 Ulrich Baumann
Ulrich Baumann John M. Routes
John M. Routes Pere Soler-Palacín
Pere Soler-Palacín Stephen Jolles
Stephen Jolles- 1Department of Paediatric Pulmonology, Allergy and Neonatology, Hannover Medical School, Hannover, Germany
- 2Division of Asthma, Allergy and Clinical Immunology, Department of Pediatrics, Medical College of Wisconsin, Milwaukee, WI, United States
- 3Pediatric Infectious Diseases and Immunodeficiencies Unit, Hospital Universitari Vall d’Hebron, Institut de Recerca Vall d’Hebron, Universitat Autònoma de Barcelona, Barcelona, Spain
- 4Immunodeficiency Centre for Wales, University Hospital of Wales, Cardiff, United Kingdom
Immunoglobulin replacement therapy (IGRT) has contributed critically to the management of primary antibody deficiencies (PAD) and the decrease in pneumonia rate. However, despite adequate IGRT and improved prognosis, patients with PAD continue to experience recurrent respiratory tract infections, leading to bronchiectasis and continuing decline in lung function with a severe impact on their quality of life. Moreover, non-infectious inflammatory and interstitial lung complications, such as granulomatous-lymphocytic interstitial lung disease, contribute substantially to the overall morbidity of PAD. These conditions develop much more often than appreciated and represent a major therapeutic challenge. Therefore, a regular assessment of the structural and functional condition of the lung and the upper airways with appropriate treatment is required to minimize the deterioration of lung function. This work summarizes the knowledge on lung complications in PAD and discusses the currently available diagnostic tools and treatment options.
Introduction
Immunoglobulin (Ig) replacement therapy (IGRT) has made a critical difference in the treatment of primary antibody deficiencies (PAD). Improvements in care have led to a decrease in severe bacterial infections, notably pneumonia. Since the introduction of modern IGRT in the 1980s, Ig doses have increased, resulting in significantly higher plasma IgG trough levels (Figure 1) (1). The incidence of bacterial pneumonia in patients with PAD has been nearly halved (2) and the major risk factors—low IgA levels, combination of low IgA and IgM levels (3, 4), low IgG levels despite replacement IGRT, chronic sinusitis, bronchiectasis, and low number of class-switched memory B cells—are known, directing risk stratification, informing monitoring, and detecting early symptoms. Over a period of 40 years, the mortality rate in patients with common variable immunodeficiency (CVID) has decreased steadily from 29% in 1971 [before the introduction of intravenous IgG (IVIG)] to 24% in 1999 and 19.6% in 2012 (5–7). An analysis of the European Society for Immunodeficiencies (ESID) registry data provided an overall mortality estimate of 15%, with 75th and 60th percentiles for survival of 25 and 41 years after diagnosis [95% confidence intervals (CI) 22–33 and 33–41 years, respectively] (8).
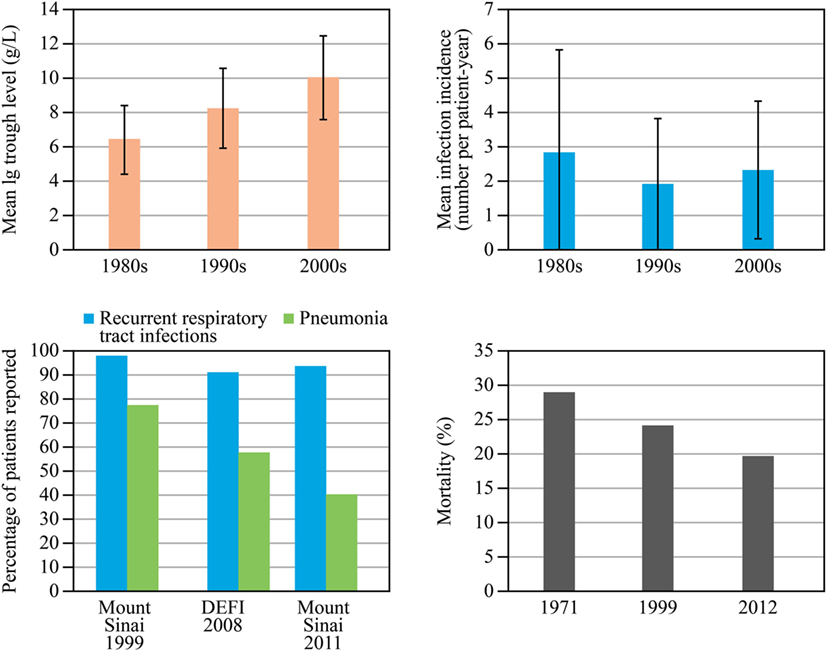
Figure 1. Development of immunoglobulin replacement therapy and infection rates since 1990s. Data for immunoglobulin (Ig) trough levels (top left) and infection rates (top right) are from a cohort of 115 patients with common variable immunodeficiency (CVID) or X-linked agammaglobulinemia reported by Lucas et al. (1). A combined analysis of data on the percentage of patients experiencing pneumonia and recurrent respiratory tract infections (bottom left) is shown in three large series of CVID patients in 1999, 2008, and 2011 (two from the same cohort in the United States 12 years apart, 248 and 252 patients, and one from a French cohort, 473 patients); reproduced with kind permission from 2014 British Society for Immunology, Clinical and Experimental Immunology, 178: 67–69 (2). Mortality data (bottom right) for 1971 are from Healy et al. (6); those for 1999 and 2012 are from a single cohort follow up reported by Resnick et al. (7). DEFI, a French national study. Abbreviation: PAD, primary antibody deficiencies.
However, despite improved therapy and prognosis, patients with PAD continue to experience non-infectious chronic lung disease and recurrent respiratory tract infections even with adequate IGRT (i.e., IGRT that raises trough IgG levels to within the range of healthy individuals), leading to an ongoing decline in lung function—in fact, the decline is greater than that in heavy smokers (9, 10). Pulmonary complications are common in a wide range of PAD and have a severe impact on patients’ quality of life (11–13). At the time of diagnosis, most patients have already suffered from recurrent bacterial pulmonary infections, leading to bronchiectasis and/or obstructive lung disease in some patients. Additionally, non-infectious complications are an increasing cause of morbidity and mortality in PAD. For example, structural and functional lung impairment is now recognized as an important risk factor for early mortality in CVID (7). In fact, these non-infectious complications—caused by immune dysregulation and not limited to the lung—have emerged as a major diagnostic and therapeutic challenge.
Therefore, active and accurate screening is mandatory even in “well-controlled” patients, but the absence of structured protocols for follow-up is a major challenge toward identifying and treating lung complications (14). This review focuses on the current challenges in the management of lung disease in patients with PAD.
Immunity and Immunodeficiency of the Respiratory System
The lung is an organ of enormous surface area combined with delicate structure that encounters a large amount and variety of pathogens capable of causing infection. Igs play a major role in the protection of the lung against infection, with specific roles for different isotypes (15). Different Ig isotypes are dominant in different parts of the lung in the airways: the respiratory surface in the upper and lower airways is covered predominantly with secretory IgA (sIgA) and IgM, while in the alveolar space, IgG is the dominant isotype (16, 17).
Both sIgA and IgM at the bronchial surface mostly derive from the mucosa associated lymphatic tissue, rather than from the systemic circulation (15, 18, 19). sIgA prevents bacterial adhesion or neutralizes toxins without causing an inflammatory response, in contrast to IgG and monomeric IgA (20). IgM activates the complement system, which enhances opsonization of pathogens. Due to its multimeric structure, IgM is highly effective for agglutination, especially of viruses (20). Alveolar IgG originates from the systemic circulation by passive diffusion and effectively prevents bacterial infections such as pneumonia (19, 21).
In most patients with PAD, both systemic IgG and local IgA are absent or dysfunctional; indeed, one of the criteria in the definition of CVID in the revised ESID and International Consensus guidelines is low IgA and/or low IgM (22, 23). sIgA seems to play a negligible role for airway defense because individuals with selective IgA deficiency are commonly healthy. However, the observation that patients with CVID suffer from airway infections more severely with very low IgA levels (i.e., less than 0.07 g/L) compared with those with higher IgA levels (24) argues that IgA can compensate for IgG deficiency in airway defense, at least in part. Combined deficiency of IgA and IgM seems to be even more detrimental if IgG is lacking (25) suggesting that IgM is also a cofactor for airway defense.
Without IGRT, the most common infection sites in patients with X-linked agammaglobulinemia (XLA) and CVID are in the airways (2, 26, 27). The same types of bacteria reside in the upper and lower airways in PAD, with analogous evidence in the setting of cystic fibrosis (CF) for Pseudomonas aeruginosa (28). The concept that the upper airways form a gateway and a reservoir of infection for the lower airways was supported by the same genetic fingerprint bacteria collected from nasal and bronchial simultaneously (29). Therefore, the optimal pulmonary management of patients with PAD must encompass the upper airway, the “gateway to the lungs.”
Respiratory Infections in PAD: What is Being Missed
The different and only partially overlapping physiological roles and locations of IgG, IgA, and IgM suggest that there are several defects in the defense of the airways, only one of which is addressed by current IGRT. This helps to explain why patients with PAD experience recurrent respiratory tract infections even with regular IGRT. The most common infections are sinusitis and upper respiratory tract infections, but the range is much broader and not limited to the lungs (Figure 2).
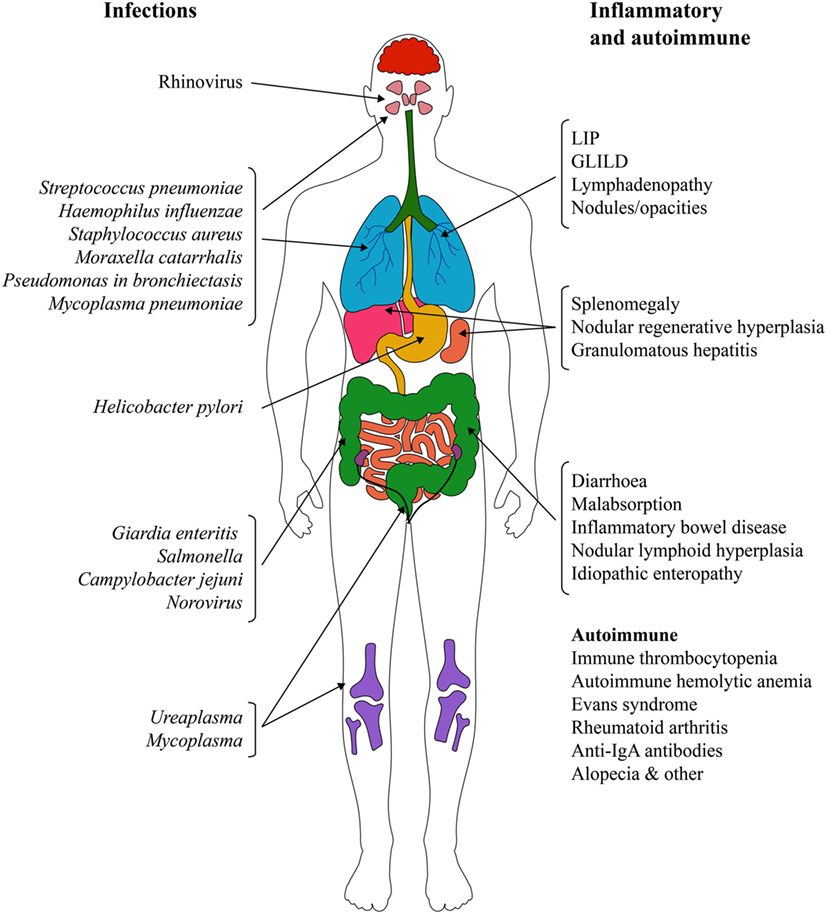
Figure 2. Infectious and non-infectious complications in primary antibody deficiencies. Abbreviations: GLILD, granulomatous-lymphocytic interstitial lung disease; IgA, immunoglobulin A; LIP, lymphocytic interstitial pneumonitis.
Encapsulated bacteria such as Haemophilus influenzae, Streptococcus pneumonia, and Moraxella catarrhalis are the most common causative agents of recurrent infections. However, non-encapsulated, non-typeable strains of these bacteria have been identified as an important cause of pneumonia, sinusitis, bronchitis, and otitis in this patient population (30). Rhinovirus is another frequent causative agent of recurrent infections that leads to prolonged airway infection and inflammation in patients with hypogammaglobulinemia (31). Viral infection may render the airway mucosa susceptible for new bacterial infection (32) or aggravate bacterial infection as shown in patients with CF (33). In chronic obstructive pulmonary disease (COPD), rhinoviral infection exacerbates the existing microbiome with outgrowth of particular bacteria (34). P. aeruginosa should always be sought particularly where there is an evidence of structural damage (26). Also, Mycoplasma spp infections seem to play a role in pulmonary and non-pulmonary disease in patients with PAD (35–38).
Subclinical Infections
Subclinical infections are well documented in patients with PAD, and a number of bacteria and viruses can be identified even in the periods when patients have no evident active infection. For example, a study of patient bronchoalveolar lavage fluid (BALF) detected bacteria in the lower respiratory tract of 9/14 asymptomatic patients, 6 of whom harbored erythromycin-resistant strains of H. influenzae (26). Adenoviruses were found in 4/14 patients; 2 of these patients had a dual infection with both adenovirus and rhinovirus (26). Similarly, a 12-month follow up study in 12 patients with PAD demonstrated the presence of respiratory viruses, most commonly rhinovirus, in the sputum in half of the 65 acute infection episodes (27). Moreover, virus elimination was much longer in patients with PAD than in healthy individuals. Rhinovirus shedding in patients with PAD lasted on average 40.9 days (95% CI: 26.4–55.4 days) compared to 11.4 (8.2–14.7) days and 10.1 (7.4–12.9) days in immunocompetent children and adults, respectively (27, 31). Not only did respiratory tract symptoms persist for the duration of virus shedding, but also new infections by another rhinovirus type appeared often soon after the first episode (31).
In the long term, recurrent infections often lead to lung damage and chronic lung disease, with bronchiectasis being the most common complication. Studies with long follow up (up to 11 years) have shown that a substantial proportion of patients present with chronic lung disease at the time of diagnosis or develop it despite IGRT maintaining IgG levels to within the normal range (24, 39, 40) (Figure 3). The cumulative risk of chronic lung disease increases with disease duration and is not dependent on the age at diagnosis (39, 40). The rate of lung decline in PAD is much faster than that predicted in healthy individuals: the average decline of forced expiratory volume in 1 s (FEV1) in patients with CVID or XLA [45 mL/year (9)] is not only more than twofold higher than the normal age-related decline in healthy non-smoking individuals [19.6 and 17.6 mL/year for males and females, respectively (10)], but also higher than the decline in continuous smokers [38.2 and 23.9 mL/year in males and females, respectively (10)]. Even in one IVIG cycle, infections appear more frequently at the end of the cycle when trough Ig levels are low. A recent study showed that the risk ratio of a new infection was higher in the last compared to the first week of the IVIG cycle (1.55 for a 4-week cycle; P = 0.0314) (41). Therefore, it is likely that the optimization of trough IgG levels by increasing the total dose or shortening the interval will limit this difference in risk of infection. The stable IgG levels achieved using SCIG would also be expected to limit this in-dosing cycle variation.
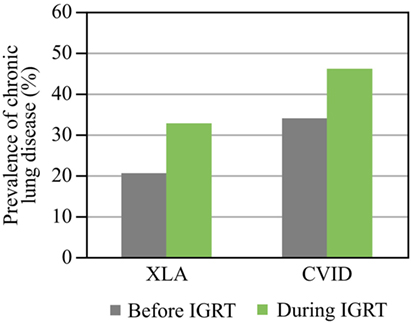
Figure 3. Prevalence of chronic lung disease before and during immunoglobulin replacement therapy (IGRT). Prevalence before and during IGRT is shown. Data for chronic lung disease are from Plebani et al. (XLA) (39) and Quinti et al. (CVID) (40). Abbreviations: CVID, common variable immunodeficiency; XLA, X-linked agammaglobulinemia.
Bronchiectasis
Bronchiectasis is the irreversible scarring of the lung tissue resulting from infection and the ensuing inflammatory response (Figure 4A). Inflammation leads to excess mucus production and impaired mucociliary clearance, the latter in turn contributes to the increased number of bacteria residing in the lung, leading to a vicious cycle of inflammation and infection (42, 43). If inflammation lasts long enough or is severe enough, it leads to irreversible ulceration of the tissue with the involvement of fibroblasts and to scaring (44). Similar to other lung complications, bronchiectasis may develop despite regular IGRT (40). Morphologically, there are three types of bronchiectasis that reflect increasing severity of bronchial disease and its progression to the more central airways: cylindrical (also called tubular), varicose, and cystic (45, 46) (Figure 4B). The key symptoms of bronchiectasis are prolonged cough, excessive production of sputum, shortness of breath and wheezing, and chest pain. Severe and/or widespread bronchiectasis can lead to respiratory failure, atelectasis, and heart failure. Diagnosis of bronchiectasis is not straightforward and requires chest tomography imaging by either computed tomography (CT) or magnetic resonance tomography. Conventional chest X-ray imaging detects only a third of bronchiectatic lesions (47).
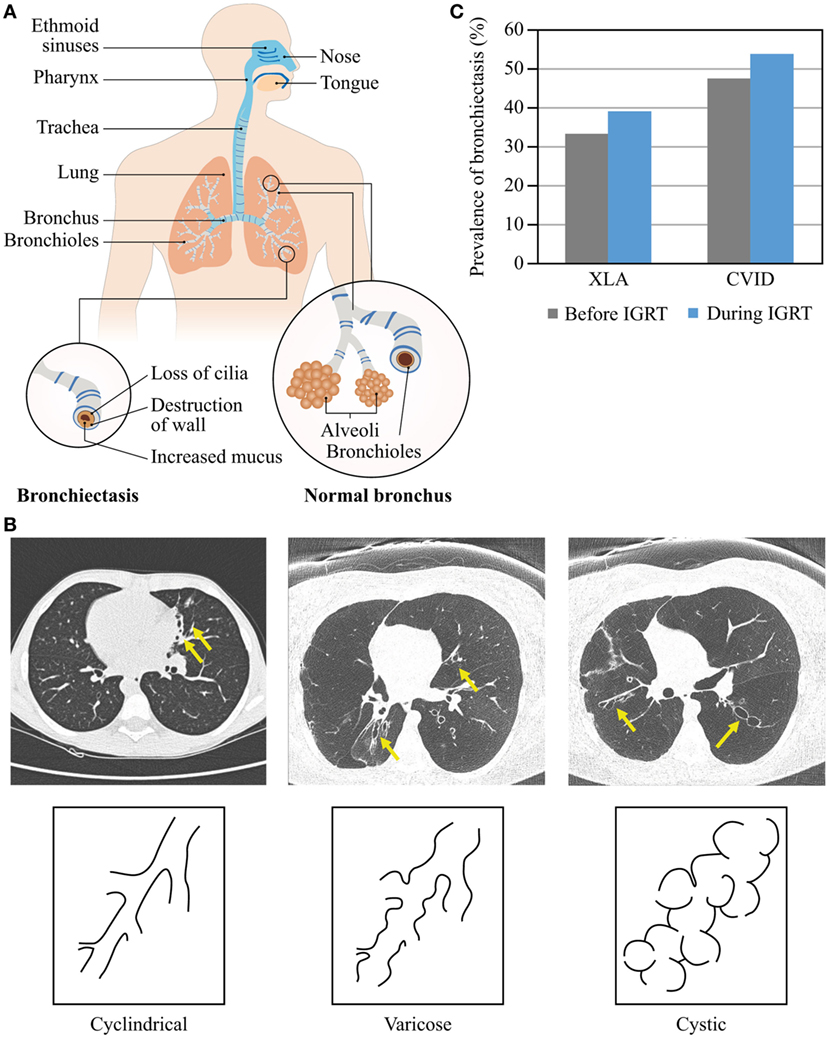
Figure 4. Morphology and prevalence of bronchiectasis. (A) Schematic presentation of normal bronchi and bronchi with bronchiectasis. The upper airways (blue) are more exposed to pathogens compared with the lower airways (orange). (B) Types of bronchiectasis. (C) Prevalence of bronchiectasis before and during IGRT. Data are from Quinti et al. (24).
The prevalence of bronchiectasis among patients with PAD rises with age regardless of IGRT (Figure 4C). Over a 5-year observation period, the prevalence of bronchiectasis in patients with CVID increased from 47.3 to 53.7%, with age and serum IgA levels lower than 0.07 g/L being independent risk factors (24). Also in patients with XLA, the prevalence of bronchiectasis increased from 33 to 39% (24). The prevalence of chronic sinusitis increased with age in CVID and XLA patients, too, with low IgA serum levels being an independent risk factor in CVID (24, 39, 40). Moreover, because prevalence of chronic sinusitis was higher than prevalence of bronchiectasis in both patient cohorts, it is likely that bronchiectasis may develop late in the course of a common airway disease in PAD as a result of a chronic bacterial infection commencing in the upper airways, or as a sequel of prolonged inflammation, or both. Etiology of bronchiectasis may differ between CVID patients, as suggested by an increasing number of conditions with immune dysregulation rather than immunodeficiency. According to our own preliminary data in a large cohort of pediatric and adult CVID patients, IGRT does not appear to ameliorate the course of lung disease; the prevalence of bronchiectasis rises no less after establishing IGRT than with duration of disease.
Because the scarring of the lung tissue is irreversible, treatment of bronchiectasis focuses on preventing further tissue damage and alleviating the consequences. Treating the primary cause of bronchiectasis and reducing the risk of recurrent infections by optimization of IGRT and use of prophylactic antibiotics to stop the vicious cycle of inflammation are the primary goals. In addition, medication helping bronchodilation, mucus thinning, and expectoration can provide relief.
New Therapeutic Approaches
New preventive and therapeutic approaches are urgently needed to alleviate lung damage in patients with PAD. Several potential treatments, including oral or inhaled antibiotics, inhaled hyperosmolar agents, mucolytic agents, airway clearance techniques, nasal irrigation, or nasal drug deposition have been tested in various settings associated with bronchiectasis (48). However, there are yet no published clinical trial results documenting their efficacy and safety in PAD.
Nasal irrigation and drug deposition in the nose or sinuses is a logical choice for delivering medication in patients with PAD similarly to immunocompetent individuals. The paranasal sinuses are poorly ventilated and often harbor chronic infections as in chronic sinusitis. Improving the sinus clearance may reduce the risk of infection in the lower airways. However, despite the fact that aerosolized drugs have been used for a long time, efficient drug delivery to the posterior nose and paranasal sinuses is still a challenge, because most aerosolized drugs are filtered by their passage through the nose (49). Irrigation of the nose using large-volume squeeze bottles has been shown to be effective (50), whereas nasal pump sprays could not deliver the aerosol to the sinuses (51). Pulsating aerosol delivery systems use a pulsating airflow to ventilate the sinuses and introduce the aerosolized medication (e.g., antibiotics and mucolytic agents) into the paranasal sinuses, which otherwise would be inaccessible (49). Studies using a paranasal nebulizer have shown that up to 6.5% of the nasally deposited drug was found in the sinuses (51). In addition, compared with nasal pump sprays, retention kinetics of the drug deposit in the nose was prolonged threefold (49, 52). This method can be used to deliver topical antibiotics, hyperosmolar agents, or even Ig directly in the airways or the lungs (53). In CF, application of tobramycin and recombinant human deoxyribonuclease (DNAse, Dornase alfa, Genentech, Inc., San Francisco, CA, USA) to the paranasal sinuses by oscillating pulse wave nebulization has proven beneficial for reduction of bacterial load and clinical symptoms (54, 55).
Antiviral treatment may be beneficial in patients who suffer from recurrent viral respiratory infections despite adequate IGRT. Preliminary studies in patients with PAD suggest that antiviral agents, such as nebulized ribavirin and pegylated interferon can reduce replication of rhinovirus at the airway site and improve symptoms (56). Patients with PAD do not appear to be prone to all viruses equally. Respiratory syncytial virus, which affects virtually all infants before they mount a specific immune response, does not appear to be a frequent pathogen in patients with PAD beyond infancy (57); this argues that IGRT affords protection against this respiratory pathogen. It is surprising that influenza infection, which regularly causes epidemics in the healthy population, has not received major interest in studies of PAD (57). Passive immunization by IGRT is likely to afford only limited protection against influenza, for donors (whose blood has been used to manufacture Ig products) have not been immunized against the most recent influenza strains. Moreover, active immunization of CVID patients induces only a weak, if any, antibody response (58). However, T-cell mediated immunity may respond better to influenza vaccination, supporting the common recommendation to immunize all patients with PAD (59). Recently, the first case of melanoma differentiation-associated protein 5 deficiency was reported in a patient with PAD; this inborn error resulted in susceptibility specifically to rhinovirus infection (60). Antiviral treatment might be useful in patients who suffer from recurrent viral respiratory infections despite adequate IGRT. Unfortunately, antiviral agents are not routinely available to treat all viral infections, including rhinovirus infection, and even if any were available there might be a risk of resistance with repeated or prolonged use.
In the past several years, research of the immune mechanisms controlling the susceptibility to chronic bacterial infection focused increasingly on the lung microbiota as a major factor informing and calibrating the immune system. The topic of the interaction of the microbiota with the host immune system is outside the scope of this work, and there are several excellent reviews summarizing the research to date (61–63). The “disappearing microbiota hypothesis” proposed by Blaser and Falkow offers an explanation for the increased burden of inflammatory disease as a result of changing hygiene, human macroecology, and clinical practice (64). The lung is a complex microbial ecosystem, in which different pathogens compete for survival not only with one another but also with the commensal microbiota (62). Animal studies have shown that not all members of the microbiota have an equal ability to influence the immune system, thus the disruption of the microbiota composition may result in a reduced host defense against bacterial and viral infection (65–67). Also, studies have shown that microbiota-depleted mice had significant loads of bacteria and viruses compared with mice with normal microbiota. In humans, the lung microbiota differs between health and inflammation, whereby in the latter situation the broad range of microbiota is substantially reduced, appearing “missing” or “simplified” (62, 64, 68). For example, subjects with asthma or COPD have a distinctive lung microbiota composition compared to healthy controls (69, 70). In patients with CVID, inflammatory complications are associated with reduced within-individual bacterial diversity, and dysbiosis (i.e., an unhealthy imbalance in the normal bacterial ecology), and elevated immune cell activation markers in the gut (71). Evaluating the pulmonary microbiome may evolve as a new diagnostic tool to assess the risk of progression of lung disease. Active modification of the pulmonary microbiome in patients with PAD may reduce ecological niches for pathogens and have a beneficial effect on host defense even with an immune system that functions only in part. Although research of the lung microbiota lags substantially behind that of the gut microbiota, modifying (or restoring) the microbiota of the airway mucosa using probiotics, similar to the approach used in the gut, may help in maintaining a favorable immunological balance (63).
Adjunctive Therapies
The recently completed first study on antibiotic prophylaxis in Italian patients with PAD evaluated the efficacy and safety of low doses of azithromycin (250 mg three times per week on three consecutive days) for 24 months vs placebo (72). The results of this study will provide much needed evidence of the efficacy of antibiotic prophylaxis. Regarding the practice of antibiotic usage for the treatment of respiratory infections, a recent prospective study reported that patients delay antibiotic treatment by a median of 5 days until they suffer specific “warning” symptoms, such as cough, shortness of breath, and purulent sputum (73). As the authors of this article point out, this finding is unexpected, as patients should have access to antibiotics for immediate use and are advised to start treatment when they have a breakthrough infection. Whether this delay is a matter of patient choice or of access to healthcare remains to be investigated. In addition, only exacerbations characterized by purulent sputum appeared to respond to use of antibiotics, while exacerbations with signs of upper respiratory tract infections did not. Moreover, exacerbations were as frequent in patients using prophylactic antibiotics, as in those using antibiotics on demand. It remains unclear whether the outcomes would have been different if antibiotic treatment was commenced earlier than 5 days after onset of symptoms. Finally, respiratory viruses were detected more frequently than bacterial pathogens. These findings argue for a careful evaluation of the causing pathogen as guidance for the use of antibiotics.
Hyperosmolar agents, most commonly hypertonic saline, have been used to break the vicious cycle of mucus retention and infection in patients with bronchiectasis (74). Another approach to improve mucus elimination is using mucolytic agents (carbocysteine orally or nebulized DNAse) to increase mucus fluidity and facilitate expectoration and, while this approach has been used in selected PAD patients, there are no controlled studies in PAD. The observation that DNAse improves lung function parameters (FEV1) in patients with CF (75), while it leads to FEV1 deterioration in patients with non-CF bronchiectasis (76), indicates that the efficacy of therapeutic interventions may differ substantially between various pulmonary conditions. Therapeutic strategies, therefore, need to be carefully assessed in controlled clinical trials in patients with PAD.
Non-Infectious Pulmonary Complication in PAD
Non-infectious pulmonary complications in PAD are common and contribute substantially to morbidity. Thus, in CVID, two independent studies have shown that patients with disease-related non-infectious complications have a significantly poorer survival prognosis than those without complications: the risk of death was estimated to be 11-fold higher than that in patients with infections only (P < 0.001) (7, 8). Approximately 30% of patients with CVID have diffuse interstitial lung disease (ILD) (51). Etiology is unknown in most cases, but multiple monogenic causes have been identified (77).
Non-infectious pulmonary complications include different forms of ILD, such as granulomatous-lymphocytic ILD (GLILD), cryptogenic organizing pneumonia (COP), lymphocytic interstitial pneumonitis (LIP), follicular bronchiolitis (FB), and/or lymphoid hyperplasia. Screening with high-resolution computed tomography (HRCT) chest scan indicates a much higher percentage of patients with ILD and bronchiectasis among those with PAD than previously appreciated (78). Establishing a correct pathological diagnosis is crucial for treatment of ILD. For instance, COP is responsive to steroids, whereas in GLILD, other treatments may be needed.
Granulomatous-Lymphocytic ILD
Granulomatous-lymphocytic ILD is the pulmonary component of a non-necrotizing, systemic disease characterized by adenopathy, splenomegaly, and granulomatous inflammation that may affect not only the lung, but also the liver, bone marrow, and lymph nodes (79, 80). The prevalence of GLILD in patients with PAD is unknown; however, it is found in CVID and an increasing number of monogenic disorders. Approximately 20% of patients with GLILD present with polyclonal lymphocytic infiltration or nonmalignant hyperplasia of the lymph nodes in addition to granuloma (80). GLILD typically occurs in the context of CVID; so far, GLILD has not been described in congenital agammaglobulinemia.
The key histopathological features of GLILD are LIP, FB, and non-necrotizing granuloma (Table 1; Figure 5). These histopathological features are numerically more frequent in the lower lung zones and are found in the same biopsy in nearly all patients. LIP often manifests as a mix of moderate to severe peribronchiolar and interstitial lymphoid infiltration. The predominant cells in the infiltrate are CD4+ T cells, but nodules of CD20+ B cells surrounded by CD4+ T cells are also found, predominantly localized to the interstitium (81). Distinct B-cell follicles and T-cell areas have been observed also in patients with CVID and lymphoid hyperplasia (82). Surprisingly, regulatory T cells are absent in the lungs in GLILD (81). This latter observation is in agreement with previous reports indicating reduced numbers and function of regulatory T cells in blood in CVID associated with GLILD (83–86). Granulomas are non-necrotizing, poorly to well-formed, and widely distributed, but with lower lung zone predominance, occasionally in association with interstitial inflammation (81).
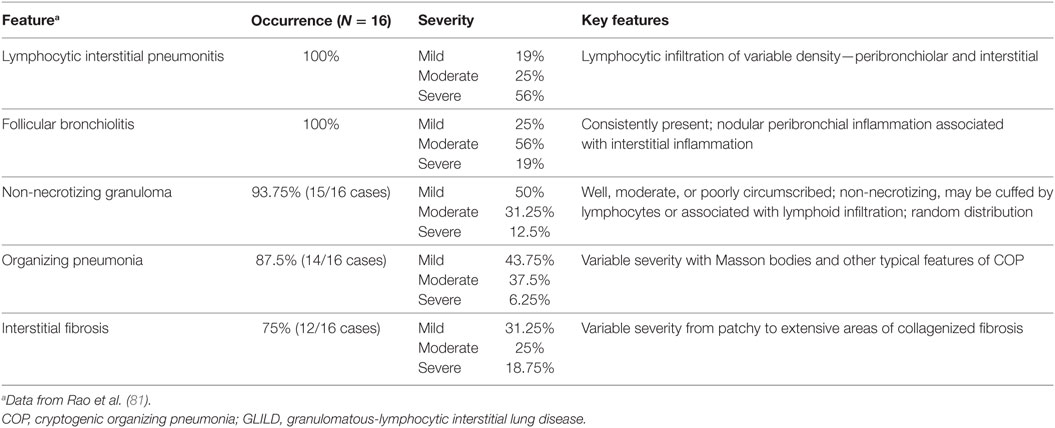
Table 1. Histopathological features of GLILD.
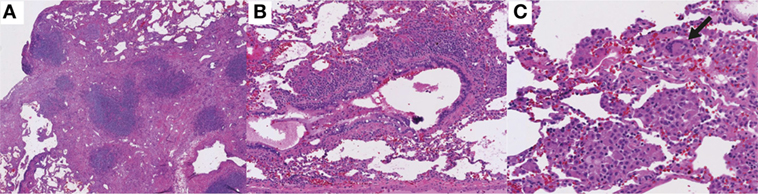
Figure 5. Histologic features of granulomatous-lymphocytic interstitial lung disease. (A) Lymphocytic interstitial pneumonitis. Diffuse, lymphocytic interstitial infiltrates obliterating the alveolar spaces in some areas (100×). (B) Follicular bronchiolitis. Nodular, lymphoid peribronchiolar aggregates (100×). (C) Non-necrotizing granuloma with multinucleated giant cells (arrow; 200×). Hematoxylin and eosin staining.
In addition to LIP, FB, and granuloma, organizing pneumonia and interstitial fibrosis are also found in most patients with GLILD and are moderate or severe in approximately half of the cases (81). Organizing pneumonia is characterized by Masson bodies amidst pale, myxoid stroma within the alveoli and adjacent interstitium. Interstitial fibrosis is characterized by areas of collagenized fibrosis, with the more severe cases being accompanied by alveolar remodeling (81). Whether GLILD causes progressive pulmonary fibrosis is somewhat controversial. However, moderate to severe pulmonary fibrosis has been found in 44% of patients with GLILD strongly suggesting this leads to progressive pulmonary fibrosis (78, 87, 88). However, the presence of moderate to severe interstitial fibrosis in the lung biopsies of nearly 50% of patients with GLILD strongly argues this is a progressive lung disease in most patients.
Suspicion for GLILD should be raised particularly with the presence of lymphadenopathy, splenomegaly, autoimmunity, or a low number of class switched memory B-cells. Diagnosis of GLILD is based on HRCT of the chest (Figure 6) and, possibly, lung biopsy. Typical radiographic features of GLILD include large nodules and small nodules, areas of consolidation, and ground glass abnormality predominantly located in the lower lung zone, and frequent mediastinal adenopathy. The differential diagnosis of diffuse parenchymal disease in patients with CVID is extensive and includes LIP, nonspecific interstitial pneumonia, usual interstitial pneumonia, sarcoidosis, hypersensitivity pneumonitis, COP, low- and high-grade lymphoma, GLILD, as well as infection. To make a definitive diagnosis in a patient with suspected GLILD, we (JR) recommend a biopsy via video-assisted thoracoscopic surgery (VATS) with sampling of at least two lobes of the lung. Several studies have demonstrated that VATS is superior to transbronchial biopsy in determining a definite diagnosis in cases of suspected ILD (89–94). As previously mentioned, most patients with GLILD have significant areas of organizing pneumonia. Therefore, a transbronchial biopsy may lead to the misdiagnosis of COP due to sampling artifact. Similarly, transbronchial biopsy may yield insufficient tissue to make the diagnosis of lymphoma, particularly low-grade lymphoma. VATS also provides additional prognostic (extent of fibrosis) and diagnostic (e.g., B-cell lymphoma of low or high grade) information that may not be obtained by transbronchial biopsy. The 30-day post-operative mortality rate is 0–6% (89–93, 95, 96), which varies according to the clinical status of the patient as well as the expertise present in a given center (91, 92, 95, 97). Complications include pneumothorax, pulmonary atelectasis, lower respiratory tract infections, poor lung expansion, and more rarely (<5%) surgical emphysema, prolonged neuropathic pain, delayed wound healing, persistent air leak, acute respiratory distress, and hemothorax (91, 92, 95, 97). In children, VATS is recommended only if the diagnosis is uncertain despite thorough clinical evaluation or therapeutic decisions cannot be made without histology (98, 99). Positron emission tomography (PET) with 2-[(18)F]-fluoro-2-deoxy-d-glucose (FDG) combined with CT allows identification of active lymphoproliferative sites early in the inflammatory process and shows the systemic nature of the condition (Figure 7A) (100). In addition, FDG PET-CT shows sites of lymphoproliferation that may be more amenable for biopsy and confirmation of granulomatous inflammation than the lung. An additional alternative approach for confirming granulomatous histology would be using the tissue obtained at splenectomy if this had been undertaken. Obtaining tissue from more accessible organs should also be considered. For example, a skin biopsy can help to confirm or exclude sarcoidosis.
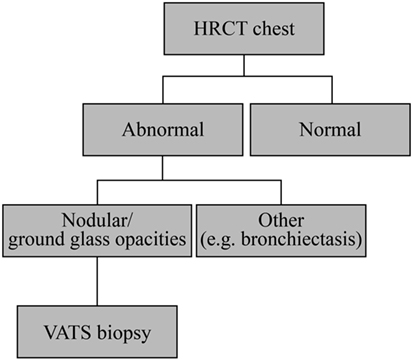
Figure 6. Granulomatous-lymphocytic interstitial lung disease diagnostic algorithm. All patients with common variable immunodeficiency should have a high-resolution computed tomography (HRCT) of the chest to screen for pulmonary disease. If there is significant interstitial lung disease (ground glass abnormalities, nodules, and areas of consolidation), one should consider lung biopsy by video-assisted thoracoscopic surgery (VATS) for definitive diagnosis. It may be worth considering if a more easily accessible organ such as a lymph node could be useful for detection of granulomas.
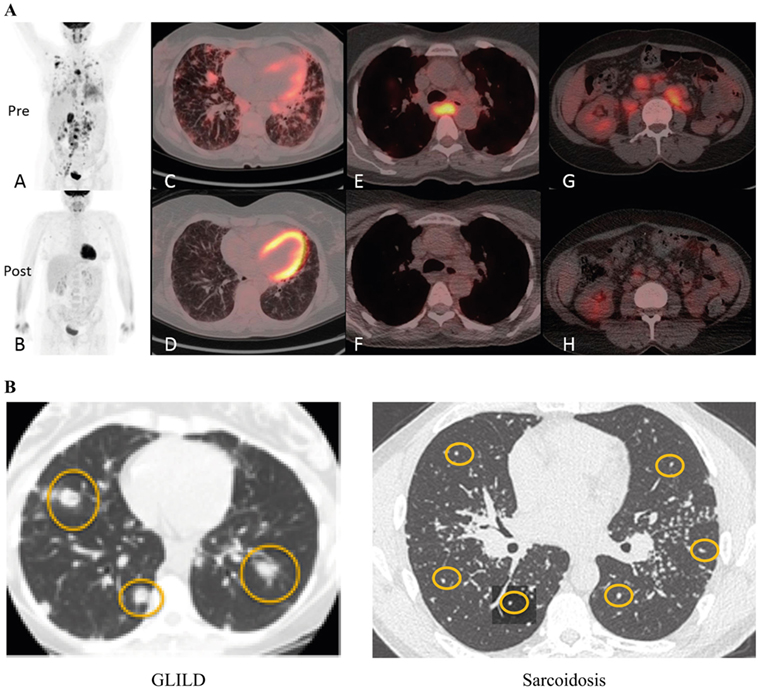
Figure 7. Diagnosis of granulomatous-lymphocytic interstitial lung disease (GLILD). (A) PET-CT before and 3 months after rituximab and mycophenolate treatment. Whole body images were acquired at 90 min following 285 MBq 2-[(18)F]-fluoro-2-deoxy-d-glucose (tracer). Maximum intensity projection whole body images before treatment show widespread abnormal uptake of tracer in the lung parenchyma and lymph nodes above and below the diaphragm (A); images following treatment show near resolution of all the areas of abnormal tracer uptake (B). (C,D) Axial fused positron emission tomography–computed tomography (PET–CT) images at the mid-thoracic level. The level of tracer uptake is represented by the intensity of color superimposed upon the CT image. There is a combination of interstitial septal thickening and ill-defined tracer-avid peri-bronchovascular nodules before treatment (C); post-treatment image shows improvement of the nodularity with near resolution of tracer uptake (D). Images at the level of the carina show an enlarged lymph node exhibiting intense abnormal tracer uptake prior to treatment (E), and a normalized lymph node with no abnormal tracer uptake following treatment (F). Pre-treatment images through the abdomen at the level of the right renal hilum demonstrate numerous enlarged tracer-avid retroperitoneal lymph nodes (G) which reduce in size and tracer uptake following treatment (H). Reproduced with kind permission from 2016 British Society for Immunology, Clinical and Experimental Immunology, 187: 138–145 (100). (B) Radiographic features of GLILD versus sarcoidosis. GLILD is characterized by macronodular (yellow circles) disease that is located predominantly in the lower lung zones. In contrast, micronodular (yellow circles) disease located in the upper lung zones and frequently accompanied by marked hilar adenopathy (stars) is characteristic of sarcoidosis. Reproduced from Ref. (101) with kind permission from European Respiratory Society.
While there is no universally accepted diagnostic process for GLILD, decisions are made individually and further studies will be needed to define the optimal approach.
Differentiating between GLILD and sarcoidosis can be potentially challenging, owing to the systemic nature of both disorders and the somewhat similar pathological findings (80). While both sarcoidosis and GLILD have non-necrotizing granuloma, FB and LIP are much more prominent in GLILD. Radiographically, the abnormalities in sarcoidosis are predominantly in the upper lung zones and consist of micronodular disease and a pronounced hilar adenopathy, whereas GLILD is characterized by micronodular and macronodular disease, diffuse adenopathy with less prominent hilar adenopathy, and lower lung zone predominance (Figure 7B). Bronchiectasis is uncommon in sarcoidosis, but occurs in 20–50% of patients with GLILD. In addition, plasma cells are present in the lung in sarcoidosis, but not in GLILD, consistent with the general absence of plasma cells in GLILD and CVID.
Monogenic disorders causing CVID-like diseases have now been well described in patients with GLILD: mutations in the transmembrane activator and calcium-modulator and cyclophilin ligand interactor (TACI), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) haploinsufficiency (102), lipopolysaccharide responsive beige-like anchor protein (LRBA) deficiency (103), mutations in the X-linked inhibitor of apoptosis protein (XIAP), hypomorphic recombinase-activating gene 1 (RAG1) mutations (104, 105), and lysine methyltransferase 2D (KMT2D) mutations (106). Patients with enteropathy, immune cytopenias, or other autoimmune or central nervous system diseases should raise the suspicion of LRBA deficiency or CTLA4 haploinsufficiency. Overall, 66% of patients with CTLA-4 deficiency were found to have GLILD (107, 108). LRBA functions to maintain the intracellular stores of CTLA-4 (109), and, therefore, LRBA deficiency and CTLA-4 deficiency have a similar clinical phenotype. The question whether all CVID patients should undergo at least targeted DNA sequencing remains to be answered (110–112). Determining the genetic cause of CVID might result in a change of treatment of various complications and improve prognosis. However, patients with a severe inflammatory/autoimmune disease, early presentation (younger than 10 years), multiple affected family members or very low numbers of B cells may have a higher likelihood of identification of a causative gene using next-generation sequencing approaches (112).
There are no guidelines and few reports on treatment of GLILD. However, the presence of both B and T cells in the infiltrate suggests a combination immunosuppressive therapy targeting both cell populations might be beneficial. Indeed, a combination of rituximab and azathioprine improved radiographic abnormalities and pulmonary functional tests (PFTs) in seven patients with CVID and GLILD (113). Rituximab (375 mg/m2/week) is administered intravenously once a week for 4 weeks, and this course is repeated 3–4 times at 4–6-month intervals. Oral azathioprine is given concomitantly for 18 months. In patients with poor tolerability for azathioprine, its active metabolite, 6-mercaptopurine, can be used. If the patient does not tolerate either of these drugs, mycophenolate mofetil is also effective. The dosage of these immunosuppressants needs to be adjusted to maintain an adequate lymphocyte and neutrophil count. Other immunosuppressive medications that have been reported to have a beneficial effect in GLILD include abatacept in patients with LRBA or CTLA4 haploinsufficiency (109, 114), tumor necrosis factor-α inhibitors (115), and corticosteroids (116). In our experience, corticosteroids do not lead to a durable remission. A double-blind placebo-controlled trial is needed to define the natural history of GLILD and document efficacy of immunosuppressive approaches in GLILD.
Assessment of the Lung in PAD
Pulmonary Function Tests
Pulmonary function tests provide useful information on lung performance and on the extent and progression of chronic lung disease in patients with PAD, in addition to the overall clinical assessment and physical examination. The most commonly performed PFT is spirometry providing data on the forced vital capacity (FVC) and FEV1: abnormal FVC and FEV1 readings may indicate restrictive and obstructive lung disease, respectively. In patients with PAD, decline in FEV1 has been shown to be a more sensitive indicator of respiratory function than FVC, as expected due to the obstructive nature of bronchiectasis (117). In addition to assessments performed by spirometry, regular assessment of diffusing capacity for carbon monoxide (DLCO) in suspected ILD is useful alone (117). This is one of the reasons why care of CVID patients should be managed by specialized centers that have access to other specialties, including a pulmonology liaison service. DLCO should also be evaluated in all patients with splenomegaly and/or lymphadenopathy.
In a study of 37 patients with CVID and XLA, the average annual decline in FEV1 was 45 ± 6 mL/year (9), which exceeds the normal rate of decline of 19.6 and 17.6 mL/year in healthy non-smoking males and females, respectively (10). In this study, lung function decline correlated inversely with IVIG dose (P = 0.004), indicating that higher IVIG doses are associated with preservation of lung function, although the correlation between trough IgG levels and FEV1 decline was not statistically significant (P = 0.485). In another study of 20 patients with PAD, a clear correlation between higher IgG trough levels and preservation of FEV1 (R2 = 0.2688; P = 0.0281) was observed (117). A significant correlation between serum IgG trough level and FVC and FEV1 values was found also in 12 patients with hypogammaglobulinemia and ILD and led to the conclusion that the minimal target serum IgG level should be 5 g/L (118). Collectively, these studies suggest higher doses of IgG may be considered for patients showing deteriorating lung function; however, maximum recommended trough IgG levels have not been determined. The decline in FEV1 also appeared slower in patients who spent longer periods of time on antibiotics; however, the correlation was not significant (P = 0.1195) (117).
Based on these rates of decline in FEV1, we recommend at least annual PFT monitoring (including DLCO if available) for patients with PAD older than 5 years of age (117). Testing more frequently may be appropriate for those experiencing a more rapid decline of lung function (119).
Recently, several clinical trials employed a new technology, the multiple breath washout technique (MBW). MBW assesses the quality of ventilation, indicated by the lung clearance index (LCI) during regular breathing. This method is not only less dependent on the cooperation of the patient, thus allowing use in children as young as age 4, but also appears more sensitive to minor changes at an early stage of lung disease and more meaningful in advanced disease, as shown in CF (120), primary ciliary dyskinesia (121), and non-CF bronchiectasis (122), in all of which LCI correlated better to the extent of bronchiectasis than FEV1. Finally, MBW appeared also more sensitive for assessing changes in lung disease in clinical trials with relatively short-term interventions. Inhalation of hypertonic saline in a small group (n = 20) of children with CF over a period of 4 weeks resulted in a significant improvement of LCI (treatment effect 1.16 ± 0.94), but not of spirometry parameters (123). However, to observe a significant change in FEV1 [% predicted] with this intervention, a sample size of 351 patients would have been required.
High-Resolution CT
Pulmonary functional tests have a low sensitivity to screen for pulmonary complications such as ILD and bronchiectasis (124) and, therefore, should be combined with imaging techniques. The sensitivity of HRCT for detection of pulmonary complications in patients with CVID is superior to both chest X-ray and PFTs (125), but the method is associated with higher costs and radiation doses. Radiation dose is a potential concern for patients with CVID, who in some cases may be more radiosensitive than healthy subjects (126–128), particularly where over time repeated imaging may be clinically necessary. For these patients, and especially for children, CT techniques that use lower radiation doses should be considered. Although low-dose computed tomography (LDCT) uses less ionizing radiation than conventional CT scans, the predictive effective doses used in LDCT of the lungs may still be 4–12 times greater than those used in conventional X-rays (129). A possibility to further reduce the radiation dose in lung CT scans would be to use the model-based iterative reconstruction (MBIR) technique for ultra-low-dose chest CT. MBIR provides an almost 80% reduction in radiation dose for chest CT without reducing sensitivity (130).
HCRT can be critical in the early diagnosis of airway abnormalities (airway disease) and ILD, which cannot be differentiated by PFTs (131). In patients with CVID, HRCT revealed airway disease in 20% of pediatric patients and 30% of adults, and ILD in 15% of children and 34% of adults, respectively (124, 131). In the adult population, the presence of ILD correlated positively with autoimmune disease and markedly lower CD4+ T cell, naïve CD4+ T cell, and naïve and switched memory B-cell counts (124). Both airway disease and ILD are treatable diseases but differ in etiology and require different treatment. While high-dose IGRT may impact progression of airway disease in patients with CVID, ILD responds to immunosuppressive therapy. In pediatric CVID patients with ILD, nodules on HCRT disappeared following treatment with steroids, suggesting that early ILD may be reversible (131). As ILD appears to be asymptomatic during the initial stage, screening of all patients with CVID should facilitate early disease detection and enable early therapy which could be less aggressive (131).
There is a need for standardized HCRT scoring methods to facilitate efficient screening and follow-up for pulmonary complications associated with CVID (131, 132). To aid the assessment of unrecognized lung disease progression in these patients, the scoring system should be based on abnormalities specific for PAD, rather than adapted from other diseases such as CF. A scoring method was developed in a cohort of 54 children with stable CVID or CVID-like disorders (132). In this pediatric study population, lung abnormalities were common (found in 85–95% of patients), although the overall extent and severity of abnormalities were mild. Bronchial pathology was the most common pathology, observed in up to half and one-third of the patients with bronchial wall thickening and bronchiectasis, respectively. However, interstitial pathology and ventilation pathology were described as affecting a third (air trapping) and a quarter (nodules) of the patients. The latter was effectively treated with corticosteroids in some cases. The Chest CT in Antibody Deficiency Group is currently developing a quantitative scoring system that can be used as a validated tool in multicenter studies and could help to endorse uniform documentation of chest CT scans (133).
Magnetic Resonance Imaging (MRI) With Diffusion Weighted Imaging (DWI)
Magnetic resonance imaging with DWI offers a radiation-free alternative to the HRCT scan. MRI has been shown to be non-inferior to HRCT in identifying bronchial and parenchymal abnormalities, although HCRT had a higher capacity in identifying peripheral airway abnormalities (134). PFT alterations correlated well with MRI bronchial abnormalities, but not with MRI parenchymal scores (134). The implementation of DWI facilitates the acquisition of information about the microstructure of the tissue and may enable detection of areas of inflammation and subclinical infection in lung parenchyma (134). However, although promising, MRI DWI is technically demanding, requires an expert radiologist, and possibly necessitates sedation in children.
Inflammatory and Clinical Biomarkers
To determine the need of anti-inflammatory drugs as adjunctive therapy in PAD, reliable biomarkers are required. Potentially useful biomarkers of infection risk are the presence of sinusitis, low IgG trough levels, low IgA, and bronchiectasis. Similarly, the following symptoms could be valuable as biomarkers of non-infectious inflammation: low class-switched memory B-cells, autoimmunity, low T cells and naïve T cells, splenomegaly, lymphadenopathy, and potentially elevated IgM and beta 2 microglobulin (100). However, data on biomarkers in peripheral blood are not available, except for low endogenous plasma IgG concentrations measured prior to IGRT and a deficiency of class-switched memory B cells (24, 135, 136). IgM levels are not always low in patients with CVID, but they were reported to be lower in patients experiencing recurrent infections compared with those with fewer infections (24). Splenomegaly can be a biomarker for GLILD, given the multi-systemic nature of the disease (81). Abnormalities in the T-cell population found in BALF, such as a low CD4/CD8 ratio, can be associated with poor functional outcomes including FEV1 and FVC (137). However, bronchoalveolar lavage for T-cell assessment is an invasive technique and thus it is usually used only in patients with severe disease. Other potential inflammatory markers in spontaneously expectorated (138) or induced (139) sputum, including cellular content, cytokines, reactive oxygen metabolites, and bacterial density (140) have been evaluated in various conditions including CVID, yet before they can become a reality in this setting, further studies are needed (43). Likewise, there are no reliable data of the usefulness of beta 2 microglobulin in patients with PAD.
Microbiological surveillance of patients with PAD is of great clinical importance. Although it is currently unclear how many episodes of bacterial infection in PAD are preceded by viral infections of the upper airway (2), appropriate microbiological surveillance may help understand and prevent infection progression. The surveillance protocol in patients with PAD may include regular chest HRCT and PFTs (including DLCO and possibly LCI) or MRI with DWI as an alternative to CT with optional sputum analysis and molecular detection with polymerase chain reaction (3, 4).
Conclusion
Pulmonary morbidity in patients with PAD advances despite apparently adequate IGRT. New preventive and therapeutic approaches are urgently needed. Clinical trials to evaluate these approaches will rely on new ways of assessing and documenting lung disease. Optimal pulmonary management must encompass the upper airway, the “gateway to the lung.” Non-infectious complications such as non-infectious lymphoproliferation, where GLILD represents the pulmonary component, contribute substantially to morbidity in these patients, and requires specific treatment. Regular assessment of the structural and functional condition of the lung as well as an improved understanding of subclinical recurrent infections will, we hope, inform therapeutic decisions aiming to reduce lung damage.
Key Take-Home Messages
• Lung disease, both infectious and non-infectious, progresses despite IGRT
• Characteristics making lung disease more likely include existing inflammatory/autoimmune disease, lymphadenopathy, splenomegaly, low counts of class-switched memory B cells, and CD4+ T cells
• Improved molecular characterization of gene defects has led to identification of several monogenic defects which appear to be associated with granulomatous-lymphocytic ILD (mutations in TACI, CTLA-4 haploinsufficiency, or LRBA deficiency, mutations in XIAP, hypomorphic RAG1 mutations, and KMT2D mutations)
• A better understanding of the natural history and pathogenesis of ILD in PAD is needed
• Early diagnosis may allow interventions to prevent or slow end organ damage
• New and improved PFT modalities now exist and warrant further study for utility in PAD
• Low-dose CT scans and radiation-free alternative scans offer an improved safety profile in PAD
• Standardized multi-center documentation of chest CT scans is one of the tools for providing better data on natural history and interventions
• Optimal pulmonary management of patients with PAD must encompass the upper airway, which serves as the “the gateway” to the lungs
• There are few clinical studies evaluating potential treatments of lung disease in PAD
• Early targeted intervention may reduce the burden of steroids currently employed, which are used over long periods of time with only partial efficacy
• New therapies are needed to relieve the burden of viral infections
Author Contributions
All authors contributed to the same extent to the development of this article.
Conflict of Interest Statement
UB has received honoraria, expenses, and consulting fees from Baxalta, now part of Shire, Biotest, CSL Behring, and Octapharma, and research grants from CSL Behring and Baxalta, now part of Shire. JR has received support from CSL Behring for participating in the CSL Behring sponsored satellite symposium at the ESID 2016 Annual Meeting. PS-P has received advisory board, speaker, or project support from Grifols; support from CSL Behring for attending the ESID 2016 Annual Meeting. SJ has received support for speaker, conference, advisory board, clinical trial, or projects from CSL Behring, Shire, Octapharma, Biotest, LFB, UCB Pharma, GlaxoSmithKline, Swedish Orphan Biovitrum, Binding Site, Grifols, Zarodex, and Weatherden, and support from LFB for attending the ESID 2016 Annual Meeting.
Acknowledgments
Part of this work was presented at the CSL Behring sponsored symposium at the 2016 European Society for Immunodeficiencies (ESID) Meeting in Edinburgh, UK. Editorial assistance was provided by Emiliana Jelezarova, Ph.D., CMPP™, Fishawack Communications GmbH, a member of the Fishawack Group of Companies, funded by CSL Behring.
Abbreviations
BALF, bronchoalveolar lavage fluid; CF, cystic fibrosis; CI, confidence interval; COP, cryptogenic organizing pneumonia; COPD, chronic obstructive pulmonary disease; CT, computed tomography; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; CVID, common variable immunodeficiency; DLCO, diffusing capacity for carbon monoxide; DNase, recombinant human deoxyribonuclease (dornase alfa); DWI, diffusion weighted imaging; ESID, European Society for immunodeficiencies; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; GLILD, granulomatous-lymphocytic interstitial lung disease; HRCT, high-resolution computed tomography; Ig, immunoglobulin; IGRT, immunoglobulin replacement therapy; ILD, interstitial lung disease; IVIG, intravenous immunoglobulin G; KMT2D, lysine methyltransferase 2D; LCI, lung clearance index; LDCT, low-dose computed tomography; LIP, lymphocytic interstitial pneumonitis; LRBA, lipopolysaccharide responsive beige-like anchor protein; MBIR, model-based iterative reconstruction; MBW, multiple breath washout technique; MDA5, melanoma differentiation-associated protein 5; MRI, magnetic resonance imaging; PAD, primary antibody deficiencies; PET, positron emission tomography; RAG1, recombinase-activating gene 1; TACI, transmembrane activator and calcium-modulator and cyclophilin ligand interactor; VATS, video-assisted thoracoscopic surgery; XIAP, X-linked inhibitor of apoptosis protein; XLA, X-linked agammaglobulinemia.
References
1. Lucas M, Lee M, Lortan J, Lopez-Granados E, Misbah S, Chapel H. Infection outcomes in patients with common variable immunodeficiency disorders: relationship to immunoglobulin therapy over 22 years. J Allergy Clin Immunol (2010) 125(6):1354–60.e4. doi:10.1016/j.jaci.2010.02.040
2. Jolles S. Subclinical infection and dosing in primary immunodeficiencies. Clin Exp Immunol (2014) 178(Suppl 1):67–9. doi:10.1111/cei.12516
3. Hodkinson JP, Bangs C, Wartenberg-Demand A, Bauhofer A, Langohr P, Buckland MS, et al. Low IgA and IgM is associated with a higher prevalence of bronchiectasis in primary antibody deficiency. J Clin Immunol (2017) 37(4):329–31. doi:10.1007/s10875-017-0381-y
4. Hodkinson JP, Bangs C, Wartenberg-Demand A, Bauhofer A, Langohr P, Buckland MS, et al. Erratum to: low IgA and IgM is associated with a higher prevalence of bronchiectasis in primary antibody deficiency. J Clin Immunol (2017) 37(4):332. doi:10.1007/s10875-017-0392-8
5. Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol (1999) 92(1):34–48. doi:10.1006/clim.1999.4725
6. Healy MJ. Hypogammaglobulinaemia in the United Kingdom. XII. Statistical analyses: prevalence, mortality and effects of treatment. Spec Rep Ser Med Res Counc (G B) (1971) 310:115–23.
7. Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood (2012) 119(7):1650–7. doi:10.1182/blood-2011-09-377945
8. Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood (2008) 112(2):277–86. doi:10.1182/blood-2007-11-124545
9. Chen Y, Stirling RG, Paul E, Hore-Lacy F, Thompson BR, Douglass JA. Longitudinal decline in lung function in patients with primary immunoglobulin deficiencies. J Allergy Clin Immunol (2011) 127(6):1414–7. doi:10.1016/j.jaci.2011.03.041
10. Kohansal R, Martinez-Camblor P, Agusti A, Buist AS, Mannino DM, Soriano JB. The natural history of chronic airflow obstruction revisited: an analysis of the Framingham offspring cohort. Am J Respir Crit Care Med (2009) 180(1):3–10. doi:10.1164/rccm.200901-0047OC
11. Ataeinia B, Montazeri A, Tavakol M, Azizi G, Kiaee F, Tavakolinia N, et al. Measurement of health-related quality of life in primary antibody-deficient patients. Immunol Invest (2017) 46(4):329–40. doi:10.1080/08820139.2016.1258710
12. Tcheurekdjian H, Palermo T, Hostoffer R. Quality of life in common variable immunodeficiency requiring intravenous immunoglobulin therapy. Ann Allergy Asthma Immunol (2004) 93(2):160–5. doi:10.1016/S1081-1206(10)61469-X
13. Titman P, Allwood Z, Gilmour C, Malcolmson C, Duran-Persson C, Cale C, et al. Quality of life in children with primary antibody deficiency. J Clin Immunol (2014) 34(7):844–52. doi:10.1007/s10875-014-0072-x
14. Jolles S, Sanchez-Ramon S, Quinti I, Soler-Palacin P, Agostini C, Florkin B, et al. Screening protocols to monitor respiratory status in primary immunodeficiency disease: findings from a European survey and subclinical infection working group. Clin Exp Immunol (2017) 190(2):226–34. doi:10.1111/cei.13012
15. Pilette C, Ouadrhiri Y, Godding V, Vaerman J-P, Sibille Y. Lung mucosal immunity: immunoglobulin-A revisited. Eur Respir J (2001) 18(3):571–88. doi:10.1183/09031936.01.00228801
16. Baumann U, Gocke K, Gewecke B, Freihorst J, von Specht BU. Assessment of pulmonary antibodies with induced sputum and bronchoalveolar lavage induced by nasal vaccination against Pseudomonas aeruginosa: a clinical phase I/II study. Respir Res (2007) 8:57. doi:10.1186/1465-9921-8-57
17. Labro MT. Immunopathology of respiratory infections. Clin Microbiol Infect (1998) 4(Suppl 2):2S1–2. doi:10.1111/j.1469-0691.1998.tb00689.x
18. Cerruti A, Cheng K, Alejo C. Immunoglobulin responses at the mucosal interface. Annu Rev Immunol (2011) 29:273–93. doi:10.1146/annurev-immunol-031210-101317
19. Twigg HL. Humoral immune defense (antibodies). Proc Am Thorac Soc (2005) 2(5):417–21. doi:10.1513/pats.200508-089JS
22. Ameratunga R, Woon ST, Gillis D, Koopmans W, Steele R. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol (2013) 174(2):203–11. doi:10.1111/cei.12178
23. Routes J, Abinun M, Al-Herz W, Bustamante J, Condino-Neto A, De La Morena MT, et al. ICON: the early diagnosis of congenital immunodeficiencies. J Clin Immunol (2014) 34(4):398–424. doi:10.1007/s10875-014-0003-x
24. Quinti I, Soresina A, Guerra A, Rondelli R, Spadaro G, Agostini C, et al. Effectiveness of immunoglobulin replacement therapy on clinical outcome in patients with primary antibody deficiencies: results from a multicenter prospective cohort study. J Clin Immunol (2011) 31(3):315–22. doi:10.1007/s10875-011-9511-0
25. Hodkinson JP. Considerations for dosing immunoglobulin in obese patients. Clin Exp Immunol (2017) 188(3):353–62. doi:10.1111/cei.12955
26. Kainulainen L, Nikoskelainen J, Vuorinen T, Tevola K, Liippo K, Ruuskanen O. Viruses and bacteria in bronchial samples from patients with primary hypogammaglobulinemia. Am J Respir Crit Care Med (1999) 159(4 Pt 1):1199–204. doi:10.1164/ajrccm.159.4.9807067
27. Kainulainen L, Vuorinen T, Rantakokko-Jalava K, Osterback R, Ruuskanen O. Recurrent and persistent respiratory tract viral infections in patients with primary hypogammaglobulinemia. J Allergy Clin Immunol (2010) 126(1):120–6. doi:10.1016/j.jaci.2010.04.016
28. Burns JL, Gibson RL, McNamara S, Yim D, Emerson J, Rosenfeld M, et al. Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. J Infect Dis (2001) 183(3):444–52. doi:10.1086/318075
29. Mainz JG, Naehrlich L, Schien M, Kading M, Schiller I, Mayr S, et al. Concordant genotype of upper and lower airways P aeruginosa and S aureus isolates in cystic fibrosis. Thorax (2009) 64(6):535–40. doi:10.1136/thx.2008.104711
30. Micol R, Kayal S, Mahlaoui N, Beaute J, Brosselin P, Dudoit Y, et al. Protective effect of IgM against colonization of the respiratory tract by nontypeable Haemophilus influenzae in patients with hypogammaglobulinemia. J Allergy Clin Immunol (2012) 129(3):770–7. doi:10.1016/j.jaci.2011.09.047
31. Peltola V, Waris M, Kainulainen L, Kero J, Ruuskanen O. Virus shedding after human rhinovirus infection in children, adults and patients with hypogammaglobulinaemia. Clin Microbiol Infect (2013) 19(7):E322–7. doi:10.1111/1469-0691.12193
32. Johansen HK, Hoiby N. Seasonal onset of initial colonisation and chronic infection with Pseudomonas aeruginosa in patients with cystic fibrosis in Denmark. Thorax (1992) 47(2):109–11. doi:10.1136/thx.47.2.109
33. Esther CR Jr, Lin FC, Kerr A, Miller MB, Gilligan PH. Respiratory viruses are associated with common respiratory pathogens in cystic fibrosis. Pediatr Pulmonol (2014) 49(9):926–31. doi:10.1002/ppul.22917
34. Molyneaux PL, Mallia P, Cox MJ, Footitt J, Willis-Owen SA, Homola D, et al. Outgrowth of the bacterial airway microbiome after rhinovirus exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2013) 188(10):1224–31. doi:10.1164/rccm.201302-0341OC
35. Franz A, Webster AD, Furr PM, Taylor-Robinson D. Mycoplasmal arthritis in patients with primary immunoglobulin deficiency: clinical features and outcome in 18 patients. Br J Rheumatol (1997) 36(6):661–8. doi:10.1093/rheumatology/36.6.661
36. Hamvas RM, Johnson M, Vlieger AM, Ling C, Sherriff A, Wade A, et al. Role for mannose binding lectin in the prevention of Mycoplasma infection. Infect Immun (2005) 73(8):5238–40. doi:10.1128/IAI.73.8.5238-5240.2005
37. Heilmann C, Jensen L, Jensen JS, Lundstrom K, Windsor D, Windsor H, et al. Treatment of resistant Mycoplasma infection in immunocompromised patients with a new pleuromutilin antibiotic. J Infect (2001) 43(4):234–8. doi:10.1053/jinf.2001.0910
38. Roifman CM, Rao CP, Lederman HM, Lavi S, Quinn P, Gelfand EW. Increased susceptibility to Mycoplasma infection in patients with hypogammaglobulinemia. Am J Med (1986) 80(4):590–4. doi:10.1016/0002-9343(86)90812-0
39. Plebani A, Soresina A, Rondelli R, Amato GM, Azzari C, Cardinale F, et al. Clinical, immunological, and molecular analysis in a large cohort of patients with X-linked agammaglobulinemia: an Italian multicenter study. Clin Immunol (2002) 104(3):221–30. doi:10.1006/clim.2002.5241
40. Quinti I, Soresina A, Spadaro G, Martino S, Donnanno S, Agostini C, et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol (2007) 27(3):308–16. doi:10.1007/s10875-007-9075-1
41. Rojavin MA, Hubsch A, Lawo JP. Quantitative evidence of wear-off effect at the end of the intravenous IgG (IVIG) dosing cycle in primary immunodeficiency. J Clin Immunol (2016) 36(3):210–9. doi:10.1007/s10875-016-0243-z
42. King P. Pathogenesis of bronchiectasis. Paediatr Respir Rev (2011) 12(2):104–10. doi:10.1016/j.prrv.2010.10.011
43. Pereira AC, Kokron CM, Romagnolo BM, Yagi CS, Saldiva PH, Lorenzi Filho G, et al. Analysis of the sputum and inflammatory alterations of the airways in patients with common variable immunodeficiency and bronchiectasis. Clinics (Sao Paulo) (2009) 64(12):1155–60. doi:10.1590/S1807-59322009001200004
44. Javidan-Nejad C, Bhalla S. Bronchiectasis. Thorac Surg Clin (2010) 20(1):85–102. doi:10.1016/j.thorsurg.2009.12.007
45. McDonnell MJ, Ward C, Lordan JL, Rutherford RM. Non-cystic fibrosis bronchiectasis. QJM (2013) 106(8):709–15. doi:10.1093/qjmed/hct109
46. Reid LM. Reduction in bronchial subdivision in bronchiectasis. Thorax (1950) 5(3):233–47. doi:10.1136/thx.5.3.233
47. Thickett KM, Kumararatne DS, Banerjee AK, Dudley R, Stableforth DE. Common variable immune deficiency: respiratory manifestations, pulmonary function and high-resolution CT scan findings. QJM (2002) 95(10):655–62. doi:10.1093/qjmed/95.10.655
48. Verma N, Grimbacher B, Hurst JR. Lung disease in primary antibody deficiency. Lancet Respir Med (2015) 3(8):651–60. doi:10.1016/S2213-2600(15)00202-7
49. Moller W, Munzing W, Canis M. Clinical potential of pulsating aerosol for sinus drug delivery. Expert Opin Drug Deliv (2010) 7(11):1239–45. doi:10.1517/17425247.2010.523078
50. Pynnonen MA, Mukerji SS, Kim HM, Adams ME, Terrell JE. Nasal saline for chronic sinonasal symptoms: a randomized controlled trial. Arch Otolaryngol Head Neck Surg (2007) 133(11):1115–20. doi:10.1001/archotol.133.11.1115
51. Moller W, Schuschnig U, Khadem Saba G, Meyer G, Junge-Hulsing B, Keller M, et al. Pulsating aerosols for drug delivery to the sinuses in healthy volunteers. Otolaryngol Head Neck Surg (2010) 142(3):382–8. doi:10.1016/j.otohns.2009.12.028
52. Jolles S, Kaveri SV, Orange J. Intravenous immunoglobulins. Current understanding and future directions. Clin Exp Immunol (2009) 158(Suppl 1):68–70. doi:10.1111/j.1365-2249.2009.04029.x
53. Moller W, Saba GK, Haussinger K, Becker S, Keller M, Schuschnig U. Nasally inhaled pulsating aerosols: lung, sinus and nose deposition. Rhinology (2011) 49(3):286–91. doi:10.4193/Rhino10.268
54. Mainz JG, Schadlich K, Schien C, Michl R, Schelhorn-Neise P, Koitschev A, et al. Sinonasal inhalation of tobramycin vibrating aerosol in cystic fibrosis patients with upper airway Pseudomonas aeruginosa colonization: results of a randomized, double-blind, placebo-controlled pilot study. Drug Des Devel Ther (2014) 8:209–17. doi:10.2147/DDDT.S54064
55. Mainz JG, Schien C, Schiller I, Schadlich K, Koitschev A, Koitschev C, et al. Sinonasal inhalation of dornase alfa administered by vibrating aerosol to cystic fibrosis patients: a double-blind placebo-controlled cross-over trial. J Cyst Fibros (2014) 13(4):461–70. doi:10.1016/j.jcf.2014.02.005
56. Ruuskanen O, Waris M, Kainulainen L. Treatment of persistent rhinovirus infection with pegylated interferon alpha2a and ribavirin in patients with hypogammaglobulinemia. Clin Infect Dis (2014) 58(12):1784–6. doi:10.1093/cid/ciu169
57. Nanishi E, Hoshina T, Takada H, Ishimura M, Nishio H, Uehara T, et al. A nationwide survey of common viral infections in childhood among patients with primary immunodeficiency diseases. J Infect (2016) 73(4):358–68. doi:10.1016/j.jinf.2016.07.018
58. Hanitsch LG, Lobel M, Mieves JF, Bauer S, Babel N, Schweiger B, et al. Cellular and humoral influenza-specific immune response upon vaccination in patients with common variable immunodeficiency and unclassified antibody deficiency. Vaccine (2016) 34(21):2417–23. doi:10.1016/j.vaccine.2016.03.091
59. Principi N, Esposito S. Vaccine use in primary immunodeficiency disorders. Vaccine (2014) 32(30):3725–31. doi:10.1016/j.vaccine.2014.05.022
60. Lamborn IT, Jing H, Zhang Y, Drutman SB, Abbott JK, Munir S, et al. Recurrent rhinovirus infections in a child with inherited MDA5 deficiency. J Exp Med (2017) 214(7):1949–72. doi:10.1084/jem.20161759
61. Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell (2014) 157(1):121–41. doi:10.1016/j.cell.2014.03.011
62. Boyton RJ, Reynolds CJ, Quigley KJ, Altmann DM. Immune mechanisms and the impact of the disrupted lung microbiome in chronic bacterial lung infection and bronchiectasis. Clin Exp Immunol (2013) 171(2):117–23. doi:10.1111/cei.12003
63. Brown RL, Clarke TB. The regulation of host defences to infection by the microbiota. Immunology (2017) 150(1):1–6. doi:10.1111/imm.12634
64. Blaser MJ, Falkow S. What are the consequences of the disappearing human microbiota? Nat Rev Microbiol (2009) 7(12):887–94. doi:10.1038/nrmicro2245
65. Chen LW, Chen PH, Hsu CM. Commensal microflora contribute to host defense against Escherichia coli pneumonia through toll-like receptors. Shock (2011) 36(1):67–75. doi:10.1097/SHK.0b013e3182184ee7
66. Fagundes CT, Amaral FA, Vieira AT, Soares AC, Pinho V, Nicoli JR, et al. Transient TLR activation restores inflammatory response and ability to control pulmonary bacterial infection in germfree mice. J Immunol (2012) 188(3):1411–20. doi:10.4049/jimmunol.1101682
67. Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, et al. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci U S A (2011) 108(13):5354–9. doi:10.1073/pnas.1019378108
68. Huang YJ, Charlson ES, Collman RG, Colombini-Hatch S, Martinez FD, Senior RM. The role of the lung microbiome in health and disease. A National Heart, Lung, and Blood Institute workshop report. Am J Respir Crit Care Med (2013) 187(12):1382–7. doi:10.1164/rccm.201303-0488WS
69. Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, et al. Disordered microbial communities in asthmatic airways. PLoS One (2010) 5(1):e8578. doi:10.1371/journal.pone.0008578
70. Huang YJ, Kim E, Cox MJ, Brodie EL, Brown R, Wiener-Kronish JP, et al. A persistent and diverse airway microbiota present during chronic obstructive pulmonary disease exacerbations. OMICS (2010) 14(1):9–59. doi:10.1089/omi.2009.0100
71. Jorgensen SF, Troseid M, Kummen M, Anmarkrud JA, Michelsen AE, Osnes LT, et al. Altered gut microbiota profile in common variable immunodeficiency associates with levels of lipopolysaccharide and markers of systemic immune activation. Mucosal Immunol (2016) 9(6):1455–65. doi:10.1038/mi.2016.18
72. Milito C, Pulvirenti F, Tabolli S, Carrabba MD, Fabio G, Pietrogrande MC, et al. Antibiotic prophylaxis in primary antibody deficiency patients: study design. 17th Biennial Meeting of the European Society for Immunodeficiencies, 21-24 Sept 2016. Conference Proceedings. Barcelona, Spain: (2016). p. 158, AB270.
73. Sperlich JM, Grimbacher B, Workman S, Haque T, Seneviratne SL, Burns SO, et al. Respiratory infections and antibiotic usage in common variable immunodeficiency. J Allergy Clin Immunol Pract (2018) 6(1):159–68.e3. doi:10.1016/j.jaip.2017.05.024
74. Kellett F, Robert NM. Nebulised 7% hypertonic saline improves lung function and quality of life in bronchiectasis. Respir Med (2011) 105(12):1831–5. doi:10.1016/j.rmed.2011.07.019
75. Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med (1994) 331(10):637–42. doi:10.1056/NEJM199409083311003
76. O’Donnell AE, Barker AF, Ilowite JS, Fick RB. Treatment of idiopathic bronchiectasis with aerosolized recombinant human DNase I. rhDNase Study Group. Chest (1998) 113(5):1329–34. doi:10.1378/chest.113.5.1329
77. Ameratunga R, Lehnert K, Woon ST, Gillis D, Bryant VL, Slade CA, et al. Review: diagnosing common variable immunodeficiency disorder in the era of genome sequencing. Clin Rev Allergy Immunol (2018) 54(2):261–8. doi:10.1007/s12016-017-8645-0
78. Bates CA, Ellison MC, Lynch DA, Cool CD, Brown KK, Routes JM. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol (2004) 114(2):415–21. doi:10.1016/j.jaci.2004.05.057
79. Sander CA, Medeiros LJ, Weiss LM, Yano T, Sneller MC, Jaffe ES. Lymphoproliferative lesions in patients with common variable immunodeficiency syndrome. Am J Surg Pathol (1992) 16(12):1170–82. doi:10.1097/00000478-199212000-00004
80. Tam JS, Routes JM. Common variable immunodeficiency. Am J Rhinol Allergy (2013) 27(4):260–5. doi:10.2500/ajra.2013.27.3899
81. Rao N, Mackinnon AC, Routes JM. Granulomatous and lymphocytic interstitial lung disease: a spectrum of pulmonary histopathologic lesions in common variable immunodeficiency – histologic and immunohistochemical analyses of 16 cases. Hum Pathol (2015) 46(9):1306–14. doi:10.1016/j.humpath.2015.05.011
82. Maglione PJ, Ko HM, Beasley MB, Strauchen JA, Cunningham-Rundles C. Tertiary lymphoid neogenesis is a component of pulmonary lymphoid hyperplasia in patients with common variable immunodeficiency. J Allergy Clin Immunol (2014) 133(2):535–42. doi:10.1016/j.jaci.2013.08.022
83. Arandi N, Mirshafiey A, Jeddi-Tehrani M, Abolhassani H, Sadeghi B, Mirminachi B, et al. Evaluation of CD4+CD25+FOXP3+ regulatory T cells function in patients with common variable immunodeficiency. Cell Immunol (2013) 281(2):129–33. doi:10.1016/j.cellimm.2013.03.003
84. Fevang B, Yndestad A, Sandberg WJ, Holm AM, Muller F, Aukrust P, et al. Low numbers of regulatory T cells in common variable immunodeficiency: association with chronic inflammation in vivo. Clin Exp Immunol (2007) 147(3):521–5. doi:10.1111/j.1365-2249.2006.03314.x
85. Xiao X, Miao Q, Chang C, Gershwin ME, Ma X. Common variable immunodeficiency and autoimmunity – an inconvenient truth. Autoimmun Rev (2014) 13(8):858–64. doi:10.1016/j.autrev.2014.04.006
86. Yu GP, Chiang D, Song SJ, Hoyte EG, Huang J, Vanishsarn C, et al. Regulatory T cell dysfunction in subjects with common variable immunodeficiency complicated by autoimmune disease. Clin Immunol (2009) 131(2):240–53. doi:10.1016/j.clim.2008.12.006
87. Maglione PJ, Overbey JR, Cunningham-Rundles C. Progression of common variable immunodeficiency interstitial lung disease accompanies distinct pulmonary and laboratory findings. J Allergy Clin Immunol Pract (2015) 3(6):941–50. doi:10.1016/j.jaip.2015.07.004
88. Mannina A, Chung JH, Swigris JJ, Solomon JJ, Huie TJ, Yunt ZX, et al. Clinical predictors of a diagnosis of common variable immunodeficiency-related granulomatous-lymphocytic interstitial lung disease. Ann Am Thorac Soc (2016) 13(7):1042–9. doi:10.1513/AnnalsATS.201511-728OC
89. Blackhall V, Asif M, Renieri A, Civitelli S, Kirk A, Jilaihawi A, et al. The role of surgical lung biopsy in the management of interstitial lung disease: experience from a single institution in the UK. Interact Cardiovasc Thorac Surg (2013) 17(2):253–7. doi:10.1093/icvts/ivt217
90. Lee YC, Wu CT, Hsu HH, Huang PM, Chang YL. Surgical lung biopsy for diffuse pulmonary disease: experience of 196 patients. J Thorac Cardiovasc Surg (2005) 129(5):984–90. doi:10.1016/j.jtcvs.2004.07.033
91. Luo Q, Han Q, Chen X, Xie J, Wu L, Chen R. The diagnosis efficacy and safety of video-assisted thoracoscopy surgery (VATS) in undefined interstitial lung diseases: a retrospective study. J Thorac Dis (2013) 5(3):283–8. doi:10.3978/j.issn.2072-1439.2013.04.12
92. Morris D, Zamvar V. The efficacy of video-assisted thoracoscopic surgery lung biopsies in patients with interstitial lung disease: a retrospective study of 66 patients. J Cardiothorac Surg (2014) 9:45. doi:10.1186/1749-8090-9-45
93. Sigurdsson MI, Isaksson HJ, Gudmundsson G, Gudbjartsson T. Diagnostic surgical lung biopsies for suspected interstitial lung diseases: a retrospective study. Ann Thorac Surg (2009) 88(1):227–32. doi:10.1016/j.athoracsur.2009.04.002
94. Lieberman S, Gleason JB, Ilyas MIM, Martinez F, Mehta JP, Savage EB. Assessing the safety and clinical impact of thoracoscopic lung biopsy in patients with interstitial lung disease. J Clin Diagn Res (2017) 11(3):OC57–9. doi:10.7860/JCDR/2017/20281.9626
95. Kreider ME, Hansen-Flaschen J, Ahmad NN, Rossman MD, Kaiser LR, Kucharczuk JC, et al. Complications of video-assisted thoracoscopic lung biopsy in patients with interstitial lung disease. Ann Thorac Surg (2007) 83(3):1140–4. doi:10.1016/j.athoracsur.2006.10.002
96. Yang W, He B. [Complications of lung biopsy in patients with idiopathic interstitial pneumonia and risk factors thereof]. Zhonghua Yi Xue Za Zhi (2009) 89(2):109–13. doi:10.3760/cma.j.issn.0376-2491.2009.02.010
97. Miller JD. Invited commentary. Ann Thorac Surg (2007) 83(3):1145. doi:10.1016/j.athoracsur.2006.11.080
98. Bush A, Cunningham S, de Blic J, Barbato A, Clement A, Epaud R, et al. European protocols for the diagnosis and initial treatment of interstitial lung disease in children. Thorax (2015) 70(11):1078–84. doi:10.1136/thoraxjnl-2015-207349
99. Clement A, Force ERST. Task force on chronic interstitial lung disease in immunocompetent children. Eur Respir J (2004) 24(4):686–97. doi:10.1183/09031936.04.00089803
100. Jolles S, Carne E, Brouns M, El-Shanawany T, Williams P, Marshall C, et al. FDG PET-CT imaging of therapeutic response in granulomatous lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID). Clin Exp Immunol (2017) 187(1):138–45. doi:10.1111/cei.12856
101. Nunes H, Uzunhan Y, Gille T, Lamberto C, Valeyre D, Brillet PY. Imaging of sarcoidosis of the airways and lung parenchyma and correlation with lung function. Eur Respir J (2012) 40(3):750–65. doi:10.1183/09031936.00025212
102. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science (2014) 345(6204):1623–7. doi:10.1126/science.1255904
103. Lopez-Herrera G, Tampella G, Pan-Hammarstrom Q, Herholz P, Trujillo-Vargas CM, Phadwal K, et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet (2012) 90(6):986–1001. doi:10.1016/j.ajhg.2012.04.015
104. Buchbinder D, Baker R, Lee YN, Ravell J, Zhang Y, McElwee J, et al. Identification of patients with RAG mutations previously diagnosed with common variable immunodeficiency disorders. J Clin Immunol (2015) 35(2):119–24. doi:10.1007/s10875-014-0121-5
105. Lawless D, Geier CB, Farmer JR, Lango Allen H, Thwaites D, Atschekzei F, et al. Prevalence and clinical challenges among adults with primary immunodeficiency and recombination-activating gene deficiency. J Allergy Clin Immunol (2018) 141(6):2303–6. doi:10.1016/j.jaci.2018.02.007
106. Lindsley AW, Saal HM, Burrow TA, Hopkin RJ, Shchelochkov O, Khandelwal P, et al. Defects of B-cell terminal differentiation in patients with type-1 Kabuki syndrome. J Allergy Clin Immunol (2016) 137(1):179–87.e10. doi:10.1016/j.jaci.2015.06.002
107. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med (2014) 20(12):1410–6. doi:10.1038/nm.3746
108. Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 CTLA-4-insufficient individuals. J Allergy Clin Immunol (2018). doi:10.1016/j.jaci.2018.02.055
109. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. Autoimmune disease. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science (2015) 349(6246):436–40. doi:10.1126/science.aaa1663
110. Bogaert DJ, Dullaers M, Lambrecht BN, Vermaelen KY, De Baere E, Haerynck F. Genes associated with common variable immunodeficiency: one diagnosis to rule them all? J Med Genet (2016) 53(9):575–90. doi:10.1136/jmedgenet-2015-103690
111. Kienzler AK, Hargreaves CE, Patel SY. The role of genomics in common variable immunodeficiency disorders. Clin Exp Immunol (2017) 188(3):326–32. doi:10.1111/cei.12947
112. Maffucci P, Filion CA, Boisson B, Itan Y, Shang L, Casanova JL, et al. Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front Immunol (2016) 7:220. doi:10.3389/fimmu.2016.00220
113. Chase NM, Verbsky JW, Hintermeyer MK, Waukau JK, Tomita-Mitchell A, Casper JT, et al. Use of combination chemotherapy for treatment of granulomatous and lymphocytic interstitial lung disease (GLILD) in patients with common variable immunodeficiency (CVID). J Clin Immunol (2013) 33(1):30–9. doi:10.1007/s10875-012-9755-3
114. Lee S, Moon JS, Lee CR, Kim HE, Baek SM, Hwang S, et al. Abatacept alleviates severe autoimmune symptoms in a patient carrying a de novo variant in CTLA-4. J Allergy Clin Immunol (2016) 137(1):327–30. doi:10.1016/j.jaci.2015.08.036
115. Franxman TJ, Howe LE, Baker JR Jr. Infliximab for treatment of granulomatous disease in patients with common variable immunodeficiency. J Clin Immunol (2014) 34(7):820–7. doi:10.1007/s10875-014-0079-3
116. Hurst JR, Verma N, Lowe D, Baxendale HE, Jolles S, Kelleher P, et al. British Lung Foundation/United Kingdom Primary Immunodeficiency Network consensus statement on the definition, diagnosis, and management of granulomatous-lymphocytic interstitial lung disease in common variable immunodeficiency disorders. J Allergy Clin Immunol Pract (2017) 5(4):938–45. doi:10.1016/j.jaip.2017.01.021
117. Rich AL, Le Jeune IR, McDermott L, Kinnear WJ. Serial lung function tests in primary immune deficiency. Clin Exp Immunol (2008) 151(1):110–3. doi:10.1111/j.1365-2249.2007.03550.x
118. Roifman CM, Levison H, Gelfand EW. High-dose versus low-dose intravenous immunoglobulin in hypogammaglobulinaemia and chronic lung disease. Lancet (1987) 1(8541):1075–7. doi:10.1016/S0140-6736(87)90494-6
119. Immune Deficiency Foundation Diagnostic & Clinical Care Guidelines for Primary Immunodeficiency Diseases. 3rd ed. (2016). Available from: https://primaryimmune.org/sites/default/files/publications/2015-Diagnostic-and-Clinical-Care-Guidelines-for-PI_1.pdf
120. Gustafsson PM, De Jong PA, Tiddens HA, Lindblad A. Multiple-breath inert gas washout and spirometry versus structural lung disease in cystic fibrosis. Thorax (2008) 63(2):129–34. doi:10.1136/thx.2007.077784
121. Boon M, Vermeulen FL, Gysemans W, Proesmans M, Jorissen M, De Boeck K. Lung structure-function correlation in patients with primary ciliary dyskinesia. Thorax (2015) 70(4):339–45. doi:10.1136/thoraxjnl-2014-206578
122. Gonem S, Scadding A, Soares M, Singapuri A, Gustafsson P, Ohri C, et al. Lung clearance index in adults with non-cystic fibrosis bronchiectasis. Respir Res (2014) 15:59. doi:10.1186/1465-9921-15-59
123. Amin R, Subbarao P, Jabar A, Balkovec S, Jensen R, Kerrigan S, et al. Hypertonic saline improves the LCI in paediatric patients with CF with normal lung function. Thorax (2010) 65(5):379–83. doi:10.1136/thx.2009.125831
124. Maarschalk-Ellerbroek LJ, de Jong PA, van Montfrans JM, Lammers JW, Bloem AC, Hoepelman AI, et al. CT screening for pulmonary pathology in common variable immunodeficiency disorders and the correlation with clinical and immunological parameters. J Clin Immunol (2014) 34(6):642–54. doi:10.1007/s10875-014-0068-6
125. Touw CM, van de Ven AA, de Jong PA, Terheggen-Lagro S, Beek E, Sanders EA, et al. Detection of pulmonary complications in common variable immunodeficiency. Pediatr Allergy Immunol (2010) 21(5):793–805. doi:10.1111/j.1399-3038.2009.00963.x
126. Aghamohammadi A, Moin M, Kouhi A, Mohagheghi MA, Shirazi A, Rezaei N, et al. Chromosomal radiosensitivity in patients with common variable immunodeficiency. Immunobiology (2008) 213(5):447–54. doi:10.1016/j.imbio.2007.10.018
127. Palanduz S, Palanduz A, Yalcin I, Somer A, Ones U, Ustek D, et al. In vitro chromosomal radiosensitivity in common variable immune deficiency. Clin Immunol Immunopathol (1998) 86(2):180–2. doi:10.1006/clin.1997.4478
128. Vorechovsky I, Scott D, Haeney MR, Webster DA. Chromosomal radiosensitivity in common variable immune deficiency. Mutat Res (1993) 290(2):255–64. doi:10.1016/0027-5107(93)90166-D
129. Gorycki T, Lasek I, Kaminski K, Studniarek M. Evaluation of radiation doses delivered in different chest CT protocols. Pol J Radiol (2014) 79:1–5. doi:10.12659/PJR.889952
130. Katsura M, Matsuda I, Akahane M, Yasaka K, Hanaoka S, Akai H, et al. Model-based iterative reconstruction technique for ultralow-dose chest CT: comparison of pulmonary nodule detectability with the adaptive statistical iterative reconstruction technique. Invest Radiol (2013) 48(4):206–12. doi:10.1097/RLI.0b013e31827efc3a
131. van de Ven AA, de Jong PA, Hoytema van Konijnenburg DP, Kessels OA, Boes M, Sanders EA, et al. Airway and interstitial lung disease are distinct entities in paediatric common variable immunodeficiency. Clin Exp Immunol (2011) 165(2):235–42. doi:10.1111/j.1365-2249.2011.04425.x
132. van de Ven AA, van Montfrans JM, Terheggen-Lagro SW, Beek FJ, Hoytema van Konijnenburg DP, Kessels OA, et al. A CT scan score for the assessment of lung disease in children with common variable immunodeficiency disorders. Chest (2010) 138(2):371–9. doi:10.1378/chest.09-2398
133. Chest CT in ADS Group [cited 2017 Nov 13]. Available from: https://esid.org/Working-Parties/Clinical-Working-Party/Resources/Chest-CT-in-antibody-deficiency-Updated-CRF
134. Milito C, Pulvirenti F, Serra G, Valente M, Pesce AM, Granata G, et al. Lung magnetic resonance imaging with diffusion weighted imaging provides regional structural as well as functional information without radiation exposure in primary antibody deficiencies. J Clin Immunol (2015) 35(5):491–500. doi:10.1007/s10875-015-0172-2
135. Sánchez-Ramón S, Radigan L, Yu JE, Bard S, Cunningham-Rundles C. Memory B cells in common variable immunodeficiency: clinical associations and sex differences. Clin Immunol (2008) 128(3):314–21. doi:10.1016/j.clim.2008.02.013
136. Ahn S, Cunningham-Rundles C. Role of B cells in common variable immune deficiency. Expert Rev Clin Immunol (2009) 5(5):557–64. doi:10.1586/eci.09.43
137. Gregersen S, Holm AM, Fevang B, Ueland T, Sikkeland LI, Aalokken TM, et al. Lung disease, T-cells and inflammation in common variable immunodeficiency disorders. Scand J Clin Lab Invest (2013) 73(6):514–22. doi:10.3109/00365513.2013.819523
138. Chalmers JD, Smith MP, McHugh BJ, Doherty C, Govan JR, Hill AT. Short- and long-term antibiotic treatment reduces airway and systemic inflammation in non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med (2012) 186(7):657–65. doi:10.1164/rccm.201203-0487OC
139. Chmiel JF, Konstan MW, Accurso FJ, Lymp J, Mayer-Hamblett N, VanDevanter DR, et al. Use of ibuprofen to assess inflammatory biomarkers in induced sputum: implications for clinical trials in cystic fibrosis. J Cyst Fibros (2015) 14(6):720–6. doi:10.1016/j.jcf.2015.03.007
Keywords: primary immunodeficiency, lung complications, immunoglobulin, comorbidity, bronchiectasis, granulomatous-lymphocytic interstitial lung disease, pulmonary functional tests, lung computed tomographic scan
Citation: Baumann U, Routes JM, Soler-Palacín P and Jolles S (2018) The Lung in Primary Immunodeficiencies: New Concepts in Infection and Inflammation. Front. Immunol. 9:1837. doi: 10.3389/fimmu.2018.01837
Received: 17 April 2018; Accepted: 25 July 2018;
Published: 08 August 2018
Edited by:
Waleed Al-Herz, Kuwait University, KuwaitReviewed by:
Bodo Grimbacher, Universitätsklinikum Freiburg, GermanyJolan Eszter Walter, University of South Florida, United States
Copyright: © 2018 Baumann, Routes, Soler-Palacín and Jolles. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ulrich Baumann, YmF1bWFubi51bHJpY2hAbWgtaGFubm92ZXIuZGU=;
John M. Routes, anJvdXRlc0BtY3cuZWR1
†These authors have contributed equally to this work.