tolerance to myocardial antigens. J Autoimmun (2002) 18:131–8. doi:10.1006/jaut.2001.0574
28. Bach-Elias M, Bahia D, Teixeira DC, Cicarelli RMB. Presence of autoantibodies against small nuclear ribonucleoprotein epitopes in Chagas’ patients’ sera. Parasitol Res (1998) 84:796–9. doi:10.1007/s004360050490
29. Kaplan D, Ferrari I, Bergami PL, Mahler E, Levitus G, Chiale P, et al. Antibodies to ribosomal P proteins of Trypanosoma cruzi in Chagas disease possess functional autoreactivity with heart tissue and differ from anti-P autoantibodies in lupus. Proc Natl Acad Sci U S A (1997) 94:10301–6. doi:10.1073/pnas.94.19.10301
30. Muñoz Saravia SG, Haberland A, Bartel S, Araujo R, Valda G, Duchen D, et al. Distinct patterns of autoantibodies against G-protein-coupled receptors in Chagas’ cardiomyopathy and megacolon: their potential impact for early risk assessment in asymptomatic Chagas’ patients. J Am Coll Cardiol (2010) 56:526–7. doi:10.1016/j.jacc.2010.02.057
31. Wallukat G, Muñoz Saravia SG, Haberland A, Bartel S, Araujo R, Valda G, et al. Distinct patterns of autoantibodies against G-protein-coupled receptors in Chagas’ cardiomyopathy and megacolon. their potential impact for early risk assessment in asymptomatic Chagas’ patients. J Am Coll Cardiol (2010) 55:463–8. doi:10.1016/j.jacc.2009.06.064
32. Giordanengo L, Gea S, Barbieri G, Rabinovich GA. Anti-galectin-1 autoantibodies in human Trypanosoma cruzi infection: differential expression of this β-galactoside-binding protein in cardiac Chagas’ disease. Clin Exp Immunol (2001) 124:266–73. doi:10.1046/j.1365-2249.2001.01512.x
33. Retondaro FC, Dos Santos Costa PC, Pedrosa RC, Kurtenbach E. Presence of antibodies against the third intracellular loop of the m2 muscarinic receptor in the sera of chronic chagasic patients. FASEB J (1999) 13:2015–20. doi:10.1096/fasebj.13.14.2015
34. Goin JC, Leiros CP, Borda E, Sterin-Borda L. Interaction of human chagasic IgG with the second extracellular loop of the human heart muscarinic acetylcholine receptor: functional and pathological implications. FASEB J (1997) 11:77–83. doi:10.1096/fasebj.11.1.9034169
35. Pereira PCM, Navarro EC. Challenges and perspectives of Chagas disease: a review. J Venom Anim Toxins Incl Trop Dis (2013) 19:34. doi:10.1186/1678-9199-19-34
36. Barrias ES, de Carvalho TMU, De Souza W. Trypanosoma cruzi: entry into mammalian host cells and parasitophorous vacuole formation. Front Immunol (2013) 4:186. doi:10.3389/fimmu.2013.00186
37. Dvorak JA, Hyde TP. Trypanosoma cruzi: interaction with vertebrate cells in vitro. 1. Individual interactions at the cellular and subcellular levels. Exp Parasitol (1973) 34:268–83. doi:10.1016/0014-4894(73)90087-8
38. Dvorak JA, Howe CL. The attraction of Trypanosoma cruzi to vertebrate cells in vitro. J Protozool (1976) 23:534–7. doi:10.1111/j.1550-7408.1976.tb03835.x
39. Michailowsky V, Luhrs K, Rocha MOC, Fouts D, Gazzinelli RT, Manning JE. Humoral and cellular immune responses to Trypanosoma cruzi-derived paraflagellar rod proteins in patients with Chagas’ disease. Infect Immun (2003) 71:3165–71. doi:10.1128/IAI.71.6.3165-3171.2003
40. Almeida IC, Milani SR, Gorin PA, Travassos LR. Complement-mediated lysis of Trypanosoma cruzi trypomastigotes by human anti-alpha-galactosyl antibodies. J Immunol (1991) 146:2394–400.
41. Evans-Osses I, Mojoli A, Beltrame MH, Da Costa DE, Darocha WD, Velavan TP, et al. Differential ability to resist to complement lysis and invade host cells mediated by MBL in R4 and 860 strains of Trypanosoma cruzi. FEBS Lett (2014) 588:956–61. doi:10.1016/j.febslet.2014.01.054
42. Alcantara A, Brener Z. The in vitro interaction of Trypanosoma cruzi bloodstream forms and mouse peritoneal macrophages. Acta Trop (1978) 35:209–19.
43. Lages-Silva E, Ramirez LE, Krettli AU, Brener Z. Effect of protective and non-protective antibodies in the phagocytosis rate of Trypanosoma cruzi blood forms by mouse peritoneal macrophages. Parasite Immunol (1987) 9:21–30. doi:10.1111/j.1365-3024.1987.tb00485.x
44. Kumar S, Tarleton RL. The relative contribution of antibody production and CD8+ T cell function to immune control of Trypanosoma cruzi. Parasite Immunol (1998) 20:207–16. doi:10.1046/j.1365-3024.1998.00154.x
45. Scott MT, Moyes L, Wood JN. Lack of identity between the 90K protectiye glycoprotein of Trypanosoma cruzi and hybridoma (CE5)-defined T. Cruzi antigen which cross reacts with mammalian neurones. Trans R Soc Trop Med Hyg (1982) 76:698–700. doi:10.1016/0035-9203(82)90242-5
46. Voltarelli JC, Falcão RP, Da Silva JS. Antibody-dependent cellular cytotoxicity in chronic human Chagas disease. Parasite Immunol (1983) 5:377–84. doi:10.1111/j.1365-3024.1983.tb00753.x
47. Laguens RP, Cabeza Meckert P, Chambo JG. Antiheart antibody-dependent cytotoxicity in the sera of mice chronically infected with Trypanosoma cruzi. Infect Immun (1988) 56:993–7.
48. Jokiranta TS, Jokipii L, Meri S. Complement resistance of parasites. Scand J Immunol (1995) 42:9–20. doi:10.1111/j.1365-3083.1995.tb03620.x
49. Cestari I, Evans-Osses I, Schlapbach LJ, de Messias-Reason I, Ramirez MI. Mechanisms of complement lectin pathway activation and resistance by trypanosomatid parasites. Mol Immunol (2013) 53:328–34. doi:10.1016/j.molimm.2012.08.015
50. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol (2010) 11:785–97. doi:10.1038/ni.1923
51. Ferreira V, Valck C, Sánchez G, Gingras A, Tzima S, Molina MC, et al. The classical activation pathway of the human complement system is specifically inhibited by calreticulin from Trypanosoma cruzi. J Immunol (2004) 172:3042–50. doi:10.4049/jimmunol.172.5.3042
52. Ramírez-Toloza G, Ferreira A. Trypanosoma cruzi evades the complement system as an efficient strategy to survive in the mammalian host: the specific roles of host/parasite molecules and Trypanosoma cruzi calreticulin. Front Microbiol (2017) 8:1667. doi:10.3389/fmicb.2017.01667
53. Tambourgi DV, Kipnis TL, da Silva WD, Joiner KA, Sher A, Heath S, et al. A partial cDNA clone of trypomastigote decay-accelerating factor (T-DAF), a developmentally regulated complement inhibitor of Trypanosoma cruzi, has genetic and functional similarities to the human complement inhibitor DAF. Infect Immun (1993) 61:3656–63.
54. Cestari I, Ansa-Addo E, Deolindo P, Inal JM, Ramirez MI. Trypanosoma cruzi immune evasion mediated by host cell-derived microvesicles. J Immunol (2012) 188:1942–52. doi:10.4049/jimmunol.1102053
55. Ramirez MI, Deolindo P, de Messias-Reason IJ, Arigi EA, Choi H, Almeida IC, et al. Dynamic flux of microvesicles modulate parasite-host cell interaction of Trypanosoma cruzi in eukaryotic cells. Cell Microbiol (2017) 19:e12672. doi:10.1111/cmi.12672
56. Carroll MV, Sim RB. Complement in health and disease. Adv Drug Deliv Rev (2011) 63:965–75. doi:10.1016/j.addr.2011.06.005
57. Engman DM, Leon JS. Pathogenesis of Chagas heart disease: role of autoimmunity. Acta Trop (2002) 81:123–32. doi:10.1016/S0001-706X(01)00202-9
58. Bonney KM, Engman DM. Autoimmune pathogenesis of chagas heart disease: looking back, looking ahead. Am J Pathol (2015) 185:1537–47. doi:10.1016/j.ajpath.2014.12.023
59. Tarleton RL. Parasite persistence in the aetiology of Chagas disease. Int J Parasitol (2001) 31:550–4. doi:10.1016/S0020-7519(01)00158-8
60. Tarleton RL. Chagas disease: a role for autoimmunity? Trends Parasitol (2003) 19:447–51. doi:10.1016/j.pt.2003.08.008
61. Cossio PM, Laguens RP, Diez C, Szarfman A, Segal A, Arana RM. Chagasic cardiopathy: antibodies reacting with plasma membrane of striated muscle and endothelial cells. Circulation (1974) 50:1252–9. doi:10.1161/01.CIR.50.6.1252
62. Kierszenbaum F. Chagas’ disease and the autoimmunity hypothesis. Clin Microbiol Rev (1999) 12:210–23.
63. Kierszenbaum F. Where do we stand on the autoimmunity hypothesis of Chagas disease? Trends Parasitol (2005) 21:513–6. doi:10.1016/j.pt.2005.08.013
64. Nussinovitch U, Shoenfeld Y. The diagnostic and clinical significance of anti-muscarinic receptor autoantibodies. Clin Rev Allergy Immunol (2012) 42:298–308. doi:10.1007/s12016-010-8235-x
65. Costa RS, Monteiro RC, Lehuen A, Joskowicz M, Noel L-H, Droz D. Immune complex-mediated glomerulopathy in experimental Chagas’ disease. Clin Immunol Immunopathol (1991) 58:102–14. doi:10.1016/0090-1229(91)90152-Z
66. Leon JS, Godsel LM, Wang K, Engman DM. Cardiac myosin autoimmunity in acute Chagas’ heart disease. Infect Immun (2001) 69:5643–9. doi:10.1128/IAI.69.9.5643-5649.2001
67. Cunha-Neto E, Teixeira PC, Nogueira LG, Kalil J. Autoimmunity. Adv Parasitol (2011) 76:129–52. doi:10.1016/B978-0-12-385895-5.00006-2
68. Tarleton R, Zhang L. Chagas disease etiology: autoimmunity or parasite persistence? Parasitol Today (1999) 15:94–9. doi:10.1016/S0169-4758(99)01398-8
69. Ben Younes-Chennoufi A, Said G, Eisen H, Durand A, Hontebeyrie-Joskowicz M. Cellular immunity to Trypanosoma cruzi is mediated by helper T cells (CD4+). Trans R Soc Trop Med Hyg (1988) 82:84–9. doi:10.1016/0035-9203(88)90271-4
70. Jiménez MAV, Nascimento JHM, Monnerat G, Maciel L, Paiva CN, Pedrosa RC, et al. Autoantibodies with beta-adrenergic activity from chronic chagasic patients induce cardiac arrhythmias and early afterdepolarization in a drug-induced LQT2 rabbit hearts. Int J Cardiol (2017) 240:354–9. doi:10.1016/j.ijcard.2017.02.066
71. Farias de Oliveira S, Pedrosa RC, Nascimento JHM, Campos de Carvalho AC, Masuda MO. Sera from chronic chagasic patients with complex cardiac arrhythmias depress electrogenesis and conduction in isolated rabbit hearts. Circulation (1997) 96:2031–7. doi:10.1161/01.CIR.96.6.2031
72. Gea S, Iosa D, Chizzolini C, Cerban F, Ordonez P, Vottero-Cima E. Chagas’ disease cardioneuropathy: association of anti-Trypanosoma Cruzi and anti-sciatic nerve antibodies. Am J Trop Med Hyg (1993) 49:581–8. doi:10.4269/ajtmh.1993.49.581
73. Van Voorhis WC, Eisen H. Fl-160. A surface antigen of Trypanosoma cruzi that mimics mammalian nervous tissue. J Exp Med (1989) 169:641–52. doi:10.1084/jem.169.3.641
74. Kerner N, Liégeard P, Levin MJ, Hontebeyrie-Joskowicz M. Trypanosoma cruzi: antibodies to a MAP-like protein in chronic Chagas’ disease cross-react with mammalian cytoskeleton. Exp Parasitol (1991) 73:451–9. doi:10.1016/0014-4894(91)90069-9
75. Vermelho AB, de Meirelles Mde N, Pereira MC, Pohlentz G, Barreto-Bergter E. Heart muscle cells share common neutral glycosphingolipids with Trypanosoma cruzi. Acta Trop (1997) 64:131–43. doi:10.1016/S0001-706X(96)00627-4
76. Labovsky V, Smulski CR, Gómez K, Levy G, Levin MJ. Anti-β1-adrenergic receptor autoantibodies in patients with chronic Chagas heart disease. Clin Exp Immunol (2007) 148:440–9. doi:10.1111/j.1365-2249.2007.03381.x
77. Goin JC, Borda E, Segovia A, Sterin-Borda L. Distribution of antibodies against β-adrenoceptors in the course of human Trypanosoma cruzi infection. Exp Biol Med (1991) 197:186–92. doi:10.3181/00379727-197-43244
78. Goin JC, Borda ES, Auger S, Storino R, Sterin-Borda L. Cardiac M2 muscarinic cholinoceptor activation by human chagasic autoantibodies: association with bradycardia. Heart (1999) 82:273–8. doi:10.1136/hrt.82.3.273
79. Santos-Buch CA, Acosta AM, Zweerink HJ, Sadigursky M, Andersen OF, von Kreuter BF, et al. Primary muscle disease: definition of a 25-kDa polypeptide myopathic specific chagas antigen. Clin Immunol Immunopathol (1985) 37:334–50. doi:10.1016/0090-1229(85)90103-5
80. Laucella SA, de Titto EH, Segura EL. Epitopes common to Trypanosoma cruzi and mammalian tissues are recognized by sera from Chagas’ disease patients: prognosis value in Chagas disease. Acta Trop (1996) 62:151–62. doi:10.1016/S0001-706X(96)00032-0
81. Laucella SA, Velazquez E, Dasso M, de Titto E. Trypanosoma cruzi and mammalian heart cross-reactive antigens. Acta Trop (1996) 61:223–38. doi:10.1016/0001-706X(96)00004-6
82. Hernández-Munaín C, De Diego JL, Alcina A, Fresno M. A Trypanosoma cruzi membrane protein shares an epitope with a lymphocyte activation antigen and induces crossreactive antibodies. J Exp Med (1992) 175:1473–82. doi:10.1084/jem.175.6.1473
83. Girones N, Rodriguez CI, Basso B, Bellon JM, Resino S, Munoz-Fernandez MA, et al. Antibodies to an epitope from the cha human autoantigen are markers of Chagas’ Disease. Clin Vaccine Immunol (2001) 8:1039–43. doi:10.1128/CDLI.8.6.1039-1043.2001
84. Bonfa E, Viana VS, Barreto AC, Yoshinari NH, Cossermelli W. Autoantibodies in Chagas’ disease. An antibody cross-reactive with human and Trypanosoma cruzi ribosomal proteins. J Immunol (1993) 150:3917–23.
85. Ternynck T, Bleux C, Gregoire J, Avrameas S, Kanellopoulos LC. Comparison between autoantibodies arising during Trypanosoma cruzi infection in mice and natural autoantibodies. J Immunol (1990) 144:1504–11.
86. Tibbetts RS, McCormick TS, Rowland EC, Miller SD, Engman DM. Cardiac antigen-specific autoantibody production is associated with cardiomyopathy in Trypanosoma cruzi-infected mice. J Immunol (1994) 152:1493–9.
87. Medei E, Pedrosa RC, Benchimol Barbosa PR, Costa PC, Hernández CC, Chaves EA, et al. Human antibodies with muscarinic activity modulate ventricular repolarization: basis for electrical disturbance. Int J Cardiol (2007) 115:373–80. doi:10.1016/j.ijcard.2006.03.022
88. Salles G. Prognostic value of QT interval parameters for mortality risk stratification in Chagas’ disease: results of a long-term follow-up study. Circulation (2003) 108:305–12. doi:10.1161/01.CIR.0000079174.13444.9C
89. Leon JS, Daniels MD, Toriello KM, Wang K, Engman DM. A cardiac myosin-specific autoimmune response is induced by immunization with Trypanosoma cruzi proteins. Infect Immun (2004) 72:3410–7. doi:10.1128/IAI.72.6.3410-3417.2004
90. Guiñazú N, Pellegrini A, Giordanengo L, Aoki MP, Rivarola HW, Cano R, et al. Immune response to a major Trypanosoma cruzi antigen, cruzipain, is differentially modulated in C57BL/6 and BALB/c mice. Microbes Infect (2004) 6:1250–8. doi:10.1016/j.micinf.2004.07.010
91. Iwai LK, Juliano MA, Juliano L, Kalil J, Cunha-Neto E. T-cell molecular mimicry in Chagas disease: identification and partial structural analysis of multiple cross-reactive epitopes between Trypanosoma cruzi B13 and cardiac myosin heavy chain. J Autoimmun (2005) 24:111–7. doi:10.1016/j.jaut.2005.01.006
92. da Costa SCG. Mouse as a model for Chagas disease: does mouse represent a good model for Chagas disease? Mem Inst Oswaldo Cruz (1999) 94:269–72. doi:10.1590/S0074-02761999000700045
93. Bonney KM, Engman DM. Chagas heart disease pathogenesis: one mechanism or many? Curr Mol Med (2008) 8:510–8. doi:10.2174/156652408785748004
94. Kipnis TL, James SL, Sher AL, David JR. Cell-mediated cytotoxicity to Trypanosoma cruzi II antibody-dependent killing of bloodstream form by mouse eosinophils and neutrophils. Am J Trop Med Hyg (1981) 30:47–53. doi:10.4269/ajtmh.1981.30.47
95. Aiello VD, Reis MM, Benvenuti LA, De Higuchi ML, Ramires JAF, Halperin JA. A possible role for complement in the pathogenesis of chronic chagasic cardiomyopathy. J Pathol (2002) 197:224–9. doi:10.1002/path.1095
96. Higuchi Mde L, Benvenuti LA, Reis MM, Metzger M. Pathophysiology of the heart in Chagas’ disease: current status and new developments. Cardiovasc Res (2003) 60:96–107. doi:10.1016/S0008-6363(03)00361-4
Keywords: Chagas disease, autoimmunity, autoantibodies, chronic Chagas disease, mimicry, bystander activation, complement system
Citation: De Bona E, Lidani KCF, Bavia L, Omidian Z, Gremski LH, Sandri TL and Messias Reason IJ (2018) Autoimmunity in Chronic Chagas Disease: A Road of Multiple Pathways to Cardiomyopathy? Front. Immunol. 9:1842. doi: 10.3389/fimmu.2018.01842
Received: 07 May 2018; Accepted: 26 July 2018;
Published: 06 August 2018
Edited by:
Marco Emilio Bianchi, Università Vita-Salute San Raffaele, ItalyReviewed by:
Janine Adele Lamb, University of Manchester, United KingdomBryce Binstadt, University of Minnesota Twin Cities, United States
Copyright: © 2018 De Bona, Lidani, Bavia, Omidian, Gremski, Sandri and Messias Reason. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thaisa Lucas Sandri, dGhhaXNhLmx1Y2FzLXNhbmRyaUB1bmktdHVlYmluZ2VuLmRl
†Shared senior authors.