 Anderson Dik Wai Luk1†Ke Ni1,2†
Anderson Dik Wai Luk1†Ke Ni1,2† Yuet Wu1Kwok-Tai Lam1Koon-Wing Chan1
Yuet Wu1Kwok-Tai Lam1Koon-Wing Chan1 Pamela P. Lee1,3Wenwei Tu1,3
Pamela P. Lee1,3Wenwei Tu1,3 Huawei Mao1,4*
Huawei Mao1,4* Yu Lung Lau1,3*
Yu Lung Lau1,3*
- 1Department of Paediatrics and Adolescent Medicine, LKS Faculty of Medicine, The University of Hong Kong, Hong Kong, Hong Kong
- 2Institute of Biology, Westlake Institute for Advanced Study, Hangzhou, Zhejiang, China
- 3Shenzhen Primary Immunodeficiency Diagnostic and Therapeutic Laboratory, Department of Paediatrics, The University of Hong Kong-Shenzhen Hospital, Shenzhen, China
- 4Department of Rheumatology and Immunology, Ministry of Education Key Laboratory of Child Development and Disorder, Children’s Hospital of Chongqing Medical University, Chongqing, China
Background: X-linked agammaglobulinemia (XLA) is a primary immunodeficiency caused by Bruton’s tyrosine kinase (BTK) mutation. Patients are susceptible to severe enterovirus infections. The underlying mechanism remains unknown. BTK is involved in toll-like receptors pathway, which initiates antiviral responses including interferon (IFN) productions.
Objective: To demonstrate type I and III IFN productions in dendritic cells of XLA patients is decreased in response to oral poliovirus vaccine (OPV) but not H1N1 virus.
Methods: Monocyte-derived dendritic cells (MoDCs) were derived from nine XLA patients aged 22–32 years old and 23 buffy coats from Hong Kong Red Cross blood donors. LFM-A13 was used to inhibit BTK. OPV Sabin type 1 and H1N1 influenza virus were used to stimulate MoDCs with RPMI as mock stimulation. The antiviral cytokine productions and phenotypic maturation of MoDCs were determined 24 h post-stimulation. OPV RNA was determined at 0, 6, 12, and 24 h post-stimulation.
Results: Upon OPV stimulation, IFN-α2, IFN-β, and IFN-λ1 productions in MoDCs from XLA patients and BTK-inhibited MoDCs of healthy controls were significantly lower than that from healthy controls. Whereas upon H1N1 stimulation, the IFN-α2, IFN-β, and IFN-λ1 productions were similar in MoDCs from XLA patients, BTK-inhibited MoDCs of healthy controls and healthy controls. The mean fluorescent intensities (MFI) of CD83, CD86, and MHC-II in MoDCs from XLA patients in response to OPV was similar to that in response to mock stimulation, while the MFI of CD83, CD86, and MHC-II were significantly higher in response to H1N1 stimulation than that in response to mock stimulation. Whereas, the MFI of CD83, CD86, and MHC-II in MoDCs of healthy controls were significantly higher in response to both OPV and H1N1 stimulation compared to that in response to mock stimulation.
Conclusion: Production of type I and III IFN in response to OPV was deficient in MoDCs from XLA patients, but was normal in response to H1N1 due to deficient BTK function. Moreover, phenotypic maturation of MoDCs from XLA patients was impaired in response to OPV but not to H1N1. These selective impairments may account for the unique susceptibility of XLA patients toward severe enterovirus infections.
Introduction
X-linked agammaglobulinemia (XLA) is a primary immunodeficiency caused by mutations of Bruton’s tyrosine kinase (BTK) (1, 2). BTK is expressed in all lineages of hematopoietic cells except T cells (3). As a result, XLA patients typically suffer from recurrent respiratory infections caused by encapsulated bacteria but are generally not susceptible to viral infections (1, 4). However, XLA patients are particularly vulnerable to severe enterovirus infections, notably chronic meningoencephalitis from Echo virus and vaccine-associated paralytic poliomyelitis (VAPP) from live-attenuated oral poliovirus vaccine (OPV) (5–9). Antibodies deficiency has been suggested to be responsible for the increase in susceptibility (10), but enterovirus infections in XLA patients with adequate immunoglobulin replacement have been reported (11, 12), indicating that antibody deficiency alone cannot fully explain the susceptibility toward severe enterovirus infections. To date the exact mechanism behind this unique susceptibility is unknown.
Enteroviruses are mainly transmitted through feco-oral route. The viruses then replicate in the upper respiratory tract, distal small bowel, and submucosal lymphoid tissues, leading to viremia and sometimes disseminated infections in the central nervous system (CNS), heart, and skin (7). Innate immunity, particularly interferons (IFN), as well as T cell-mediated immune responses are critical in protecting hosts from enterovirus infections (13).
Type I and III IFN play important roles in mucosal immunity and are often among the first cytokines produced in response to viral infections, restricting viral replications. Majority of the antiviral actions of type I IFN are exerted through the expression of interferon-stimulated genes (ISGs) (14, 15). ISGs are heterogeneous proteins with myriad antiviral mechanisms that target the different stages of viral life cycles (16), from blocking nuclear import of viral nucleocapsids by MX dynamin like GTPase 1 [MX dynamin like GTPase 1 (MX1)] to inhibiting translations by 2′-5′ oligoadenylate synthetase (OAS) and protein kinase R (PKR) (14, 17, 18). Production of ISGs such as interferon regulatory factor 3 (IRF3), IRF7, and toll-like receptor 3 (TLR3) sensitize pathogen detection and enhance IFN production to amplify antiviral responses (14). In addition, type I IFN has a broad range of effects on innate and adaptive immune cells that eventually result in clearance of viral infections (14). Type III IFN has similar effects as type I IFN but its actions are restricted to mucosal epithelium, liver, and a handful of immune cells including plasmacytoid dendritic cells (pDCs) and monocyte-derived macrophages (15).
Innate immune cells sense pathogens by recognizing the pathogen-associated molecular patterns (PAMPs) via the pattern recognition receptors (PRRs) (14, 15). Toll-like receptors (TLRs) and retinoic acid-inducible gene I (RIG)-I-like receptors (RLRs) are the two main PRRs to detect viral infections. TLR3, 7, 8, 9, and melanoma differentiation-associated protein 5 (MDA5) are utilized to detect enterovirus infections (2, 19) while TLR3, 7, 8, and RIG-I are utilized to detect influenza virus infections (20).
Activation of PRRs by viral PAMPs triggers downstream signaling cascades which lead to the production of type I and III IFN, along with other cytokines (14, 15). The influence of the TLRs and RLRs pathways on initiating IFNs productions varies from virus to virus (Figures 1 and 2). It has been shown that the TLR pathways, but not the MDA5 pathway, play the essential role in initiating IFN productions in response to enterovirus infections (Figure 1) (2, 21). On the other hand, it has been shown either TLRs or RLRs on its own is sufficient to initiate the production of type I IFN in response to influenza virus infection (Figure 2) (22).
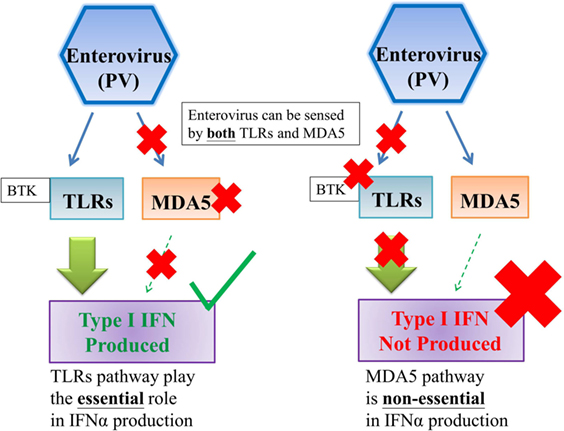
Figure 1. TLRs pathway, but not MDA5 pathway, is essential to the production of type I interferon against enterovirus infections. Enterovirus can be sensed by both TLRs and MDA5; however, TLRs pathway, but not MDA5 pathway, plays the essential role on type I interferon production against enterovirus infections (2). Abbreviations: PV, poliovirus; TLRs, toll-like receptors; MDA5, melanoma differentiation-associated protein 5; IFN, interferon.
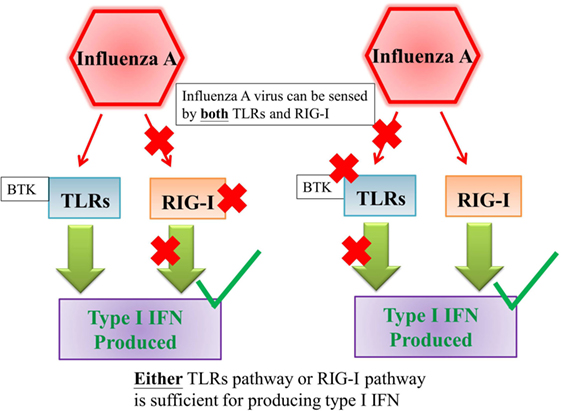
Figure 2. Either TLRs pathway or RIG-I pathway is sufficient for producing type I interferon against influenza A virus infection. Influenza A virus can be sensed by both TLRs and RIG-I and either TLRs pathway or RIG-I pathway is sufficient for producing type I interferon against influenza A virus infection (22). Abbreviations: TLRs, toll-like receptors; RIG-I, retinoic acid-inducible gene I; IFN, interferon.
Recent studies have revealed specific roles of BTK in TLR signaling pathways, from directly phosphorylating the TLR (23) to interacting with the adapters of TLRs (24–27). We, therefore, hypothesized that XLA patients have impaired type I and III IFN productions in response to enteroviruses but not to other viruses in a BTK-dependent manner. In this study, we sought to demonstrate type I and III IFN productions are decreased in response to OPV, but normal to H1N1 virus in monocyte-derived dendritic cells (MoDCs) of XLA patients.
Materials and Methods
Subjects
Nine XLA patients aged 22–32 years old were recruited for the study (Table 1). All of the nine patients have received OPV vaccination before and none had a history of acute flaccid paralysis before or excreting vaccine-derived poliovirus (VDPV). 40 mL of heparinized fresh blood was drawn for the study before the commencement of their regular intravenous immunoglobulin replacement therapy in Queen Mary Hospital. Twenty-three donor buffy coats from Hong Kong Red Cross were obtained as healthy control. This study was approved by the Institutional Review Board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster (UW 08-002). All subjects gave written informed consent in accordance with the Declaration of Helsinki.
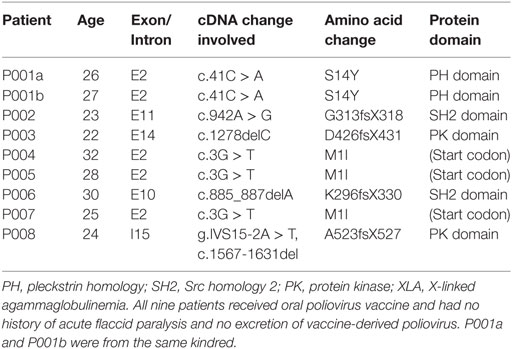
Table 1. Bruton’s tyrosine kinase mutations of the nine XLA patients.
Generation of Monocyte-Derived Dendritic Cells
Monocytes of patients and healthy controls were obtained from peripheral blood mononuclear cells (PBMC) using CD14 MicroBeads (Miltenyi Biotec, Germany), and were cultured in RPMI with 10% fetal bovine serum (FBS), 50 ng/mL GM-CSF, and 10 ng/mL IL-4 for 6 days to obtain monocyte-derived dendritic cells (MoDCs) as described previously (4).
Bruton’s tyrosine kinase-inhibited MoDCs were generated by pretreating MoDCs from healthy control with LFM-A13 (Calbiochem, USA) at 150 µM (Figure S1 in Supplementary Material) 2 h prior to viral stimulation (23, 28).
Viral Stimulation of MoDCs
In our previous study, we have optimized the conditions of stimulating MoDCs of XLA patients with H1N1 (4). In our current study, OPV Sabine type 1 and influenza A virus H1N1 were used to stimulate MoDCs on day 6. H1N1 virus was incubated at 56°C for 30 min before used to stimulate MoDCs as described previously (4). RPMI was used as mock stimulation. MoDCs and BTK-inhibited MoDCs from healthy controls, and MoDCs from XLA patients were incubated with OPV and H1N1 at multiplicity of infection (MOI) of 1 in the absence of FBS at 37°C for 2 h. 10% FBS was then supplemented to MoDCs.
All procedures involving viruses were conducted in biosafety level 2 (BSL 2) laboratory and in accordance with the World Health Organization polio laboratory manual (29).
Phenotypic and Cytokine Analysis
At 24 h post-stimulation, supernatant and MoDCs were collected for analysis. Antiviral cytokines levels in the supernatant were measured using cytometric bead array [LEGENDplex™ Human Anti-Virus Response Panel (13-plex), BioLegend, San Diego, CA, USA].
Phenotypic maturation of MoDCs was analyzed by flow cytometry using anti-CD14 PB, anti-CD86 PE, anti-MHC II FITC, anti-CD83 FITC, and anti-CD155 APC (Biolegend, San Diego, CA, USA).
RNA Analysis
Viral RNA of OPV was determined at 0, 6, 12, and 24 h post-stimulation in MoDCs from healthy controls and patients. IRF3, IRF7, TLR3, PKR, MX1, OAS1, and IFN-α2 RNA were determined at 0, 24, and 48 h post-stimulation in MoDCs from healthy controls and XLA patients by OPV.
Total RNA was extracted from MoDCs and supernatant using TaKaRa MiniBEST Universal RNA Extraction Kit (TaKaRa, Japan). cDNA conversion was performed using TaKaRa PrimeScript RT reagent Kit (TaKaRa, Japan). Quantitative PCR for OPV (Custom TaqMan® Gene Expression Assay PN4331348, Assay ID: AIY9Z0P, ThermoFisher, USA), IRF3 (TaqMan® Gene Expression Assay 4331182, Assay ID: Hs01547283_m1), IRF7 (TaqMan® Gene Expression Assay 4331182, Assay ID: Hs00185375_m1), TLR3 (TaqMan® Gene Expression Assay 4331182, Assay ID: Hs00152933_m1), PKR (TaqMan® Gene Expression Assay 4331182, Assay ID: Hs00169345_m1), MX1 (TaqMan® Gene Expression Assay 4331182, Assay ID: Hs00182073_m1), OAS1 (TaqMan® Gene Expression Assay 4331182, Assay ID: Hs00973635_m1), and IFN- α2 (TaqMan® Gene Expression Assay 4331182, Assay ID: Hs00265051_s1) was performed using ABI 7900 sequence detection system (Applied Biosystems). The amplification was performed with denaturation for 20 s at 95°C followed by 40 cycles of 95°C for 2 s and 60°C for 30 s. β-actin (Hs99999903_m1, TaqMan Gene Expression Assays, ThermoFisher, USA) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Hs02758991_g1, TaqMan Gene Expression Assays, ThermoFisher, USA) were used as internal control. Arbitrary threshold cycle of 40 was set for measurements under the detection limit of qPCR. Results were normalized to β-actin or GAPDH expression and presented as fold increase in RNA expression at 6, 12, 24, and 48 h post-stimulation compared to that at 0 h using the comparative threshold cycle method (ΔΔCt).
Statistics
All data were expressed in mean ± SEM. Statistics were analyzed by one-way ANOVA with Dunnett’s multiple comparison test, unpaired t-test, or Wilcoxon matched-pairs signed rank test using Prism 7 (GraphPad Software). Statistical significance was defined as p < 0.05.
Results
Production of IFN-α2, IFN-β, and IFN-λ1 Were Impaired in a BTK-Dependent Manner in MoDCs of XLA Patients Upon OPV Stimulation
Cytokines produced upon OPV stimulation from the MoDCs of XLA patients, BTK-inhibited MoDCs and MoDCs of healthy controls were measured at 24 h post-stimulation.
IFN-α2, IFN-β, IFN-λ1, and interferon gamma-induced protein 10 (IP-10) productions were lower in MoDCs from XLA patients compared to that from healthy controls upon OPV stimulation (Figure 3).
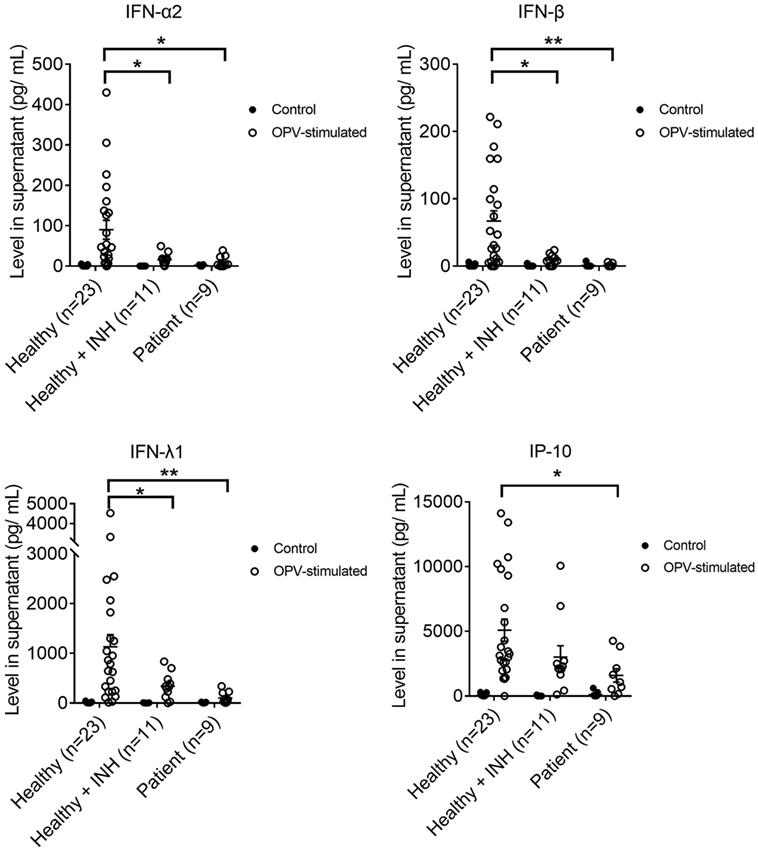
Figure 3. IFN-α2, IFN-β, IFN-λ1, and interferon gamma-induced protein 10 productions in MoDCs from healthy controls (n = 23), Bruton’s tyrosine kinase (BTK)-inhibited MoDCs from healthy controls (n = 11), and MoDCs from X-linked agammaglobulinemia patients (n = 9) upon OPV stimulation. MoDCs were stimulated with OPV at multiplicity of infection of 1 for 24 h. Open symbols represent MoDCs stimulated with OPV; filled symbols represent MoDCs that were mock-stimulated with RPMI. Healthy + INH, BTK-inhibited MoDCs from healthy controls. Data represented as mean ± SEM. *p < 0.05 and **p < 0.01.
IFN-α2, IFN-β, and IFN-λ1 productions were lower in BTK-inhibited MoDCs of healthy controls compared to that from MoDCs of healthy controls upon OPV stimulation (Figure 3).
There was no difference in the production of IFN-α2, IFN-β, IFN-λ1, and IP-10 between MoDCs from patients and healthy controls upon mock stimulation (Figure 3). There was no difference in the production of interleukin-1 beta (IL-1β), tumor necrosis factor alpha (TNF-α), and IL-8 between MoDCs from patients and healthy control upon OPV stimulation (Figure S2 in Supplementary Material).
Production of IFN-α2, IFN-β, and IFN-λ1 Were Normal in MoDCs of XLA Patients Upon H1N1 Stimulation
Cytokines produced upon H1N1 stimulation from the MoDCs of XLA patients, BTK-inhibited MoDCs and MoDCs of healthy controls were measured at 24 h post-stimulation.
There was no difference in IFN-α2, IFN-β, IFN-λ1, and IP-10 productions among the MoDCs of XLA patients, BTK-inhibited MoDCs, and MoDCs of healthy controls upon H1N1 stimulation (Figure 4). There was no difference in the production of IL-1β, TNF-α, and IL-8 among the three groups upon H1N1 stimulation (Figure S3 in Supplementary Material).
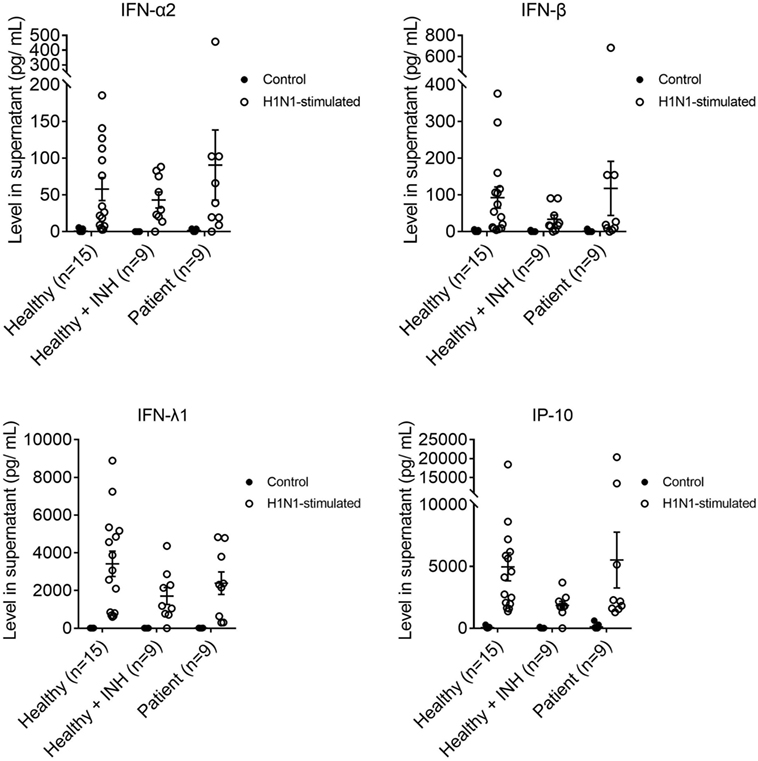
Figure 4. IFN-α2, IFN-β, IFN-λ1, and interferon gamma-induced protein 10 productions in MoDCs from healthy controls (n = 15), Bruton’s tyrosine kinase (BTK)-inhibited MoDCs from healthy controls (n = 9), and MoDCs from X-linked agammaglobulinemia (XLA) patients (n = 9) upon H1N1 stimulation. MoDCs were stimulated with H1N1 at multiplicity of infection of 1 for 24 h. Open symbols represent MoDCs stimulated with H1N1; filled symbols represent MoDCs that were mock-stimulated with RPMI. Controls of healthy controls, BTK-inhibited healthy controls, and XLA patients were subsets of controls in Figure 3. Healthy + INH, BTK-inhibited MoDCs from healthy controls. Data represented as mean ± SEM.
Expression of IRF7, TLR3, PKR, MX1, OAS1, and IFN-α2 Were Impaired in XLA Patients Following OPV Stimulation
RNA expressions of IRF3, IRF7, TLR3, PKR, MX1, OAS1, and IFN-α2 in MoDCs of XLA patients and healthy controls were measured at 24 and 48 h post-stimulation with OPV.
There was no increase in RNA expressions of IRF3, IRF7, TLR3, PKR, MX1, OAS1, and IFN-α2 in MoDCs of XLA patients upon OPV stimulation (Figure 5).
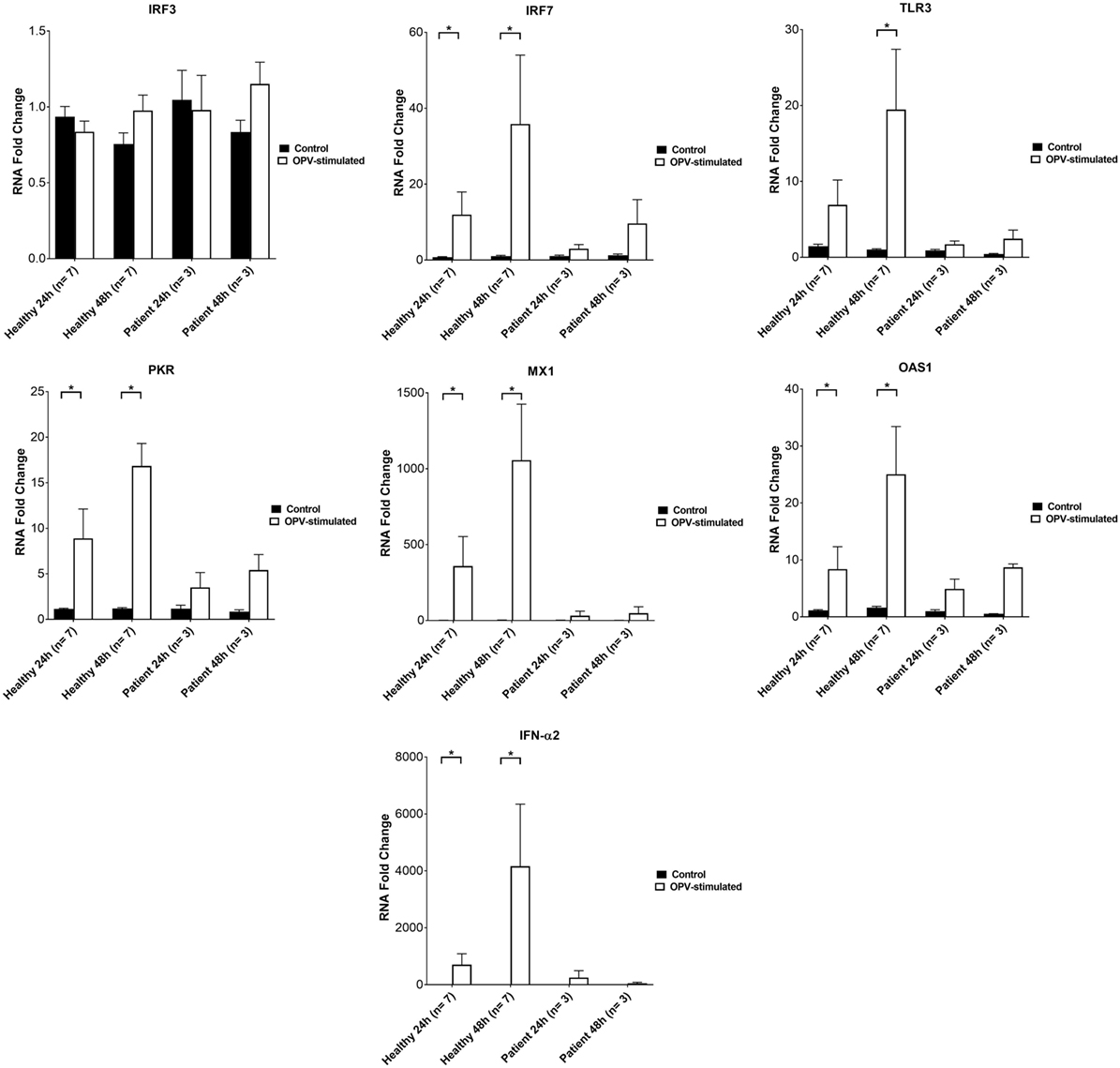
Figure 5. IRF3, IRF7, TLR3, PKR, MX1, OAS, and IFN-α2 RNA levels in MoDCs from healthy controls (n = 7) and X-linked agammaglobulinemia patients (n = 3) after 0 to 48 h post-infection. MoDCs were treated with OPV at multiplicity of infection of 1. Open bars represent RNA fold changes in MoDCs treated with OPV; filled bars represent RNA fold changes in MoDCs treated with RPMI. Fold change of RNA at 24 and 48 h compared to 0 h post-infection was calculated by the “ΔΔCt” method using GAPDH as endogenous control. Abbreviations: IRF, interferon regulatory factor; TLR, toll-like receptor; PKR, protein kinase R; MX1, MX dynamin-like GTPase 1; OAS1, 2′-5′ oligoadenylate synthetase 1; IFN, interferon; GAPDH, glyceraldehyde-3-phosphate dehydrogenase. Data represented as mean ± SEM. Statistics were analyzed by Wilcoxon matched-pairs signed rank test. *p < 0.05.
RNA expressions of IRF7, PKR, MX1, OAS1, and IFN-α2 were increased in MoDCs of healthy controls at 24 and 48 h post-stimulation with OPV (Figure 5). RNA expression of TLR3 was increased in MoDCs of healthy control at 48 h post-stimulation with OPV (Figure 5).
There was no increase in RNA expression of IRF3 in MoDCs of healthy control upon OPV stimulation (Figure 5).
Phenotypic Maturation of XLA Patients DC Was Impaired Upon OPV but Not H1N1 Stimulation
MoDCs from patients, BTK-inhibited MoDCs, and MoDCs of healthy controls were collected at 24 h post-stimulation with OPV and H1N1 for flow cytometry.
Mean fluorescent intensity (MFI) of CD83, MHC-II, and CD86 of MoDCs and BTK-inhibited MoDCs of healthy controls were higher upon both OPV and H1N1 stimulation when compared to mock stimulation (Figure 6).
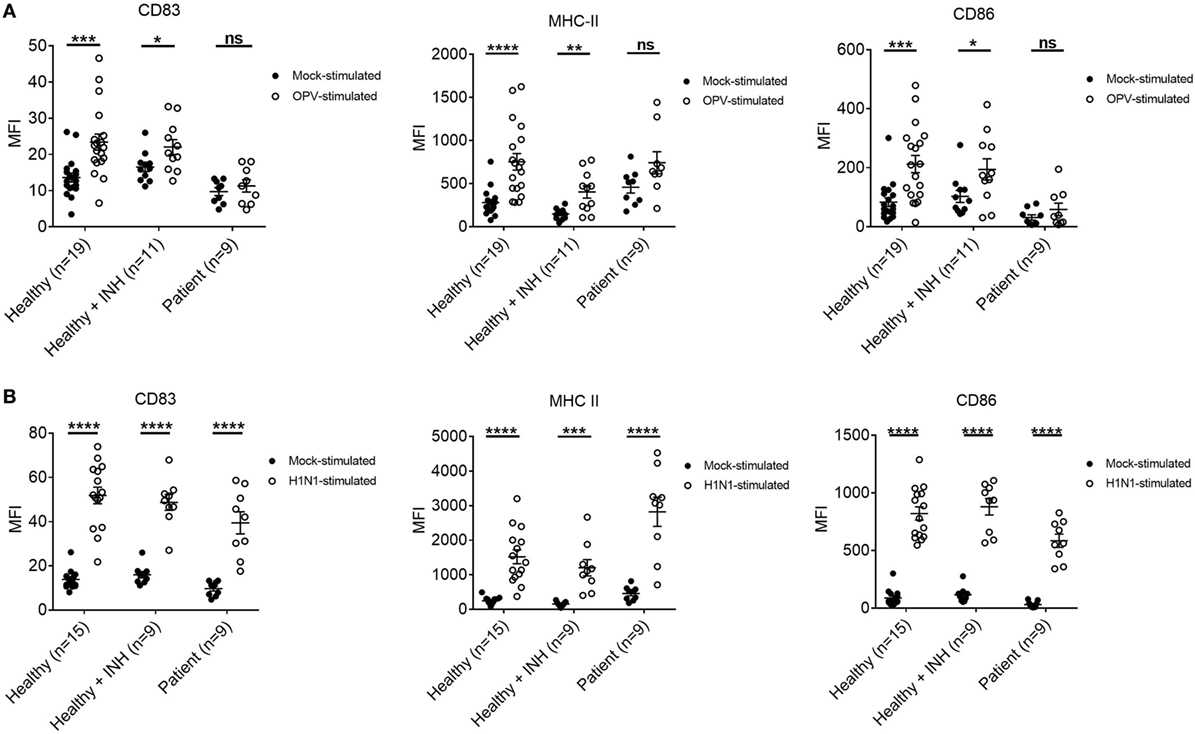
Figure 6. Surface marker changes in MoDCs from healthy controls, Bruton’s tyrosine kinase (BTK)-inhibited MoDCs from healthy controls, and MoDCs from X-linked agammaglobulinemia (XLA) patients upon OPV and H1N1 stimulation. MoDCs were stimulated with OPV and H1N1 at multiplicity of infection of 1 for 24 h. Open symbols represent MoDCs stimulated with OPV or H1N1; filled symbols represent MoDCs that were mock-stimulated with RPMI. Healthy + INH, BTK-inhibited MoDCs from healthy controls. Data represented as mean ± SEM. ns, p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001; and ****p < 0.0001. (A) MFI of CD83, MHC-II, and CD86 in MoDCs from healthy controls (n = 19), BTK-inhibited MoDCs from healthy controls (n = 11), and MoDCs from XLA patients (n = 9) upon OPV stimulation. (B) MFI of CD83, MHC-II, and CD86 in MoDCs from healthy controls (n = 15), BTK-inhibited MoDCs from healthy controls (n = 9), and MoDCs from XLA patients (n = 9) upon H1N1 stimulation. Controls of MoDCs from healthy controls, BTK-inhibited MoDCs from healthy controls, and MoDCs from XLA patients were subsets of controls in Figure 6A.
There was no difference in MFI of CD83, MHC-II, and CD86 of MoDCs from XLA patients upon OPV stimulation when compared to mock stimulation (Figure 6A).
However, MFI of CD83, MHC-II, and CD86 were higher in MoDCs from XLA patients upon H1N1 stimulation when compared to mock stimulation. Interestingly the MFI of MHC-II in MoDCs from patients was higher than that in MoDCs from healthy control (Figure 6B).
No Difference of Viral Replication in MoDCs of XLA Patients and Healthy Control
The fold increase of viral RNA at 6, 12, and 24 h were similar in MoDCs of XLA patients and healthy control (Figure 7).
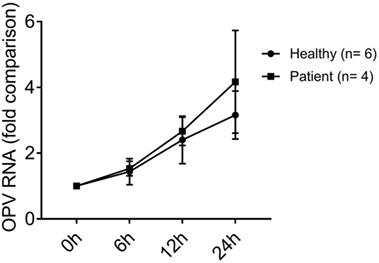
Figure 7. OPV RNA level in MoDCs from healthy controls and X-linked agammaglobulinemia (XLA) patients from 0 to 24 h post-infection. MoDCs from healthy controls (n = 6) and XLA patients (n = 4) were treated with OPV at multiplicity of infection of 1. Circles represent healthy control; squares represent patients. Fold change of viral RNA compared to 0 h post-infection was calculated by the “ΔΔCt” method using beta-actin as endogenous control. Data represented as mean ± SEM.
Discussion
Type I and III IFN productions were found to be near absent in MoDCs from XLA patients upon OPV stimulation. The deficits were replicated when BTK of healthy control MoDCs was inhibited, indicating the deficits were BTK-dependent. This is the first time such deficits were observed, which may be attributed to impairment of TLR signaling. Previous studies have shown that activation of TLR3, 4, 7, 8, and 9 can signal type I and III IFN productions (14, 15, 30). BTK has been shown to interact with the TLRs responsible for detecting OPV, phosphorylating the toll/interleukin-1 receptor (TIR) domain of TLR3 (23) as well as interacting with TIR domain of TLR 8 and 9 (24). In addition, BTK has been shown to interact with myeloid differentiation primary response gene 88, TIR-domain-containing adapter-inducing interferon-β, and TIR domain-containing adaptor protein (25, 26), which are the adapter molecules for TLRs. The impaired type I and III IFN responses in XLA patients could be due to interruption of TLR signaling by loss of BTK functions.
The near absence of type I and III IFN production in XLA patients impaired their antiviral responses toward OPV. MoDCs of XLA patients failed to express PKR, MX1, and OAS1 in response to OPV (Figure 5), which are well-known ISGs to directly restrict viral replication and translation (14, 17, 18).
In addition to the above ISGs, MoDCs of XLA patients failed to up-regulate the expression of IRF7 and TLR3 in response to OPV, while OPV failed to induce IRF3 expression in MoDCs of both XLA patients and healthy controls (Figure 5), possibly because IRF3 is constitutively expressed in all dendritic cell types and its expression cannot be further stimulated by IFN-α (31, 32). Apart from dendritic cells, previous study has also reported on the failure to induce IRF3 expression in neuronal cells by OPV (33). IRF7 is not constitutively expressed in majority of the dendritic cell types except pDCs, but its expression can be strongly induced by type I IFN in MoDCs (34). IRF3 and IRF7 are integral components of the TLR signaling pathway for type I and III IFN production (14, 15). IRF3 is more important than IRF7 in inducing IFN-β (35, 36). On the other hand, IRF7 can induce a wider range of type I and III IFN (34–36) and is critical in inducing IFN-α (36). Furthermore, IRF7 is required to amplify IFN production to mount an adequate antiviral response through the following mechanism (37). Upon recognition of PAMPs, the constitutively expressed IRF3 is activated, rapidly produces a small amount of IFN-β, which then stimulates the expression of IRF7 to amplify and diversify the IFN production in an autocrine and a paracrine fashion (34, 35, 37). This positive feedback loop was absent in MoDCs of XLA patients, resulting in prolonged impairment of IFN production (Figure 5). Such impairment may impede the clearance of viral infection and may further increase the susceptibility of XLA patients toward OPV infection.
Interferon gamma-induced protein 10 production was found to be impaired in MoDCs from XLA patients in response to OPV stimulation, but such impairment was not demonstrated by inhibiting BTK in healthy controls. This may be due to the IP-10 production is regulated by other cytokines apart from type I IFN (38, 39), including TNF-α and IL-1 (40, 41), which were shown to be normal in XLA patients (Figure S2 in Supplementary Material). IP-10 is a chemokine attracting activated T lymphocytes, macrophages and natural killer (NK) cells (42). Impaired IP-10 production in XLA patients may result in impaired recruitment of T cells to attack enterocytes infected with OPV during vaccination, increasing the risk of developing complications (43).
Type I and III IFN productions were normal in MoDCs from XLA patients upon H1N1 stimulation, which was similar as reported previously (4). The IFN productions were normal even with the inhibition of BTK in healthy controls. This indicates XLA patients have a normal capacity to produce type I and III IFN, supporting the deficit of producing type I and III IFN in response to OPV resides in the difference in initiating IFN productions between enterovirus and influenza virus. OPV is sensed by TLRs and MDA5, which is a type of RLR (2), while H1N1 is sensed by TLRs and RIG-I (22). It has been shown that TLRs, but not MDA5, play the essential role in initiating IFN productions in response to poliovirus infection (Figure 1) (2, 21). On the other hand, it has been shown either TLRs or RLRs on its own is sufficient to initiate the production of type I IFN in response to influenza infection (Figure 2) (22). This difference in utilizing PRRs to initiate IFN productions between OPV and H1N1 maybe behind the selective type I and III IFN production impairments upon OPV but not H1N1 stimulation in XLA patients.
Production of IL-1β, IL-8, and TNF-α were not impaired in MoDCs of XLA patients upon OPV stimulation. Instead of using pathogens, many previous studies used TLR agonists as stimulants. To date, at least 19 such studies using TLR agonists have been performed to investigate the effect of BTK deficiency on cytokine productions (Table 2) (23, 24, 44–60). They reported conflicting results on the effect of BTK deficiency on these cytokine productions upon TLR agonist stimulations (61), which may be due to the differences in experimental designs, including TLR agonists and BTK-deficient models used in these studies.
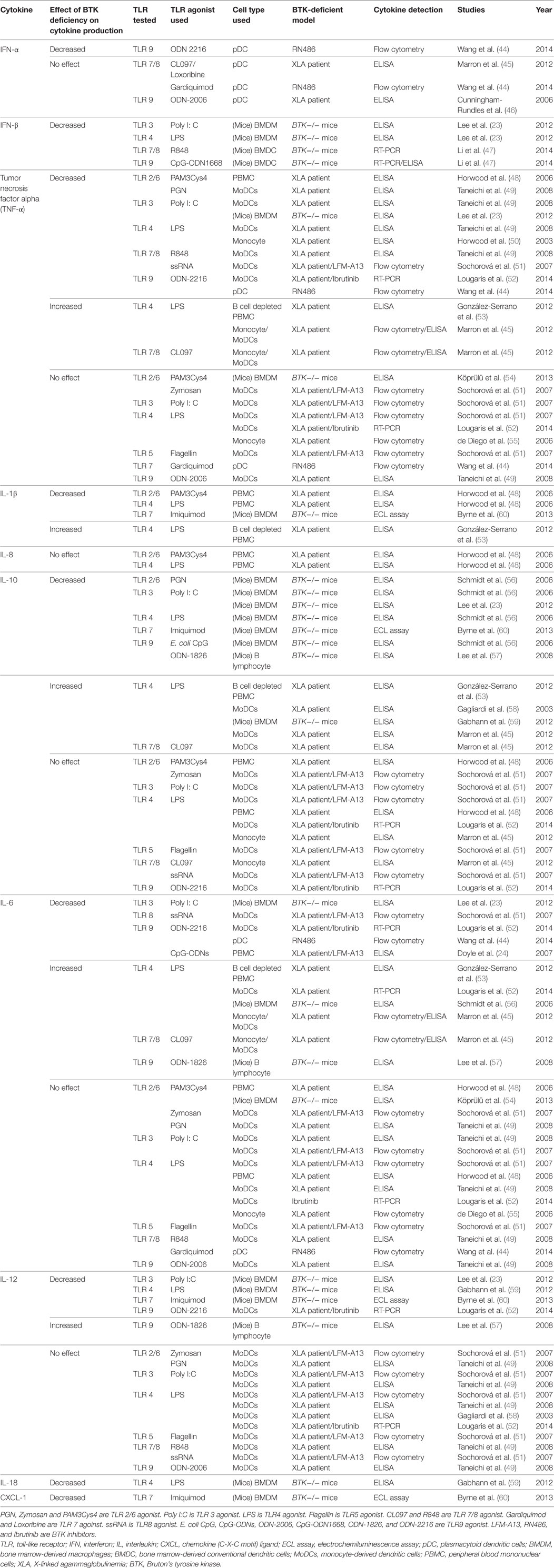
Table 2. Previous studies on the effect of BTK deficiency on cytokine productions in response to TLR agonists.
In our current study, using OPV instead of TLR agonists as stimulation resulted in more complex interactions between virus and host, as OPV can be recognized by various PRRs. To date there are at least 3 more studies in addition to our current study that used pathogen instead of TLR agonists to stimulate cytokine productions (Table 3) (4, 23, 54). Cytokine productions in response to live or dead pathogen stimulation are different (54) as live pathogen may have mechanisms to evade immunity (62, 63), which further complicate the virus–host interactions.
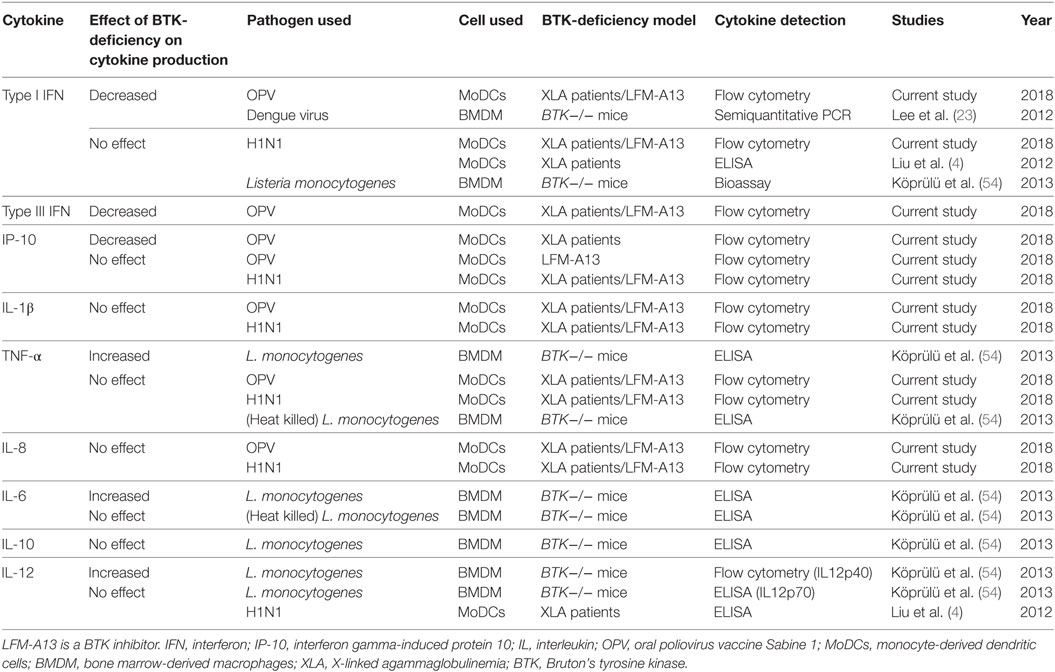
Table 3. Studies of the effect of BTK deficiency on cytokine productions in response to pathogen stimulation.
In our current study, we used XLA patients and LFM-A13 inhibited healthy control as BTK-deficiency groups. LFM-A13 has been used to generate BTK-deficiency models in previous studies (24, 51, 60), along with Ibrutinib and RN486 (Table 2) (44, 52). LFM-A13 is a dual kinase inhibitor for BTK and Polo-like kinase 3 (PLK3) (64, 65). PLK3 is a serine/threonine protein kinase that regulates cell cycle and apoptosis (66). Since PLK3 is not involved in TLRs signaling or IFN productions (67), therefore using LFM-A13 in our current study would demonstrate the impairment of type I and III IFN productions were through BTK inhibition.
Phenotypic maturation of MoDCs, characterized by increased expressions of CD83, CD86, and MHC-II, was impaired in MoDCs of XLA patients upon OPV but not H1N1 stimulation. However, such pattern was not demonstrated by inhibiting BTK in MoDCs of healthy controls. It has been shown that MoDCs of XLA patients have partial impairment of phenotypic maturation when stimulated with TLRs agonists (49). Such partial impairment could be explained by the maturation of dendritic cells not only depends on TLR engagement but also on RLRs, type I IFN, and TNF-α (68–70).
There was no difference in the rise of OPV RNA between MoDCs of XLA patients and healthy controls in spite of the near absence of type I and III IFN productions in MoDCs of XLA patients. With the use of MOI of 1 during viral stimulation, all MoDCs were simultaneously exposed to OPV before type I and III IFN were produced to limit OPV replication. In addition, with the replication capacity of OPV attenuated (71), the protection effect of IFN may not be critical, resulting in no difference in replication between the two groups. More importantly, the lack of difference in OPV RNA level indicated the difference in type I and III IFN productions between MoDCs of XLA patients and healthy controls were independent of the titers of OPV.
All patients in our current study received OPV and none developed complications, namely VAPP and VDPV excretion (Table 1) (5, 6, 72). The risk of VAPP in the world is approximately four cases per million births (73). Patients with antibody deficiencies, including XLA patients, are 3,000 to 10,000 times more susceptible to have VAPP compared to general population (8, 9) (i.e., with a risk of 1.2–4%). In addition, XLA patients can have prolonged VDPV excretion for as long as 5 years (5), thus posing a significant obstacle for global eradication of poliovirus.
Apart from antibody deficiency, impaired type I IFN response has been hypothesized before to be responsible for developing VAPP in general population by allowing unchecked multiplication of OPV in extraneural tissues (74). However, IFN or antibody deficiency alone cannot explain the susceptibility of having VAPP and VDPV excretion in XLA patients. Hyper IgM syndrome patients, who have a deficiency of IgG, IgA, and IgE, are not susceptible to VAPP or VDPV excretion. In addition, they are less susceptible to severe enterovirus infections compared to XLA patients (75). Similarly, IFN deficiency patients, including those with TLR3 and UNC-93B deficiency, are not susceptible to OPV vaccination complications or severe enterovirus infections (76, 77). Therefore, it may require a combined impairment of both IFN and antibody to render XLA patients susceptible toward severe enterovirus infections.
Therefore, based on our findings and previous reports, we postulated the increased risk of VAPP in XLA patients is due to combined deficiency of IFN and antibody which impairs defenses at various checkpoints against the development of VAPP after OPV vaccination (Figure 8). The lack of secretory Ig A on mucosa promotes the adherence and invasion of OPV (78). Decreased type I and III IFN and impaired maturation of antigen presentation cells may permit unrestricted viral replication and impair T cell recruitment as well as activation (14, 15). In addition, our previous study has shown that BTK-deficient NK cells have impaired IFN-γ production and reduced cytotoxicity in response to TLR3 stimulation (79). Together these defects result in prolonged replication and shedding of OPV. Moreover, decreased type I IFN and antibody deficiency result in enhanced tissue tropism of OPV which may lead to more effective infection of extraneural tissue and OPV replication (74, 80). Defects in these checkpoints allow OPV replication which may result in reversion of neurovirulence through mutations, thus increasing risk of having VAPP and excreting VDPV (5, 6, 73, 81). Since type I and III IFN offers some protection against CNS invasion by maintaining the integrity of blood–brain barrier (82), the integrity of blood–brain barrier in XLA patients may be compromised, becoming more vulnerable to neuroinvasion.
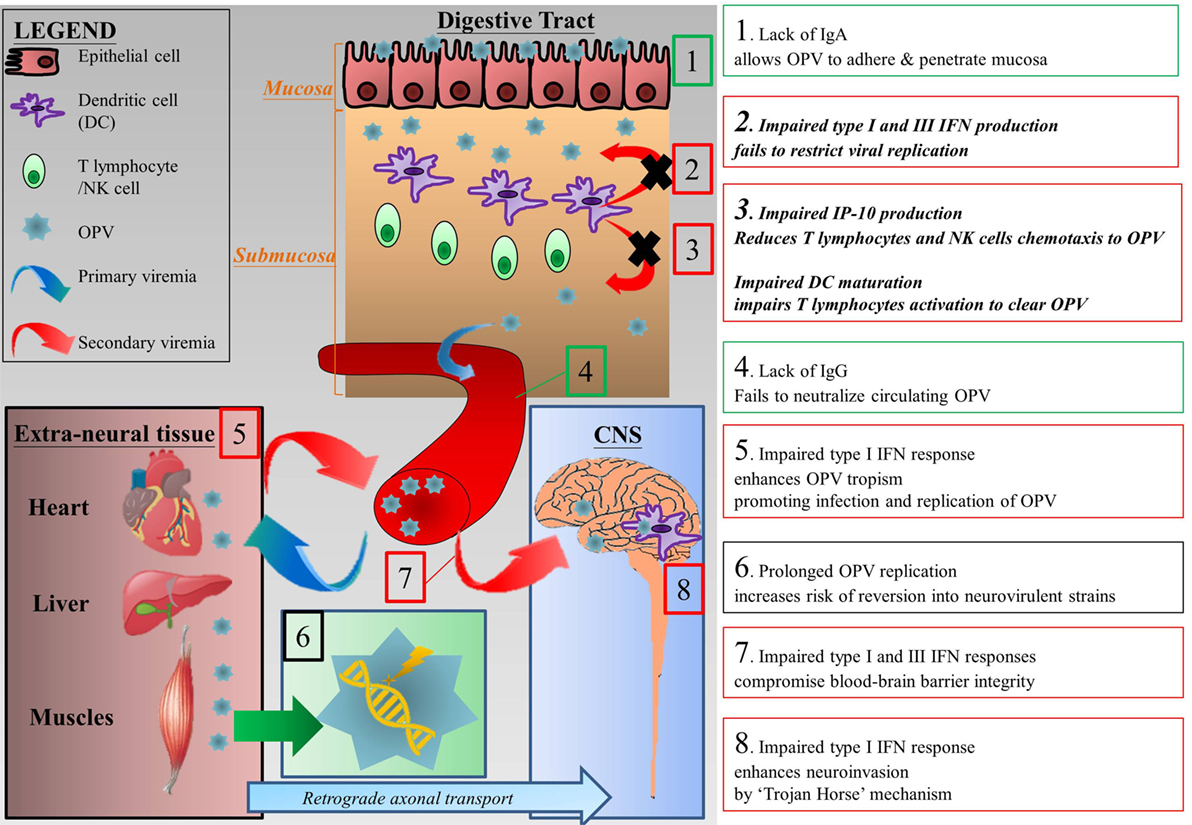
Figure 8. Postulated mechanisms that increase the risk of vaccine-associated paralytic poliomyelitis in XLA patients. (1) Lack of secretory Ig A allows OPV to adhere and penetrate digestive tract mucosa (78). (2) At the mucosa, restriction of viral replication is impaired due to impaired type I and III IFN productions (14, 15). (3) In addition, impaired IP-10 production and phenotypic maturation of dendritic cells reduce T lymphocytes migration and activation which in turns impair clearance of OPV infection (42, 43). (4) The lack of neutralizing antibodies in XLA patients results in prolonged viremia. (5) The lack of type I IFN enhances tropism of OPV, allowing more extensive infection of extraneural tissues by OPV and thus enhancing replication of OPV, which leads to sustained secondary viremia (74, 80). (6) Prolonged replication of OPV increases the chance of mutation which may revert the vaccine strain to a neurovirulent one (5, 6, 73, 81). (7) Type III IFN protects the epithelium component of blood–brain barrier, while type I IFN protects the endothelium component. Combined impairment of both type I and III IFN compromises blood–brain barrier integrity (82). (8) Finally, OPV can enter central nervous system (CNS) via “Trojan horse” mechanism in which they cross the blood–brain barrier by hiding in immune cells such as macrophages. Type I IFN response may limit CNS invasion by “Trojan horse” mechanism (82). Green textboxes represent humoral immunodeficiency and its consequences in XLA patients and red textboxes represent innate immunodeficiencies and their consequences in XLA patients. Textboxes with italic and bolded text represent our findings and their consequences. Abbreviations: CNS, central nervous system; IFN, interferon; IP-10, interferon gamma-induced protein 10; OPV, oral polio vaccine.
In conclusion, selective impairment of type I and III IFN productions in response to OPV but not to H1N1 was demonstrated in XLA patients, implicating BTK-dependent impairments are responsible for the unique susceptibility of these patients to severe enterovirus infections.
Ethics Statement
This study was carried out in accordance with the recommendations of the Institutional Review Board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster (UW 08-002) with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the Institutional Review Board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster (UW 08-002).
Author Contributions
WT and YL designed the study and discussed with HM who then wrote the research proposal. PL and YL recruited the patients for this study. AL, KN, YW, KL, and KC performed the experiments. WT and YL supervised the experiments and interpreted the data. AL and YL wrote the manuscript with extensive appraisal from PL, WT, and HM.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was mainly supported by the General Research Fund from the Research Grants Council of Hong Kong (HKU 770313M), and in part by the Society for the Relief of Disabled Children (YL), the Shenzhen Development and Reform Commission ([2015]164) (YL, HM), Shenzhen Science, Technology and Innovation Commission (JCYJ20140411175241066) (HM), and the National Natural Science Foundation of China (81671626) (HM).
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.01826/full#supplementary-material.
Figure S1. IFN-β production in MoDCs from healthy controls (n = 4) and Bruton’s tyrosine kinase-inhibited MoDCs from healthy controls (n = 4) upon OPV and H1N1 stimulation. LFM-A13 at 0, 100, and 150 µM were added to MoDCs from healthy controls 2 h prior to viral stimulation. Data represented as mean + SEM. *p < 0.05; ns, p > 0.05.
Figure S2. IL-1β, tumor necrosis factor alpha, and IL-8 productions in MoDCs from healthy controls (n = 23), Bruton’s tyrosine kinase (BTK)-inhibited MoDCs from healthy controls (n = 11), and MoDCs from XLA patients (n = 9) upon OPV stimulation. MoDCs were stimulated with OPV at multiplicity of infection of 1 for 24 h. Open symbols represent MoDCs stimulated with OPV; filled symbols represent MoDCs that were mock-stimulated with RPMI. Healthy + INH, BTK-inhibited MoDCs from healthy controls. Data represented as mean ± SEM.
Figure S3. IL-1β, tumor necrosis factor alpha, and IL-8 productions in MoDCs from healthy controls (n = 15), Bruton’s tyrosine kinase (BTK)-inhibited MoDCs from healthy controls (n = 9), and MoDCs from XLA patients (n = 9) upon H1N1 stimulation. MoDCs were stimulated with H1N1 at multiplicity of infection of 1 for 24 h. Open symbols represent MoDCs stimulated with H1N1; filled symbols represent MoDCs that were mock-stimulated with RPMI. Controls of healthy controls, BTK-inhibited healthy controls, and XLA patients were subsets of controls in Figure S2 in Supplementary Material. Healthy + INH, BTK-inhibited MoDCs from healthy controls. Data represented as mean ± SEM.
Abbreviations
BMDC, bone marrow-derived conventional dendritic cells; BMDM, bone marrow-derived macrophages; BTK, Bruton’s tyrosine kinase; CNS, central nervous system; CXCL, chemokine (C-X-C motif) ligand; MoDCs, monocyte-derived dendritic cells; ECL, electrochemiluminescence; FBS, fetal bovine serum; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IFN, interferon; IL, interleukin; IP-10, interferon gamma-induced protein 10; IRF, interferon regulatory factor; IVIG, intravenous immunoglobulin; MDA5, melanoma differentiation-associated protein 5; MFI, mean fluorescent intensity; MOI, multiplicity of infection; MX1, MX dynamin like GTPase 1; MyD88, myeloid differentiation primary response gene 88; NK, natural killer; OAS1, 2′-5′ oligoadenylate synthetase 1; OPV, oral poliovirus vaccine; PAMPs, pathogen-associated molecular patterns; PBMC, peripheral blood mononuclear cells; pDCs, plasmacytoid dendritic cells; PH, pleckstrin homology; PID, primary immunodeficiency; PK, protein kinase; PKR, protein kinase R; PLK3, Polo-like kinase 3; PRRs, pattern recognition receptors; RIG-I, retinoic acid-inducible gene I; RLRs, RIG-I-like receptors; SEM, standard error of mean; SH2, Src homology 2; TIR, toll/interleukin-1 receptor; TIRAP, TIR domain-containing adaptor protein; TLRs, toll-like receptors; TNF-α, tumor necrosis factor alpha; TRIF, TIR-domain-containing adapter-inducing interferon-β; VAPP, vaccine-associated paralytic poliomyelitis; VDPV, vaccine-derived poliovirus; XLA, X-linked agammaglobulinemia.
References
1. Suri D, Rawat A, Singh S. X-linked agammaglobulinemia. Indian J Pediatr (2016) 83(4):331–7. doi:10.1007/s12098-015-2024-8
2. Abe Y, Fujii K, Nagata N, Takeuchi O, Akira S, Oshiumi H, et al. The toll-like receptor 3-mediated antiviral response is important for protection against poliovirus infection in poliovirus receptor transgenic mice. J Virol (2012) 86(1):185–94. doi:10.1128/JVI.05245-11
3. de Weers M, Verschuren M, Kraakman M, Mensink R, Schuurman R, van Dongen J, et al. The Bruton’s tyrosine kinase gene is expressed throughout B cell differentiation, from early precursor B cell stages preceding immunoglobulin gene rearrangement up to mature B cell stages. Eur J Immunol (1993) 23(12):3109–14. doi:10.1002/eji.1830231210
4. Liu Y, Wu Y, Lam KT, Lee PP, Tu W, Lau YL. Dendritic and T cell response to influenza is normal in the patients with X-linked agammaglobulinemia. J Clin Immunol (2012) 32(3):421–9. doi:10.1007/s10875-011-9639-y
5. Macklin G, Liao Y, Takane M, Dooling K, Gilmour S, Mach O, et al. Prolonged excretion of poliovirus among individuals with primary immunodeficiency disorder: an analysis of the World Health Organization Registry. Front Immunol (2017) 8(1103):1103. doi:10.3389/fimmu.2017.01103
6. Aghamohammadi A, Abolhassani H, Kutukculer N, Wassilak SG, Pallansch MA, Kluglein S, et al. Patients with primary immunodeficiencies are a reservoir of poliovirus and a risk to polio eradication. Front Immunol (2017) 8:685. doi:10.3389/fimmu.2017.00685
7. Noor A, Krilov LR. Enterovirus infections. Pediatr Rev (2016) 37(12):505–15. doi:10.1542/pir.2016-0103
8. Sutter R, Prevots D. Vaccine-associated paralytic poliomyelitis among immunodeficient persons. Infect Med (1994) 11:426,9–30,35–8.
9. Wyatt H. Poliomyelitis in hypogammaglobulinemics. J Infect Dis (1973) 128(6):802–6. doi:10.1093/infdis/128.6.802
10. Alexander JJ, Gary HJ, Pallansch M. Duration of poliovirus excretion and its implications for acute flaccid paralysis surveillance: a review of the literature. J Infect Dis (1997) 175(Suppl 1):S176–82. doi:10.1093/infdis/175.Supplement_1.S176
11. Misbah S, Spickett G, Ryba P, Hockaday J, Kroll J, Sherwood C, et al. Chronic enteroviral meningoencephalitis in agammaglobulinemia: case report and literature review. J Clin Immunol (1992) 12(4):266–70. doi:10.1007/BF00918150
12. Quartier P, Foray S, Casanova J, Hau-Rainsard I, Blanche S, Fischer A. Enteroviral meningoencephalitis in X-linked agammaglobulinemia: intensive immunoglobulin therapy and sequential viral detection in cerebrospinal fluid by polymerase chain reaction. Pediatr Infect Dis J (2000) 19(11):1106–8. doi:10.1097/00006454-200011000-00020
13. Rhoades RE, Tabor-Godwin JM, Tsueng G, Feuer R. Enterovirus infections of the central nervous system. Virology (2011) 411(2):288–305. doi:10.1016/j.virol.2010.12.014
14. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15(2):87–103. doi:10.1038/nri3787
15. Wack A, Terczynska-Dyla E, Hartmann R. Guarding the frontiers: the biology of type III interferons. Nat Immunol (2015) 16(8):802–9. doi:10.1038/ni.3212
16. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol (2014) 32:513–45. doi:10.1146/annurev-immunol-032713-120231
17. Villalon-Letelier F, Brooks AG, Saunders PM, Londrigan SL, Reading PC. Host cell restriction factors that limit influenza A infection. Viruses (2017) 9(12):376. doi:10.3390/v9120376
18. Raftery N, Stevenson NJ. Advances in anti-viral immune defence: revealing the importance of the IFN JAK/STAT pathway. Cell Mol Life Sci (2017) 74(14):2525–35. doi:10.1007/s00018-017-2520-2
19. Harris KG, Coyne CB. Enter at your own risk: how enteroviruses navigate the dangerous world of pattern recognition receptor signaling. Cytokine (2013) 63(3):230–6. doi:10.1016/j.cyto.2013.05.007
20. Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol (2014) 14(5):315–28. doi:10.1038/nri3665
21. Wang C, Ji L, Yuan X, Jin Y, Cardona CJ, Xing Z. Differential regulation of TLR signaling on the induction of antiviral interferons in human intestinal epithelial cells infected with enterovirus 71. PLoS One (2016) 11(3):e0152177. doi:10.1371/journal.pone.0152177
22. Koyama S, Ishii KJ, Kumar H, Tanimoto T, Coban C, Uematsu S, et al. Differential role of TLR- and RLR-signaling in the immune responses to influenza A virus infection and vaccination. J Immunol (2007) 179(7):4711–20. doi:10.4049/jimmunol.179.7.4711
23. Lee KG, Xu S, Kang ZH, Huo J, Huang M, Liu D, et al. Bruton’s tyrosine kinase phosphorylates toll-like receptor 3 to initiate antiviral response. Proc Natl Acad Sci U S A (2012) 109(15):5791–6. doi:10.1073/pnas.1119238109
24. Doyle SL, Jefferies CA, Feighery C, O’Neill LA. Signaling by toll-like receptors 8 and 9 requires Bruton’s tyrosine kinase. J Biol Chem (2007) 282(51):36953–60. doi:10.1074/jbc.M707682200
25. Jefferies C, Doyle S, Brunner C, Dunne A, Brint E, Wietek C, et al. Bruton’s tyrosine kinase is a toll/interleukin-1 receptor domain-binding protein that participates in nuclear factor kappaB activation by toll-like receptor 4. J Biol Chem (2003) 278(28):26258–64. doi:10.1074/jbc.M301484200
26. Liu X, Zhan Z, Li D, Xu L, Ma F, Zhang P, et al. Intracellular MHC class II molecules promote TLR-triggered innate immune responses by maintaining activation of the kinase Btk. Nat Immunol (2011) 12(5):416–24. doi:10.1038/ni.2015
27. Weber ANR, Bittner Z, Liu X, Dang TM, Radsak MP, Brunner C. Bruton’s tyrosine kinase: an emerging key player in innate immunity. Front Immunol (2017) 8:1454. doi:10.3389/fimmu.2017.01454
28. Lee KG, Kim SS, Kui L, Voon DC, Mauduit M, Bist P, et al. Bruton’s tyrosine kinase phosphorylates DDX41 and activates its binding of dsDNA and STING to initiate type 1 interferon response. Cell Rep (2015) 10(7):1055–65. doi:10.1016/j.celrep.2015.01.039
29. World Health Organization. Polio Laboratory Manual. 4th ed. Geneva: World Health Organization, Department of Immunization, Vaccines and Biologicals (2004).
30. Zhang SY, Jouanguy E, Sancho-Shimizu V, von Bernuth H, Yang K, Abel L, et al. Human toll-like receptor-dependent induction of interferons in protective immunity to viruses. Immunol Rev (2007) 220:225–36. doi:10.1111/j.1600-065X.2007.00564.x
31. DeFilippis VR, Robinson B, Keck TM, Hansen SG, Nelson JA, Fruh KJ. Interferon regulatory factor 3 is necessary for induction of antiviral genes during human cytomegalovirus infection. J Virol (2006) 80(2):1032–7. doi:10.1128/JVI.80.2.1032-1037.2006
32. Izaguirre A, Barnes BJ, Amrute S, Yeow WS, Megjugorac N, Dai J, et al. Comparative analysis of IRF and IFN-alpha expression in human plasmacytoid and monocyte-derived dendritic cells. J Leukoc Biol (2003) 74(6):1125–38. doi:10.1189/jlb.0603255
33. Mohanty MC, Deshpande JM. Differential induction of toll-like receptors & type 1 interferons by Sabin attenuated & wild type 1 polioviruses in human neuronal cells. Indian J Med Res (2013) 138:209–18.
34. Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity (2006) 25(3):349–60. doi:10.1016/j.immuni.2006.08.009
35. Tailor P, Tamura T, Ozato K. IRF family proteins and type I interferon induction in dendritic cells. Cell Res (2006) 16(2):134–40. doi:10.1038/sj.cr.7310018
36. Hatesuer B, Hoang HT, Riese P, Trittel S, Gerhauser I, Elbahesh H, et al. Deletion of Irf3 and Irf7 genes in mice results in altered interferon pathway activation and granulocyte-dominated inflammatory responses to influenza A infection. J Innate Immun (2017) 9(2):145–61. doi:10.1159/000450705
37. Levy DE, Marie I, Smith E, Prakash A. Enhancement and diversification of IFN induction by IRF-7-mediated positive feedback. J Interferon Cytokine Res (2002) 22(1):87–93. doi:10.1089/107999002753452692
38. Padovan E, Spagnoli G, Ferrantini M, Heberer M. IFN-α2a induces IP-10/CXCL10 and MIG/CXCL9 production in monocyte-derived dendritic cells and enhances their capacity to attract and stimulate CD8+ effector T cells. J Leukoc Biol (2002) 71(4):669–76.
39. Bolen CR, Ding S, Robek MD, Kleinstein SH. Dynamic expression profiling of type I and type III interferon-stimulated hepatocytes reveals a stable hierarchy of gene expression. Hepatology (2014) 59(4):1262–72. doi:10.1002/hep.26657
40. Narumi S, Yoneyama H, Inadera H, Nishioji K, Itoh Y, Okanoue T, et al. TNF-alpha is a potent inducer for IFN-inducible protein-10 in hepatocytes and unaffected by GM-CSF in vivo, in contrast to IL-1beta and IFN-gamma. Cytokine (2000) 12(7):1007–16. doi:10.1006/cyto.1999.0672
41. Harikumar KB, Yester JW, Surace MJ, Oyeniran C, Price MM, Huang WC, et al. K63-linked polyubiquitination of transcription factor IRF1 is essential for IL-1-induced production of chemokines CXCL10 and CCL5. Nat Immunol (2014) 15(3):231–8. doi:10.1038/ni.2810
42. Liu M, Guo S, Stiles JK. The emerging role of CXCL10 in cancer (review). Oncol Lett (2011) 2(4):583–9. doi:10.3892/ol.2011.300
43. Wahid R, Cannon MJ, Chow M. Virus-specific CD4+ and CD8+ cytotoxic T-cell responses and long-term T-cell memory in individuals vaccinated against polio. J Virol (2005) 79(10):5988–95. doi:10.1128/JVI.79.10.5988-5995.2005
44. Wang J, Lau KY, Jung J, Ravindran P, Barrat FJ. Bruton’s tyrosine kinase regulates TLR9 but not TLR7 signaling in human plasmacytoid dendritic cells. Eur J Immunol (2014) 44(4):1130–6. doi:10.1002/eji.201344030
45. Marron TU, Martinez-Gallo M, Yu JE, Cunningham-Rundles C. Toll-like receptor 4-, 7-, and 8-activated myeloid cells from patients with X-linked agammaglobulinemia produce enhanced inflammatory cytokines. J Allergy Clin Immunol (2012) 129(1):184–90.e1–4. doi:10.1016/j.jaci.2011.10.009
46. Cunningham-Rundles C, Radigan L, Knight AK, Zhang L, Bauer L, Nakazawa A. TLR9 activation is defective in common variable immune deficiency. J Immunol (2006) 176(3):1978–87. doi:10.4049/jimmunol.176.3.1978
47. Li YF, Lee KG, Ou X, Lam KP. Bruton’s tyrosine kinase and protein kinase C micro are required for TLR7/9-induced IKKalpha and IRF-1 activation and interferon-beta production in conventional dendritic cells. PLoS One (2014) 9(8):e105420. doi:10.1371/journal.pone.0105420
48. Horwood N, Page T, McDaid J, Palmer C, Campbell J, Mahon T, et al. Bruton’s tyrosine kinase is required for TLR2 and TLR4-induced TNF, but not IL-6, production. J Immunol (2006) 176(6):3635–41. doi:10.4049/jimmunol.176.6.3635
49. Taneichi H, Kanegane H, Sira M, Futatani T, Agematsu K, Sako M, et al. Toll-like receptor signaling is impaired in dendritic cells from patients with X-linked agammaglobulinemia. Clin Immunol (2008) 126(2):148–54. doi:10.1016/j.clim.2007.10.005
50. Horwood NJ, Mahon T, McDaid JP, Campbell J, Mano H, Brennan FM, et al. Bruton’s tyrosine kinase is required for lipopolysaccharide-induced tumor necrosis factor alpha production. J Exp Med (2003) 197(12):1603–11. doi:10.1084/jem.20021845
51. Sochorová KHR, Rozková D, Litzman J, Bartunková J, Sedivá A, Spísek R. Impaired Toll-like receptor 8-mediated IL-6 and TNF-alpha production in antigen-presenting cells from patients with X-linked agammaglobulinemia. Blood (2007) 109(6):2553–6. doi:10.1182/blood-2006-07-037960
52. Lougaris V, Baronio M, Vitali M, Tampella G, Cattalini M, Tassone L, et al. Bruton tyrosine kinase mediates TLR9-dependent human dendritic cell activation. J Allergy Clin Immunol (2014) 133(6):1644–50.e4. doi:10.1016/j.jaci.2013.12.1085
53. Gonzalez-Serrano ME, Estrada-Garcia I, Mogica-Martinez D, Gonzalez-Garay A, Lopez-Herrera G, Berron-Ruiz L, et al. Increased pro-inflammatory cytokine production after lipopolysaccharide stimulation in patients with X-linked agammaglobulinemia. J Clin Immunol (2012) 32(5):967–74. doi:10.1007/s10875-012-9706-z
54. Koprulu AD, Kastner R, Wienerroither S, Lassnig C, Putz EM, Majer O, et al. The tyrosine kinase Btk regulates the macrophage response to Listeria monocytogenes infection. PLoS One (2013) 8(3):e60476. doi:10.1371/journal.pone.0060476
55. Perez de Diego R, Lopez-Granados E, Pozo M, Rodriguez C, Sabina P, Ferreira A, et al. Bruton’s tyrosine kinase is not essential for LPS-induced activation of human monocytes. J Allergy Clin Immunol (2006) 117(6):1462–9. doi:10.1016/j.jaci.2006.01.037
56. Schmidt NW, Thieu VT, Mann BA, Ahyi ANN, Kaplan MH. Bruton’s tyrosine kinase is required for TLR-induced IL-10 production. J Immunol (2006) 177(10):7203–10. doi:10.4049/jimmunol.177.10.7203
57. Lee KG, Xu S, Wong ET, Tergaonkar V, Lam KP. Bruton’s tyrosine kinase separately regulates NFkappaB p65RelA activation and cytokine interleukin (IL)-10/IL-12 production in TLR9-stimulated B Cells. J Biol Chem (2008) 283(17):11189–98. doi:10.1074/jbc.M708516200
58. Gagliardi M, Finocchi A, Orlandi P, Cursi L, Cancrini C, Moschese V, et al. Bruton’s tyrosine kinase defect in dendritic cells from X-linked agammaglobulinaemia patients does not influence their differentiation, maturation and antigen-presenting cell function. Clin Exp Immunol (2003) 133(1):115–22. doi:10.1046/j.1365-2249.2003.t01-1-02178.x
59. Ni Gabhann J, Spence S, Wynne C, Smith S, Byrne JC, Coffey B, et al. Defects in acute responses to TLR4 in Btk-deficient mice result in impaired dendritic cell-induced IFN-gamma production by natural killer cells. Clin Immunol (2012) 142(3):373–82. doi:10.1016/j.clim.2011.12.009
60. Byrne JC, Ni Gabhann J, Stacey KB, Coffey BM, McCarthy E, Thomas W, et al. Bruton’s tyrosine kinase is required for apoptotic cell uptake via regulating the phosphorylation and localization of calreticulin. J Immunol (2013) 190(10):5207–15. doi:10.4049/jimmunol.1300057
61. Maglione PJ, Simchoni N, Cunningham-Rundles C. Toll-like receptor signaling in primary immune deficiencies. Ann N Y Acad Sci (2015) 1356:1–21. doi:10.1111/nyas.12763
62. Lei X, Xiao X, Wang J. Innate immunity evasion by enteroviruses: insights into virus-host interaction. Viruses (2016) 8(1):22. doi:10.3390/v8010022
63. Dotzauer A, Kraemer L. Innate and adaptive immune responses against picornaviruses and their counteractions: an overview. World J Virol (2012) 1(3):91–107. doi:10.5501/wjv.v1.i3.91
64. Uckun FM, Dibirdik I, Qazi S, Vassilev A, Ma H, Mao C, et al. Anti-breast cancer activity of LFM-A13, a potent inhibitor of Polo-like kinase (PLK). Bioorg Med Chem (2007) 15(2):800–14. doi:10.1016/j.bmc.2006.10.050
65. Akinleye A, Chen Y, Mukhi N, Song Y, Liu D. Ibrutinib and novel BTK inhibitors in clinical development. J Hematol Oncol (2013) 6:59. doi:10.1186/1756-8722-6-59
66. Helmke C, Becker S, Strebhardt K. The role of Plk3 in oncogenesis. Oncogene (2016) 35(2):135–47. doi:10.1038/onc.2015.105
67. Chevrier N, Mertins P, Artyomov MN, Shalek AK, Iannacone M, Ciaccio MF, et al. Systematic discovery of TLR signaling components delineates viral-sensing circuits. Cell (2011) 147(4):853–67. doi:10.1016/j.cell.2011.10.022
68. Berthier-Vergnes O, Bermond F, Flacher V, Massacrier C, Schmitt D, Péguet-Navarro J. TNF-α enhances phenotypic and functional maturation of human epidermal Langerhans cells and induces IL-12 p40 and IP-10/CXCL-10 production. FEBS Lett (2005) 579(17):3660–8. doi:10.1016/j.febslet.2005.04.087
69. Pantel A, Teixeira A, Haddad E, Wood EG, Steinman RM, Longhi MP. Direct type I IFN but not MDA5/TLR3 activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly IC stimulation. PLoS Biol (2014) 12(1):e1001759. doi:10.1371/journal.pbio.1001759
70. Boullart AC, Aarntzen EH, Verdijk P, Jacobs JF, Schuurhuis DH, Benitez-Ribas D, et al. Maturation of monocyte-derived dendritic cells with toll-like receptor 3 and 7/8 ligands combined with prostaglandin E2 results in high interleukin-12 production and cell migration. Cancer Immunol Immunother (2008) 57(11):1589–97. doi:10.1007/s00262-008-0489-2
71. Freistadt MS, Eberle KE. Correlation between poliovirus type 1 Mahoney replication in blood cells and neurovirulence. J Virol (1996) 70(9):6486–92.
72. Mamishi S, Shahmahmoudi S, Tabatabaie H, Teimourian S, Pourakbari B, Gheisari Y, et al. Novel BTK mutation presenting with vaccine-associated paralytic poliomyelitis. Eur J Pediatr (2008) 167(11):1335–8. doi:10.1007/s00431-008-0674-5
73. Platt LR, Estivariz CF, Sutter RW. Vaccine-associated paralytic poliomyelitis: a review of the epidemiology and estimation of the global burden. J Infect Dis (2014) 210(Suppl 1):S380–9. doi:10.1093/infdis/jiu184
74. Racaniello VR. One hundred years of poliovirus pathogenesis. Virology (2006) 344(1):9–16. doi:10.1016/j.virol.2005.09.015
75. Carneiro-Sampaio M, Coutinho A. Immunity to microbes: lessons from primary immunodeficiencies. Infect Immun (2007) 75(4):1545–55. doi:10.1128/IAI.00787-06
77. Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science (2006) 314(5797):308–12. doi:10.1126/science.1128346
78. Corthesy B. Multi-faceted functions of secretory IgA at mucosal surfaces. Front Immunol (2013) 4:185. doi:10.3389/fimmu.2013.00185
79. Bao Y, Zheng J, Han C, Jin J, Han H, Liu Y, et al. Tyrosine kinase Btk is required for NK cell activation. J Biol Chem (2012) 287(28):23769–78. doi:10.1074/jbc.M112.372425
80. Ida-Hosonuma M, Iwasaki T, Yoshikawa T, Nagata N, Sato Y, Sata T, et al. The alpha/beta interferon response controls tissue tropism and pathogenicity of poliovirus. J Virol (2005) 79(7):4460–9. doi:10.1128/JVI.79.7.4460-4469.2005
81. Zhang Y, Yan D, Zhu S, Nishimura Y, Ye X, Wang D, et al. An insight into recombination with enterovirus species C and nucleotide G-480 reversion from the viewpoint of neurovirulence of vaccine-derived polioviruses. Sci Rep (2015) 5:17291. doi:10.1038/srep17291
Keywords: type I interferon, type III interferon, X-linked agammaglobulinemia, oral poliovirus vaccine, H1N1 influenza virus, innate immunity, dendritic cells
Citation: Luk ADW, Ni K, Wu Y, Lam K-T, Chan K-W, Lee PP, Tu W, Mao H and Lau YL (2018) Type I and III Interferon Productions Are Impaired in X-Linked Agammaglobulinemia Patients Toward Poliovirus but Not Influenza Virus. Front. Immunol. 9:1826. doi: 10.3389/fimmu.2018.01826
Received: 22 December 2017; Accepted: 24 July 2018;
Published: 10 August 2018
Edited by:
Sudhir Gupta, University of California, Irvine, United StatesReviewed by:
Tomohiro Morio, Tokyo Medical and Dental University, JapanMike Recher, Universität Basel, Switzerland
Copyright: © 2018 Luk, Ni, Wu, Lam, Chan, Lee, Tu, Mao and Lau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huawei Mao, bWFvaHdlaUBxcS5jb20=;
Yu Lung Lau, bGF1eWx1bmdAaGt1Lmhr
†These authors are co-first authors and contributed equally to this work.