 Kavita M. Dhodapkar
Kavita M. Dhodapkar- Aflac Cancer Center of Children’s Healthcare of Atlanta, Department of Pediatrics, Emory University, Atlanta, GA, United States
Tissue-resident memory T (TRM) cells are a distinct subset of memory T cells that reside in non-lymphoid tissues for prolonged periods of time without significant recirculation providing continued immune surveillance at these sites. Recent studies suggest that TRM cells are also enriched within tumor tissue. Expression of inhibitory immune checkpoints (ICPs) is particularly enriched on this subset of tumor-infiltrating T cells, suggesting that they are major targets for newer therapies targeting ICPs such as the programmed death-1 pathway. Recent studies suggest that tissue restriction of these cells without recirculation may also lead to heterogeneity of TRM cells within individual metastatic lesions, ultimately leading to inter-lesional diversity. Thus, individual metastatic lesions may contain genomically distinct immune microenvironments that impact both evolution of tumors as well as the mechanisms underlying response and resistance to immune therapies. Understanding the biology of TRM cells infiltrating tumors will be essential to improving immune-based approaches in diverse settings.
Immune-based approaches, particularly those based on the blockade of inhibitory immune checkpoints (ICPs) on T cells have emerged as among the most promising new strategies to treat cancer (1). An important aspect of immune therapies is their potential ability to mediate long-term control of tumors. The capacity of the immune system to mediate long-term protection, particularly against pathogens, such as in the context of vaccines, is mediated in large part by immunologic memory (2). Therefore, understanding immunologic memory mediated by T cells is likely to be important for deeper understanding of immune-mediated long-term control of tumors. It is thought that uptake of antigens from dying tumor cells by antigen-presenting cells leads to activation of anti-tumor T cells in the lymph nodes, and resultant effector memory T cells traffic back to the tumor to mediate anti-tumor effects, creating a tumor-immunity cycle (3). Several studies have shown that infiltration of primary and metastatic lesions by immune cells, particularly T cells and myeloid cells impacts outcome (4). Studies in both mice and humans suggest that there are differences in the memory T cell subsets that provide immune surveillance within lymphoid and non-lymphoid tissues (NLTs). As tumor-related mortality in most solid tumors is not due to growth of primary tumors, but rather due to the growth of metastatic tumor cells in NLTs, it is the immune surveillance in NLTs that may be critical for protective tumor immunity. In this review, we discuss newer insights into spatial aspect of immunologic memory and particularly memory T cells within NLTs in the context of tumor immunity. We will discuss emerging evidence suggesting that the biology of these tissue-resident memory (TRM) T cells may not only be critical for understanding and improving clinical responses to ICP blockade, but may also contribute to the complexity of immune microenvironment by creating inter-lesional heterogeneity in the setting of metastatic cancer.
TRM T Cells: Local Policemen
Initial models of T cell memory classified effector/central memory (TEM/TCM) T cells, with the effector subset implicated in surveying NLTs (5). Recent studies have identified a third subset, termed TRM T cells that reside for prolonged periods in NLTs and play an important role in protective immunity (6). Mouse TRM cells have been described in diverse tissues, including lung, liver, brain, as well as barrier tissues (6, 7). Murine TRM cells haven been shown to mediate rapid in situ protection against viral, bacterial, and parasitic infections and are more effective in this regard than their circulating counterparts, including central memory T cells (7, 8). An important aspect of TRM-mediated immune surveillance is its regional nature. Thus in parabiotic mice that share systemic circulation, TRM cells remain localized within tissues and do not cross over to equilibrate in the paired mouse carrying antigenic stimulus (6). TRM cells express CD69, which is implicated in tissue retention by sequestration of the sphingosine-1-phosphate receptor (9).
Tissue-resident memory cells have also been identified in several human tissues and implicated in tissue-restricted pathology particularly in the skin, such as fixed drug eruptions (10–12). As in the mouse, human TRM cells have been identified by the expression of CD69 on memory T cells within tissues, which is generally lacking in blood memory T cells (13). In humans, CD103 is expressed only in a subset of CD69+CD8+ memory T cells in some barrier tissues, but not by CD4+ memory T cells in any tissue, indicating that CD69 may be a more universal marker distinguishing both CD4+ and CD8+ memory T cells in tissues from their blood counterparts. It is notable that the proportion of TRM cells differs in different tissues, with enrichment in some barrier tissues such as skin. Recent studies have also characterized transcriptional profiles of human TRM cells, which resemble their murine counterparts and also illustrate that these are a distinct subset of human memory T cells (14, 15).
The pathways that regulate generation, recruitment, retention, and long-term maintenance of these T cells in NLTs remain an active area of research. New insights into transcriptional regulation of the TRM differentiation are emerging and may differ between humans and mice. For example, the transcription factor Hobit/ZNF683 is exclusively expressed and required for the generation of murine TRM cells after infection, but expressed at low/negligible levels on human TRM cells (14, 16). In recent studies, we have shown that human and murine TRM cells express NR4A1/nur77, which is also essential for TRM differentiation in several murine tissues (17). Runx3 is another transcription factor that promotes the differentiation of T cells with TRM phenotype (18). Retention and maintenance of TRM cells may also depend on the availability of local antigen, interactions with myeloid cells as well as cytokines like TGFβ and IL-15 in NLTs (19, 20). Tissue distribution of TRM cells, at least against pathogens may depend on the site of initial exposure. For example, human influenza-specific TRM cells are preferentially found in the lung (21) and hepatitis-B specific TRM cells particularly in the liver (22). Human bone marrow may also be a particularly interesting compartment for long-lived memory T cells with phenotype of TRM cells (17, 23, 24).
TRM Cells in Tumors
Several studies have now documented that a large proportion of T cells infiltrating human tumors have TRM phenotype, at least based on the expression of CD69 and CD103 (11, 12, 25–27). In some studies, these T cells were also shown to have genomic signatures consistent with those described for TRM cells (11, 25, 26). This includes altered expression of genes involved in tissue retention/homing (such as downregulation of S1PR1, S1PR5, and KLF2; increase in CD69 and CD103) as well as transcription factors now functionally implicated in this phenotype (such as NR4A1, NR4A2, and Runx3) in several tissues. It is notable that some of the genes (such as Hobit) critically implicated in the biology of murine TRM cells are not expressed at high levels in their human counterparts. It is notable that in mouse models of viral infections such as lymphocytic choriomeningitis virus (LCMV), T cell memory has been largely studied when the underlying viral antigen is depleted. However, the biology in human tumors or other states of persistent viral infection may differ from LCMV models and local antigen may have important implications for TRM biology. Indeed, recent studies suggest that local antigen may drive proliferation of TRM cells in situ (28, 29).
While the infiltration of tumors by T cells has in general emerged as a strong indicator of improved prognosis, the presence of TRM cells within tumor-infiltrating lymphocytes (TILs) may be a particular driver of this correlation. The proportion of TILs that have TRM phenotype differs between studies (for example, from 25 to 75%) and may depend in part on the nature of specific markers utilized to identify these cells as well as the specific tissue/organ studied. This subset of cells may also be enriched for tumor reactivity, which is also consistent with other studies showing enrichment of tumor reactivity such as against tumor-associated neoantigens in CD8+ memory T cells with PD1+ phenotype (26, 30). Recent studies in murine models also suggest that these cells are important contributors to protective tumor immunity (31). In this study, the presence of TRM cells was modeled in the setting of autoimmune vitiligo and melanoma-specific TRM cells infiltrating these lesions were shown to mediate strong tumor protection. To date, most of the data relating to the biology of TRM cells in human tumor tissues are largely based on patients with solid tumors. Further studies are needed to better characterize this subset of T cells within hematologic malignancies. Below, we particularly focus on two aspects of the biology of tumor-associated TRM cells, their contribution to clinical responses to ICP blockade therapies and emergence of inter-lesional heterogeneity.
Are TRM Cells a Critical Target for ICP Blockade?
Antibody-mediated blockade of inhibitory ICPs such as programmed death-1 (PD-1) have led to impressive and durable clinical regressions in several cancers (32). This is remarkable as the expression of ICPs such as PD-1 is limited to only a subset of TILs (33). The principle of ICP blockade is based on the concept of unleashing the activity of pre-existing anti-tumor T cells against the tumor (34). Studies of T cell receptor (TCR) sequencing of T cells from patients receiving anti-PD1 therapy suggests that this therapy leads to in situ proliferation of CD8+ T cells within tumors of patients who respond to therapy (35). The ICP expressing T cells were found to include most of the tumor reactive T cells. While such tumor-reactive T cells can be detected in peripheral blood, these cells are predominantly present within the tumor tissue. In recent studies, we and others have shown that TRM cells are the dominant T cell subset expressing ICPs within the tumor microenvironment (11, 25). While most studies have described the presence of TRM cells within adult tumors recent data suggest that TRM cells are also enriched within pediatric tumors like glioma and are the T cell subset within these tumors that predominantly expresses ICPs (36). While TRM cells were initially identified in the tumor tissue based on the expression of classic TRM markers such as CD69 or CD103, gene expression studies confirmed that these T cells are a distinct subset of TILs with a genomic signature overlapping with TRM signature. Importantly, although CD69 is well studied as a T cell activation marker, the genomic profiles of CD69+ TRM cells are distinct from activated T cells and instead enriched for tissue retention genes (25). Therefore, while tumor tissue contains antigens recognized by these cells, and TRM cells express CD45RO consistent with memory T cells, they are genomically distinct from simply activated effector memory T cells. Recent studies in murine tumor models also support the importance of tumor-infiltrating TRM cells in mediating long-term control of melanoma tumors (31). The relationships between TRM cells and other populations such as stem memory cells implicated as targets of proliferative burst after PD-1 blockade need further study (37). Further studies are also needed to better characterize the proportion of tumor infiltrating TRM cells that are truly tumor specific.
The concept that TRM cells may be major targets of ICP blockade therapies is consistent with emerging insights into their functional properties. TRM cells seem to provide a dual role that encompasses both protection and regulation. Thus, while human TRM cells in NLTs can produce higher levels of effector cytokines, such as IFNγ, IL2, and TNF, they also produce higher levels of immune regulatory cytokines such as IL10 (14, 15). Moreover, TRM cells also express higher levels of ICPs, such as CTLA4, PD-1, TIM-3, and LAG-3 (14, 25). TRM cells also seem to have a quiescent phenotype, which may be essential for their ability to survive long-term in tissues, being poised for activation but not harming tissues (17). Antibody-mediated blockade of ICPs such as PD-1, therefore, provides a mechanism for activation of these T cells in situ. The precise nature of the activation signal may differ between CTLA4 and PD-1 blockade (or combination thereof) (38).
The concept that TRM cells within tumors may be major targets of ICP blockade has several implications for immune therapies. Vaccines that foster the generation of TRM cells may be best suited for combination with ICP blockade (39). The ability of TRM cells to mediate long-term residence in tissues may help to explain why clinical responses to ICP blockade have been durable. Along these lines, strategies that help to maintain or even enrich these TRM pools may allow enhanced durability of responses. It would also be important to better understand the nature of antigenic targets on tumors recognized by these T cells, and the impact of tumor genetics as well as other cells in the tumor microenvironment on the functional properties and retention of these cells.
Do TRM Cells Contribute to Intra-Tumor Heterogeneity of Tumors?
Advances in cancer genomics and particularly the capacity to sequence multiple lesions in the same patient or even different parts of the same tumor have demonstrated a complex and heterogeneous landscape with varying sub-clonal architecture; studies have also suggested a potential impact of such intra-tumoral heterogeneity on clinical outcome (40, 41). However, the degree to which the genetics of the microenvironment contributes to intra-tumoral heterogeneity is less clear. Diversity within the immune microenvironment may in principle not only impact the mechanisms underlying response or resistance to immune therapies but also evolution of tumors in individual metastases. Advances in TCR sequencing provide an opportunity to gain some insights into the nature and genetics of T cells infiltrating tumor lesions. While the same antigenic epitope may in principle be recognized by different TCRs, they are likely to differ in terms of their affinity or functional properties.
In the setting of advanced or metastatic cancer, tumor cells grow as discrete lesions in diverse NLTs. These lesions by definition share the systemic circulation of the host and could in principle be likened to the situation in parabiotic mice that share systemic circulation. As discussed earlier, a characteristic feature of TRM cells is tissue residence without recirculation, revealed by lack of equilibration in parabiotic mice. We hypothesized that if TRM cells within individual tissues (e.g., lung or liver or skin lesions) indeed remain local, then dominant TCRs within individual metastatic lesions in the same patient would not equilibrate even if the oncogenic mutations or neoantigen-load were largely shared between these lesions (Figure 1). Concurrent sequencing of tumor cells as well as TCRs from individual lesions in patients with advanced melanoma supported this hypothesis; as expected, the inter-lesional diversity of TCRs was mostly accounted for by TCRs from TRM subset of TILs (25). Differences in dominant TCRs between individual lesions from the same patient is consistent with lack of equilibration of TCRs between individual metastatic lesions even though they may share a major component of neoantigen load. However, the mechanisms that limit this equilibration need to be better defined; our current hypothesis is that it may relate to the lack of recirculation of tissue-resident TCRs, or their relative tissue retention, both consistent with TRM biology.
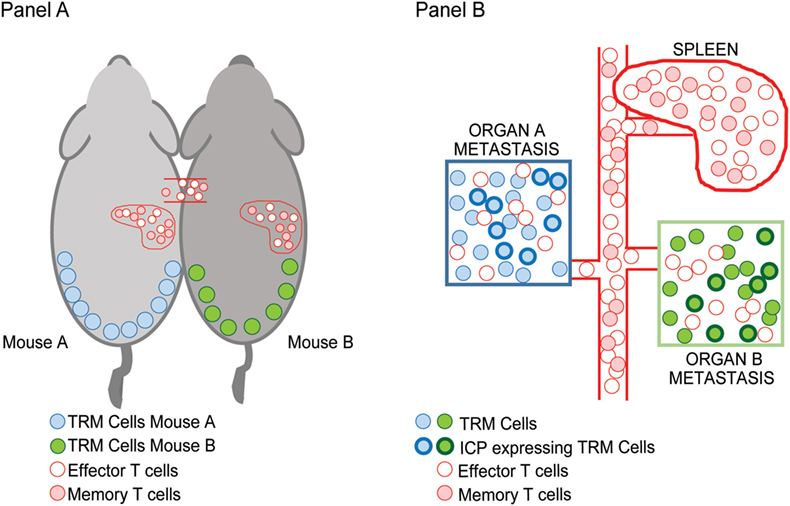
Figure 1. Inter-lesional heterogeneity in metastatic cancer and biology of tissue-resident memory (TRM) cells. TRM T cells were identified in mice based on their restriction to non-lymphoid tissues and lack of recirculation. This was demonstrated using parabiotic mice (A) that share the same systemic circulation. Figure shows that TRM cells in the skin (blue/green) do not equilibrate between mice, while other effector/memory T cells (pink/red) do. In the setting of advanced cancer in humans (B), individual metastatic lesions can be observed in diverse tissues that share systemic circulation analogous to the parabiotic mice. Sequencing of T cell receptors (TCRs) in individual lesions from the same patient demonstrated that dominant TCRs in each of the lesions were non-overlapping and that the inter-lesional heterogeneity of TCRs exceeded differences in neoantigens. Importantly, TRM cells were the major contributors to this heterogeneity suggesting that they do not equilibrate between lesions as in parabiotic mice in Ref. (25). A subset of TRM cells that infiltrate these tumors express inhibitory immune checkpoints such as PD1 (shown by bolded outlines).
The concept that TRM cells infiltrating tumor tissues may exhibit local residence and little recirculation has several implications for immune therapies, immune monitoring, and cancer biology. If the individual metastatic lesions are established early, and carry different TCRs, then the level of immune pressure in individual lesions may differ and provide a pathway for divergent genomic evolution (42). Along the same lines, it may be important to carefully consider the specific site of tissue biopsy when evaluating the results of immune monitoring. It should be noted, however, that the impact of ICP blockade on TRM homeostasis and redistribution in vivo in humans remains understudied and may add additional layers of complexity. Studies harvesting TILs for adoptive transfer are now entering the clinic in diverse cancers. If the dominant TCRs differ between individual lesions, it may be desirable to harvest and pool T cells from more than one lesion to optimize efficacy of such cell therapies. Finally, if the T cells in individual lesions differ, then it raises the potential that multiple mechanisms of immune resistance may be simultaneously operative in the same patient (43); along these lines, isolated progression at a single site in the face of continued regression at other sites may not reflect systemic loss of tumor control in the context of immune therapies. Clinicians have already come to appreciate this difference between immune therapies as compared to chemotherapies and often utilize localized therapies to tackle such lesions.
Summary
In summary, TRM cells within tumor lesions are likely to gain increasing importance as targets of immune therapies as well as deeper understanding of cancer biology and evolution. It is likely that optimal integration of these immune therapies will require attention to the unique biology of these immune cells and exploit their regional nature of enhance tumor immunity with reduced systemic toxicity.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi:10.1038/nrc3239
2. Farber DL, Netea MG, Radbruch A, Rajewsky K, Zinkernagel RM. Immunological memory: lessons from the past and a look to the future. Nat Rev Immunol (2016) 16(2):124–8. doi:10.1038/nri.2016.13
3. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39(1):1–10. doi:10.1016/j.immuni.2013.07.012
4. Bindea G, Mlecnik B, Angell HK, Galon J. The immune landscape of human tumors: implications for cancer immunotherapy. Oncoimmunology (2014) 3(1):e27456. doi:10.4161/onci.27456
5. Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol (2013) 31:137–61. doi:10.1146/annurev-immunol-032712-095954
6. Schenkel JM, Masopust D. Tissue-resident memory T cells. Immunity (2014) 41(6):886–97. doi:10.1016/j.immuni.2014.12.007
7. Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol (2009) 10(5):524–30. doi:10.1038/ni.1718
8. Schenkel JM, Fraser KA, Masopust D. Cutting edge: resident memory CD8 T cells occupy frontline niches in secondary lymphoid organs. J Immunol (2014) 192(7):2961–4. doi:10.4049/jimmunol.1400003
9. Mackay LK, Braun A, Macleod BL, Collins N, Tebartz C, Bedoui S, et al. Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J Immunol (2015) 194(5):2059–63. doi:10.4049/jimmunol.1402256
10. Sathaliyawala T, Kubota M, Yudanin N, Turner D, Camp P, Thome JJ, et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity (2013) 38(1):187–97. doi:10.1016/j.immuni.2012.09.020
11. Djenidi F, Adam J, Goubar A, Durgeau A, Meurice G, de Montpreville V, et al. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol (2015) 194(7):3475–86. doi:10.4049/jimmunol.1402711
12. Webb JR, Milne K, Nelson BH. Location, location, location: CD103 demarcates intraepithelial, prognostically favorable CD8 tumor-infiltrating lymphocytes in ovarian cancer. Oncoimmunology (2014) 3:e27668. doi:10.4161/onci.27668
13. Clark RA. Resident memory T cells in human health and disease. Sci Transl Med (2015) 7(269):269rv1. doi:10.1126/scitranslmed.3010641
14. Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, et al. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep (2017) 20(12):2921–34. doi:10.1016/j.celrep.2017.08.078
15. Hombrink P, Helbig C, Backer RA, Piet B, Oja AE, Stark R, et al. Programs for the persistence, vigilance and control of human CD8(+) lung-resident memory T cells. Nat Immunol (2016) 17(12):1467–78. doi:10.1038/ni.3589
16. Mackay LK, Minnich M, Kragten NA, Liao Y, Nota B, Seillet C, et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science (2016) 352(6284):459–63. doi:10.1126/science.aad2035
17. Boddupalli CS, Nair S, Gray SM, Nowyhed HN, Verma R, Gibson JA, et al. ABC transporters and NR4A1 identify a quiescent subset of tissue-resident memory T cells. J Clin Invest (2016) 126(10):3905–16. doi:10.1172/JCI85329
18. Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, et al. Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature (2017) 552(7684):253–7. doi:10.1038/nature24993
19. Iijima N, Iwasaki A. T cell memory. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science (2014) 346(6205):93–8. doi:10.1126/science.1257530
20. Mackay LK, Wynne-Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM, et al. T-box transcription factors combine with the cytokines TGF-beta and IL-15 to control tissue-resident memory T cell fate. Immunity (2015) 43(6):1101–11. doi:10.1016/j.immuni.2015.11.008
21. Purwar R, Campbell J, Murphy G, Richards WG, Clark RA, Kupper TS. Resident memory T cells (T(RM)) are abundant in human lung: diversity, function, and antigen specificity. PLoS One (2011) 6(1):e16245. doi:10.1371/journal.pone.0016245
22. Pallett LJ, Davies J, Colbeck EJ, Robertson F, Hansi N, Easom NJW, et al. IL-2(high) tissue-resident T cells in the human liver: sentinels for hepatotropic infection. J Exp Med (2017) 214(6):1567–80. doi:10.1084/jem.20162115
23. Okhrimenko A, Grun JR, Westendorf K, Fang Z, Reinke S, von Roth P, et al. Human memory T cells from the bone marrow are resting and maintain long-lasting systemic memory. Proc Natl Acad Sci U S A (2014) 111(25):9229–34. doi:10.1073/pnas.1318731111
24. Sercan Alp O, Durlanik S, Schulz D, McGrath M, Grun JR, Bardua M, et al. Memory CD8(+) T cells colocalize with IL-7(+) stromal cells in bone marrow and rest in terms of proliferation and transcription. Eur J Immunol (2015) 45(4):975–87. doi:10.1002/eji.201445295
25. Boddupalli CS, Bar N, Kadaveru K, Krauthammer M, Pornputtapong N, Mai Z, et al. Interlesional diversity of T cell receptors in melanoma with immune checkpoints enriched in tissue-resident memory T cells. JCI Insight (2016) 1(21):e88955. doi:10.1172/jci.insight.88955
26. Ganesan AP, Clarke J, Wood O, Garrido-Martin EM, Chee SJ, Mellows T, et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat Immunol (2017) 18(8):940–50. doi:10.1038/ni.3775
27. Edwards J, Wilmott JS, Madore J, Gide TN, Quek C, Tasker A, et al. CD103+ tumor-resident CD8+ T cells are associated with improved survival in immunotherapy naive melanoma patients and expand significantly during anti-PD1 treatment. Clin Cancer Res (2018) 24(13):3036–45. doi:10.1158/1078-0432.CCR-17-2257
28. Park SL, Zaid A, Hor JL, Christo SN, Prier JE, Davies B, et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat Immunol (2018) 19(2):183–91. doi:10.1038/s41590-017-0027-5
29. Muschaweckh A, Buchholz VR, Fellenzer A, Hessel C, Konig PA, Tao S, et al. Antigen-dependent competition shapes the local repertoire of tissue-resident memory CD8+ T cells. J Exp Med (2016) 213(13):3075–86. doi:10.1084/jem.20160888
30. Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J Clin Invest (2014) 124(5):2246–59. doi:10.1172/JCI73639
31. Malik BT, Byrne KT, Vella JL, Zhang P, Shabaneh TB, Steinberg SM, et al. Resident memory T cells in the skin mediate durable immunity to melanoma. Sci Immunol (2017) 2(10):eaam6346. doi:10.1126/sciimmunol.aam6346
32. Sznol M, Chen L. Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer. Clin Cancer Res (2013) 19(5):1021–34. doi:10.1158/1078-0432.CCR-12-2063
33. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi:10.1056/NEJMoa1200690
34. Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv Immunol (2006) 90:297–339. doi:10.1016/S0065-2776(06)90008-X
35. Ribas A, Shin DS, Zaretsky J, Frederiksen J, Cornish A, Avramis E, et al. PD-1 blockade expands intratumoral memory T cells. Cancer Immunol Res. (2016) 4(3):194–203. doi:10.1158/2326-6066.CIR-15-0210
36. Vasquez JC, Huttner A, Zhang L, Marks A, Chan A, Baehring JM, et al. SOX2 immunity and tissue resident memory in children and young adults with glioma. J Neurooncol (2017) 134(1):41–53. doi:10.1007/s11060-017-2515-8
37. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature (2016) 537(7620):417–21. doi:10.1038/nature19330
38. Das R, Verma R, Sznol M, Boddupalli CS, Gettinger SN, Kluger H, et al. Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J Immunol (2015) 194(3):950–9. doi:10.4049/jimmunol.1401686
39. Nizard M, Roussel H, Diniz MO, Karaki S, Tran T, Voron T, et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat Commun (2017) 8:15221. doi:10.1038/ncomms15221
40. Jamal-Hanjani M, Quezada SA, Larkin J, Swanton C. Translational implications of tumor heterogeneity. Clin Cancer Res (2015) 21(6):1258–66. doi:10.1158/1078-0432.CCR-14-1429
41. Sanborn JZ, Chung J, Purdom E, Wang NJ, Kakavand H, Wilmott JS, et al. Phylogenetic analyses of melanoma reveal complex patterns of metastatic dissemination. Proc Natl Acad Sci U S A (2015) 112(35):10995–1000. doi:10.1073/pnas.1508074112
42. Gerlinger M, McGranahan N, Dewhurst SM, Burrell RA, Tomlinson I, Swanton C. Cancer: evolution within a lifetime. Annu Rev Genet (2014) 48:215–36. doi:10.1146/annurev-genet-120213-092314
Keywords: tissue-resident memory cells, immune checkpoint blockade, tumor heterogeneity, cancer immunotherapy, immunity to cancer
Citation: Dhodapkar KM (2018) Role of Tissue-Resident Memory in Intra-Tumor Heterogeneity and Response to Immune Checkpoint Blockade. Front. Immunol. 9:1655. doi: 10.3389/fimmu.2018.01655
Received: 19 April 2018; Accepted: 04 July 2018;
Published: 16 July 2018
Edited by:
Fathia Mami-Chouaib, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Kimberly Sue Schluns, University of Texas MD Anderson Cancer Center, United StatesKim Klonowski, University of Georgia, United States
Copyright: © 2018 Dhodapkar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kavita M. Dhodapkar, a2F2aXRhLmRob2RhcGthckBlbW9yeS5lZHU=