 Yiliang Li
Yiliang Li Sapna Pradyuman Patel
Sapna Pradyuman Patel Jason Roszik
Jason Roszik Yong Qin
Yong Qin- 1Key Laboratory of Radiation Medicine and Molecular Nuclear Medicine, Institute of Radiation Medicine, Peking Union Medical College & Chinese Academy of Medical Sciences, Tianjin, China
- 2Department of Melanoma Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
Hypoxia is not only a prominent contributor to the heterogeneity of solid tumors but also a crucial stressor in the microenvironment to drive adaptations for tumors to evade immunosurveillance. Herein, we discuss the potential role of hypoxia within the microenvironment contributing to immune resistance and immune suppression of tumor cells. We outline recent discoveries of hypoxia-driven adaptive mechanisms that diminish immune cell response via skewing the expression of important immune checkpoint molecules (e.g., cluster of differentiation 47, programmed death ligand 1, and human leukocyte antigen G), altered metabolism and metabolites, and pH regulation. Importantly, inhibition of hypoxic stress-relevant pathways can collectively enhance T-cell-mediated tumor cell killing. Furthermore, we discuss how manipulation of hypoxia stress may pose a promising new strategy for a combinational therapeutic intervention to enhance immunotherapy of solid tumors.
Introduction
To survive and grow, tumors insulate themselves with various layers of immunosuppressive stroma to locally disable and/or evade the immune system. It is known that the tumor microenvironment contributes to the heterogeneity of the tumor and supports tumor growth and resistance to systemic therapies (1). In solid tumors, a hypoxic area is a common structural characteristic and some tumor cells exist in a hypoxic environment, whereas some exist in a vascularized area with sufficient oxygen supply (2). Numerous studies have shown that hypoxia is strongly associated with lower overall survival and disease-free survival of various tumor types (3–5). Notably, hypoxic tumor cells are considered to be more aggressive and more resistant to conventional systemic therapies and radiotherapy than non-hypoxic tumors (6, 7). Besides increasing the expression of various genes involved in angiogenesis and drug-resistance, hypoxia also promotes the selection of apoptosis-resistant clones and induces metastasis (8). Although the roles of hypoxic stress in the crosstalk among immune cells, stroma components, and tumor cells are not fully elucidated, it is widely appreciated that the hypoxic zone in solid tumors induces immune tolerance by impeding the homing of immune effector cells into tumors. Several regulatory mechanisms related to redundant levels of immune suppression and functional heterogeneity driven by hypoxia in the tumor microenvironment have been identified (9, 10).
Hypoxic Regulation of the Expressions of Immunosuppressive Molecules in the Tumor
The key mediators of hypoxic signaling are hypoxia-inducible factors (HIFs). HIFs are a family of transcription factors consisting of three alpha subunits, HIF-1α, HIF-2α, and HIF-3α that can heterodimerize with HIF-1β (11). HIF transcriptional activity is known to be oxygen-dependent. Under normoxic conditions, conserved proline residues on HIF-1α are hydroxylated by proly-4-hydroxylase (PHD), and the hydroxylated HIF-1α can be downregulated by ubiquitination and proteasomal degradation mediated by von Hippel−Lindau protein (11, 12) (Figure 1). Under hypoxic stress, induction and stabilization of HIF-1α and/or HIF-2α lead to upregulation of transcription of numerous hypoxia-responsive genes related to metabolic and immune pathways, and resulting in modulation of both metabolism and immunity of tumor and stromal cells (12, 13) (Figure 1). Particularly, hypoxia has been proven to contribute to increased angiogenesis through upregulation of interleukin 8 (IL-8), osteopontin, and vascular endothelial growth factor (13–17). Notably, hypoxic tumor microenvironment was correlated to a high expression of genes that promote epithelial–mesenchymal transition (EMT), including inhibitor of differentiation 2, snail 1 and 2 (SNAI1 and SNAI2), transcription factor 3, transforming growth factor alpha, twist transcription factor (TWIST), vimentin (VIM), and zinc finger E-box-binding homeobox 1 and 2 (ZEB1 and ZEB2) (18–22). Moreover, hypoxia has been shown to downregulate E-cadherin expression in tumors affecting cell–cell adhesion (22, 23). Recent evidence shows that the hypoxia-HIF-EMT niche is considered to support maintenance of cancer stem cells (24, 25). Hypoxic regions in tumors can lead to long-lasting HIF signaling, which is known to function as an oncogenic stimulus in some settings driving cancer development, invasion, immune suppression, and metastasis (25–27) (Figure 1).
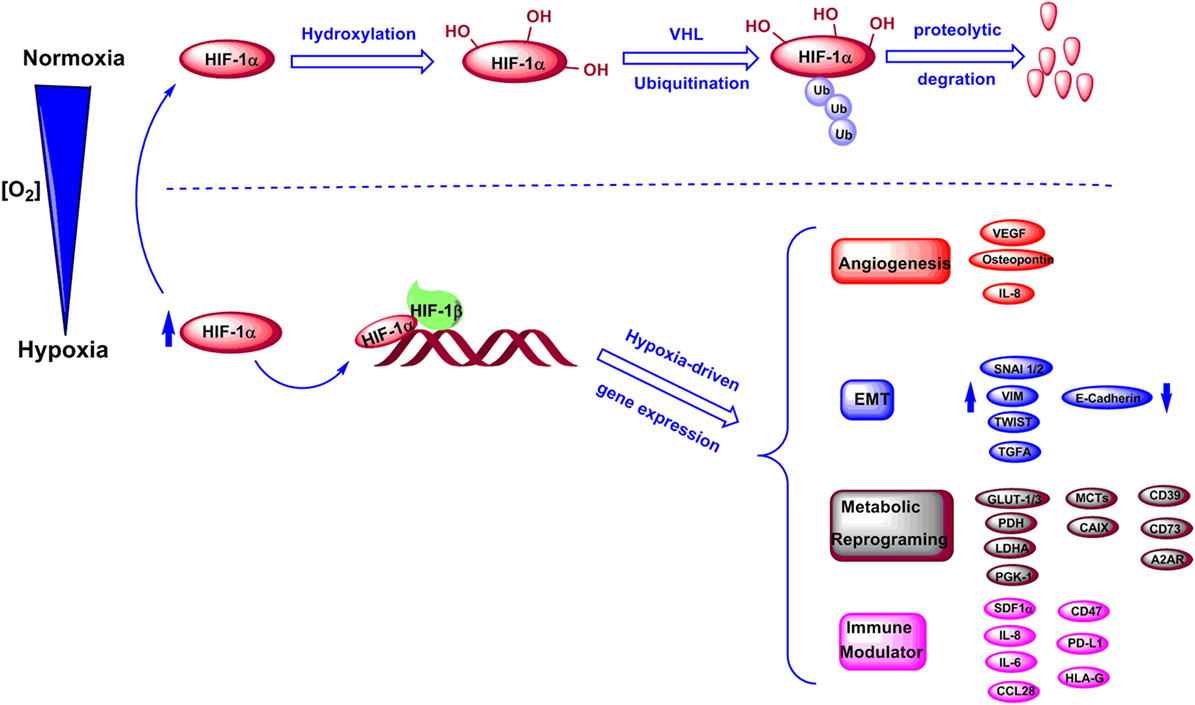
Figure 1. Regulation of HIF-1α levels and downstream genes under normoxic and hypoxic conditions. Under normoxic conditions, HIF-1α is hydroxylated and further undergoes degradation through an ubiquitination-dependent process mediated by VHL. Under hypoxic conditions, HIF-1α is stabilized and forms a complex with HIF-1β, which induces transcriptions of various genes involved in angiogenesis, epithelial–mesenchymal transition (EMT), metabolic reprogramming, and immune regulation.
In addition to tumor cells and stromal cells, the hypoxic regions in solid tumors have been found to be infiltrated by high levels of immunosuppressive cells, such as myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and T-regulatory (Treg) cells (10, 28). The mechanisms of recruitment and function of these immunosuppressive cells within the hypoxic tumor microenvironment have also been studied (29). Hypoxia has been shown to induce the production of stromal cell-derived factor 1α (SDF1α) by hypoxic tumor cells, which binds to C-X-C chemokine receptor type, IL-8, and IL-6, and directly regulates the function and differentiation of MDSCs within the tumor microenvironment (30, 31). Moreover, MDSCs derived from hypoxic tumor regions show stronger immunosuppressive function than splenic MDSCs. The phenomenon is mostly due to the HIF-1α-driven increased arginase activity and nitric oxide production of tumor MDSCs (32). Several studies have confirmed that hypoxia is strongly associated with the selective accumulation of Tregs in tumors, which not only suppress antitumor response but also promote neo-angiogenesis (33–35). The most direct evidence for hypoxia providing a tolerogenic tumor microenvironment for Tregs is the fact that hypoxia-driven HIF-1α strongly increases the expression of forkhead box P3 (Foxp3), which is a distinct marker and a master regulator in the development and function of Tregs (36–40). Tumor cells under hypoxia can also produce high levels of C-C motif chemokine ligand 28, which is an important chemokine to selectively attract CXCR10-positive Tregs in tumors resulting in antigen tolerance and angiogenesis (34).
Beside regulation of immunosuppressive MDSCs and Tregs within the tumor, recent studies showed that hypoxia promotes immune evasion through HIF-1α-dependent upregulation of immune checkpoint proteins in tumors (26). Of interest, three important checkpoint molecules, such as cluster of differentiation 47 (CD47), programmed death ligand 1 (PD-L1), and human leukocyte antigen G (HLA-G), have been shown by multiple reports to be modulated by hypoxic conditions and to contribute to an immunosuppressive microenvironment in tumors.
Hypoxia-Driven CD47 Expression in Tumors
Cluster of differentiation 47, also known as integrin-associated protein, has been identified as a membrane protein interacting with β3 integrin, thrombospondin-1, and signal regulatory protein-alpha (SIRPα) to regulate various important cellular functions, including cell migration, cytokine production, T cell activation, and cancer metastasis (41–45). Overexpression of CD47 has been found in a number of tumors, such as acute myeloid leukemia, non-small cell lung cancer, bladder cancer, non-Hodgkin’s lymphoma, and breast cancer (46–53). Based on the analysis of datasets derived from thousands of primary tumors of breast cancer patients, CD47 expression was not only significantly correlated with patient survival but also correlated with the expression of HIF-1α targeting genes (53). A recent study from Semenza’s group showed that HIF-1α activated transcription of CD47 in mesenchymal triple-negative primary breast cancer cells (SUM159) and promoted the breast cancer stem cell phenotype, which further protected cancer cells from phagocytosis by bone marrow-derived macrophages (53). Thus, hypoxic cancer cells evade innate immunity through HIF-1α-dependent expression of CD47. CD47 is emerging as a negative checkpoint for innate immunity and subsequent adaptive immunity in tumors (54). One important mechanism for CD47-mediated immune evasion is that binding of CD47 to SIRPα, which is abundantly expressed on myeloid-linage hematopoietic cells such as TAMs and MDSCs, causes phosphorylation of the SIRPα ultimately resulting in delivering an anti-phagocytic signal (46, 48, 54, 55). The CD47–SIRPα axis not only directly functions as a negative checkpoint of innate immunity but also affects adaptive immunity. In a series of studies, anti-CD47 blockade significantly increased the presence of IFN-γ-expressing antigen-specific CD8+ T cells and promoted T cell-mediated destruction of tumor cells (56, 57). Thus, the CD47–SIRPα axis has become an attractive target for developing novel cancer immunotherapies, and anti-CD47 blockades are currently investigated by several clinical trials in various solid tumors (54) (Table 1).
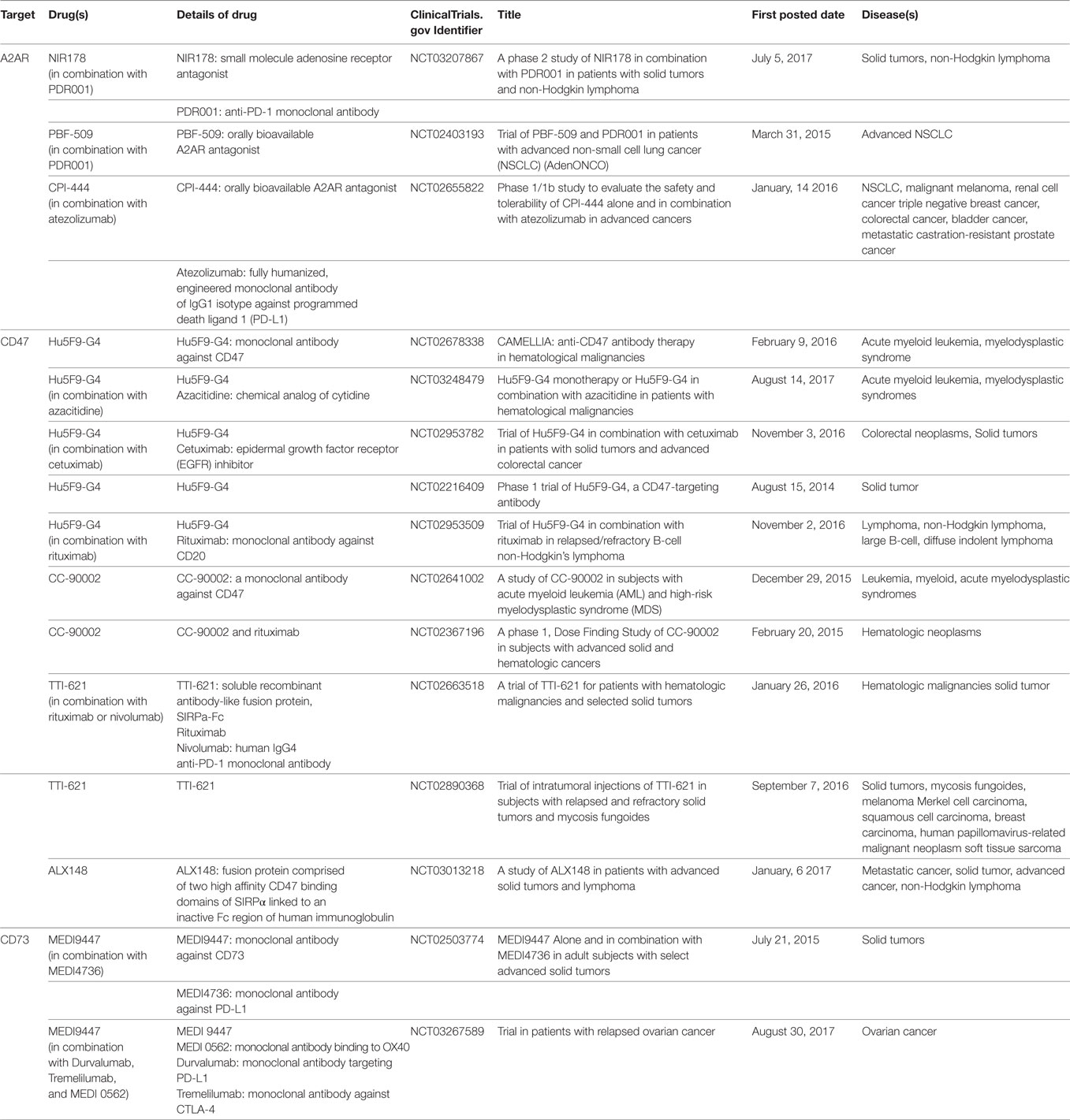
Table 1. List of drugs targeting A2A adenosine receptor (A2AR), cluster of differentiation 47 (CD47), and CD73 currently being investigated in clinical trials.
Hypoxia-Driven PD-L1 Expression in Tumors
One crucial mechanism by which cancer cells block antitumor immunity is through expression of PD-L1, which binds to the cell surface checkpoint receptor PD-1 on effector T cells to inhibit their activation (58). Accumulated studies confirm that hypoxia can strongly induce HIF-1α-dependent PD-L1 expression on tumor cells, macrophages, and dendritic cells (59–61). In human DU145 metastatic prostate and MDA-MB-231 metastatic breast carcinoma cells, hypoxia-induced expression of PD-L1 has been confirmed to be HIF-1α-dependent (59). The elevated expression of PD-L1 in cancer cells under hypoxic conditions leads to increased apoptosis of cultured cytotoxic T lymphocytes (CTLs) and Jurkat leukemia T cells (59). These observations indicate a mechanism by which hypoxic tumors upregulate PD-L1 expression on tumor cells to promote immune escape from CTLs. A study of B16-F10 melanoma-bearing mice models showed that hypoxia also selectively caused a rapid up-regulation of PD-L1 on splenic MDSCs (60). The up-regulation of PD-L1 in MDSCs under hypoxia was confirmed to be dependent on HIF-1α but not HIF-2α (60). Blockade of PD-L1 under hypoxia enhanced MDSC-mediated T cell activation and was accompanied by decreased production of IL-6 and IL-10 by MDSCs (60). The potential mechanism of hypoxic stress upregulating PD-L1 in tumors may rely on simultaneous binding of HIF-1α and pyruvate kinase M2 (PKM2) to hypoxia response elements (HRE) in the PD-L1 promoter (61). As shown in the study of O’Neill’s group, the inhibition of PKM2 by a small reagent or specific siRNA could lead to downregulation of PD-L1 expression on macrophages, MDSCs, and tumor cells (61). Thus, hypoxia can promote an immunosuppressive microenvironment by recruiting MDSCs to hypoxic regions and increase checkpoint PD-L1 expression on MDSCs and tumor cells. According to the study of a large number of malignant primary tumor tissues from pheochromocytomas and paragangliomas, PD-L2 expression but not PD-L1 expression is significantly associated with stronger hypoxia-driven HIF-1α and carbonic anhydrase 9 (CAIX) (62). These studies suggest that simultaneous blockade of PD-L1/PD-L2 along with inhibition of HIF-1α may represent a promising approach to enhance the activity of cytotoxic T cells.
Hypoxia-Driven HLA-G Expression in Tumors
The non-classic major histocompatibility complex (MHC) class I molecule is known as an immune checkpoint molecule with specific relevance in cancer immunotherapy (63, 64). HLA-G is a crucial MHC-I molecule and plays an essential role in maintaining immune tolerance and inhibiting the functions of immunocompetent cells to support tumor cells escape from immunosurveillance (64). The immunosuppressive function of HLA-G is mediated by the direct binding of HLA-G to relevant inhibitory receptors. Three HLA-G receptors have been identified: immunoglobulin-like transcript 2 (ILT2, CD85j) expressed by B cells, T cells, natural killer cells (NK cells), and myelomonocytic cells, ILT4 (CD85d) expressed by dendritic cells, monocytes, and macrophages, and KIR2DL4 (CD158d) expressed by NK cells (65, 66). Through these inhibitory receptors, HLA-G can interact with B cells, T cells, NK cells, and antigen-presenting cells, and exert its immunosuppressive functions at different stages of the immune response (65, 66). HLA-G expression is very restricted in adult normal tissues, but is frequently induced in numerous malignant tumors, such as glioblastoma, melanoma, and cervical tumors, contributing to their immune escape (67–70). The expression of HLA-G on cancer cells is also found to associate with a higher tumor grade and poor prognosis, such as in primary and metastatic ovarian tumors and primary colorectal tumors (71–73). Several HREs have been identified in HLA-G promoter and non-promoter regions (74). Recent studies showed that the expressions of HLA-G mRNA and protein were upregulated in HLA-G-negative cancer cells via HIF-1α under hypoxic conditions (74–76). However, in cancer cells expressing HLA-G constitutively, hypoxia decreases HLA-G gene expression (73). It is still unclear why the expression patterns of HLA-G in HLA-G-negative and HLA-G-positive cancer cells are so different upon hypoxic stress. Since HLA-G is considered an immune checkpoint molecule, augmentation of HLA-G expression in hypoxic tumor cells may contribute to immunosuppression in tumors. Up till now, several reports showed that hypoxia upregulates HLA-G expression in human cancer cells, but very few studies have been published on the effects of hypoxia on other MHC-I molecules in tumors. A recent study showed that combining hypoxic stress and glucose deprivation increased surface expression of HLA-E in human and Qa-1 in mouse tumor cells (77). Further studies are needed to address how hypoxic microenvironment modulates MHC-I and MHC-II molecules in tumors.
Hypoxia-Driven Immunosuppressive Metabolites
In order to support rapid growth of tumor cells, hypoxic signaling permits tumor cells to sense and adapt to low O2 stress and carbon source availability by re-programming their metabolism and gene expression via HIF’s transcriptional regulation. Under hypoxia, tumor cells switch to glycolysis to continue ATP production and prevent O2-dependent oxidative phosphorylation (78). Also, metabolic intermediates from glycolysis can be utilized for the biosynthesis of other macromolecules. HIF-1α plays a critical role in the glycolytic switch to increase glucose utilization in hypoxic tumor cells by upregulating the expression of glucose transporters and glycolytic enzymes, such as glucose transporters 1 and 3 (GLUT1 and GLUT3), pyruvate dehydrogenase, lactate dehydrogenase A (LDHA), phosphoglycerate kinase 1, and hexokinases 1 (HK1) (78). The increase of these gene’s expressions alters glucose metabolism and prevents glucose entry into the tricarboxylic acid (TCA) cycle and reducing acetyl coenzyme A (CoA) production from pyruvate (78). Reprogramming glucose flux is considered as a major factor to shape the tumor microenvironment via increase of HIF-1α levels in rapidly growing tumor cells within hypoxic regions.
Hypoxia-Driven Acid–Base Regulation and Production of Immuno-Modulatory Lactate
A notable feature of solid tumors is the presence of an acidic extracellular tumor microenvironment mainly due to the production of large amounts of acidic metabolites by glycolytic tumor cells (79). As a consequence of glycolysis, hypoxic tumor cells upregulate LDHA and convert pyruvate into lactic acid, which results in increased tumor acidosis (79–81). At the same time, tumor cells also adapt to intracellular acidification by enhancing export of lactate/H+. MCT1 and MCT4 are major players from the monocarboxylate transporter (MCT) family to preferentially transport lactate/H+ across the plasma membrane (82, 83). Noteworthy, MCTs are among those genes that are upregulated under hypoxic condition (82–84). The export of lactate/H+ by MCT has been found to not only contribute to the acidosis of tumor microenvironment but also to promote tumor cell metastasis, angiogenesis, and suppressing immunosurveillance (82, 83).
In order to provide sufficient energy for rapidly growing tumors, cancer cells use substitute carbon sources, like glutamine, to promote the TCA cycle and maintain oxidative phosphorylation under hypoxic microenvironment (85, 86). Besides lactate, increased levels of CO2 generated by oxidative metabolism are another major source for tumor acidity. Indeed, CO2 produced by the TCA cycle and the pentose phosphate pathway under hypoxia can be hydrated by carbonic anhydrases (CA) and converted into bicarbonate (HCO3−) and protons (H+) (87, 88). To balance intracellular acidosis in hypoxic tumor cells, the HCO3– are imported back into cells through bicarbonate transporters and anion exchange, but the H+ remains extracellularly and contributes to an increasingly acidic tumor microenvironment (88, 89). Carbonic anhydrase IX (CAIX) plays a key role in pH regulation in hypoxic cancer cells as its expression is induced by hypoxia via HIF-1α (89, 90). Together, the key pH regulatory components, such as MCT4 and CAIX, are upregulated in hypoxic tumor cells leading to the acidification of the tumor microenvironment (Figure 2).
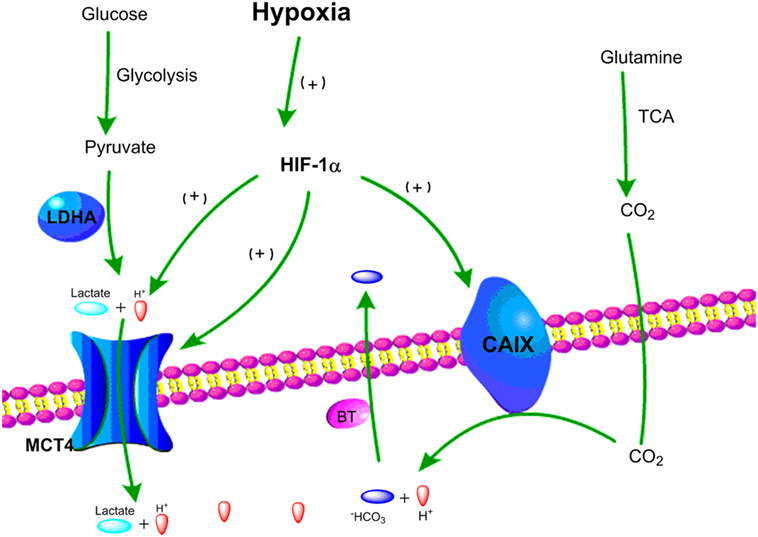
Figure 2. Schematic representation of two major pathways of hypoxia-driven extracellular acidity by tumor cells. (1) Under hypoxic conditions, glucose goes through glycolysis and produces pyruvate, which is mainly converted to lactic acid by lactate dehydrogenase A (LDHA). Lactic acid is exported by MCT4 resulting in acidification of the tumor microenvironment. (2) Under hypoxic conditions, glutamine goes through tricarboxylic acid (TCA) cycle and releases CO2, which can be converted into HCO3− and H+. HCO3− is transported back to tumor cells by bicarbonate transporters (BT), and accumulation of extracellular H+ leads to tumor acidosis.
Due to the limitation of technology to directly and accurately measure the pH within the hypoxic region of tumors, our current knowledge on the effects of cancer acidity on T cells is based on studies applying in vitro cultures to stimulate immune cells at low pH. Under low pH conditions (≤6.6), the secretion of IL-2, tumor necrosis factors, and IFN-γ was impaired in T lymphocytes upon stimulation with anti-CD3 antibody and phytohemagglutinin (PHA) (79, 91, 92). However, IFN-γ R2 chain (IFNR2) and CTLA-4 expressions were upregulated under the same condition which rendered tumor-infiltrating T cells sensitive to negative regulatory signaling. Moreover, the cytotoxicity of antigen-specific T cells appeared to be highly sensitive to low pH. In the in vitro study by Takahashi’s group, the cytotoxic activity of CD8+ CTLs decreased in a pH-dependent manner and the induction of functional CTLs were markedly inhibited under low pH (93). Tumor acidity also plays a critical role in assisting tumor cells escape from NK cell-mediated cytolysis. Several in vitro studies showed that the cytotoxic activities of NK cells and lymphokine-activated killer (LAK) cells were markedly reduced under acidic conditions (79, 94–96). Indeed, NK activation and LAK generation by IL-2 were inhibited at an extracellular culture condition below pH 7.2 (97). Notably, the low environmental pH caused irreversible damage to NK and LAK cells resulting in a permanent decrease of the cytotoxic activity of these cells, which could not be recovered upon switching to higher pH culture medium (97). A handful studies showed that tumor acidity contributed to the direct induction of a series of pro-inflammatory molecules in tumor-associated myeloid cells (79, 98). Extracellular acidosis has been shown to activate PI3K/Akt and ERK pathways and led to the stimulation of human neutrophils, which resembled the functional profile of MDSCs (99).
Although lactic acid is not the major contributor to the extracellular acidification of tumors, more and more data underline the important role of lactate as a “signaling molecule” involved in regulating cancer cell survival, proliferation, and metastasis (100, 101). Importantly, current studies indicate that lactate is emerging as an important immunosuppressive metabolite promoting escape of immune surveillance in hypoxic tumors (101). Lactate generated by hypoxic tumor cells was found to strongly inhibit the anti-tumor immune response via attenuating the cytotoxic activity of human CTLs (102, 103) and NK cells (104, 105). Previous studies also showed that lactate not only inhibited dendritic cells releasing cytokines but also impeded the differentiation and activation of monocyte-derived dendritic cells (106–109). A study using a pancreatic cancer mouse model further demonstrated that tumor-derived lactate could directly inhibit cytolytic function of NK cells. Moreover, lactate could recruit and increase the number of MDSCs in tumor to indirectly inhibit NK cytotoxicity (104). Lactate also acts as an important signaling molecule to promote the production of cytokines such as IL-23 and IL-6 contributing to tumor-associated inflammation (104, 109). Recently, lactate has been identified as a valuable prognostic marker of disease progression and poor patient survival especially in primary carcinomas, including cervical cancer, rectal adenocarcinoma, glioblastoma, and prostate cancer (110–117).
Hypoxia-Driven Production of Immunosuppressive Adenosine
One important immunomodulatory metabolite which accumulates in hypoxic tumors is adenosine. It is known that hypoxia can upregulate the expression of CD39 and CD73. Under hypoxia, the nucleotide metabolism mainly undergoes phosphohydrolysis via ectonucleoside triphosphate diphosphohydrolase CD39 that converts ATP/ADP to AMP. Then, 5′-ectonucleotidase CD73 converts AMP to adenosine (118–120). The CD39–CD73-adenosine signaling represents an important pathway to generate extracellular adenosine. Moreover, HIF-1α inhibits the intracellular adenosine kinase to prevent re-phosphorylating adenosine to AMP resulting in elevated levels of intracellular adenosine. The high level intracellular adenosine is subsequently transported into extracellular space (120, 121). Hypoxia also enhances adenosine signaling by increasing the expression of A2A adenosine receptor (A2AR), which is a key G-protein coupled receptors conducting adenosine singling (121, 122). The adenosine accumulated in the tumor microenvironment acts as a negative regulator for both the activation and effector phases of the anti-tumor T cell response (123–126). The binding of adenosine to A2AR on T cells can lead to T cells apoptosis, which contributes to tumor immune evasion (125–127). Thus, the adenosine–CD73 axis represents a hypoxia-driven immunosuppressive mechanism in solid tumors (Figure 3). The high concentration of extracellular adenosine is usually present in cancer tissues, which is an important mediator in the alteration of immune cell functions to drive the immunosuppressive microenvironment in tumors (125, 128).
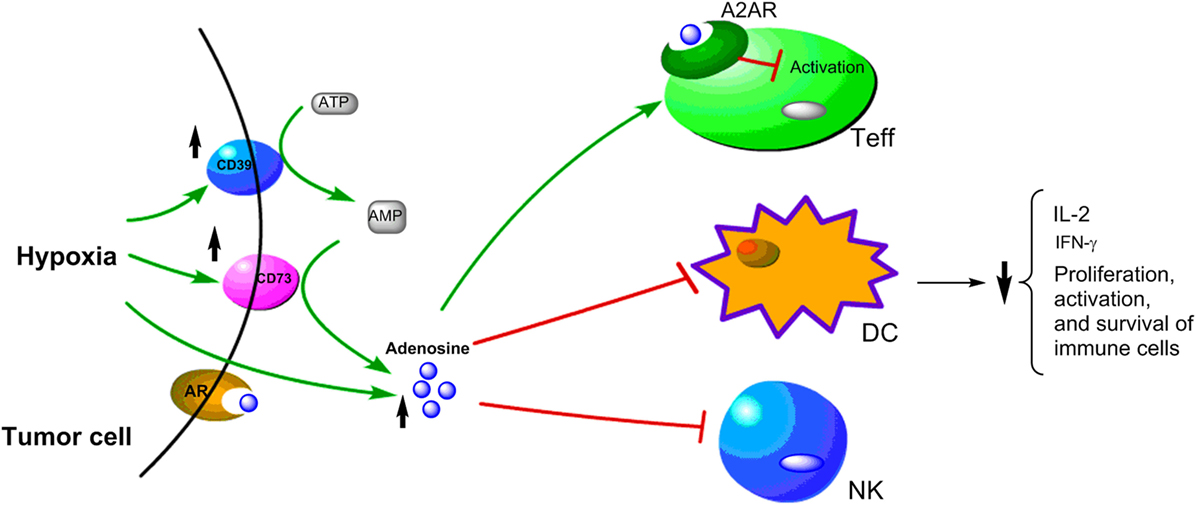
Figure 3. Schematic representation of hypoxia upregulating immunosuppressive adenosine signaling pathway in cancer cells. Under hypoxic conditions, the upregulation of CD39 and CD79 expressions lead to increase of adenosine, which has been shown to have immunosuppressive effects on Teff, DC, and NK cells.
Targeting Hypoxia-Driven Metabolic Pathways to Enhance the Efficacy of Immunotherapy of Tumors
As a hallmark of solid tumors, hypoxia is known to mediate aggressive, metastatic, and resistant characteristics. For a long time, hypoxia-relevant signaling pathways have been among the most attractive therapeutic targets in cancer drug development. Various approaches have been proposed to target hypoxic tumor cells, including hypoxia-activate prodrugs, gene therapy, recombinant anaerobic bacteria, small inhibitors specifically targeting HIFs, or targeting important downstream components of hypoxic pathways such as, mTOR and URP pathways (129–131). These approaches have been comprehensively discussed in multiple reviews (129–131). Although much effort has been put into investigating drugs targeting hypoxic pathways in clinical trials; results have been generally disappointing (132, 133). There is an urgent need to improve our understanding of the complexity of hypoxic pathways and their roles in solid tumors for drug development.
To date, there are no approved drugs that directly inhibit the HIF pathway. It is known that hypoxia-driven HIF-1 regulates a highly complex network involving multiple signaling cascades and overlapping mechanisms. The failure of clinical studies targeting hypoxic pathways in tumors may be partially due to the lack of specificity of inhibitors and redundancy in hypoxic signaling/metabolism, which impede the efficacy of drugs. Most of the reported HIF-1 inhibitors were originally designed to target other molecules, and they were found to have HIF-1 inhibitory effect later. The development of specific inhibitors of HIF-1 represents a challenge, which is mainly due to the difficulty of targeting transcription factors to selectively interrupt protein–DNA or protein–protein interactions without affecting other pathways. Although it is assumed that hypoxic regions exist in most solid tumors, the inhibitors of HIF-1 or other hypoxia-relevant molecules may be less effective in the patients that do not have high levels of HIF-1. Therefore, the lack of specific patient selection may also contribute to the failure of those clinical trials of HIF-1 inhibitors, in which the selection of patients were not directly based on the HIF levels in tumors (129–133). The hypoxic microenvironment is considered to be a major contributor but not a driving force for tumor progression and metastasis. Thus, understanding the mechanism of hypoxic pathways and their interaction with other pathways in tumors is of particular importance. Future directions would be directed toward developing potent and more specific inhibitors targeting hypoxia-relevant molecules, which can be used in combination therapies and will hopefully overcome hypoxia-driven resistance.
Currently, one of the most promising treatments for metastatic melanoma and several other cancers is checkpoint blockade immunotherapy. In contrast to the direct cytotoxic effects of chemotherapy, checkpoint blockade relies on antigen-specific T cell responses by blunting tumor-induced immunoregulatory mechanisms. This form of treatment has provided durable, long-lasting responses in many patients, largely due to the persistence and adaptability of the immune system. As summarized in previous sections, hypoxia plays a crucial role in immunoregulatory networks to promote an immunosuppressive tumor microenvironment. Interventions in several critical hypoxic axes emerge as promising adjuvants for a variety of immunotherapies.
One crucial enzyme mediating hypoxia-driven immunosuppression in solid tumor is CAIX. CAIX is upregulated by HIF-1α and represents a prototypic tumor-associated antigen. Overexpression of CAIX was found in metastatic renal cell carcinoma (RCC), and decreased CAIX levels are independently associated with poor survival in advanced RCC (134). A monoclonal antibody specifically targeting CAIX has been developed. In the study from Marasco’s group, human anti-CAIX mAbs not only inhibited CAIX enzymatic activity but also promoted immune-mediated killing of RCC by NK cell-mediated antibody-dependent cell-mediated cytotoxicity, complement-dependent cytotoxicity, and macrophage-mediated antibody-dependent cell-mediated cytotoxicity. This study demonstrated that targeting CAIX could induce an immune response to inhibit CAIX-positive tumor growth in vivo through tumor infiltration of NK cells and activation of T cells (135). Thus, the anti-CAIX reagent presents a therapeutic potential for the unmet medical need of targeted killing of HIF-1α-driven CAIX-positive RCC. Girentuximab has been developed as a chimeric monoclonal antibody drug specifically against CAIX. The current report from the phase 3 clinical trial of girentuximab in clear cell renal cell carcinoma (ccRCC) showed that participants treated with girentuximab had no statistically significant disease-free survival (hazard ratio, 0.97; 95% CI, 0.79–1.18) or OS advantage (hazard ratio, 0.99; 95% CI, 0.74–1.32) compared to the placebo group (136). Although girentuximab had no clinical benefit as adjuvant treatment for patients with high-risk ccRCC, it is still not clear whether girentuximab facilitates releasing the immunosuppression driven by hypoxic CAIX in tumors.
Although immune checkpoint blockade, such as anti-PD-1 therapy, has led to dramatic responses in some cancer patients, overall response rates are still less than 30% (58), which likely reflects the fact there are multiple layers and redundant mechanisms of immune evasion in solid tumors. Thus a single targeted therapy or immunotherapy is insufficient to restore antitumor immunity to clear various highly heterogeneous tumor cells. The hypoxia-dependent metabolic reprogramming also contributes to immune evasion, as T cells subjected to glucose deprivation (due to increased glucose uptake by hypoxic cancer cells) have diminished antitumor effector functions (26).
The binding of hypoxia-driven adenosine to A2AR could protect tumor cells from immune clearance by inhibiting T cells response. High level of A2AR expression has been confirmed in primary tumor tissues of head and neck squamous cell carcinoma (HNSCC), and it was significantly correlated with HIF-1α, CD73, CD8, and Foxp3 (137). Moreover, increased expression of A2AR on tumor infiltrating immune cells has been shown to correlate with advanced pathological grade, larger tumor size and positive lymph node status in primary HNSCC (137). The in vivo study of HNSCC mouse model showed that the A2AR antagonist, SCH58261, not only delayed the tumor growth but also significantly reduced the population of CD4+ Foxp3+ Tregs and increased the anti-tumor response of CD8+ T cells in HNSCC tumors (137). This preclinical study indicates that A2AR blockade can be a potential strategy to enhance immunotherapy in HNSCC. Other key mediators in hypoxia-driven immunosuppression that also draw a great attention for drug development include CD73 and CD47. Multiple therapeutic approaches targeting A2AR, CD73, or CD47 are currently being investigated in clinical trials (Table 1). Given the unique roles of these targets in modulating immunosuppression within hypoxic tumors, the specific therapeutics targeting these are expected to work as potential boosters to synergize with other immunotherapies and offer opportunities to enhance anti-tumor activity of immune effector cells.
Author Contributions
YQ, SP, JR, and YL wrote the review. All authors approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We thank Leyuan Chen (Institute of Radiation Medicine, Peking Union Medical College) for support on the edition of figures in this article.
Funding
This work has been supported by generous philanthropic contributions to The University of Texas MD Anderson Cancer Center Moon Shots Program, the MD Anderson Institutional Research Grant Award #40080 (PI: SP), the NIH/NCI 1R21CA208609 Grant Award (PI: SP), and the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (CIFMS 2016-12M-3-022 and 2017-12M-3-019, PI: YL).
References
1. Qin Y, Roszik J, Chattopadhyay C, Hashimoto Y, Liu C, Cooper ZA, et al. Hypoxia-driven mechanism of vemurafenib resistance in melanoma. Mol Cancer Ther (2016) 15:2442–54. doi:10.1158/1535-7163.MCT-15-0963
2. Jiang L, Greenwood TR, Artemov D, Raman V, Winnard PT Jr, Heeren RM, et al. Localized hypoxia results in spatially heterogeneous metabolic signatures in breast tumor models. Neoplasia (2012) 14:732–41. doi:10.1593/neo.12858
3. Evans SM, Koch CJ. Prognostic significance of tumor oxygenation in humans. Cancer Lett (2003) 195:1–16. doi:10.1016/S0304-3835(03)00012-0
4. Höckel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst (2001) 93:266–76. doi:10.1093/jnci/93.4.266
5. Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev (2007) 26:225–39. doi:10.1007/s10555-007-9055-1
6. Nordsmark M, Overgaard J. Overgaard, tumor hypoxia is independent of hemoglobin and prognostic for loco-regional tumor control after primary radiotherapy in advanced head and neck cancer. Acta Oncol (2004) 43:396–403. doi:10.1080/02841860410026189
7. Brizel DM, Dodge RK, Clough RW, Dewhirst MW. Oxygenation of head and neck cancer: changes during radiotherapy and impact on treatment outcome. Radiother Oncol (1999) 53:113–7. doi:10.1016/S0167-8140(99)00102-4
8. Casazza A, Di Conza G, Wenes M, Finisguerra V, Deschoemaeker S, Mazzone M. Tumor stroma: a complexity dictated by the hypoxic tumor microenvironment. Oncogene (2014) 33:1743–54. doi:10.1038/onc.2013.121
9. Noman MZ, Chouaib S. Targeting hypoxia at the forefront of anticancer immune responses. Oncoimmunology (2014) 3:e954463. doi:10.4161/21624011.2014.954463
10. Noman MZ, Hasmim M, Messai Y, Terry S, Kieda C, Janji B, et al. Hypoxia: a key player in antitumor immune response. A review in the theme: cellular responses to hypoxia. Am J Physiol Cell Physiol (2015) 309:C569–79. doi:10.1152/ajpcell.00207.2015
11. Bracken CP, Whitelaw ML, Peet DJ. The hypoxia-inducible factors: key transcriptional regulators of hypoxic responses. Cell Mol Life Sci (2003) 60:1376–93. doi:10.1007/s00018-003-2370-y
12. Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity (2014) 41:518–28. doi:10.1016/j.immuni.2014.09.008
13. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell (2012) 148:399–408. doi:10.1016/j.cell.2012.01.021
14. Kwon WK, Moon HJ, Kwon TH, Park YK, Kim JH. The role of hypoxia in angiogenesis and extracellular matrix regulation of intervertebral disc cells during inflammatory reactions. Neurosurgery (2017) 81:867–75. doi:10.1093/neuros/nyx149
15. Potier E, Ferreira E, Andriamanalijaona R, Pujol JP, Oudina K, Logeart-Avramoglou D, et al. Hypoxia affects mesenchymal stromal cell osteogenic differentiation and angiogenic factor expression. Bone (2007) 40:1078–87. doi:10.1016/j.bone.2006.11.024
16. Zhang J, Sattler M, Tonon G, Grabher C, Lababidi S, Zimmerhackl A, et al. Targeting angiogenesis via a c-Myc/hypoxia-inducible factor-1alpha-dependent pathway in multiple myeloma. Cancer Res (2009) 69:5082–90. doi:10.1158/0008-5472
17. Takahashi F, Akutagawa S, Fukumoto H, Tsukiyama S, Ohe Y, Takahashi K, et al. Osteopontin induces angiogenesis of murine neuroblastoma cells in mice. Int J Cancer (2002) 98:707–12. doi:10.1002/ijc.10261
18. Wu MZ, Tsai YP, Yang MH, Huang CH, Chang SY, Chang CC, et al. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol Cell (2011) 43(5):811–22. doi:10.1016/j.molcel.2011.07.012
19. Joseph JP, Harishankar MK, Pillai AA, Devi A. Hypoxia induced EMT: a review on the mechanism of tumor progression and metastasis in OSCC. Oral Oncol (2018) 80:23–32. doi:10.1016/j.oraloncology.2018.03.004
20. Gunaratnam L, Morley M, Franovic A, de Paulsen N, Mekhail K, Parolin DA, et al. Hypoxia inducible factor activates the transforming growth factor-alpha/epidermal growth factor receptor growth stimulatory pathway in VHL(-/-) renal cell carcinoma cells. J Biol Chem (2003) 278(45):44966–74. doi:10.1074/jbc.M305502200
21. Katoh M, Katoh M. Integrative genomic analyses of ZEB2: transcriptional regulation of ZEB2 based on SMADs, ETS1, HIF1alpha, POU/OCT, and NF-kappaB. Int J Oncol (2009) 34(6):1737–42. doi:10.3892/ijo_00000304
22. Krishnamachary B, Zagzag D, Nagasawa H, Rainey K, Okuyama H, Baek JH, et al. Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res (2006) 66(5):2725–31. doi:10.1158/0008-5472.CAN-05-3719
23. Esteban MA, Tran MG, Harten SK, Hill P, Castellanos MC, Chandra A, et al. Regulation of E-cadherin expression by VHL and hypoxia-inducible factor. Cancer Res (2006) 66(7):3567–75. doi:10.1158/0008-5472.CAN-05-2670
24. Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell (2007) 129(3):465–72. doi:10.1016/j.cell.2007.04.019
25. Vadde R, Vemula S, Jinka R, Merchant N, Bramhachari PV, Nagaraju GP. Role of hypoxia-inducible factors (HIF) in the maintenance of stemness and malignancy of colorectal cancer. Crit Rev Oncol Hematol (2017) 113:22–7. doi:10.1016/j.critrevonc.2017.02.025
26. Schito L, Semenza GL. Hypoxia-inducible factors: master regulators of cancer progression. Trends Cancer (2016) 2:758–70. doi:10.1016/j.trecan.2016.10.016
27. Wellmann S, Guschmann M, Griethe W, Eckert C, von Stackelberg A, Lottaz C, et al. Activation of the HIF pathway in childhood ALL, prognostic implications of VEGF. Leukemia (2004) 18:926–33. doi:10.1038/sj.leu.2403332
28. Noman MZ, Messai Y, Carre T, Akalay I, Meron M, Janji B, et al. Microenvironmental hypoxia orchestrating the cell stroma cross talk, tumor progression and antitumor response. Crit Rev Immunol (2011) 31:357–77. doi:10.1615/CritRevImmunol.v31.i5.10
29. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9:162–74. doi:10.1038/nri2506
30. Alfaro C, Suárez N, Martínez-Forero I, Palazón A, Rouzaut A, Solano S, et al. Carcinoma-derived interleukin-8 disorients dendritic cell migration without impairing T-cell stimulation. PLoS One (2011) 6:e17922. doi:10.1371/journal.pone.0017922
31. Hong J, Tobin NP, Rundqvist H, Li T, Lavergne M, García-Ibáñez Y, et al. Role of tumor pericytes in the recruitment of myeloid-derived suppressor cells. J Natl Cancer Inst (2015) 107:djv209. doi:10.1093/jnci/djv209
32. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med (2010) 207:2439–53. doi:10.1084/jem.20100587
33. Ren L, Yu Y, Wang L, Zhu Z, Lu R, Yao Z. Hypoxia-induced CCL28 promotes recruitment of regulatory T cells and tumor growth in liver cancer. Oncotarget (2016) 7:75763–73. doi:10.18632/oncotarget.12409
34. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature (2011) 475:226–30. doi:10.1038/nature10169
35. Facciabene A, Motz GT, Coukos G. T-regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res (2012) 72:2162–71. doi:10.1158/0008-5472.CAN-11-3687
36. Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci U S A (2012) 109:E2784–93. doi:10.1073/pnas.1202366109
37. Lee JH, Elly C, Park Y, Liu YC. E3 Ubiquitin Ligase VHL regulates hypoxia-inducible factor-1α to maintain regulatory T cell stability and suppressive capacity. Immunity (2015) 42:1062–74. doi:10.1016/j.immuni.2015.05.016
38. Pollizzi KN, Powell JD. Integrating canonical and metabolic signaling programmes in the regulation of T cell responses. Nat Rev Immunol (2014) 14:435–46. doi:10.1038/nri3701
39. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299:1057–61. doi:10.1126/science.1079490
40. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4:330–6. doi:10.1038/ni904
41. Gao AG, Lindberg FP, Finn MB, Blystone SD, Brown EJ, Frazier WA. Integrin-associated protein is a receptor for the C-terminal domain of thrombospondin. J Biol Chem (1996) 271:21–4. doi:10.1074/jbc.271.1.21
42. Liu Y, Merlin D, Burst SL, Pochet M, Madara JL, Parkos CA. The role of CD47 in neutrophil transmigration. Increased rate of migration correlates with increased cell surface expression of CD47. J Biol Chem (2001) 276:40156–66. doi:10.1074/jbc.M104138200
43. Miyashita M, Ohnishi H, Okazawa H, Tomonaga H, Hayashi A, Fujimoto TT, et al. Promotion of neurite and filopodium formation by CD47: roles of integrins, Rac, and Cdc42. Mol Biol Cell (2004) 15:3950–63. doi:10.1091/mbc.E04-01-0019
44. Reinhold MI, Lindberg FP, Kersh GJ, Allen PM, Brown EJ. Costimulation of T cell activation by integrin-associated protein (CD47) is an adhesion-dependent, CD28-independent signaling pathway. J Exp Med (1997) 185:1–11. doi:10.1084/jem.185.1.1
45. Zhao H, Wang J, Kong X, Li E, Liu Y, Du X, et al. CD47 promotes tumor invasion and metastasis in non-small cell lung cancer. Sci Rep (2016) 6:29719. doi:10.1038/srep29719
46. Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell (2009) 138:271–85. doi:10.1016/j.cell.2009.05.046
47. Barrera L, Montes-Servín E, Hernandez-Martinez JM, García-Vicente MLÁ, Montes-Servín E, Herrera-Martínez M, et al. CD47 overexpression is associated with decreased neutrophil apoptosis/phagocytosis and poor prognosis in non-small-cell lung cancer patients. Br J Cancer (2017) 117:385–97. doi:10.1038/bjc.2017.173
48. Kim MJ, Lee JC, Lee JJ, Kim S, Lee SG, Park SW, et al. Association of CD47 with natural killer cell-mediated cytotoxicity of head-and-neck squamous cell carcinoma lines. Tumour Biol (2008) 29:28–34. doi:10.1159/000132568
49. Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell (2010) 142:699–713. doi:10.1016/j.cell.2010.07.044
50. Chan KS, Espinosa I, Chao M, Wong D, Ailles L, Diehn M, et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc Natl Acad Sci U S A (2009) 106:14016–21. doi:10.1073/pnas.0906549106
51. Manna PP, Frazier WA. CD47 mediates killing of breast tumor cells via Gi-dependent inhibition of protein kinase A. Cancer Res (2004) 64:1026–36. doi:10.1158/0008-5472.CAN-03-1708
52. Fan D, Li Z, Zhang X, Yang Y, Yuan X, Zhang X, et al. AntiCD3Fv fused to human interleukin-3 deletion variant redirected T cells against human acute myeloid leukemic stem cells. J Hematol Oncol (2015) 8:18. doi:10.1186/s13045-015-0109-5
53. Zhang H, Lu H, Xiang L, Bullen JW, Zhang C, Samanta D, et al. HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc Natl Acad Sci U S A (2015) 112:E6215–23. doi:10.1073/pnas.1520032112
54. Liu X, Kwon H, Li Z, Fu YX. Is CD47 an innate immune checkpoint for tumor evasion? J Hematol Oncol (2017) 10:12. doi:10.1186/s13045-016-0381-z
55. Okazawa H, Motegi S, Ohyama N, Ohnishi H, Tomizawa T, Kaneko Y, et al. Negative regulation of phagocytosis in macrophages by the CD47-SHPS-1 system. J Immunol (2005) 174:2004–11. doi:10.4049/jimmunol.174.4.2004
56. Liu X, Pu Y, Cron K, Deng L, Kline J, Frazier WA, et al. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat Med (2015) 21:1209–15. doi:10.1038/nm.3931
57. Soto-Pantoja DR, Terabe M, Ghosh A, Ridnour LA, DeGraff WG, Wink DA, et al. CD47 in the tumor microenvironment limits cooperation between antitumor T-cell immunity and radiotherapy. Cancer Res (2014) 74:6771–83. doi:10.1158/0008-5472.CAN-14-0037-T
58. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell (2015) 27:450–61. doi:10.1016/j.ccell.2015.03.001
59. Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res (2014) 74:665–74. doi:10.1158/0008-5472.CAN-13-0992
60. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med (2014) 211:781–90. doi:10.1084/jem.20131916
61. Palsson-McDermott EM, Dyck L, Zasłona Z, Menon D, McGettrick AF, Mills KHG, et al. Pyruvate kinase M2 is required for the expression of the immune checkpoint PD-L1 in immune cells and tumors. Front Immunol (2017) 8:1300. doi:10.3389/fimmu.2017.01300
62. Pinato DJ, Black JR, Trousil S, Dina RE, Trivedi P, Mauri FA, et al. Programmed cell death ligands expression in phaeochromocytomas and paragangliomas: relationship with the hypoxic response, immune evasion and malignant behavior. Oncoimmunology (2017) 6:e1358332. doi:10.1080/2162402X.2017
63. Carosella ED, Moreau P, Le Maoult J, Le Discorde M, Dausset J, Rouas-Freiss N. HLA-G molecules: from maternal-fetal tolerance to tissue acceptance. Adv Immunol (2003) 81:199–252. doi:10.1016/S0065-2776(03)81006-4
64. Rouas-Freiss N, Moreau P, Ferrone S, Carosella ED. HLA-G proteins in cancer: do they provide tumor cells with an escape mechanism? Cancer Res (2005) 65:10139–44. doi:10.1158/0008-5472.CAN-05-0097
65. Carosella ED, Favier B, Rouas-Freiss N, Moreau P, Lemaoult J. Beyond the increasing complexity of the immunomodulatory HLA-G molecule. Blood (2008) 111:4862–70. doi:10.1182/blood-2007-12-127662
66. Amodio G, Sales de Albuquerque R, Gregori S. New insights into HLA-G mediated tolerance. Tissue Antigens (2014) 84:255–63. doi:10.1111/tan.12427
67. Kren L, Slaby O, Muckova K, Lzicarova E, Sova M, Vybihal V, et al. Expression of immune-modulatory molecules HLA-G and HLA-E by tumor cells in glioblastomas: an unexpected prognostic significance? Neuropathology (2011) 31:129–34. doi:10.1111/j.1440-1789.2010.01149.x
68. Ugurel S, Reinhold U, Tilgen W. HLA-G in melanoma: a new strategy to escape from immunosurveillance? Onkologie (2002) 25:129–34. doi:10.1159/000055222
69. Li XJ, Zhang X, Lin A, Ruan YY, Yan WH. Human leukocyte antigen-G (HLA-G) expression in cervical cancer lesions is associated with disease progression. Hum Immunol (2012) 73:946–9. doi:10.1016/j.humimm.2012.07.041
70. Yan WH. HLA-G expression in cancers: potential role in diagnosis, prognosis and therapy. Endocr Metab Immune Disord Drug Targets (2011) 11:76–89. doi:10.2174/187153011794982059
71. Andersson E, Poschke I, Villabona L, Carlson JW, Lundqvist A, Kiessling R, et al. Non-classical HLA-class I expression in serous ovarian carcinoma: correlation with the HLA-genotype, tumor infiltrating immune cells and prognosis. Oncoimmunology (2015) 5:e1052213. doi:10.1080/2162402X.2015.1052213
72. Guo ZY, Lv YG, Wang L, Shi SJ, Yang F, Zheng GX, et al. Predictive value of HLA-G and HLA-E in the prognosis of colorectal cancer patients. Cell Immunol (2015) 293:10–6. doi:10.1016/j.cellimm.2014.10.003
73. Mouillot G, Marcou C, Zidi I, Guillard C, Sangrouber D, Carosella ED, et al. Hypoxia modulates HLA-G gene expression in tumor cells. Hum Immunol (2007) 68:277–85. doi:10.1016/j.humimm.2006.10.016
74. Yaghi L, Poras I, Simoes RT, Donadi EA, Tost J, Daunay A, et al. Hypoxia inducible factor-1 mediates the expression of the immune checkpoint HLA-G in glioma cells through hypoxia response element located in exon 2. Oncotarget (2016) 7:63690–707. doi:10.18632/oncotarget.11628
75. Ferns DM, Heeren AM, Samuels S, Bleeker MCG, de Gruijl TD, Kenter GG, et al. Classical and non-classical HLA class I aberrations in primary cervical squamous- and adenocarcinomas and paired lymph node metastases. J Immunother Cancer (2016) 4:78. doi:10.1186/s40425-016-0184-3
76. Garziera M, Scarabel L, Toffoli G. Hypoxic modulation of HLA-G expression through the metabolic sensor HIF-1 in human cancer cells. J Immunol Res (2017) 2017:4587520. doi:10.1155/2017/4587520
77. Sasaki T, Kanaseki T, Shionoya Y, Tokita S, Miyamoto S, Saka E, et al. Microenvironmental stresses induce HLA-E/Qa-1 surface expression and thereby reduce CD8(+) T-cell recognition of stressed cells. Eur J Immunol (2016) 46(4):929–40. doi:10.1002/eji.201545835
78. Xie H, Simon MC. Oxygen availability and metabolic reprogramming in cancer. J Biol Chem (2017) 292:16825–32. doi:10.1074/jbc.R117.799973
79. Huber V, Camisaschi C, Berzi A, Ferro S, Lugini L, Triulzi T, et al. Cancer acidity: an ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin Cancer Biol (2017) 43:74–89. doi:10.1016/j.semcancer.2017.03.001
80. Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer (2011) 11:671–7. doi:10.1038/nrc3110
81. Damaghi M, Wojtkowiak JW, Gillies RJ. pH sensing and regulation in cancer. Front Physiol (2013) 4:370. doi:10.3389/fphys.2013.00370
82. Parks SK, Cormerais Y, Marchiq I, Pouyssegur J. Hypoxia optimises tumour growth by controlling nutrient import and acidic metabolite export. Mol Aspects Med (2016) 47-48:3–14. doi:10.1016/j.mam.2015.12.001
83. Kim Y, Choi JW, Lee JH, Kim YS. Expression of lactate/H+ symporters MCT1 and MCT4 and their chaperone CD147 predicts tumor progression in clear cell renal cell carcinoma: immunohistochemical and The Cancer Genome Atlas data analyses. Hum Pathol (2015) 46:104–12. doi:10.1016/j.humpath.2014.09.013
84. Halestrap AP. Monocarboxylic acid transport. Compr Physiol (2013) 3:1611–43. doi:10.1002/cphy.c130008
85. Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T, et al. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol (2013) 9:712. doi:10.1038/msb.2013.65
86. Corbet C, Feron O. Metabolic and mind shifts: from glucose to glutamine and acetate addictions in cancer. Curr Opin Clin Nutr Metab Care (2015) 18:346–53. doi:10.1097/MCO.0000000000000178
87. Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov (2008) 7:168–81. doi:10.1038/nrd2467
88. Sedlakova O, Svastova E, Takacova M, Kopacek J, Pastorek J, Pastorekova S. Carbonic anhydrase IX, a hypoxia-induced catalytic component of the pH regulating machinery in tumors. Front Physiol (2014) 4:400. doi:10.3389/fphys.2013.00400
89. McDonald PC, Dedhar S. Carbonic anhydrase IX (CAIX) as a mediator of hypoxia-induced stress response in cancer cells. Subcell Biochem (2014) 75:255–69. doi:10.1007/978-94-007-7359-2_13
90. Pastorek J, Pastorekova S. Hypoxia-induced carbonic anhydrase IX as a target for cancer therapy: from biology to clinical use. Semin Cancer Biol (2015) 31:52–64. doi:10.1016/j.semcancer.2014.08.002
91. Bosticardo M, Ariotti S, Losana G, Bernabei P, Forni G, Novelli F. Biased activation of human T lymphocytes due to low extracellular pH is antagonized by B7/CD28 costimulation. Eur J Immunol (2001) 31:2829–38. doi:10.1002/1521-4141(200109)31:9<2829::AID-IMMU2829>3.0.CO;2-U
92. Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res (2012) 72:2746–56. doi:10.1158/0008-5472.CAN-11-1272
93. Nakagawa Y, Negishi Y, Shimizu M, Takahashi M, Ichikawa M, Takahashi H. Effects of extracellular pH and hypoxia on the function and development of antigen-specific cytotoxic T lymphocytes. Immunol Lett (2015) 167(2):72–86. doi:10.1016/j.imlet.2015.07.003
94. Severin T, Müller B, Giese G, Uhl B, Wolf B, Hauschildt S, et al. pH-dependent LAK cell cytotoxicity. Tumour Biol (1994) 15(5):304–10. doi:10.1159/000217905
95. Fischer B, Müller B, Fischer KG, Baur N, Kreutz W. Acidic pH inhibits non-MHC-restricted killer cell functions. Clin Immunol (2000) 96:252–63. doi:10.1006/clim.2000.4904
96. Fischer B, Müller B, Fisch P, Kreutz W. An acidic microenvironment inhibits antitumoral non-major histocompatibility complex-restricted cytotoxicity: implications for cancer immunotherapy. J Immunother (2000) 23:196–207. doi:10.1097/00002371-200003000-00004
97. Müller B, Fischer B, Kreutz W. An acidic microenvironment impairs the generation of non-major histocompatibility complex-restricted killer cells. Immunology (2000) 99:375–84. doi:10.1046/j.1365-2567.2000.00975.x
98. Bellocq A, Suberville S, Philippe C, Bertrand F, Perez J, Fouqueray B, et al. Low environmental pH is responsible for the induction of nitric-oxide synthase in macrophages. Evidence for involvement of nuclear factor-kappaB activation. J Biol Chem (1998) 273(9):5086–92. doi:10.1074/jbc.273.9.5086
99. Martínez D, Vermeulen M, Trevani A, Ceballos A, Sabatté J, Gamberale R, et al. Extracellular acidosis induces neutrophil activation by a mechanism dependent on activation of phosphatidylinositol 3-kinase/Akt and ERK pathways. J Immunol (2006) 176:1163–71. doi:10.4049/jimmunol.176.2.1163
100. Marchiq I, Pouysségur J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J Mol Med (Berl) (2016) 94:155–71. doi:10.1007/s00109-015-1307-x
101. Hirschhaeuser F, Sattler UG, Mueller-Klieser W. Lactate: a metabolic key player in cancer. Cancer Res (2011) 71(22):6921–5. doi:10.1158/0008-5472.CAN-11-1457
102. Feder-Mengus C, Ghosh S, Weber WP, Wyler S, Zajac P, Terracciano L, et al. Multiple mechanisms underlie defective recognition of melanoma cells cultured in three-dimensional architectures by antigen specific cytotoxic T lymphocytes. Br J Cancer (2007) 96(7):1072–82. doi:10.1038/sj.bjc.6603664
103. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood (2007) 109(9):3812–9. doi:10.1182/blood-2006-07-035972
104. Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid derived suppressor cells and NK cells. J Immunol (2013) 191:1486–95. doi:10.4049/jimmunol.1202702
105. Lv LH, Yu JD, Li GL, Long TZ, Zhang W, Chen YJ, et al. Functional distinction of rat liver natural killer cells from spleen natural killer cells under normal and acidic conditions in vitro. Hepatobiliary Pancreat Dis Int (2012) 11:285–93. doi:10.1016/S1499-3872(12)60162-3
106. Dietl K, Renner K, Dettmer K, Timischl B, Eberhart K, Dorn C, et al. Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J Immunol (2010) 184:1200–9. doi:10.4049/jimmunol.0902584
107. Gottfried E, Kunz-Schughart LA, Ebner S, Mueller-Klieser W, Hoves S, Andreesen R, et al. Tumor derived lactic acid modulates dendritic cell activation and antigen expression. Blood (2006) 107:2013–21. doi:10.1182/blood-2005-05-1795
108. Puig-Kroger A, Pello OM, Selgas R, Criado G, Bajo MA, Sanchez-Tomero JA, et al. Peritoneal dialysis solutions inhibit the differentiation and maturation of human monocyte-derived dendritic cells: effect of lactate and glucose-degradation products. J Leukoc Biol (2003) 73(4):482–92. doi:10.1189/jlb.0902451
109. Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, et al. IL-23 promotes tumour incidence and growth. Nature (2006) 442:461–5. doi:10.1038/nature04808
110. Walenta S, Schroeder T, Mueller-Klieser W. Lactate in solid malignant tumors: potential basis of a metabolic classification in clinical oncology. Curr Med Chem (2004) 11:2195–204. doi:10.2174/0929867043364711
111. Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfør K, Rofstad EK, et al. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res (2000) 60:916–21.
112. Ziebart T, Walenta S, Kunkel M, Reichert TE, Wagner W, Mueller-Klieser W. Metabolic and proteomic differentials in head and neck squamous cell carcinomas and normal gingival tissue. J Cancer Res Clin Oncol (2011) 137:193–9. doi:10.1007/s00432-010-0875-y
113. Walenta S, Mueller-Klieser WF. Lactate: mirror and motor of tumor malignancy. Semin Radiat Oncol (2004) 14:267–74. doi:10.1016/j.semradonc.2004.04.004
114. Park I, Larson PE, Zierhut ML, Hu S, Bok R, Ozawa T, et al. Hyperpolarized 13C magnetic resonance metabolic imaging: application to brain tumors. Neuro Oncol (2010) 12:133–44. doi:10.1093/neuonc/nop043
115. Saraswathy S, Crawford FW, Lamborn KR, Pirzkall A, Chang S, Cha S, et al. Evaluation of MR markers that predict survival in patients with newly diagnosed GBM prior to adjuvant therapy. J Neurooncol (2009) 91:69–81. doi:10.1007/s11060-008-9685-3
116. Keshari KR, Sriram R, Van Criekinge M, Wilson DM, Wang ZJ, Vigneron DB, et al. Metabolic reprogramming and validation of hyperpolarized 13C lactate as a prostate cancer biomarker using a human prostate tissue slice culture bioreactor. Prostate (2013) 73:1171–81. doi:10.1002/pros.22665
117. Yaligar J, Thakur SB, Bokacheva L, Carlin S, Thaler HT, Rizwan A, et al. Lactate MRSI and DCE MRI as surrogate markers of prostate tumor aggressiveness. NMR Biomed (2012) 25:113–22. doi:10.1002/nbm.1723
118. Eltzschig HK, Thompson LF, Karhausen J, Cotta RJ, Ibla JC, Robson SC, et al. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood (2004) 104:3986–92. doi:10.1182/blood-2004-06-2066
119. Kaczmarek E, Koziak K, Sévigny J, Siegel JB, Anrather J, Beaudoin AR, et al. Identification and characterization of CD39/vascular ATP diphosphohydrolase. J Biol Chem (1996) 271:33116–22. doi:10.1074/jbc.271.51.33116
120. Decking UK, Schlieper G, Kroll K, Schrader J. Hypoxia induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ Res (1997) 81:154–64. doi:10.1161/01.RES.81.2.154
121. Morote-Garcia JC, Rosenberger P, Kuhlicke J, Eltzschig HK. HIF-1-dependent repression of adenosine kinase attenuates hypoxia-induced vascular leak. Blood (2008) 111:5571–80. doi:10.1182/blood-2007-11-126763
122. Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, et al. Ecto-5’-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest (2002) 110:993–1002. doi:10.1172/JCI15337
123. Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5’-adenosine monophosphate to adenosine. J Immunol (2006) 177:6780–6. doi:10.4049/jimmunol.177.10.6780
124. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204:1257–65. doi:10.1084/jem.20062512
125. Antonioli L, Blandizzi C, Pacher P, Haskó G. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer (2013) 13:842–57. doi:10.1038/nrc3613
126. Jin D, Fan J, Wang L, Thompson LF, Liu A, Daniel BJ, et al. CD73 on tumor cells impairs antitumor T-cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res (2010) 70:2245–55. doi:10.1158/0008-5472.CAN-09-3109
127. Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, et al. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci U S A (2010) 107:1547–52. doi:10.1073/pnas.0908801107
128. Blay J, White TD, Hoskin DW. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res (1997) 57(13):2602–5.
129. Wigerup C, Påhlman S, Bexell D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol Ther (2016) 164:152–69. doi:10.1016/j.pharmthera.2016.04.009
130. Chan MC, Holt-Martyn JP, Schofield CJ, Ratcliffe PJ. Pharmacological targeting of the HIF hydroxylases – a new field in medicine development. Mol Aspects Med (2016) 47-48:54–75. doi:10.1016/j.mam.2016.01.001
131. Haase VH. Therapeutic targeting of the HIF oxygen-sensing pathway: lessons learned from clinical studies. Exp Cell Res (2017) 356:160–5. doi:10.1016/j.yexcr.2017.05.004
132. Yu T, Tang B, Sun X. Development of inhibitors targeting hypoxia-inducible factor 1 and 2 for cancer therapy. Yonsei Med J (2017) 58:489–96. doi:10.3349/ymj.2017.58.3.489
133. Onnis B, Rapisarda A, Melillo G. Development of HIF-1 inhibitors for cancer therapy. J Cell Mol Med (2009) 13:2780–6. doi:10.1111/j.1582-4934.2009.00876.x
134. Bui MH, Seligson D, Han KR, Pantuck AJ, Dorey FJ, Huang Y, et al. Carbonic anhydrase IX is an independent predictor of survival in advanced renal clear cell carcinoma: implications for prognosis and therapy. Clin Cancer Res (2003) 9(2):802–11.
135. Chang DK, Moniz RJ, Xu Z, Sun J, Signoretti S, Zhu Q, et al. Human anti-CAIX antibodies mediate immune cell inhibition of renal cell carcinoma in vitro and in a humanized mouse model in vivo. Mol Cancer (2015) 14:119. doi:10.1186/s12943-015-0384-3
136. Chamie K, Donin NM, Klöpfer P, Bevan P, Fall B, Wilhelm O, et al. Adjuvant weekly girentuximab following nephrectomy for high-risk renal cell carcinoma: the ARISER randomized clinical trial. JAMA Oncol (2017) 3:913–20. doi:10.1001/jamaoncol.2016.4419
Keywords: hypoxia, immunosuppression, metabolism, immunotherapy, microenvironment
Citation: Li Y, Patel SP, Roszik J and Qin Y (2018) Hypoxia-Driven Immunosuppressive Metabolites in the Tumor Microenvironment: New Approaches for Combinational Immunotherapy. Front. Immunol. 9:1591. doi: 10.3389/fimmu.2018.01591
Received: 14 December 2017; Accepted: 27 June 2018;
Published: 16 July 2018
Edited by:
Anahid Jewett, University of California, Los Angeles, United StatesReviewed by:
Amedeo Amedei, Università degli Studi di Firenze, ItalyCarlos Alfaro, Universidad de Navarra, Spain
Copyright: © 2018 Li, Patel, Roszik and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong Qin, eW9uZ3FpbkBtZGFuZGVyc29uLm9yZw==