 Russell B. Hawkins1
Russell B. Hawkins1 Steven L. Raymond1
Steven L. Raymond1 Julie A. Stortz1
Julie A. Stortz1 Hiroyuki Horiguchi1
Hiroyuki Horiguchi1 Scott C. Brakenridge1
Scott C. Brakenridge1 Anna Gardner2
Anna Gardner2 Philip A. Efron1Azra Bihorac1,3Mark Segal1,3Frederick A. Moore1
Philip A. Efron1Azra Bihorac1,3Mark Segal1,3Frederick A. Moore1 Lyle L. Moldawer1*
Lyle L. Moldawer1*
- 1Sepsis and Critical Illness Research Center, Department of Surgery, University of Florida College of Medicine, Gainesville, FL, United States
- 2Department of Aging and Geriatric Research, Institute on Aging, University of Florida College of Medicine, Gainesville, FL, United States
- 3Division of Nephrology, Department of Medicine, University of Florida College of Medicine, Gainesville, FL, United States
Dysregulated host immune responses to infection often occur, leading to sepsis, multiple organ failure, and death. Some patients rapidly recover from sepsis, but many develop chronic critical illness (CCI), a debilitating condition that impacts functional outcomes and long-term survival. The “Persistent Inflammation, Immunosuppression, and Catabolism Syndrome” (PICS) has been postulated as the underlying pathophysiology of CCI. We propose that PICS is initiated by an early genomic and cytokine storm in response to microbial invasion during the early phase of sepsis. However, once source control, antimicrobial coverage, and supportive therapies have been initiated, we propose that the persistent inflammation in patients developing CCI is a result of ongoing endogenous alarmin release from damaged organs and loss of muscle mass. This ongoing alarmin and danger-associated molecular pattern signaling causes chronic inflammation and a shift in bone marrow stem cell production toward myeloid cells, contributing to chronic anemia and lymphopenia. We propose that therapeutic interventions must target the chronic organ injury and lean tissue wasting that contribute to the release of endogenous alarmins and the expansion and deposition of myeloid progenitors that are responsible for the propagation and persistence of CCI.
Introduction
Normal protective host responses to infection often become excessive, resulting in the systemic inflammatory response syndrome (SIRS) that can cause a clinical trajectory of refractory shock, fulminant multiple organ failure (MOF), and early in-hospital death. Until recently, this was a common clinical scenario (occurring in >35% of those with sepsis), and a tremendous effort has been directed at understanding and treating this response. Unfortunately, despite dramatic increases in our understanding of sepsis, more than 150 clinical trials testing biological response modifiers directed at SIRS have failed to improve sepsis-associated mortality (1–3). This undoubtedly occurred because the complexity of the human response to sepsis was underestimated, and previous preclinical (bacterial administration and high-mortality peritonitis) (4–9) and early clinical [endotoxin lipopolysaccharide (LPS) administration] (10–13) models did not accurately recapitulate human pathobiology (14–16).
Despite these efforts, as the result of unprecedented quality improvement to identify sepsis early and provide rapid evidence-based care (17–19), early in-hospital deaths after sepsis have decreased substantially over the last decade. The acute phase of sepsis has been characterized by both a “genomic storm” and “cytokine storm,” the activation of a plethora of genes that encode inflammatory cytokines, signal transducers, and cell adhesion molecules (20), and subsequent spike of inflammatory cytokines (21). In modern ICUs (Figure 1), severely septic patients are resuscitated through their “genomic storm” characterized by SIRS with organ dysfunction, but now relatively few (<10%) progress into the “MOF/early death” trajectory (18, 22). Our recent studies have shown that as SIRS resolves, roughly half of the sepsis survivors exhibit “rapid recovery” (RAP) from their organ dysfunction and achieve “immune homeostasis.” Unfortunately, the remainder develop “chronic critical illness” (CCI), characterized by prolonged ICU stays (>14 days) and low-grade organ dysfunction (especially kidney injury). CCI encompasses multiple phenotypes, including chronic long-term inflammation as well as immunosuppression and catabolism. Chronically critically ill patients may exhibit either a predominantly pro-inflammatory phenotype or the immunosuppressed phenotype or a combination of the two. CCI patients are often observed to have (a) persistent inflammation [elevated C-reactive protein and increased inflammatory cytokines interleukin (IL)-6 and IL-8] with myeloid-derived suppressor cell (MDSC) expansion, (b) immunosuppression (increased secondary and nosocomial infections and reactivation of latent viral reactivation), and (c) protein catabolism with muscle wasting and cachexia similar to cancer and other chronic inflammatory diseases. MDSCs are a heterogeneous population of immature myeloid cells that accumulate during pathologic conditions such as cancer or sepsis (23). In sepsis survivors, MDSCs are persistently increased, functionally immunosuppressive, and are associated with adverse clinical outcomes (24). CCI patients are commonly discharged to long-term acute care facilities (LTACs) for expensive care because there are no effective interventions (25) and their profound disabilities preclude home care. Here, they experience accelerated aging, induced frailty, sepsis recidivism (requiring re-hospitalization), physical and cognitive disabilities (resulting in dismal life quality), and a high rate of ongoing indolent death (~40% at 1 year) (26, 27). The personal and economic burdens to these patients and their families, as well as the immense costs of this exploding population to society, are immense (1, 28).
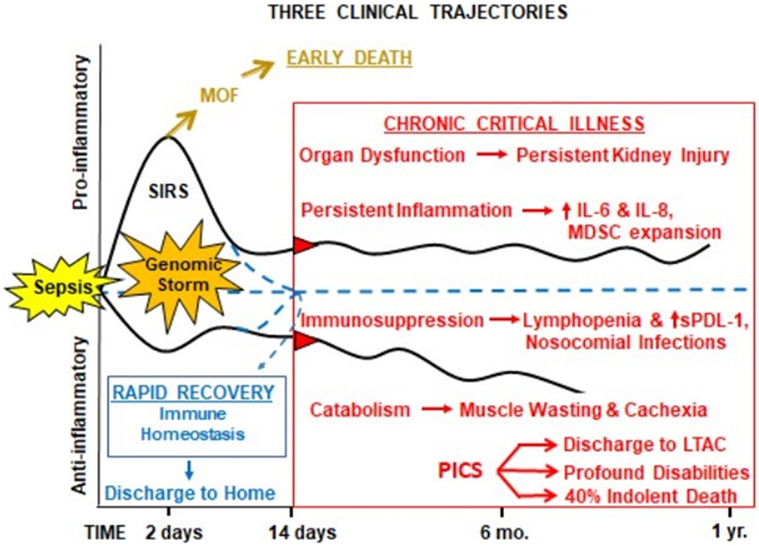
Figure 1. Proposed hypothesis for the PICS. Abbreviations: SIRS, systemic inflammatory response syndrome; MDSC, myeloid-derived suppressor cell; sPDL-1, soluble programmed death ligand-1; LTAC, long-term acute care facility; PICS, persistent inflammation, immunosuppression, and catabolism syndrome.
Persistent Inflammation, Immunosuppression, and Catabolism Syndrome (PICS)
In 2012, our group proposed the hypothesis that the underlying pathobiology of CCI in sepsis survivors was a “Persistent Inflammation, Immunosuppression, and Catabolism Syndrome” (29). Since 2012, the PICS hypothesis has been validated (30, 31). We now propose, based on new information, that the underlying pathobiology which drives PICS and CCI is a maladaptive self-perpetuating cycle of persistent inflammation involving reduced host protective immunity, continued organ injury and its sequelae, loss of muscle mass and function, changes in bone marrow (BM) function predominated by “emergency myelopoiesis,” and failure of metabolic adaptation (Figure 2) (22, 24, 27, 32, 33). Organ injury results in the release of alarmins, which perpetuates expansion of immunosuppressive myeloid cells, which play a role in ongoing inflammation and muscle wasting which all contribute to the progression of CCI. Recent studies by Bihorac and Segal from our center have shown that new or ongoing acute kidney injury (AKI) is a significant independent predictor of adverse outcomes in sepsis survivors. Not only is the kidney the most commonly injured organ in sepsis (34) but it is by far the most problematic for long-term recovery (29, 35, 36). AKI increases the likelihood of chronic kidney disease (CKD), which is both catabolic and inflammatory. These patients also experience profound muscle wasting with loss of up to 30% of lean body mass within weeks (37, 38).
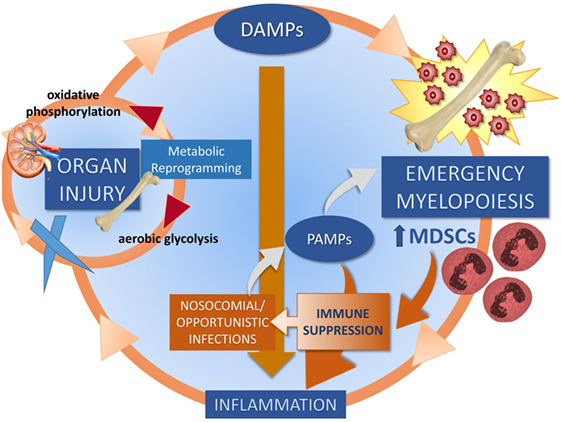
Figure 2. Proposed self-perpetuating cycle of persistent inflammation driving organ injury through a failure of metabolic adaptation, leading to release of endogenous alarmins and bone marrow changes. Three cycles drive CCI: muscle wasting (discussed in Section “Skeletal Muscle as a Target for Oxidant Injury and DAMP Release”), organ injury (discussed in Section “Role of AKI in CCI”), and emergency myelopoiesis (discussed in Section “Abnormal Myelopoiesis and MDSCs”).
Since proposing PICS in 2012, several observations have been notable. In regards to the three clinical trajectories (Figure 3), only 6% of septic patients died early (<14 days), 46% experienced rapid recovery (with 6-month survival of 98%), while a notably high 49% progressed into CCI (with 6-month survival of only 63%) (27). Sepsis survivors who developed CCI were significantly older and had more hospital-acquired (rather than community-acquired) infections (27). Importantly, these subjects exhibited not only persistent inflammation, as demonstrated by elevated plasma cytokine concentrations, but also increased immunosuppressive proteins (sPD-L1 or IL-10) (Figure 4) (27). Most (~80%) were discharged to LTACs, developed recurrent infections, and had significant ongoing functional disabilities at 6 months. CCI patients reported worse quality of life and had significantly worse functional status at 6-month follow-up compared with patients who experienced rapid recovery. These results were based on detailed long-term follow-up of CCI patients using the EuroQol-5D weighted utility index (0 = Death, 1 = Full health), the Short Physical Performance Battery (0 = worst performance, 12 = best performance), and the Eastern Cooperative Oncology Group/World Health Organization/Zubrod Scale (0 = Asymptomatic, 5 = Death) (Gardner and Brakenridge, in review).
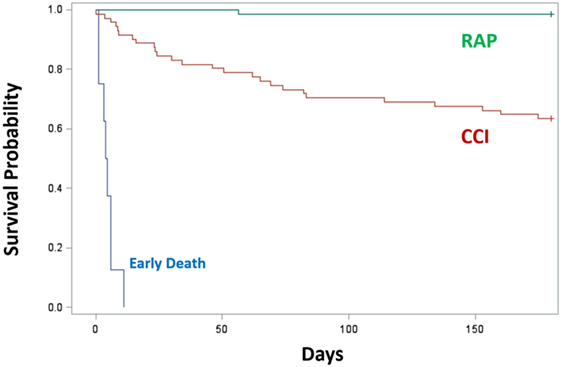
Figure 3. Survival of CCI (n = 71) or RAP (n = 66) patients 6 months after sepsis. Trajectories were classified as early death (blue), RAP (green), and CCI (red). Kaplan–Meier analysis demonstrated significant differences (p < 0.01) in survival between groups. Abbreviations: CCI, chronic critical illness; RAP, rapid recovery.
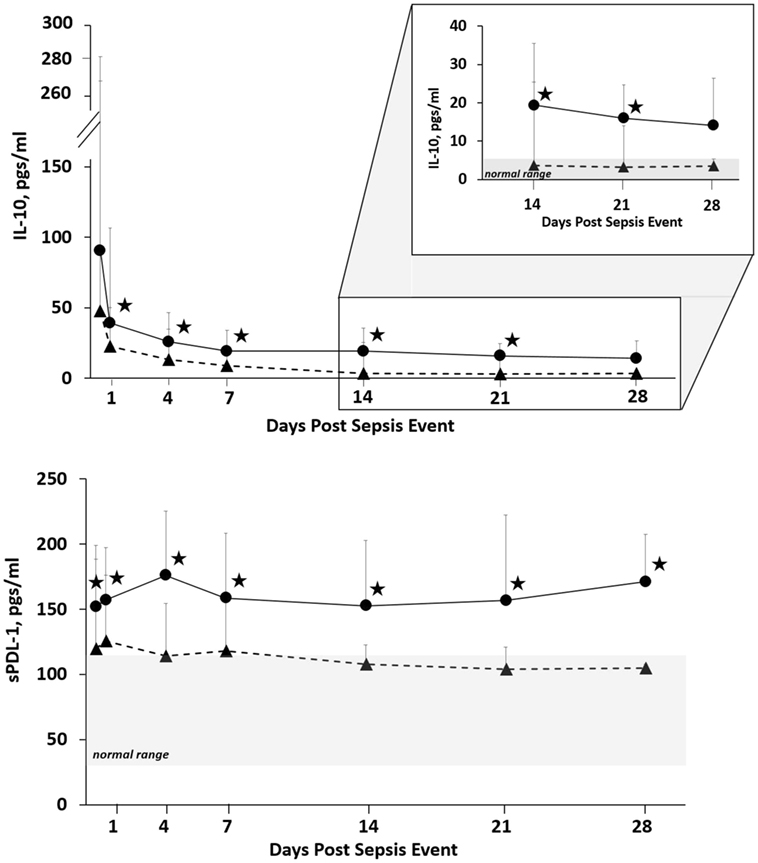
Figure 4. Biomarker concentrations in patients with CCI and RAP. Blood samples were collected at 0.5, 1, 4, 7, 14, 21, and 28 days after sepsis onset. Differences in concentrations between CCI (filled circles) and RAP (filled triangles) cohorts at individual time points are identified with an asterisk at a p value less than 0.05 using nonparametric tests. Patients with CCI had evidence of prolonged immunosuppression. Abbreviations: CCI, chronic critical illness; RAP rapid recovery; IL-10, interleukin-10; sPDL-1, soluble programmed death ligand-1.
The Challenge of CCI
Traditionally, the lean tissue wasting and cachexia frequently observed in CCI has been viewed as a macro-endocrine and cytokine-driven injury stress response (39), and recent studies in CCI survivors have shown biopsy-proven defective mitochondrial biogenesis and myocyte necrosis associated with leukocyte infiltration (38). Inflammation, therefore, is likely causing direct mitochondrial and skeletal muscle myocyte injury, which in turn releases breakdown products that amplify ongoing inflammation. A number of known mitochondrial DAMPs (mitoDAMPs) from skeletal muscle include mitochondrial DNA (mtDNA), HMGB1, and transcription factor A, mitochondrial (TFAM) (40–42). We envision this as a complex, futile, self-perpetuating cycle driven by an inability of organs and tissues to adapt to persistent inflammation, fueled by a continuous release of endogenous alarmins and expansion of MDSCs that infiltrate not only secondary lymphoid organs but also tissues of the reticuloendothelial system. These complex interactions have not been studied systematically.
The current CCI epidemic represents a new and seemingly insurmountable scientific challenge. Sepsis-induced CCI and its bleak long-term outcomes occur in a diverse patient population whose characterization requires both in-hospital and long-term post-hospital discharge follow-up. While these detailed assessments are essential in elucidating the debilitating outcomes associated with CCI, CCI’s natural history is confounded by multiple co-morbid diseases, making a definitive, clinical, mechanistic investigation difficult.
This has made the study of sepsis-associated CCI in animal models challenging. Appropriate, validated laboratory models are required to develop a mechanistic framework describing the variable effects on outcomes in different organs and subgroups of patients (14, 43). Historically, most animal models of sepsis have focused on the early host immune responses in the first few days. This reflected the challenge of treating human sepsis during that acute phase (5, 44). To better study the underlying mechanisms that drive CCI’s progression, continual calibration/validation and improvement of more chronic animal models through ongoing bidirectional bench-to-bedside research will be required. Several groups including ourselves have used a semi-lethal cecal ligation and puncture (CLP) model because it recapitulates the persistent inflammation, weight loss, and immune suppression seen in sepsis survivors (45–50). However, the mechanism(s) driving the process in mice are likely different from those seen in CCI and PICS, because survivors of CLP have a necrotic cecum in addition to an indeterminate nidus of infection. High-dose antibiotics have been attempted to sterilize the peritoneal cavity, but accomplishment of sterility has rarely been demonstrated (51). With that said, some investigators have looked at the long-term outcomes in murine survivors of CLP sepsis, and demonstrating myeloid cell expansion, inflammation, and immune suppression (52–54).
In addition, most investigators have used juvenile or young adult in-bred mice; however, sepsis is a disease of the elderly. Efron et al. have used aged mice and clearly shown that not only do they differ from young mice at baseline but they also respond differently to sepsis (14). We are also beginning to understand the effects of comorbidities on the sepsis response. Delano has shown that preexisting diabetes exaggerates the host response to sepsis (55). This type of information will be crucial in the future development of multimodal interventional trials.
Alarmins and Danger-Associated Molecular Patterns (DAMPs) Drive the Persistent Inflammation
Alarmins have been identified as important mediators of persistent inflammation in CCI. There are two sources of alarmins and both are recognized by the same pattern-recognition receptors on immune and parenchymal cells that perpetuate ongoing inflammation. The first source is exogenous pathogen-associated molecular patterns elaborated during nosocomial infections and reactivation of latent viral infections (43). Our recent studies show that 61% of sepsis survivors suffering from CCI experience one or more secondary nosocomial infections compared with 18% in RAP survivors (33). Hotchkiss reported that 100% of patients spending 14 days in the ICU show evidence of viral reactivation (56). The second source of alarmins is the constant release of endogenous DAMPs from injured organs and inflammatory cells (57). These danger signals represent both nucleic acids and cellular proteins that are released upon cell death, as well as proteins or nucleic acids that are actively secreted in response to cellular stress (57, 58). Multiple studies have demonstrated that several of these DAMPs, including nuclear (nuc) DNA, HMGB1, and S100 are significantly elevated in sepsis survivors, especially during their entire hospitalization (59–62). We hypothesize that the two primary sources of these endogenous DAMPs are the kidney (with AKI progressing to CKD) and wasting of skeletal muscle, the largest and most labile protein reserve in the body.
Abnormal Myelopoiesis and MDSCs
Sepsis initiates an emergency myelopoiesis response (24, 63). Cytokine and chemokine release (64–66), along with adrenergic stimulation (67, 68), promotes the rapid release of myeloid populations from BM and secondary lymphoid organs. This release of both mature and immature neutrophils (PMNs) is an essential requirement for the initial control of an invading pathogen, but it can also cause collateral organ injury. This release also creates a void in the BM niche that stimulates expansion of hematopoietic stem cells and other early progenitors (69). Myelopoiesis is favored at the expense of both lymphopoiesis and hematopoiesis, which explains in part the persistent lymphopenia and chronic anemia seen in sepsis survivors (66, 70).
In cases of CCI in which alarmin release continues unabated, however, emergency myelopoiesis persists and myeloid cells are released with an immature phenotype (71, 72). We have seen in a chronic murine CLP model that by 7 days post-sepsis, up to 95% of BM cells are myeloid cells, mostly immature and functional like MDSCs (53). Interestingly, McCall has shown that the phenotype of these cells evolves over time, and the myeloid cells become more immunosuppressive with time (73). These cells not only overwhelm the BM but also significantly infiltrate the spleen, lymph nodes, reticuloendothelial tissues (such as lung and liver), and likely also skeletal muscle and brain (53, 74). MDSCs are generally sorted into two phenotypes based on cell surface markers: granulocytic MDSCs are CD11b+Ly6G+Ly6Clow and monocytic MDSCs are CD11b+Ly6G−Ly6Chigh (75). MDSCs have been implicated in immunosuppression in sepsis via IL-10 production and inhibition of T-cell response and proliferation (76). Not only are MDSCs potently immunosuppressive toward macrophages and CD4+ and CD8+ T-cells, they are also pro-inflammatory, producing oxidation, and peroxidation products, as well as nitric oxide, all of which are potentially damaging to parenchymal cells and promote inflammation (66, 76). We have recently demonstrated that in sepsis survivors, there is rapid and sustained appearance of MDSCs in the blood (24). More importantly, we have shown that CCI sepsis survivors have persistently elevated MDSCs that are strong independent predictors of nosocomial infection and hospital discharge to LTACs (24, 27, 33). Others have confirmed our results and demonstrated that sepsis survivors have elevated levels of MDSCs and similarly have a high incidence of secondary nosocomial infections (77, 78).
Role of AKI in CCI
The kidney is likely the most critical organ related to long-term recovery from sepsis. Our previous work found a strong association between sepsis and AKI. Not only are patients with AKI more likely to develop sepsis (34, 79–81), but AKI progressing to CKD is a major factor that perpetuates organ dysfunction in sepsis, leading to CCI and decreased survival (29, 35, 36). Renal tubule epithelial cells are highly susceptible to intrinsic oxidative stress. During sepsis, necrotic tubule epithelial cells and PMNs release DAMPs that activate PPR toll-like receptors (TLRs). Others have shown that patients with sepsis and AKI have increased levels of urinary DAMPs (82). In addition, urinary cellular RNA from sepsis patients reveals overexpression of several DAMP receptors. The local and systemic influx of DAMPs leads to secretion of chemokines by renal parenchymal cells and dendritic cells (DCs) which promotes a further local PMN-dependent inflammatory response (83–85) as well as distant systemic effects on other organs (36, 86–90).
In addition to being constitutively expressed in renal tubule epithelial cells, TLRs are further upregulated in AKI via epigenetic remodeling which leads to an exaggerated cytokine production in response to LPS and lipoteichoic acid, causing a renal “hyper-responsive” state (91). The cytokine efflux into the systemic circulation, together with the decrease in renal clearance, leads to exaggerated systemic inflammation and systemic organ injury. In trauma patients with AKI, we observed an increase in plasma levels of IL-8, chemokine (C-X-C motif) ligand 1 (CXCL1), monocyte chemoattractant protein-1, and macrophage inflammatory protein 1-beta (92). In both animal models and human studies, distant lung injury after AKI was associated with an increase in plasma levels of CXCL1, IL-6, and IL-8 within 2 h after AKI onset (93, 94). Our data demonstrate persistent elevation of both pro-inflammatory and immunosuppressive cytokines among patients with persistent AKI.
There is currently a dearth of knowledge explaining why the kidney is so susceptible to inflammatory injury and why CCI patients fail to recover from renal dysfunction. Large numbers of immune cells (such as DCs, macrophages, and few lymphocytes) reside in the kidney (95). We propose that during sepsis and CCI, the kidney is infiltrated by MDSCs that bring with them oxidative and immunosuppressive properties (24, 30, 76). Each nephron selectively filters small molecules such as DAMPs and pathogenic antigens and together the kidneys filter approximately 180 l of fluid per day, filtering the entire blood volume over 30 times daily (96). Thus, renal DCs and renal lymph nodes are exposed to DAMPS, pathogens, and antigens in the blood over 30 times more frequently than any other tissue. The kidney hosts many different cell types expressing a subset of TLRs (1–6) and can thus respond to DAMPs to induce innate immune responses (97).
Skeletal Muscle as a Target for Oxidant Injury and DAMP Release
The skeletal muscle system is the largest, most labile protein reserve in the body. Sepsis induces catabolism characterized by profound muscle wasting, reflecting breakdown of myofibrillar proteins, decreased protein synthesis, increased mitochondrial dysfunction, and the release of potential pro-inflammatory degradation products from the large numbers of myocyte mitochondria (40, 98, 99). In long-term follow-up studies, muscle atrophy has been shown to cause severe functional disabilities in CCI survivors (100, 101). Currently, it is unknown what the precise role is of mitochondrial dysfunction and inflammation in sepsis on skeletal muscle wasting and long-term outcomes.
A novel emerging role of skeletal muscle is its ability to regulate inflammation not only locally but also systemically. Increased catabolism of skeletal muscle during sepsis, either through oxidant injury-induced cellular apoptosis or autophagy, can stimulate an immune response via cellular constituents being released into the circulation and acting as DAMPs. Infiltration of skeletal muscle with myeloid cell populations, including potentially MDSCs, is recognized in skeletal muscle injury and sepsis-associated skeletal muscle wasting (38, 102–105). Fragments such as mtDNA, ATP, TFAM, N-formyl peptides, HMGB1, succinate, and cardiolipin are known mitoDAMPs and can function as endogenous alarmins to propagate chronic inflammation (40). These factors, which can act systemically, may be released during skeletal muscle injury or critical illness-associated muscle wasting. TFAM, one of the potential alarmins released during skeletal muscle damage, has divergent local and systemic functions (106). Acting locally within tissues, TFAM binding to the D-loop in mtDNA is essential for upregulation of mtDNA replication, resulting in increased mtDNA copy number. In striking contrast, if mtDNA or TFAM is released into the cytosol or circulation, it can activate TLR9 pathway factors (41, 107). Animal models of sepsis-induced cardiac inflammation show that at least partially mtDNA–TLR9–RAGE pathway is involved and activated but can be inhibited by a TLR9 inhibitor (41).
Conclusion
Sepsis induces a genomic and cytokine storm that can cause variable host responses in the long term. A substantial portion of sepsis survivors go on to develop CCI, a debilitating condition with profound personal and social costs. CCI is multifactorial and complex, and better understanding is necessary to improve long-term outcomes from sepsis. We hypothesize that CCI is initiated by an early genomic storm, organ injury, and skeletal muscle wasting that leads to a DAMP-driven pro-inflammatory expansion of immature myeloid cells, and infiltration of MDSCs in organs and tissues. The continual release of DAMPs from CKD or muscle wasting propagates the PICS response in sepsis survivors. We propose further work in developing appropriate animal models to study CCI, AKI, and CKD, and targeted interventions to alter the disrupted homeostasis of metabolic reprogramming after sepsis.
Author Contributions
RH, SR, JS, HH, AG, and LL drafted the manuscript. SB, PE, AB, MS, FM, and LM provided critical revisions. All the authors made substantial contributions to the conception and design of the work, approved the submitted version of the manuscript, and agreed to be accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported in part by grants R01 GM-40586, R01 GM-104181, and P50 GM-111182. RH and JS were funded by an NIGMS training grant in burns, trauma and perioperative injury (T32 GM-008721).
References
1. Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med (2001) 29(7):1303–10. doi:10.1097/00003246-200107000-00002
2. Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med (2013) 369(21):2063. doi:10.1056/NEJMra1208623
3. Stevenson EK, Rubenstein AR, Radin GT, Wiener RS, Walkey AJ. Two decades of mortality trends among patients with severe sepsis: a comparative meta-analysis*. Crit Care Med (2014) 42(3):625–31. doi:10.1097/CCM.0000000000000026
4. Lewis AJ, Rosengart MR. Bench-to-bedside: a translational perspective on murine models of sepsis. Surg Infect (Larchmt) (2018) 19(2):137–41. doi:10.1089/sur.2017.308
5. Chiswick EL, Mella JR, Bernardo J, Remick DG. Acute-phase deaths from murine polymicrobial sepsis are characterized by innate immune suppression rather than exhaustion. J Immunol (2015) 195(8):3793–802. doi:10.4049/jimmunol.1500874
6. Remick DG, Newcomb DE, Bolgos GL, Call DR. Comparison of the mortality and inflammatory response of two models of sepsis: lipopolysaccharide vs. cecal ligation and puncture. Shock (2000) 13(2):110–6. doi:10.1097/00024382-200013020-00004
7. Rogy MA, Moldawer LL, Oldenburg HS, Thompson WA, Montegut WJ, Stackpole SA, et al. Anti-endotoxin therapy in primate bacteremia with HA-1A and BPI. Ann Surg (1994) 220(1):77–85. doi:10.1097/00000658-199407000-00011
8. van der Poll T, Van Zee KJ, Endert E, Coyle SM, Stiles DM, Pribble JP, et al. Interleukin-1 receptor blockade does not affect endotoxin-induced changes in plasma thyroid hormone and thyrotropin concentrations in man. J Clin Endocrinol Metab (1995) 80(4):1341–6. doi:10.1210/jcem.80.4.7714108
9. Van Zee KJ, Moldawer LL, Oldenburg HS, Thompson WA, Stackpole SA, Montegut WJ, et al. Protection against lethal Escherichia coli bacteremia in baboons (Papio anubis) by pretreatment with a 55-kDa TNF receptor (CD120a)-Ig fusion protein, Ro 45-2081. J Immunol (1996) 156(6):2221–30.
10. Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, et al. A network-based analysis of systemic inflammation in humans. Nature (2005) 437(7061):1032–7. doi:10.1038/nature03985
11. Fong YM, Marano MA, Moldawer LL, Wei H, Calvano SE, Kenney JS, et al. The acute splanchnic and peripheral tissue metabolic response to endotoxin in humans. J Clin Invest (1990) 85(6):1896–904. doi:10.1172/JCI114651
12. Granowitz EV, Santos AA, Poutsiaka DD, Cannon JG, Wilmore DW, Wolff SM, et al. Production of interleukin-1-receptor antagonist during experimental endotoxaemia. Lancet (1991) 338(8780):1423–4. doi:10.1016/0140-6736(91)92725-H
13. Michie HR, Manogue KR, Spriggs DR, Revhaug A, O’Dwyer S, Dinarello CA, et al. Detection of circulating tumor necrosis factor after endotoxin administration. N Engl J Med (1988) 318(23):1481–6. doi:10.1056/NEJM198806093182301
14. Efron PA, Mohr AM, Moore FA, Moldawer LL. The future of murine sepsis and trauma research models. J Leukoc Biol (2015) 98(6):945–52. doi:10.1189/jlb.5MR0315-127R
15. Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A (2013) 110(9):3507–12. doi:10.1073/pnas.1222878110
16. Warren HS, Tompkins RG, Moldawer LL, Seok J, Xu W, Mindrinos MN, et al. Mice are not men. Proc Natl Acad Sci U S A (2015) 112(4):E345. doi:10.1073/pnas.1414857111
17. Gaieski DF, Edwards JM, Kallan MJ, Carr BG. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med (2013) 41(5):1167–74. doi:10.1097/CCM.0b013e31827c09f8
18. Raymond SL, Holden DC, Mira JC, Stortz JA, Loftus TJ, Mohr AM, et al. Microbial recognition and danger signals in sepsis and trauma. Biochim Biophys Acta (2017) 1863(10 Pt B):2564–73. doi:10.1016/j.bbadis.2017.01.013
19. Yende S, Austin S, Rhodes A, Finfer S, Opal S, Thompson T, et al. Long-term quality of life among survivors of severe sepsis: analyses of two international trials. Crit Care Med (2016) 44(8):1461–7. doi:10.1097/CCM.0000000000001658
20. DiGiandomenico A, Veach RA, Zienkiewicz J, Moore DJ, Wylezinski LS, Hutchens MA, et al. The “genomic storm” induced by bacterial endotoxin is calmed by a nuclear transport modifier that attenuates localized and systemic inflammation. PLoS One (2014) 9(10):e110183. doi:10.1371/journal.pone.0110183
21. Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, Vincent JL. Sepsis and septic shock. Nat Rev Dis Primers (2016) 2:16045. doi:10.1038/nrdp.2016.45
22. Mira JC, Gentile LF, Mathias BJ, Efron PA, Brakenridge SC, Mohr AM, et al. Sepsis pathophysiology, chronic critical illness, and persistent inflammation-immunosuppression and catabolism syndrome. Crit Care Med (2017) 45(2):253–62. doi:10.1097/CCM.0000000000002074
23. Chesney JA, Mitchell RA, Yaddanapudi K. Myeloid-derived suppressor cells—a new therapeutic target to overcome resistance to cancer immunotherapy. J Leukoc Biol (2017) 102(3):727–40. doi:10.1189/jlb.5VMR1116-458RRR
24. Mathias B, Delmas AL, Ozrazgat-Baslanti T, Vanzant EL, Szpila BE, Mohr AM, et al. Human myeloid-derived suppressor cells are associated with chronic immune suppression after severe sepsis/septic shock. Ann Surg (2017) 265(4):827–34. doi:10.1097/SLA.0000000000001783
25. Prescott HC, Angus DC. Enhancing recovery from sepsis: a review. JAMA (2018) 319(1):62–75. doi:10.1001/jama.2017.17687
26. Guirgis FW, Brakenridge S, Sutchu S, Khadpe JD, Robinson T, Westenbarger R, et al. The long-term burden of severe sepsis and septic shock: sepsis recidivism and organ dysfunction. J Trauma Acute Care Surg (2016) 81(3):525–32. doi:10.1097/TA.0000000000001135
27. Stortz JA, Mira JC, Raymond SL, Loftus TJ, Ozrazgat-Baslanti T, Wang Z, et al. Benchmarking clinical outcomes and the immunocatabolic phenotype of chronic critical illness after sepsis in surgical intensive care unit patients. J Trauma Acute Care Surg (2018) 84(2):342–9. doi:10.1097/TA.0000000000001758
28. Torio CM, Andrews RM. National Inpatient Hospital Costs: The Most Expensive Conditions by Payer, 2011: Statistical Brief #160. Rockville, MD: Healthcare Cost and Utilization Project (HCUP) Statistical Briefs (2006).
29. Gentile LF, Cuenca AG, Efron PA, Ang D, Bihorac A, McKinley BA, et al. Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg (2012) 72(6):1491–501. doi:10.1097/TA.0b013e318256e000
30. Darcy CJ, Minigo G, Piera KA, Davis JS, McNeil YR, Chen Y, et al. Neutrophils with myeloid derived suppressor function deplete arginine and constrain T cell function in septic shock patients. Crit Care (2014) 18(4):R163. doi:10.1186/cc14003
31. Hu D, Ren J, Wang G, Gu G, Chen J, Zhou B, et al. Persistent inflammation-immunosuppression catabolism syndrome, a common manifestation of patients with enterocutaneous fistula in intensive care unit. J Trauma Acute Care Surg (2014) 76(3):725–9. doi:10.1097/TA.0b013e3182aafe6b
32. Mira JC, Cuschieri J, Ozrazgat-Baslanti T, Wang Z, Ghita GL, Loftus TJ, et al. The epidemiology of chronic critical illness after severe traumatic injury at two level-one trauma centers. Crit Care Med (2017) 45(12):1989–96. doi:10.1097/CCM.0000000000002697
33. Stortz JA, Murphy TJ, Raymond SL, Mira JC, Ungaro R, Dirain ML, et al. Evidence for persistent immune suppression in patients WHO develop chronic critical illness after sepsis. Shock (2017) 49(3):9. doi:10.1097/SHK.0000000000000981
34. White LE, Hassoun HT, Bihorac A, Moore LJ, Sailors RM, McKinley BA, et al. Acute kidney injury is surprisingly common and a powerful predictor of mortality in surgical sepsis. J Trauma Acute Care Surg (2013) 75(3):432–8. doi:10.1097/TA.0b013e31829de6cd
35. Matejovic M, Chvojka J, Radej J, Ledvinova L, Karvunidis T, Krouzecky A, et al. Sepsis and acute kidney injury are bidirectional. Contrib Nephrol (2011) 174:78–88. doi:10.1159/000329239
36. White LE, Chaudhary R, Moore LJ, Moore FA, Hassoun HT. Surgical sepsis and organ crosstalk: the role of the kidney. J Surg Res (2011) 167(2):306–15. doi:10.1016/j.jss.2010.11.923
37. Dos Santos C, Hussain SN, Mathur S, Picard M, Herridge M, Correa J, et al. Mechanisms of chronic muscle wasting and dysfunction after an intensive care unit stay. A pilot study. Am J Respir Crit Care Med (2016) 194(7):821–30. doi:10.1164/rccm.201512-2344OC
38. Puthucheary ZA, Rawal J, McPhail M, Connolly B, Ratnayake G, Chan P, et al. Acute skeletal muscle wasting in critical illness. JAMA (2013) 310(15):1591–600. doi:10.1001/jama.2013.278481
39. Jeschke MG, Chinkes DL, Finnerty CC, Kulp G, Suman OE, Norbury WB, et al. Pathophysiologic response to severe burn injury. Ann Surg (2008) 248(3):387–401. doi:10.1097/SLA.0b013e3181856241
40. Picca A, Lezza AMS, Leeuwenburgh C, Pesce V, Calvani R, Bossola M, et al. Circulating mitochondrial DNA at the crossroads of mitochondrial dysfunction and inflammation during aging and muscle wasting disorders. Rejuvenation Res (2018). doi:10.1089/rej.2017.1989
41. Yao X, Carlson D, Sun Y, Ma L, Wolf SE, Minei JP, et al. Mitochondrial ROS induces cardiac inflammation via a pathway through mtDNA damage in a pneumonia-related sepsis model. PLoS One (2015) 10(10):e0139416. doi:10.1371/journal.pone.0139416
42. Zhang W, Lavine KJ, Epelman S, Evans SA, Weinheimer CJ, Barger PM, et al. Necrotic myocardial cells release damage-associated molecular patterns that provoke fibroblast activation in vitro and trigger myocardial inflammation and fibrosis in vivo. J Am Heart Assoc (2015) 4(6):e001993. doi:10.1161/JAHA.115.001993
43. Stortz JA, Raymond SL, Mira JC, Moldawer LL, Mohr AM, Efron PA. Murine models of sepsis and trauma: can we bridge the gap? ILAR J (2017) 58(1):90–105. doi:10.1093/ilar/ilx007
44. Craciun FL, Schuller ER, Remick DG. Early enhanced local neutrophil recruitment in peritonitis-induced sepsis improves bacterial clearance and survival. J Immunol (2010) 185(11):6930–8. doi:10.4049/jimmunol.1002300
45. Shindo Y, McDonough JS, Chang KC, Ramachandra M, Sasikumar PG, Hotchkiss RS. Anti-PD-L1 peptide improves survival in sepsis. J Surg Res (2017) 208:33–9. doi:10.1016/j.jss.2016.08.099
46. Jung E, Perrone EE, Liang Z, Breed ER, Dominguez JA, Clark AT, et al. Cecal ligation and puncture followed by methicillin-resistant Staphylococcus aureus pneumonia increases mortality in mice and blunts production of local and systemic cytokines. Shock (2012) 37(1):85–94. doi:10.1097/SHK.0b013e3182360faf
47. Gentile LF, Nacionales DC, Lopez MC, Vanzant E, Cuenca A, Cuenca AG, et al. Protective immunity and defects in the neonatal and elderly immune response to sepsis. J Immunol (2014) 192(7):3156–65. doi:10.4049/jimmunol.1301726
48. Gentile LF, Nacionales DC, Lopez MC, Vanzant E, Cuenca A, Szpila BE, et al. Host responses to sepsis vary in different low-lethality murine models. PLoS One (2014) 9(5):e94404. doi:10.1371/journal.pone.0094404
49. Kelly-Scumpia KM, Scumpia PO, Delano MJ, Weinstein JS, Cuenca AG, Wynn JL, et al. Type I interferon signaling in hematopoietic cells is required for survival in mouse polymicrobial sepsis by regulating CXCL10. J Exp Med (2010) 207(2):319–26. doi:10.1084/jem.20091959
50. Nacionales DC, Szpila B, Ungaro R, Lopez MC, Zhang J, Gentile LF, et al. A detailed characterization of the dysfunctional immunity and abnormal myelopoiesis induced by severe shock and trauma in the aged. J Immunol (2015) 195(5):2396–407. doi:10.4049/jimmunol.1500984
51. Halbach JL, Wang AW, Hawisher D, Cauvi DM, Lizardo RE, Rosas J, et al. Why antibiotic treatment is not enough for sepsis resolution: an evaluation in an experimental animal model. Infect Immun (2017) 85(12): e00664-17. doi:10.1128/IAI.00664-17
52. Busch D, Kapoor A, Rademann P, Hildebrand F, Bahrami S, Thiemermann C, et al. Delayed activation of PPAR-beta/delta improves long-term survival in mouse sepsis: effects on organ inflammation and coagulation. Innate Immun (2018) 24(4):262–73. doi:10.1177/1753425918771748
53. Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM, et al. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J Exp Med (2007) 204(6):1463–74. doi:10.1084/jem.20062602
54. McPeak MB, Youssef D, Williams DA, Pritchett CL, Yao ZQ, McCall CE, et al. Frontline science: myeloid cell-specific deletion of Cebpb decreases sepsis-induced immunosuppression in mice. J Leukoc Biol (2017) 102(2):191–200. doi:10.1189/jlb.4HI1216-537R
55. Frydrych LM, Fattahi F, He K, Ward PA, Delano MJ. Diabetes and sepsis: risk, recurrence, and ruination. Front Endocrinol (2017) 8:271. doi:10.3389/fendo.2017.00271
56. Walton AH, Muenzer JT, Rasche D, Boomer JS, Sato B, Brownstein BH, et al. Reactivation of multiple viruses in patients with sepsis. PLoS One (2014) 9(2):e98819. doi:10.1371/journal.pone.0098819
57. Kang JW, Kim SJ, Cho HI, Lee SM. DAMPs activating innate immune responses in sepsis. Ageing Res Rev (2015) 24(Pt A):54–65. doi:10.1016/j.arr.2015.03.003
58. Fleshner M, Crane CR. Exosomes, DAMPs and miRNA: features of stress physiology and immune homeostasis. Trends Immunol (2017) 38(10):768–76. doi:10.1016/j.it.2017.08.002
59. Achouiti A, Foll D, Vogl T, van Till JW, Laterre PF, Dugernier T, et al. S100A12 and soluble receptor for advanced glycation end products levels during human severe sepsis. Shock (2013) 40(3):188–94. doi:10.1097/SHK.0b013e31829fbc38
60. Hu Q, Ren J, Wu J, Li G, Wu X, Liu S, et al. Elevated levels of plasma mitochondrial DNA are associated with clinical outcome in intra-abdominal infections caused by severe trauma. Surg Infect (Larchmt) (2017) 18(5):610–8. doi:10.1089/sur.2016.276
61. Ingels C, Derese I, Wouters PJ, Van den Berghe G, Vanhorebeek I. Soluble RAGE and the RAGE ligands HMGB1 and S100A12 in critical illness: impact of glycemic control with insulin and relation with clinical outcome. Shock (2015) 43(2):109–16. doi:10.1097/SHK.0000000000000278
62. Timmermans K, Kox M, Scheffer GJ, Pickkers P. Plasma nuclear and mitochondrial DNA levels, and markers of inflammation, shock, and organ damage in patients with septic shock. Shock (2016) 45(6):607–12. doi:10.1097/SHK.0000000000000549
63. Cuenca AG, Delano MJ, Kelly-Scumpia KM, Moreno C, Scumpia PO, Laface DM, et al. A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol Med (2011) 17(3–4):281–92. doi:10.2119/molmed.2010.00178
64. Chiba Y, Mizoguchi I, Hasegawa H, Ohashi M, Orii N, Nagai T, et al. Regulation of myelopoiesis by proinflammatory cytokines in infectious diseases. Cell Mol Life Sci (2017) 75(8):1363–76. doi:10.1007/s00018-017-2724-5
65. Herault A, Binnewies M, Leong S, Calero-Nieto FJ, Zhang SY, Kang YA, et al. Myeloid progenitor cluster formation drives emergency and leukaemic myelopoiesis. Nature (2017) 544(7648):53–8. doi:10.1038/nature21693
66. Loftus TJ, Mohr AM, Moldawer LL. Dysregulated myelopoiesis and hematopoietic function following acute physiologic insult. Curr Opin Hematol (2018) 25(1):37–43. doi:10.1097/MOH.0000000000000395
67. Cosentino M, Marino F, Maestroni GJ. Sympathoadrenergic modulation of hematopoiesis: a review of available evidence and of therapeutic perspectives. Front Cell Neurosci (2015) 9:302. doi:10.3389/fncel.2015.00302
68. Hasan S, Mosier MJ, Szilagyi A, Gamelli RL, Muthumalaiappan K. Discrete beta-adrenergic mechanisms regulate early and late erythropoiesis in erythropoietin-resistant anemia. Surgery (2017) 162(4):901–16. doi:10.1016/j.surg.2017.06.001
69. Scumpia PO, Kelly-Scumpia KM, Delano MJ, Weinstein JS, Cuenca AG, Al-Quran S, et al. Cutting edge: bacterial infection induces hematopoietic stem and progenitor cell expansion in the absence of TLR signaling. J Immunol (2010) 184(5):2247–51. doi:10.4049/jimmunol.0903652
70. Fraenkel PG. Anemia of inflammation: a review. Med Clin North Am (2017) 101(2):285–96. doi:10.1016/j.mcna.2016.09.005
71. Boettcher S, Manz MG. Sensing and translation of pathogen signals into demand-adapted myelopoiesis. Curr Opin Hematol (2016) 23(1):5–10. doi:10.1097/MOH.0000000000000201
72. Buechler MB, Akilesh HM, Hamerman JA. Cutting edge: direct sensing of TLR7 ligands and type I IFN by the common myeloid progenitor promotes mTOR/PI3K-dependent emergency myelopoiesis. J Immunol (2016) 197(7):2577–82. doi:10.4049/jimmunol.1600813
73. Brudecki L, Ferguson DA, McCall CE, El Gazzar M. Myeloid-derived suppressor cells evolve during sepsis and can enhance or attenuate the systemic inflammatory response. Infect Immun (2012) 80(6):2026–34. doi:10.1128/IAI.00239-12
74. Gielen PR, Schulte BM, Kers-Rebel ED, Verrijp K, Bossman SA, Ter Laan M, et al. Elevated levels of polymorphonuclear myeloid-derived suppressor cells in patients with glioblastoma highly express S100A8/9 and arginase and suppress T cell function. Neuro Oncol (2016) 18(9):1253–64. doi:10.1093/neuonc/now034
75. Lai D, Qin C, Shu Q. Myeloid-derived suppressor cells in sepsis. Biomed Res Int (2014) 2014:598654. doi:10.1155/2014/598654
76. Zhou J, Nefedova Y, Lei A, Gabrilovich D. Neutrophils and PMN-MDSC: their biological role and interaction with stromal cells. Semin Immunol (2017) 35:19–28. doi:10.1016/j.smim.2017.12.004
77. Guerin E, Orabona M, Raquil MA, Giraudeau B, Bellier R, Gibot S, et al. Circulating immature granulocytes with T-cell killing functions predict sepsis deterioration*. Crit Care Med (2014) 42(9):2007–18. doi:10.1097/CCM.0000000000000344
78. Janols H, Bergenfelz C, Allaoui R, Larsson AM, Ryden L, Bjornsson S, et al. A high frequency of MDSCs in sepsis patients, with the granulocytic subtype dominating in gram-positive cases. J Leukoc Biol (2014) 96(5):685–93. doi:10.1189/jlb.5HI0214-074R
79. Bihorac A, Brennan M, Ozrazgat-Baslanti T, Bozorgmehri S, Efron PA, Moore FA, et al. National surgical quality improvement program underestimates the risk associated with mild and moderate postoperative acute kidney injury. Crit Care Med (2013) 41(11):2570–83. doi:10.1097/CCM.0b013e31829860fc
80. Bihorac A, Delano MJ, Schold JD, Lopez MC, Nathens AB, Maier RV, et al. Incidence, clinical predictors, genomics, and outcome of acute kidney injury among trauma patients. Ann Surg (2010) 252(1):158–65. doi:10.1097/SLA.0b013e3181deb6bc
81. Bihorac A, Efron P, Ang D, Maier R, Moldawer L. Acute kidney injury is associated with nosocomial infections and surgical site infections after trauma. Surg Infect (Larchmt) (2011) 12:S017. doi:10.1097/TA.0b013e31828586ec
82. Jansen MPB, Pulskens WP, Butter LM, Florquin S, Juffermans NP, Roelofs J, et al. Mitochondrial DNA is released in urine of SIRS patients with acute kidney injury and correlates with severity of renal dysfunction. Shock (2017) 49(3):301–10. doi:10.1097/SHK.0000000000000967
83. Allam R, Scherbaum CR, Darisipudi MN, Mulay SR, Hagele H, Lichtnekert J, et al. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol (2012) 23(8):1375–88. doi:10.1681/ASN.2011111077
84. Leemans JC, Stokman G, Claessen N, Rouschop KM, Teske GJ, Kirschning CJ, et al. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J Clin Invest (2005) 115(10):2894–903. doi:10.1172/JCI22832
85. Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest (2007) 117(10):2847–59. doi:10.1172/JCI31008
86. Grigoryev DN, Liu M, Hassoun HT, Cheadle C, Barnes KC, Rabb H. The local and systemic inflammatory transcriptome after acute kidney injury. J Am Soc Nephrol (2008) 19(3):547–58. doi:10.1681/ASN.2007040469
87. Hassoun HT, Grigoryev DN, Lie ML, Liu M, Cheadle C, Tuder RM, et al. Ischemic acute kidney injury induces a distant organ functional and genomic response distinguishable from bilateral nephrectomy. Am J Physiol Renal Physiol (2007) 293(1):F30–40. doi:10.1152/ajprenal.00023.2007
88. Hoste EA, De Corte W. Clinical consequences of acute kidney injury. Contrib Nephrol (2011) 174:56–64. doi:10.1159/000329236
89. Lee DW, Faubel S, Edelstein CL. Cytokines in acute kidney injury (AKI). Clin Nephrol (2011) 76(3):165–73. doi:10.5414/CN106491
90. White LE, Cui Y, Shelak CM, Lie ML, Hassoun HT. Lung endothelial cell apoptosis during ischemic acute kidney injury. Shock (2012) 38(3):320–7. doi:10.1097/SHK.0b013e31826359d0
91. Zager RA, Johnson AC, Lund S, Hanson S. Acute renal failure: determinants and characteristics of the injury-induced hyperinflammatory response. Am J Physiol Renal Physiol (2006) 291(3):F546–56. doi:10.1152/ajprenal.00072.2006
92. Bihorac A, Baslanti TO, Cuenca AG, Hobson CE, Ang D, Efron PA, et al. Acute kidney injury is associated with early cytokine changes after trauma. J Trauma Acute Care Surg (2013) 74(4):1005–13. doi:10.1097/TA.0b013e31828586ec
93. Hoke TS, Douglas IS, Klein CL, He Z, Fang W, Thurman JM, et al. Acute renal failure after bilateral nephrectomy is associated with cytokine-mediated pulmonary injury. J Am Soc Nephrol (2007) 18(1):155–64. doi:10.1681/ASN.2006050494
94. Liu KD, Altmann C, Smits G, Krawczeski CD, Edelstein CL, Devarajan P, et al. Serum interleukin-6 and interleukin-8 are early biomarkers of acute kidney injury and predict prolonged mechanical ventilation in children undergoing cardiac surgery: a case-control study. Crit Care (2009) 13(4):R104. doi:10.1186/cc7940
95. Kurts C, Panzer U, Anders HJ, Rees AJ. The immune system and kidney disease: basic concepts and clinical implications. Nat Rev Immunol (2013) 13(10):738–53. doi:10.1038/nri3523
96. Tryggvason K, Wartiovaara J. How does the kidney filter plasma? Physiology (Bethesda) (2005) 20:96–101. doi:10.1152/physiol.00045.2004
97. Mulay SR, Kulkarni OP, Rupanagudi KV, Migliorini A, Darisipudi MN, Vilaysane A, et al. Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1beta secretion. J Clin Invest (2013) 123(1):236–46. doi:10.1172/JCI63679
98. Picca A, Lezza AMS, Leeuwenburgh C, Pesce V, Calvani R, Landi F, et al. Fueling inflamm-aging through mitochondrial dysfunction: mechanisms and molecular targets. Int J Mol Sci (2017) 18(5). doi:10.3390/ijms18050933
99. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464(7285):104–7. doi:10.1038/nature08780
100. Batt J, dos Santos CC, Cameron JI, Herridge MS. Intensive care unit-acquired weakness: clinical phenotypes and molecular mechanisms. Am J Respir Crit Care Med (2013) 187(3):238–46. doi:10.1164/rccm.201205-0954SO
101. Puthucheary Z, Harridge S, Hart N. Skeletal muscle dysfunction in critical care: wasting, weakness, and rehabilitation strategies. Crit Care Med (2010) 38(10 Suppl):S676–82. doi:10.1097/CCM.0b013e3181f2458d
102. Tidball JG, Villalta SA. Regulatory interactions between muscle and the immune system during muscle regeneration. Am J Physiol Regul Integr Comp Physiol (2010) 298(5):R1173–87. doi:10.1152/ajpregu.00735.2009
103. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell (2013) 155(6):1282–95. doi:10.1016/j.cell.2013.10.054
104. Flores RR, Clauson CL, Cho J, Lee BC, McGowan SJ, Baker DJ, et al. Expansion of myeloid-derived suppressor cells with aging in the bone marrow of mice through a NF-kappaB-dependent mechanism. Aging Cell (2017) 16(3):480–7. doi:10.1111/acel.12571
105. Matthias N, Hunt SD, Wu J, Lo J, Smith Callahan LA, Li Y, et al. Volumetric muscle loss injury repair using in situ fibrin gel cast seeded with muscle-derived stem cells (MDSCs). Stem Cell Res (2018) 27:65–73. doi:10.1016/j.scr.2018.01.008
106. Picca A, Pesce V, Fracasso F, Joseph AM, Leeuwenburgh C, Lezza AM. A comparison among the tissue-specific effects of aging and calorie restriction on TFAM amount and TFAM-binding activity to mtDNA in rat. Biochim Biophys Acta (2014) 1840(7):2184–91. doi:10.1016/j.bbagen.2014.03.004
Keywords: sepsis, inflammation, critical illness, PICS, immunosuppression
Citation: Hawkins RB, Raymond SL, Stortz JA, Horiguchi H, Brakenridge SC, Gardner A, Efron PA, Bihorac A, Segal M, Moore FA and Moldawer LL (2018) Chronic Critical Illness and the Persistent Inflammation, Immunosuppression, and Catabolism Syndrome. Front. Immunol. 9:1511. doi: 10.3389/fimmu.2018.01511
Received: 15 February 2018; Accepted: 18 June 2018;
Published: 02 July 2018
Edited by:
Christoph Thiemermann, Queen Mary University of London, United KingdomReviewed by:
Anja Fuchs, Washington University in St. Louis, United StatesSinisa Savic, University of Leeds, United Kingdom
Copyright: © 2018 Hawkins, Raymond, Stortz, Horiguchi, Brakenridge, Gardner, Efron, Bihorac, Segal, Moore and Moldawer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lyle L. Moldawer, bW9sZGF3ZXJAc3VyZ2VyeS51ZmwuZWR1