 Cristiane de Jesus Nunes-Santos1,2
Cristiane de Jesus Nunes-Santos1,2 Sergio D. Rosenzweig2*
Sergio D. Rosenzweig2*
- 1Faculdade de Medicina, Instituto da Crianca, Universidade de São Paulo, São Paulo, Brazil
- 2Immunology Service, Department of Laboratory Medicine, NIH Clinical Center, National Institutes of Health (NIH), Bethesda, MD, United States
Bacille Calmette–Guerin (BCG) vaccine is widely used as a prevention strategy against tuberculosis. BCG is a live vaccine, usually given early in life in most countries. While safe to most recipients, it poses a risk to immunocompromised patients. Several primary immunodeficiency diseases (PIDD) have been classically associated with complications related to BCG vaccine. However, a number of new inborn errors of immunity have been described lately in which little is known about adverse reactions following BCG vaccination. The aim of this review is to summarize the existing data on BCG-related complications in patients diagnosed with PIDD described since 2010. When BCG vaccination status or complications were not specifically addressed in those manuscripts, we directly contacted the corresponding authors for further clarification. We also analyzed data on other mycobacterial infections in these patients. Based on our analysis, around 8% of patients with gain-of-function mutations in STAT1 had mycobacterial infections, including localized complications in 3 and disseminated disease in 4 out of 19 BCG-vaccinated patients. Localized BCG reactions were also frequent in activated PI3Kδ syndrome type 1 (3/10) and type 2 (2/18) vaccinated children. Also, of note, no BCG-related complications have been described in either CTLA4 or LRBA protein-deficient patients; and not enough information on BCG-vaccinated NFKB1 or NFKB2-deficient patients was available to drive any conclusions about these diseases. Despite the high prevalence of environmental mycobacterial infections in GATA2-deficient patients, only one case of BCG reaction has been reported in a patient who developed disseminated disease. In conclusion, BCG complications could be expected in some particular, recently described PIDD and it remains a preventable risk factor for pediatric PIDD patients.
Introduction
Based on the World Health Organization (WHO) Global Tuberculosis Report, tuberculosis remains a public health global problem: it is the ninth leading cause of death worldwide, and the leading cause of death from a single pathogen (1). Bacille Calmette–Guerin (BCG) vaccine is widely used as a prevention strategy against tuberculosis. The BCG vaccine was developed between 1908 and 1921 by Albert Calmette and Camille Guerín in France by culturing and attenuating a live strain of Mycobacterium bovis. BCG was first administered to humans in 1921 and has been used for more than 95 years until now (2).
Vaccination policies vary around the world, linked mostly to tuberculosis disease prevalence. While tuberculosis endemic areas (mainly in developing countries) adopt universal vaccination, tuberculosis low-prevalence countries either restrict BCG vaccine to high-risk groups or choose not to administer it at all (3). Controversies surrounding the vaccine’s efficacy account for variations in vaccination policies. While BCG vaccine is believed to provide a somehow consistent protection against severe forms of tuberculosis (i.e., miliary, meningeal) in childhood, most adult individuals remain susceptible to pulmonary tuberculosis despite vaccination (4). As previous exposure to nontuberculous mycobacteria (NTM) seems to influence vaccine efficacy, and to assure full coverage, BCG is usually given right after birth in the first months of life (5).
Even in the context of its questionable efficacy, BCG vaccine is considered safe in immunocompetent subjects (6). However, being a live vaccine, it can result in serious illness or even fatal disease in immunocompromised hosts (7). For instance, WHO guidelines recommend holding BCG vaccination in high-risk infants until assessment of HIV status (8). Patients with primary immunodeficiency diseases (PIDD) are at equal or even greater risk of complications and represent a challenging group regarding live vaccines in general and BCG vaccination in particular (7, 9).
Primary immunodeficiency diseases are inborn errors of immunity which commonly lead to increased susceptibility to infection (10). Defects impairing cellular immunity, phagocytic function, and interferon-γ-mediated immunity have been classically associated with BCG vaccine complications (11). The advent of next-generation sequencing technology boosted discoveries in the field of PIDD. To date, 354 different disorders affecting 344 genes have been described, nearly one-third of them after 2010 (12). Meanwhile, the potential impact of recently described PIDD on BCG immune response remains blurry, as our understanding of natural history and detailed molecular mechanisms of these defects are still limited and continuously expanding.
The aim of this review is to update and summarize the published data on BCG-related complications in patients diagnosed with PIDD described after 2010. We arbitrarily selected defects affecting either innate (i.e., monocytes, macrophages, or dendritic cells) or adaptive (i.e., T cells) arms of antimycobacterial immunity that had been described in at least 20 patients or 10 unrelated kindreds. Diseases unequivocally classified as having Mendelian susceptibility to mycobacterial diseases (MSMD), were not reanalyzed in this review. Finally, 18 genetic defects or allelic variants associated with particular PIDD were analyzed. Considering the timing of vaccine administration, most patients’ PIDD were undiagnosed when vaccinated. As BCG vaccination history is not described in all publications and many reports come from countries where BCG vaccine is not universally applied, we also collected data on any mycobacterial infection, highlighting the occurrence of weakly virulent strains. For those reports where BCG vaccination status was not specifically described, we directly contacted the corresponding author/s for further clarification in terms of patients’ BCG vaccination status and complications associated with it. The overall retrieve rate for extra information requests was 17%, range 0.8–100%, depending on the specific PIDD analyzed (Table 1).
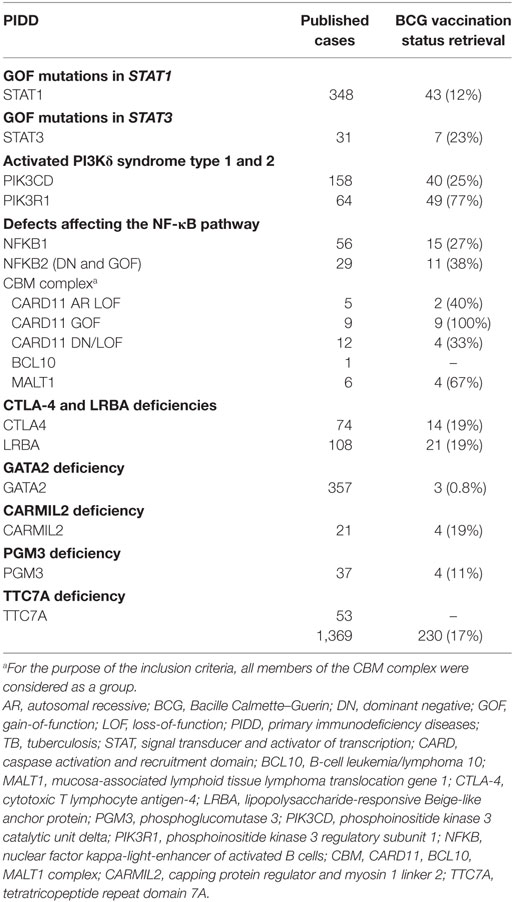
Table 1. Retrieval rate of BCG vaccination status among patients with recently described PIDD.
Gain-of-Function (GOF) Mutations in Signal Transducer and Activator of Transcription 1 (STAT1)
STAT1 is a transcription factor involved in several cytokine-dependent signaling pathways, notably IFN-α/β, IFN-γ, and IL-27 (13, 14). Both biallelic and heterozygous loss-of-function (LOF) mutations of STAT1 have been previously described and associated with either susceptibility to intracellular bacterial/viral infections, or Mendelian susceptibility to mycobacterial disease, respectively (15–17). In 2011, Liu et al. and van de Veerdonk et al. demonstrated that an enhanced STAT1 activity, as a result of heterozygous GOF mutations of STAT1, was also deleterious, presenting with a chronic mucocutaneous candidiasis (CMC) phenotype due to impaired IL-17 immunity (18, 19). Afterward, several studies confirmed that these mutations caused a delay in nuclear dephosphorylation of activated STAT1 resulting in reduced production of Th17 cells (20–22), which are pivotal for candida-specific immune responses (23).
While CMC is still the hallmark of this disease—it was present in 98% of patients from an international cohort of 274 STAT1 GOF patients (24)—as more patients were described, the phenotype was proven to be much more diverse than initially assumed (22, 24). These patients are at higher risk of other infections (bacterial/viral/fungal) (25–27), autoimmunity (28, 29), aneurisms (30, 31), and malignancies (32, 33). Many of these manifestations cannot be solely explained by a reduction of Th17 cells, implying that excessive activation of STAT1 potentially impairs immunity through other mechanisms not yet fully understood (29, 34, 35).
Twenty-seven reports of mycobacterial infections could be retrieved from more than 350 published STAT1 GOF patients (20, 24, 25, 27, 29, 31, 33, 34, 36–40). Vaccination status was not addressed in most descriptions (3, 18–22, 24–34, 36–39, 41–72). We were able to retrieve BCG vaccination history from 43 patients, of whom 19 received the vaccine (Tables 1 and 2). BCG complications were seen in seven patients. Three patients had local reactions [(29, 37, 38), and Acknowledgments] and four experienced disseminated disease (24, 25, 31). In addition to BCG, other mycobacteria also seem to threaten a subset of these patients (Table 2). Disseminated disease was observed in eight more patients (six cases of Mycobacterium tuberculosis and two of NTM) (24, 31, 34, 38, 39), four patients had pulmonary infections caused by environmental mycobacteria (24, 29, 33), and two patients had lymphadenitis caused by Mycobacterium fortuitum (27, 37). M. tuberculosis caused pulmonary disease in five patients (24, 36, 37) and extrapulmonary localized disease in one patient (20).
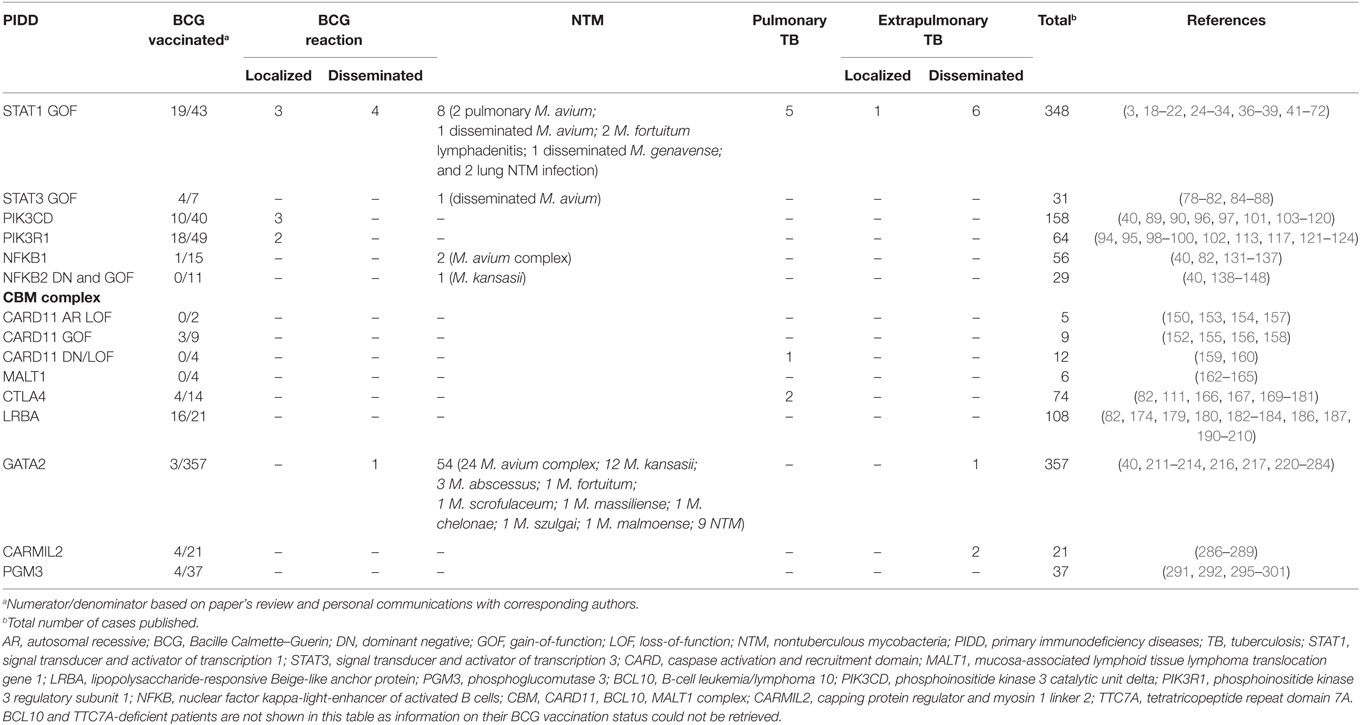
Table 2. BCG and other mycobacterial complications in recently described PIDD.
Overall, our own literature review shows an estimated 8% prevalence of mycobacterial infections among patients carrying STAT1 GOF mutations. Mycobacterial susceptibility is a classical presentation for LOF STAT1 mutations (15–17) as decreased STAT1-mediated IFN-γ responses impair immunity against mycobacteria (73). Published data demonstrate that excessive activation of STAT1 also impairs IFN-γ responses and, therefore, increases the risk of infection (27, 38). IFN-γ tachyphylaxis has been proposed as one possible explanation to this effect (27). Tight regulation of STAT1 phosphorylation, and in turn its activity, is likely required to mount a protective response against mycobacteria (73).
GOF Mutations in Signal Transducer and Activator of Transcription 3 (STAT3)
Signal transducer and activator of transcription 3 is another member of the STATs protein family of transcription factors. STAT3 is activated by various cytokines and growth factors and plays critical roles in several cell processes, such as cell growth, differentiation, apoptosis, as well as inflammation and oncogenesis (74). LOF-dominant negative (DN) mutations in STAT3, cause autosomal-dominant hyper-IgE syndrome, characterized by CMC, bacterial infections, eczema, and connective tissue abnormalities (75, 76). GOF somatic mutations in STAT3 have been associated with some particular types of lymphoproliferative diseases, being found in 40% of large granular lymphocytic leukemia patients (77).
Heterozygous activating germline mutations in STAT3 were first described in 2014, in a cohort of five patients presenting early-onset autoimmunity (78). One-year later, two groups simultaneously described 14 additional cases, providing informative data on this PIDD phenotype (79, 80). Early-onset autoimmunity was prominent, alongside short stature and lymphoproliferative disease. Recurrent infections were frequent and two patients had malignancies, one adult patient had Hodgkin lymphoma, and one pediatric patient had a T-cell large granular lymphocytic leukemia (79, 80). Immunologically, most patients showed hypogammaglobulinemia, and also decreased numbers of T regulatory cells (79–81).
A NTM infection was reported in one patient with a germline STAT3 GOF mutation. The patient had received BCG vaccine as a child without any reported complication. However, at the age of 19 years she developed Mycobacterium avium pneumonia, followed by dissemination (M. avium was isolated from lymph node, feces, and bone marrow samples). The IL-12/IFN-γ pathway was evaluated and found to be normal. The authors hypothesized that the mycobacterial infection could be due to the lack of plasmacytoid dendritic cells detected in the patient (80). A total of four patients received BCG vaccination without complications [(78–80, 82), and Acknowledgments] (Table 2).
The underlying molecular mechanisms of increased transcriptional activity of STAT3 are not yet fully elucidated (83, 84). Although BCG complications have not been reported in these patients [(78–82, 84–88), and Acknowledgments], the occurrence of disseminated environmental mycobacterial infection in one patient raises awareness (80).
Activated PI3Kδ Syndrome Type 1 and 2
Activated phosphoinositide 3-kinase δ syndrome (APDS) in an immunodeficiency and immune dysregulation syndrome, first described in 2013 (89, 90). It results from pathological hyper activation of PI3Kδ, a lipid kinase responsible for the conversion of phosphatidylinositol 4,5-bisphosphate (PIP2) to the second messenger phosphatidylinositol 3,4,5-trisphosphate (PIP3). PI3Kδ is a class IA PI3K, composed of a catalytic (p110δ) and a regulatory subunit (p85). It is predominantly expressed in leukocytes and can be induced by several transmembrane receptors, such as antigen receptors, cytokine receptors, toll-like receptors, and costimulatory molecules. It is important for cell growth, proliferation, motility, and survival (91–93).
Germline heterozygous GOF mutations in PIK3CD, that encode the catalytic subunit p110δ, were initially described as the genetic cause of APDS (89, 90). Less than a year later, heterozygous GOF mutations in PIK3R1, which encodes the regulatory subunit p85, were identified as a phenocopy of this PIDD, being designated APDS type 2 (94, 95). The vast majority of patients present with recurrent sinopulmonary infections commonly leading to bronchiectasis. Other clinical manifestations are highly variable and comprise herpesvirus infections, autoimmune disease (mainly cytopenias), benign lymphoproliferation, and an increased risk of lymphoma (96–99). Immunologically, CD4+ T cell and B cell lymphopenia, progressive loss of naïve CD4+ and CD8+ T cells, expansion of senescent CD8+ T cells, reduced class-switched memory B cells, and poor antibody responses are common findings. A subset of patients presents with hyper IgM (96–101).
In a cohort study of 36 APDS2 patients, Elkaim et al. reported two cases of persistent local skin lesions at BCG vaccination injection site, out of 17 patients who had been BCG vaccinated (99). We gathered information regarding BCG vaccination history from 32 more APDS2 patients and only 1 received the vaccine [(94, 95, 100, 102), and Acknowledgments]. In total, 2/18 BCG-vaccinated APDS2 patients developed local reactions to the vaccine. Likewise, Coulter et al. reported two additional cases of persistent granulomatous local skin reactions to BCG vaccine in a large cohort study of 53 APDS1 patients (97). Through our literature search we identified another local reaction to BCG in an APDS1 patient (103). Collectively, among 10 APDS1 BCG vaccinated patients, three of them developed local reactions to BCG (Table 2) [(40, 90, 97, 101, 103–109), and Acknowledgments].
The ability to control BCG infection was assessed in one APDS1 patient by Chiriaco et al. Monocyte-derived macrophages from the patient failed to restrict intracellular mycobacterial growth in vitro, compared to a healthy control. Treatment with a PI3Kδ inhibitor restored patient’s cells ability to kill BCG, suggesting that normal PI3Kδ activity is important to control BCG infection. Despite the failure in vitro, this patient received BCG vaccine without complications (104).
There were no reports of disseminated BCG reactions or other mycobacterial infections in the APDS patients reviewed (40, 89, 90, 94–124).
Defects Affecting the NF-κB Pathway
Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) represents a protein complex that plays pivotal roles in immune and inflammatory responses, cell development, and survival. It is composed of five transcription factors, NFκB1 (p50/p105), NFκB2 (p52/p100), RelA, RelB, and c-Rel that bind to form homodimers or heterodimers. In resting state these dimers are sequestrated in the cytoplasm, tightly controlled by inhibitory proteins. Once activated, they are released after phosphorylation, ubiquitination, and proteasome degradation of their inhibitors and translocated to the nucleus where they control the transcription of a large set of genes (125, 126).
Mutations that affect the NF-kappa-B inhibitor alpha (NFκBIA or IKBA) or the kinases responsible for its inactivation (IKK-β or IKK-γ/NF-κB essential modifier) are known causes of combined immunodeficiency with increased susceptibility to mycobacteria (11, 127, 128). Several patients carrying these mutations present with disseminated disease after receiving BCG vaccine (129, 130).
More recently, germline mutations in two genes encoding transcription factors of the NF-κB family have been described as new causes of PIDD.
NFKB1 encodes the transcription factor p50 and its precursor p105, which are activated via canonical NF-κB signaling pathway (125, 126). Germline heterozygous mutations in NFKB1 were first described in 2015 (131). They led to haploinsufficiency of NFκB1 and penetrance was shown to be incomplete. The first symptomatic patients reported presented a common variable immunodeficiency (CVID)-like phenotype, suggesting a primarily B-cell disorder. Hypogammaglobulinemia, recurrent infections, autoimmunity, and lymphoproliferation were common findings (82, 131, 132). Finally, new reports expanded the phenotype of this PIDD, highlighting the occurrence of recurrent EBV infection and EBV-driven lymphoproliferative disease, pointing that this defect likely underlies a combined immunodeficiency (133). More than 50 patients carrying NFKB1 mutations have been published (40, 82, 131–137). No BCG-related complications have been reported but BCG vaccination history from these patients was not described in the literature (40, 82, 131–137). We had access to vaccination status of 15 patients, of whom only one received BCG vaccine, uneventfully (Table 2) [(40, 82, 135, 137), and Acknowledgments]. Interestingly, one group reported M. avium intracellulare infections in two patients who were not BCG vaccinated (82).
The second gene, NFKB2, encodes p52 and its precursor p100, the transcription factor that mediates the noncanonical NF-κB signaling pathway (125, 126). Initial reports described an autosomal-dominant inheritance and DN effect (138–140). Patients presented with a phenotype of early-onset CVID (hypogammaglobulinemia, impaired antigen response, recurrent infections) associated with unique autoimmune manifestations, ectodermal dysplasia (alopecia areata or totalis and trachyonychia) and endocrine defects, most notably central adrenal insufficiency (138–140). While humoral immunity was always affected, impairment of T lymphocytes and NK cells varied among published cases (138–142). Neither BCG vaccination status nor vaccine adverse reactions were mentioned in the reports (40, 138–148), however, one patient had Mycobacterium kansasii infection (146). In 2017, Kuehn et al., described three additional patients with novel mutations in NFKB2 that resulted in constitutive NFκB2 activation because of a GOF effect (144). Clinically, these patients presented manifestations consistent with a combined immunodeficiency, such as Pneumocystis jirovecii pneumonia and severe viral infections. Of notice, no endocrine, ectodermal, or autoimmune manifestations were found. None of these patients were BCG vaccinated, but one patient received anti-tuberculous treatment due to pulmonary nodules and caseating granulomas in a lung biopsy, although a mycobacterial infection was not documented [(144), and Acknowledgments].
In the canonical NF-κB signaling pathway, defects in a protein complex upstream NF-κB have also been described as new causes of PIDD. This complex is composed of three proteins: B-cell leukemia/lymphoma 10 (BCL10), mucosa-associated lymphoid tissue lymphoma translocation gene 1 (MALT1), both ubiquitously expressed, and a member of the caspase activation and recruitment domain (CARD) protein family, which is expressed in a cell-type specific manner (149, 150). In lymphocytes, CARD11, BCL10, and MALT1 bind to form the CBM signalosome complex, which is responsible for triggering NF-κB activation following antigen binding to either T- or B-cell receptor (151).
Germline mutations in CARD11 have been reported in 26 patients (150, 152–160) leading to three distinct phenotypes. Patients with biallelic LOF mutations presented with a combined immunodeficiency, similarly to what is seen in other mutations affecting the CBM complex (150, 153, 154, 157). Patients with heterozygous GOF mutations showed a lymphoproliferative condition known as “B cell expansion with NF-KB and T-cell anergy” (BENTA) disease (152). This disease is characterized by childhood-onset polyclonal B-cell lymphocytosis, splenomegaly, recurrent bacterial and viral infections, along with impaired vaccine responses (152, 155, 156, 158). In 2017, two groups identified both LOF and dominant-negative heterozygous defects in CARD11 causing severe atopy and recurrent infections (159, 160). One of these patients had pulmonary tuberculosis (159). BCG-related complications were not mentioned in the reports and BCG vaccination history was not addressed either (150, 152–160). Three BENTA patients were BCG vaccinated without complications (Table 2) [(152, 155, 156, 158), and Acknowledgments].
Concerning the other members of the CBM complex, BCL10 deficiency has been identified in one patient (161) and MALT1 deficiency in six (162–165). These two defects, together with CARD11 LOF mutations, share a similar phenotype of combined immunodeficiency without T cell lymphopenia, poor growth, severe infections, and gastrointestinal disease. Despite the limited number of BCL10 and MALT1 deficiency cases reported, we chose to include them in this review as an exception to our inclusion criteria as they all are a constitutive part of the CBM complex, and help us to expand our understanding on BCG reactions related to new immunodeficiencies in general and CBM complex defects in particular. Neither BCG vaccination status nor BCG-related complications were described in any of the reports (161–165). From our direct contact with the corresponding authors, we got information on the vaccination status of four MALT1 deficient patients, none of them were BCG vaccinated (Table 2) [(163, 164), and Acknowledgments].
Cytotoxic T Lymphocyte Antigen-4 (CTLA-4) and Lipopolysaccharide-Responsive Beige-Like Anchor Protein (LRBA) Deficiencies
CTLA4 haploinsufficiency is an immune dysregulation syndrome characterized by hypogammaglobulinemia, progressive B-cell lymphopenia, autoimmunity, and lymphocytic organ infiltration (166, 167). CTLA-4 is as an inhibitory receptor (168) and, once defective, immune tolerance is disrupted due to impairment of regulatory T cell suppressor function along with an increase of autoreactive B cells, among other immunological abnormalities. It is inherited in an autosomal-dominant manner with incomplete penetrance (166, 167). Seventy-six patients carrying pathogenic mutations in CTLA4 have been published to date (82, 111, 166, 167, 169–181), of whom 62 are symptomatic and 14 asymptomatic mutation carriers due to incomplete disease penetrance. No adverse reactions to BCG vaccine have been reported in their past medical history, although patient’s BCG vaccination status information was not available in the publications. We retrieved information on BCG vaccination status of 14 CTLA4-deficient patients, 4 of whom received BCG vaccine without complications (Table 2) [(82, 166, 170, 174, 177, 179), and Acknowledgments]. Moreover, there were no reports of infections caused by weakly virulent mycobacteria in this cohort (82, 111, 166, 167, 169–181). Two patients did have pulmonary tuberculosis in their early twenties (167). They both progressed to significant lung morbidity, but recurrent bacterial pneumonias also accounted for their unfavorable pulmonary outcomes.
LRBA protein deficiency is closely related to CLTA-4 haploinsufficiency in terms of its pathophysiology (182). It is an autosomal recessive disease with almost complete penetrance, also characterized by recurrent infections, hypogammaglobulinemia, autoimmunity, and lymphocytic organ infiltration (183, 184). Autoimmune disease is particularly severe in the gut, presenting with inflammatory bowel disease-like manifestations (179, 185, 186). LRBA protein protects CTLA-4 from lysosomal degradation, maintaining its intracellular stores (182). Although symptoms are similar to CTLA-4 haploinsufficiency, LRBA deficiency is usually more severe and presents at an earlier age (184, 187). One possible explanation to these differences is that when biallelic mutations of LRBA occur, CTLA-4 surface levels can be even lower than those seen in CTLA-4 haploinsufficiency (188). Also, LRBA protein is present in more cell types than CTLA-4 and not all of its functions are completely known (189). Despite the broader phenotype, over 100 LRBA-deficient patients have been described so far and no adverse reactions to BCG vaccine or any mycobacterial disease have been highlighted in their reports (82, 174, 179, 180, 182–184, 186, 187, 190–210). Out of 21 LRBA-deficient patients that we had access to BCG vaccination history, 16 received BCG vaccine without adverse reactions (Table 2) [(82, 174, 179, 185, 191, 193, 202, 205, 210), and Acknowledgments].
GATA2 Deficiency
GATA2 deficiency was described as a new PIDD in 2011 (211–214). Heterozygous germline mutations in GATA2, a highly pleiotropic gene that encodes the hematopoietic transcription factor GATA2 (215), revealed to be the unifying genetic cause of four apparently distinct syndromes: monocytopenia with M. avium complex (MonoMAC) (216), dendritic cell, monocyte, B and NK lymphoid deficiency (DCML) (217), primary lymphedema with myelodysplasia (Emberger syndrome) (218), and familial myelodysplastic syndrome/acute myeloid leukemia (219).
As more patients were described, the phenotype was broadened and significant overlap among previously known syndromes was noticed (220, 221). Childhood-onset cases have been reported (222), but most patients start manifesting symptoms later in life (220, 221). Patients present increased susceptibility to infections, mainly viral (particularly human papillomavirus), mycobacterial, and fungal (220, 221). Lung involvement is common, and a subset of patients present with pulmonary alveolar proteinosis (223, 224). Lymphedema and hearing loss are also common (221, 222). Most patients progress to myelodysplasia and are at increased risk of acute myeloid leukemia (221, 225).
Immunologically, progressive monocytopenia, B and NK lymphocytopenia, and absence of dendritic cells are hallmarks of the disease. CD4+ lymphopenia and neutropenia can also be seen but are less pronounced (220, 221). Bone marrow usually shows multilineage dysplasia and atypical megakaryocytes (221, 226).
Susceptibility to NTM is a remarkable finding in GATA2 deficiency. Fifty-four cases of NTM (212, 213, 216, 217, 220, 221, 224, 227–243) and only one case of disseminated M. tuberculosis (244) have been described in more than 350 GATA2-deficient patients published (Table 2) (40, 211–214, 216, 217, 220–284).
Information on BCG vaccination was available only for three patients in the reports (217, 241, 245). While two of them did not experience vaccine-related complications, one developed disseminated BCG infection at the age of 12 years, as the initial presentation of his disease. Four years later, he underwent a successful bone marrow transplantation from a matched unrelated donor (285). In the first month post-transplant, the patient had immune reconstitution syndrome (fever and rash) treated with a short course of systemic corticosteroids. Antimycobacterial treatment was stopped within a year.
CARMIL2 Deficiency
Recently, biallelic LOF mutations in RLTPR, also known as CARMIL2, were described as a new PIDD (286–289). Capping protein regulator and myosin 1 linker 2 (CARMIL2) is a cytosolic protein found to be important for CD28-co-stimulation pathway and migration in T cells, as well as BCR-mediated activation of B cells (289, 290).
CARMIL2-deficient patients presented with recurrent bacterial respiratory and cutaneous infections, widespread warts alongside other viral infections (varicella zoster virus, molluscum contagiosum, and EBV), persistent dermatitis (eczema or psoriasiform hyperkeratotic lesions), and CMC. Inflammatory bowel disease and chronic esophagitis were seen in some patients. Four patients had disseminated EBV+ smooth muscle tumors (286–289).
Immunologically, patients shared a significant reduction in regulatory T cells, CD4+ memory and follicular helper cells. Th1 and Th17 cytokine production were impaired. Switched memory B cells counts were low and antibody responses to vaccines were poor (286–289).
Mycobacterial infections were seen in 2 out of 21 published cases. They both had disseminated tuberculosis (described as multifocal disease in one patient and miliary tuberculosis in the other). BCG vaccine had been given to both patients, uneventfully (289). Two other CARMIL2-deficient patients received BCG vaccine, without complications (287) (Table 2). Information on BCG vaccination status of the other CARMIL2-deficient patients was not available (286–289).
Phosphoglucomutase 3 (PGM3) Deficiency
Recently, biallelic hypomorphic mutations in PGM3 have been described as a new congenital disorder of glycosylation resulting in PIDD (291, 292). PGM3 is an enzyme that catalyzes the conversion of N-acetylglucosamine-6-phosphate (GlcNAc-6-P) to 1-phosphate (GlcNAc-1-P) which is necessary for the generation of uridine diphosphate N-acetylglucosamine, an important precursor to multiple glycosylation pathways. Glycosylation is a complex posttranslational enzymatic process responsible for the attachment and trimming of glycans to proteins and lipids, critically affecting their structure and function (293, 294).
Congenital disorders of glycosylation usually manifest with broad, multisystemic symptoms (294). Two distinct phenotypes have been described for PGM3 mutations, possibly correlated with levels of residual enzymatic activity (291, 292, 295). The majority of patients presented with an AR hyper-IgE syndrome phenotype of eczema and multiple manifestations of atopy, recurrent sinopulmonary and skin infections, failure to thrive, and varying degrees of neurological impairment. Dysmorphic features were present in some patients. Serum IgE levels and eosinophils counts were elevated and T cell lymphopenia was frequent (291, 292, 296). A subset of patients manifested a severe combined immunodeficiency phenotype with profound T- and B-cell lymphopenia but normal IgE levels. Neutropenia was also present. Skeletal dysplasia and multiple dysmorphisms were common findings among these patients (295, 297, 298).
There were no reports of BCG-related complications in the PGM3-deficient patients (291, 292, 295–301). Information on vaccination status was not provided in most reports, including all patients with severe combined immunodeficiency (SCID) presentation. Four patients were BCG vaccinated uneventfully (Table 2) (301).
TTC7A Deficiency
The hereditary association of multiple intestinal atresia (MIA) and immunodeficiency has been long reported in the literature (302). However, the discovery of mutations in TTC7A as its underlying cause was only possible in 2013, after the advent of whole exome sequencing (303, 304).
Tetratricopeptide repeat domain 7A (TTC7A) protein function was not clear until the description of TTC7A-deficient patients (305). To date, more than 50 cases of biallelic mutations in TTC7A have been reported (303, 304, 306–317), and this protein was found critical to gut and immune system development and homeostasis (305).
The initial phenotype recognized is shared by the majority of patients reported. MIA requiring surgical interventions is associated with profound lymphopenia, hypogammaglobulinemia, severe and recurrent infections, with high incidence of sepsis caused by intestinal microbes (303, 304, 306–309, 313). Some patients present MIA alone (304, 308), and others manifest very early onset inflammatory bowel disease (204, 307). The prognosis is poor and most patients die at a young age. More recently, a milder phenotype of enteropathy and predominantly humoral immunodeficiency has been reported (315).
None of the reports of TTC7A-deficient patients had information regarding BCG vaccination status. Neither BCG vaccine-related complications nor mycobacterial infections were reported (Table 2) (303, 304, 306–317).
Conclusion
The prevalence of BCG-associated complications in the general population can vary widely depending on the reporting country, the vaccine strain used, and the age at vaccination. Reports of 1 in 2,500 vaccines presenting with localized BCG-associated complications, and 1 in 100,000 presenting with disseminated complications represent a fair estimate of the general prevalence of such side effects. However, when focused on patients with SCID, the most severe forms of PIDD, ~1 in 2 (51%) develop BCG-associated complications after vaccination, 2/3 presenting with disseminated disease, and the remaining 1/3 as localized disease (9). In a recent cohort of 71 chronic granulomatous disease (CGD) patients, 75% presented with BCG-related complications (318). Moreover, among patients with MSMD due to mutations in IL12Rβ1, ~3 in 4 (77%) present with BCG-associated disease, 4/5 as disseminated and 1/5 as localized complications (319). While these diseases are well known for their increased susceptibility to BCG-related side effects, less specific information is available regarding other or more recently described PIDD.
In this review, we focused on PIDD first reported since 2010. In order to assure a fair patient representation, we limited our analysis to those diseases affecting 20 or more unrelated patients belonging to 10 or more families, with defects impairing areas of the immune system known to be involved in the control of mycobacterial infections.
Not surprisingly and as previously shown by Toubiana et al. (24), we found that patients with GOF STAT1 mutations showed an increased susceptibility to mycobacterial diseases, including BCG. Of note, our analysis of the published data also showed that patients with APDS 1 and 2 appear to have an increased risk for BCG-related localized complications. These findings per se can involve actionable recommendations as to ban BCG vaccination in STAT1 GOF, APDS1, and APDS2 patients and in their genetically untested newborn relatives until their status is clarified.
Among other newly described PIDD, no BCG-related complications were reported in the relatively frequent LRBA and CTLA4-deficient patients, or in NFKB1 and NFKB2-mutated individuals. As a limitation of this review, and despite our efforts to try to retrieve as much information as possible regarding BCG vaccination status and complications in these diseases, this negative data have to be taken with a grain of salt as the n of vaccinated patients might not have been sufficient to capture low but still relevant BCG complication rates in these diseases.
In summary, despite its debatable efficacy against tuberculosis, BCG remains one of the most popular and with higher coverage rate vaccines in many parts of the world. Interestingly, certain vaccines—BCG included—have been recently suggested to provide a significantly positive impact on overall health through heterologous immunity, sometimes even surpassing the specific protection originally intended (320). In any case, as clinicians taking care of a vulnerable population of individuals with inborn errors of their immune system, we should remain vigilant of any preventable medical interventions that can detrimentally impact our patients. As already well described, BCG vaccination should be avoided not only in SCID, MSMD, and CGD patients (321) but also in other particular newly described PIDD (e.g., STAT1 GOF, APDS1, and APDS2) in which its complication rates have shown to markedly exceed when compared to the general population.
Author Contributions
CJNS contributed to the research design and wrote the first draft. SDR contributed to the research design and supervised the whole project.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We want to specially thank our colleagues and friends who reviewed their published data for BCG-vaccinated patients (in alphabetical order): Dr. Juan Carlos Aldave, Dr. Sanem Eren Akarcan, Dr. Fowzan S. Alkuraya, Dr. Fayhan J. Alroqi, Dr. Magdalena Avbelj Stefanija, Dr. Safa Baris, Dr. Moshe Ben-Shoshan, Dr. Thierry Brue, Dr. Rebecca H. Buckley, Dr. Jean-Laurent Casanova, Dr. Anita Chandra, Dr. Talal Chatila, Dr. Alison Condliffe, Dr. Megan A. Cooper, Dr. Tanya Coulter, Dr. Charlotte Cunningham-Rundles, Dr. Virgil A. S. H. Dalm, Dr. Anjan Dhar, Dr. Oscar de la Calle-Martin, Dr. Anne Durandy, Dr. Stephan Ehl, Dr. Saul N. Faust, Dr. Sarah E. Flanagan, Dr. Maria Francisca Fontes, Dr. Raif S. Geha, Dr. Bodo Grimbacher, Dr. Mais O. Hashem, Dr. Andrew T. Hattersley, Dr. Merja Helminen, Dr. Alan D. Irvine, Dr. Christian Klemann, Dr. Sven Kracker, Dr. Michael Lenardo, Dr. Francesco Licciardi, Dr. Vassilios Lougaris, Dr. Isabelle Meyts, Dr. Davide Montin, Dr. Mihai G. Netea, Dr. Jennifer Puck, Dr. Anne Puel, Dr. Refik Pul, Dr. Satoshi Okada, Dr. William Rae, Dr. V. Koneti Rao, Dr. Chaim M. Roifman, Dr. Janna Saarela, Dr. David Sansom, Dr. Markus G. Seidel, Dr. Mary A. Slatter, Dr. Andrew L. Snow, Dr. Yuki Tsujita, Dr. Stuart Turvey, Dr. Gulbu Uzel, and Dr. Katja G. Weinacht. This work was supported by the Intramural Research Program, National Institutes of Health Clinical Center. The content of this article does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
References
1. World Health Organization. Global Tuberculosis Report 2017. Geneva: World Health Organization (2017).
2. Ritz N, Hanekom WA, Robins-Browne R, Britton WJ, Curtis N. Influence of BCG vaccine strain on the immune response and protection against tuberculosis. FEMS Microbiol Rev (2008) 32(5):821–41. doi:10.1111/j.1574-6976.2008.00118.x
3. Zwerling A, Behr MA, Verma A, Brewer TF, Menzies D, Pai M. The BCG World Atlas: a database of global BCG vaccination policies and practices. PLoS Med (2011) 8(3):e1001012. doi:10.1371/journal.pmed.1001012
4. Dockrell HM, Smith SG. What have we learnt about BCG Vaccination in the last 20 years? Front Immunol (2017) 8:1134. doi:10.3389/fimmu.2017.01134
5. Mangtani P, Abubakar I, Ariti C, Beynon R, Pimpin L, Fine PE, et al. Protection by BCG vaccine against tuberculosis: a systematic review of randomized controlled trials. Clin Infect Dis (2014) 58(4):470–80. doi:10.1093/cid/cit790
6. World Health Organization. Meeting of the Global Advisory Committee on Vaccine Safety, June 2017. (2017) July, 2017. Report No.: Contract No.: 28.
7. Bonilla FA. Update: vaccines in primary immunodeficiency. J Allergy Clin Immunol (2018) 141(2):474–81. doi:10.1016/j.jaci.2017.12.980
8. World Health Organization. Revised BCG vaccination guidelines for infants at risk for HIV infection. Wkly Epidemiol Rec (2007) 82(21):193–6.
9. Marciano BE, Huang CY, Joshi G, Rezaei N, Carvalho BC, Allwood Z, et al. BCG vaccination in patients with severe combined immunodeficiency: complications, risks, and vaccination policies. J Allergy Clin Immunol (2014) 133(4):1134–41. doi:10.1016/j.jaci.2014.02.028
10. Casanova JL. Severe infectious diseases of childhood as monogenic inborn errors of immunity. Proc Natl Acad Sci U S A (2015) 112(51):E7128–37. doi:10.1073/pnas.1521651112
11. Boisson-Dupuis S, Bustamante J, El-Baghdadi J, Camcioglu Y, Parvaneh N, El Azbaoui S, et al. Inherited and acquired immunodeficiencies underlying tuberculosis in childhood. Immunol Rev (2015) 264(1):103–20. doi:10.1111/imr.12272
12. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International union of immunological societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol (2018) 38(1):96–128. doi:10.1007/s10875-017-0464-9
13. Ramana CV, Chatterjee-Kishore M, Nguyen H, Stark GR. Complex roles of Stat1 in regulating gene expression. Oncogene (2000) 19(21):2619–27. doi:10.1038/sj.onc.1203525
14. Iwasaki Y, Fujio K, Okamura T, Yamamoto K. Interleukin-27 in T cell immunity. Int J Mol Sci (2015) 16(2):2851–63. doi:10.3390/ijms16022851
15. Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet (2003) 33(3):388–91. doi:10.1038/ng1097
16. Chapgier A, Kong XF, Boisson-Dupuis S, Jouanguy E, Averbuch D, Feinberg J, et al. A partial form of recessive STAT1 deficiency in humans. J Clin Invest (2009) 119(6):1502–14. doi:10.1172/JCI37083
17. Chapgier A, Boisson-Dupuis S, Jouanguy E, Vogt G, Feinberg J, Prochnicka-Chalufour A, et al. Novel STAT1 alleles in otherwise healthy patients with mycobacterial disease. PLoS Genet (2006) 2(8):e131. doi:10.1371/journal.pgen.0020131
18. Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med (2011) 208(8):1635–48. doi:10.1084/jem.20110958
19. van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med (2011) 365(1):54–61. doi:10.1056/NEJMoa1100102
20. Lee PP, Mao H, Yang W, Chan KW, Ho MH, Lee TL, et al. Penicillium marneffei infection and impaired IFN-gamma immunity in humans with autosomal-dominant gain-of-phosphorylation STAT1 mutations. J Allergy Clin Immunol (2014) 133(3):894–6.e5. doi:10.1016/j.jaci.2013.08.051
21. Zheng J, van de Veerdonk FL, Crossland KL, Smeekens SP, Chan CM, Al Shehri T, et al. Gain-of-function STAT1 mutations impair STAT3 activity in patients with chronic mucocutaneous candidiasis (CMC). Eur J Immunol (2015) 45(10):2834–46. doi:10.1002/eji.201445344
22. Depner M, Fuchs S, Raabe J, Frede N, Glocker C, Doffinger R, et al. The extended clinical phenotype of 26 patients with chronic mucocutaneous candidiasis due to gain-of-function mutations in STAT1. J Clin Immunol (2016) 36(1):73–84. doi:10.1007/s10875-015-0214-9
23. Mengesha BG, Conti HR. The role of IL-17 in protection against mucosal candida infections. J Fungi (Basel) (2017) 3(4):E52. doi:10.3390/jof3040052
24. Toubiana J, Okada S, Hiller J, Oleastro M, Lagos Gomez M, Aldave Becerra JC, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood (2016) 127(25):3154–64. doi:10.1182/blood-2015-11-679902
25. Sharfe N, Nahum A, Newell A, Dadi H, Ngan B, Pereira SL, et al. Fatal combined immunodeficiency associated with heterozygous mutation in STAT1. J Allergy Clin Immunol (2014) 133(3):807–17. doi:10.1016/j.jaci.2013.09.032
26. Toth B, Mehes L, Tasko S, Szalai Z, Tulassay Z, Cypowyj S, et al. Herpes in STAT1 gain-of-function mutation [corrected]. Lancet (2012) 379(9835):2500. doi:10.1016/S0140-6736(12)60365-1
27. Sampaio EP, Hsu AP, Pechacek J, Bax HI, Dias DL, Paulson ML, et al. Signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations and disseminated coccidioidomycosis and histoplasmosis. J Allergy Clin Immunol (2013) 131(6):1624–34. doi:10.1016/j.jaci.2013.01.052
28. Hori T, Ohnishi H, Teramoto T, Tsubouchi K, Naiki T, Hirose Y, et al. Autosomal-dominant chronic mucocutaneous candidiasis with STAT1-mutation can be complicated with chronic active hepatitis and hypothyroidism. J Clin Immunol (2012) 32(6):1213–20. doi:10.1007/s10875-012-9744-6
29. Uzel G, Sampaio EP, Lawrence MG, Hsu AP, Hackett M, Dorsey MJ, et al. Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like syndrome. J Allergy Clin Immunol (2013) 131(6):1611–23. doi:10.1016/j.jaci.2012.11.054
30. Tanimura M, Dohi K, Hirayama M, Sato Y, Sugiura E, Nakajima H, et al. Recurrent inflammatory aortic aneurysms in chronic mucocutaneous candidiasis with a gain-of-function STAT1 mutation. Int J Cardiol (2015) 196:88–90. doi:10.1016/j.ijcard.2015.05.183
31. Dadak M, Jacobs R, Skuljec J, Jirmo AC, Yildiz O, Donnerstag F, et al. Gain-of-function STAT1 mutations are associated with intracranial aneurysms. Clin Immunol (2017) 178:79–85. doi:10.1016/j.clim.2017.01.012
32. Koo S, Kejariwal D, Al-Shehri T, Dhar A, Lilic D. Oesophageal candidiasis and squamous cell cancer in patients with gain-of-function STAT1 gene mutation. United European Gastroenterol J (2017) 5(5):625–31. doi:10.1177/2050640616684404
33. Romberg N, Morbach H, Lawrence MG, Kim S, Kang I, Holland SM, et al. Gain-of-function STAT1 mutations are associated with PD-L1 overexpression and a defect in B-cell survival. J Allergy Clin Immunol (2013) 131(6):1691–3. doi:10.1016/j.jaci.2013.01.004
34. Kataoka S, Muramatsu H, Okuno Y, Hayashi Y, Mizoguchi Y, Tsumura M, et al. Extrapulmonary tuberculosis mimicking Mendelian susceptibility to mycobacterial disease in a patient with signal transducer and activator of transcription 1 (STAT1) gain-of-function mutation. J Allergy Clin Immunol (2016) 137(2):619–22.e1. doi:10.1016/j.jaci.2015.06.028
35. Zimmerman O, Rosler B, Zerbe CS, Rosen LB, Hsu AP, Uzel G, et al. Risks of ruxolitinib in STAT1 gain-of-function-associated severe fungal disease. Open Forum Infect Dis (2017) 4(4):ofx202. doi:10.1093/ofid/ofx202
36. Baris S, Alroqi F, Kiykim A, Karakoc-Aydiner E, Ogulur I, Ozen A, et al. Severe early-onset combined immunodeficiency due to heterozygous gain-of-function mutations in STAT1. J Clin Immunol (2016) 36(7):641–8. doi:10.1007/s10875-016-0312-3
37. Leiding JW, Okada S, Hagin D, Abinun M, Shcherbina A, Balashov DN, et al. Hematopoietic stem cell transplantation in patients with gain-of-function signal transducer and activator of transcription 1 mutations. J Allergy Clin Immunol (2018) 141(2):704–17.e5. doi:10.1016/j.jaci.2017.03.049
38. Pedraza-Sanchez S, Lezana-Fernandez JL, Gonzalez Y, Martinez-Robles L, Ventura-Ayala ML, Sadowinski-Pine S, et al. Disseminated tuberculosis and chronic mucocutaneous candidiasis in a patient with a gain-of-function mutation in signal transduction and activator of transcription 1. Front Immunol (2017) 8:1651. doi:10.3389/fimmu.2017.01651
39. Zerbe CS, Marciano BE, Katial RK, Santos CB, Adamo N, Hsu AP, et al. Progressive multifocal leukoencephalopathy in primary immune deficiencies: Stat1 gain of function and review of the literature. Clin Infect Dis (2016) 62(8):986–94. doi:10.1093/cid/civ1220
40. Rae W, Ward D, Mattocks CJ, Gao Y, Pengelly RJ, Patel SV, et al. Autoimmunity/inflammation in a monogenic primary immunodeficiency cohort. Clin Transl Immunology (2017) 6(9):e155. doi:10.1038/cti.2017.38
41. Al Rushood M, McCusker C, Mazer B, Alizadehfar R, Grimbacher B, Depner M, et al. Autosomal dominant cases of chronic mucocutaneous candidiasis segregates with mutations of signal transducer and activator of transcription 1, but not of toll-like receptor 3. J Pediatr (2013) 163(1):277–9. doi:10.1016/j.jpeds.2013.02.040
42. Aldave Becerra JC, Cachay Rojas E. A 3-year-old girl with recurrent infections and autoimmunity due to a STAT1 gain-of-function mutation: the expanding clinical presentation of primary immunodeficiencies. Front Pediatr (2017) 5:55. doi:10.3389/fped.2017.00055
43. Aldave JC, Cachay E, Nunez L, Chunga A, Murillo S, Cypowyj S, et al. A 1-year-old girl with a gain-of-function STAT1 mutation treated with hematopoietic stem cell transplantation. J Clin Immunol (2013) 33(8):1273–5. doi:10.1007/s10875-013-9947-5
44. Breuer O, Daum H, Cohen-Cymberknoh M, Unger S, Shoseyov D, Stepensky P, et al. Autosomal dominant gain of function STAT1 mutation and severe bronchiectasis. Respir Med (2017) 126:39–45. doi:10.1016/j.rmed.2017.03.018
45. Dhalla F, Fox H, Davenport EE, Sadler R, Anzilotti C, van Schouwenburg PA, et al. Chronic mucocutaneous candidiasis: characterization of a family with STAT-1 gain-of-function and development of an ex-vivo assay for Th17 deficiency of diagnostic utility. Clin Exp Immunol (2016) 184(2):216–27. doi:10.1111/cei.12746
46. Dotta L, Scomodon O, Padoan R, Timpano S, Plebani A, Soresina A, et al. Clinical heterogeneity of dominant chronic mucocutaneous candidiasis disease: presenting as treatment-resistant candidiasis and chronic lung disease. Clin Immunol (2016) 164:1–9. doi:10.1016/j.clim.2015.12.010
47. Eren Akarcan S, Ulusoy Severcan E, Edeer Karaca N, Isik E, Aksu G, Migaud M, et al. Gain-of-function mutations in STAT1: a recently defined cause for chronic mucocutaneous candidiasis disease mimicking combined immunodeficiencies. Case Reports Immunol (2017) 2017:2846928. doi:10.1155/2017/2846928
48. Eslami N, Tavakol M, Mesdaghi M, Gharegozlou M, Casanova JL, Puel A, et al. A gain-of-function mutation of STAT1: a novel genetic factor contributing to chronic mucocutaneous candidiasis. Acta Microbiol Immunol Hung (2017) 64(2):191–201. doi:10.1556/030.64.2017.014
49. Frans G, Moens L, Schaballie H, Van Eyck L, Borgers H, Wuyts M, et al. Gain-of-function mutations in signal transducer and activator of transcription 1 (STAT1): chronic mucocutaneous candidiasis accompanied by enamel defects and delayed dental shedding. J Allergy Clin Immunol (2014) 134(5):1209–13.e6. doi:10.1016/j.jaci.2014.05.044
50. Giardino G, Somma D, Cirillo E, Ruggiero G, Terrazzano G, Rubino V, et al. Novel STAT1 gain-of-function mutation and suppurative infections. Pediatr Allergy Immunol (2016) 27(2):220–3. doi:10.1111/pai.12496
51. Higgins E, Al Shehri T, McAleer MA, Conlon N, Feighery C, Lilic D, et al. Use of ruxolitinib to successfully treat chronic mucocutaneous candidiasis caused by gain-of-function signal transducer and activator of transcription 1 (STAT1) mutation. J Allergy Clin Immunol (2015) 135(2):551–3. doi:10.1016/j.jaci.2014.12.1867
52. Kilic SS, Puel A, Casanova JL. Orf infection in a patient with Stat1 gain-of-function. J Clin Immunol (2015) 35(1):80–3. doi:10.1007/s10875-014-0111-7
53. Kobbe R, Kolster M, Fuchs S, Schulze-Sturm U, Jenderny J, Kochhan L, et al. Common variable immunodeficiency, impaired neurological development and reduced numbers of T regulatory cells in a 10-year-old boy with a STAT1 gain-of-function mutation. Gene (2016) 586(2):234–8. doi:10.1016/j.gene.2016.04.006
54. Kumar N, Hanks ME, Chandrasekaran P, Davis BC, Hsu AP, Van Wagoner NJ, et al. Gain-of-function signal transducer and activator of transcription 1 (STAT1) mutation-related primary immunodeficiency is associated with disseminated mucormycosis. J Allergy Clin Immunol (2014) 134(1):236–9. doi:10.1016/j.jaci.2014.02.037
55. Martinez-Martinez L, Martinez-Saavedra MT, Fuentes-Prior P, Barnadas M, Rubiales MV, Noda J, et al. A novel gain-of-function STAT1 mutation resulting in basal phosphorylation of STAT1 and increased distal IFN-gamma-mediated responses in chronic mucocutaneous candidiasis. Mol Immunol (2015) 68(2 Pt C):597–605. doi:10.1016/j.molimm.2015.09.014
56. Meesilpavikkai K, Dik WA, Schrijver B, Nagtzaam NM, van Rijswijk A, Driessen GJ, et al. A novel heterozygous mutation in the STAT1 SH2 domain causes chronic mucocutaneous candidiasis, atypically diverse infections, autoimmunity, and impaired cytokine regulation. Front Immunol (2017) 8:274. doi:10.3389/fimmu.2017.00274
57. Mekki N, Ben-Mustapha I, Liu L, Boussofara L, Okada S, Cypowyj S, et al. IL-17 T cells’ defective differentiation in vitro despite normal range ex vivo in chronic mucocutaneous candidiasis due to STAT1 mutation. J Invest Dermatol (2014) 134(4):1155–7. doi:10.1038/jid.2013.480
58. Mizoguchi Y, Tsumura M, Okada S, Hirata O, Minegishi S, Imai K, et al. Simple diagnosis of STAT1 gain-of-function alleles in patients with chronic mucocutaneous candidiasis. J Leukoc Biol (2014) 95(4):667–76. doi:10.1189/jlb.0513250
59. Mossner R, Diering N, Bader O, Forkel S, Overbeck T, Gross U, et al. Ruxolitinib induces interleukin 17 and ameliorates chronic mucocutaneous candidiasis caused by STAT1 gain-of-function mutation. Clin Infect Dis (2016) 62(7):951–3. doi:10.1093/cid/ciw020
60. Nielsen J, Kofod-Olsen E, Spaun E, Larsen CS, Christiansen M, Mogensen TH. A STAT1-gain-of-function mutation causing Th17 deficiency with chronic mucocutaneous candidiasis, psoriasiform hyperkeratosis and dermatophytosis. BMJ Case Rep (2015) 2015:1–5. doi:10.1136/bcr-2015-211372
61. Sampaio EP, Ding L, Rose SR, Cruz P, Hsu AP, Kashyap A, et al. Novel signal transducer and activator of transcription 1 mutation disrupts small ubiquitin-related modifier conjugation causing gain of function. J Allergy Clin Immunol (2017) 141(5):1844–53.e2. doi:10.1016/j.jaci.2017.07.027
62. Second J, Korganow AS, Jannier S, Puel A, Lipsker D. Rosacea and demodicidosis associated with gain-of-function mutation in STAT1. J Eur Acad Dermatol Venereol (2017) 31(12):e542–4. doi:10.1111/jdv.14413
63. Smeekens SP, Plantinga TS, van de Veerdonk FL, Heinhuis B, Hoischen A, Joosten LA, et al. STAT1 hyperphosphorylation and defective IL12R/IL23R signaling underlie defective immunity in autosomal dominant chronic mucocutaneous candidiasis. PLoS One (2011) 6(12):e29248. doi:10.1371/journal.pone.0029248
64. Sobh A, Chou J, Schneider L, Geha RS, Massaad MJ. Chronic mucocutaneous candidiasis associated with an SH2 domain gain-of-function mutation that enhances STAT1 phosphorylation. J Allergy Clin Immunol (2016) 138(1):297–9. doi:10.1016/j.jaci.2015.12.1320
65. Soltesz B, Toth B, Shabashova N, Bondarenko A, Okada S, Cypowyj S, et al. New and recurrent gain-of-function STAT1 mutations in patients with chronic mucocutaneous candidiasis from Eastern and Central Europe. J Med Genet (2013) 50(9):567–78. doi:10.1136/jmedgenet-2013-101570
66. Tabellini G, Vairo D, Scomodon O, Tamassia N, Ferraro RM, Patrizi O, et al. Impaired natural killer cell functions in patients with signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations. J Allergy Clin Immunol (2017) 140(2):553–64.e4. doi:10.1016/j.jaci.2016.10.051
67. Takezaki S, Yamada M, Kato M, Park MJ, Maruyama K, Yamazaki Y, et al. Chronic mucocutaneous candidiasis caused by a gain-of-function mutation in the STAT1 DNA-binding domain. J Immunol (2012) 189(3):1521–6. doi:10.4049/jimmunol.1200926
68. Wang X, Lin Z, Gao L, Wang A, Wan Z, Chen W, et al. Exome sequencing reveals a signal transducer and activator of transcription 1 (STAT1) mutation in a child with recalcitrant cutaneous fusariosis. J Allergy Clin Immunol (2013) 131(4):1242–3. doi:10.1016/j.jaci.2012.11.005
69. Weinacht KG, Charbonnier LM, Alroqi F, Plant A, Qiao Q, Wu H, et al. Ruxolitinib reverses dysregulated T helper cell responses and controls autoimmunity caused by a novel signal transducer and activator of transcription 1 (STAT1) gain-of-function mutation. J Allergy Clin Immunol (2017) 139(5):1629–40.e2. doi:10.1016/j.jaci.2016.11.022
70. Wildbaum G, Shahar E, Katz R, Karin N, Etzioni A, Pollack S. Continuous G-CSF therapy for isolated chronic mucocutaneous candidiasis: complete clinical remission with restoration of IL-17 secretion. J Allergy Clin Immunol (2013) 132(3):761–4. doi:10.1016/j.jaci.2013.04.018
71. Yamazaki Y, Yamada M, Kawai T, Morio T, Onodera M, Ueki M, et al. Two novel gain-of-function mutations of STAT1 responsible for chronic mucocutaneous candidiasis disease: impaired production of IL-17A and IL-22, and the presence of anti-IL-17F autoantibody. J Immunol (2014) 193(10):4880–7. doi:10.4049/jimmunol.1401467
72. Faitelson Y, Bates A, Shroff M, Grunebaum E, Roifman CM, Naqvi A. A mutation in the STAT1 DNA-binding domain associated with hemophagocytic lymphohistiocytosis. LymphoSign J (2014) 1(2):87–95. doi:10.14785/lpsn-2014-0004
73. Boisson-Dupuis S, Kong XF, Okada S, Cypowyj S, Puel A, Abel L, et al. Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr Opin Immunol (2012) 24(4):364–78. doi:10.1016/j.coi.2012.04.011
74. Qi QR, Yang ZM. Regulation and function of signal transducer and activator of transcription 3. World J Biol Chem (2014) 5(2):231–9. doi:10.4331/wjbc.v5.i2.231
75. Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature (2007) 448(7157):1058–62. doi:10.1038/nature06096
76. Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med (2007) 357(16):1608–19. doi:10.1056/NEJMoa073687
77. Koskela HL, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmaki H, Andersson EI, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med (2012) 366(20):1905–13. doi:10.1056/NEJMoa1114885
78. Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Allen HL, De Franco E, et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet (2014) 46(8):812–4. doi:10.1038/ng.3040
79. Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood (2015) 125(4):591–9. doi:10.1182/blood-2014-09-602763
80. Haapaniemi EM, Kaustio M, Rajala HL, van Adrichem AJ, Kainulainen L, Glumoff V, et al. Autoimmunity, hypogammaglobulinemia, lymphoproliferation, and mycobacterial disease in patients with activating mutations in STAT3. Blood (2015) 125(4):639–48. doi:10.1182/blood-2014-04-570101
81. Weinreich MA, Vogel TP, Rao VK, Milner JD. Up, down, and all around: diagnosis and treatment of novel STAT3 variant. Front Pediatr (2017) 5:49. doi:10.3389/fped.2017.00049
82. Maffucci P, Filion CA, Boisson B, Itan Y, Shang L, Casanova JL, et al. Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front Immunol (2016) 7:220. doi:10.3389/fimmu.2016.00220
83. Consonni F, Dotta L, Todaro F, Vairo D, Badolato R. Signal transducer and activator of transcription gain-of-function primary immunodeficiency/immunodysregulation disorders. Curr Opin Pediatr (2017) 29(6):711–7. doi:10.1097/MOP.0000000000000551
84. Gutierrez M, Scaglia P, Keselman A, Martucci L, Karabatas L, Domene S, et al. Partial growth hormone insensitivity and dysregulatory immune disease associated with de novo germline activating STAT3 mutations. Mol Cell Endocrinol (2018). doi:10.1016/j.mce.2018.01.016
85. Khoury T, Molho-Pessach V, Ramot Y, Ayman AR, Elpeleg O, Berkman N, et al. Tocilizumab promotes regulatory T-cell alleviation in STAT3 gain-of-function-associated multi-organ autoimmune syndrome. Clin Ther (2017) 39(2):444–9. doi:10.1016/j.clinthera.2017.01.004
86. Nabhani S, Schipp C, Miskin H, Levin C, Postovsky S, Dujovny T, et al. STAT3 gain-of-function mutations associated with autoimmune lymphoproliferative syndrome like disease deregulate lymphocyte apoptosis and can be targeted by BH3 mimetic compounds. Clin Immunol (2017) 181:32–42. doi:10.1016/j.clim.2017.05.021
87. Sediva H, Dusatkova P, Kanderova V, Obermannova B, Kayserova J, Sramkova L, et al. Short stature in a boy with multiple early-onset autoimmune conditions due to a STAT3 activating mutation: could intracellular growth hormone signalling be compromised? Horm Res Paediatr (2017) 88(2):160–6. doi:10.1159/000456544
88. Velayos T, Martinez R, Alonso M, Garcia-Etxebarria K, Aguayo A, Camarero C, et al. An activating mutation in STAT3 results in neonatal diabetes through reduced insulin synthesis. Diabetes (2017) 66(4):1022–9. doi:10.2337/db16-0867
89. Angulo I, Vadas O, Garçon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase δ gene mutation predisposes to respiratory infection and airway damage. Science (2013) 342(6160):866–71. doi:10.1126/science.1243292
90. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol (2014) 15(1):88–97. doi:10.1038/ni.2771
91. Vanhaesebroeck B, Welham MJ, Kotani K, Stein R, Warne PH, Zvelebil MJ, et al. P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc Natl Acad Sci U S A (1997) 94(9):4330–5. doi:10.1073/pnas.94.9.4330
92. Fung-Leung WP. Phosphoinositide 3-kinase delta (PI3Kdelta) in leukocyte signaling and function. Cell Signal (2011) 23(4):603–8. doi:10.1016/j.cellsig.2010.10.002
93. Fruman DA, Bismuth G. Fine tuning the immune response with PI3K. Immunol Rev (2009) 228(1):253–72. doi:10.1111/j.1600-065X.2008.00750.x
94. Deau MC, Heurtier L, Frange P, Suarez F, Bole-Feysot C, Nitschke P, et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest (2014) 124(9):3923–8. doi:10.1172/JCI75746
95. Lucas CL, Zhang Y, Venida A, Wang Y, Hughes J, McElwee J, et al. Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med (2014) 211(13):2537–47. doi:10.1084/jem.20141759
96. Elgizouli M, Lowe DM, Speckmann C, Schubert D, Hulsdunker J, Eskandarian Z, et al. Activating PI3Kdelta mutations in a cohort of 669 patients with primary immunodeficiency. Clin Exp Immunol (2016) 183(2):221–9. doi:10.1111/cei.12706
97. Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase δ syndrome: a large patient cohort study. J Allergy Clin Immunol (2017) 139(2):597–606.e4. doi:10.1016/j.jaci.2016.06.021
98. Olbrich P, Lorenz M, Cura Daball P, Lucena JM, Rensing-Ehl A, Sanchez B, et al. Activated PI3Kdelta syndrome type 2: two patients, a novel mutation, and review of the literature. Pediatr Allergy Immunol (2016) 27(6):640–4. doi:10.1111/pai.12585
99. Elkaim E, Neven B, Bruneau J, Mitsui-Sekinaka K, Stanislas A, Heurtier L, et al. Clinical and immunologic phenotype associated with activated phosphoinositide 3-kinase delta syndrome 2: a cohort study. J Allergy Clin Immunol (2016) 138(1):210–8.e9. doi:10.1016/j.jaci.2016.03.022
100. Lougaris V, Faletra F, Lanzi G, Vozzi D, Marcuzzi A, Valencic E, et al. Altered germinal center reaction and abnormal B cell peripheral maturation in PI3KR1-mutated patients presenting with HIGM-like phenotype. Clin Immunol (2015) 159(1):33–6. doi:10.1016/j.clim.2015.04.014
101. Crank MC, Grossman JK, Moir S, Pittaluga S, Buckner CM, Kardava L, et al. Mutations in PIK3CD can cause hyper IgM syndrome (HIGM) associated with increased cancer susceptibility. J Clin Immunol (2014) 34(3):272–6. doi:10.1007/s10875-014-0012-9
102. Petrovski S, Parrott RE, Roberts JL, Huang H, Yang J, Gorentla B, et al. Dominant splice site mutations in PIK3R1 cause hyper IgM syndrome, lymphadenopathy and short stature. J Clin Immunol (2016) 36(5):462–71. doi:10.1007/s10875-016-0281-6
103. Tsujita Y, Mitsui-Sekinaka K, Imai K, Yeh TW, Mitsuiki N, Asano T, et al. Phosphatase and tensin homolog (PTEN) mutation can cause activated phosphatidylinositol 3-kinase delta syndrome-like immunodeficiency. J Allergy Clin Immunol (2016) 138(6):1672–80.e10. doi:10.1016/j.jaci.2016.03.055
104. Chiriaco M, Brigida I, Ariganello P, Di Cesare S, Di Matteo G, Taus F, et al. The case of an APDS patient: defects in maturation and function and decreased in vitro anti-mycobacterial activity in the myeloid compartment. Clin Immunol (2017) 178:20–8. doi:10.1016/j.clim.2015.12.008
105. Hartman HN, Niemela J, Hintermeyer MK, Garofalo M, Stoddard J, Verbsky JW, et al. Gain of function mutations of PIK3CD as a cause of primary sclerosing cholangitis. J Clin Immunol (2015) 35(1):11–4. doi:10.1007/s10875-014-0109-1
106. Rae W, Gao Y, Ward D, Mattocks CJ, Eren E, Williams AP. A novel germline gain-of-function variant in PIK3CD. Clin Immunol (2017) 181:29–31. doi:10.1016/j.clim.2017.05.020
107. Heurtier L, Lamrini H, Chentout L, Deau MC, Bouafia A, Rosain J, et al. Mutations in the adaptor-binding domain and associated linker region of p110delta cause activated PI3K-delta syndrome 1 (APDS1). Haematologica (2017) 102(7):e278–81. doi:10.3324/haematol.2017.167601
108. Rae W, Ramakrishnan KA, Gao Y, Ashton-Key M, Pengelly RJ, Patel SV, et al. Precision treatment with sirolimus in a case of activated phosphoinositide 3-kinase delta syndrome. Clin Immunol (2016) 171:38–40. doi:10.1016/j.clim.2016.07.017
109. Rao VK, Webster S, Dalm VASH, Šedivá A, van Hagen PM, Holland S, et al. Effective “activated PI3Kδ syndrome”-targeted therapy with the PI3Kδ inhibitor leniolisib. Blood (2017) 130(21):2307–16. doi:10.1182/blood-2017-08-801191
110. Kracker S, Curtis J, Ibrahim MA, Sediva A, Salisbury J, Campr V, et al. Occurrence of B-cell lymphomas in patients with activated phosphoinositide 3-kinase δ syndrome. J Allergy Clin Immunol (2014) 134(1):233–6. doi:10.1016/j.jaci.2014.02.020
111. Buchbinder D, Seppanen M, Rao VK, Uzel G, Nugent D. Clinical challenges: identification of patients with novel primary immunodeficiency syndromes. J Pediatr Hematol Oncol (2017). doi:10.1097/MPH.0000000000001003
112. Dulau Florea AE, Braylan RC, Schafernak KT, Williams KW, Daub J, Goyal RK, et al. Abnormal B-cell maturation in the bone marrow of patients with germline mutations in PIK3CD. J Allergy Clin Immunol (2017) 139(3):1032–5.e6. doi:10.1016/j.jaci.2016.08.028
113. Nademi Z, Slatter MA, Dvorak CC, Neven B, Fischer A, Suarez F, et al. Hematopoietic stem cell transplant in patients with activated PI3K delta syndrome. J Allergy Clin Immunol (2017) 139(3):1046–9. doi:10.1016/j.jaci.2016.09.040
114. Mettman D, Thiffault I, Dinakar C, Saunders C. Immunodeficiency-associated lymphoid hyperplasia as a cause of intussusception in a case of activated PI3K-δ syndrome. Front Pediatr (2017) 5:71. doi:10.3389/fped.2017.00071
115. Saettini F, Pelagatti MA, Sala D, Moratto D, Giliani S, Badolato R, et al. Early diagnosis of PI3Kδ syndrome in a 2 years old girl with recurrent otitis and enlarged spleen. Immunol Lett (2017) 190:279–81. doi:10.1016/j.imlet.2017.08.021
116. Takeda AJ, Zhang Y, Dornan GL, Siempelkamp BD, Jenkins ML, Matthews HF, et al. Novel PIK3CD mutations affecting N-terminal residues of p110δ cause activated PI3Kδ syndrome (APDS) in humans. J Allergy Clin Immunol (2017) 140(4):1152–6.e10. doi:10.1016/j.jaci.2017.03.026
117. Wentink M, Dalm V, Lankester AC, van Schouwenburg PA, Schölvinck L, Kalina T, et al. Genetic defects in PI3Kδ affect B-cell differentiation and maturation leading to hypogammaglobulineamia and recurrent infections. Clin Immunol (2017) 176:77–86. doi:10.1016/j.clim.2017.01.004
118. Jou ST, Chien YH, Yang YH, Wang TC, Shyur SD, Chou CC, et al. Identification of variations in the human phosphoinositide 3-kinase p110delta gene in children with primary B-cell immunodeficiency of unknown aetiology. Int J Immunogenet (2006) 33(5):361–9. doi:10.1111/j.1744-313X.2006.00627.x
119. Kannan JA, Davila-Saldana BJ, Zhang K, Filipovich AH, Kucuk ZY. Activated phosphoinositide 3-kinase delta syndrome in a patient with a former diagnosis of common variable immune deficiency, bronchiectasis, and lymphoproliferative disease. Ann Allergy Asthma Immunol (2015) 115(5):452–4. doi:10.1016/j.anai.2015.08.009
120. Goto F, Uchiyama T, Nakazawa Y, Imai K, Kawai T, Onodera M. Persistent impairment of T-cell regeneration in a patient with activated PI3K delta syndrome. J Clin Immunol (2017) 37(4):347–50. doi:10.1007/s10875-017-0393-7
121. Kuhlen M, Honscheid A, Loizou L, Nabhani S, Fischer U, Stepensky P, et al. De novo PIK3R1 gain-of-function with recurrent sinopulmonary infections, long-lasting chronic CMV-lymphadenitis and microcephaly. Clin Immunol (2016) 162:27–30. doi:10.1016/j.clim.2015.10.008
122. Martinez-Saavedra MT, Garcia-Gomez S, Dominguez Acosta A, Mendoza Quintana JJ, Paez JP, Garcia-Reino EJ, et al. Gain-of-function mutation in PIK3R1 in a patient with a narrow clinical phenotype of respiratory infections. Clin Immunol (2016) 173:117–20. doi:10.1016/j.clim.2016.09.011
123. Bravo Garcia-Morato M, Garcia-Minaur S, Molina Garicano J, Santos Simarro F, Del Pino Molina L, Lopez-Granados E, et al. Mutations in PIK3R1 can lead to APDS2, SHORT syndrome or a combination of the two. Clin Immunol (2017) 179:77–80. doi:10.1016/j.clim.2017.03.004
124. Hauck F, Magg T, Krolo A, Bilic I, Hirschmugl T, Laass M, et al. Variant PIK3R1 hypermorphic mutation and clinical phenotypes in a family with short statures, mild immunodeficiency and lymphoma. Klin Padiatr (2017) 229(3):113–7. doi:10.1055/s-0043-104218
125. Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev (2012) 26(3):203–34. doi:10.1101/gad.183434.111
126. Mitchell S, Vargas J, Hoffmann A. Signaling via the NFκB system. Wiley Interdiscip Rev Syst Biol Med (2016) 8(3):227–41. doi:10.1002/wsbm.1331
127. Puel A, Picard C, Ku CL, Smahi A, Casanova JL. Inherited disorders of NF-kappaB-mediated immunity in man. Curr Opin Immunol (2004) 16(1):34–41. doi:10.1016/j.coi.2003.11.013
128. Paciolla M, Pescatore A, Conte MI, Esposito E, Incoronato M, Lioi MB, et al. Rare mendelian primary immunodeficiency diseases associated with impaired NF-kappaB signaling. Genes Immun (2015) 16(4):239–46. doi:10.1038/gene.2015.3
129. Karaca NE, Aksu G, Ulusoy E, Cavusoglu C, Oleaga-Quintas C, Nieto-Patlan A, et al. Disseminated BCG infectious disease and hyperferritinemia in a patient with a novel NEMO mutation. J Investig Allergol Clin Immunol (2016) 26(4):268–71. doi:10.18176/jiaci.0068
130. Imamura M, Kawai T, Okada S, Izawa K, Takachi T, Iwabuchi H, et al. Disseminated BCG infection mimicking metastatic nasopharyngeal carcinoma in an immunodeficient child with a novel hypomorphic NEMO mutation. J Clin Immunol (2011) 31(5):802–10. doi:10.1007/s10875-011-9568-9
131. Fliegauf M, Bryant VL, Frede N, Slade C, Woon ST, Lehnert K, et al. Haploinsufficiency of the NF-kappaB1 subunit p50 in common variable immunodeficiency. Am J Hum Genet (2015) 97(3):389–403. doi:10.1016/j.ajhg.2015.07.008
132. Keller B, Cseresnyes Z, Stumpf I, Wehr C, Fliegauf M, Bulashevska A, et al. Disturbed canonical nuclear factor of kappa light chain signaling in B cells of patients with common variable immunodeficiency. J Allergy Clin Immunol (2017) 139(1):220–31.e8. doi:10.1016/j.jaci.2016.04.043
133. Boztug H, Hirschmugl T, Holter W, Lakatos K, Kager L, Trapin D, et al. NF-kappaB1 haploinsufficiency causing immunodeficiency and EBV-driven lymphoproliferation. J Clin Immunol (2016) 36(6):533–40. doi:10.1007/s10875-016-0306-1
134. Kaustio M, Haapaniemi E, Goos H, Hautala T, Park G, Syrjanen J, et al. Damaging heterozygous mutations in NFKB1 lead to diverse immunologic phenotypes. J Allergy Clin Immunol (2017) 140(3):782–96. doi:10.1016/j.jaci.2016.10.054
135. Lougaris V, Patrizi O, Baronio M, Tabellini G, Tampella G, Damiati E, et al. NFKB1 regulates human NK cell maturation and effector functions. Clin Immunol (2017) 175:99–108. doi:10.1016/j.clim.2016.11.012
136. Schipp C, Nabhani S, Bienemann K, Simanovsky N, Kfir-Erenfeld S, Assayag-Asherie N, et al. Specific antibody deficiency and autoinflammatory disease extend the clinical and immunological spectrum of heterozygous NFKB1 loss-of-function mutations in humans. Haematologica (2016) 101(10):e392–6. doi:10.3324/haematol.2016.145136
137. Lougaris V, Moratto D, Baronio M, Tampella G, van der Meer JWM, Badolato R, et al. Early and late B-cell developmental impairment in nuclear factor kappa B, subunit 1-mutated common variable immunodeficiency disease. J Allergy Clin Immunol (2017) 139(1):349–52.e1. doi:10.1016/j.jaci.2016.05.045
138. Chen K, Coonrod EM, Kumánovics A, Franks ZF, Durtschi JD, Margraf RL, et al. Germline mutations in NFKB2 implicate the noncanonical NF-κB pathway in the pathogenesis of common variable immunodeficiency. Am J Hum Genet (2013) 93(5):812–24. doi:10.1016/j.ajhg.2013.09.009
139. Lee CE, Fulcher DA, Whittle B, Chand R, Fewings N, Field M, et al. Autosomal-dominant B-cell deficiency with alopecia due to a mutation in NFKB2 that results in nonprocessable p100. Blood (2014) 124(19):2964–72. doi:10.1182/blood-2014-06-578542
140. Lindsley AW, Qian Y, Valencia CA, Shah K, Zhang K, Assa’ad A. Combined immune deficiency in a patient with a novel NFKB2 mutation. J Clin Immunol (2014) 34(8):910–5. doi:10.1007/s10875-014-0095-3
141. Lougaris V, Tabellini G, Vitali M, Baronio M, Patrizi O, Tampella G, et al. Defective natural killer-cell cytotoxic activity in NFKB2-mutated CVID-like disease. J Allergy Clin Immunol (2015) 135(6):1641–3. doi:10.1016/j.jaci.2014.11.038
142. Montin D, Licciardi F, Giorgio E, Ciolfi A, Pizzi S, Mussa A, et al. Functional evaluation of natural killer cell cytotoxic activity in NFKB2-mutated patients. Immunol Lett (2018) 194:40–3. doi:10.1016/j.imlet.2017.12.006
143. Brue T, Quentien MH, Khetchoumian K, Bensa M, Capo-Chichi JM, Delemer B, et al. Mutations in NFKB2 and potential genetic heterogeneity in patients with DAVID syndrome, having variable endocrine and immune deficiencies. BMC Med Genet (2014) 15:139. doi:10.1186/s12881-014-0139-9
144. Kuehn HS, Niemela JE, Sreedhara K, Stoddard JL, Grossman J, Wysocki CA, et al. Novel nonsense gain-of-function NFKB2 mutations associated with a combined immunodeficiency phenotype. Blood (2017) 130(13):1553–64. doi:10.1182/blood-2017-05-782177
145. Lal RA, Bachrach LK, Hoffman AR, Inlora J, Rego S, Snyder MP, et al. A case report of hypoglycemia and hypogammaglobulinemia: DAVID syndrome in a patient with a novel NFKB2 mutation. J Clin Endocrinol Metab (2017) 102(7):2127–30. doi:10.1210/jc.2017-00341
146. Liu Y, Hanson S, Gurugama P, Jones A, Clark B, Ibrahim MA. Novel NFKB2 mutation in early-onset CVID. J Clin Immunol (2014) 34(6):686–90. doi:10.1007/s10875-014-0064-x
147. Ramakrishnan KA, Rae W, Barcenas-Morales G, Gao Y, Pengelly RJ, Patel SV, et al. Anticytokine autoantibodies in a patient with a heterozygous NFKB2 mutation. J Allergy Clin Immunol (2017) 141(4):1479–82.e6. doi:10.1016/j.jaci.2017.11.014
148. Shi C, Wang F, Tong A, Zhang XQ, Song HM, Liu ZY, et al. NFKB2 mutation in common variable immunodeficiency and isolated adrenocorticotropic hormone deficiency: a case report and review of literature. Medicine (Baltimore) (2016) 95(40):e5081. doi:10.1097/MD.0000000000005081
149. Perez de Diego R, Sanchez-Ramon S, Lopez-Collazo E, Martinez-Barricarte R, Cubillos-Zapata C, Ferreira Cerdan A, et al. Genetic errors of the human caspase recruitment domain-B-cell lymphoma 10-mucosa-associated lymphoid tissue lymphoma-translocation gene 1 (CBM) complex: molecular, immunologic, and clinical heterogeneity. J Allergy Clin Immunol (2015) 136(5):1139–49. doi:10.1016/j.jaci.2015.06.031
150. Turvey SE, Durandy A, Fischer A, Fung SY, Geha RS, Gewies A, et al. The CARD11-BCL10-MALT1 (CBM) signalosome complex: stepping into the limelight of human primary immunodeficiency. J Allergy Clin Immunol (2014) 134(2):276–84. doi:10.1016/j.jaci.2014.06.015
151. Meininger I, Krappmann D. Lymphocyte signaling and activation by the CARMA1-BCL10-MALT1 signalosome. Biol Chem (2016) 397(12):1315–33. doi:10.1515/hsz-2016-0216
152. Snow AL, Xiao W, Stinson JR, Lu W, Chaigne-Delalande B, Zheng L, et al. Congenital B cell lymphocytosis explained by novel germline CARD11 mutations. J Exp Med (2012) 209(12):2247–61. doi:10.1084/jem.20120831
153. Greil J, Rausch T, Giese T, Bandapalli OR, Daniel V, Bekeredjian-Ding I, et al. Whole-exome sequencing links caspase recruitment domain 11 (CARD11) inactivation to severe combined immunodeficiency. J Allergy Clin Immunol (2013) 131(5):1376–83.e3. doi:10.1016/j.jaci.2013.02.012
154. Stepensky P, Keller B, Buchta M, Kienzler AK, Elpeleg O, Somech R, et al. Deficiency of caspase recruitment domain family, member 11 (CARD11), causes profound combined immunodeficiency in human subjects. J Allergy Clin Immunol (2013) 131(2):477–85.e1. doi:10.1016/j.jaci.2012.11.050
155. Buchbinder D, Stinson JR, Nugent DJ, Heurtier L, Suarez F, Sukumar G, et al. Mild B-cell lymphocytosis in patients with a CARD11 C49Y mutation. J Allergy Clin Immunol (2015) 136(3):819–21.e1. doi:10.1016/j.jaci.2015.03.008
156. Brohl AS, Stinson JR, Su HC, Badgett T, Jennings CD, Sukumar G, et al. Germline CARD11 mutation in a patient with severe congenital B cell lymphocytosis. J Clin Immunol (2015) 35(1):32–46. doi:10.1007/s10875-014-0106-4
157. Fuchs S, Rensing-Ehl A, Pannicke U, Lorenz MR, Fisch P, Jeelall Y, et al. Omenn syndrome associated with a functional reversion due to a somatic second-site mutation in CARD11 deficiency. Blood (2015) 126(14):1658–69. doi:10.1182/blood-2015-03-631374
158. Outinen T, Syrjanen J, Rounioja S, Saarela J, Kaustio M, Helminen M. Constant B cell lymphocytosis since early age in a patient with CARD11 mutation: a 20-year follow-up. Clin Immunol (2016) 165:19–20. doi:10.1016/j.clim.2016.02.002
159. Ma CA, Stinson JR, Zhang Y, Abbott JK, Weinreich MA, Hauk PJ, et al. Germline hypomorphic CARD11 mutations in severe atopic disease. Nat Genet (2017) 49(8):1192–201. doi:10.1038/ng.3898
160. Dadi H, Jones TA, Merico D, Sharfe N, Ovadia A, Schejter Y, et al. Combined immunodeficiency and atopy caused by a dominant negative mutation in caspase activation and recruitment domain family member 11 (CARD11). J Allergy Clin Immunol (2017) 141(5):1818–30.e2. doi:10.1016/j.jaci.2017.06.047
161. Torres JM, Martinez-Barricarte R, Garcia-Gomez S, Mazariegos MS, Itan Y, Boisson B, et al. Inherited BCL10 deficiency impairs hematopoietic and nonhematopoietic immunity. J Clin Invest (2014) 124(12):5239–48. doi:10.1172/JCI77493
162. Jabara HH, Ohsumi T, Chou J, Massaad MJ, Benson H, Megarbane A, et al. A homozygous mucosa-associated lymphoid tissue 1 (MALT1) mutation in a family with combined immunodeficiency. J Allergy Clin Immunol (2013) 132(1):151–8. doi:10.1016/j.jaci.2013.04.047
163. McKinnon ML, Rozmus J, Fung SY, Hirschfeld AF, Del Bel KL, Thomas L, et al. Combined immunodeficiency associated with homozygous MALT1 mutations. J Allergy Clin Immunol (2014) 133(5):1458–62, 62.e1–7. doi:10.1016/j.jaci.2013.10.045
164. Punwani D, Wang H, Chan AY, Cowan MJ, Mallott J, Sunderam U, et al. Combined immunodeficiency due to MALT1 mutations, treated by hematopoietic cell transplantation. J Clin Immunol (2015) 35(2):135–46. doi:10.1007/s10875-014-0125-1
165. Charbit-Henrion F, Jeverica AK, Begue B, Markelj G, Parlato M, Avcin SL, et al. Deficiency in mucosa-associated lymphoid tissue lymphoma translocation 1: a novel cause of IPEX-like syndrome. J Pediatr Gastroenterol Nutr (2017) 64(3):378–84. doi:10.1097/MPG.0000000000001262
166. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science (2014) 345(6204):1623–7. doi:10.1126/science.1255904
167. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med (2014) 20(12):1410–6. doi:10.1038/nm.3746
168. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity (1995) 3(5):541–7. doi:10.1016/1074-7613(95)90125-6
169. Zeissig S, Petersen BS, Tomczak M, Melum E, Huc-Claustre E, Dougan SK, et al. Early-onset Crohn’s disease and autoimmunity associated with a variant in CTLA-4. Gut (2015) 64(12):1889–97. doi:10.1136/gutjnl-2014-308541
170. Hayakawa S, Okada S, Tsumura M, Sakata S, Ueno Y, Imai K, et al. A patient with CTLA-4 haploinsufficiency presenting gastric cancer. J Clin Immunol (2016) 36(1):28–32. doi:10.1007/s10875-015-0221-x
171. Lee S, Moon JS, Lee CR, Kim HE, Baek SM, Hwang S, et al. Abatacept alleviates severe autoimmune symptoms in a patient carrying a de novo variant in CTLA-4. J Allergy Clin Immunol (2016) 137(1):327–30. doi:10.1016/j.jaci.2015.08.036
172. Shields CL, Say EA, Mashayekhi A, Garg SJ, Dunn JP, Shields JA. Assessment of CTLA-4 deficiency-related autoimmune choroidopathy response to abatacept. JAMA Ophthalmol (2016) 134(7):844–6. doi:10.1001/jamaophthalmol.2016.1013
173. Slatter MA, Engelhardt KR, Burroughs LM, Arkwright PD, Nademi Z, Skoda-Smith S, et al. Hematopoietic stem cell transplantation for CTLA4 deficiency. J Allergy Clin Immunol (2016) 138(2):615–9.e1. doi:10.1016/j.jaci.2016.01.045
174. Alroqi FJ, Charbonnier LM, Baris S, Kiykim A, Chou J, Platt CD, et al. Exaggerated follicular helper T-cell responses in patients with LRBA deficiency caused by failure of CTLA4-mediated regulation. J Allergy Clin Immunol (2017) 141(3):1050–9.e10. doi:10.1016/j.jaci.2017.05.022
175. Greil C, Roether F, La Rosee P, Grimbacher B, Duerschmied D, Warnatz K. Rescue of cytokine storm due to HLH by hemoadsorption in a CTLA4-deficient patient. J Clin Immunol (2017) 37(3):273–6. doi:10.1007/s10875-017-0377-7
176. Kucuk ZY, Charbonnier LM, McMasters RL, Chatila T, Bleesing JJ. CTLA-4 haploinsufficiency in a patient with an autoimmune lymphoproliferative disorder. J Allergy Clin Immunol (2017) 140(3):862–4.e4. doi:10.1016/j.jaci.2017.02.032
177. Moraes-Fontes MF, Hsu AP, Caramalho I, Martins C, Araujo AC, Lourenco F, et al. Fatal CTLA-4 heterozygosity with autoimmunity and recurrent infections: a de novo mutation. Clin Case Rep (2017) 5(12):2066–70. doi:10.1002/ccr3.1257
178. Navarini AA, Hruz P, Berger CT, Hou TZ, Schwab C, Gabrysch A, et al. Vedolizumab as a successful treatment of CTLA-4-associated autoimmune enterocolitis. J Allergy Clin Immunol (2017) 139(3):1043–6.e5. doi:10.1016/j.jaci.2016.08.042
179. Tegtmeyer D, Seidl M, Gerner P, Baumann U, Klemann C. Inflammatory bowel disease caused by primary immunodeficiencies-clinical presentations, review of literature, and proposal of a rational diagnostic algorithm. Pediatr Allergy Immunol (2017) 28(5):412–29. doi:10.1111/pai.12734
180. Besnard C, Levy E, Aladjidi N, Stolzenberg MC, Magerus-Chatinet A, Alibeu O, et al. Pediatric-onset Evans syndrome: heterogeneous presentation and high frequency of monogenic disorders including LRBA and CTLA4 mutations. Clin Immunol (2018) 188:52–7. doi:10.1016/j.clim.2017.12.009
181. Hou TZ, Olbrich P, Soto JML, Sanchez B, Moreno PS, Borte S, et al. Study of an extended family with CTLA-4 deficiency suggests a CD28/CTLA-4 independent mechanism responsible for differences in disease manifestations and severity. Clin Immunol (2018) 188:94–102. doi:10.1016/j.clim.2018.01.001
182. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science (2015) 349(6246):436–40. doi:10.1126/science.aaa1663
183. Lopez-Herrera G, Tampella G, Pan-Hammarstrom Q, Herholz P, Trujillo-Vargas CM, Phadwal K, et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet (2012) 90(6):986–1001. doi:10.1016/j.ajhg.2012.04.015
184. Gamez-Diaz L, August D, Stepensky P, Revel-Vilk S, Seidel MG, Noriko M, et al. The extended phenotype of LPS-responsive beige-like anchor protein (LRBA) deficiency. J Allergy Clin Immunol (2016) 137(1):223–30. doi:10.1016/j.jaci.2015.09.025
185. Alangari A, Alsultan A, Adly N, Massaad MJ, Kiani IS, Aljebreen A, et al. LPS-responsive beige-like anchor (LRBA) gene mutation in a family with inflammatory bowel disease and combined immunodeficiency. J Allergy Clin Immunol (2012) 130(2):481–8.e2. doi:10.1016/j.jaci.2012.05.043
186. Serwas NK, Kansu A, Santos-Valente E, Kuloglu Z, Demir A, Yaman A, et al. Atypical manifestation of LRBA deficiency with predominant IBD-like phenotype. Inflamm Bowel Dis (2015) 21(1):40–7. doi:10.1097/MIB.0000000000000266
187. Alkhairy OK, Abolhassani H, Rezaei N, Fang M, Andersen KK, Chavoshzadeh Z, et al. Spectrum of phenotypes associated with mutations in LRBA. J Clin Immunol (2016) 36(1):33–45. doi:10.1007/s10875-015-0224-7
188. Lo B, Fritz JM, Su HC, Uzel G, Jordan MB, Lenardo MJ. CHAI and LATAIE: new genetic diseases of CTLA-4 checkpoint insufficiency. Blood (2016) 128(8):1037–42. doi:10.1182/blood-2016-04-712612
189. Park MY, Sudan R, Srivastava N, Neelam S, Youngs C, Wang JW, et al. LRBA is essential for allogeneic responses in bone marrow transplantation. Sci Rep (2016) 6:36568. doi:10.1038/srep36568
190. Burns SO, Zenner HL, Plagnol V, Curtis J, Mok K, Eisenhut M, et al. LRBA gene deletion in a patient presenting with autoimmunity without hypogammaglobulinemia. J Allergy Clin Immunol (2012) 130(6):1428–32. doi:10.1016/j.jaci.2012.07.035
191. Charbonnier LM, Janssen E, Chou J, Ohsumi TK, Keles S, Hsu JT, et al. Regulatory T-cell deficiency and immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like disorder caused by loss-of-function mutations in LRBA. J Allergy Clin Immunol (2015) 135(1):217–27. doi:10.1016/j.jaci.2014.10.019
192. Revel-Vilk S, Fischer U, Keller B, Nabhani S, Gamez-Diaz L, Rensing-Ehl A, et al. Autoimmune lymphoproliferative syndrome-like disease in patients with LRBA mutation. Clin Immunol (2015) 159(1):84–92. doi:10.1016/j.clim.2015.04.007
193. Seidel MG, Hirschmugl T, Gamez-Diaz L, Schwinger W, Serwas N, Deutschmann A, et al. Long-term remission after allogeneic hematopoietic stem cell transplantation in LPS-responsive beige-like anchor (LRBA) deficiency. J Allergy Clin Immunol (2015) 135(5):1384–90.e1–8. doi:10.1016/j.jaci.2014.10.048
194. Bakhtiar S, Ruemmele F, Charbit-Henrion F, Lévy E, Rieux-Laucat F, Cerf-Bensussan N, et al. Atypical manifestation of LPS-responsive beige-like anchor deficiency syndrome as an autoimmune endocrine disorder without enteropathy and immunodeficiency. Front Pediatr (2016) 4:98. doi:10.3389/fped.2016.00098
195. Lévy E, Stolzenberg MC, Bruneau J, Breton S, Neven B, Sauvion S, et al. LRBA deficiency with autoimmunity and early onset chronic erosive polyarthritis. Clin Immunol (2016) 168:88–93. doi:10.1016/j.clim.2016.03.006
196. Schreiner F, Plamper M, Dueker G, Schoenberger S, Gámez-Díaz L, Grimbacher B, et al. Infancy-onset T1DM, short stature, and severe immunodysregulation in two siblings with a homozygous LRBA mutation. J Clin Endocrinol Metab (2016) 101(3):898–904. doi:10.1210/jc.2015-3382
197. Shokri S, Nabavi M, Hirschmugl T, Aghamohammadi A, Arshi S, Bemanian MH, et al. LPS-responsive beige-like anchor gene mutation associated with possible bronchiolitis obliterans organizing pneumonia associated with hypogammaglobulinemia and normal IgM phenotype and low number of B cells. Acta Med Iran (2016) 54(10):620–3.
198. Tesi B, Priftakis P, Lindgren F, Chiang SC, Kartalis N, Löfstedt A, et al. Successful hematopoietic stem cell transplantation in a patient with LPS-responsive beige-like anchor (LRBA) gene mutation. J Clin Immunol (2016) 36(5):480–9. doi:10.1007/s10875-016-0289-y
199. Azizi G, Abolhassani H, Yazdani R, Mohammadikhajehdehi S, Parvaneh N, Negahdari B, et al. New therapeutic approach by sirolimus for enteropathy treatment in patients with LRBA deficiency. Eur Ann Allergy Clin Immunol (2017) 49(5):235–9. doi:10.23822/EurAnnACI.1764-1489.22
200. Bakhtiar S, Gámez-Díaz L, Jarisch A, Soerensen J, Grimbacher B, Belohradsky B, et al. Treatment of infantile inflammatory bowel disease and autoimmunity by allogeneic stem cell transplantation in LPS-responsive beige-like anchor deficiency. Front Immunol (2017) 8:52. doi:10.3389/fimmu.2017.00052
201. Kostel Bal S, Haskologlu S, Serwas NK, Islamoglu C, Aytekin C, Kendirli T, et al. Multiple presentations of LRBA deficiency: a single-center experience. J Clin Immunol (2017) 37(8):790–800. doi:10.1007/s10875-017-0446-y
202. Bratanič N, Kovač J, Pohar K, Trebušak Podkrajšek K, Ihan A, Battelino T, et al. Multifocal gastric adenocarcinoma in a patient with LRBA deficiency. Orphanet J Rare Dis (2017) 12(1):131. doi:10.1186/s13023-017-0682-5
203. Johnson MB, De Franco E, Lango Allen H, Al Senani A, Elbarbary N, Siklar Z, et al. Recessively inherited LRBA mutations cause autoimmunity presenting as neonatal diabetes. Diabetes (2017) 66(8):2316–22. doi:10.2337/db17-0040
204. Kammermeier J, Dziubak R, Pescarin M, Drury S, Godwin H, Reeve K, et al. Phenotypic and genotypic characterisation of inflammatory bowel disease presenting before the age of 2 years. J Crohns Colitis (2017) 11(1):60–9. doi:10.1093/ecco-jcc/jjw118
205. Seidel MG, Bohm K, Dogu F, Worth A, Thrasher A, Florkin B, et al. Treatment of severe forms of LPS-responsive beige-like anchor protein deficiency with allogeneic hematopoietic stem cell transplantation. J Allergy Clin Immunol (2018) 141(2):770–5.e1. doi:10.1016/j.jaci.2017.04.023
206. Şıklar Z, de Franco E, Johnson MB, Flanagan SE, Ellard S, Ceylaner S, et al. Monogenic diabetes not caused by mutations in mody genes: a very heterogenous group of diabetes. Exp Clin Endocrinol Diabetes (2017). doi:10.1055/s-0043-120571
207. Al Sukaiti N, AbdelRahman K, AlShekaili J, Al Oraimi S, Al Sinani A, Al Rahbi N, et al. Agammaglobulinaemia despite terminal B-cell differentiation in a patient with a novel LRBA mutation. Clin Transl Immunology (2017) 6(5):e144. doi:10.1038/cti.2017.20
208. Vairo FP, Boczek NJ, Cousin MA, Kaiwar C, Blackburn PR, Conboy E, et al. The prevalence of diseases caused by lysosome-related genes in a cohort of undiagnosed patients. Mol Genet Metab Rep (2017) 13:46–51. doi:10.1016/j.ymgmr.2017.08.001
209. Ye Z, Zhou Y, Huang Y, Wang Y, Lu J, Tang Z, et al. Phenotype and management of infantile-onset inflammatory bowel disease: experience from a tertiary care center in China. Inflamm Bowel Dis (2017) 23(12):2154–64. doi:10.1097/MIB.0000000000001269
210. Sari S, Dogu F, Hwa V, Haskologlu S, Dauber A, Rosenfeld R, et al. A successful HSCT in a girl with novel LRBA mutation with refractory celiac disease. J Clin Immunol (2016) 36(1):8–11. doi:10.1007/s10875-015-0220-y
211. Dickinson RE, Griffin H, Bigley V, Reynard LN, Hussain R, Haniffa M, et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood (2011) 118(10):2656–8. doi:10.1182/blood-2011-06-360313
212. Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, Li XC, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet (2011) 43(10):1012–7. doi:10.1038/ng.913
213. Hsu AP, Sampaio EP, Khan J, Calvo KR, Lemieux JE, Patel SY, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood (2011) 118(10):2653–5. doi:10.1182/blood-2011-05-356352
214. Ostergaard P, Simpson MA, Connell FC, Steward CG, Brice G, Woollard WJ, et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat Genet (2011) 43(10):929–31. doi:10.1038/ng.923
215. Bresnick EH, Katsumura KR, Lee HY, Johnson KD, Perkins AS. Master regulatory GATA transcription factors: mechanistic principles and emerging links to hematologic malignancies. Nucleic Acids Res (2012) 40(13):5819–31. doi:10.1093/nar/gks281
216. Vinh DC, Patel SY, Uzel G, Anderson VL, Freeman AF, Olivier KN, et al. Autosomal dominant and sporadic monocytopenia with susceptibility to mycobacteria, fungi, papillomaviruses, and myelodysplasia. Blood (2010) 115(8):1519–29. doi:10.1182/blood-2009-03-208629
217. Bigley V, Haniffa M, Doulatov S, Wang XN, Dickinson R, McGovern N, et al. The human syndrome of dendritic cell, monocyte, B and NK lymphoid deficiency. J Exp Med (2011) 208(2):227–34. doi:10.1084/jem.20101459
218. Mansour S, Connell F, Steward C, Ostergaard P, Brice G, Smithson S, et al. Emberger syndrome-primary lymphedema with myelodysplasia: report of seven new cases. Am J Med Genet A (2010) 152A(9):2287–96. doi:10.1002/ajmg.a.33445
219. Owen C, Barnett M, Fitzgibbon J. Familial myelodysplasia and acute myeloid leukaemia – a review. Br J Haematol (2008) 140(2):123–32. doi:10.1111/j.1365-2141.2007.06909.x
220. Dickinson RE, Milne P, Jardine L, Zandi S, Swierczek SI, McGovern N, et al. The evolution of cellular deficiency in GATA2 mutation. Blood (2014) 123(6):863–74. doi:10.1182/blood-2013-07-517151
221. Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood (2014) 123(6):809–21. doi:10.1182/blood-2013-07-515528
222. Kazenwadel J, Secker GA, Liu YJ, Rosenfeld JA, Wildin RS, Cuellar-Rodriguez J, et al. Loss-of-function germline GATA2 mutations in patients with MDS/AML or MonoMAC syndrome and primary lymphedema reveal a key role for GATA2 in the lymphatic vasculature. Blood (2012) 119(5):1283–91. doi:10.1182/blood-2011-08-374363
223. Ballerie A, Nimubona S, Meunier C, Gutierrez FL, Desrues B, Delaval P, et al. Association of pulmonary alveolar proteinosis and fibrosis: patient with GATA2 deficiency. Eur Respir J (2016) 48(5):1510–4. doi:10.1183/13993003.00252-2016
224. Sanges S, Prévotat A, Fertin M, Terriou L, Lefèvre G, Quesnel B, et al. Haemodynamically proven pulmonary hypertension in a patient with GATA2 deficiency-associated pulmonary alveolar proteinosis and fibrosis. Eur Respir J (2017) 49(5):1700407. doi:10.1183/13993003.00178-2017
225. Wlodarski MW, Hirabayashi S, Pastor V, Starý J, Hasle H, Masetti R, et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood (2016) 127(11):1387–97; quiz 1518. doi:10.1182/blood-2015-09-669937
226. Ganapathi KA, Townsley DM, Hsu AP, Arthur DC, Zerbe CS, Cuellar-Rodriguez J, et al. GATA2 deficiency-associated bone marrow disorder differs from idiopathic aplastic anemia. Blood (2015) 125(1):56–70. doi:10.1182/blood-2014-06-580340
227. Cuellar-Rodriguez J, Gea-Banacloche J, Freeman AF, Hsu AP, Zerbe CS, Calvo KR, et al. Successful allogeneic hematopoietic stem cell transplantation for GATA2 deficiency. Blood (2011) 118(13):3715–20. doi:10.1182/blood-2011-06-365049
228. Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics (2012) 28(21):2747–54. doi:10.1093/bioinformatics/bts526
229. Pasquet M, Bellanné-Chantelot C, Tavitian S, Prade N, Beaupain B, Larochelle O, et al. High frequency of GATA2 mutations in patients with mild chronic neutropenia evolving to MonoMac syndrome, myelodysplasia, and acute myeloid leukemia. Blood (2013) 121(5):822–9. doi:10.1182/blood-2012-08-447367
230. Camargo JF, Lobo SA, Hsu AP, Zerbe CS, Wormser GP, Holland SM. MonoMAC syndrome in a patient with a GATA2 mutation: case report and review of the literature. Clin Infect Dis (2013) 57(5):697–9. doi:10.1093/cid/cit368
231. Hsu AP, Johnson KD, Falcone EL, Sanalkumar R, Sanchez L, Hickstein DD, et al. GATA2 haploinsufficiency caused by mutations in a conserved intronic element leads to MonoMAC syndrome. Blood (2013) 121(19):3830–7, S1–7. doi:10.1182/blood-2012-08-452763
232. Chou J, Lutskiy M, Tsitsikov E, Notarangelo LD, Geha RS, Dioun A. Presence of hypogammaglobulinemia and abnormal antibody responses in GATA2 deficiency. J Allergy Clin Immunol (2014) 134(1):223–6. doi:10.1016/j.jaci.2014.01.041
233. Fujiwara T, Fukuhara N, Funayama R, Nariai N, Kamata M, Nagashima T, et al. Identification of acquired mutations by whole-genome sequencing in GATA-2 deficiency evolving into myelodysplasia and acute leukemia. Ann Hematol (2014) 93(9):1515–22. doi:10.1007/s00277-014-2090-4
234. West ES, Kingsbery MY, Mintz EM, Hsu AP, Holland SM, Rady PL, et al. Generalized verrucosis in a patient with GATA2 deficiency. Br J Dermatol (2014) 170(5):1182–6. doi:10.1111/bjd.12794
235. Griese M, Zarbock R, Costabel U, Hildebrandt J, Theegarten D, Albert M, et al. GATA2 deficiency in children and adults with severe pulmonary alveolar proteinosis and hematologic disorders. BMC Pulm Med (2015) 15:87. doi:10.1186/s12890-015-0083-2
236. Brondfield S, Reid M, Patel K, Ten R, Dhaliwal G. Disseminated mycobacterial infection and scabies infestation. Am J Med (2015) 128(10):e41–2. doi:10.1016/j.amjmed.2015.06.008
237. Maciejewski-Duval A, Meuris F, Bignon A, Aknin ML, Balabanian K, Faivre L, et al. Altered chemotactic response to CXCL12 in patients carrying GATA2 mutations. J Leukoc Biol (2016) 99(6):1065–76. doi:10.1189/jlb.5MA0815-388R
238. Crall C, Morley KW, Rabinowits G, Schmidt B, Dioun Broyles A, Huang JT. Merkel cell carcinoma in a patient with GATA2 deficiency: a novel association with primary immunodeficiency. Br J Dermatol (2016) 174(1):169–71. doi:10.1111/bjd.14062
239. Koegel AK, Hofmann I, Moffitt K, Degar B, Duncan C, Tubman VN. Acute lymphoblastic leukemia in a patient with MonoMAC syndrome/GATA2 haploinsufficiency. Pediatr Blood Cancer (2016) 63(10):1844–7. doi:10.1002/pbc.26084
240. Cohen JI, Dropulic L, Hsu AP, Zerbe CS, Krogmann T, Dowdell K, et al. Association of GATA2 deficiency with severe primary Epstein-Barr virus (EBV) infection and EBV-associated cancers. Clin Infect Dis (2016) 63(1):41–7. doi:10.1093/cid/ciw160
241. Vila A, Dapás JI, Rivero CV, Bocanegra F, Furnari RF, Hsu AP, et al. Multiple opportunistic infections in a woman with GATA2 mutation. Int J Infect Dis (2017) 54:89–91. doi:10.1016/j.ijid.2016.11.408
242. Shah NN, Parta M, Baird K, Rafei H, Cole K, Holland SM, et al. Monozygotic twins with GATA2 deficiency: same haploidentical-related donor, different severity of GvHD. Bone Marrow Transplant (2017) 52(11):1580–2. doi:10.1038/bmt.2017.180
243. Schlums H, Jung M, Han H, Theorell J, Bigley V, Chiang SC, et al. Adaptive NK cells can persist in patients with. Blood (2017) 129(14):1927–39. doi:10.1182/blood-2016-08-734236
244. Johnson KD, Hsu AP, Ryu MJ, Wang J, Gao X, Boyer ME, et al. Cis-element mutated in GATA2-dependent immunodeficiency governs hematopoiesis and vascular integrity. J Clin Invest (2012) 122(10):3692–704. doi:10.1172/JCI61623
245. Ishida H, Imai K, Honma K, Tamura S, Imamura T, Ito M, et al. GATA-2 anomaly and clinical phenotype of a sporadic case of lymphedema, dendritic cell, monocyte, B- and NK-cell (DCML) deficiency, and myelodysplasia. Eur J Pediatr (2012) 171(8):1273–6. doi:10.1007/s00431-012-1715-7
246. Bödör C, Renneville A, Smith M, Charazac A, Iqbal S, Etancelin P, et al. Germ-line GATA2 p.THR354MET mutation in familial myelodysplastic syndrome with acquired monosomy 7 and ASXL1 mutation demonstrating rapid onset and poor survival. Haematologica (2012) 97(6):890–4. doi:10.3324/haematol.2011.054361
247. Holme H, Hossain U, Kirwan M, Walne A, Vulliamy T, Dokal I. Marked genetic heterogeneity in familial myelodysplasia/acute myeloid leukaemia. Br J Haematol (2012) 158(2):242–8. doi:10.1111/j.1365-2141.2012.09136.x
248. Mutsaers PG, van de Loosdrecht AA, Tawana K, Bödör C, Fitzgibbon J, Menko FH. Highly variable clinical manifestations in a large family with a novel GATA2 mutation. Leukemia (2013) 27(11):2247–8. doi:10.1038/leu.2013.105
249. Mace EM, Hsu AP, Monaco-Shawver L, Makedonas G, Rosen JB, Dropulic L, et al. Mutations in GATA2 cause human NK cell deficiency with specific loss of the CD56(bright) subset. Blood (2013) 121(14):2669–77. doi:10.1182/blood-2012-09-453969
250. Maeurer M, Magalhaes I, Andersson J, Ljungman P, Sandholm E, Uhlin M, et al. Allogeneic hematopoietic cell transplantation for GATA2 deficiency in a patient with disseminated human papillomavirus disease. Transplantation (2014) 98(12):e95–6. doi:10.1097/TP.0000000000000520
251. Gao J, Gentzler RD, Timms AE, Horwitz MS, Frankfurt O, Altman JK, et al. Heritable GATA2 mutations associated with familial AML-MDS: a case report and review of literature. J Hematol Oncol (2014) 7:36. doi:10.1186/1756-8722-7-36
252. Lübking A, Vosberg S, Konstandin NP, Dufour A, Graf A, Krebs S, et al. Young woman with mild bone marrow dysplasia, GATA2 and ASXL1 mutation treated with allogeneic hematopoietic stem cell transplantation. Leuk Res Rep (2015) 4(2):72–5. doi:10.1016/j.lrr.2015.10.001
253. Wang X, Muramatsu H, Okuno Y, Sakaguchi H, Yoshida K, Kawashima N, et al. GATA2 and secondary mutations in familial myelodysplastic syndromes and pediatric myeloid malignancies. Haematologica (2015) 100(10):e398–401. doi:10.3324/haematol.2015.127092
254. Cortés-Lavaud X, Landecho MF, Maicas M, Urquiza L, Merino J, Moreno-Miralles I, et al. GATA2 germline mutations impair GATA2 transcription, causing haploinsufficiency: functional analysis of the p.Arg396Gln mutation. J Immunol (2015) 194(5):2190–8. doi:10.4049/jimmunol.1401868
255. Mir MA, Kochuparambil ST, Abraham RS, Rodriguez V, Howard M, Hsu AP, et al. Spectrum of myeloid neoplasms and immune deficiency associated with germline GATA2 mutations. Cancer Med (2015) 4(4):490–9. doi:10.1002/cam4.384
256. Churpek JE, Pyrtel K, Kanchi KL, Shao J, Koboldt D, Miller CA, et al. Genomic analysis of germ line and somatic variants in familial myelodysplasia/acute myeloid leukemia. Blood (2015) 126(22):2484–90. doi:10.1182/blood-2015-04-641100
257. Svobodova T, Mejstrikova E, Salzer U, Sukova M, Hubacek P, Matej R, et al. Diffuse parenchymal lung disease as first clinical manifestation of GATA-2 deficiency in childhood. BMC Pulm Med (2015) 15:8. doi:10.1186/s12890-015-0006-2
258. Zhang MY, Keel SB, Walsh T, Lee MK, Gulsuner S, Watts AC, et al. Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity. Haematologica (2015) 100(1):42–8. doi:10.3324/haematol.2014.113456
259. Spinner MA, Ker JP, Stoudenmire CJ, Fadare O, Mace EM, Orange JS, et al. GATA2 deficiency underlying severe blastomycosis and fatal herpes simplex virus-associated hemophagocytic lymphohistiocytosis. J Allergy Clin Immunol (2016) 137(2):638–40. doi:10.1016/j.jaci.2015.07.043
260. Delgado-Márquez AM, Zarco C, Ruiz R, Simarro A, Vanaclocha F. Severe disseminated primary herpes simplex infection as skin manifestation of GATA2 deficiency. J Eur Acad Dermatol Venereol (2016) 30(7):1248–50. doi:10.1111/jdv.13183
261. Mallhi K, Dix DB, Niederhoffer KY, Armstrong L, Rozmus J. Successful umbilical cord blood hematopoietic stem cell transplantation in pediatric patients with MDS/AML associated with underlying GATA2 mutations: two case reports and review of literature. Pediatr Transplant (2016) 20(7):1004–7. doi:10.1111/petr.12764
262. Seo SK, Kim KY, Han SA, Yoon JS, Shin SY, Sohn SK, et al. First Korean case of Emberger syndrome (primary lymphedema with myelodysplasia) with a novel GATA2 gene mutation. Korean J Intern Med (2016) 31(1):188–90. doi:10.3904/kjim.2016.31.1.188
263. Saida S, Umeda K, Yasumi T, Matsumoto A, Kato I, Hiramatsu H, et al. Successful reduced-intensity stem cell transplantation for GATA2 deficiency before progression of advanced MDS. Pediatr Transplant (2016) 20(2):333–6. doi:10.1111/petr.12667
264. Buchbinder D, Sassoon A. You GATA look at the marrow! Blood (2016) 128(4):603. doi:10.1182/blood-2016-04-712547
265. Keel SB, Scott A, Sanchez-Bonilla M, Ho PA, Gulsuner S, Pritchard CC, et al. Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients. Haematologica (2016) 101(11):1343–50. doi:10.3324/haematol.2016.149476
266. Ciullini Mannurita S, Vignoli M, Colarusso G, Tucci F, Veltroni M, Frenos S, et al. Timely follow-up of a GATA2 deficiency patient allows successful treatment. J Allergy Clin Immunol (2016) 138(5):1480–3.e4. doi:10.1016/j.jaci.2016.06.004
267. Nováková M, Žaliová M, Suková M, Wlodarski M, Janda A, Froňková E, et al. Loss of B cells and their precursors is the most constant feature of GATA-2 deficiency in childhood myelodysplastic syndrome. Haematologica (2016) 101(6):707–16. doi:10.3324/haematol.2015.137711
268. Ding LW, Ikezoe T, Tan KT, Mori M, Mayakonda A, Chien W, et al. Mutational profiling of a MonoMAC syndrome family with GATA2 deficiency. Leukemia (2017) 31(1):244–5. doi:10.1038/leu.2016.256
269. Kurata T, Shigemura T, Muramatsu H, Okuno Y, Nakazawa Y. A case of GATA2-related myelodysplastic syndrome with unbalanced translocation der(1;7)(q10;p10). Pediatr Blood Cancer (2017) 64(8):e26419. doi:10.1002/pbc.26419
270. Rastogi N, Abraham RS, Chadha R, Thakkar D, Kohli S, Nivargi S, et al. Successful nonmyeloablative allogeneic stem cell transplant in a child with emberger syndrome and GATA2 mutation. J Pediatr Hematol Oncol (2017). doi:10.1097/MPH.0000000000000995
271. Kuriyama Y, Hattori M, Mitsui T, Nakano H, Oikawa D, Tokunaga F, et al. Generalized verrucosis caused by various human papillomaviruses in a patient with GATA2 deficiency. J Dermatol (2017) 45(5):e108–9. doi:10.1111/1346-8138.14149
272. Álvarez-Chinchilla P, Poveda I, Marco FM, López-Fernández JA, Peiro G, Illán F, et al. Vulvar lymphedema and refractory VIN-III heralding GATA2 deficiency syndrome. Eur J Obstet Gynecol Reprod Biol (2017) 218:138–40. doi:10.1016/j.ejogrb.2017.09.016
273. Hamadou WS, Mani R, Besbes S, Bourdon V, Youssef YB, Eisinger F, et al. GATA2 gene analysis in several forms of hematological malignancies including familial aggregations. Ann Hematol (2017) 96(10):1635–9. doi:10.1007/s00277-017-3076-9
274. Polat A, Dinulescu M, Fraitag S, Nimubona S, Toutain F, Jouneau S, et al. Skin manifestations among GATA2-deficient patients. Br J Dermatol (2017) 178(3):781–5. doi:10.1111/bjd.15548
275. Dorn JM, Patnaik MS, Van Hee M, Smith MJ, Lagerstedt SA, Newman CC, et al. WILD syndrome is GATA2 deficiency: a novel deletion in the GATA2 gene. J Allergy Clin Immunol Pract (2017) 5(4):1149–52.e1. doi:10.1016/j.jaip.2017.02.010
276. Brambila-Tapia AJL, García-Ortiz JE, Brouillard P, Nguyen HL, Vikkula M, Ríos-González BE, et al. GATA2 null mutation associated with incomplete penetrance in a family with Emberger syndrome. Hematology (2017) 22(8):467–71. doi:10.1080/10245332.2017.1294551
277. Perez Botero J, Rodriguez V. Primary lymphedema and viral warts in GATA2 haploinsufficiency. Mayo Clin Proc (2017) 92(3):482. doi:10.1016/j.mayocp.2016.12.003
278. Berry D, Fekrat S. Central retinal vein occlusion in GATA2 deficiency. Retin Cases Brief Rep (2017). doi:10.1097/ICB.0000000000000558
279. Ramzan M, Lowry J, Courtney S, Krueger J, Schechter Finkelstein T, Ali M. Successful myeloablative matched unrelated donor hematopoietic stem cell transplantation in a young girl with GATA2 deficiency and Emberger syndrome. J Pediatr Hematol Oncol (2017) 39(3):230–2. doi:10.1097/MPH.0000000000000737
280. González-Lara MF, Wisniowski-Yáñez A, Pérez-Patrigeon S, Hsu AP, Holland SM, Cuellar-Rodríguez JM. Pneumocystis jiroveci pneumonia and GATA2 deficiency: expanding the spectrum of the disease. J Infect (2017) 74(4):425–7. doi:10.1016/j.jinf.2017.01.005
281. Burroughs LM, Shimamura A, Talano JA, Domm JA, Baker KK, Delaney C, et al. Allogeneic hematopoietic cell transplantation using treosulfan-based conditioning for treatment of marrow failure disorders. Biol Blood Marrow Transplant (2017) 23(10):1669–77. doi:10.1016/j.bbmt.2017.06.002
282. Ruiz-García R, Rodríguez-Vigil C, Marco FM, Gallego-Bustos F, Castro-Panete MJ, Diez-Alonso L, et al. Acquired senescent T-cell phenotype correlates with clinical severity in GATA binding protein 2-deficient patients. Front Immunol (2017) 8:802. doi:10.3389/fimmu.2017.00802
283. Kazamel M, Klein CJ, Benarroch EE, Patnaik MM, Tracy JA. Subacute demyelinating polyradiculoneuropathy complicating Epstein-Barr virus infection in GATA2 haploinsufficiency. Muscle Nerve (2018) 57(1):150–6. doi:10.1002/mus.25581
284. Azevedo L, Jay A, Lorenzana A, Keel S, Abraham RS, Horwitz M, et al. Case report of an adolescent male with unexplained pancytopenia: GATA-2 associated bone marrow failure and genetic testing. Glob Pediatr Health (2017) 4:1–4. doi:10.1177/2333794X17744947
285. Tholouli E, Sturgess K, Dickinson RE, Gennery A, Cant AJ, Jackson G, et al. In vivo T-depleted reduced-intensity transplantation for. Blood (2018) 131(12):1383–7. doi:10.1182/blood-2017-10-811489
286. Alazami AM, Al-Helale M, Alhissi S, Al-Saud B, Alajlan H, Monies D, et al. Novel CARMIL2 mutations in patients with variable clinical dermatitis, infections, and combined immunodeficiency. Front Immunol (2018) 9:203. doi:10.3389/fimmu.2018.00203
287. Schober T, Magg T, Laschinger M, Rohlfs M, Linhares ND, Puchalka J, et al. A human immunodeficiency syndrome caused by mutations in CARMIL2. Nat Commun (2017) 8:14209. doi:10.1038/ncomms14209
288. Sorte HS, Osnes LT, Fevang B, Aukrust P, Erichsen HC, Backe PH, et al. A potential founder variant in CARMIL2/RLTPR in three Norwegian families with warts, molluscum contagiosum, and T-cell dysfunction. Mol Genet Genomic Med (2016) 4(6):604–16. doi:10.1002/mgg3.237
289. Wang Y, Ma CS, Ling Y, Bousfiha A, Camcioglu Y, Jacquot S, et al. Dual T cell- and B cell-intrinsic deficiency in humans with biallelic RLTPR mutations. J Exp Med (2016) 213(11):2413–35. doi:10.1084/jem.20160576
290. Roncagalli R, Cucchetti M, Jarmuzynski N, Gregoire C, Bergot E, Audebert S, et al. The scaffolding function of the RLTPR protein explains its essential role for CD28 co-stimulation in mouse and human T cells. J Exp Med (2016) 213(11):2437–57. doi:10.1084/jem.20160579
291. Sassi A, Lazaroski S, Wu G, Haslam SM, Fliegauf M, Mellouli F, et al. Hypomorphic homozygous mutations in phosphoglucomutase 3 (PGM3) impair immunity and increase serum IgE levels. J Allergy Clin Immunol (2014) 133(5):e1–13. doi:10.1016/j.jaci.2014.02.025
292. Zhang Y, Yu X, Ichikawa M, Lyons JJ, Datta S, Lamborn IT, et al. Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J Allergy Clin Immunol (2014) 133(5):1400–9, 9.e1–5. doi:10.1016/j.jaci.2014.02.013
293. Pang H, Koda Y, Soejima M, Kimura H. Identification of human phosphoglucomutase 3 (PGM3) as N-acetylglucosamine-phosphate mutase (AGM1). Ann Hum Genet (2002) 66(Pt 2):139–44. doi:10.1046/j.1469-1809.2002.00103.x
294. Monticelli M, Ferro T, Jaeken J, Dos Reis Ferreira V, Videira PA. Immunological aspects of congenital disorders of glycosylation (CDG): a review. J Inherit Metab Dis (2016) 39(6):765–80. doi:10.1007/s10545-016-9954-9
295. Stray-Pedersen A, Backe PH, Sorte HS, Morkrid L, Chokshi NY, Erichsen HC, et al. PGM3 mutations cause a congenital disorder of glycosylation with severe immunodeficiency and skeletal dysplasia. Am J Hum Genet (2014) 95(1):96–107. doi:10.1016/j.ajhg.2014.05.007
296. Ben-Khemis L, Mekki N, Ben-Mustapha I, Rouault K, Mellouli F, Khemiri M, et al. A founder mutation underlies a severe form of phosphoglutamase 3 (PGM3) deficiency in Tunisian patients. Mol Immunol (2017) 90:57–63. doi:10.1016/j.molimm.2017.06.248
297. Pacheco-Cuellar G, Gauthier J, Desilets V, Lachance C, Lemire-Girard M, Rypens F, et al. A novel PGM3 mutation is associated with a severe phenotype of bone marrow failure, severe combined immunodeficiency, skeletal dysplasia, and congenital malformations. J Bone Miner Res (2017) 32(9):1853–9. doi:10.1002/jbmr.3173
298. Bernth-Jensen JM, Holm M, Christiansen M. Neonatal-onset T(-)B(-)NK(+) severe combined immunodeficiency and neutropenia caused by mutated phosphoglucomutase 3. J Allergy Clin Immunol (2016) 137(1):321–4. doi:10.1016/j.jaci.2015.07.047
299. Lundin KE, Hamasy A, Backe PH, Moens LN, Falk-Sorqvist E, Elgstoen KB, et al. Susceptibility to infections, without concomitant hyper-IgE, reported in 1976, is caused by hypomorphic mutation in the phosphoglucomutase 3 (PGM3) gene. Clin Immunol (2015) 161(2):366–72. doi:10.1016/j.clim.2015.10.002
300. Moens LN, Falk-Sorqvist E, Asplund AC, Bernatowska E, Smith CI, Nilsson M. Diagnostics of primary immunodeficiency diseases: a sequencing capture approach. PLoS One (2014) 9(12):e114901. doi:10.1371/journal.pone.0114901
301. Bjorksten B, Lundmark KM. Recurrent bacterial infections in four siblings with neutropenia, eosinophilia, hyperimmunoglobulinemia A, and defective neutrophil chemotaxis. J Infect Dis (1976) 133(1):63–71. doi:10.1093/infdis/133.1.63
302. Ali YA, Rahman S, Bhat V, Al Thani S, Ismail A, Bassiouny I. Hereditary multiple intestinal atresia (HMIA) with severe combined immunodeficiency (SCID): a case report of two siblings and review of the literature on MIA, HMIA and HMIA with immunodeficiency over the last 50 years. BMJ Case Rep (2011) 2011:1–7. doi:10.1136/bcr.05.2010.3031
303. Chen R, Giliani S, Lanzi G, Mias GI, Lonardi S, Dobbs K, et al. Whole-exome sequencing identifies tetratricopeptide repeat domain 7A (TTC7A) mutations for combined immunodeficiency with intestinal atresias. J Allergy Clin Immunol (2013) 132(3):656–64.e17. doi:10.1016/j.jaci.2013.06.013
304. Samuels ME, Majewski J, Alirezaie N, Fernandez I, Casals F, Patey N, et al. Exome sequencing identifies mutations in the gene TTC7A in French-Canadian cases with hereditary multiple intestinal atresia. J Med Genet (2013) 50(5):324–9. doi:10.1136/jmedgenet-2012-101483
305. Notarangelo LD. Multiple intestinal atresia with combined immune deficiency. Curr Opin Pediatr (2014) 26(6):690–6. doi:10.1097/MOP.0000000000000159
306. Agarwal NS, Northrop L, Anyane-Yeboa K, Aggarwal VS, Nagy PL, Demirdag YY. Tetratricopeptide repeat domain 7A (TTC7A) mutation in a newborn with multiple intestinal atresia and combined immunodeficiency. J Clin Immunol (2014) 34(6):607–10. doi:10.1007/s10875-014-0067-7
307. Avitzur Y, Guo C, Mastropaolo LA, Bahrami E, Chen H, Zhao Z, et al. Mutations in tetratricopeptide repeat domain 7A result in a severe form of very early onset inflammatory bowel disease. Gastroenterology (2014) 146(4):1028–39. doi:10.1053/j.gastro.2014.01.015
308. Bigorgne AE, Farin HF, Lemoine R, Mahlaoui N, Lambert N, Gil M, et al. TTC7A mutations disrupt intestinal epithelial apicobasal polarity. J Clin Invest (2014) 124(1):328–37. doi:10.1172/JCI71471
309. Fernandez I, Patey N, Marchand V, Birlea M, Maranda B, Haddad E, et al. Multiple intestinal atresia with combined immune deficiency related to TTC7A defect is a multiorgan pathology: study of a French-Canadian-based cohort. Medicine (Baltimore) (2014) 93(29):e327. doi:10.1097/MD.0000000000000327
310. Guanà R, Garofano S, Teruzzi E, Vinardi S, Carbonaro G, Cerrina A, et al. The complex surgical management of the first case of severe combined immunodeficiency and multiple intestinal atresias surviving after the fourth year of life. Pediatr Gastroenterol Hepatol Nutr (2014) 17(4):257–62. doi:10.5223/pghn.2014.17.4.257
311. Lemoine R, Bigorgne A, Farin H, Saint Basile G. [TTC7A, a critical effector for the intestinal and immune system homeostasis]. Med Sci (Paris) (2014) 30(6–7):616–8. doi:10.1051/medsci/20143006006
312. Woutsas S, Aytekin C, Salzer E, Conde CD, Apaydin S, Pichler H, et al. Hypomorphic mutation in TTC7A causes combined immunodeficiency with mild structural intestinal defects. Blood (2015) 125(10):1674–6. doi:10.1182/blood-2014-08-595397
313. Yang W, Lee PP, Thong MK, Ramanujam TM, Shanmugam A, Koh MT, et al. Compound heterozygous mutations in TTC7A cause familial multiple intestinal atresias and severe combined immunodeficiency. Clin Genet (2015) 88(6):542–9. doi:10.1111/cge.12553
314. Kammermeier J, Lucchini G, Pai SY, Worth A, Rampling D, Amrolia P, et al. Stem cell transplantation for tetratricopeptide repeat domain 7A deficiency: long-term follow-up. Blood (2016) 128(9):1306–8. doi:10.1182/blood-2016-01-696385
315. Lawless D, Mistry A, Wood PM, Stahlschmidt J, Arumugakani G, Hull M, et al. Bialellic mutations in tetratricopeptide repeat domain 7A (TTC7A) cause common variable immunodeficiency-like phenotype with enteropathy. J Clin Immunol (2017) 37(7):617–22. doi:10.1007/s10875-017-0427-1
316. Lien R, Lin YF, Lai MW, Weng HY, Wu RC, Jaing TH, et al. Novel mutations of the tetratricopeptide repeat domain 7A gene and phenotype/genotype comparison. Front Immunol (2017) 8:1066. doi:10.3389/fimmu.2017.01066
317. Neves JF, Afonso I, Borrego L, Martins C, Cordeiro AI, Neves C, et al. Missense mutation of TTC7A mimicking tricho-hepato-enteric (SD/THE) syndrome in a patient with very-early onset inflammatory bowel disease. Eur J Med Genet (2018) 61(4):185–8. doi:10.1016/j.ejmg.2017.11.014
318. Conti F, Lugo-Reyes SO, Blancas Galicia L, He J, Aksu G, Borges de Oliveira E Jr, et al. Mycobacterial disease in patients with chronic granulomatous disease: a retrospective analysis of 71 cases. J Allergy Clin Immunol (2016) 138(1):241–8.e3. doi:10.1016/j.jaci.2015.11.041
319. de Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, Feinberg J, et al. Revisiting human IL-12Rbeta1 deficiency: a survey of 141 patients from 30 countries. Medicine (Baltimore) (2010) 89(6):381–402. doi:10.1097/MD.0b013e3181fdd832
320. Aaby P, Roth A, Ravn H, Napirna BM, Rodrigues A, Lisse IM, et al. Randomized trial of BCG vaccination at birth to low-birth-weight children: beneficial nonspecific effects in the neonatal period? J Infect Dis (2011) 204(2):245–52. doi:10.1093/infdis/jir240
321. Medical Advisory Committee of the Immune Deficiency Foundation, Shearer WT, Fleisher TA, Buckley RH, Ballas Z, Ballow M, et al. Recommendations for live viral and bacterial vaccines in immunodeficient patients and their close contacts. J Allergy Clin Immunol (2014) 133(4):961–6. doi:10.1016/j.jaci.2013.11.043
Keywords: mycobacteria, bacille Calmette–Guerin, primary immunodeficiency, live vaccines, complications, adverse reactions
Citation: Nunes-Santos CJ and Rosenzweig SD (2018) Bacille Calmette–Guerin Complications in Newly Described Primary Immunodeficiency Diseases: 2010–2017. Front. Immunol. 9:1423. doi: 10.3389/fimmu.2018.01423
Received: 09 April 2018; Accepted: 07 June 2018;
Published: 22 June 2018
Edited by:
Menno C. van Zelm, Monash University, AustraliaReviewed by:
Ahmed Aziz Bousfiha, University of Hassan II Casablanca, MoroccoSaul Oswaldo Lugo Reyes, National Institute of Pediatrics, United States
Copyright: © 2018 Nunes-Santos and Rosenzweig. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sergio D. Rosenzweig, c3Jvc2VuendlaWdAY2MubmloLmdvdg==