 Georgina Bowyer1*
Georgina Bowyer1* Amy Grobbelaar1
Amy Grobbelaar1 Tommy Rampling1Navin Venkatraman1Danielle Morelle2Ripley W. Ballou2Adrian V. S. Hill1
Tommy Rampling1Navin Venkatraman1Danielle Morelle2Ripley W. Ballou2Adrian V. S. Hill1 Katie J. Ewer1
Katie J. Ewer1
- 1The Jenner Institute, University of Oxford, Oxford, United Kingdom
- 2GSK Vaccines, Rixensart, Belgium
A malaria vaccine strategy targeting multiple lifecycle stages may be required to achieve a high level of efficacy. In two Phase IIa clinical trials, we tested immunogenicity and efficacy of RTS,S/AS01B administered alone, in a staggered regimen with viral-vectored vaccines or co-administered with viral-vectored vaccines. RTS,S/AS01B induces high titers of antibody against sporozoites and viral-vectored vaccines ChAd63 ME-TRAP and MVA ME-TRAP induce potent T cell responses against infected hepatocytes. By combining these two strategies, we aimed to improve efficacy by inducing immune responses targeting multiple parasite antigens. Vaccination with RTS,S/AS01B alone or in a staggered regimen with viral vectors produced strong immune responses and demonstrated high levels of protection against controlled human malaria infection. However, concomitant administration of these vaccines significantly reduced humoral immunogenicity and protective efficacy. Strong Th1-biased cytokine responses induced by MVA ME-TRAP were associated with a skew in circulating T follicular helper cells toward a CXCR3+ phenotype and a reduction in antibody quantity and quality. This study illustrates that while a multistage-targeting vaccine strategy could provide high-level efficacy, the regimen design will require careful optimization.
Introduction
Despite years of remarkable success in reducing malaria morbidity and mortality, progress appears to have stalled with 216 million new cases in 2016, 5 million more than 2015 (1). An efficacious vaccine could be an essential tool to enable any further reduction in morbidity and mortality, and for the ultimate goal of eradication (2). The most advanced vaccine candidate, RTS,S, has shown significant short-term protective efficacy and has completed testing in a large Phase III trial (3–7). However, there remains a need to improve efficacy to achieve the goals laid out in the Malaria Vaccine Technology Roadmap (8). To improve efficacy, it may be necessary to develop a vaccine regimen targeting multiple stages of the parasite lifecycle (9). In addition to RTS,S, which targets the pre-liver stage, vaccines are being developed to target liver- and blood-stage parasites, or block parasite transmission and these could be combined into a multistage malaria vaccine program (9–12). It is likely that for a multistage vaccine regimen to provide high-level efficacy, it will need to induce both potent T cell and antibody responses (9).
The primary mechanism by which RTS,S induces protection appears to be antibody responses against the NANP repeat region of the circumsporozoite protein (CSP) on the sporozoite surface (13–16). The viral-vectored vaccines used in this study were chimpanzee adenovirus serotype 63 (ChAd63) and modified vaccinia virus Ankara (MVA), both expressing a multi-epitope (ME) string fused to the Plasmodium falciparum protein thrombospondin-related adhesive protein (TRAP). The ME string contains 17 epitopes from potentially protective P. falciparum and bacille Calmette–Guérin antigens in addition to epitopes from tetanus toxoid. TRAP is expressed on the surface of sporozoites and contains a thrombospondin domain that binds to heparin sulfate proteoglycans to facilitate sporozoite entry into host hepatocytes. ChAd63 ME-TRAP and MVA ME-TRAP provide protection by inducing CD8+ T cell responses against infected hepatocytes (17). When RTS,S/AS01B and viral-vectored vaccines were tested in a combination regimen, efficacy against controlled human malaria infection (CHMI) was higher for volunteers receiving the combination of vaccines (14/17 subjects protected; vaccine efficacy (VE) 82.4% [95% CI 64–100]) than for those receiving RTS,S/AS01B alone (12/16 subjects protected; VE 75% [95% CI 54–96]), suggesting that TRAP-specific T cell responses could add to the protective effect of RTS,S-induced antibody responses (18). In that study, NANP IgG titers in the combination group were comparable to those in the group given RTS,S/AS01B alone and were significantly higher in protected individuals. Titers of IgG against NANP were negatively correlated with parasitemia at day 7.5, indicating a reduced liver to blood inoculum. However, as viral-vectored vaccinations were given at staggered time points, a minimum of 2 weeks after RTS,S/AS01B, this regimen required five separate clinic visits over a period of 10 weeks. For a vaccine regimen to be logistically and economically feasible for deployment in malaria-endemic regions, the number of clinic visits should be reduced. For this reason, we conducted a further Phase I/IIa clinical trial to assess concomitant administration of RTS,S/AS01B with viral-vectored vaccines (Rampling et al. manuscript under review). On the basis of high efficacy in two previous trials, additional groups were included to test a reduced third dose of RTS,S/AS01B (1/5th, 10 μg) (14, 19). In this study, co-administration of these two vaccine platforms resulted in a significant reduction in humoral immunogenicity and efficacy with only 11/19 volunteers protected (VE 57.9% 95% CI [33.2–76.3]), compared with 14/17 (82.4% 95% CI [54.7–93.9]) in the group receiving RTS,S/AS01B alone.
Durable, high-affinity IgG is generated in germinal center (GC) reactions in secondary lymphoid organs, during which B cells undergo class-switching, somatic hypermutation, and differentiation into memory B cells and plasma cells. T follicular helper cells (Tfh) expressing CD4 and CXCR5 and the transcription factor Bcl-6 provide essential help to B cells for this process in the form of cytokine production (IL-21) and the expression of costimulatory molecules (CD40L, ICOS) (20, 21). Circulating PD1+CXCR5+CD45RA− CD4+ T cells appear to be a peripheral counterpart of conventional lymphoid resident Tfh, may represent GC responses, and are a useful tool for clinical trials in which lymphoid tissue is rarely available for analysis (22–24). Circulating T follicular helper cells (cTfh) can be further defined by differential expression of chemokine receptors CXCR3 and CCR6: Th17-like (cTfh17) CXCR3−CCR6+, double-positive CXCR3+CCR6+, Th1-like (cTfh1) CXCR3+CCR6−, and Th2-like (cTfh2) CXCR3−CCR6−. These subsets have been associated with varying degrees of helper activity in different contexts (25). In particular, CXCR3− Tfh have been associated with the production of broadly neutralizing antibodies against HIV and Tfh17 induced by rVSV-ZEBOV vaccination were associated with antibody responses against Ebola (26, 27). Therefore, the type of cTfh induced by vaccination may be an indicator of the quality of the GC reaction and the resulting antibodies produced.
To investigate the mechanisms underlying the reduction in humoral immunogenicity after co-administration of ChAd63-MVA ME-TRAP and RTS,S, we conducted a thorough analysis of the differences in antibody quality and cTfh responses in volunteers receiving RTS,S/AS01B alone (R), RTS,S/AS01B given with viral vectors in a staggered regimen (R2V) or co-administered (R + V). Trial regimens are summarized in Table 1. This is the first study to assess the impact of vaccine co-administration on the cTfh response in humans and also defines a functional antibody quality that may explain the improved efficacy observed in RTS,S regimens with a reduced third dose.
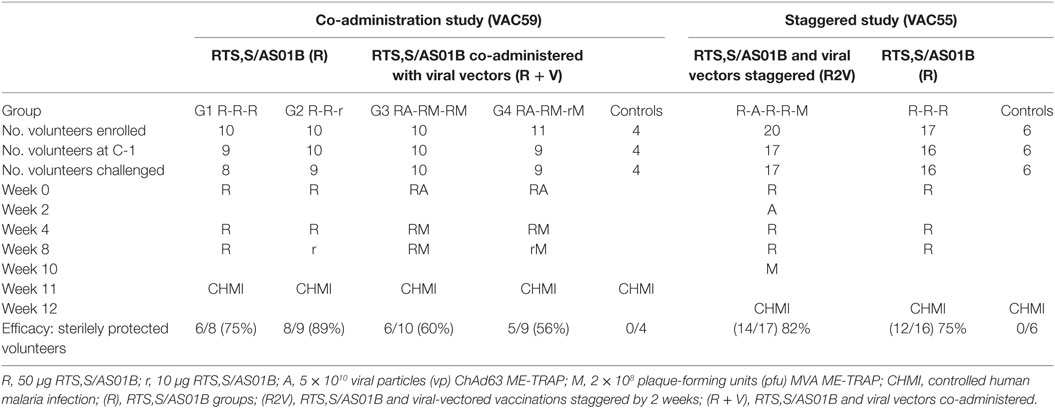
Table 1. Vaccination schedules.
Materials and Methods
Samples and Study Details
Full details of these studies are available in the clinical trial reports [(18), Rampling et al. manuscript under review]. Healthy adult volunteers were recruited and vaccinated at four UK sites, in Oxford, Southampton, London, and Surrey. The CHMI procedure was performed as previously described using five infectious bites from P. falciparum 3D7-strain infected Anopheles stephensi mosquitoes at Imperial College, London (28). All subjects were infected with a single batch of infected mosquitoes for each trial, supplied by the Department of Entomology, Walter Reed Army Institute of Research, Washington DC, USA. All vaccinations were administered intramuscularly into the deltoid region of the arm. For participants who received concomitant vaccinations, RTS,S/AS01B was administered first followed by the viral-vectored vaccine in the same site no longer than 5 min later.
Ethics Statement
All volunteers gave written informed consent prior to participation, and the studies were conducted according to the principles of the Declaration of Helsinki and in accordance with Good Clinical Practice. The trials were registered with ClinicalTrials.gov (Ref: NCT01883609 and NCT02252640). The study protocols were approved by the UK National Research Ethics Service, Committee South Central—Oxford A (Refs: 13/SC/0208 and 14/SC/0227), the Western Institution Review Board (Ref: 20130698), and the UK Medicines and Healthcare products Regulatory Agency (Refs: 21584/0317/001-0001 and 21584/0333/001-0001). The Local Safety Committee provided safety oversight for both trials and GCP compliance was monitored by the Clinical Trials and Research Governance Team (CTRG) of the University of Oxford.
Total IgG ELISA
ELISA 96-well plates were coated with a synthetic peptide (Eurogentec, Liège, Belgium) based on the PfCSP repeat region ((NANP)6C) diluted to 0.2 µg/mL in 100 µL dPBS per well and incubated overnight at room temperature (RT). Plates were washed six times with PBS containing 0.5% Tween-20 (PBS/T) and blocked with casein for 1 h at RT. Plates were washed again and serum samples diluted in casein at 1:100, 1:500, 1:1,000, or 1:5,000, were added for 2 h at RT. After washing again, secondary antibody (goat anti-human IgG conjugated to alkaline phosphatase, Sigma) was added at 1:1,000 in casein for 1 h at RT. Plates were washed a final time and developed using 4-nitrophenyl phosphate in diethanolamine buffer (Pierce, Rockford, IL, USA). Optical density (OD) was read at 405 nm using an ELx800 microplate reader (Bio-Rad, Hercules, CA, USA). A reference pool of positive serum formed a standard curve on each plate and was used to calculate ELISA units for each sample. An internal control was included on each plate to standardize between assays. All samples were tested in triplicate.
Isotype ELISA
Isotype ELISAs were conducted as described above, except that all serum samples were diluted to 1:100 and added to the plate in duplicate wells on each of six plates. One of six secondary antibodies against IgG1, IgG2, IgG3, IgG4, IgM, or IgA was added to each plate at 1:1,000 in casein before developing as above. Blank wells and internal development controls were included on each plate. A “seropositive cut-off” value was calculated for each isotype or subclass using the mean plus 3 SDs of 36 UK malaria-naïve serum samples.
Indirect Immunofluorescence Assay (IFA)
Chambered microscope slides coated with P. falciparum sporozoites were stored at −80°C until use. Slides were brought to RT and then fixed for 15 min in 4% paraformaldehyde. After washing twice in PBS for 5 min, slides were blocked for 1 h in casein. Slides were washed as before and 10 µL of serum sample diluted 1:100 in casein was added to each well. Slides were incubated for 30 min at RT in a humidity chamber then wells were individually washed with PBS three times for 5 min. Secondary antibody (anti-IgG-AlexaFluor488) was diluted 1:800 in casein and 15 µL was added to each well for 30–45 min in a humidity chamber at RT protected from the light. Slides were washed a final time, rinsed in distilled water and left to dry before mounting with DAPI-containing media. Slides were left to set overnight at 4°C before being examined under a Leica DMI3000 B microscope. Images were captured in QCapturePro software using brightfield illumination, GFP and DAPI filters at set exposure levels. ImageJ software was used to measure the median fluorescence intensity (MFI) for five sporozoites in each well and an average was taken.
Inhibition of Sporozoite Invasion (ISI) Assay
The ISI assay was carried out as previously described (29). Human hepatoma cells (HC04) cultured in R10 medium (RPMI 1640 with 10% FCS, 1% penicillin/streptomycin, and 1% l-glutamine) were added to 96-well culture plates at 30,000 cells/well and left to settle overnight at 37°C, 5% CO2. Viable GFP-labeled Plasmodium berghei sporozoites expressing P. falciparum CSP at the P. berghei CSP locus (P. berghei PfCSP@CSP) were obtained by dissecting infected A. stephensi mosquitoes. Salivary glands were pooled into RPMI 1640 medium and homogenized. Sporozoites were counted and diluted to 100,000/mL in RPMI 1640. Culture medium was aspirated from the hepatoma cells then 100 µL of serum diluted 1:5 in R10 and 100 µL of sporozoite dilution (10,000 sporozoites, 10% final serum concentration) were added to each well. Samples were tested in duplicate and an average calculated. “Hepatoma only” wells and infectivity control wells that contained hepatoma cells and sporozoites but no serum were included. Pre-vaccination and C-1 samples were run for each volunteer. After incubation for 20–26 h at 37°C, medium was aspirated, and plates were washed with 90 µL/well dPBS. Cells were trypsinized, re-suspended in 65 µL dPBS with 1% bovine serum albumin (BSA), and acquired immediately using a BD LSRII. DAPI stain was added to each sample just before acquisition. Data were analyzed in Flow Jo software v10.6 (Tree Star Inc., Ashland, OR, USA) according to a predefined gating strategy. The percentage of sporozoite inhibition was calculated for each sample (average of duplicate wells) based on the reduction in the percentage of infected cells compared with the infectivity controls (average of 4–6 wells).
Ex Vivo IFNγ ELISpot
Ex vivo ELISpot assays were performed for TRAP-specific T cell responses as previously described (10). Average responses were taken across triplicate wells, background subtracted and then responses in individual pools were summed.
cTfh Phenotyping and ICS
Surface phenotyping of cTfh was carried out using cryopreserved peripheral blood mononucleocytes (PBMCs). Thawing was performed rapidly in a water bath and cells were rested for 2 h at 37°C with 5% CO2 and Benzonase at 25U/106 PBMC (Novagen, Madison, WI, USA) before staining. For surface phenotyping 1–2 million PBMC were stained. Cells were washed in FACS buffer (PBS containing 0.1% BSA and 0.01% sodium azide) and stained with LIVE/DEAD aqua amine reactive dye (Life Technologies Ltd., Carlsbad, CA, USA) for 20 min at RT in the dark. Cells were washed in FACS buffer and a cocktail of antibodies for Tfh surface staining (Table 2) was added for 30 min at RT. Cells were washed again in FACS Buffer and re-suspended in PBS containing 1% paraformaldehyde, prior to acquisition on a BD LSR II using FACSDiva v6.2 (BD Biosciences, Franklin Lakes, NJ, USA) on the day of staining. Compensation control beads (OneComp Beads, eBioscience, San Diego, CA, USA, ArC Amine Reactive Beads, Invitrogen, Carlsbad, CA, USA) were stained according to the manufacturer’s instructions for compensation between parameters. A median of 100,000 live CD4+ cells were acquired [inter-quartile range (IQR) 25% = 68,406, 75% = 143,000] per sample. Data analysis was performed using Flow Jo v9.6.2 (Tree Star Inc.). IFNγ production was measured after overnight stimulation with 2 µg/mL of a pool of 31 peptides spanning the CSP antigen (15mers overlapping by 11 amino acids, at 2 µg/mL, all volunteers) or 10 µg/mL superantigen Staphylococcus enterotoxin B (SEB). Brefeldin A and monensin were added at 10 µg/mL after 2 h. The staining protocol was the same as for cTfh phenotyping, except that after surface staining, cells were permeabilized with fix/perm buffer (BD biosciences) then stained intracellularly at RT for 30 min with IFNγ-FITC (1:250, eBioscience), washed and fixed in 1% paraformaldehyde. Acquisition and analysis was performed as for cTfh phenotyping.
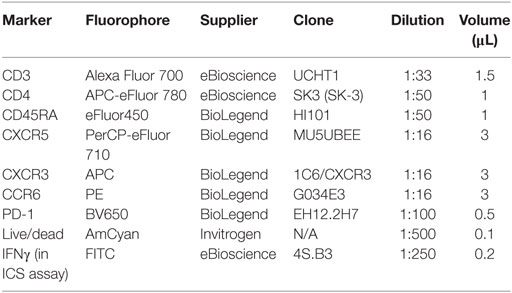
Table 2. Circulating T follicular helper cell phenotyping panel.
Multiplex Cytokine Assay
Between 1 and 2 million PBMC from the C-1 time point were plated per well in a 96-well plate and stimulated for 21 h at 37°C. Cells were stimulated either with a pool of 31 peptides spanning the CSP antigen (15mers overlapping by 11 amino acids, at 2 µg/mL, all volunteers) or 106 pfu of MVA. Supernatants were taken and stored at −20°C in 96-well U-bottom polypropylene plates until use. Cytokine concentrations in the supernatants were measured using the LEGENDplex human Th cytokine panel 13-plex assay (BioLegend, San Diego, CA, USA) according to the manufacturer’s instructions. The samples were read on the same day on a BD LSR II using FACSDiva v6.2 (BD Biosciences) with 5,000 beads acquired per sample. Data analysis was conducted using the LEGENDplex data analysis software.
Statistical Analysis
Data tested negative for a normal distribution by D’Agostina–Pearson omnibus normality test; therefore, non-parametric tests were used and medians with IQRs are presented. Mann–Whitney analysis was used to compare differences between two groups. Kruskal–Wallis with Dunn’s multiple comparisons post-test was used to compare responses across multiple groups at a given time point. Wilcoxon matched-pairs analysis was used to compare responses at two time points within a group. Spearman’s rank was calculated for correlations. P < 0.05 was considered significant and all P values are two-tailed. Analyses were performed in GraphPad Prism, version 7.
Results
Reduced Quantity and Quality of Antibody Responses When RTS,S/AS01B Is Co-Administered With Viral-Vectored Vaccines
Antibody responses against the NANP repeat region of CSP were measured in each of the clinical trials and reported separately (18) (Rampling et al. manuscript under review). Total NANP-specific IgG was measured by ELISA at baseline (day 0, D0), D28, D42, D56, D76 (the day before CHMI, C-1, in the co-administration trial) and 35 and 90 days after CHMI (C + 35, C + 90). In the staggered regimen trial, anti-NANP-specific IgG was also measured at D83 (C-1 for that trial) as the CHMI was 1 week later to accommodate the additional vaccinations (Figure 1A). Titers were comparable in all regimens after the first two vaccinations but failed to re-boost after the third vaccination in the co-administration regimen, resulting in significantly lower titers in these groups at D76 (Figure 1B, median ELISA units, R: 1,102 IQR [757–2,035], R + V: 533 IQR [394–790], R2V: 1,969 IQR [983–2,724] Kruskal–Wallis P < 0.0001). There were no significant differences in NANP IgG titers between groups receiving a full or reduced third dose of RTS,S/AS01B either alone or co-administered with viral-vectored vaccines. The highest titers were seen in the staggered regimen at D83 (4 weeks after the third dose of RTS,S/AS01B), although there was no comparable time point in the co-administration study.
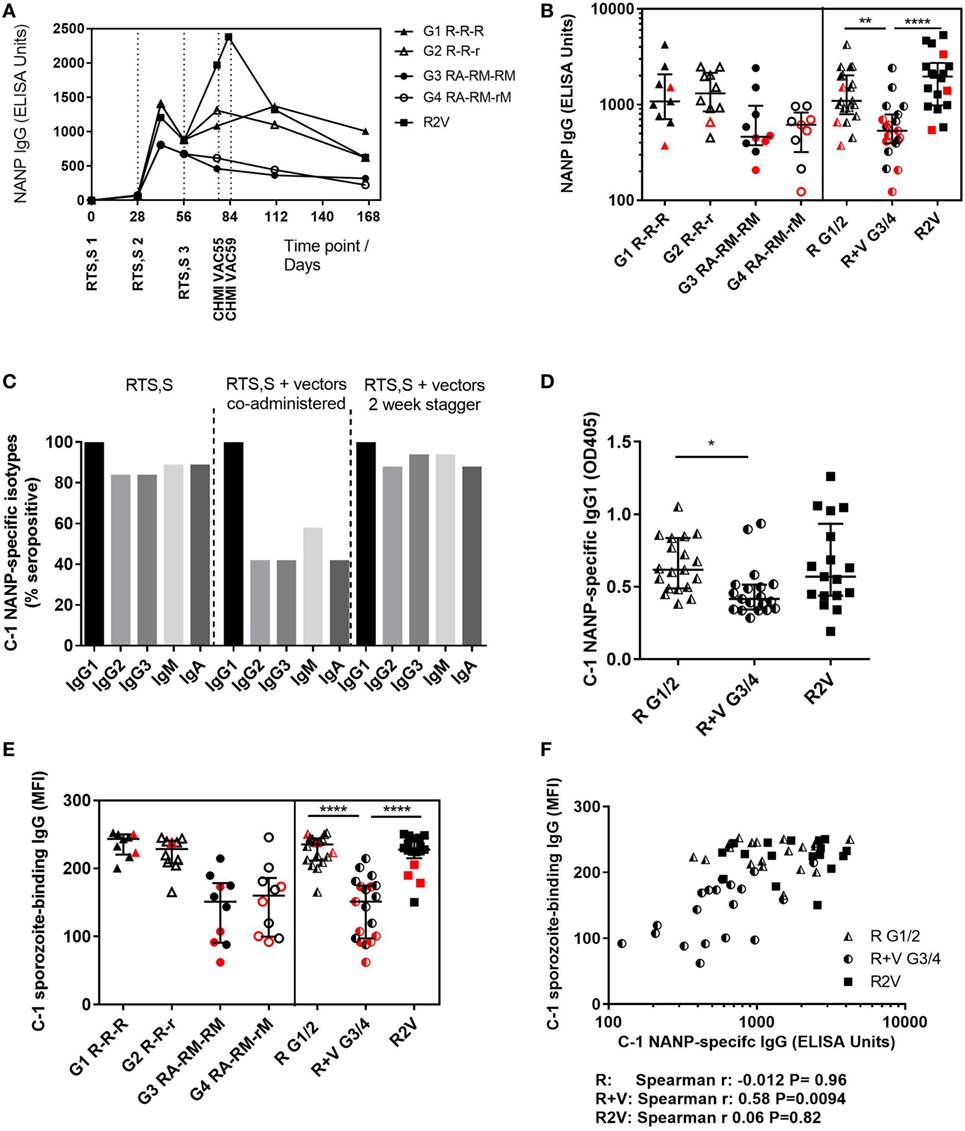
Figure 1. Antibody quantity and quality. (A) Median NANP-specific IgG time courses. (B) Total IgG titers against the CSP repeat region NANP in each group at D76 (2 weeks after third dose of RTS,S/AS01B), in G1/2 combined (R) and G3/4 combined (R + V) or in the staggered regimen (R2V), Kruskal–Wallis with Dunn’s post-test P < 0.0001. (C) NANP-specific isotype and IgG subclass responses at C-1. (D) NANP-specific IgG1 at C-1, Kruskal–Wallis P = 0.0062. (E) MFI of sporozoite-binding IgG at C-1 measured by IFA, Kruskal–Wallis P < 0.0001. (F) Relationship between NANP IgG titer and level of sporozoite-binding at C-1. Volunteers receiving RTS,S/AS01B alone (G1/2, R) Spearman r: −0.012, P = 0.96, RTS,S/AS01B co-administered with vectors (G3/4, R + V) Spearman r: 0.58, P = 0.0094 or RTS,S/AS01B and viral vectors given in a staggered regimen Spearman r: 0.06, P = 0.82. Medians + IQRs shown and non-protected volunteers highlighted in red for all column graphs. Abbreviations: R G1/2, RTS,S/AS01B vaccinated; R + V G3/4, RTS,S/AS01B and viral-vectored vaccines co-administered; R2V, RTS,S/AS01B and viral-vectored vaccines at a 2-week stagger; A, ChAd63 ME-TRAP; M, MVA ME-TRAP; R, 50 µg third dose of RTS,S/AS01B; r, 10 µg third dose of RTS,S/AS01B; CHMI, controlled human malaria infection; C-1, day before CHMI; CSP, circumsporozoite protein; TRAP, thrombospondin-related adhesive protein; IQRs, inter-quartile ranges; IFA, immunofluorescence assay; MFI, median fluorescence intensity.
Isotype and subclass responses were measured by ELISA against the NANP repeat region at C-1 in both trials (Figure 1C). Titers were measured for IgG1–4, IgM, and IgA. No NANP-specific IgG4 was detected in any volunteers (data not shown). Over 80% of volunteers given RTS,S/AS01B alone were seropositive for NANP-specific IgG2, IgG3, IgM, and IgA. There were significant reductions in seroconversion for these isotypes/subclasses in groups that received concomitant viral-vectored vaccinations, but not in the staggered regimen. All volunteers were positive for NANP IgG1 and titers were comparable in the RTS,S/AS01B only groups (R) and the staggered administration group (R2V), but were significantly reduced in the co-administration regimen (R + V) (Figure 1D, median OD, R: 0.617 IQR [0.488–0.835], R + V: 0.415 [0.343–0.514], R2V: 0.570 [0.438–0.935] Kruskal–Wallis P = 0.0062). There were no significant differences in IgG1 titers between groups receiving full or reduced third doses of RTS,S/AS01B (G1 R-R-R vs G2 R-R-r and G3 RA-RM-RM vs G4 RA-RM-rM, data not shown).
Antibody binding to fixed whole sporozoites was measured for all volunteers at C-1 using an indirect IFA. Sporozoite-binding IgG was significantly lower in the co-administration regimen but the staggered regimen was comparable to RTS,S/AS01B alone (Figure 1E, MFI, R: 238 IQR [213–245] R + V: 151 [98–175], R2V: 228 [215–245] Kruskal–Wallis P < 0.0001). There were no differences between groups receiving full or reduced third doses of RTS,S/AS01B. Sporozoite-binding was significantly associated with NANP IgG titers in the co-administration groups (Figure 1F, Spearman r: 0.58, P = 0.0094). There was no association in the RTS,S/AS01B alone or staggered regimens (Spearman r: −0.012, P = 0.96 and r: 0.058, P = 0.83, respectively), in which sporozoite-binding was higher for a given NANP IgG titer than in the co-administration regimen.
ISI Is Associated With Protection From CHMI
The functional quality of vaccine-induced antibodies was assessed using an in vitro assay measuring the ability of serum to block sporozoite infection of hepatoma cells (29). A defined gating strategy was used to identify infected hepatoma cells (Figure 2). The percentage of infection blocked by vaccine-induced antibody was significantly lower in the co-administration regimen compared with RTS,S/AS01B alone (Figure 3A, median percentage infection blocked R, 90% IQR [76–98], R + V 80% [52–89], Mann–Whitney P = 0.016). However, blocking ability was significantly higher in groups receiving a reduced third dose of RTS,S/AS01B than those receiving three standard doses, even when this dose was co-administered with viral-vectored vaccines (Figure 3A, medians + IQRs G1 R-R-R: 80% [71.5–91.5], G2 R-R-r: 96% [89.3–98], G3 RA-RM-RM: 55% [50–73.5], G4 RA-RM-rM: 89% [85–95], Mann–Whitney analyses G1 R-R-R vs G2 R-R-r P = 0.014, G3 RA-RM-RM vs G4 RA-RM-rM P < 0.0001, G1 R-R-R and G3 RA-RM-RM vs G2 R-R-r and G4 RA-RM-rM P < 0.0001). ISI was significantly associated with C-1 NANP IgG titer for groups receiving three standard doses of RTS,S/AS01B (Figure 3B, G1 R-R-R and G3 RA-RM-RM Spearman r: 0.78, P < 0.0001) but not in groups receiving a reduced third dose, in which blocking ability was higher even at lower NANP titers (G2 R-R-r and G4 RA-RM-rM Spearman r: 0.44, P = 0.061). The relationship between blocking ability and the NANP-specific isotype/subclass titers was also assessed (Figure S1 in Supplementary Material). Relationships between all subclasses and isotypes tested and blocking ability demonstrated a similar pattern to total IgG, with a positive correlation in G1/3 and high blocking regardless of titer in G2/4. However, IgG1 was the only isotype that showed an association with blocking ability in the data set as a whole (G1–4, Spearman r: 0.42, P = 0.009).
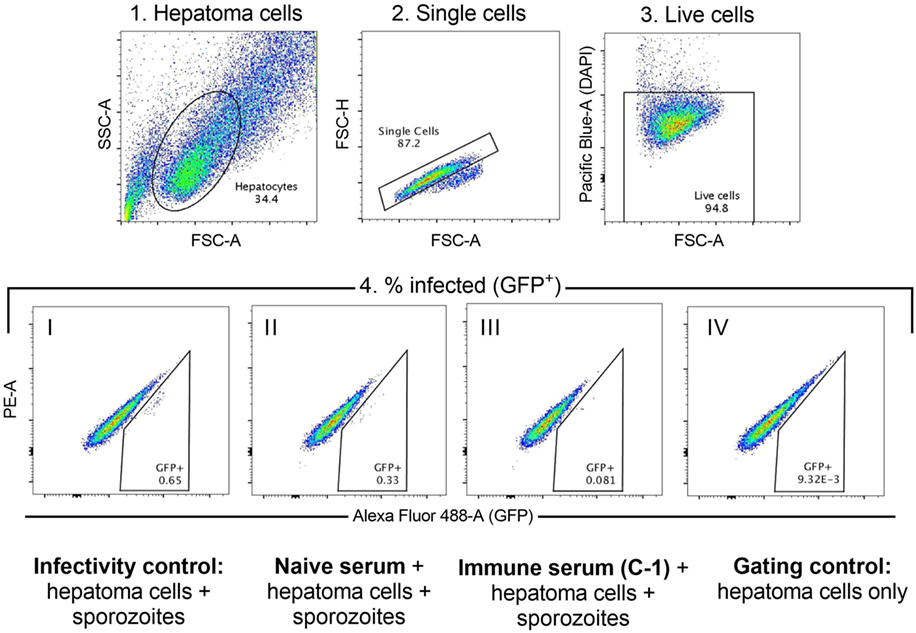
Figure 2. Inhibition of sporozoite invasion assay. Hierarchical gating strategy used to determine the percentage of hepatoma cells infected with GFP-expressing sporozoites. 1. Hepatoma cells are gated based on size to exclude debris and large cell clumps. 2. Singlets are gated to exclude smaller cell clumps. 3. Live cells (DAPI-negative) are gated to exclude dead cells. 4. Cells infected with the GFP-expressing sporozoites are GFP+. Cells are gated against both GFP (Alexa Fluor-AF488 channel, GFP+) and the adjacent channel (PE−) to exclude signal that is caused by autofluorescence. Representative populations in each of the conditions are shown: I. Infectivity control—sporozoite infection of hepatoma cells in the absence of any serum; II. Sporozoite infection of hepatoma cells in the presence of naïve serum (D0); III. Sporozoite infection of hepatoma cells in the presence of immune serum (C-1); IV. Gating control—hepatoma cells only, no sporozoites or serum added.
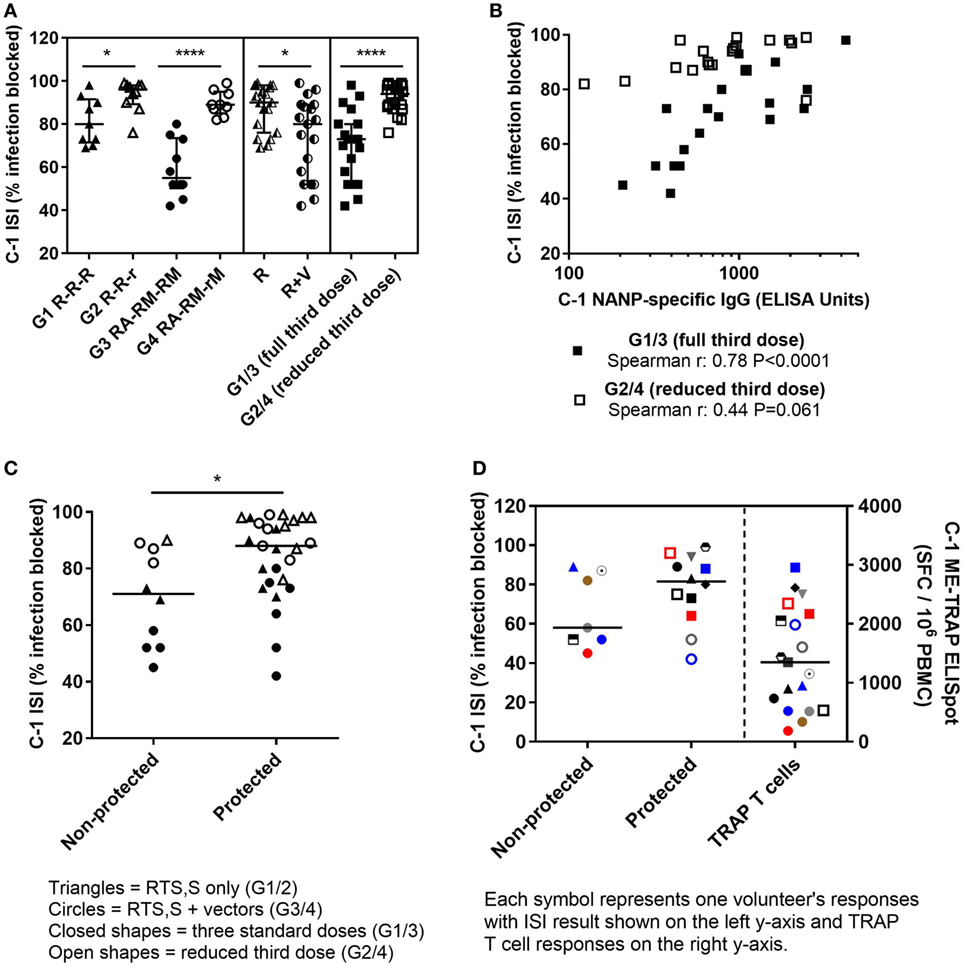
Figure 3. ISI results. (A) Percentage of sporozoite invasion into hepatoma cells that was blocked by serum at C-1. Comparison of groups given three standard doses of RTS,S/AS01B or a reduced (1/5th) third dose of RTS,S/AS01B. Mann–Whitney analyses G1 R-R-R vs G2 R-R-r P = 0.014, G3 RA-RM-RM vs G4 RA-RM-rM P < 0.0001, G1/2 (R) vs G3/4 (R + V) P = 0.016, G1/3 (full third dose of RTS,S/AS01B) vs G2/4 (reduced third dose of RTS,S/AS01B) P < 0.0001, medians + IQRs. (B) Relationship between C-1 NANP IgG titers and sporozoite-blocking ability in individuals who received three full doses of RTS,S/AS01B with or without vectored vaccines (G1 and G3, closed squares, Spearman r: 0.78, P < 0.0001) or a reduced third dose of RTS,S/AS01B with, or without vectored vaccines (G2 and G4, open squares, Spearman r: 0.44, P = 0.061). (C) Percentage of sporozoite invasion blocked by C-1 serum protected and non-protected individuals, Mann–Whitney P = 0.017. Closed triangles: G1, open triangles: G2, closed circles: G3, open circles: G4. (D) Percentage of sporozoite invasion blocked by serum at C-1 in protected and non-protected volunteers in G3/4, R + V (left Y-axis) and corresponding TRAP T cell responses (right Y-axis) at C-1 for each volunteer measured by IFNγ ELISpot, summed responses from T9/96 TRAP pools + ME, spot-forming cells per million PBMC (SFC/106 PBMC), lines at medians. Abbreviations: R G1/2, RTS,S/AS01B vaccinated; R + V G3/4, RTS,S/AS01B and viral-vectored vaccines co-administered; R2V, RTS,S/AS01B and viral-vectored vaccines at a 2-week stagger; A, ChAd63 ME-TRAP; M, MVA ME-TRAP; R, 50 µg third dose of RTS,S/AS01B; r, 10 µg third dose of RTS,S/AS01B; CHMI, controlled human malaria infection; C-1: day before CHMI; ISI, inhibition of sporozoite invasion; PBMCs, peripheral blood mononucleocytes; TRAP, thrombospondin-related adhesive protein; IQRs, inter-quartile ranges.
Blocking ability was associated with protection from malaria after CHMI, with significantly higher percentages of infection blocked in protected than non-protected volunteers (Figure 3C, medians + IQRs protected: 88% [75–97], non-protected: 71% [52–87.5] Mann–Whitney P = 0.019). TRAP-specific T cell responses elicited by the viral-vectored vaccines were measured by IFNγ ELISpot and previously reported (Rampling et al. manuscript under review). T cell responses (IFNγ responses against summed T9/96 TRAP pools + the multi-epitope, ME) were not reduced by co-administration and responses were significantly higher in protected than non-protected individuals in these groups (Rampling et al. manuscript under review). Volunteers in the co-administration groups that were protected despite having low levels of sporozoite-blocking antibody had high TRAP-specific T cell responses (Figure 3D).
Proportion of CXCR3+ cTfh Increases When RTS,S/AS01B Is Co-Administered With Viral-Vectored Vaccines and Negatively Correlates With Antibody Responses
To determine whether cellular differences associated with the reduction in antibody responses in the co-administration regimen could be detected in the blood, cTfh were phenotyped at C-1 by surface staining and flow cytometry using a defined gating strategy (Figure 4A). Total cTfh were analyzed for all volunteers in the co-administration study (except 1 volunteer in G4 for which there were no cryopreserved cells remaining) and 10 volunteers in the staggered administration study with enough cryopreserved cells remaining for the experiment (Figure 4B). The proportion of cTfh (PD1+CXCR5+) within memory CD4+ T cells ranged from 0.1 to 4.8%, was comparable across groups and did not correlate with CSP- or TRAP-specific antibody responses. Subsets within cTfh were identified using CXCR3 and CCR6: cTfh17 (CXCR3−CCR6+), CXCR3+ [including double-positive (CXCR3+CCR6+) and cTfh1 (CXCR3+CCR6−)] and cTfh2 (CXCR3−CCR6−). Volunteers who received RTS,S/AS01B co-administered with viral-vectored vaccines had significantly higher frequencies of CXCR3+ cTfh and significantly lower frequencies of cTfh2 than those who received RTS,S/AS01B alone, while the staggered administration group had an intermediate phenotype which was not significantly different to either of the other regimens (Figure 4C). There were no significant differences in frequencies of any population between G1 R-R-R and G2 R-R-r or G3 RA-RM-RM and G4 RA-RM-rM (data not shown). In the co-administration regimen, the percentage of CXCR3+ cTfh was negatively correlated with antibody responses to both vaccines (Figure 4D, NANP Spearman r: −0.78, P = 0.0001, TRAP Spearman r: −0.50, P = 0.036). For groups that received RTS,S/AS01B alone, the frequency of CXCR3+ cTfh was lower, and there was no association with antibody responses (Figure 4E, Spearman r: 0.32, P = 0.18). Although in the staggered regimen the proportion of CXCR3+ cTfh was comparable to that in the co-administration regimen, they were not associated with a reduction in antibody responses in this regimen (Spearman r: 0.26, P = 0.47). The proportion of CXCR3+ in CXCR5− memory CD4+ T cells was not associated with antibody responses in any regimen (data not shown).
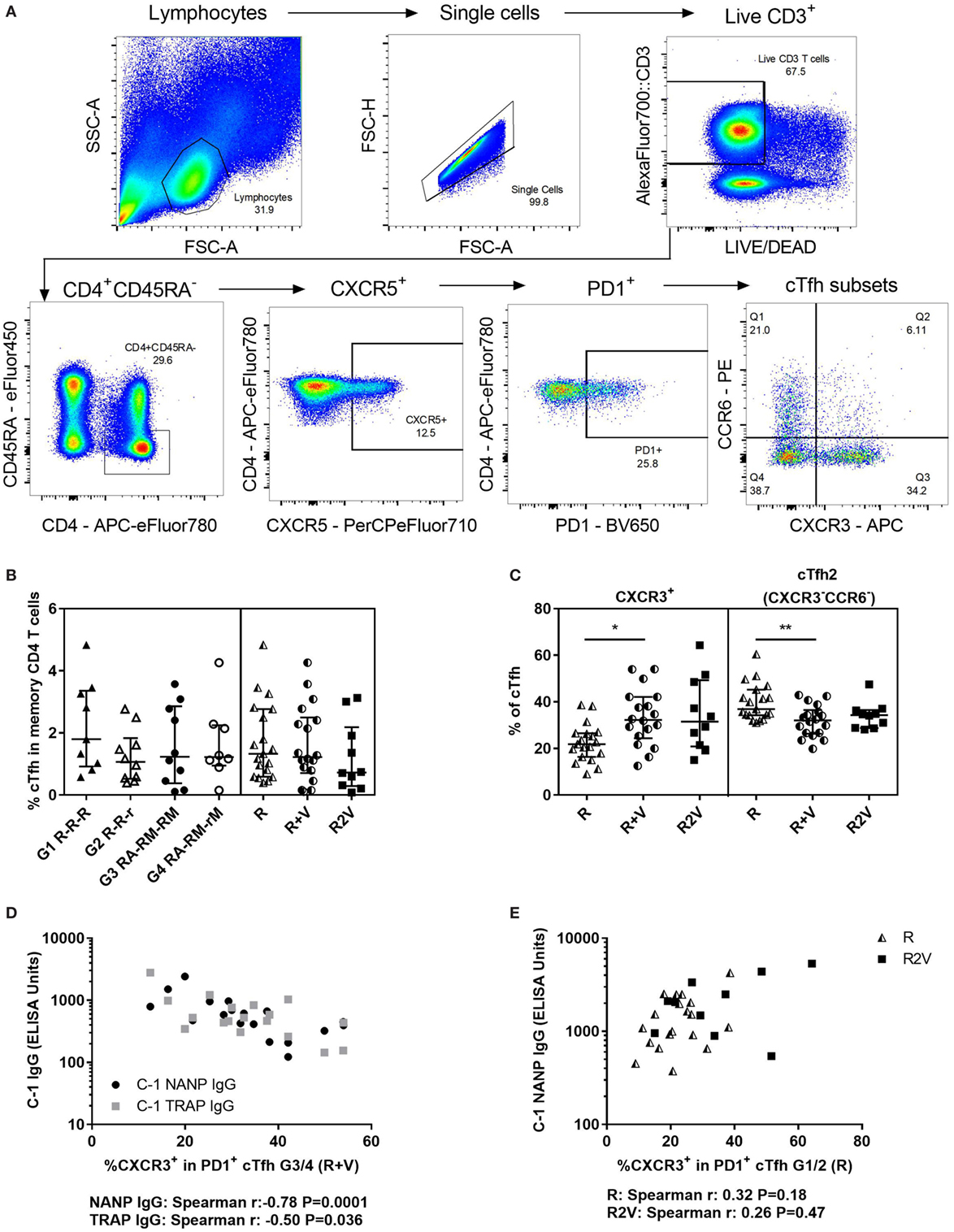
Figure 4. Total cTfhs. (A) Gating strategy for cTfh phenotyping using cell surface staining and flow cytometry. (B) Percentage of cTfh (PD1+CXCR5+) within memory CD4 T cells (CD45RA−) at C-1. (C) Subsets within cTfh (at C-1) defined by chemokine receptor expression: cTfh2 (CXCR3−CCR6−), or CXCR3+; including double-positive, dp (CXCR3+CCR6+), and cTfh1 (CXCR3+CCR6−). Kruskal–Wallis analyses; cTfh2 P = 0.009, CXCR3+ P = 0.01. (D) Relationship between percentage of CXCR3+ cTfh and antibody responses at C-1 in individuals who received RTS,S/AS01B co-administered with viral-vectored vaccines (G3/4). NANP IgG (Spearman r: −0.78, P = 0.0001), TRAP IgG (Spearman r: −0.50, P = 0.036). (E) Relationship between percentage of CXCR3+ cTfh and C-1 NANP IgG in individuals vaccinated with RTS,S/AS01B alone (R, G1/2, Spearman r: 0.32, P = 0.18) or RTS,S/AS01B and viral-vectored vaccines in a staggered regimen (R2V, Spearman r: 0.26, P = 0.47). Abbreviations: R G1/2, RTS,S/AS01B vaccinated; R + V G3/4, RTS,S/AS01B and viral-vectored vaccines co-administered; R2V, RTS,S/AS01B and viral-vectored vaccines at a 2-week stagger; A, ChAd63 ME-TRAP; M, MVA ME-TRAP; R, 50 µg third dose of RTS,S/AS01B; r, 10 µg third dose of RTS,S/AS01B; CHMI, controlled human malaria infection; C-1, day before CHMI; cTfh, circulating T follicular helper cell; TRAP, thrombospondin-related adhesive protein.
Co-Administration of Viral-Vectored Vaccines With RTS,S/AS01B Drives Th1-Biased Cytokine Responses Which Are Associated With the Increase in CXCR3+ cTfh and Reduction in Antibody Responses
Concentrations of a range of T-helper cytokines in the supernatant of C-1 PBMC from the co-administration study were measured using a cytometric bead array (LEGENDplex, BioLegend). PBMCs were stimulated with CSP (all groups) or MVA (G3&4) and concentrations of IL-5, IL-13, IL-2, IL-6, IL-9, IL-10, IFNγ, TNFα, IL-17A, IL17-F, IL-4, IL-21, and IL-22 were measured (Figure 5A). High concentrations of IL-2, IFNγ, TNFα, IL-6, and IL-22 were detected. The concentration of IFNγ was particularly high in the supernatant from MVA-stimulated PBMCs where all samples produced >3,000 pg/mL. In comparison, IFNγ responses were significantly lower after CSP stimulation, with no detectable IFNγ in 9/34 samples and less than 1,000 pg/mL in most where responses were detected. However, PBMCs from volunteers in the co-administration groups produced more IFNγ in response to CSP stimulation than those from volunteers who received RTS,S/AS01B alone (Figure 5B, median pg/mL + IQR, R CSP: 114 [1–348], R + V CSP: 311 [96–610], R + V MVA: 16,796 [11,409–20,462], Kruskal–Wallis P < 0.0001). In addition, IFNγ was a greater proportion of the cytokine response to CSP stimulation in PBMC from G3/4 (R + V) volunteers who were not sterilely protected after CHMI than those who were protected (Figure 5C).
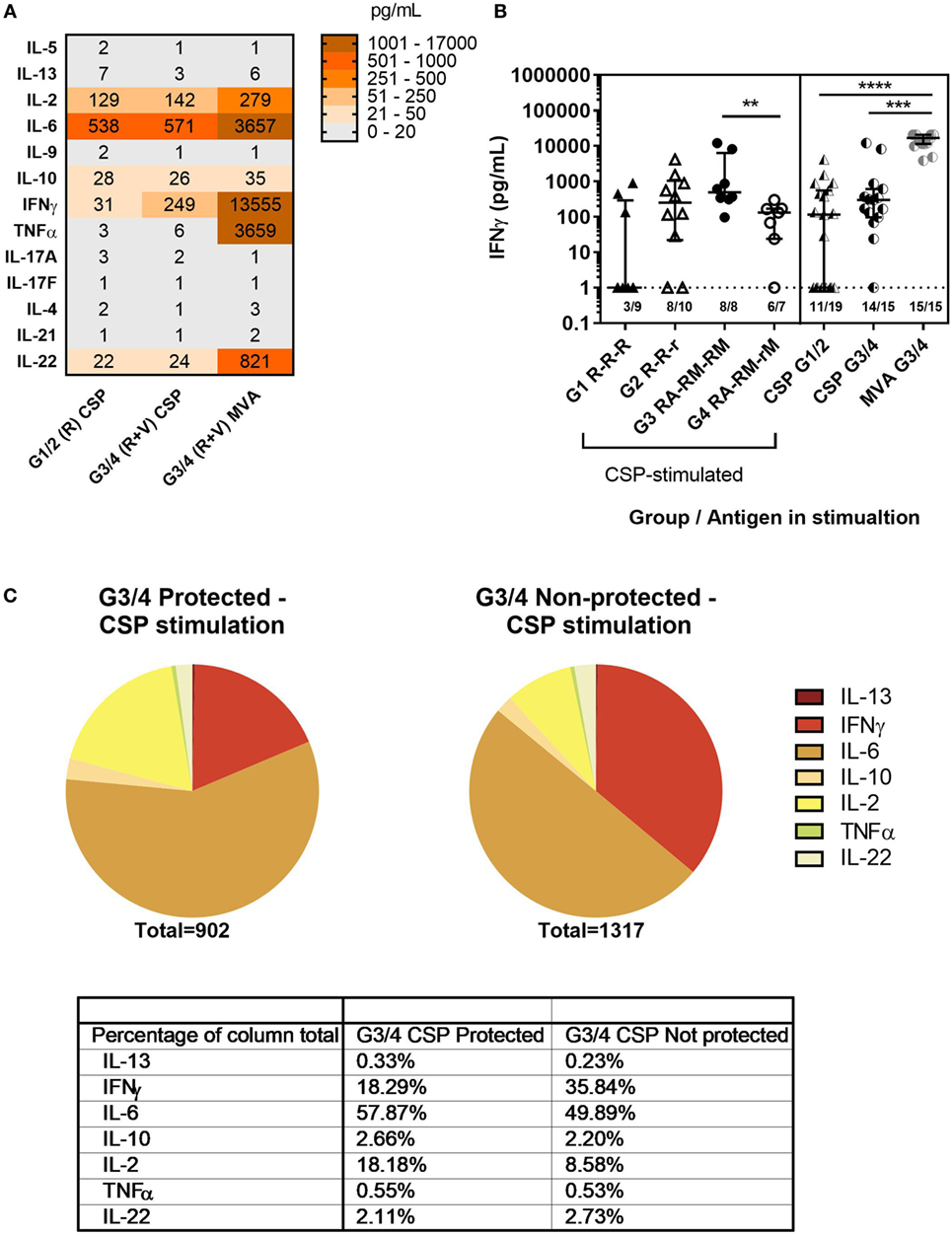
Figure 5. Cytokine responses to CSP and MVA. Cytokine responses measured in supernatant after stimulation of 1–2 × 106 PBMC at C-1 with CSP or MVA. A multiplex cytokine bead assay (Legendplex, BioLegend) was used to measure a panel of T-helper cytokines. Responses were measured for 19 G1/2 samples stimulated with CSP and 15 G3/4 samples for which there were enough cells to run both CSP and MVA stimulations. (A) Heatmap of geomean cytokine concentrations in pg/mL in supernatant of PBMC from G1/2 stimulated with CSP and G3/4 stimulated with CSP or MVA. (B) Concentration of IFNγ in supernatant of PBMC stimulated with CSP or MVA, Kruskal–Wallis with Dunn’s multiple comparisons P < 0.0001. Mann–Whitney analysis between G3 RA-RM-RM and G4 RA-RM-rM P = 0.0037. Lower limit of detection for all cytokines in this assay was 1 pg/mL (indicated by the dashed line). (C) Proportions of cytokines produced in response to CSP stimulation (geomeans) in G3/4 (R + V) volunteers who were protected compared with those who were not. Table shows the level of each cytokine produced as a percentage of the total cytokine response to CSP in each group. Abbreviations: R G1/2, RTS,S/AS01B vaccinated; R + V G3/4, RTS,S/AS01B and viral-vectored vaccines co-administered; R2V, RTS,S/AS01B and viral-vectored vaccines at a 2-week stagger; A, ChAd63 ME-TRAP; M, MVA ME-TRAP; R, 50 µg third dose of RTS,S/AS01B; r, 10 µg third dose of RTS,S/AS01B; CHMI, controlled human malaria infection; C-1, day before CHMI; CSP, circumsporozoite protein; PBMCs, peripheral blood mononucleocytes; TRAP, thrombospondin-related adhesive protein.
The concentration of IFNγ in the CSP supernatant was positively associated with the proportion of CXCR3+ cTfh (Figure 6A, Spearman r: 0.41, P = 0.01) and negatively with the proportion of cTfh17 within cTfh (Figure 6B, Spearman r: −0.63, P < 0.0001). In addition, there was a negative association between the concentration of IFNγ in the CSP supernatant and the ability of antibody to block sporozoite entry into hepatocytes (Figure 6C, Spearman r: −0.79, P = 0.0001). Analysis of IFNγ production by ICS of cTfh after stimulation with CSP or the superantigen SEB showed very low frequencies of these cells expressing IFNγ [Figure 6D, less than 2% in G1/2 (R) and less than 3% in G3/4 (R + V) after CSP stimulation].
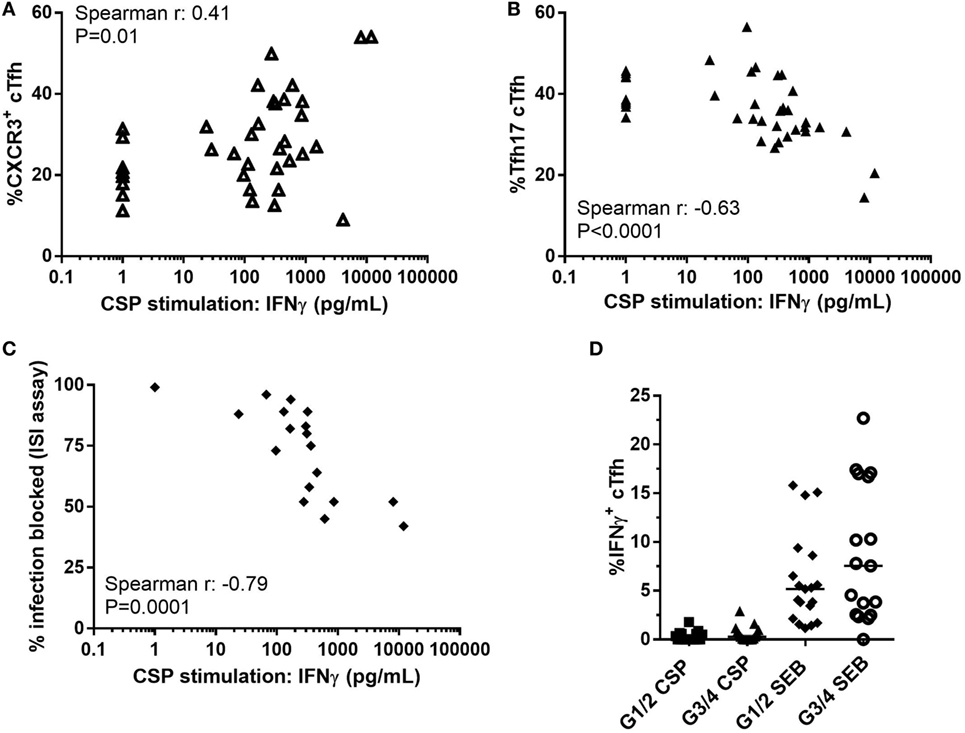
Figure 6. Association between Th1-biased cytokine responses and suppressed humoral immunity. Relationship between concentration of IFNγ in supernatant of PBMCs stimulated with CSP in co-administration trial (G1–4, R, and R + V combined) and: (A) Proportion of CXCR3+ cTfh (Spearman r: 0.41, P = 0.01); (B) Proportion of cTfh17 (CXCR3+CCR6−) within cTfh (Spearman r: -0.63, P < 0.0001); (C) Percentage of infection blocked in the ISI assay (Spearman r: -0.79, P = 0.0001). (D) Proportion of IFNγ+ cTfh after stimulation with CSP or SEB, Mann–Whitney analyses between G1/2 (R) and G3/4 (R + V) P = 0.14 and P = 0.26 for CSP and SEB stimulations, respectively. Abbreviations: R G1/2, RTS,S/AS01B vaccinated; R + V G3/4, RTS,S/AS01B and viral-vectored vaccines co-administered; R2V, RTS,S/AS01B and viral-vectored vaccines at a 2-week stagger; A, ChAd63 ME-TRAP; M, MVA ME-TRAP; R, 50 µg third dose of RTS,S/AS01B; r, 10 µg third dose of RTS,S/AS01B; CHMI, controlled human malaria infection; C-1, day before CHMI; CSP, circumsporozoite protein; cTfh, circulating T follicular helper cell; ISI, inhibition of sporozoite invasion; PBMCs, peripheral blood mononucleocytes; TRAP, thrombospondin-related adhesive protein; SEB, Staphylococcus enterotoxin B.
Discussion
Viral-vectored vaccines ChAd63 ME-TRAP and MVA ME-TRAP given with RTS,S/AS01B in a staggered regimen induced high titers of antibodies against sporozoites and potent T cell responses against infected liver cells (18). This regimen required five separate vaccinations and would likely be impractical and uneconomical for deployment in malaria-endemic regions. One way to overcome this obstacle would be to co-administer the RTS,S/AS01B and viral-vectored vaccines. Co-administration of vaccines can lead to interactions between the immune responses, which may be beneficial, enhancing the response as seen after co-administration of the live and attenuated polio vaccines or may result in a reduction of immunogenicity as is the case for multivalent inactivated or live viral vaccines (30, 31).
We examined the immune responses induced by both vaccines when co-administered to assess the extent of the immunological interaction between RTS,S/AS01B and viral-vectored vaccines. An in vitro assay measuring the ability of vaccine-induced antibody to block sporozoite invasion of hepatocytes was used to assess the functional quality of antibody. Infection-blocking ability was associated with protection. However, some volunteers who received viral-vectored vaccines were protected despite having antibodies with only low levels of infection-blocking activity. These volunteers had some of the highest TRAP-specific T cell responses, suggesting that cellular responses may provide protection by killing infected hepatocytes in volunteers who do not produce sufficient anti-NANP titers to block sporozoite entry. This demonstrates the potential of a multistage targeting regimen to provide high-level efficacy if each vaccine can be given without interfering with the immunogenicity of the other. However, co-administration of viral-vectored vaccines with RTS,S/AS01B in this study induced a strong Th1 cytokine response and increased proportions of CXCR3+ cTfh, which were associated with reduced antibody quantity and quality and lower efficacy in these groups.
In addition, we observed qualitative differences in RTS,S-induced antibody responses when a reduced third (1/5th, 10 µg) dose was given compared with three standard doses (50 µg). Although NANP IgG titers were comparable, administration of a reduced third dose of RTS,S/AS01B-induced antibodies that were capable of blocking a significantly higher level of sporozoite infection in vitro—a measure which was associated with protection from CHMI. Previously, a reduced third dose of RTS,S/AS01B was shown to provide higher levels of protection (14, 19) and in a more recent study, a fractional third dose boost induced antibody with increased somatic hypermutation and higher avidity (19). However, it was unclear whether this effect was due to the delayed boost (0, 1, and 7-month regimen) or the fractional dose. Our study is the first to demonstrate a functional difference in the antibodies induced by the reduced third dose regimen that is associated with protection. However, it is unclear whether the quality of the antibody response is enhanced by a reduced third dose in particular or whether this could be achieved with a reduction of all three doses, which would also have economical and practical advantages. Lower vaccine doses decrease the availability of antigen and therefore could result in greater affinity maturation through increased competition between B cells for T cell help and preferential expansion of B cell clones with the highest affinity B cell receptors (32, 33). Lower doses of antigen at priming also preferentially drive the induction of memory, while higher antigen doses drive differentiation of plasma cells (34). The preferential induction of memory by reducing the priming dose could enhance responses to the subsequent vaccinations and also potentially generate more durable protection. This suggests that the dosing regimen should also be carefully tested to ensure the optimal type of immune response is achieved.
Previous studies have demonstrated that cTfh may be useful biomarkers for GC responses in the absence of lymphoid tissue (35). However, cTfh are a heterogeneous population composed of a number of different subsets, some of which appear to more closely resemble bona fide GC Tfh than others (22, 36). The proportions of these subsets have been associated with different diseases: increases in cTfh2/cTfh17 subsets are associated with the production of autoantibodies and disease severity in various autoimmune diseases (22, 37, 38), the development of allergy (39, 40) and the production of broadly neutralizing antibody in HIV+ individuals (26, 41). By contrast, increases in CXCR3+ cTfh have been implicated in the poor development of humoral immunity against malaria (42, 43) and are proportionally increased in patients with primary immunodeficiencies (44). In addition, a study that observed CXCR3+ cTfh to positively correlate with antibody responses after influenza vaccination also showed that CXCR3+ Tfh that were localized to tonsillar GCs, expressed Fas-L, secreted IFNγ, lacked CD154 expression, and suppressed the activity of GC B cells. Therefore, although this subset was correlated with antibody responses, they were not optimal for their induction (45). In our study, co-administration of RTS,S/AS01B with viral-vectored vaccines led to an increased frequency of CXCR3+ total cTfh compared with RTS,S/AS01B administered alone, and this phenotype was associated with the observed reduction in antibody quantity and quality. Although the observed association was for total cTfh, it would be useful to profile the antigen-specificity of these cells to determine if the increase in CXCR3+ cTfh was due to the induction of cTfh specific for the vector or whether this was a change in the phenotype of the CSP-specific cTfh. There are several methods used to look at antigen-specific cTfh, including cytokine production or CD154 expression after overnight antigen stimulation or the use of antigen-induced markers (45–48). Unfortunately, we found that staining for the chemokine receptors CXCR3 and CCR6 could not reliably be incorporated into these assays (unpublished data). Therefore, we were unable to determine the specificity of the CXCR3+ cTfh.
Circulating Tfh have been shown to produce cytokines, with CXCR3+ cTfh in particular producing IFNγ (49). However, the association we saw between IFNγ in the supernatant and CXCR3+ cTfh was not likely due to production of IFNγ by the CXCR3+ cTfh themselves as only very low percentages of cTfh were observed to produce IFNγ after CSP or SEB stimulation. This suggests that exogenous sources of IFNγ are associated with the polarization of cTfh toward a CXCR3+ phenotype, although vector-specific CXCR3+ cTfh could be a source IFNγ in G3/4 volunteers. MVA has previously been shown to drive a strong IFNγ response, particularly in CD8+ T cells and IFNγ enhances CXCR3 expression on T cells through STAT1 signaling (50–53). It is perhaps therefore unsurprising that MVA induced a CXCR3-skewed cTfh response. However, the impact of this skew on the antibody responses was less predictable, given that CXCR3+ cTfh have in some contexts been positively associated with antibody responses (45, 54) while in other studies they have been associated with suboptimal GC responses and poor humoral immunity (42, 55). In our study when the two different vaccine platforms were co-administered, the IFNγ-dominated cytokine responses driven by viral-vectored vaccines were associated with an increase in CXCR3+ cTfh and a reduction in humoral immunity and protective efficacy. However, if given 2 weeks after RTS,S/AS01B the extent of this CXCR3+ cTfh skew, although only slightly reduced, is no longer associated with a reduction in humoral immunogenicity.
IFNγ induces the production of chemokines CXCL9 (MIG, monokine induced by gamma-interferon), CXCL10 (IP-10, interferon-induced protein of 10 kDa), and CXCL11 (I-TAC, interferon inducible T cell alpha chemoattractant), which all bind CXCR3. This chemokine system mediates the migration of Th1 CD4+ T cells and cytotoxic T lymphocytes to sites of Th1 inflammation in the periphery (56). MVA has been shown to induce high systemic levels of IP-10 (57). The systemic induction of CXCR3 ligands, in combination with the CXCR3+ cTfh polarization, may result in the reduction of antibody responses by causing an egress of these cTfh from the draining lymph node, preventing them from providing help to B cells in the GC response. A staggered regimen may reduce this effect by allowing time for the RTS,S/AS01B-induced GC response to occur before MVA-induced inflammation begins. Alternative strategies to reduce or avoid the observed immune interference without increasing the number of clinic visits could be to reduce the dose of MVA or to exclude the additional MVA vaccination at week 4, which was included for practical reasons to simplify the vaccination protocol and is not required to induce potent T cell responses (17, 18).
Producing an effective vaccine against malaria will likely depend on a combination of vaccines targeting multiple stages of the parasite lifecycle. In resource-poor settings, mixing, or co-administering, the vaccines will be necessary to reduce the number of clinic visits required, particularly in infants to fit in with the established Expanded Program on Immunisation vaccine schedule. However, the effects of co-administration on immunogenicity and protective efficacy of each vaccine must be carefully examined. Ideally, a combination regimen could be designed to elicit antibody and T cell responses with an additive protective effect and without the immune interference observed here. Examining the cellular mechanisms underlying these differences in antibody responses will be critical for determining how effective, long-lived antibody responses can be induced by vaccination and for informing rational design of vaccine regimens.
Ethics Statement
All volunteers gave written informed consent prior to participation, and the studies were conducted according to the principles of the Declaration of Helsinki and in accordance with Good Clinical Practice. The trials were registered with ClinicalTrials.gov (Ref: NCT01883609 and NCT02252640). The study protocols were approved by the UK National Research Ethics Service, Committee South Central—Oxford A (Refs: 13/SC/0208 and 14/SC/0227), the Western Institution Review Board (Ref: 20130698) and the UK Medicines and Healthcare products Regulatory Agency (Refs: 21584/0317/001-0001 and 21584/0333/001-0001). The Local Safety Committee provided safety oversight for both trials and GCP compliance was monitored by the Clinical Trials and Research Governance Team (CTRG) of the University of Oxford.
Author Contributions
GB designed laboratory study, interpreted data, and wrote the manuscript. GB and AG acquired and analyzed data. DM, RB, AH, and KE conceived clinical studies and provided project oversight. TR, NV, AH, and KE designed clinical studies. TR and NV conducted clinical work. All the authors revised the manuscript.
Disclaimer
The views expressed are those of the author(s) and not necessarily those of the PATH Malaria Vaccine Initiative, the United Kingdom National Health Service, the United Kingdom NIHR, or the Department of Health.
Conflict of Interest Statement
AH is a named inventor on patent applications covering malaria vectored vaccines and immunization regimens. DM and RB are employees of GSK, which is developing vaccines for malaria and other diseases. All other authors report no potential conflicts.
Acknowledgments
We thank the VAC55/59 clinical and laboratory team—N. Edwards, D. Wright, S. Sridhar, R. Payne, J. Powlson, C. Bliss, N. Venkatraman, I. Poulton, R. Roberts, B. Angus, A. Lawrie, R. Lopez-Ramon, O. Griffiths, N. Lella, and C. Tyson for logistical support; the study volunteers; J. Furze for laboratory assistance; J. Illingworth and A. Douglas for production of recombinant TRAP protein; the staff at the NIHR Wellcome Trust Clinical Research Facilities at the Southampton, Guildford and Hammersmith Hospital sites; S. Davidson and colleagues at the WRAIR Entomology laboratories for supply of infected mosquitoes for CHMI; GSK for supply of RTS,S/AS01B vaccine and scientific input; A. Spencer, M. Ulaszewska and the Jenner insectary for A. stephensi mosquitoes; A. Salman for P. berghei PfCSP@CSP sporozoites; K. Collins and F. Halstead for preparation of IFA slides; N. Obeng-Adjei and P. Crompton for their advice and training on the Tfh phenotyping flow cytometry; PATH for funding.
Funding
This work was supported by the PATH Malaria Vaccine Initiative and by the United Kingdom NIHR, through the NIHR Oxford Biomedical Research Centre, the Southampton NIHR Wellcome Trust Clinical Research Facility, and the Imperial College NIHR Wellcome Trust Clinical Research Facility. Support was also received from the UK National Institute for Health Research Oxford Biomedical Research Centre (Vaccines Theme). AVS Hill is supported by a Wellcome Trust Senior Investigator Award (104750/Z/14).
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.01660/full#supplementary-material.
Figure S1. Relationship between NANP-specific isotypes/subclasses and inhibition of sporozoite invasion (ISI). Associations between NANP-specific isotypes and subclass titers at C-1 and ISI (A) IgG1, (B) IgG2, (C) IgG3, (D) IgM, and (E) IgA.
Abbreviations
ChAd, chimpanzee adenovirus; CHMI, controlled human malaria infection; CSP, circumsporozoite protein; cTfh, circulating T follicular helper cell; GC, germinal center; ISI, inhibition of sporozoite invasion; ME, multi-epitope; MVA, modified vaccinia virus Ankara; PBMCs, peripheral blood mononucleocytes; pfu, plaque-forming units; TRAP, thrombospondin-related adhesive protein; VE, vaccine efficacy; VP, viral particles.
References
1. World Health Organization. World Malaria Report [Online]. (2016). Available from: http://www.who.int/malaria/publications/world-malaria-report-2016/report/en/ [Accessed: July 15, 2018].
2. malERA Consultative Group on Vaccines. A research agenda for malaria eradication: vaccines. PLoS Med (2011) 8:e1000398. doi:10.1371/journal.pmed.1000398
3. RTS,S Clinical Trials Partnership, Agnandji ST, Lell B, Soulanoudjingar SS, Fernandes JF, Abossolo BP, et al. First results of phase 3 trial of RTS,S/AS01 malaria vaccine in African children. N Engl J Med (2011) 365:1863–75. doi:10.1056/NEJMoa1102287
4. RTS,S Clinical Trials Partnership, Agnandji ST, Lell B, Fernandes JF, Abossolo BP, Methogo BG, et al. A phase 3 trial of RTS,S/AS01 malaria vaccine in African infants. N Engl J Med (2012) 367:2284–95. doi:10.1056/NEJMoa1208394
5. Olotu A, Fegan G, Wambua J, Nyangweso G, Leach A, Lievens M, et al. Seven-year efficacy of RTS,S/AS01 malaria vaccine among young African children. N Engl J Med (2016) 374:2519–29. doi:10.1056/NEJMoa1515257
6. RTS,S Clinical Trials Partnership. Efficacy and safety of the RTS,S/AS01 malaria vaccine during 18 months after vaccination: a phase 3 randomized, controlled trial in children and young infants at 11 African sites. PLoS Med (2014) 11:e1001685. doi:10.1371/journal.pmed.1001685
7. RTS,S Clinical Trials Partnership. Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial. Lancet (2015) 386:31–45. doi:10.1016/S0140-6736(15)60721-8
8. Malaria Vaccine Funders Group. Malaria, Vaccine Technology Roadmap [Online]. (2013). Available from: http://www.who.int/immunization/topics/malaria/vaccine_roadmap/TRM_update_nov13.pdf [Accessed: July 15, 2018].
9. Ewer KJ, Sierra-Davidson K, Salman AM, Illingworth JJ, Draper SJ, Biswas S, et al. Progress with viral vectored malaria vaccines: a multi-stage approach involving “unnatural immunity”. Vaccine (2015) 33:7444–51. doi:10.1016/j.vaccine.2015.09.094
10. Ogwang C, Kimani D, Edwards NJ, Roberts R, Mwacharo J, Bowyer G, et al. Prime-boost vaccination with chimpanzee adenovirus and modified vaccinia Ankara encoding TRAP provides partial protection against Plasmodium falciparum infection in Kenyan adults. Sci Transl Med (2015) 7:286re285. doi:10.1126/scitranslmed.aaa2373
11. Hodgson SH, Choudhary P, Elias SC, Milne KH, Rampling TW, Biswas S, et al. Combining viral vectored and protein-in-adjuvant vaccines against the blood-stage malaria antigen AMA1: report on a phase 1a clinical trial. Mol Ther (2014) 22:2142–54. doi:10.1038/mt.2014.157
12. Kapulu MC, Da DF, Miura K, Li Y, Blagborough AM, Churcher TS, et al. Comparative assessment of transmission-blocking vaccine candidates against Plasmodium falciparum. Sci Rep (2015) 5:11193. doi:10.1038/srep11193
13. Moorthy VS, Ballou WR. Immunological mechanisms underlying protection mediated by RTS,S: a review of the available data. Malar J (2009) 8:312–312. doi:10.1186/1475-2875-8-312
14. Stoute JA, Slaoui M, Heppner DG, Momin P, Kester KE, Desmons P, et al. A preliminary evaluation of a recombinant circumsporozoite protein vaccine against Plasmodium falciparum malaria. RTS,S Malaria Vaccine Evaluation Group. N Engl J Med (1997) 336:86–91. doi:10.1056/NEJM199701093360202
15. White MT, Bejon P, Olotu A, Griffin JT, Riley EM, Kester KE, et al. The relationship between RTS,S vaccine-induced antibodies, CD4+ T cell responses and protection against Plasmodium falciparum infection. PLoS One (2013) 8:e61395. doi:10.1371/journal.pone.0061395
16. White MT, Verity R, Griffin JT, Asante KP, Owusu-Agyei S, Greenwood B, et al. Immunogenicity of the RTS,S/AS01 malaria vaccine and implications for duration of vaccine efficacy: secondary analysis of data from a phase 3 randomised controlled trial. Lancet Infect Dis (2015) 15:1450–8. doi:10.1016/S1473-3099(15)00239-X
17. Ewer KJ, O’Hara GA, Duncan CJ, Collins KA, Sheehy SH, Reyes-Sandoval A, et al. Protective CD8+ T-cell immunity to human malaria induced by chimpanzee adenovirus-MVA immunisation. Nat Commun (2013) 4:2836. doi:10.1038/ncomms3836
18. Rampling T, Ewer KJ, Bowyer G, Bliss CM, Edwards NJ, Wright D, et al. Safety and high level efficacy of the combination malaria vaccine regimen of RTS,S/AS01B with chimpanzee adenovirus 63 and modified vaccinia Ankara vectored vaccines expressing ME-TRAP. J Infect Dis (2016) 214:772–81. doi:10.1093/infdis/jiw244
19. Regules JA, Cicatelli SB, Bennett JW, Paolino KM, Twomey PS, Moon JE, et al. Fractional third and fourth dose of RTS,S/AS01 malaria candidate vaccine: a phase 2a controlled human malaria parasite infection and immunogenicity study. J Infect Dis (2016) 214:762–71. doi:10.1093/infdis/jiw237
20. Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med (2000) 192:1545–52. doi:10.1084/jem.192.11.1545
21. Ma CS, Deenick EK, Batten M, Tangye SG. The origins, function, and regulation of T follicular helper cells. J Exp Med (2012) 209:1241–53. doi:10.1084/jem.20120994
22. Morita R, Schmitt N, Bentebibel SE, Ranganathan R, Bourdery L, Zurawski G, et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity (2011) 34:108–21. doi:10.1016/j.immuni.2010.12.012
23. Chevalier N, Jarrossay D, Ho E, Avery DT, Ma CS, Yu D, et al. CXCR5 expressing human central memory CD4 T cells and their relevance for humoral immune responses. J Immunol (2011) 186:5556–68. doi:10.4049/jimmunol.1002828
24. Vinuesa CG, Cook MC. Blood relatives of follicular helper T cells. Immunity (2011) 34:10–2. doi:10.1016/j.immuni.2011.01.006
25. Schmitt N, Bentebibel S-E, Ueno H. Phenotype and functions of memory Tfh cells in human blood. Trends Immunol (2014) 35:436–42. doi:10.1016/j.it.2014.06.002
26. Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, Arlehamn CL, et al. Human circulating PD-1+CXCR3-CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity (2013) 39:758–69. doi:10.1016/j.immuni.2013.08.031
27. Farooq F, Beck K, Paolino KM, Phillips R, Waters NC, Regules JA, et al. Circulating follicular T helper cells and cytokine profile in humans following vaccination with the rVSV-ZEBOV Ebola vaccine. Sci Rep (2016) 6:27944. doi:10.1038/srep27944
28. Thompson FM, Porter DW, Okitsu SL, Westerfeld N, Vogel D, Todryk S, et al. Evidence of blood stage efficacy with a virosomal malaria vaccine in a phase IIa clinical trial. PLoS One (2008) 3:e1493. doi:10.1371/journal.pone.0001493
29. Rodríguez-Galán A, Salman AM, Bowyer G, Collins KA, Longley RJ, Brod F, et al. An in vitro assay to measure antibody-mediated inhibition of P. berghei sporozoite invasion against P. falciparum antigens. Sci Rep (2017) 7:17011. doi:10.1038/s41598-017-17274-5
30. WHO collaborative Study Group on Oral and Inactivated Poliovirus Vaccines. Combined immunization of infants with oral and inactivated poliovirus vaccines: results of a randomized trial in The Gambia, Oman, and Thailand/WHO Collaborative Study Group on oral and inactivated poliovirus vaccines. Bull World Health Organ (1996) 74:253–68.
31. Migasena S, Simasathien S, Samakoses R, Pitisuttitham P, Sangaroon P, van Steenis G, et al. Simultaneous administration of oral rhesus-human reassortant tetravalent (RRV-TV) rotavirus vaccine and oral poliovirus vaccine (OPV) in Thai infants. Vaccine (1995) 13:168–74. doi:10.1016/0264-410X(95)93131-R
32. Eisen HN, Siskind GW. Variations in affinities of antibodies during the immune response. Biochemistry (1964) 3:996–1008.
33. González-Fernández A, Milstein C. Low antigen dose favours selection of somatic mutants with hallmarks of antibody affinity maturation. Immunology (1998) 93:149–53. doi:10.1046/j.1365-2567.1998.00423.x
34. Ahman H, Käyhty H, Vuorela A, Leroy O, Eskola J. Dose dependency of antibody response in infants and children to pneumococcal polysaccharides conjugated to tetanus toxoid. Vaccine (1999) 17:2726–32.
35. He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5+ CD4+ T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity (2013) 39:770–81. doi:10.1016/j.immuni.2013.09.007
36. Heit A, Schmitz F, Gerdts S, Flach B, Moore MS, Perkins JA, et al. Vaccination establishes clonal relatives of germinal center T cells in the blood of humans. J Exp Med (2017) 214:2139–52. doi:10.1084/jem.20161794
37. Li XY, Wu ZB, Ding J, Zheng ZH, Li XY, Chen LN, et al. Role of the frequency of blood CD4(+) CXCR5(+) CCR6(+) T cells in autoimmunity in patients with Sjögren’s syndrome. Biochem Biophys Res Commun (2012) 422:238–44. doi:10.1016/j.bbrc.2012.04.133
38. Romme Christensen J, Börnsen L, Ratzer R, Piehl F, Khademi M, Olsson T, et al. Systemic inflammation in progressive multiple sclerosis involves follicular T-helper, Th17- and activated B-cells and correlates with progression. PLoS One (2013) 8:e57820. doi:10.1371/journal.pone.0057820
39. Gong F, Qian C, Zhu H, Zhu J, Pan Y, Dong Q, et al. Circulating follicular T-helper cell subset distribution in patients with asthma. Allergy Asthma Proc (2016) 37:154–61. doi:10.2500/aap.2016.37.3982
40. Kamekura R, Shigehara K, Miyajima S, Jitsukawa S, Kawata K, Yamashita K, et al. Alteration of circulating type 2 follicular helper T cells and regulatory B cells underlies the comorbid association of allergic rhinitis with bronchial asthma. Clin Immunol (2015) 158:204–11. doi:10.1016/j.clim.2015.02.016
41. Schmitt N, Ueno H. Human T follicular helper cells: development and subsets. Adv Exp Med Biol (2013) 785:87–94. doi:10.1007/978-1-4614-6217-0_10
42. Obeng-Adjei N, Portugal S, Tran TM, Yazew TB, Skinner J, Li S, et al. Circulating Th1-cell-type Tfh cells that exhibit impaired B cell help are preferentially activated during acute malaria in children. Cell Rep (2015) 13:425–39. doi:10.1016/j.celrep.2015.09.004
43. Ryg-Cornejo V, Ioannidis LJ, Ly A, Chiu CY, Tellier J, Hill DL, et al. Severe malaria infections impair germinal center responses by inhibiting T follicular helper cell differentiation. Cell Rep (2016) 14:68–81. doi:10.1016/j.celrep.2015.12.006
44. Cunill V, Clemente A, Lanio N, Barceló C, Andreu V, Pons J, et al. Follicular T cells from smB(-) common variable immunodeficiency patients are skewed toward a Th1 phenotype. Front Immunol (2017) 8:174. doi:10.3389/fimmu.2017.00174
45. Bentebibel SE, Lopez S, Obermoser G, Schmitt N, Mueller C, Harrod C, et al. Induction of ICOS+CXCR3+CXCR5+ TH cells correlates with antibody responses to influenza vaccination. Sci Transl Med (2013) 5:176ra132. doi:10.1126/scitranslmed.3005191
46. Chattopadhyay PK, Yu J, Roederer M. Live-cell assay to detect antigen-specific CD4+ T-cell responses by CD154 expression. Nat Protoc (2006) 1:1–6. doi:10.1038/nprot.2006.1
47. Dan JM, Lindestam Arlehamn CS, Weiskopf D, da Silva Antunes R, Havenar-Daughton C, Reiss SM, et al. A cytokine-independent approach to identify antigen-specific human germinal center t follicular helper cells and rare antigen-specific CD4+ T cells in blood. J Immunol (2016) 197:983–93. doi:10.4049/jimmunol.1601321
48. Havenar-Daughton C, Reiss SM, Carnathan DG, Wu JE, Kendric K, Torrents de la Peña A, et al. Cytokine-independent detection of antigen-specific germinal center T follicular helper cells in immunized nonhuman primates using a live cell activation-induced marker technique. J Immunol (2016) 197:994–1002. doi:10.4049/jimmunol.1600320
49. Ma CS, Wong N, Rao G, Avery DT, Torpy J, Hambridge T, et al. Monogenic mutations differentially affect the quantity and quality of T follicular helper cells in patients with human primary immunodeficiencies. J Allergy ClinImmunol (2015) 136:993–1006.e1. doi:10.1016/j.jaci.2015.05.036
50. Remy-Ziller C, Germain C, Spindler A, Hoffmann C, Silvestre N, Rooke R, et al. Immunological characterization of a modified vaccinia virus Ankara vector expressing the human papillomavirus 16 E1 protein. Clin Vaccine Immunol (2014) 21:147–55. doi:10.1128/CVI.00678-13
51. Kimani D, Jagne YJ, Cox M, Kimani E, Bliss CM, Gitau E, et al. Translating the immunogenicity of prime-boost immunization with ChAd63 and MVA ME-TRAP from malaria naive to malaria-endemic populations. Mol Ther (2014) 22:1992–2003. doi:10.1038/mt.2014.109
52. Reyes-Sandoval A, Berthoud T, Alder N, Siani L, Gilbert SC, Nicosia A, et al. Prime-boost immunization with adenoviral and modified vaccinia virus Ankara vectors enhances the durability and polyfunctionality of protective malaria CD8+ T-cell responses. Infect Immun (2010) 78:145–53. doi:10.1128/IAI.00740-09
53. Nakajima C, Mukai T, Yamaguchi N, Morimoto Y, Park WR, Iwasaki M, et al. Induction of the chemokine receptor CXCR3 on TCR-stimulated T cells: dependence on the release from persistent TCR-triggering and requirement for IFN-gamma stimulation. Eur J Immunol (2002) 32:1792–801. doi:10.1002/1521-4141(200206)32:6<1792::AID-IMMU1792>3.0.CO;2-0
54. Bentebibel SE, Khurana S, Schmitt N, Kurup P, Mueller C, Obermoser G, et al. ICOS(+)PD-1(+)CXCR3(+) T follicular helper cells contribute to the generation of high-avidity antibodies following influenza vaccination. Sci Rep (2016) 6:26494. doi:10.1038/srep26494
55. Keitany GJ, Kim KS, Krishnamurty AT, Hondowicz BD, Hahn WO, Dambrauskas N, et al. Blood stage malaria disrupts humoral immunity to the pre-erythrocytic stage circumsporozoite protein. Cell Rep (2016) 17:3193–205. doi:10.1016/j.celrep.2016.11.060
56. Groom JR, Luster AD. CXCR3 in T cell function. Exp Cell Res (2011) 317:620–31. doi:10.1016/j.yexcr.2010.12.017
Keywords: malaria, vaccine, T follicular helper cell, antibody, efficacy, CXCR3, RTSS viral-vectored vaccines
Citation: Bowyer G, Grobbelaar A, Rampling T, Venkatraman N, Morelle D, Ballou RW, Hill AVS and Ewer KJ (2018) CXCR3+ T Follicular Helper Cells Induced by Co-Administration of RTS,S/AS01B and Viral-Vectored Vaccines Are Associated With Reduced Immunogenicity and Efficacy Against Malaria. Front. Immunol. 9:1660. doi: 10.3389/fimmu.2018.01660
Received: 10 May 2018; Accepted: 04 July 2018;
Published: 25 July 2018
Edited by:
Julius Clemence Hafalla, London School of Hygiene & Tropical Medicine, United KingdomReviewed by:
Robin Stephens, The University of Texas Medical Branch at Galveston, United StatesUrszula Krzych, Walter Reed Army Institute of Research, United States
Jason Scott Stumhofer, University of Arkansas for Medical Sciences, United States
Copyright: © 2018 Bowyer, Grobbelaar, Rampling, Venkatraman, Morelle, Ballou, Hill and Ewer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Georgina Bowyer, Z2VvcmdpbmEuYm93eWVyQG5kbS5veC5hYy51aw==