 Shih-Feng Cho1,2,3,4
Shih-Feng Cho1,2,3,4 Yu-Tzu Tai
Yu-Tzu Tai- 1LeBow Institute for Myeloma Therapeutics and Jerome Lipper Multiple Myeloma Center, Dana-Farber Cancer Institute, Boston, MA, United States
- 2Harvard Medical School, Boston, MA, United States
- 3Division of Hematology and Oncology, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung, Taiwan
- 4Faculty of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
The approval of the first two monoclonal antibodies targeting CD38 (daratumumab) and SLAMF7 (elotuzumab) in late 2015 for treating relapsed and refractory multiple myeloma (RRMM) was a critical advance for immunotherapies for multiple myeloma (MM). Importantly, the outcome of patients continues to improve with the incorporation of this new class of agents with current MM therapies. However, both antigens are also expressed on other normal tissues including hematopoietic lineages and immune effector cells, which may limit their long-term clinical use. B cell maturation antigen (BCMA), a transmembrane glycoprotein in the tumor necrosis factor receptor superfamily 17 (TNFRSF17), is expressed at significantly higher levels in all patient MM cells but not on other normal tissues except normal plasma cells. Importantly, it is an antigen targeted by chimeric antigen receptor (CAR) T-cells, which have already shown significant clinical activities in patients with RRMM who have undergone at least three prior treatments, including a proteasome inhibitor and an immunomodulatory agent. Moreover, the first anti-BCMA antibody–drug conjugate also has achieved significant clinical responses in patients who failed at least three prior lines of therapy, including an anti-CD38 antibody, a proteasome inhibitor, and an immunomodulatory agent. Both BCMA targeting immunotherapies were granted breakthrough status for patients with RRMM by FDA in Nov 2017. Other promising BCMA-based immunotherapeutic macromolecules including bispecific T-cell engagers, bispecific molecules, bispecific or trispecific antibodies, as well as improved forms of next generation CAR T cells, also demonstrate high anti-MM activity in preclinical and even early clinical studies. Here, we focus on the biology of this promising MM target antigen and then highlight preclinical and clinical data of current BCMA-targeted immunotherapies with various mechanisms of action. These crucial studies will enhance selective anti-MM response, transform the treatment paradigm, and extend disease-free survival in MM.
Introduction
Multiple myeloma (MM), the second most common hematologic malignancy in the United States, accounts for 1% of malignancies and 10% of hematologic cancers (1). This tumor is characterized by the expansion of malignant plasma cells (PCs) in the bone marrow (BM), associated with excessive production of monoclonal immunoglobulins in blood and urine in patients. In addition, MM patients develop significant osteolytic bone lesions and have immunodeficiency that compromises both longevity and quality of life (2, 3). For the past two decades, the clinical outcome of MM patients has shown remarkable improvements primarily due to the incorporation of novel therapeutic agents into conventional treatments. Specifically, the addition of proteasome inhibitors (PI) and immumomodulatory drugs (IMiDs) has significantly increased response rate, progression-free, and overall survival in both relapsed and newly diagnosed MM patients, compared with conventional therapies (4–7). The addition of monoclonal antibodies (MoAbs) elotuzumab and daratumumab as immunotherapies in MM has further improved patient outcome. The use of autologous stem cell transplantation also results in better outcome. However, MM remains incurable for most patients, since drug-resistant clones constantly emerge and evolve (8). Persistence of minimal residual disease (MRD) is often seen and patients with MRD-negativity also relapse. Particularly, the overall survival of patients with relapsed disease after PIs IMiDs, and MoAbs treatment is extremely low. Thus, more efficacious therapies and novel strategies are urgently needed if we are to develop curative therapies.
Multiple myeloma develops from a premalignant precursor condition monoclonal gammopathy of undetermined significance, progressing to smoldering MM, then active MM, majorities of which ultimately advancing to end-stage PC leukemia. Genetic and epigenetic processes are present initially and underlie this progression, including hyperdiploidy of chromosomes, translocation of immunoglobulin heavy chain, deregulation of cell cycle genes, alteration of NFκB pathways, and abnormal DNA methylation patterns (9–11). Besides complex molecular aberrations, MM cells are heavily dependent on their BM microenvironment to support their growth, survival, and the development of drug resistance. Tumor cells closely interact with BM accessory cells in bidirectional fashions via cell–cell contact and/or production of a variety of factors, which ultimately promotes MM cell expansion, while impairing immune surveillance and effector function against MM cells. These MM-supporting cells include BM stromal cells (BMSCs) (12, 13), osteoclasts (14), endothelial cells (15), macrophages (16), T regulatory cells (17–19), dendritic cells (20), plasmacytoid DCs (pDCs) (21), myeloid-derived suppressor cells (22), and mesenchymal cells (13, 23). These accessory cells secrete various cytokines including interleukin-6 (IL-6) (24), tumor growth factor β (TGFβ) (25, 26), macrophage inflammatory protein-1α (MIP-1α) (27), insulin-like growth factor (28), vascular endothelial growth factor (29), hepatocyte growth factor (30), B cell activating factor (BAFF) (31, 32), and a proliferation-inducing ligand (APRIL) (31, 33), which further maintain an MM-supporting or immunosuppressive BM microenvironment (34). For example, the key myeloma growth factor IL-6 and the critical immune inhibitory factor TGFβ are detected at high levels in the BM of MM patients. The interplay of these two cytokines may affect generation of Th17 cells both directly or via other pro-inflammatory cytokines, and thereby downregulate antitumor immune responses (35). Increased Th17 cells and decreased regulatory T cells (Tregs) with less immune suppression is noted in MM patients with long-term survival (36). Since Tregs can inhibit function of antigen-presenting cells and effector T cells (37), increased Treg number allows MM cells to escape from immune surveillance. In fact, immune-suppressive Treg markers Foxp3 and CTLA-4 are significantly upregulated in the BM aspirates of MM patients compared with normal donor controls (17), and increased Tregs are correlated with worse outcomes in MM (36, 38, 39). These studies indicate that molecular and cellular components suppress immune BM milieu, further enhancing MM progression.
Successful targeted anti-MM immunotherapies should both target MM cells and simultaneously restore antitumor activity of immune effector cells (40). Ideally, targets for effective immunotherapies should be selectively and strongly expressed on the surface of MM cells relative to normal cells. Compared with CD38 and SLAMF7, B cell maturation antigen (BCMA) demonstrates highly restricted expression on PCs but no other tissues, is, therefore, an excellent target for immunotherapy in MM (41, 42).
BCMA is an Important Surface Protein Supporting the Survival of MM Cells
B cell maturation antigen, also termed tumor necrosis factor receptor superfamily member 17 (TNFRS17), is a type III transmembrane protein without a signal-peptide and containing cysteine-rich extracellular domains (43–45). Alignment of the human (44, 45) and murine BCMA protein sequences (43) revealed a conserved motif of six cysteines in the N-terminal part, which strongly suggests that the BCMA protein belongs to the tumor necrosis factor receptor (TNFR) superfamily. BCMA, along with two related TNFR superfamily B-cell activation factor receptor (BAFF-R) and transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI), critically regulate B cell proliferation and survival, as well as maturation and differentiation into PCs. These three functionally related receptors support long-term survival of B cells at different stages of development by binding to BAFF and/or APRIL (46–49), their cognate ligands. Specifically, BCMA is only induced in late memory B cells committed to the PC differentiation and is present on all PCs (46, 50, 51). Expression of BCMA is induced, while BAFF-R is decreased, during PC differentiation from B cells. Studies from BCMA-knockdown mice further indicate that BCMA is most important for long-lived PC survival but is dispensable for overall B cell homeostasis (50, 52). A recent study showed that an enzyme, γ-secretase can cleave membrane BCMA, leading to decreased in membrane form BCMA and formation of soluble form BCMA (sBCMA) (53) (Figure 1).
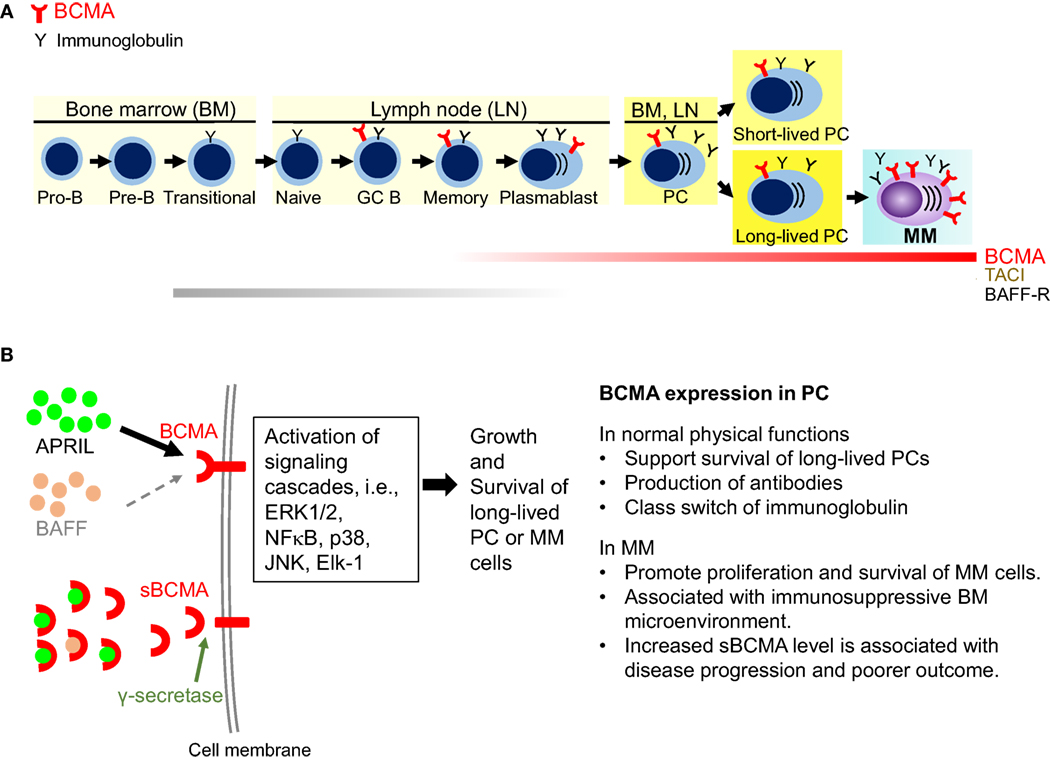
Figure 1. Biological significance of B cell maturation antigen (BCMA) in plasma cells (PCs). (A) BCMA is selectively induced during PC differentiation, associated with loss of BAFF-R. It is expressed on late-stage B-cells, short-lived proliferating plasmablasts, and long-lived PCs. BCMA does not maintain normal B-cell homeostasis but is required for the survival of long-lived PCs. In multiple myeloma (MM), expression of BCMA is significantly increased on malignant vs normal PCs. (B) A proliferation-inducing ligand (APRIL) and BAFF are two natural ligands for BCMA. Specifically, APRIL binds to BCMA with a significantly higher affinity than BAFF. Activation of BCMA supports growth and survival of PCs via activating MEK/ERK, AKT, NFκB, JNK, p38 kinase, and Elk-1. In MM cells, overexpression of BCMA or binding of APRIL to BCMA activates AKT, ERK1/2, and NFκB pathways and upregulate antiapoptotic proteins, i.e., Mcl-1, Bcl-2, Bcl-xL to protect MM cells from dexamethasone- and interleukin-6 deprivation induced apoptosis. Furthermore, BCMA upregulates genes associated with activation of osteoclast, adhesion, and angiogenesis/metastasis. Moreover, overexpressed BCMA can induce the expression immunosuppressive molecules such as PD-L1 in MM cells. Membrane BCMA can be cleaved by γ-secretase, resulting in reduced number of membrane-bound BCMA molecules and increased soluble BCMA. Soluble BCMA can bind to APRIL and BAFF, which may interfere downstream BCMA signaling cascades. TACI, transmembrane activator and calcium modulator and cyclophilin ligand interactor; GC, germinal center.
Earlier studies show that overexpression of BCMA in 293 cells activates the mitogen-activated protein kinase pathway, especially JNK and p38 kinase, the nuclear factors NFκB and Elk-1, without stimulation of BAFF or APRIL (54). BCMA expression is positively regulated by B-lymphocyte-induced maturation protein 1 (Blimp-1), a gene controlling proliferation of PCs (55). In KMS12 MM cell line, BCMA co-immunoprecipitates with interferon regulatory factor-4, a master transcription factor mediating survival of MM cells (56). Importantly, BCMA overexpression or APRIL binding to BCMA in MM cells significantly promotes MM cell growth and survival in vivo (33, 57). Conversely, BCMA knockdown blocks MM cell proliferation and viability via downregulation of cell cycle progression and antiapoptosis molecules. APRIL and BAFF, via binding to BCMA and TACI, further activate NFκB pathways and upregulate antiapoptotic proteins (Mcl-1, Bcl-2, Bcl-xL) to protect MM cells against dexamethasone- and serum deprivation-induced cell death (31, 58, 59) (Figure 1). These studies establish a pathophysiological role of BCMA and APRIL in MM.
Rationale to Targeting BCMA in MM
B cell maturation antigen is exclusively expressed on the surface of plasmablasts and differentiated PCs, but not on memory B, naive B cells, CD34+ hematopoietic stem cells, and other normal tissue cells (41, 50, 51, 60–64). BCMA mRNA and protein are more highly expressed on malignant than normal PCs, as validated by multiple gene expression profiling (41, 42, 65, 66) and immunohistochemistry (IHC) studies (41). In the study by Carpenter et al. (41), cDNA copies of BCMA were detected by qPCR in several hematologic tissues including white blood cells, BM, lymph node, spleen, and tonsil. In normal tissues, low levels of BCMA cDNA copies were detected in the samples of testis, trachea and samples from gastrointestinal organs like duodenum, rectum, and stomach. When the expression was evaluated by IHC, BCMA protein expression was only detected on MM cells, lymphoid cells, or PCs from normal human organs such as duodenum, rectum, and stomach. However, BCMA protein expression was not detected on the other cell types in these organs (41). Another study examined BCMA expression on various blood cells and Hodgkin lymphoma cells using flow cytometry (63). BCMA expression was negative on naive and memory B cells, weak on founder B cells from germinal center (GC) and Reed–Sternberg cells, positive on GC B cells, but highly positive on plasmacytoid B cells. Based on these findings, BCMA protein is highly and specifically expressed on PCs, low levels of BCMA RNA detected in these normal organs would be due to existence of PCs.
Thus far, majorities of studies indicate that BCMA transcript, protein, and the serum BCMA level are significantly higher in MM cell lines and patient MM cells, when compared with normal donors. One recent study reported that median BCMA expression on patient MM cells was not to be higher compared to normal BMPC as shown in large patient cohorts (66). At protein levels, shedding of BCMA by γ-secretase controls PCs in the BM and may represent a potential biomarker for B-cell involvement in human autoimmune diseases (53). Significantly, sBCMA levels are increased in MM patients vs healthy individuals (67–69). Its level in patient serum is further correlated with disease status and prognosis. Furthermore, anti-BCMA antibodies are detected in MM patients in remission after donor lymphocyte infusion with graft-vs-tumor response, suggesting that antibody responses to cell-surface BCMA may directly contribute to tumor elimination (70). Moreover, low BCMA is detected in pDCs (42), which support survival and drug resistance of MM cells (21). In fact, pDCs are the only cell type other than PCs with detectable BCMA at significantly lower levels (>1-log lower) compared to matched PCs (42). These data confirm BCMA as a very promising MM antigen for targeted immunotherapy.
Both BCMA ligands APRIL and BAFF, to a lesser extent, are critical BM factors supporting growth and survival of malignant PCs in MM (31, 62). The levels of both ligands are significantly increased in serum samples of MM patients vs normal controls (31, 71). APRIL, which does not bind to BAFF-R, preferably binds to BCMA with much higher affinity than BAFF (nM vs μM), whereas BAFF has an approximate 100-fold selectivity for binding to BAFF receptor (BAFF-R) over BCMA (72, 73). Coupled with the fact that APRIL also binds to TACI on PCs via interaction with CD138/syndecan-1, APRIL is more specific to PCs than BAFF (57, 74–76). Importantly, APRIL directly promotes MM cell growth and survival in vivo, since APRIL knockout mice injected with human MM cell lines live longer than wild-type mice under similar conditions (77). These results strongly supporting targeting BCMA for novel MM treatments.
As described below, GSK2857916, the first therapeutic anti-BCMA antibody–drug conjugates (ADCs) with multiple mechanisms of action against MM cells, used alone and with MM-protecting BM components, rapidly eliminates MM cells in two murine models and significantly prolongs survival of mice (42). These promising data further support clinical development of BCMA-targeted immunotherapies in MM.
BCMA-Based Immunotherapies
The development of novel agents targeting BCMA is ongoing rapidly, especially following impressive clinical responses in relapsed MM patients using the first chimeric antigen receptor (CAR) T cell therapy (78). Currently, there are multiple BCMA-based treatment modalities including: ADC, bispecific T-cell engager (BiTE), CAR T cell (CAR T), bispecific molecule, and bi/trispecific Abs (Figure 2) (summarized in Table 1), as well as cancer vaccines.
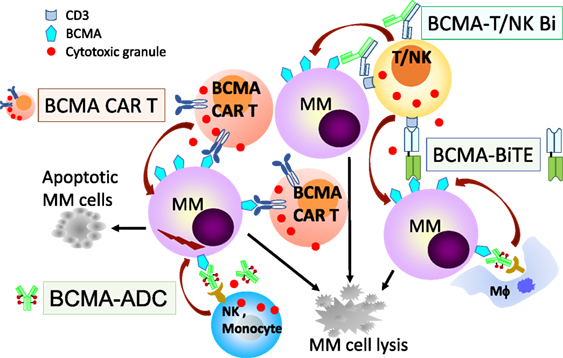
Figure 2. B cell maturation antigen (BCMA)-based immunotherapies with multiple mechanisms of action against MM cells. Various BCMA-based treatment modalities are under clinical development are listed in Table 1 and shown here. BCMA-NK Bi or Tri Ab, not shown here, can also specifically induce effector cell-mediated lysis of MM cells. ADC, antibody drug conjugate; Bi, bispecific full-length immunoglobulin; BiTE, bispecific T-cell engager; CAR T, chimeric antigen receptor T cell; MM, multiple myeloma cell; NK, natural killer cell; Mϕ, macrophage.
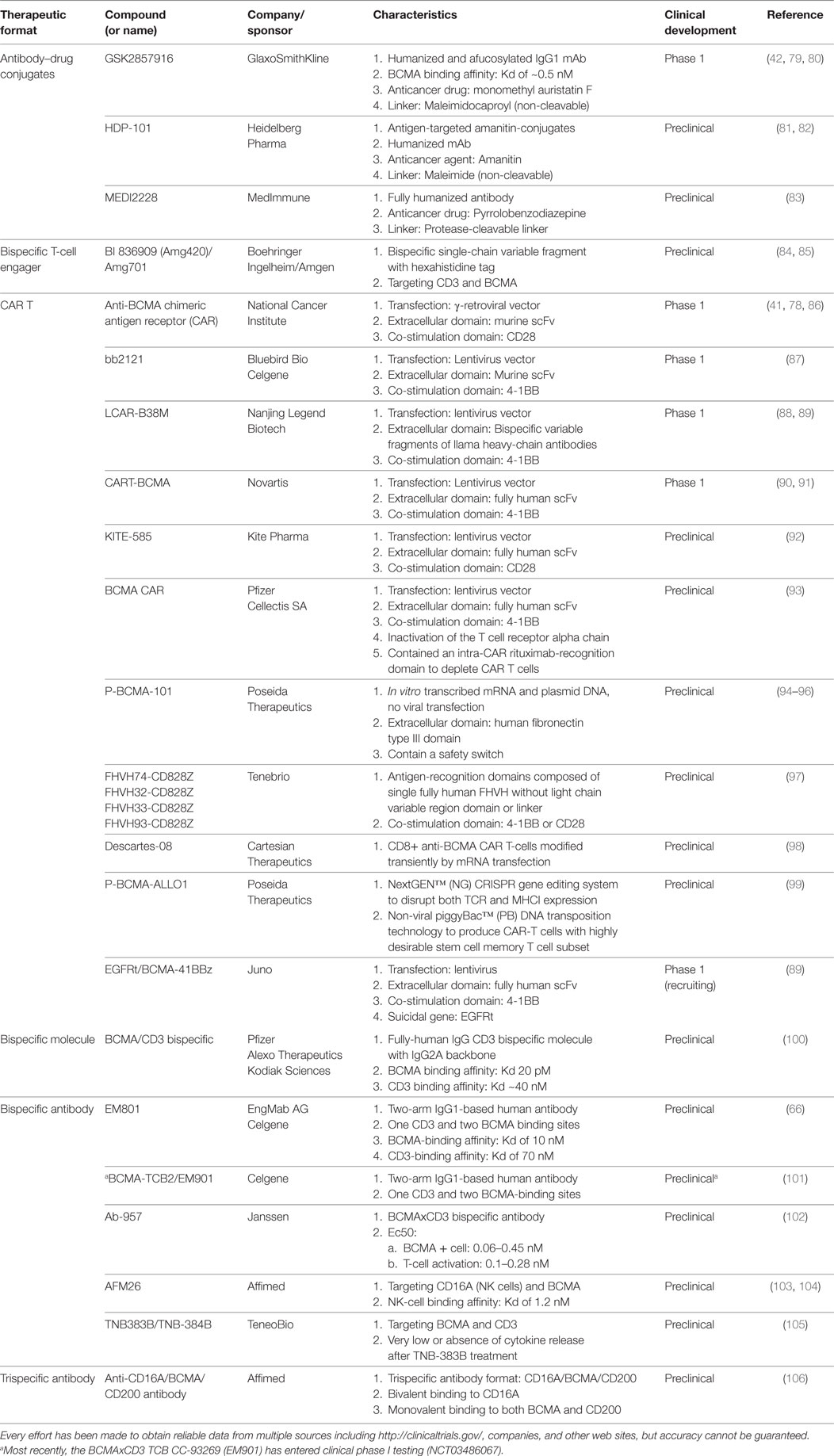
Table 1. List of Anti-B cell maturation antigen (BCMA) formats.
Antibody–Drug Conjugates
Antibody–drug conjugate, one of the fastest growing class of cancer therapeutics, is composed of recombinant mAbs covalently bound to cytotoxic chemicals (payload) via synthetic chemical linkers (107). The mAbs first identify and bind to the antigen on the surface of tumor cells, and then is absorbed or internalized, together with the payload. After the ADC is internalized, the cytotoxic chemicals are released in the lysosomes and transported to cytosol to kill the tumor cells.
GSK2857916
GSK285791 is a humanized and IgG1 mAb with high affinity to BCMA (Kd of ~0.5 nM) (42), which uses non-cleavable linker, maleimidocaproyl (mc), and a new class of antimitotic agents, monomethyl auristatin F, as payload. This structure is characterized by high stability and high antitumor potency, with low by-stander toxicity.
GSK2857916 binds to all CD138+ and BCMA+ MM cell lines and patient MM cells. MM cell proliferation is inhibited via G2/M arrest in a dose-dependent manner, and apoptosis is induced by activation of caspase 3/7 and 8. There are minimal effects on surrounding BCMA-negative normal cells. GSK2857916 also triggers ADCC and antibody-dependent cellular-mediated phagocytosis against patient MM cells. The cytotoxicity against MM cells is further enhanced when GSK2857916 is combined with lenalidomide via effector-dependent and -independent manners. Most importantly, in both disseminated and subcutaneous human MM xenograft models in mice, GSK2857916 rapidly eliminates MM cells and generated little toxicity in mice treated with continuous dosing for nine times at 4 mg/kg, with tumor-free survival up to 3.5 months in mice (42).
GSK2857916 was evaluated in a phase 1 study of patients with relapsed and refractory multiple myeloma (RRMM), including dose-escalating and expansion parts (79, 80). GSK2857916 monotherapy has demonstrated a 60% response rate and a median progression-free survival of 7.9 months in a group of hard to treat and heavily pretreated RRMM (80). It has recently been awarded Breakthrough Therapy designation from FDA and received PRIME designation from the European Medicines Agency (EMA).
HDP-101
HDP-1, an antibody-targeted amanitin conjugate, is an anti-BCMA ADC with a novel payload amanitin, which binds to the RNA polymerase II in eukaryotic cells and inhibits cellular transcription at very low concentrations (108). HDP-1 was synthesized with the conjugation of maleimide-amanitin compounds and engineered cysteine residues in the heavy chain of the humanized anti-BCMA Thiomab (109, 110).
HDP-101 demonstrated potent in vitro cytotoxicity against BCMA-expressing MM cell lines at picomolar range, without effects on BCMA-negative cells. Significant tumor regression including complete remission was observed in the mouse xenograft model in a dose-dependent manner. The tolerability and therapeutic index were good after a series of HDP-101 administrations at different concentrations in Cynomolgus monkeys. Mild-to-moderate elevation of liver enzymes and lactic dehydrogenase were noted, but these abnormalities were transient. HDP-101 has a long half-life in serum (about 12 days) (81, 82).
MEDI2228
The structure of MEDI2228 includes a fully human antibody site-specifically conjugated to a pyrrolobenzodiazepine dimer via a protease-cleavable linker. This ADC is rapidly internalized into MM cells and trafficked to lysosomes.
MEDI2228 was highly active in 8 of 10 MM cell lines (IC50 range 6 to 210 ng/mL) including cell lines regardless of BCMA levels (83). MEDI2228 was also active in the presence of BMSCs. A single injection of MEDI2228 induced human MM xenograft regression in mice at very low doses (0.1 mg/kg). MEDI2228 was characterized by weak binding capacity to recombinant monomeric human BCMA, but strong binding to membrane-bound BCMA. It kills an average of 95% of tumor cells in the presence of sBCMA at levels up to 720 ng/mL, without impact on IC50. Clinical trials of this new anti-BCMA will be starting in mid-2018.
Bispecific T-Cell Engager
Bispecific T-cell engager is a single-chain variable fragment (scFv), composed of two linked mAbs (bispecific antibodies) targeting mainly CD3 on the surface of T-cells and tumor-associated antigens. This unique structure allows BiTE to engage T-cells with tumor cells (111). After the binding, antitumor cytotoxicity and cytokine production of T cells are activated, and the formation of cytolytic immunological synapses are induced (112, 113). BiTE is also characterized by its small size (55 kDa), which makes it a highly potent and efficacious molecule to against cancer (114). However, the small size of BiTE is unstable due to short serum half-life, thus continuous infusion is required.
BI 836909
BI 836909 is the first bispecific scFv with two linked scFvs in MM (84). The scFv targeting BCMA is positioned in N-terminal, and the scFv targeting CD3ε is in C-terminal, followed by a hexa-histidine (His6-tag). BI 836909 simultaneously bind to CD3+ T cell and BCMA-expressing MM cells. This makes a cross-link between both cells to induce formation of cytolytic synapse, ultimately leading to activation of T cells and lysis of BCMA+ MM cells. These cytotoxic activities were not observed in BCMA-negative cells. When cocultured with BM stromal cells, BI 836909 retains potent anti-MM activity. Additionally, soluble APRIL and BCMA have only a mild effect on the anti-MM activity of BI 836909.
In mouse xenograft studies, BI 836909 led to tumor shrinkage in a subcutaneous NCI-H929 xenograft model and prolonged survival in an orthotopic L-363 xenograft model. In a cynomolgus monkey study, administration of BI 836909 resulted in significant depletion of BCMA + PCs in the BM of monkeys (84).
A half-life extended anti-BCMA BiTE base on BI 836909 was recently reported to be effective in vitro and in vivo and is suitable for once-weekly dosing in MM patients (85).
CAR T Cell Therapy
Adoptive transfer of T cells genetically modified to recognize tumor-associated antigens is a promising cancer treatment (115). By using techniques of genetic modification, T cells can express CAR, which are fusion proteins that have an antigen recognition region, usually scFv derived from antibody on the surface, and a costimulation domain in the cell. Unlike T cell receptor modified T cells, CAR T cells are not restricted by major histocompatibility complex (40).
In MM, several anti-BCMA CAR T cell therapies have shown impressive clinical activities (some reaching 90–100%) with more are developed and under preclinical and/or clinical investigations (see Table 1).
Other Trials of Anti-BCMA CAR-T Therapy
The combined infusion of CD19 and BCMA-specific CAR T Cells for RRMM was investigated in an early phase study (NCT 03196414) (116). The cells contained respective anti-BCMA or anti-CD19 scFv transduced by lentivirus, OX40 and CD28 costimulatory moiety, and CD3z T-cell activation domain. Clinical efficacy was evaluated in five patients monitored for more than 4 weeks, and showed that ORR was 100%, including 1 sCR, 1 VGRP, 2 PR, and 1 SD (116).
Anti-BCMA CD3 BI- or Trispecific Molecules
A Fully Human IgG CD3 Bispecific Molecule Targeting BCMA
This fully-human IgG bispecific molecule is characterized by its long half-life (about 3 days in mice) (100). The molecule utilizes hinge mutation technology to pair anti-BCMA and anti-CD3 targeting arms and places them in an IgG2A backbone. The anti-MM cytotoxicity was observed in MM patient samples at very low concentration (EC50 = 0.093 ± 0.1 nM), lower than ADC. This molecule also effectively depleted low BCMA-expressing normal plasma B cells. The evolution of toxicity in cynomologus monkeys model showed favorable safety profile.
EM801
EM801 is asymmetric two-arm IgG1-based human antibody with two binding sites for BCMA and 1 binding site for CD3 (66). EM801 promotes activation of CD4+ and CD8+ T-cells accompanied with release of IFN-γ, granzyme B, and perforin, and CD3+ T cell-dependent killing of MM cell lines. EM801 also induced significant cell death in malignant PCs by autologous T cells in BM samples of previously untreated and RRMM patients at very low concentrations (from 10 pM to 30 nM).
BCMA-TCB2
B cell maturation antigen-TCB2 is a bispecific antibody, which shares similar structure of EM801, but with higher affinity to BCMA (101). BCMA-TCB2 induces lysis of MM cells, activation of T cells, and natural emergence of the checkpoint inhibitor PD-1 on T cells at very low concentration. Combination of BCMA-TCB2 with lenalidomide or daratumumab significantly enhanced antimyeloma efficacy. NK cells were also activated after BCMA-TCB2 treatment.
Ab-957
Ab-957 is bispecific IgG-like Ab generated by Genmab DuoBody® technology to target CD3 on T cells and BCMA on MM cells (102). Preclinical studies also show that Ab-957 potently induces specific cytotoxicity of BCMA + MM cells in vitro and in vivo, with a concomitant activation of T cells at very low concentration.
AFM26
AFM26 is a bispecific antibody, which targets BCMA on MM cells and CD16A on NK cells (103, 104). AFM26 induces potent NK-cell-medicated cytotoxicity in BCMA+ MM, even when BCMA expression of BCMA was low. AFM26 does not induce NK-cell depletion. It shows similar anti-MM activity, but less inflammatory cytokine secretion, than BiTEs.
TNB383B and TNB-384B
TNB383B and TNB-384B are bispecific antibodies targeting BCMA on MM cells and CD3 on T cells, which are generated based on the basis of in silico analysis of heavy chain only/fixed light chain antibody sequences (105). Both Abs showed significant anti-MM cytotoxicity at very low concentration (nano- or pico-molar) and eradicated MM cell growth in mice. In addition, markedly reduced or absence of cytokine release is observed after TNB-383B treatment.
Anti-CD16A/BCMA/CD200 Antibody
This trispecific antibody is characterized by bivalent binding to CD16A on NK cells and monovalent binding to BCMA and CD200 on MM cells (106). This dual-targeting structure may increase selectivity of MM cells coexpressing both antigens and improve safety.
Therapeutic Agents Targeting APRIL
Therapeutic agents blocking APRIL/BCMA are under investigated as well. A novel mouse anti-human APRIL antibody hAPRIL01A (01A) inhibits the binding of APRIL to BCMA and TACI (117). Importantly, 01A inhibited APRIL- and osteoclast-induced proliferation of MM cells and further induced apoptosis of MM cells in cocultures (33). 01A also enhances the cytotoxicity mediated by IMiDs and PI in the cocultures of MM cells with BCMA-negative BM accessory cells and effector cells. Furthermore, APRIL induces expression of genes involved in immunosuppression, such as PD-L1, TGF-β, and IL-10, are decreased in MM cells following 01A treatment (33). The early phase clinical trial of BION-1301, a fully humanized 01A mAb, is ongoing (118).
Perspectives and Conclusion
Since its discovery in 1992, accumulating evidence has demonstrated that BCMA is a promising target for immunotherapy in MM (Table 2). CAR T therapy first demonstrated promising clinical efficacy in several phase 1 clinical trials in which high response rates are seen in heavily pretreated RRMM patients. GSK2857916, the first therapeutic BCMA-ADC, also shows impressive clinical efficacy and acceptable safety profile in RRMM resistant to multiple lines of current anti-MM treatments (Table 3). Similar efficacy in clinical trials can be anticipated for other anti-BCMA formats demonstrating highly selective anti-MM activity in preclinical studies.
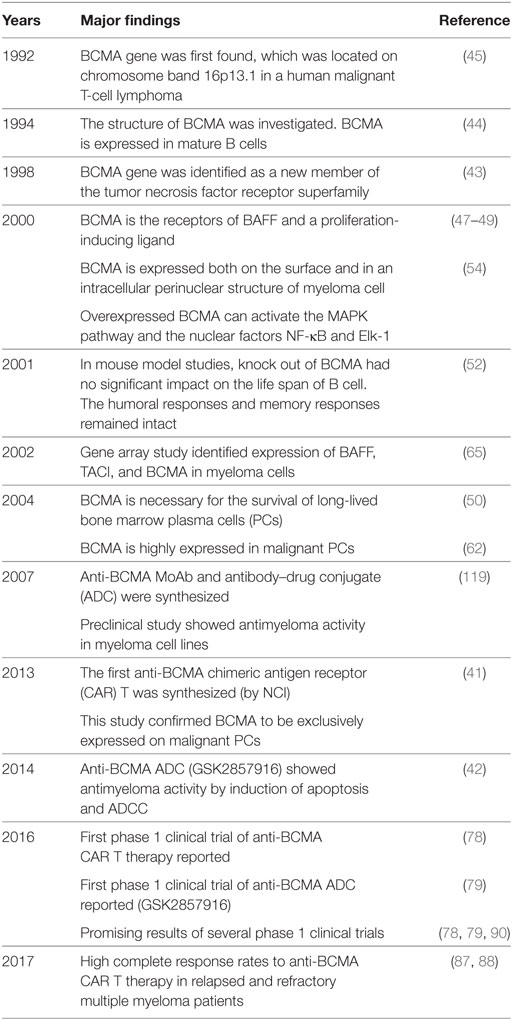
Table 2. Important milestone of anti-B cell maturation antigen (BCMA) immunotherapy for MM.
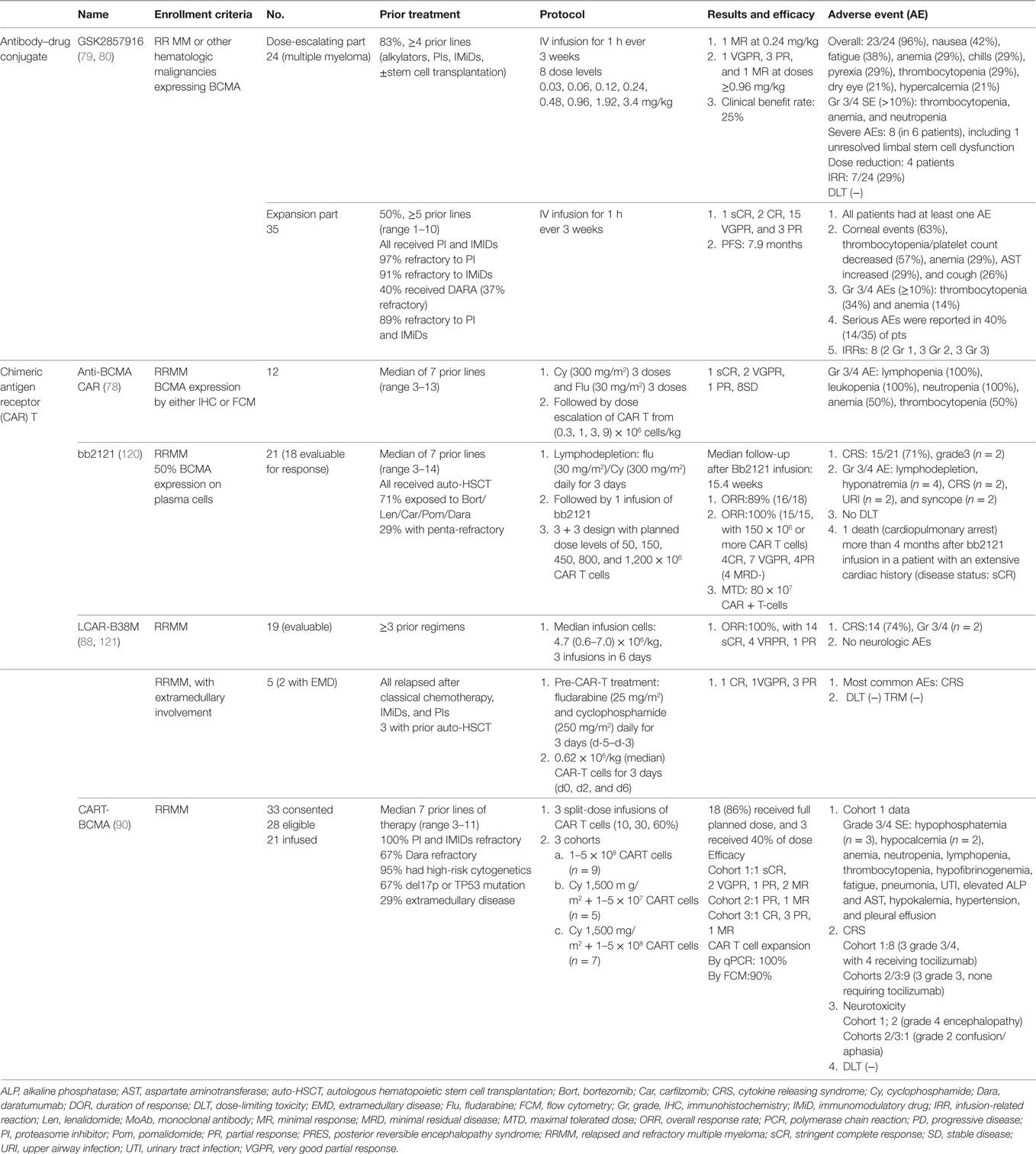
Table 3. Summary of phase 1 clinical trials of anti-B cell maturation antigen (BCMA) agents.
Ongoing efforts are attempting to make BCMA CAR T therapy more potent, safe, and affordable for patients. To improve clinical efficacy, novel CAR T therapies are being developed to overcome relapse due to reduced tumor antigen, including modification of T cells with two distinct CAR molecules with two different binding domains, or one CAR molecule with two different binding domains in tandem (122–124). To reduce toxicities of conditioning chemotherapy, possible approaches include usage of less toxic conditioning chemotherapy, treating earlier in the disease course with less tumor burden, and improved supportive care (125). For prediction of severe cytokine releasing syndrome (CRS), several inflammation cytokines (especially IL-6) have been evaluated, and models have been established (125, 126). For the treatment of CRS, cytokine-directed therapy with anti-IL6 receptor inhibitor tocilizumab can abrogate toxicities (127, 128). Other strategies to reduce side effects include modification of CAR structure, such as incorporation of suicide genes into the engineered T cells (129–131); adding an inhibitory CAR on engineered T cells to reduce off-target immune response (132); or usage of a small molecule system to control CARs (133, 134). More cost-effective, time-saving, and more accessible CAR T cell therapies are being developed, including allogeneic CAR T cells or CAR T cells utilizing novel manufacturing processes (93, 94).
For BCMA ADC, the first clinical trial has demonstrated efficacy and safety. ADC delivering highly toxic chemicals into the tumor cells is a highly selective therapy, which is critical since, the conjugated toxic chemicals are extremely deadly. Currently, several novel promising payloads are under development, including α-amanitin, tubulysins, hizoxin, or spliceostatins (135–137). To improve penetration, novel ADC formats such as non-IgG scaffolds or non-internalizing mAb scaffolds, may be applied to anti-BCMA ADC (138). Besides modification of ADC structure, combinations of ADC with other antitumor agents with different mechanisms of action are also under further investigation. Given that immune checkpoint inhibitors have clinical efficacy in several cancers, studies evaluating the clinical efficacy of combining immune checkpoint inhibitors with BCMA ADC are also warranted in MM (139).
Bispecific T-cell engagers are currently evaluated in preclinical studies. These anti-BCMA agents with excellent anti-MM effect will soon be investigated in clinical trials. Unlike CAR T cell therapy, BiTEs have a relatively short serum half-life and may not stimulate persistent immunity against cancer cells (140). Because it is difficult to maintain serum levels with bolus or intermittent infusion, continuous intravenous infusion may be needed (141). Importantly, long half-life molecules of BCMA BiTEs have been generated (85) and are currently being tested in a clinical trial. As CRS and neurotoxicity are also observed after BiTE treatment, close monitoring and adequate management for these side effects is very important (142). BCMA BiTEs mainly mediate their anti-MM effect by recruiting nearby cytotoxic T-cells to MM cells. However, the function of T cells is severely impaired in heavily pretreated MM patients (143, 144). To optimize BiTE anti-MM activity, studies are evaluating combination therapy with other anti-MM agents or immune checkpoint blockers.
Besides MM, anti-BCMA therapies may have therapeutic potential in other BCMA-expressing malignancies. For example, BAFF-R, BCMA, and TACI are all expressed on primary cells from patients with precursor B-cell acute lymphoblastic leukemia. Moreover, survival of leukemia cells is promoted by binding of BAFF and APRIL to their receptors, suggesting the therapeutic potential of targeting this signaling pathway (145). Other malignancies, such as Waldenstrom macroglobulinemia and glioblastoma/astrocytomas, also express BCMA on their cell surface (146, 147) and may benefit from these BCMA targeted therapies.
As BCMA is exclusively expressed on PCs, anti-BCMA treatment will reduce the number of long-lived PCs. Since long-lived PCs play a critical role in maintaining humoral immunity, the impact of anti-BCMA therapy on immune function needs to be carefully and serially evaluated. To address this issue, more clinical observation and correlative studies are warranted. Another potential complicating factor in anti-BCMA immunotherapy is high serum level of sBCMA, cleaved from BCMA by γ-secretase. In MM patients, high levels of sBCMA have been detected, especially in the setting of progressive disease (68). In preclinical studies, sBCMA slightly influenced the potency (shift in EC50 values) but not the maximal lysis mediated by BI 836909 (84). GSK2857916 still induced significant MM1S cell lysis in the presence of MM1S culture supernatant (42). On the other hand, sBCMA level is markedly decreased in patients after successful CAR T cell therapy (78). More clinical studies are needed to determine whether the level of sBCMA can potentially interfere with efficacy of anti-BCMA treatment. Inhibition of γ-secretase to reduce the formation of sBCMA and enhance the expression of BCMA on MM cells is another novel treatment approach.
In conclusion, BCMA-based immunotherapy is a promising in MM. It is anticipated that most of these anti-BCMA approaches, alone and in combinations with immune checkpoint inhibitors, and as well as cancer vaccines, will be evaluated in clinical studies and offer the promise of more selective, better tolerated, anti-MM therapy.
Author Contributions
Y-TT and S-FC reviewed literature and designed and wrote this paper. Y-TT and KA critically reviewed and edited the paper.
Conflict of Interest Statement
KA serves on advisory boards Celgene, Millennium, and Gilead Sciences and is a Scientific Founder of OncoPep and C4 Therapeutics. The other authors have no competing interests to declare.
Acknowledgments
We thank all laboratory and clinical research teams at the LeBow Institute for Myeloma Therapeutics and the Jerome Lipper Multiple Myeloma Center of the Dana-Farber Cancer Institute for their continuous encouragement, help, and support.
Funding
This work was supported in part by grants from the National Institutes of Health Grants P50-100007, PO1-155258, and RO1-50947. KA is an American Cancer Society Clinical Research Professor.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin (2018) 68(1):7–30. doi:10.3322/caac.21442
2. Kyle RA, Gertz MA, Witzig TE, Lust JA, Lacy MQ, Dispenzieri A, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc (2003) 78(1):21–33. doi:10.4065/78.1.21
3. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med (2011) 364(11):1046–60. doi:10.1056/NEJMra1011442
4. Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med (2005) 352(24):2487–98. doi:10.1056/NEJMoa043445
5. Kumar SK, Dispenzieri A, Lacy MQ, Gertz MA, Buadi FK, Pandey S, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia (2014) 28(5):1122–8. doi:10.1038/leu.2013.313
6. San Miguel JF, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med (2008) 359(9):906–17. doi:10.1056/NEJMoa0801479
7. Rosinol L, Oriol A, Teruel AI, Hernandez D, Lopez-Jimenez J, de la Rubia J, et al. Superiority of bortezomib, thalidomide, and dexamethasone (VTD) as induction pretransplantation therapy in multiple myeloma: a randomized phase 3 PETHEMA/GEM study. Blood (2012) 120(8):1589–96. doi:10.1182/blood-2012-02-408922
8. Laubach JP, Voorhees PM, Hassoun H, Jakubowiak A, Lonial S, Richardson PG. Current strategies for treatment of relapsed/refractory multiple myeloma. Expert Rev Hematol (2014) 7(1):97–111. doi:10.1586/17474086.2014.882764
9. Kuehl WM, Bergsagel PL. Molecular pathogenesis of multiple myeloma and its premalignant precursor. J Clin Invest (2012) 122(10):3456–63. doi:10.1172/JCI61188
10. Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer (2012) 12(5):335–48. doi:10.1038/nrc3257
11. Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol (2017) 14(2):100–13. doi:10.1038/nrclinonc.2016.122
12. Fowler JA, Mundy GR, Lwin ST, Edwards CM. Bone marrow stromal cells create a permissive microenvironment for myeloma development: a new stromal role for Wnt inhibitor Dkk1. Cancer Res (2012) 72(9):2183–9. doi:10.1158/0008-5472.CAN-11-2067
13. Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer (2007) 7(8):585–98. doi:10.1038/nrc2189
14. An G, Acharya C, Feng X, Wen K, Zhong M, Zhang L, et al. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: therapeutic implication. Blood (2016) 128(12):1590–603. doi:10.1182/blood-2016-03-707547
15. Vacca A, Ria R, Semeraro F, Merchionne F, Coluccia M, Boccarelli A, et al. Endothelial cells in the bone marrow of patients with multiple myeloma. Blood (2003) 102(9):3340–8. doi:10.1182/blood-2003-04-1338
16. Zheng Y, Cai Z, Wang S, Zhang X, Qian J, Hong S, et al. Macrophages are an abundant component of myeloma microenvironment and protect myeloma cells from chemotherapy drug-induced apoptosis. Blood (2009) 114(17):3625–8. doi:10.1182/blood-2009-05-220285
17. Braga WM, da Silva BR, de Carvalho AC, Maekawa YH, Bortoluzzo AB, Rizzatti EG, et al. FOXP3 and CTLA4 overexpression in multiple myeloma bone marrow as a sign of accumulation of CD4(+) T regulatory cells. Cancer Immunol Immunother (2014) 63(11):1189–97. doi:10.1007/s00262-014-1589-9
18. Feng X, Zhang L, Acharya C, An G, Wen K, Qiu L, et al. Targeting CD38 suppresses induction and function of T regulatory cells to mitigate immunosuppression in multiple myeloma. Clin Cancer Res (2017) 23(15):4290–300. doi:10.1158/1078-0432.CCR-16-3192
19. Kawano Y, Zavidij O, Park J, Moschetta M, Kokubun K, Mouhieddine TH, et al. Blocking IFNAR1 inhibits multiple myeloma-driven Treg expansion and immunosuppression. J Clin Invest (2018) 128(6):2487–99. doi:10.1172/JCI88169
20. Leone P, Berardi S, Frassanito MA, Ria R, De Re V, Cicco S, et al. Dendritic cells accumulate in the bone marrow of myeloma patients where they protect tumor plasma cells from CD8+ T-cell killing. Blood (2015) 126(12):1443–51. doi:10.1182/blood-2015-01-623975
21. Chauhan D, Singh AV, Brahmandam M, Carrasco R, Bandi M, Hideshima T, et al. Functional interaction of plasmacytoid dendritic cells with multiple myeloma cells: a therapeutic target. Cancer Cell (2009) 16(4):309–23. doi:10.1016/j.ccr.2009.08.019
22. Gorgun GT, Whitehill G, Anderson JL, Hideshima T, Maguire C, Laubach J, et al. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood (2013) 121(15):2975–87. doi:10.1182/blood-2012-08-448548
23. Corre J, Mahtouk K, Attal M, Gadelorge M, Huynh A, Fleury-Cappellesso S, et al. Bone marrow mesenchymal stem cells are abnormal in multiple myeloma. Leukemia (2007) 21(5):1079–88. doi:10.1038/sj.leu.2404621
24. Urashima M, Ogata A, Chauhan D, Vidriales MB, Teoh G, Hoshi Y, et al. Interleukin-6 promotes multiple myeloma cell growth via phosphorylation of retinoblastoma protein. Blood (1996) 88(6):2219–27.
25. Urashima M, Ogata A, Chauhan D, Hatziyanni M, Vidriales MB, Dedera DA, et al. Transforming growth factor-beta1: differential effects on multiple myeloma versus normal B cells. Blood (1996) 87(5):1928–38.
26. Hayashi T, Hideshima T, Nguyen AN, Munoz O, Podar K, Hamasaki M, et al. Transforming growth factor beta receptor I kinase inhibitor down-regulates cytokine secretion and multiple myeloma cell growth in the bone marrow microenvironment. Clin Cancer Res (2004) 10(22):7540–6. doi:10.1158/1078-0432.CCR-04-0632
27. Abe M, Hiura K, Wilde J, Moriyama K, Hashimoto T, Ozaki S, et al. Role for macrophage inflammatory protein (MIP)-1alpha and MIP-1beta in the development of osteolytic lesions in multiple myeloma. Blood (2002) 100(6):2195–202.
28. Sprynski AC, Hose D, Caillot L, Reme T, Shaughnessy JD Jr, Barlogie B, et al. The role of IGF-1 as a major growth factor for myeloma cell lines and the prognostic relevance of the expression of its receptor. Blood (2009) 113(19):4614–26. doi:10.1182/blood-2008-07-170464
29. Le Gouill S, Podar K, Amiot M, Hideshima T, Chauhan D, Ishitsuka K, et al. VEGF induces Mcl-1 up-regulation and protects multiple myeloma cells against apoptosis. Blood (2004) 104(9):2886–92. doi:10.1182/blood-2004-05-1760
30. Derksen PW, de Gorter DJ, Meijer HP, Bende RJ, van Dijk M, Lokhorst HM, et al. The hepatocyte growth factor/Met pathway controls proliferation and apoptosis in multiple myeloma. Leukemia (2003) 17(4):764–74. doi:10.1038/sj.leu.2402875
31. Moreaux J, Legouffe E, Jourdan E, Quittet P, Reme T, Lugagne C, et al. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood (2004) 103(8):3148–57. doi:10.1182/blood-2003-06-1984
32. Tai YT, Li XF, Breitkreutz I, Song W, Neri P, Catley L, et al. Role of B-cell-activating factor in adhesion and growth of human multiple myeloma cells in the bone marrow microenvironment. Cancer Res (2006) 66(13):6675–82. doi:10.1158/0008-5472.CAN-06-0190
33. Tai YT, Acharya C, An G, Moschetta M, Zhong MY, Feng X, et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood (2016) 127(25):3225–36. doi:10.1182/blood-2016-01-691162
34. Kawano Y, Moschetta M, Manier S, Glavey S, Gorgun GT, Roccaro AM, et al. Targeting the bone marrow microenvironment in multiple myeloma. Immunol Rev (2015) 263(1):160–72. doi:10.1111/imr.12233
35. Braga WM, Atanackovic D, Colleoni GW. The role of regulatory T cells and TH17 cells in multiple myeloma. Clin Dev Immunol (2012) 2012:293479. doi:10.1155/2012/293479
36. Bryant C, Suen H, Brown R, Yang S, Favaloro J, Aklilu E, et al. Long-term survival in multiple myeloma is associated with a distinct immunological profile, which includes proliferative cytotoxic T-cell clones and a favourable Treg/Th17 balance. Blood Cancer J (2013) 3:e148. doi:10.1038/bcj.2013.34
37. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4(4):330–6. doi:10.1038/ni904
38. Joshua D, Suen H, Brown R, Bryant C, Ho PJ, Hart D, et al. The T cell in myeloma. Clin Lymphoma Myeloma Leuk (2016) 16(10):537–42. doi:10.1016/j.clml.2016.08.003
39. Muthu Raja KR, Rihova L, Zahradova L, Klincova M, Penka M, Hajek R. Increased T regulatory cells are associated with adverse clinical features and predict progression in multiple myeloma. PLoS One (2012) 7(10):e47077. doi:10.1371/journal.pone.0047077
40. Hoyos V, Borrello I. The immunotherapy era of myeloma: monoclonal antibodies, vaccines, and adoptive T-cell therapies. Blood (2016) 128(13):1679–87. doi:10.1182/blood-2016-05-636357
41. Carpenter RO, Evbuomwan MO, Pittaluga S, Rose JJ, Raffeld M, Yang S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res (2013) 19(8):2048–60. doi:10.1158/1078-0432.CCR-12-2422
42. Tai YT, Mayes PA, Acharya C, Zhong MY, Cea M, Cagnetta A, et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood (2014) 123(20):3128–38. doi:10.1182/blood-2013-10-535088
43. Madry C, Laabi Y, Callebaut I, Roussel J, Hatzoglou A, Le Coniat M, et al. The characterization of murine BCMA gene defines it as a new member of the tumor necrosis factor receptor superfamily. Int Immunol (1998) 10(11):1693–702. doi:10.1093/intimm/10.11.1693
44. Laabi Y, Gras MP, Brouet JC, Berger R, Larsen CJ, Tsapis A. The BCMA gene, preferentially expressed during B lymphoid maturation, is bidirectionally transcribed. Nucleic Acids Res (1994) 22(7):1147–54. doi:10.1093/nar/22.7.1147
45. Laabi Y, Gras MP, Carbonnel F, Brouet JC, Berger R, Larsen CJ, et al. A new gene, BCM, on chromosome 16 is fused to the interleukin 2 gene by a t(4;16)(q26;p13) translocation in a malignant T cell lymphoma. EMBO J (1992) 11(11):3897–904.
46. Mackay F, Schneider P, Rennert P, Browning J. BAFF AND APRIL: a tutorial on B cell survival. Annu Rev Immunol (2003) 21:231–64. doi:10.1146/annurev.immunol.21.120601.141152
47. Marsters SA, Yan M, Pitti RM, Haas PE, Dixit VM, Ashkenazi A. Interaction of the TNF homologues BLyS and APRIL with the TNF receptor homologues BCMA and TACI. Curr Biol (2000) 10(13):785–8. doi:10.1016/S0960-9822(00)00566-2
48. Gross JA, Johnston J, Mudri S, Enselman R, Dillon SR, Madden K, et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature (2000) 404(6781):995–9. doi:10.1038/35010115
49. Thompson JS, Schneider P, Kalled SL, Wang L, Lefevre EA, Cachero TG, et al. BAFF binds to the tumor necrosis factor receptor-like molecule B cell maturation antigen and is important for maintaining the peripheral B cell population. J Exp Med (2000) 192(1):129–35. doi:10.1084/jem.192.1.129
50. O’Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med (2004) 199(1):91–8. doi:10.1084/jem.20031330
51. Benson MJ, Dillon SR, Castigli E, Geha RS, Xu S, Lam KP, et al. Cutting edge: the dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J Immunol (2008) 180(6):3655–9. doi:10.4049/jimmunol.180.6.3655
52. Xu S, Lam KP. B-cell maturation protein, which binds the tumor necrosis factor family members BAFF and APRIL, is dispensable for humoral immune responses. Mol Cell Biol (2001) 21(12):4067–74. doi:10.1128/MCB.21.12.4067-4074.2001
53. Laurent SA, Hoffmann FS, Kuhn PH, Cheng Q, Chu Y, Schmidt-Supprian M, et al. gamma-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat Commun (2015) 6:7333. doi:10.1038/ncomms8333
54. Hatzoglou A, Roussel J, Bourgeade MF, Rogier E, Madry C, Inoue J, et al. TNF receptor family member BCMA (B cell maturation) associates with TNF receptor-associated factor (TRAF) 1, TRAF2, and TRAF3 and activates NF-kappa B, elk-1, c-Jun N-terminal kinase, and p38 mitogen-activated protein kinase. J Immunol (2000) 165(3):1322–30. doi:10.4049/jimmunol.165.3.1322
55. Deng S, Yuan T, Cheng X, Jian R, Jiang J. B-lymphocyte-induced maturation protein1 up-regulates the expression of B-cell maturation antigen in mouse plasma cells. Mol Biol Rep (2010) 37(8):3747–55. doi:10.1007/s11033-010-0028-z
56. Shaffer AL, Emre NC, Lamy L, Ngo VN, Wright G, Xiao W, et al. IRF4 addiction in multiple myeloma. Nature (2008) 454(7201):226–31. doi:10.1038/nature07064
57. Matthes T, Dunand-Sauthier I, Santiago-Raber ML, Krause KH, Donze O, Passweg J, et al. Production of the plasma-cell survival factor a proliferation-inducing ligand (APRIL) peaks in myeloid precursor cells from human bone marrow. Blood (2011) 118(7):1838–44. doi:10.1182/blood-2011-01-332940
58. Neri P, Kumar S, Fulciniti MT, Vallet S, Chhetri S, Mukherjee S, et al. Neutralizing B-cell activating factor antibody improves survival and inhibits osteoclastogenesis in a severe combined immunodeficient human multiple myeloma model. Clin Cancer Res (2007) 13(19):5903–9. doi:10.1158/1078-0432.CCR-07-0753
59. Shen X, Guo Y, Qi J, Shi W, Wu X, Ju S. Binding of B-cell maturation antigen to B-cell activating factor induces survival of multiple myeloma cells by activating Akt and JNK signaling pathways. Cell Biochem Funct (2016) 34(2):104–10. doi:10.1002/cbf.3169
60. Yang M, Hase H, Legarda-Addison D, Varughese L, Seed B, Ting AT. B cell maturation antigen, the receptor for a proliferation-inducing ligand and B cell-activating factor of the TNF family, induces antigen presentation in B cells. J Immunol (2005) 175(5):2814–24. doi:10.4049/jimmunol.175.5.2814
61. Avery DT, Kalled SL, Ellyard JI, Ambrose C, Bixler SA, Thien M, et al. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J Clin Invest (2003) 112(2):286–97. doi:10.1172/JCI18025
62. Novak AJ, Darce JR, Arendt BK, Harder B, Henderson K, Kindsvogel W, et al. Expression of BCMA, TACI, and BAFF-R in multiple myeloma: a mechanism for growth and survival. Blood (2004) 103(2):689–94. doi:10.1182/blood-2003-06-2043
63. Chiu A, Xu W, He B, Dillon SR, Gross JA, Sievers E, et al. Hodgkin lymphoma cells express TACI and BCMA receptors and generate survival and proliferation signals in response to BAFF and APRIL. Blood (2007) 109(2):729–39. doi:10.1182/blood-2006-04-015958
64. Lee L, Draper B, Chaplin N, Philip B, Chin M, Galas-Filipowicz D, et al. An APRIL based chimeric antigen receptor for dual targeting of BCMA and TACI in multiple myeloma. Blood (2018) 131(7):746–58. doi:10.1182/blood-2017-05-781351
65. Claudio JO, Masih-Khan E, Tang H, Goncalves J, Voralia M, Li ZH, et al. A molecular compendium of genes expressed in multiple myeloma. Blood (2002) 100(6):2175–86. doi:10.1182/blood-2002-01-0008
66. Seckinger A, Delgado JA, Moser S, Moreno L, Neuber B, Grab A, et al. Target expression, generation, preclinical activity, and pharmacokinetics of the BCMA-T cell bispecific antibody EM801 for multiple myeloma treatment. Cancer Cell (2017) 31(3):396–410. doi:10.1016/j.ccell.2017.02.002
67. Ghermezi M, Li M, Vardanyan S, Harutyunyan NM, Gottlieb J, Berenson A, et al. Serum B-cell maturation antigen: a novel biomarker to predict outcomes for multiple myeloma patients. Haematologica (2017) 102(4):785–95. doi:10.3324/haematol.2016.150896
68. Sanchez E, Li M, Kitto A, Li J, Wang CS, Kirk DT, et al. Serum B-cell maturation antigen is elevated in multiple myeloma and correlates with disease status and survival. Br J Haematol (2012) 158(6):727–38. doi:10.1111/j.1365-2141.2012.09241.x
69. Sanchez E, Smith EJ, Yashar MA, Patil S, Li M, Porter AL, et al. The role of B-cell maturation antigen in the biology and management of, and as a potential therapeutic target in, multiple myeloma. Target Oncol (2018) 13(1):39–47. doi:10.1007/s11523-017-0538-x
70. Bellucci R, Alyea EP, Chiaretti S, Wu CJ, Zorn E, Weller E, et al. Graft-versus-tumor response in patients with multiple myeloma is associated with antibody response to BCMA, a plasma-cell membrane receptor. Blood (2005) 105(10):3945–50. doi:10.1182/blood-2004-11-4463
71. Fragioudaki M, Boula A, Tsirakis G, Psarakis F, Spanoudakis M, Papadakis IS, et al. B cell-activating factor: its clinical significance in multiple myeloma patients. Ann Hematol (2012) 91(9):1413–8. doi:10.1007/s00277-012-1470-x
72. Day ES, Cachero TG, Qian F, Sun Y, Wen D, Pelletier M, et al. Selectivity of BAFF/BLyS and APRIL for binding to the TNF family receptors BAFFR/BR3 and BCMA. Biochemistry (2005) 44(6):1919–31. doi:10.1021/bi048227k
73. Wallweber HJ, Compaan DM, Starovasnik MA, Hymowitz SG. The crystal structure of a proliferation-inducing ligand, APRIL. J Mol Biol (2004) 343(2):283–90. doi:10.1016/j.jmb.2004.08.040
74. Moreaux J, Sprynski AC, Dillon SR, Mahtouk K, Jourdan M, Ythier A, et al. APRIL and TACI interact with syndecan-1 on the surface of multiple myeloma cells to form an essential survival loop. Eur J Haematol (2009) 83(2):119–29. doi:10.1111/j.1600-0609.2009.01262.x
75. Tai YT, Anderson KC. Targeting B-cell maturation antigen in multiple myeloma. Immunotherapy (2015) 7(11):1187–99. doi:10.2217/imt.15.77
76. Hengeveld PJ, Kersten MJ. B-cell activating factor in the pathophysiology of multiple myeloma: a target for therapy? Blood Cancer J (2015) 5:e282. doi:10.1038/bcj.2015.3
77. Matthes T, McKee T, Dunand-Sauthier I, Manfroi B, Park S, Passweg J, et al. Myelopoiesis dysregulation associated to sustained APRIL production in multiple myeloma-infiltrated bone marrow. Leukemia (2015) 29(9):1901–8. doi:10.1038/leu.2015.68
78. Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood (2016) 128(13):1688–700. doi:10.1182/blood-2016-04-711903
79. Cohen AD, Popat R, Trude S, Richardson PG, Libby IIIEN, Lendvai N, et al. First in human study with GSK2857916, an antibody drug conjugated to microtubule-disrupting agent directed against b-cell maturation antigen (BCMA) in patients with relapsed/refractory multiple myeloma. Blood (2016) 128(22):1148.
80. Trudel S, Lendvai N, Popat R, Voorhees PM, Reeves B, Libby EN, et al. Deep and durable responses in patients (Pts) with relapsed/refractory multiple myeloma (MM) treated with monotherapy GSK2857916, an antibody drug conjugate against B-cell maturation antigen (BCMA): preliminary results from Part 2 of Study BMA117159. Blood (2017) 130(Suppl 1):741.
81. Hechler T, Palfi A, Müller C, Lutz C, Pahl A, Kulke M. Preclinical evaluation of HDP-101, an anti-BCMA antibody-drug conjugate. Cancer Res (2017) 77(13 Suppl):77. doi:10.1158/1538-7445.AM2017-77
82. Ko J, Breunig C, Figueroa V, Lehners N, Baumann A, Pálfi A, et al. Preclinical evaluation of Hdp-101, a novel anti-Bcma antibody-drug conjugate, in multiple myeloma. Blood (2017) 130(Suppl 1):3070.
83. Kinneer K, Meekin J, Varkey R, Xiao X, Zhong H, Breen S, et al. Preclinical evaluation of MEDI2228, a BCMA-targeting pyrrolobenzodiazepine-linked antibody drug conjugate for the treatment of multiple myeloma. Blood (2017) 130(Suppl 1):3153.
84. Hipp S, Tai YT, Blanset D, Deegen P, Wahl J, Thomas O, et al. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. Leukemia (2017) 31(8):1743–51. doi:10.1038/leu.2016.388
85. Goyos A, Li CM, Deegen P, Bogner P, Thomas O, Matthias K, et al. Generation of half-life extended anti-BCMA Bite® antibody construct compatible with once-weekly dosing for treatment of multiple myeloma (MM). Blood (2017) 130:5389.
86. Brudno J, Lam N, Wang M, Stroncek D, Maric I, Stetler-Stevenson M, et al. T cells genetically modified to express an anti-B-cell maturation antigen chimeric antigen receptor with a CD28 costimulatory moiety cause remissions of poor-prognosis relapsed multiple myeloma. Blood (2017) 130(Suppl 1):524.
87. Berdeja JG, Lin Y, Raje NS, Siegel DSD, Munshi NC, Liedtke M, et al. First-in-human multicenter study of bb2121 anti-BCMA CAR T-cell therapy for relapsed/refractory multiple myeloma: updated results. J Clin Oncol (2017) 35(15 Supplal):3010.
88. Fan F, Zhao W, Liu J, He A, Chen Y, Cao X, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified t cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol (2017) 35(18 Supplal):LBA3001. doi:10.1200/JCO.2017.35.18_suppl.LBA3001
89. Dolgin E. CAR-Ts move beyond B-cell cancers to myeloma. Nat Biotechnol (2017) 35(7):599–601. doi:10.1038/nbt0717-599
90. Cohen AD, Garfall AL, Stadtmauer EA, Lacey SF, Lancaster E, Vogl DT, et al. B-cell maturation antigen (BCMA)-specific chimeric antigen receptor T cells (CART-BCMA) for multiple myeloma (MM): initial safety and efficacy from a Phase I Study. Blood (2016) 128(22):1147.
91. Cohen AD, Garfall AL, Stadtmauer EA, Lacey SF, Lancaster E, Vogl DT, et al. Safety and efficacy of B-cell maturation antigen (BCMA)-specific chimeric antigen receptor T cells (CART-BCMA) with cyclophosphamide conditioning for refractory multiple myeloma (MM). Blood (2017) 130(Suppl 1):505.
92. Adams GB, Feng J, Ghogha A, Mardiros A, Murakami J, Phung T, et al. Development of KITE-585: a fully human BCMA CAR T-cell therapy for the treatment of multiple myeloma. Cancer Res (2017) 77(13 Suppl):4979. doi:10.1158/1538-7445.AM2017-4979
93. Boldajipour B, Galetto R, Sommer C, Pertel T, Valton J, Park Y, et al. Preclinical evaluation of allogeneic anti-bcma chimeric antigen receptor t cells with safety switch domains and lymphodepletion resistance for the treatment of multiple myeloma. Blood (2016) 128(22):381.
94. Barnett BE, Wang X, Hermanson DL, Tan Y, Osertag EM, Shedlock DJ. Development of novel non-immunoglobulin centyrin-based cars (CARTyrins) targeting human Bcma. Blood (2016) 128(22):4557.
95. Hermanson DL, Barnett BE, Rengarajan S, Codde R, Wang X, Tan Y, et al. A novel BCMA-specific, centyrin-based CAR-T product for the treatment of multiple myeloma. Blood (2016) 128(22):2127.
96. Mathur R, Barnett BE, Hermanson D, He J, Zhang Z, Rengarajan S, et al. B-cell maturation antigen (BCMA)-specific, Centyrin™-based, PiggyBac™-transposed CAR-T memory stem cells are effective against p53–/– and patient-derived multiple myeloma tumors. Blood (2017) 130(Suppl 1):3068.
97. Lam N, Alabanza L, Trinklein N, Buelow B, Kochenderfer JN. T cells expressing anti-B-cell maturation antigen (BCMA) chimeric antigen receptors with antigen recognition domains made up of only single human heavy chain variable domains specifically recognize Bcma and eradicate tumors in mice. Blood (2017) 130(Suppl 1):504.
98. Lin L, Xing L, Acharya CM, Wen K, Liu J, Hsieh P, et al. CD8+ anti-Bcma mRNA CAR T-cells effectively kill human multiple myeloma cells in vitro and in vivo. Blood (2017) 130(Suppl 1):3067.
99. Wang X, Barnett BE, Martin C, Hermanson D, Li X, Smith J, et al. Production of universal anti-Bcma CAR-T cells with reduced alloreactivity, but potent effector function for the treatment of multiple myeloma. Blood (2017) 130(Suppl 1):503.
100. Panowski SH, Kuo T, Chen A, Geng T, Van Blarcom TJ, Lindquist K, et al. Preclinical evaluation of a potent anti-BCMA CD3 bispecific molecule for the treatment of multiple myeloma. Blood (2016) 128(22):383.
101. Moreno L, Zabaleta A, Alignani D, Lasa M, Maiso P, Jelinek T, et al. New insights into the mechanism of action (MoA) of first-in-class IgG-based Bcma T-cell bispecific antibody (TCB) for the treatment of multiple myeloma (MM). Blood (2016) 128(22):2096.
102. Pillarisetti K, Baldwin E, Babich A, Majewski N, Barone L, Li Y, et al. Development of a new BCMAxCD3 Duobody ® antibody for multiple myeloma. Blood (2016) 128(22):2116.
103. Gantke T, Reusch U, Ellwanger K, Fucek I, Weichel M, Treder M. AFM26 – a novel CD16a-directed bispecific TandAb targeting bcma for multiple myeloma. Cancer Res (2017) 77(13 Suppl):5671. doi:10.1158/1538-7445.AM2017-5671
104. Gantke T, Reusch U, Kellner C, Klausz K, Haneke T, Knackmuss S, et al. AFM26 – targeting B cell maturation antigen (BCMA) for NK cell-mediated immunotherapy of multiple myeloma. Blood (2017) 130(Suppl 1):3082.
105. Buelow B, Pham D, Choudhry P, Dang K, Pratap P, Clarke S, et al. T Cell engagement without cytokine storm: a novel Bcma x CD3 antibody killing myeloma cells with minimal cytokine secretion. Blood (2017) 130(Suppl 1):501.
106. Gantke T, Weichel M, Reusch U, Ellwanger K, Fucek I, Griep R, et al. Trispecific antibodies for selective CD16A-directed NK-cell engagement in multiple myeloma. Blood (2016) 128(22):4513.
107. McCombs JR, Owen SC. Antibody drug conjugates: design and selection of linker, payload and conjugation chemistry. AAPS J (2015) 17(2):339–51. doi:10.1208/s12248-014-9710-8
108. Rudd MD, Luse DS. Amanitin greatly reduces the rate of transcription by RNA polymerase II ternary complexes but fails to inhibit some transcript cleavage modes. J Biol Chem (1996) 271(35):21549–58. doi:10.1074/jbc.271.35.21549
109. Bhakta S, Raab H, Junutula JR. Engineering THIOMABs for site-specific conjugation of thiol-reactive linkers. Methods Mol Biol (2013) 1045:189–203. doi:10.1007/978-1-62703-541-5_11
110. Panowski S, Bhakta S, Raab H, Polakis P, Junutula JR. Site-specific antibody drug conjugates for cancer therapy. MAbs (2014) 6(1):34–45. doi:10.4161/mabs.27022
111. Baeuerle PA, Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res (2009) 69(12):4941–4. doi:10.1158/0008-5472.CAN-09-0547
112. Brischwein K, Parr L, Pflanz S, Volkland J, Lumsden J, Klinger M, et al. Strictly target cell-dependent activation of T cells by bispecific single-chain antibody constructs of the BiTE class. J Immunother (2007) 30(8):798–807. doi:10.1097/CJI.0b013e318156750c
113. Offner S, Hofmeister R, Romaniuk A, Kufer P, Baeuerle PA. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol Immunol (2006) 43(6):763–71. doi:10.1016/j.molimm.2005.03.007
114. Klein JS, Gnanapragasam PN, Galimidi RP, Foglesong CP, West AP Jr, Bjorkman PJ. Examination of the contributions of size and avidity to the neutralization mechanisms of the anti-HIV antibodies b12 and 4E10. Proc Natl Acad Sci U S A (2009) 106(18):7385–90. doi:10.1073/pnas.0811427106
115. Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer (2008) 8(4):299–308. doi:10.1038/nrc2355
116. Yan L, Shang J, Kang L, Shi X, Zhou J, Jin S, et al. Combined infusion of CD19 and Bcma-specific chimeric antigen receptor T cells for RRMM: initial safety and efficacy report from a Clinical Pilot Study. Blood (2017) 130(Suppl 1):506.
117. Guadagnoli M, Kimberley FC, Phan U, Cameron K, Vink PM, Rodermond H, et al. Development and characterization of APRIL antagonistic monoclonal antibodies for treatment of B-cell lymphomas. Blood (2011) 117(25):6856–65. doi:10.1182/blood-2011-01-330852
118. Dulos J, Driessen L, van Zandvoort P, van de Crommert J, Skoble J, Nair N, et al. Bion-1301, a first-in-class APRIL neutralizing antibody for the treatment of multiple myeloma: preclinical safety, and analysis of pharmacokinetics – pharmacodynamics relationship. Blood (2017) 130(Suppl 1):1827.
119. Ryan MC, Hering M, Peckham D, McDonagh CF, Brown L, Kim KM, et al. Antibody targeting of B-cell maturation antigen on malignant plasma cells. Mol Cancer Ther (2007) 6(11):3009–18. doi:10.1158/1535-7163.MCT-07-0464
120. Berdeja JG, Lin Y, Raje N, Munshi N, Siegel D, Liedtke M, et al. Durable clinical responses in heavily pretreated patients with relapsed/refractory multiple myeloma: updated results from a Multicenter Study of bb2121 Anti-Bcma CAR T cell therapy. Blood (2017) 130(Suppl 1):740.
121. Mi J-Q, Fan X, Xu J, Liu Y, Zhuang Y, Yang S, et al. Effective treatment of relapsed/refractory multiple myeloma including extramedullary involvement by BCMA-specific chimeric antigen receptor-modified T cells. Blood (2017) 130(Suppl 1):3115.
122. Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest (2016) 126(10):3814–26. doi:10.1172/JCI87366
123. Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res (2016) 4(6):498–508. doi:10.1158/2326-6066.CIR-15-0231
124. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Invest (2016) 126(8):3036–52. doi:10.1172/JCI83416
125. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood (2016) 127(26):3321–30. doi:10.1182/blood-2016-04-703751
126. Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov (2016) 6(6):664–79. doi:10.1158/2159-8290.CD-16-0040
127. Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood (2013) 121(26):5154–7. doi:10.1182/blood-2013-02-485623
128. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med (2014) 371(16):1507–17. doi:10.1056/NEJMoa1407222
129. Quintarelli C, Vera JF, Savoldo B, Giordano Attianese GM, Pule M, Foster AE, et al. Co-expression of cytokine and suicide genes to enhance the activity and safety of tumor-specific cytotoxic T lymphocytes. Blood (2007) 110(8):2793–802. doi:10.1182/blood-2007-02-072843
130. Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med (2011) 365(18):1673–83. doi:10.1056/NEJMoa1106152
131. Ramos CA, Asgari Z, Liu E, Yvon E, Heslop HE, Rooney CM, et al. An inducible caspase 9 suicide gene to improve the safety of mesenchymal stromal cell therapies. Stem Cells (2010) 28(6):1107–15. doi:10.1002/stem.433
132. Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med (2013) 5(215):215ra172. doi:10.1126/scitranslmed.3006597
133. Juillerat A, Marechal A, Filhol JM, Valton J, Duclert A, Poirot L, et al. Design of chimeric antigen receptors with integrated controllable transient functions. Sci Rep (2016) 6:18950. doi:10.1038/srep18950
134. Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science (2015) 350(6258):aab4077. doi:10.1126/science.aab4077
135. Murray BC, Peterson MT, Fecik RA. Chemistry and biology of tubulysins: antimitotic tetrapeptides with activity against drug resistant cancers. Nat Prod Rep (2015) 32(5):654–62. doi:10.1039/c4np00036f
136. Prota AE, Bargsten K, Diaz JF, Marsh M, Cuevas C, Liniger M, et al. A new tubulin-binding site and pharmacophore for microtubule-destabilizing anticancer drugs. Proc Natl Acad Sci U S A (2014) 111(38):13817–21. doi:10.1073/pnas.1408124111
137. Wang J, Xiao H, Qian ZG, Zhong JJ. Bioproduction of antibody-drug conjugate payload precursors by engineered cell factories. Trends Biotechnol (2017) 35(5):466–78. doi:10.1016/j.tibtech.2017.03.001
138. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov (2017) 16(5):315–37. doi:10.1038/nrd.2016.268
139. Gerber HP, Sapra P, Loganzo F, May C. Combining antibody-drug conjugates and immune-mediated cancer therapy: what to expect? Biochem Pharmacol (2016) 102:1–6. doi:10.1016/j.bcp.2015.12.008
140. Klinger M, Brandl C, Zugmaier G, Hijazi Y, Bargou RC, Topp MS, et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood (2012) 119(26):6226–33. doi:10.1182/blood-2012-01-400515
141. Topp MS, Kufer P, Gokbuget N, Goebeler M, Klinger M, Neumann S, et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol (2011) 29(18):2493–8. doi:10.1200/JCO.2010.32.7270
142. Teicher BA, Chari RV. Antibody conjugate therapeutics: challenges and potential. Clin Cancer Res (2011) 17(20):6389–97. doi:10.1158/1078-0432.CCR-11-1417
143. Suen H, Brown R, Yang S, Weatherburn C, Ho PJ, Woodland N, et al. Multiple myeloma causes clonal T-cell immunosenescence: identification of potential novel targets for promoting tumour immunity and implications for checkpoint blockade. Leukemia (2016) 30(8):1716–24. doi:10.1038/leu.2016.84
144. Zelle-Rieser C, Thangavadivel S, Biedermann R, Brunner A, Stoitzner P, Willenbacher E, et al. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J Hematol Oncol (2016) 9(1):116. doi:10.1186/s13045-016-0345-3
145. Maia S, Pelletier M, Ding J, Hsu YM, Sallan SE, Rao SP, et al. Aberrant expression of functional BAFF-system receptors by malignant B-cell precursors impacts leukemia cell survival. PLoS One (2011) 6(6):e20787. doi:10.1371/journal.pone.0020787
146. Elsawa SF, Novak AJ, Grote DM, Ziesmer SC, Witzig TE, Kyle RA, et al. B-lymphocyte stimulator (BLyS) stimulates immunoglobulin production and malignant B-cell growth in Waldenstrom macroglobulinemia. Blood (2006) 107(7):2882–8. doi:10.1182/blood-2005-09-3552
Keywords: multiple myeloma, B-cell maturation antigen, targeted immunotherapy, monoclonal antibody, chimeric antigen receptor T cell, monoclonal antibody drug conjugate, bi-specific antibody
Citation: Cho S-F, Anderson KC and Tai Y-T (2018) Targeting B Cell Maturation Antigen (BCMA) in Multiple Myeloma: Potential Uses of BCMA-Based Immunotherapy. Front. Immunol. 9:1821. doi: 10.3389/fimmu.2018.01821
Received: 04 June 2018; Accepted: 24 July 2018;
Published: 10 August 2018
Edited by:
Nicola Giuliani, Università degli Studi di Parma, ItalyReviewed by:
Dirk Hose, Universitätsklinikum Heidelberg, GermanyCatherine Pellat-Deceunynck, Centre national de la recherche scientifique (CNRS), France
Copyright: © 2018 Cho, Anderson and Tai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu-Tzu Tai, eXUtdHp1X3RhaUBkZmNpLmhhcnZhcmQuZWR1