 Mickael Vourc’h
Mickael Vourc’h Antoine Roquilly
Antoine Roquilly Karim Asehnoune
Karim Asehnoune- 1Laboratoire UPRES EA3826 “Thérapeutiques cliniques et expérimentales des infections”, IRS2 – Nantes Biotech, Université de Nantes, Nantes, France
- 2Intensive Care Unit, Anesthesia and Critical Care Department, Hôtel Dieu, University Hospital of Nantes, Nantes, France
Trauma is one of the leading causes of death and disability in the world. Multiple trauma or isolated traumatic brain injury are both indicative of human tissue damage. In the early phase after trauma, damage-associated molecular patterns (DAMPs) are released and give rise to sterile systemic inflammatory response syndrome (SIRS) and organ failure. Later, protracted inflammation following sepsis will favor hospital-acquired infection and will worsen patient’s outcome through immunosuppression. Throughout medical care or surgical procedures, severe trauma patients will be subjected to endogenous or exogenous DAMPs. In this review, we summarize the current knowledge regarding DAMP-mediated SIRS or immunosuppression and the clinical consequences in terms of organ failure and infections.
Introduction
Trauma represents the most extreme form of human tissue damage and is one of the leading causes of mortality and disabilities in the world (1). Ischemia-reperfusion, crush syndrome, surgical procedures, acidosis, hypoxemia, blood loss, or massive transfusion can all give rise to secondary tissue damage. Following trauma, a number of mediators, also called damage-associated molecular patterns (DAMPs), are released in the bloodstream by injured tissues. The recognition of DAMPs by immune cells initiates a systemic inflammatory response syndrome (SIRS) that induces physiological changes such as hypo or hyperthermia, elevated heart rate and leukocytosis/leukocytopenia (2). Early uncontrolled and protracted SIRS has been reported to be a risk factor of “sterile” organ failure (3) and of delayed hospital-acquired infection (mainly pneumonia) (3, 4). The high susceptibility to secondary infection after the initial phase of sepsis or trauma is attributable to an intense and long-term compensatory anti-inflammatory response (CARS) (5–7) leading to posttraumatic immunosuppression (IS). Initially, IS corresponds to a homeostatic phenomenon to avoid the remote organ injuries caused by the initial SIRS. It becomes deleterious when it persists, rendering patients prone to secondary bacterial (8) or fungal infections and viral reactivation.
Damage-associated molecular patterns are the origins of both SIRS and IS and are, therefore, promising targets as biomarkers for patient stratification and as therapeutic targets.
DAMPs Definition
Damage-associated molecular patterns are endogen nuclear, mitochondrial, or cytosolic molecules that have physiological functions inside the cell. They activate innate and adaptive immunity when released into the extracellular milieu. Innate immunity cells, mainly antigen-presenting cells (APC) such as dendritic cells (DCs) and neutrophils (PMNs) recognize DAMPs via pattern recognition receptors (PRRs). After PRR activation, PMNs and APC give rise to the local production of cytokines, chemokines, and other soluble factors. Local inflammatory response aims to ensure adequate tissue repair and may also generate a systemic and uncontrolled inflammatory response inducing remote organ failure.
Damage-associated molecular patterns released after trauma are also called alarmins. Since any molecule expelled in the microenvironment after tissue damage may be considered as an alarmin, it is of major importance to detect those that are clinically relevant and immunologically active. Clinically pertinent alarmins during trauma were defined in a consensus in 2006 (9) as substances:
• immediately released after trauma,
• responsible for immune cell activation whose concentration reflects the severity of trauma,
• giving rise to pro-inflammatory response on cultured cells with a clearly elucidated mechanism of activation,
• with plasma levels that correlate with the extent of the inflammatory response.
Finally, trauma alarmins have redundant activity on several receptors with highly variable effects depending on the microenvironment (10).
Characterization of DAMPs Released After Trauma
Nucleic Acids
All human cells contain nucleic acids [desoxynucleic acid (DNA) or messenger RNA] (11) or mitochondrial DNA (mtDNA) and mature erythrocytes may also retain some residual non-functional mtDNA (12). During severe trauma, nuclear and mitochondrial nucleic acids are released into the cytosol and in the bloodstream. The plasma levels of DNA increase parallel to the severity of the trauma and the concentration of mtDNA is correlated with the occurrence of acute lung injury (ALI), the severity of SIRS and the occurrence of multiple organ dysfunction syndrome (MODS) (13–17). Nuclear DNA recognition by monocytes triggers the same inflammatory response as microbial nucleic acids (also called PAMPs or pathogen-associated molecular pattern). In particular, monocytes produce IL-6 after stimulation by nuclear DNA (18) or IL-8 after exposition to messenger RNA (19).
High-Mobility Group Box 1 (HMGB1)
High-mobility group box 1 is a nuclear chaperone protein that regulates DNA transcription. In physiological settings, HMGB1 binds to DNA and bends it to facilitate gene transcription. After severe trauma, HMGB1 is either secreted by activated or stressed immune and non-immune cells or can leak out from dead cells (20). In the same way, hypoxia or ischemia-reperfusion in hepatocytes or cardiomyocytes cause time-dependent extracellular release of HMGB1 (21, 22). Its plasma concentration peaks within 6 h after injury and the concentration remains elevated for at least 24 h (23). Its level 30 min after trauma correlates with the injury severity score (ISS), SIRS, MODS, death, and with the amplitude of immune activation (24, 25). For example, a high level of HMGB1 after trauma was associated with lung dysfunction and longer duration of mechanical ventilation (26). Moreover, HMGB1 release after trauma with bone fracture can exert remote effects on several organs. For example, HMGB1 can worsen cerebral lesion after ischemic brain injury (BI) (27) or lead to lung injury (28). Interestingly, the variations of redox conditions influence HMGB1 activity. HMGB1 contains two redox sensitive sites that deeply impact its function (29). During severe trauma, excessive production of radical oxygen species enhances oxidative stress and leads to multiple redox reactions. HMGB1 function shifts to promote severe inflammation when oxidative stress increased (30). At the opposite, the reduced form rather enhances chemotactic signaling. Finally, this protein has a poor pro-inflammatory activity but acts as a cofactor of inflammation with lipopolysaccharide (LPS), nuclear DNA, or IL-1β.
Heat-Shock Proteins (HSPs)
Heat-shock proteins are molecular chaperones that control intracellular trafficking and prevent the misfolding of polypeptide chains. These proteins are named according to their molecular weight and are expressed both constitutively and under stressful conditions (31). HSPs are present in the bloodstream of healthy volunteers (32). Their circulating levels decrease with aging (33) and increase under several pathological conditions. HSPs are released into the extracellular compartment after trauma (34–36). In particular, in severe trauma with an ISS score higher than 16, HSP72 levels were significantly higher compared with healthy controls (37). Interestingly, severe trauma patients with the highest HSP72 circulating levels on hospital admission had better survival rates (37). HSPs activate both innate and adaptive immunity and can trigger pro-inflammatory response. In particular, HSPs promote antigen presentation and the maturation of DCs as demonstrated by the upregulation of the major histocompatibility complex (MHC) class II molecules (38) and co-stimulatory signals, such as CD80 and CD86 (39, 40).
S100 Proteins
This family includes more than 20 distinct proteins. S100 proteins are calcium-binding proteins and are mainly expressed by myeloid cells. Their release in extracellular compartments is secondary to cell damage or phagocytosis (41). Three S100 proteins are specifically linked to innate immune functions: S100A8, S100A9, and S100A12. Protein S100A8, also called Calgranulin A or Myeloid-related protein 8 (MRP8), and S100A9, also called Calgranulin B or MRP14 are found in monocytes and macrophages (42). Protein S100A12 (Calgranulin C) is mostly expressed in granulocytes (43). During inflammatory response, monocytes or macrophages release MRP8 (S100A8) and MRP14 (S100A9) into the circulation. They then form a heterodimer (44) and are recognized by PRRs. After severe burn injury, myeloid-related protein (MRP) levels in bloodstream are correlated with poor outcome (45). S100β protein is specifically released after acute BI (46, 47). Similar to HMGB1, MRP8 and MRP14 levels in the bloodstream increase early in the acute phase of trauma or BI (48).
DAMPs Activate Several Receptors, Signaling Pathways, and Cellular Subsets
Damage-associated molecular patterns released by injured organs, tissues, or cells can be detected by several receptors including PRRs and activate several pathways (Figure 1). Of interest, a given DAMP may stimulate several receptors and engage different pathways—a phenomenon called receptor redundancy.
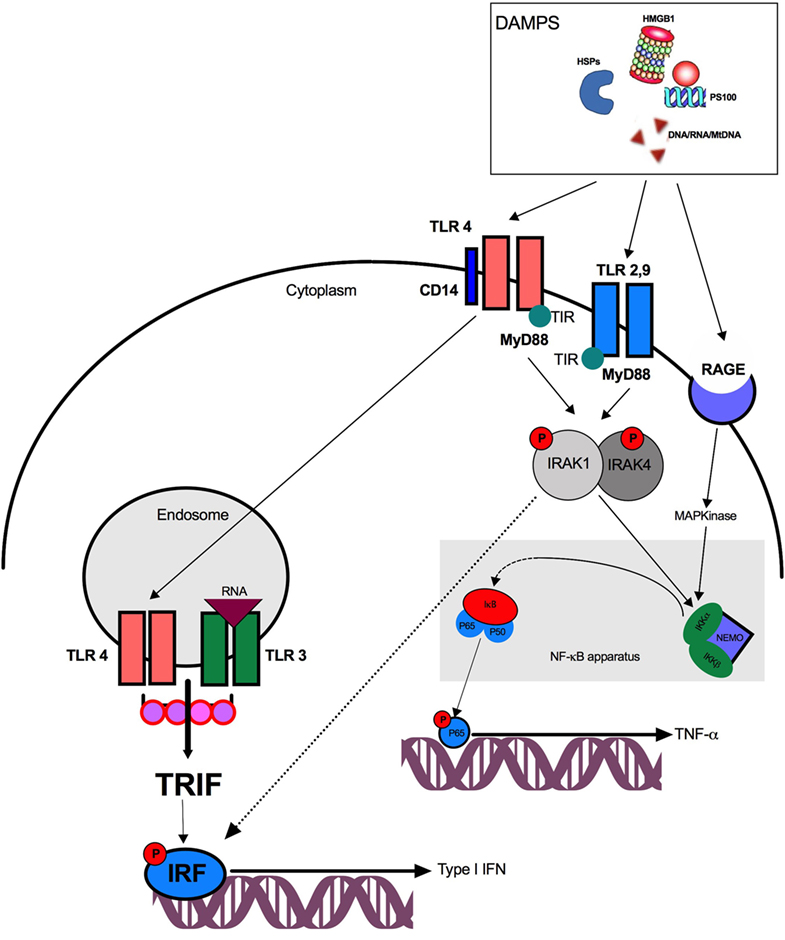
Figure 1. Pathway activation after damage-associated molecular patterns (DAMPs) stimulation. DAMPs are recognized by surface or endosomal TLRs and trigger either TRIF and NF-κB activation. TLR4, toll-like receptor 4; TIR, toll/interleukin-1 receptor; DNA, desoxyribonucleic acid; RNA, ribonucleic acid; IRF, interferon regulatory factor; TRIF, TIR domain-containing adaptor protein inducing interferon β; NF-κB, nuclear factor κB; NEMO, NF-κB essential modulator; IRAK, IL-1 receptor-associated kinase-4; MyD88, myeloid differentiation protein 88; RAGE, receptor for advanced glycation end products; HMGB1, high-mobility group box 1; HSP, heat-shock protein; IKK, Iκ-B kinase.
Pathogen Recognition Receptors
Pattern recognition receptors are expressed by a large variety of innate immune cells including PMNs, natural killer lymphocytes (NK), macrophages and DCs. Among PRRs, the main receptors involved after trauma are nucleotide oligomerization domain (NOD)-like receptors (NLRs) and toll-like receptors (TLRs). NLRs are intracellular PRRs that are localized in the cytoplasm. They usually sense degradation products from bacterial cell wall components. NOD receptors activate the nuclear transcription factor NF-κB and the MAP kinases leading to pro-inflammatory cytokine production.
Toll-like receptors, initially described for PAMP recognition, also play a key role in the recognition of DAMPs such as: nucleic acids, HMGB1, HSPs, and S100 proteins. TLRs are localized on the cell surface and/or within the intracellular endosomal compartment. After DAMPs binding, TLRs dimerize and recruit adapter proteins through their intracellular TIRs (Toll/Interleukin-1 receptor) domains. All TLRs except TLR3 signal through the adapter myeloid differentiation protein 88 (MyD88). Downstream, and depending on the TLR understudy, MyD88 can activate two distinct signaling pathways. The first requires IL-1 receptor-associated kinase (IRAK) recruitment which activates the NF-κB apparatus and leads to TNF-α, IL-6, and IL-8 production. The second involves TIR domain-containing adaptor protein inducing interferon β (TRIF) activation that induces interferon regulatory factor (IRF) phosphorylation allowing the synthesis of type I interferon (IFN) (49, 50). TLR4 plays a singular role in DAMP detection and (51) activates either NF-κB via MyD88–IRAK or TRIF after TLR4 translocation to the endosome through a CD14-dependent mechanism. Interestingly, TLR3 exclusively triggers the TRIF-dependent pathway.
The IRF pathway leads to type I IFN production and is critical for human leukocyte antigen (HLA)-DR regulation and co-stimulatory molecules (CD80/CD86) expression on APCs. The NF-κB pathway leads to the production of several pro-inflammatory mediators (including TNF-α) and may be activated by extracellular signals present in the blood and organs after trauma, such as reactive oxygen species, cytokines, and complement fragments (52). NF-κB activation requires a complex of kinases which are called IκB Kinase (IKK). IKK induces the degradation (phosphorylation and ubiquitination) of the cytoplasmic inhibitor of NF-κB: IκB-α. This degradation enables the translocation of NF-κB sub-units into the nucleus. After binding to the promoter of the gene, the heterodimer form of NF-κB (P65/P50) enhances whereas the homodimer P50/P50 inhibits the transcription of proinflammatory cytokines. NF-κB activation also gives rise to inflammasome formation and caspase-1 release and promotes IL-1β synthesis (53).
Nucleic acids trigger TLR3 (mRNA) or TLR9 (DNA and mtDNA) activation. Among mitochondrial damage-associated molecular patterns, N-formyl peptides (F-MIT), and mtDNA can give rise to sterile inflammation. The former activates formyl peptide receptor-1 (FRP1) and the latter binds TLR9 and promotes the formation of NOD-like receptor pyrin domain containing-3 (NLRP3) inflammasome (13, 54).
High-mobility group box 1 activates NF-κB transcription factor through TLR2, TLR4, or TLR9 and leads to TNF-α, IL-1β, and IL-6 production (55–57). The recognition of HMGB1 by TLR4 requires myeloid differentiation factor 2 (58). HMGB1 can also link to LPS, thereby strengthening its ability to activate TLR4 through CD14 (59). Finally, HMGB1 can complex with CpG-ODN and bind TLR9 to enhance cytokine production of APCs (60).
Heat-shock proteins promote antigen presentation, the maturation of DCs and activate the TLR-MyD88-NF-κB pathway (38). MRP8 (PS100A8), MRP14 (PS100A9), and PS100A12 are recognized by TLR4 or TLR2 (45, 61).
Receptor for Advanced Glycation End Product (RAGE) and Triggering Receptor Expressed on Myeloid Cells-1 (TREM-1)
Triggering receptor expressed on myeloid cells-1 is a member of the immunoglobulin superfamily expressed on monocytes and PMNs that synergize with TLR4 to mediate the effects of HMGB1 (62). TREM-1 is associated with an immunoreceptor tyrosine-based activation motif and an adaptor molecule called DAP12 for signal transduction. In fine, TREM-1 pathway phosphorylates extracellular signal-regulated kinase and phosphoinositide-3 kinase leading to NF-κB activation (63). HMGB1 is also detected by the RAGE (64), an immunoglobulin superfamily cell surface receptor. Contrary to TLRs, RAGE is a specific receptor for DAMP that is not activated by PAMP. It activates NF-κB pathway through RAS family proteins and MAP kinase phosphorylation (65). Finally, among S100 proteins, RAGE recognize S100A12 and S100β (46, 47) and subsequently engage the NF-κB pathway.
Overall, DAMPs triggers massive cytokine relapse including TNF-α, IL-1, IL-6, IL-8, IL-12 and IFN types I and II. These mediators amplify the activation, maturation, proliferation, and recruitment of immune cells at the site of trauma, causing indirect activation of innate and adaptive immune cells such as DCs or T cells (66).
DAMPs Activate Neutrophils and DCs
During severe trauma, F-MIT activate PMNs via FRP1, a G protein-coupled surface receptors (GPCR) (67) and trigger their chemotaxis and phagocytosis (68). In parallel, HMGB1/RAGE enhances PMNs recruitment toward injured tissues and amplifies the inflammatory response via NF-κB transcription factor activation (69). HMGB1 can also activate PMNs through TLR4 or TLR7 during ischemia-reperfusion injury (70). Besides, extracellular adenosine 5’-triphosphate (ATP) release enhances PMNs adhesion to blood vessel wall upon sterile injury (71) and promotes cell migration to injury site (72). Finally, PMNs activation contributes to SIRS, cardiovascular collapse, and remote organ injury (73). Notably, the release of metalloproteinase 9 (MMP9) after traumatic brain injury (TBI) is involved in secondary brain damage (74).
Damage-associated molecular patterns will also activate DCs and trigger both the innate and the adaptive immune response (75). Among the most described DAMPs, activation and maturation of DCs are mainly driven by HMGB1 binding to TLR2, TLR4 (55), or RAGE (76, 77). HMGB1 also acts as a chemokine for DCs when binding to CXCR4. Then, HSPs bind TLR4 (78) and participate in antigen processing by DC before presentation (79) to T cells. Moreover, mtDNA and especially mitochondrial transcription factor A can synergize with CpG to induce type I IFN release (80, 81). DCs also recognize extracellular ATP by high affinity purinergic receptors P2Z/P2X7 (82) leading to inflammasome activation (83).
Novel Insights
DAMPs Participate in Posttraumatic IS
Following trauma, DAMPs participate in IS that renders patients prone to secondary infections. Timmermans et al. recently evaluated the role of DAMPs in IS in 166 adult trauma patients. The authors reported that plasma nuclear DNA and HSP70 levels correlated negatively with HLA-DR expression. Moreover, higher levels of circulating nuclear DNA and a further decrease in HLA-DR expression on blood monocytes were associated with infections. Overall, the role of DAMPs in the induction of an overwhelming inflammatory process leading to remote organ failure is well known. This study is one of the first descriptions that associates plasma levels of DAMPs with IS and with secondary infections (34).
DAMPs Induce Endotoxin Tolerance
After massive DAMP release, the early overwhelming inflammatory response can further leave an immunological scar that will be responsible for protracted immune alterations (IS) (84) and will worsen patient outcome. This IS decreases the capacity of monocytes to produce inflammatory cytokines after TLR stimulation (52, 85, 86) and APC to prime antigen on type 2 MHC molecules. Austermann et al. highlighted that MRP linkage with TLRs induced phagocyte hyporesponsiveness to subsequent TLR agonist stimulation and was correlated with poor outcome (45). In the same way, HSPs can also induce a tolerogenic response. After HSP70 recognition by monocytes through CD14/TLR4 complex, subsequent LPS stimulation fails to activate IKK. As a result, the phosphorylation of P65 is altered and prevents the NF-κB activation (35).
Damage-associated molecular patterns may also induce epigenetic alterations including chromatin modifications leading to a transient silencing of the transcription of pro-inflammatory mediators (87). These epigenetic modifications induce hyporesponsiveness (tolerance) to subsequent stimulation of TLRs. After trauma, Austermann et al. reported that after TLR–MyD88–NF-κB activation by MRPs, RelB nuclear translocation increases the methylation of the promoter gene of TNF-α and reduces its transcription (45).
Finally, DAMPs release in severe trauma patients can induce tolerance similarly to post-septic scenarios (52). After trauma, although early tolerance attenuates the severity of the organ failures caused by ischemia-reperfusion, long-term tolerance can also increase susceptibility to secondary infection.
Immunosuppressive Properties of HMGB1
The rate of infections after severe TBI is very high—up to 40%—and is largely correlated with IS (8). TBI-induced IS consists in monocytic deactivation along with decreased expression of HLA-DR on monocytes and lymphopenia (88–90). In TBI patients, our team confirmed that the downregulation of HLA-DR on monocytes leads to an increased susceptibility to secondary infection (91). At the same time, cytotoxic response of NK cells is altered (91), CD4+Th1-type cell response is suppressed (92) whereas Th2-type response is enhanced (93). As a result, TBI appears to affect both innate and adaptive immunity.
A strong correlation between the severity of BI and the intensity of immune depression has been previously reported (94). It is generally believed that the autonomic nervous system plays a major role in IS after BI because catecholamines are elevated and alter immunity. However, recent clinical (95) and experimental (96) studies have challenged this paradigm and there is evidence that alarmins are a cornerstone of IS after TBI.
High-mobility group box 1 is released in the early phase after BI and has been identified as a key player in overwhelming sterile inflammatory response (97). HMGB1 can also trigger the proliferation of suppressive cells (89, 90). For example, after BI HMGB1 induces the expansion of altered monocytes in the spleen. This was characterized by a low level of type II MHC molecule expression and reduced cytokine production (TNF-α and IL-12) in response to TLR agonists (97). These altered monocytes can suppress lymphocytes and lead to lymphopenia, a hallmark of IS after BI.
Finally, HMGB1 may alter the prognosis of severe BI patients not only through the initial pro-inflammatory response but also because it may induce IS which is a major cause of secondary bacterial infections.
DAMPs Can Induce IS Without Prior Overwhelming Inflammatory Response
Recently published data suggest that DAMPs may directly induce IS without the need for a first overwhelming inflammatory response (98). Endogenous purine nucleosides are major regulators of the inflammatory response. In this setting, adenosine, which is a catabolite of ATP, signals through the binding and activation of GPCR such as A2AR, a purinergic receptor. High amount of adenosine released after cell injuries including severe trauma may induce a major inflammatory response. More recently, it was shown that adenosine also promotes Th2 immunity, a critical component of posttraumatic IS. Indeed, when binding to GPCR, adenosine may activate DCs to promote the production of IL-4 by CD4 T cells (99, 100). In the study by Patel et al. (98), the authors suggested that the specific GPCR for adenosine (A2AR) initiates Th2 immune response. In their model, A2AR induced expression of the alarmin IL-33, which subsequently triggered ILC-2 (Type 2 Innate lymphoid cells) and Th2 cell activation and production of IL-4 and IL-13—cytokines which are known to participate in posttraumatic IS. The results of Patel et al. are important because they suggest that a single DAMP such as adenosine may directly induce a Th2 response. There is another way by which endogenous purine molecules may alter immunity. The release of ATP from necrotic cells is increased in inflamed tissues like after severe trauma. This alarmin activates DCs and cellular immunity in general via P2 receptors (99–101). CD39 degrades ATP to adenosine (a trigger of the Th2 response, see above), and CD39 expression on CD4+Foxp3+ Treg is known to contribute to the suppressive functions of these cells (102–104). This is important since CD39 is constitutionally expressed on Treg cells and is a critical pathway for negative regulation of inflammation (Figure 2).
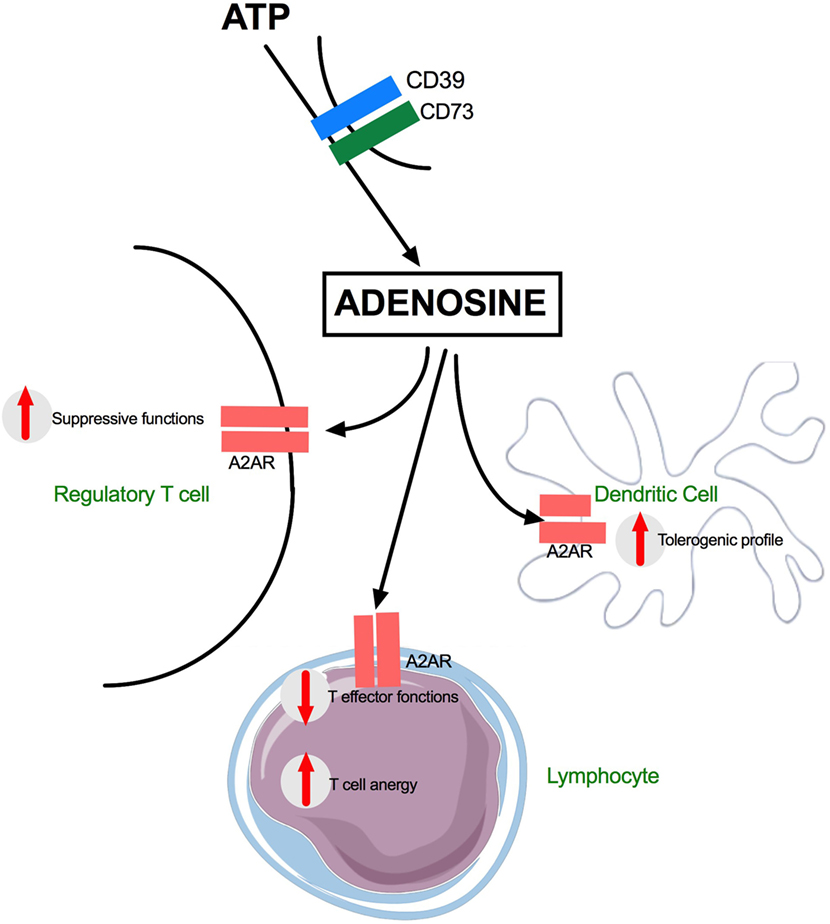
Figure 2. Adenosine effects on dendritic and T cells during trauma. G protein-coupled surface receptors (GPCR) are key actors of Adenosine mediated inflammatory after trauma. A2AR receptors are expressed on many subsets including Regulatory T cells, effector T cells, and dendritic cells. ATP, adenosine 5’-triphosphate; CD39, nucleoside triphosphate diphosphohydrolase-1; CD73, surface-expressed ecto-5′-nucleotidase; A2AR, adenosine A2A receptor.
Overall, these data show that alarmins play a role in the production of IL-4 which is mainly produced by activated T cells. The antagonistic nature of IL-4 on Th1 response makes it an important potential factor in IS. Activated Treg (CD39+) further alters immunity by increasing the levels of Adenosine.
Immune Dysfunction Following Transfusion Is Triggered by DAMP
Transfusion is reported to suppress innate immunity (105) and is associated with an increased risk of death during surgery. Multiple trauma patients often require massive transfusion (106) and transfusion correlates with infection in a dose-dependent manner (107, 108). Packed red blood cells (PRBC) contain immunologically active compounds. Several studies have reported changes in supernatant composition during storage of red blood cell (RBC) and hypothesized that these modifications could explain morbidity (109, 110). However, large randomized studies have assessed the impact of the storage duration of PRBC in blood banks and provided conflicting results regarding patient outcome (111–113). Up to 25% of RBC can undergo hemolysis within 24 h following transfusion regardless of group incompatibility or immunologic reactions (114). RBC contains most of the DAMPs described above. As a result, hemolyzed RBC release high amounts of DAMPs into the circulation including Heme, HSP70, Retinol-Binding Protein-4 (RBP-4), IL-33, S100 proteins, and adenosine (115). Heme acts as a DAMP and promotes the formation of the inflammasome in LPS-primed macrophages after TLR4 stimulation (116, 117). RBP4, which was recently identified in PRBC (118), is a TLR4 agonist and can be considered as a DAMP (119). Most RBC DAMPs will trigger NF-κB or IRF activation leading to TNF-α or type I IFN production influencing cell survival, differentiation, and proliferation (120). RBC lysis has also been reported to be responsible for IL-33 relapse (115). IL-33 is a nuclear-associated IL-1 family cytokine inducing type 2 cytokine response (121) and promoting regulatory T-cell response (122). ATP intracellular concentration in RBC is very high (123). After extracellular release, ATP is cleaved in adenosine which also acts as a DAMP (see above). Adenosine either triggers inflammatory response through NLRP3 activation and inflammasome formation or promotes Th2 response though GPCR activation (121). RBC lysis can release HSP70 (115) which stimulates monocytes and DCs via TLR2 and TLR4/CD14 pathways. This DAMP could account for transfusion-related acute renal failure (124). Finally, non-functional mtDNA can be retained in mature RBC and participate in the sterile inflammation response after transfusion via TLR9 stimulation and NLRP3 inflammasome formation.
Our team is conducting a prospective study on transfusion-induced immunomodulation and organ failure with the aim to identify DAMPs in the supernatant of labile blood products (NCT02763410). Our preliminary results have shown that the composition of each PRBC is highly variable even between 2 PRBC with the same storage duration. Our results may, therefore, account for controversial results in the last large randomized studies. For example, we have noted major differences in terms of RBP-4 or SDF1-α, stromal cell-derived factor 1 (CXCL12) concentrations (unpublished data), see Figures S1A,B in Supplementary Material. Even if CXCL12 is not strictly a DAMP, it can complex and synergize with HMGB1 and enhance CXCR4 activation (125). Finally, further studies are required to focus on RBC supernatant composition to study patient outcome and to better delineate the exact role of the DAMPs that are released after blood transfusion.
Clinical Consequences: Damp-Related Organ Dysfunction
The ISS predicts further organ failure and patient outcome (126). MODS incidence can reach 60% of patients (127). If DAMP-induced remote organ dysfunction is now a widely accepted phenomenon, its exact pathophysiology is only partially understood. Organ dysfunction after trauma follows a biphasic pattern. Early MODS is usually driven by an uncontrolled SIRS, and late MODS involves IS-induced nosocomial infections (mainly pneumonia).
Early MODS
Despite well-conducted resuscitation, hypotension, transfusion support and the combined effects of rhabdomyolysis, ischemia-reperfusion, acidosis, anemia, hypoxemia, and hypovolemia induce cell injuries leading to remote organ dysfunction [i.e., acute renal failure, disseminated intravascular coagulation (DIC), or ALI]. After cell injuries, massive DAMPs release leads to SIRS and when the response is overwhelming, the risk of MODS is high. The role of DAMPs in the occurrence of MODS is illustrated by the high level of circulating HMGB1 after trauma which has been associated with the magnitude of SIRS, MODS and death (24, 128, 129). Moreover, a high amount of HMGB1 in patient serum will give rise to lung injury characterized by tight junction alteration and an increased permeability leading to interstitial edema (25, 64, 130). Plasma mtDNA has also been associated with MODS as well as mortality in severe trauma (16).
Late MODS
In addition to SIRS, as mentioned above, DAMPs may lead to an IS that is directly responsible for increased susceptibility to secondary infection, mainly hospital-acquired pneumonia (HAP). Pneumonia in the intensive care unit (ICU) threatens patient prognosis. Of note, after severe trauma or TBI, the HAP rate can reach 40% (8). HAP can give rise to ALI, acute respiratory distress syndrome, severe sepsis and even MODS (131, 132). As a result, HAP increases ventilation duration, length of ICU stays and worsens prognosis in ICU trauma patients (133).
Disseminated Intravascular Coagulation
Disseminated intravascular coagulation is a frequent issue following severe trauma. DIC can alter microcirculation leading to MODS following severe trauma. Among DIC determinants, many DAMPs were reported to disturb the procoagulant-anticoagulant balance. For instance, the level of circulating serum HMGB1 or DNA–histone complex are two independent prognostic factors of DIC (134, 135). Specifically, nucleic acids and F-MIT activate coagulation pathways (73) whereas histones activate thrombin formation and platelets aggregation (136). In the same way, HMGB1 promotes coagulation by enhancing tissue factor expression on monocytes and neutrophil extracellular trap release (134). At the opposite, histones and HMGB1 inhibit Protein C activation and impaired coagulation (137, 138).
Conclusion
In conclusion, during severe injury, DAMPs are responsible for multiple innate and adaptive immune subset activation (Figure 3). The early stage of trauma is characterized by an inflammatory response leading to organ failure whose intensity differs between patients. Five to 15 days after trauma, a CARS occurs, leading to IS and increased susceptibility to nosocomial infection. This immunosuppressive phenotype results from the multiple aspects of immunity impairment (Figure 3).
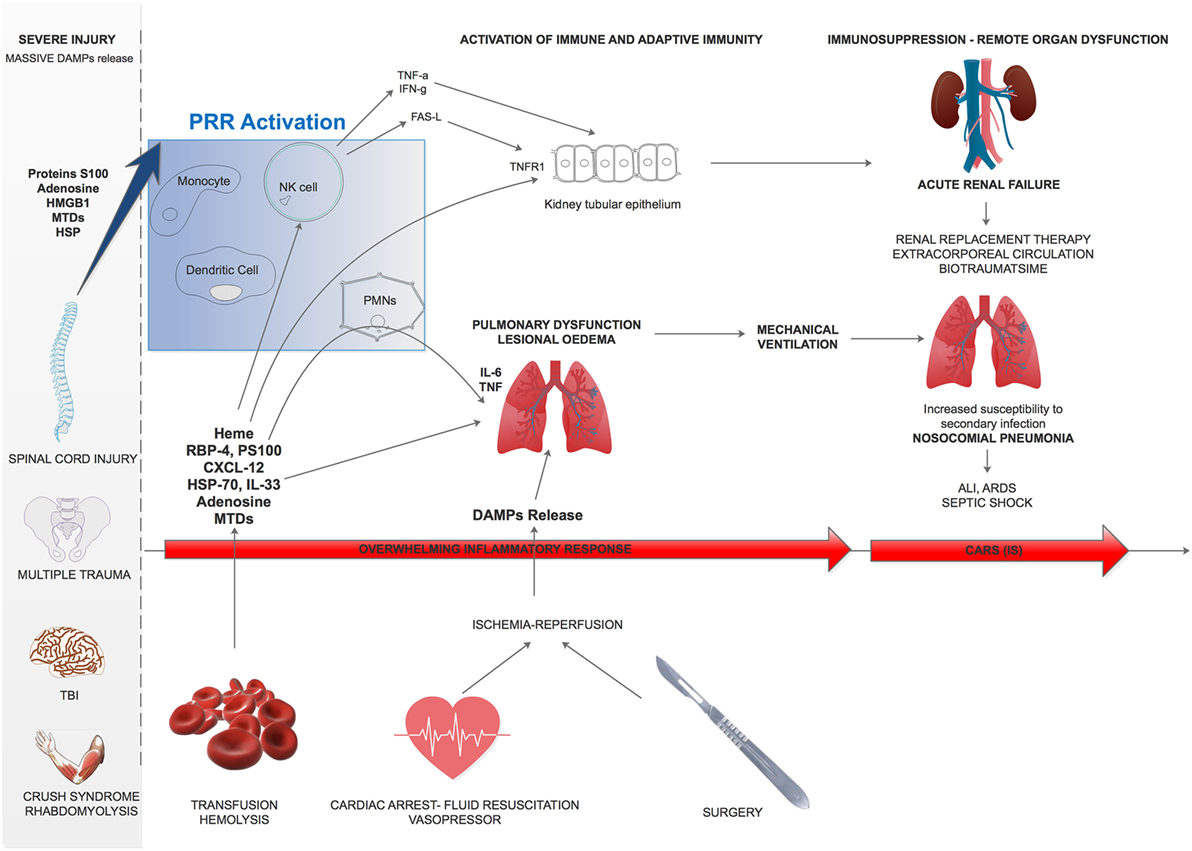
Figure 3. Damage-associated molecular patterns (DAMPs) effect overview on remote organ dysfunction after trauma. After severe trauma, multiple DAMPs are released and can trigger lung edema or kidney tubular epithelium activation. Besides, subsequent transfusion, fluid resuscitation and surgery will worsen these phenomenon and lead to remote acute organ failure. ALI, acute lung injury; ARDS, acute respiratory distress syndrome; CARS, compensatory anti-inflammatory response; PMN, polymorphonuclear leukocytes; MTD, mitochondrial damage-associated molecular patterns; RBP4, retinol-binding protein-4; PS100, protein S100; HSP, heat-shock protein; TNFR1, Type 1-TNF receptor; TNF-α, tumor necrosis factor-α; HMGB1, high-mobility group box 1.
Author Contributions
MV, AR, and KA wrote the main manuscript. All authors reviewed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The EA3826 Laboratory and the Surgical ICU of Nantes University Hospital.
Funding
Experiments provided in online-only material were funded by the “Agence nationale de la sécurité du medicament” (ANSM- AAP-2015-014).
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.01330/full#supplementary-material.
Abbreviations
ALI, acute lung injury; APC, antigen-presenting cells; ARDS, acute respiratory distress syndrome; ATP, adenosine 5′-triphosphate; CARS, compensatory anti-inflammatory response syndrome; CXCL12, SDF1-α (stromal cell-derived factor 1); DAMPs, damage-associated molecular patterns; DIC, Disseminated intravascular coagulation; DNA, desoxynucleic acid; ERK, extracellular signal-regulated kinase; F-MIT, N-formyl peptides; FRP1, formyl peptide receptor-1; GPCR, G protein-coupled surface receptors; HAP, hospital-acquired pneumonia; HLA, human leukocyte antigen; HMGB1, high-mobility group box 1; HSP, heat-shock protein; ICU, intensive care unit; IKK, IκB kinase; IFN, interferon; IRAK, IL-1 receptor-associated kinase-4; IRF, interferon regulatory factor; IS, Immunosuppression; ISS, injury severity score; ITAM, immunoreceptor tyrosine-based activation motif; LPS, lipopolysaccharide; MD-2, myeloid differentiation factor 2; MHC, major histocompatibility complex; MMP9, metalloproteinase 9; MTD, mitochondrial damage-associated molecular patterns; mtDNA, mitochondrial DNA; MODS, multiple organ dysfunction syndrome; MRPs, myeloid-related proteins; MyD88, myeloid differentiation protein 88; NET, neutrophil extracellular trap; NK, natural killer; NLRP3, NOD-like receptors pyrin domain containing 3; NOD, nucleotide oligomerization domain; PD-1, program death-1; PI3K, phosphoinositide-3 kinase; PRR, pattern recognition receptor; PRBC, packed red blood cells; RAGE, receptor for advanced glycation end products; RBP4, retinol-binding protein 4; ROS, radical oxygen species; SIRS, systemic inflammatory response syndrome; TBI, traumatic brain injury; TREM-1, triggering receptor expressed on myeloid cells-1; TRIF, TIR domain-containing adaptor protein inducing interferon β.
References
1. Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990-2020: global burden of disease study. Lancet (1997) 349:1498–504. doi:10.1016/S0140-6736(96)07492-2
2. Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest (1992) 101:1644–55. doi:10.1378/chest.101.6.1644
3. Hoover L, Bochicchio GV, Napolitano LM, Joshi M, Bochicchio K, Meyer W, et al. Systemic inflammatory response syndrome and nosocomial infection in trauma. J Trauma (2006) 61:310–7. doi:10.1097/01.ta.0000229052.75460.c2
4. Bochicchio GV, Napolitano LM, Joshi M, Knorr K, Tracy JK, Ilahi O, et al. Persistent systemic inflammatory response syndrome is predictive of nosocomial infection in trauma. J Trauma (2002) 53:245–51. doi:10.1097/00005373-200208000-00010
5. Asehnoune K, Roquilly A, Abraham E. Innate immune dysfunction in trauma patients. Anesthesiology (2012) 117:411–6. doi:10.1097/ALN.0b013e31825f018d
6. Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, et al. A genomic storm in critically injured humans. J Exp Med (2011) 208:2581–90. doi:10.1084/jem.20111354
7. Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis (2013) 13:260–8. doi:10.1016/S1473-3099(13)70001-X
8. Roquilly A, Mahe PJ, Seguin P, Guitton C, Floch H, Tellier AC, et al. Hydrocortisone therapy for patients with multiple trauma: the randomized controlled HYPOLYTE study. JAMA (2011) 305:1201–9. doi:10.1001/jama.2011.360
9. Harris HE, Raucci A. Alarmin(g) news about danger: workshop on innate danger signals and HMGB1. EMBO Rep (2006) 7:774–8. doi:10.1038/sj.embor.7400759
10. Manson J, Thiemermann C, Brohi K. Trauma alarmins as activators of damage-induced inflammation. Br J Surg (2012) 99(Suppl 1):12–20. doi:10.1002/bjs.7717
11. Ma KC, Schenck EJ, Pabon MA, Choi AMK. The role of danger signals in the pathogenesis and perpetuation of critical illness. Am J Respir Crit Care Med (2018) 197(3):300–9. doi:10.1164/rccm.201612-2460PP
12. Lee YL, King M, Gillespie MN, Gonzalez RP, Brevard SB, Frotan MA, et al. Blood transfusion products contain mitochondrial DNA damps: a potential effector of transfusion related acute lung injury. J Surg Res (2014) 186:509. doi:10.1016/j.jss.2013.11.280
13. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464:104–7. doi:10.1038/nature08780
14. Gan L, Chen X, Sun T, Li Q, Zhang R, Zhang J, et al. Significance of serum mtDNA concentration in lung injury induced by hip fracture. Shock (2015) 44:52–7. doi:10.1097/SHK.0000000000000366
15. Lo YM, Rainer TH, Chan LY, Hjelm NM, Cocks RA. Plasma DNA as a prognostic marker in trauma patients. Clin Chem (2000) 46:319–23.
16. Simmons JD, Lee Y-L, Mulekar S, Kuck JL, Brevard SB, Gonzalez RP, et al. Elevated levels of plasma mitochondrial DNA DAMPs are linked to clinical outcome in severely injured human subjects. Ann Surg (2013) 258:591–6. doi:10.1097/SLA.0b013e3182a4ea46
17. Gu X, Yao Y, Wu G, Lv T, Luo L, Song Y. The plasma mitochondrial DNA is an independent predictor for post-traumatic systemic inflammatory response syndrome. PLoS One (2013) 8:e72834. doi:10.1371/journal.pone.0072834
18. Atamaniuk J, Kopecky C, Skoupy S, Säemann MD, Weichhart T. Apoptotic cell-free DNA promotes inflammation in haemodialysis patients. Nephrol Dial Transplant (2012) 27:902–5. doi:10.1093/ndt/gfr695
19. Karikó K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for toll-like receptor 3. J Biol Chem (2004) 279:12542–50. doi:10.1074/jbc.M310175200
20. He Z-W, Qin Y-H, Wang Z-W, Chen Y, Shen Q, Dai S-M. HMGB1 acts in synergy with lipopolysaccharide in activating rheumatoid synovial fibroblasts via p38 MAPK and NF-κB signaling pathways. Mediators Inflamm (2013) 2013:1–10. doi:10.1155/2013/596716
21. Andrassy M, Volz HC, Igwe JC, Funke B, Eichberger SN, Kaya Z, et al. High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation (2008) 117:3216–26. doi:10.1161/CIRCULATIONAHA.108.769331
22. Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med (2005) 201:1135–43. doi:10.1084/jem.20042614
23. Yang R, Harada T, Mollen KP, Prince JM, Levy RM, Englert JA, et al. Anti-HMGB1 neutralizing antibody ameliorates gut barrier dysfunction and improves survival after hemorrhagic shock. Mol Med (2006) 12:105–14. doi:10.2119/2006-00010.Yang
24. Cohen MJ, Brohi K, Calfee CS, Rahn P, Chesebro BB, Christiaans SC, et al. Early release of high mobility group box nuclear protein 1 after severe trauma in humans: role of injury severity and tissue hypoperfusion. Crit Care (2009) 13:R174. doi:10.1186/cc8152
25. Kim JY, Park JS, Strassheim D, Douglas I, Diaz del Valle F, Asehnoune K, et al. HMGB1 contributes to the development of acute lung injury after hemorrhage. Am J Physiol Lung Cell Mol Physiol (2005) 288:L958–65. doi:10.1152/ajplung.00359.2004
26. van Zoelen MAD, Ishizaka A, Wolthuis EK, Choi G, van der Poll T, Schultz MJ. Pulmonary levels of high-mobility group box 1 during mechanical ventilation and ventilator-associated pneumonia. Shock (2008) 29(4):441–5. doi:10.1097/SHK.0b013e318157eddd
27. Degos V, Maze M, Vacas S, Hirsch J, Guo Y, Shen F, et al. Bone fracture exacerbates murine ischemic cerebral injury. Anesthesiology (2013) 118:1362–72. doi:10.1097/ALN.0b013e31828c23f8
28. Sodhi CP, Jia H, Yamaguchi Y, Lu P, Good M, Egan C, et al. Intestinal epithelial TLR-4 activation is required for the development of acute lung injury after trauma/hemorrhagic shock via the release of HMGB1 from the gut. J Immunol (2015) 194:4931–9. doi:10.4049/jimmunol.1402490
29. Tang D, Kang R, Zeh HJ, Lotze MT. High-mobility group box 1, oxidative stress, and disease. Antioxid Redox Signal (2011) 14:1315–35. doi:10.1089/ars.2010.3356
30. Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med (2012) 209:1519–28. doi:10.1084/jem.20120189
31. Gaston JSH. Heat shock proteins and innate immunity. Clin Exp Immunol (2002) 127:1–3. doi:10.1046/j.1365-2249.2002.01759.x
32. Xu Q. Role of heat shock proteins in atherosclerosis. Arterioscler Thromb Vasc Biol (2002) 22:1547–59. doi:10.1161/01.ATV.0000029720.59649.50
33. Rea IM, McNerlan S, Pockley AG. Serum heat shock protein and anti-heat shock protein antibody levels in aging. Exp Gerontol (2001) 36:341–52. doi:10.1016/S0531-5565(00)00215-1
34. Timmermans K, Kox M, Vaneker M, van den Berg M, John A, van Laarhoven A, et al. Plasma levels of danger-associated molecular patterns are associated with immune suppression in trauma patients. Intensive Care Med (2016) 42:551–61. doi:10.1007/s00134-015-4205-3
35. Aneja R, Odoms K, Dunsmore K, Shanley TP, Wong HR. Extracellular heat shock protein-70 induces endotoxin tolerance in THP-1 cells. J Immunol (2006) 177:7184–92. doi:10.4049/jimmunol.177.10.7184
36. Pockley AG, Shepherd J, Corton JM. Detection of heat shock protein 70 (Hsp70) and anti-Hsp70 antibodies in the serum of normal individuals. Immunol Invest (1998) 27:367–77. doi:10.3109/08820139809022710
37. Pittet J-F, Lee H, Morabito D, Howard MB, Welch WJ, Mackersie RC. Serum levels of Hsp 72 measured early after trauma correlate with survival. J Trauma (2002) 52:611–7; discussion 617.
38. Murphy TJ, Paterson HM, Mannick JA, Lederer JA. Injury, sepsis, and the regulation of Toll-like receptor responses. J Leukoc Biol (2004) 75:400–7. doi:10.1189/jlb.0503233
39. Wang Y, Kelly CG, Singh M, McGowan EG, Carrara AS, Bergmeier LA, et al. Stimulation of Th1-polarizing cytokines, C-C chemokines, maturation of dendritic cells, and adjuvant function by the peptide binding fragment of heat shock protein 70. J Immunol (2002) 169:2422–9. doi:10.4049/jimmunol.169.5.2422
40. Singh-Jasuja H, Scherer HU, Hilf N, Arnold-Schild D, Rammensee H-G, Toes REM, et al. The heat shock protein gp96 induces maturation of dendritic cells and down-regulation of its receptor. Eur J Immunol (2000) 30:2211. doi:10.1002/1521-4141(2000)30:8<2211::AID-IMMU2211>3.0.CO;2-0
41. Foell D, Wittkowski H, Vogl T, Roth J. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol (2007) 81:28–37. doi:10.1189/jlb.0306170
42. Roth J, Teigelkamp S, Wilke M, Grün L, Tümmler B, Sorg C. Complex pattern of the myelo-monocytic differentiation antigens MRP8 and MRP14 during chronic airway inflammation. Immunobiology (1992) 186:304–14. doi:10.1016/S0171-2985(11)80259-7
43. Vogl T, Pröpper C, Hartmann M, Strey A, Strupat K, van den Bos C, et al. S100A12 is expressed exclusively by granulocytes and acts independently from MRP8 and MRP14. J Biol Chem (1999) 274:25291–6. doi:10.1074/jbc.274.36.25291
44. Zwadlo G, Brüggen J, Gerhards G, Schlegel R, Sorg C. Two calcium-binding proteins associated with specific stages of myeloid cell differentiation are expressed by subsets of macrophages in inflammatory tissues. Clin Exp Immunol (1988) 72:510–5.
45. Austermann J, Friesenhagen J, Fassl SK, Petersen B, Ortkras T, Burgmann J, et al. Alarmins MRP8 and MRP14 induce stress tolerance in phagocytes under sterile inflammatory conditions. Cell Rep (2014) 9:2112–23. doi:10.1016/j.celrep.2014.11.020
46. Raabe A, Grolms C, Sorge O, Zimmermann M, Seifert V. Serum S-100B protein in severe head injury. Neurosurgery (1999) 45:477–83. doi:10.1097/00006123-199909000-00012
47. Goyal A, Failla MD, Niyonkuru C, Amin K, Fabio A, Berger RP, et al. S100b as a prognostic biomarker in outcome prediction for patients with severe traumatic brain injury. J Neurotrauma (2013) 30:946–57. doi:10.1089/neu.2012.2579
48. Vos PE, Jacobs B, Andriessen TMJC, Lamers KJB, Borm GF, Beems T, et al. GFAP and S100B are biomarkers of traumatic brain injury: an observational cohort study. Neurology (2010) 75:1786–93. doi:10.1212/WNL.0b013e3181fd62d2
49. Murphy MB, Medvedev AE. Long noncoding RNAs as regulators of Toll-like receptor signaling and innate immunity. J Leukoc Biol (2016) 99:839–50. doi:10.1189/jlb.2RU1215-575R
50. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity (2011) 34:637–50. doi:10.1016/j.immuni.2011.05.006
51. Kagan JC, Medzhitov R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell (2006) 125:943–55. doi:10.1016/j.cell.2006.03.047
52. Adib-Conquy M, Asehnoune K, Moine P, Cavaillon JM. Long-term-impaired expression of nuclear factor-kappa B and I kappa B alpha in peripheral blood mononuclear cells of trauma patients. J Leukoc Biol (2001) 70:30–8. doi:10.1189/jlb.70.1.30
53. Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol (2010) 28:321–42. doi:10.1146/annurev-immunol-030409-101311
54. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity (2012) 36:401–14. doi:10.1016/j.immuni.2012.01.009
55. Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem (2004) 279:7370–7. doi:10.1074/jbc.M306793200
56. Wang H, Vishnubhakat JM, Bloom O, Zhang M, Ombrellino M, Sama A, et al. Proinflammatory cytokines (tumor necrosis factor and interleukin 1) stimulate release of high mobility group protein-1 by pituicytes. Surgery (1999) 126:389–92. doi:10.1016/S0039-6060(99)70182-0
57. Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science (1999) 285:248–51. doi:10.1126/science.285.5425.248
58. Yang H, Wang H, Ju Z, Ragab AA, Lundbäck P, Long W, et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J Exp Med (2015) 212:5–14. doi:10.1084/jem.20141318
59. Youn JH, Oh YJ, Kim ES, Choi JE, Shin JS. High mobility group box 1 protein binding to lipopolysaccharide facilitates transfer of lipopolysaccharide to CD14 and enhances lipopolysaccharide-mediated TNF-production in human monocytes. J Immunol (2008) 180:5067–74. doi:10.4049/jimmunol.180.7.5067
60. Tian J, Avalos AM, Mao S-Y, Chen B, Senthil K, Wu H, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol (2007) 8:487–96. doi:10.1038/ni0707-780b
61. Coveney AP, Wang W, Kelly J, Liu JH, Blankson S, Wu QD, et al. Myeloid-related protein 8 induces self-tolerance and cross-tolerance to bacterial infection via TLR4- and TLR2-mediated signal pathways. Sci Rep (2015) 5:13694. doi:10.1038/srep13694
62. Sharif O, Knapp S. From expression to signaling: roles of TREM-1 and TREM-2 in innate immunity and bacterial infection. Immunobiology (2008) 213:701–13. doi:10.1016/j.imbio.2008.07.008
63. Tammaro A, Derive M, Gibot S, Leemans JC, Florquin S, Dessing MC. TREM-1 and its potential ligands in non-infectious diseases: from biology to clinical perspectives. Pharmacol Ther (2017) 177:81–95. doi:10.1016/j.pharmthera.2017.02.043
64. Weber DJ, Gracon ASA, Ripsch MS, Fisher AJ, Cheon BM, Pandya PH, et al. The HMGB1-RAGE axis mediates traumatic brain injury-induced pulmonary dysfunction in lung transplantation. Sci Transl Med (2014) 6:252ra124. doi:10.1126/scitranslmed.3009443
65. van Beijnum JR, Buurman WA, Griffioen AW. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis (2008) 11:91–9. doi:10.1007/s10456-008-9093-5
66. Vega-Ramos J, Roquilly A, Zhan Y, Young LJ, Mintern JD, Villadangos JA. Inflammation conditions mature dendritic cells to retain the capacity to present new antigens but with altered cytokine secretion function. J Immunol (2014) 193:3851–9. doi:10.4049/jimmunol.1303215
67. Hauser CJ, Sursal T, Rodriguez EK, Appleton PT, Zhang Q, Itagaki K. Mitochondrial damage associated molecular patterns from femoral reamings activate neutrophils through formyl peptide receptors and P44/42 MAP kinase. J Orthop Trauma (2010) 24:534–8. doi:10.1097/BOT.0b013e3181ec4991
68. Wenceslau CF, Szasz T, McCarthy CG, Baban B, NeSmith E, Webb RC. Mitochondrial N-formyl peptides cause airway contraction and lung neutrophil infiltration via formyl peptide receptor activation. Pulm Pharmacol Ther (2016) 37:49–56. doi:10.1016/j.pupt.2016.02.005
69. Huebener P, Pradere J-P, Hernandez C, Gwak G-Y, Caviglia JM, Mu X, et al. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J Clin Invest (2015) 125:539–50. doi:10.1172/JCI76887
70. Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology (2015) 62:600–14. doi:10.1002/hep.27841
71. McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CCM, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science (2010) 330:362–6. doi:10.1126/science.1195491
72. Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, et al. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science (2006) 314:1792–5. doi:10.1126/science.1132559
73. Wenceslau CF, McCarthy CG, Szasz T, Goulopoulou S, Webb RC. Mitochondrial N-formyl peptides induce cardiovascular collapse and sepsis-like syndrome. Am J Physiol Heart Circ Physiol (2015) 308:H768–77. doi:10.1152/ajpheart.00779.2014
74. Hadass O, Tomlinson BN, Gooyit M, Chen S, Purdy JJ, Walker JM, et al. Selective inhibition of matrix metalloproteinase-9 attenuates secondary damage resulting from severe traumatic brain injury. PLoS One (2013) 8:e76904. doi:10.1371/journal.pone.0076904
75. Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med (1999) 5:1249–55. doi:10.1038/15200
76. Kokkola R, Andersson A, Mullins G, Ostberg T, Treutiger C-J, Arnold B, et al. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol (2005) 61:1–9. doi:10.1111/j.0300-9475.2005.01534.x
77. Han J, Zhong J, Wei W, Wang Y, Huang Y, Yang P, et al. Extracellular high-mobility group box 1 acts as an innate immune mediator to enhance autoimmune progression and diabetes onset in NOD mice. Diabetes (2008) 57:2118–27. doi:10.2337/db07-1499
78. Flohé SB, Brüggemann J, Lendemans S, Nikulina M, Meierhoff G, Flohé S, et al. Human heat shock protein 60 induces maturation of dendritic cells versus a Th1-promoting phenotype. J Immunol (2003) 170:2340–8. doi:10.4049/jimmunol.170.5.2340
79. Srivastava P. Roles of heat-shock proteins in innate and adaptive immunity. Nat Rev Immunol (2002) 2:185–94. doi:10.1038/nri749
80. Julian MW, Shao G, Bao S, Knoell DL, Papenfuss TL, VanGundy ZC, et al. Mitochondrial transcription factor A serves as a danger signal by augmenting plasmacytoid dendritic cell responses to DNA. J Immunol (2012) 189:433–43. doi:10.4049/jimmunol.1101375
81. Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, et al. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med (2005) 202:1131–9. doi:10.1084/jem.20050914
82. Marriott I, Inscho EW, Bost KL. Extracellular uridine nucleotides initiate cytokine production by murine dendritic cells. Cell Immunol (1999) 195:147–56. doi:10.1006/cimm.1999.1531
83. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature (2004) 430:213–8. doi:10.1038/nature02664
84. Roquilly A, McWilliam HEG, Jacqueline C, Tian Z, Cinotti R, Rimbert M, et al. Local modulation of antigen-presenting cell development after resolution of pneumonia induces long-term susceptibility to secondary infections. Immunity (2017) 47:135–47.e5. doi:10.1016/j.immuni.2017.06.021
85. Adib-Conquy M, Moine P, Asehnoune K, Edouard A, Espevik T, Miyake K, et al. Toll-like receptor-mediated tumor necrosis factor and interleukin-10 production differ during systemic inflammation. Am J Respir Crit Care Med (2003) 168:158–64. doi:10.1164/rccm.200209-1077OC
86. Roquilly A, Braudeau C, Cinotti R, Dumonte E, Motreul R, Josien R, et al. Impaired blood dendritic cell numbers and functions after aneurysmal subarachnoid hemorrhage. PLoS One (2013) 8:e71639. doi:10.1371/journal.pone.0071639
87. Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature (2007) 447:972–8. doi:10.1038/nature05836
88. Chamorro A, Meisel A, Planas AM, Urra X, van de Beek D, Veltkamp R. The immunology of acute stroke. Nat Rev Neurol (2012) 8:401–10. doi:10.1038/nrneurol.2012.98
89. Liesz A, Hagmann S, Zschoche C, Adamek J, Zhou W, Sun L, et al. The spectrum of systemic immune alterations after murine focal ischemia: immunodepression versus immunomodulation. Stroke (2009) 40:2849–58. doi:10.1161/STROKEAHA.109.549618
90. Urra X, Cervera A, Obach V, Climent N, Planas AM, Chamorro A. Monocytes are major players in the prognosis and risk of infection after acute stroke. Stroke (2009) 40:1262–8. doi:10.1161/STROKEAHA.108.532085
91. Roquilly A, David G, Cinotti R, Vourc’h M, Morin H, Rozec B, et al. Role of IL-12 in overcoming the low responsiveness of NK cells to missing self after traumatic brain injury. Clin Immunol (2017) 177:87–94. doi:10.1016/j.clim.2015.08.006
92. Murphy T, Paterson H, Rogers S, Mannick JA, Lederer JA. Use of intracellular cytokine staining and bacterial superantigen to document suppression of the adaptive immune system in injured patients. Ann Surg (2003) 238:401–10. doi:10.1097/01.sla.0000086661.45300.14
93. O’Sullivan ST, Lederer JA, Horgan AF, Chin DHL, Mannick JA, Rodrick ML. Major injury leads to predominance of the T helper-2 lymphocyte phenotype and diminished interleukin-12 production associated with decreased resistance to infection. Ann Surg (1995) 222:482–92. doi:10.1097/00000658-199522240-00006
94. Hug A, Dalpke A, Wieczorek N, Giese T, Lorenz A, Auffarth G, et al. Infarct volume is a major determiner of post-stroke immune cell function and susceptibility to infection. Stroke (2009) 40:3226–32. doi:10.1161/STROKEAHA.109.557967
95. Liesz A, Rüger H, Purrucker J, Zorn M, Dalpke A, Möhlenbruch M, et al. Stress mediators and immune dysfunction in patients with acute cerebrovascular diseases. PLoS One (2013) 8:e74839. doi:10.1371/journal.pone.0074839
96. Mracsko E, Liesz A, Karcher S, Zorn M, Bari F, Veltkamp R. Differential effects of sympathetic nervous system and hypothalamic-pituitary-adrenal axis on systemic immune cells after severe experimental stroke. Brain Behav Immun (2014) 41:200–9. doi:10.1016/j.bbi.2014.05.015
97. Liesz A, Dalpke A, Mracsko E, Antoine DJ, Roth S, Zhou W, et al. DAMP signaling is a key pathway inducing immune modulation after brain injury. J Neurosci (2015) 35:583–98. doi:10.1523/JNEUROSCI.2439-14.2015
98. Patel N, Wu W, Mishra PK, Chen F, Millman A, Csóka B, et al. A2B adenosine receptor induces protective antihelminth type 2 immune responses. Cell Host Microbe (2014) 15:339–50. doi:10.1016/j.chom.2014.02.001
99. Ryzhov S, Goldstein AE, Matafonov A, Zeng D, Biaggioni I, Feoktistov I. Adenosine-activated mast cells induce IgE synthesis by B lymphocytes: an A2B-mediated process involving Th2 cytokines IL-4 and IL-13 with implications for asthma. J Immunol (2004) 172:7726–33. doi:10.4049/jimmunol.172.12.7726
100. Yang G, Wan M, Zhang Y, Sun L, Sun R, Hu D, et al. Inhibition of a C-rich oligodeoxynucleotide on activation of immune cells in vitro and enhancement of antibody response in mice. Immunology (2010) 131:501–12. doi:10.1111/j.1365-2567.2010.03322.x
101. Noble A, Mehta H, Lovell A, Papaioannou E, Fairbanks L. IL-12 and IL-4 activate a CD39-dependent intrinsic peripheral tolerance mechanism in CD8(+) T cells. Eur J Immunol (2016) 46:1438–48. doi:10.1002/eji.201545939
102. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204:1257–65. doi:10.1084/jem.20062512
103. Boer MC, van Meijgaarden KE, Bastid J, Ottenhoff THM, Joosten SA. CD39 is involved in mediating suppression by Mycobacterium bovis BCG-activated human CD8(+) CD39(+) regulatory T cells. Eur J Immunol (2013) 43:1925–32. doi:10.1002/eji.201243286
104. Li P, Gao Y, Cao J, Wang W, Chen Y, Zhang G, et al. CD39+ regulatory T cells attenuate allergic airway inflammation. Clin Exp Allergy (2015) 45:1126–37. doi:10.1111/cea.12521
105. Karam O, Tucci M, Toledano BJ, Robitaille N, Cousineau J, Thibault L, et al. Length of storage and in vitro immunomodulation induced by prestorage leukoreduced red blood cells. Transfusion (2009) 49:2326–34. doi:10.1111/j.1537-2995.2009.02319.x
106. Pham HP, Shaz BH. Update on massive transfusion. Br J Anaesth (2013) 111(Suppl 1):i71–82. doi:10.1093/bja/aet376
107. Claridge JA, Sawyer RG, Schulman AM, McLemore EC, Young JS. Blood transfusions correlate with infections in trauma patients in a dose-dependent manner. Am Surg (2002) 68:566–72.
108. Chelemer SB, Prato BS, Cox PM Jr, O’Connor GT, Morton JR. Association of bacterial infection and red blood cell transfusion after coronary artery bypass surgery. Association of bacterial infection and red blood cell transfusion after coronary artery bypass surgery. Ann Thorac Surg (2001) 73:138–42. doi:10.1016/S0003-4975(01)03308-2
109. Ghio M, Contini P, Mazzei C, Brenci S, Barberis G, Filaci G, et al. Soluble HLA class I, HLA class II, and Fas ligand in blood components: a possible key to explain the immunomodulatory effects of allogeneic blood transfusions. Blood (1999) 93:1770–7.
110. MacConmara MP, Maung AA, Fujimi S, McKenna AM, Delisle A, Lapchak PH, et al. Increased CD4+ CD25+ T regulatory cell activity in trauma patients depresses protective Th1 immunity. Ann Surg (2006) 244:514–23. doi:10.1097/01.sla.0000239031.06906.1f
111. Koch CG, Li L, Sessler DI, Figueroa P, Hoeltge GA, Mihaljevic T, et al. Duration of red-cell storage and complications after cardiac surgery. Duration of red-cell storage and complications after cardiac surgery. N Engl J Med (2008) 358:1229–39. doi:10.1056/NEJMoa070403
112. Cooper DJ, McQuilten ZK, Nichol A, Ady B, Aubron C, Bailey M, et al. Age of red cells for transfusion and outcomes in critically ill adults. N Engl J Med (2017) 377(19):1858–67. doi:10.1056/NEJMoa1707572
113. Lacroix J, Hebert PC, Fergusson DA, Tinmouth A, Cook DJ, Marshall JC, et al. Age of transfused blood in critically ill adults. N Engl J Med (2015) 372:1410–8. doi:10.1056/NEJMoa1500704
114. European Directorate for the Quality of Medicines & HealthCare. Guide for the Preparation, Use and Quality Assurance of Blood Component. Strasbourg: European Directorate for the Quality of Medicines & HealthCare (2007).
115. Mendonça R, Silveira AAA, Conran N. Red cell DAMPs and inflammation. Inflamm Res (2016) 65:665–78. doi:10.1007/s00011-016-0955-9
116. Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, et al. Characterization of heme as activator of toll-like receptor 4. J Biol Chem (2007) 282:20221–9. doi:10.1074/jbc.M610737200
117. Wegiel B, Hauser CJ, Otterbein LE. Heme as a danger molecule in pathogen recognition. Free Radic Biol Med (2015) 89:651–61. doi:10.1016/j.freeradbiomed.2015.08.020
118. Dzieciatkowska M, Silliman CC, Moore EE, Kelher MR, Banerjee A, Land KJ, et al. Proteomic analysis of the supernatant of red blood cell units: the effects of storage and leucoreduction. Vox Sang (2013) 105:210–8. doi:10.1111/vox.12042
119. Du M, Martin A, Hays F, Johnson J, Farjo RA, Farjo KM. Serum retinol-binding protein-induced endothelial inflammation is mediated through the activation of toll-like receptor 4. Mol Vis (2017) 23:185–97.
120. Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta (2014) 1843:2563–82. doi:10.1016/j.bbamcr.2014.05.014
121. Lott JM, Sumpter TL, Turnquist HR. New dog and new tricks: evolving roles for IL-33 in type 2 immunity. J Leukoc Biol (2015) 97:1037–48. doi:10.1189/jlb.3RI1214-595R
122. Molofsky AB, Savage AK, Locksley RM. Interleukin-33 in tissue homeostasis, injury, and inflammation. Immunity (2015) 42:1005–19. doi:10.1016/j.immuni.2015.06.006
123. Sprague RS, Ellsworth ML. Erythrocyte-derived ATP and perfusion distribution: role of intracellular and intercellular communication. Microcirculation (2012) 19:430–9. doi:10.1111/j.1549-8719.2011.00158.x
124. Jheng H-F, Tsai P-J, Chuang Y-L, Shen Y-T, Tai T-A, Chen W-C, et al. Albumin stimulates renal tubular inflammation through an HSP70-TLR4 axis in mice with early diabetic nephropathy. Dis Model Mech (2015) 8:1311–21. doi:10.1242/dmm.019398
125. Bertheloot D, Latz E. HMGB1, IL-1α, IL-33 and S100 proteins: dual-function alarmins. Cell Mol Immunol (2017) 14:43–64. doi:10.1038/cmi.2016.34
126. Rendy L, Sapan HB, Kalesaran LTB. Multiple organ dysfunction syndrome (MODS) prediction score in multi-trauma patients. Int J Surg Open (2017) 8:1–6. doi:10.1016/j.ijso.2017.05.003
127. Sauaia A, Moore FA, Moore EE, Moser KS, Brennan R, Read RA, et al. Epidemiology of trauma deaths: a reassessment. J Trauma (1995) 38:185–93. doi:10.1097/00005373-199502000-00006
128. Peltz ED, Moore EE, Eckels PC, Damle SS, Tsuruta Y, Johnson JL, et al. HMGB1 is markedly elevated within 6 hours of mechanical trauma in humans. Shock (2009) 32:17–22. doi:10.1097/SHK.0b013e3181997173
129. Wang X-W, Karki A, Zhao X-J, Xiang X-Y, Lu Z-Q. High plasma levels of high mobility group box 1 is associated with the risk of sepsis in severe blunt chest trauma patients: a prospective cohort study. J Cardiothorac Surg (2014) 9:133. doi:10.1186/s13019-014-0133-5
130. Deng Y, Yang Z, Gao Y, Xu H, Zheng B, Jiang M, et al. Toll-like receptor 4 mediates acute lung injury induced by high mobility group box-1. PLoS One (2013) 8:e64375. doi:10.1371/journal.pone.0064375
131. Kalanuria A, Zai W, Mirski M. Ventilator-associated pneumonia in the ICU. Crit Care (2014) 18:208. doi:10.1186/cc13775
132. de Lange DW, Bonten MJM. Can we predict septic shock in patients with hospital-acquired pneumonia? Crit Care (2005) 9:640–1. doi:10.1186/cc3919
133. Kesinger MR, Kumar RG, Wagner AK, Puyana JC, Peitzman AP, Billiar TR, et al. Hospital-acquired pneumonia is an independent predictor of poor global outcome in severe traumatic brain injury up to 5 years after discharge. J Trauma Acute Care Surg (2015) 78:396–402. doi:10.1097/TA.0000000000000526
134. Ito T. PAMPs and DAMPs as triggers for DIC. J Intensive Care (2014) 2:67. doi:10.1186/s40560-014-0065-0
135. Kim J-E, Lee N, Gu J-Y, Yoo HJ, Kim HK. Circulating levels of DNA-histone complex and dsDNA are independent prognostic factors of disseminated intravascular coagulation. Thromb Res (2015) 135:1064–9. doi:10.1016/j.thromres.2015.03.014
136. Altincicek B, Stötzel S, Wygrecka M, Preissner KT, Vilcinskas A. Host-derived extracellular nucleic acids enhance innate immune responses, induce coagulation, and prolong survival upon infection in insects. J Immunol (2008) 181:2705–12. doi:10.4049/jimmunol.181.4.2705
137. Ammollo CT, Semeraro F, Xu J, Esmon NL, Esmon CT. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J Thromb Haemost (2011) 9:1795–803. doi:10.1111/j.1538-7836.2011.04422.x
Keywords: damage-associated molecular pattern, posttraumatic immunosuppression, remote organ dysfunction, hospital-acquired infection, critical care, transfusion
Citation: Vourc’h M, Roquilly A and Asehnoune K (2018) Trauma-Induced Damage-Associated Molecular Patterns-Mediated Remote Organ Injury and Immunosuppression in the Acutely Ill Patient. Front. Immunol. 9:1330. doi: 10.3389/fimmu.2018.01330
Received: 30 January 2018; Accepted: 28 May 2018;
Published: 15 June 2018
Edited by:
Julien Pottecher, Hôpitaux Universitaires de Strasbourg, FranceReviewed by:
Angelo A. Manfredi, Università Vita-Salute San Raffaele, ItalyBeate E. Kehrel, University Hospital Muenster, Germany
Copyright: © 2018 Vourc’h, Roquilly and Asehnoune. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karim Asehnoune, a2FyaW0uYXNlaG5vdW5lQGNodS1uYW50ZXMuZnI=
Hosted by Drs. Julien Pottecher, Walter Gottlieb Land, Carl Jeffrey Hauser, Alain Meyer, Camilla Ferreira Wenceslau, Kim Timmermans, Frontiers in Immunology, Section Inflammation.