 Nicolas Gaudenzio
Nicolas Gaudenzio Thomas Marichal
Thomas Marichal Stephen J. Galli5,6,7
Stephen J. Galli5,6,7 Laurent L. Reber
Laurent L. Reber- 1Unité de Différenciation Epithéliale et Autoimmunité Rhumatoïde (UDEAR), UMR 1056, INSERM, Université de Toulouse, Toulouse, France
- 2Laboratory of Cellular and Molecular Immunology, GIGA Institute, Liege University, Liège, Belgium
- 3Faculty of Veterinary Medicine, Liege University, Liège, Belgium
- 4WELBIO, Walloon Excellence in Life Sciences and Biotechnology, Wallonia, Belgium
- 5Department of Pathology, Stanford University School of Medicine, Stanford, CA, United States
- 6Department of Immunology and Microbiology, Stanford University School of Medicine, Stanford, CA, United States
- 7Sean N. Parker Center for Allergy and Asthma Research, Stanford University School of Medicine, Stanford, CA, United States
- 8Unit of Antibodies in Therapy and Pathology, INSERM Unit 1222, Department of Immunology, Institut Pasteur, Paris, France
Contact hypersensitivity (CHS) is a common T cell-mediated skin disease induced by epicutaneous sensitization to haptens. Mast cells (MCs) are widely deployed in the skin and can be activated during CHS responses to secrete diverse products, including some with pro-inflammatory and anti-inflammatory functions. Conflicting results have been obtained regarding pathogenic versus protective roles of MCs in CHS, and this has been attributed in part to the limitations of certain models for studying MC functions in vivo. This review discusses recent advances in the development and analysis of mouse models to investigate the roles of MCs and MC-associated products in vivo. Notably, fluorescent avidin-based two-photon imaging approaches enable in vivo selective labeling and simultaneous tracking of MC secretory granules (e.g., during MC degranulation) and MC gene activation by real-time longitudinal intravital microscopy in living mice. The combination of such genetic and imaging tools has shed new light on the controversial role played by MCs in mouse models of CHS. On the one hand, they can amplify CHS responses of mild severity while, on the other hand, can limit the inflammation and tissue injury associated with more severe or chronic models, in part by representing an initial source of the anti-inflammatory cytokine IL-10.
Introduction
Allergic contact dermatitis (ACD), and its animal model contact hypersensitivity (CHS), are T cell-mediated skin inflammatory diseases caused by delayed-type hypersensitivity responses to environmental allergens (1–3). An important group of contact allergens are small organic compounds that rapidly penetrate into the skin and bind to proteins in the dermis, a process called ‘‘haptenization’’ that alters self-proteins and renders them antigenic. Sensitization to haptens is characterized by the activation of dendritic cells (DCs), which migrate to the skin-draining lymph nodes, and by the priming of allergen specific T cells. Re-exposure of the skin to the same hapten leads to development of a type of delayed-type hypersensitivity reaction to that antigen mediated by T cells (3). Besides T cells, other immune cell populations, including neutrophils (4), natural killer (NK) cells (5), and innate lymphoid cells (6), can influence the magnitude and duration of CHS responses. This can be particularly important during severe CHS reactions, in which the immune system must dampen the intensity of the pathology in order to maintain optimal tissue integrity.
Mast cells (MCs) are widely deployed in many tissues, including the skin, and can represent powerful sentinels of the immune system (7–10). MCs can be stimulated via the high-affinity receptor for IgE (FcεRI), or by any of multiple other mechanisms [including activation by the KIT ligand stem cell factor (SCF), immune complexes of IgG, various complement peptides, cytokines and chemokines], leading to the release a diverse spectrum of biologically active mediators, including some with pro- or anti-inflammatory functions (9, 11). As a result, MCs can have potentially important effector or immunoregulatory functions during inflammatory processes, including during the sensitization and effector phases of CHS responses.
Different sophisticated mouse models and fluorescent avidin-based imaging tools can now be used to study MC functions in vivo and to visualize the dynamics of MCs, the release of MC granules, and MC gene activation using intravital two-photon microscopy. The combined use of such genetic and imaging tools has shed new light on how skin MCs, on the one hand, can amplify CHS responses of mild severity while, on the other hand, can limit the inflammation and tissue injury associated with more severe or chronic models, in part by representing an initial source of the anti-inflammatory cytokine IL-10.
Mouse Models to Investigate the Roles of MCs and Mast Cell-Associated Products In Vivo
In order to investigate the potential contributions of MCs or MC products in particular biological settings, the “holy grail” would be to be able to solely and selectively deplete MCs or MC products in vivo (12, 13). Enormous progress has been made to reach this goal since the discovery by Kitamura and colleagues that WBB6F1-KitW/KitW-v mice, hereafter named KitW/W-v mice, were profoundly deficient in MCs (14). KitW/W-v and C57BL/6-KitW-sh/KitW-sh mice, hereafter named KitW-sh/W-sh mice, are the two most common strains of MC-deficient mice with abnormalities affecting KIT, the receptor for the main MC growth and survival factor, SCF (15, 16). These mice are also generally called “Kit mutant” mice. Kit-mutant mice not only exhibit a profound MC deficiency but also a variety of other phenotypic abnormalities and have been widely used to analyze the functions of MCs in vivo (11, 17–20). Since differences in the biological responses in Kit mutant mice compared with wild-type (WT) mice may not be solely due to their deficiency in MCs, we and others have used “MC knock-in mice” to assess the importance of MCs in regulating the expression of biological responses in Kit-mutant mice. Such an approach consists of restoring the MC deficiency in Kit-mutant mice by adoptively transferring genetically compatible, in vitro-derived WT or mutant MCs (10, 18, 19, 21, 22). This approach can be helpful, but it may not be possible to prove that such adoptively transferred MCs are fully identical (in either anatomical location, phenotype, or function) to those in the same anatomical location as in the corresponding WT mice (13, 18, 23).
In addition to these Kit-mutant mice, newer models in which the MC deficiency is not dependent on mutations affecting the structure or expression of KIT have emerged (Table 1). They consist of genetically modified mice that exhibit constitutive deficiencies (Cpa3Cre/+, Cpa3-Cre+; Mcl-1fl/fl, Mcpt5-Cre+; DTAfl/+) or diphtheria toxin-inducible deficiencies [Mcpt5-Cre; iDTRfl/+, “Mas-TRECK,” “RMB” (these mice are described in detail in the section entitled The Red Mast cell and Basophil mouse)] in populations of MCs. In some of such models, there are also abnormalities in numbers and functions of basophils (24–29).
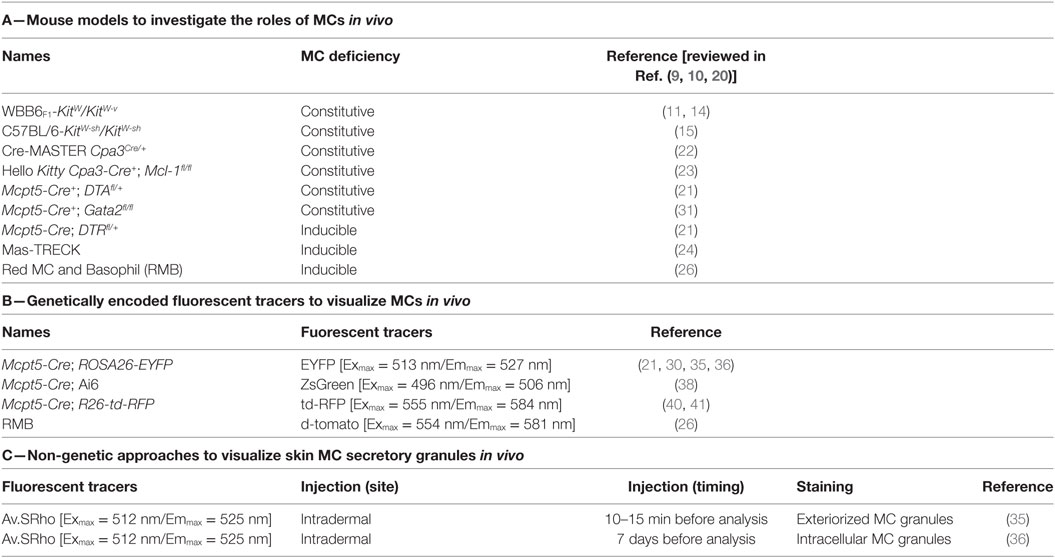
Table 1. Genetic and imaging approaches to study mast cell (MC) functions in vivo.
As reviewed in detail elsewhere (12, 13, 23), each model currently available has limitations that must be kept in mind when interpreting the results of studies using such mice, and the importance of particular MC (or basophil) roles in individual biological responses may vary both according to the details of the model used to study that biological response and based on the strain background of the mice. Analyzing to what extent MC-associated products contribute to a particular biological response requires that the gene coding for such molecule would be specifically deleted/inactivated in MCs. In this regard, “MC-specific CRE” mice, which express CRE recombinase under the control of a promoter specific for MCs (26, 30–33), may allow for specific deletion of floxed genes in MCs. Interestingly, Li et al. recently used this approach to delete the transcription factor GATA2 in connective tissue-type MCs (CTMCs) by crossing Gata2fl/fl mice with Mcpt5-Cre mice [which express the CRE recombinase in connective tissue MCs (these mice are discussed in detail in part 4.1, below)] (34). The authors found that Mcpt5-Cre+; Gata2fl/fl mice exhibit a nearly complete deficiency in CTMCs in the skin, stomach, trachea, and peritoneal cavity, while having comparable number of basophils, T cells, B cells, NK cells, neutrophils, and macrophages (34). These mice thus represent another useful model of constitutive deficiency in CTMCs.
The Use of Genetically Encoded Fluorescent Tracers to Visualize MCs In Vivo
The Mcpt5 Cre/loxP Reporter System
The Mcpt5 gene encodes the mouse MC protease 5, also known as α-chymase, that is predominantly detected in connective tissue MCs [i.e., mostly skin and peritoneal MCs (PMCs)] (35). In 2010, Scholten and coworkers reported the generation of the Mcpt5-Cre mouse strain in which a modified iCre gene [i.e., encoding an improved CRE recombinase (36)] cassette was strategically inserted into the Mcpt5 gene (33). Importantly, compared to the Cpa3-Cre mouse strain reported by Feyerabend et al. in 2011 in which the targeted insertion of iCre gene into the carboxypeptidase A3 (Cpa3) locus deleted MCs via a genotoxic mechanism (25), the Mcpt5-Cre mouse did not show any signs of CRE-mediated genotoxicity. The Mcpt5-Cre mice were bred with a ROSA26_Enhanced Yellow Fluorescent Protein (EYFP) reporter strain, in which the gene encoding EYFP [a yellow fluorescent tracer (Exmax = 513 nm/Emmax = 527 nm)] has been placed under the control of the ubiquitous ROSA26 promoter flanked by loxP stop elements (37). The resulting Mcpt5-Cre; ROSA26-EYFP, thereafter named Mcpt5-EYFP, double transgenic mice expressed EYFP fluorescence signal specifically in peritoneal and skin MCs (33). These Mcpt5-EYFP mice then were used in different studies by our (38, 39) and other (24) groups in order to monitor the behavior of dermal MCs during inflammatory reactions using intravital two-photon systems in living mice. As a first example of such an application, Dudeck et al. used the Mcpt5-EYFP mice to visualize and monitor simultaneously perivascular EYFP+ dermal MCs and Qdot-labeled blood vessels by intravital two-photon microscopy in a model of moderate CHS. Upon induction of the disease, they reported that EYFP+ perivascular MCs exhibited changes in their morphology in parallel with the dilatation of blood vessels and vascular leakage of Qdots into the dermis (24).
Mcpt5-Cre mice were also bred with ROSA26-ZsGreen mice (40) [i.e., also called Ai6 mice, expressing the Zoanthus sp. Green fluorescent protein (Exmax = 496 nm/Emmax = 506 nm) with a targeted insertion of a construct containing the strong and ubiquitous CAG promoter in the ROSA26 locus] (41). Compared to the previously described ROSA-EYFP mouse, the Ai6 mouse has been reported to express a stronger fluorescence signal and is thought to be more appropriate to visualize discrete cellular projections in vivo. Using the Mcpt5-Cre; Ai6 double transgenic mice and intravital two-photon microscopy, the authors showed that ZsGreen+ skin MCs can “sample” circulating IgE by extending cell processes across the vessel wall (41).
Recently, Dudeck and colleagues mated Mcpt5-Cre mice with R26-td-RFP reporter mice (42), i.e., expressing the tandem dimer_Red Fluorescent Protein [Exmax = 555 nm/Emmax = 584 nm], under the control of the ROSA26 promotor. The resulting Mcpt5-Cre; R26-td-RFP, also called Mcpt5-RFP, double transgenic mice were expressing the RFP specifically in peritoneal and skin MCs (43). In this study, the authors then bred the Mcpt5-RFP mice with the DC reporter strain Cd11c-EGFP (44), i.e., expressing the Enhanced Green Fluorescent Protein [Exmax = 488 nm/Emmax = 509 nm] under the control of the Cd11c gene promotor, and used the Mcpt5-RFP; Cd11c-EGFP triple transgenic mice to track simultaneously RFP+ MCs and EGFP+ DCs (the emission wavelengths of RFP and EGFP fluorescence being far enough apart to be analyzed simultaneously) in the skin of living mice. Using such conditions, they reported that MCs and DCs can exchange membrane proteins in a model of moderate hapten-induced CHS, as discussed in more detail in the section Pro-inflammatory functions of MCs in CHS (43). We will discuss below a potential alternative to the time-consuming and costly approach consisting of the generation of double or triple transgenic animals. This approach permits labeling and monitoring of exteriorized or intracellular skin MC granules using intravital imaging.
The Red MC and Basophil Mouse
In 2014, Dahdah and coworkers described a new knock-in mouse model named the “Red MC and Basophil” (RMB) mouse (29). The RMB mouse is based on the insertion of a cassette composed of an internal ribosomal entry site, a sequence coding for the protein red tandem dimer-Tomato, i.e., tdT, a genetically encoded red fluorescent tracer [Exmax = 554 nm/Emmax = 581 nm] (45), a 2A cleavage sequence, and the human diphtheria toxin receptor (DTR) in the 3′-UTR of the Ms4a2 gene [encoding the FcεRIβ chain that is reported to be specifically expressed in MCs and basophils (46, 47)]. This mouse model allows the tracking of MCs and basophils based on their expression of tdT red fluorescence and render both cell types vulnerable to diphtheria toxin treatment. Using ex vivo flow cytometry, the authors reported that FcεRI+KIT+ PMCs and FcεRI+KIT-CD49b+ blood and splenic basophils were highly positive for the tdT and that two intraperitoneal injections of diphtheria toxin were sufficient to deplete both cell types, including FcεRI+KIT+ skin MCs. Importantly, diphtheria toxin treatment of RMB mice apparently did not affect other tested blood cell populations, including red blood cells, CD19+ B cells, CD4+ and CD8+ T cells, neutrophils, eosinophils, and monocytes (29). They also showed that tdT+ MCs and basophils exhibited distinct kinetics of repopulation in tissue upon diphtheria toxin treatment. While the percentage of blood tdT+ basophils was back to normal at day 12 posttreatment, only 50% of the tdT+ PMC population was restored at 6 months posttreatment. Therefore, the RMB mouse could be used either as a model of both basophil- and MC-deficient mouse when used at day 6 posttreatment with diphtheria toxin, or as a model of basophil-sufficient but MC-deficient mouse when used at day 12 posttreatment. In this study, the authors reported that MCs were detrimental to survival in a model of severe sepsis by impairing peritoneal macrophages phagocytosis (29). Finally, this model could also potentially be used to monitor either basophil or MC behavior in different tissues, including the skin, using intravital imaging microscopy.
The Use of New Non-Genetic Approaches to Visualize MC Secretory Granules In Vivo
Dynamic Analysis of Exteriorized MC Granules In Vitro and In Vivo
A principal characteristic of MCs is their capacity to respond to a broad panel of activation signals and rapidly exteriorize intracellular secretory granules enriched in pre-stored bioactive molecules, e.g., histamine, proteases, TNF, etc., during inflammatory reactions and influence ongoing immune response (7, 9, 48, 49). Although important progress has been made in efforts to localize and track MCs in vivo, i.e., Cre/LoxP or RMB reporter mice, the lack of in vivo MC granule-specific tracking systems has limited the analysis and the understanding of the dynamics and quantitative characteristics of MC granule exteriorization strategies.
To circumvent those constraints, we developed a new approach that permits the monitoring, in real-time and at the single-cell level, of the spatially complex, rapidly evolving features of MCs undergoing activation using both real-time confocal microscopy in vitro or non-invasive two-photon microscopy in living mice (38, 50) (Figure 1A). The matrix of MC granules is composed of abundant proteoglycans, in CTMC consisting mostly of heparin and its core peptide serglycin, a highly anionic macromolecular complex in which MC bioactive mediators are embedded (51, 52). Fluorochrome-labeled avidin [a cationic molecule with a very high-affinity for heparin (53)] can be used to detect exteriorized MC granule structures in vitro and in vivo (38, 50, 54). As an example of the in vivo application of this method, we found that substance P [predominately via the MC receptor for cationic molecules MrgprB2 (55)] induced dermal MCs rapidly to secrete small and relatively spherical granule structures, a pattern consistent with the secretion of individual granules (38). Conversely, activating dermal MCs with IgE/antigen (via FcεRI) increased the time partition between signaling and secretion, which was associated with the formation of larger and more heterogeneously shaped granule structures that underwent prolonged exteriorization. IgE/antigen- and substance P-dependent activation in vivo were also associated with distinct local and systemic pathophysiological responses (38).
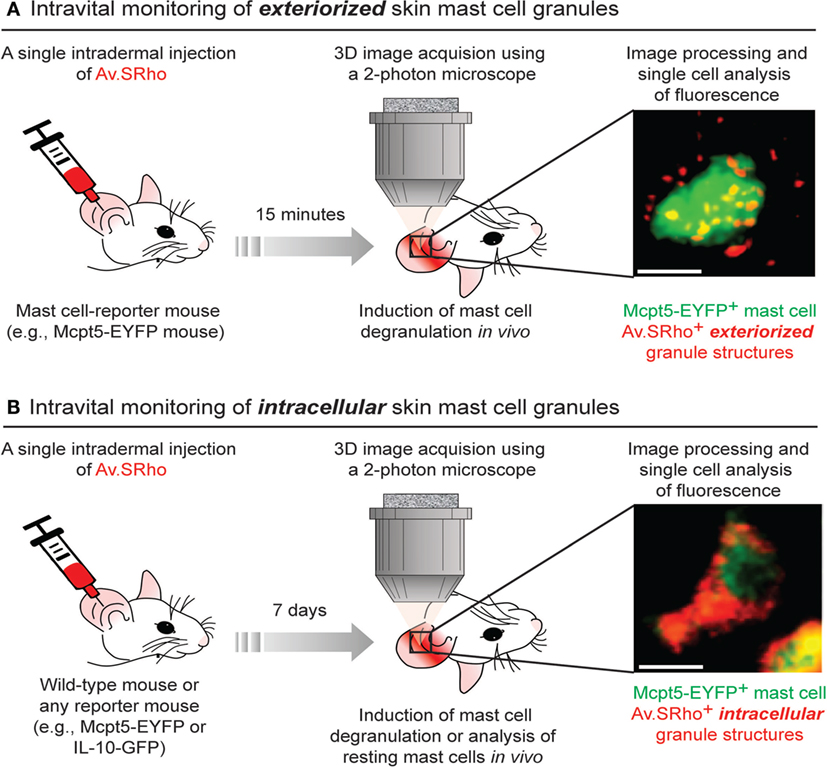
Figure 1. Fluorochrome-labeled avidin can be used to probe either exteriorized or intracellular mast cell (MC) granule structures in living mice. (A) 5 µg of sulforhodamine 101-coupled avidin (Av.SRho) is injected intradermally (i.d.) into the ear pinna of a MC-reporter mouse (e.g., Mcpt5-EYFP). 15 min later, the mouse is anesthetized and placed under a two-photon microscope; MC degranulation is induced by i.d. injection of stimulus; 3D high-resolution images of single cells are taken and Av.SRho fluorescence signal is assessed. The photograph shows a single dermal EYFP+ MC activated upon i.d. injection of substance P exhibiting Av.SRho + exteriorized small granule structures. Image extracted from Gaudenzio et al. (38). (B) 5 µg of Av.SRho is injected i.d. into the ear pinna of a mouse. 7 days later, the mouse is anesthetized and placed under a two-photon microscope; 3D high-resolution single-cell images are taken and Av.SRho fluorescence signal is assessed. The photograph shows a single EYFP+ MC exhibiting Av.SRho+ intracellular granule structures. Image extracted from Reber et al. (39).
Using Fluorochrome-Labeled Avidin to Probe Intracellular MC Granules in Living Mice
We next investigated whether we could also use fluorochrome-labeled avidin to specifically label intracellular MC granules in vivo and use this probe as a non-genetically encoded MC tracer in the skin of living mice. To this end, we injected fluorochrome-labeled avidin into the ear dermis of mice and analyzed the location and intensity of the avidin fluorescence signal 7 days after the injection. Surprisingly, we found that fluorochrome-labeled avidin was located, apparently nearly exclusively, within dermal MC intracellular secretory granules and that such “fluorescent MC granules” could be visualized for many weeks in the same animal using longitudinal intravital two-photon microscopy (Figure 1B).
Therefore, this approach can be employed regardless of the mouse genetic background, as long as the studied mouse strain exhibits negatively charged proteoglycans in MC granules, and enables the direct visualization of skin MC granule content for a prolonged period of time. If combined with a reporter strain, it also permits the longitudinal monitoring of gene activation in such labeled MCs in living mice (39) (Figure 1B).
Role of MCs in CHS: A Long Controversy
The potential role of MCs in CHS has been assessed for almost 30 years, and most of the results found in the literature might make one conclude that their actual contribution remains to be clarified. Indeed, the use of different MC-deficient animals in several experimental conditions has suggested either a positive immunomodulatory role (24, 27, 39, 56–60), a negative immunomodulatory role (24, 39, 60–63), or no contribution (64–66) for MCs in the CHS models and conditions tested.
Although the reasons for such discrepant results are not yet fully understood, it is clear that the outcome of the CHS response and the subsequent interpretation of the results might be influenced by numerous factors, including the type of mutant MC-deficient mice, hapten and protocol tested. Last but not least, unreported and more unpredictable variables can come from the animals, e.g., gender, age, and microbiome/pathogen status, and the housing conditions. Several studies indeed suggest that the commensal microbiota may substantially influence both the immune homeostasis and the behavior of particular hematopoietic cells (67–69).
Dudeck et al. first assessed the influence of the type of MC-deficient mice used (24). Consistent with a previous report (61), they observed that DNFB-sensitized KitW-sh/W-sh and KitW/W-v MC-deficient mice exhibited enhanced ear swelling after DNFB challenge. However, they found that, in the same model, the ear swelling was significantly reduced when MCs were depleted by injection of DT in Mcpt5-Cre; iDTR mice (24). Since Kit mutant mice have many phenotypical abnormalities beside their MC deficiency (18, 20, 70–73), the authors concluded that the exaggerated CHS responses in these mice is not caused by the absence of MCs but is related to the KIT deficiency (24). In striking contrast with this, we recently reported that both KitW-sh/W-sh mice and the Kit-independent Cpa3-cre+; Mcl1fl/fl; and Mcpt5-Cre+; DTA MC-deficient strains have significantly enhanced ear swelling and epidermal hyperplasia compared to the values in their respective littermate controls in a severe CHS model induced by DNFB (39). However, the KitW-sh/W-sh mice exhibited a more pronounced increase in ear swelling in this model as compared to Kit-independent MC-deficient mice.
Using a chronic model of oxazolone-induced CHS, Gimenez-Rivera and colleagues also recently reported increased CHS responses in both KitW-sh/W-sh mice and Kit-independent Mcpt5-Cre+; iDTR MC-deficient mice (63). Similarly to us, the authors noticed that the responses were more pronounced in KitW-sh/W-sh mice, since these mice but not the Kit-independent strain, showed skin scaling and necrosis at the site of chronic CHS reactions (63). We think that the simplest interpretation of these results is that MCs can have effects that can significantly limit the assessed features of these severe and chronic CHS models in each of the examined mouse strains, but that additional phenotypic abnormalities in KitW-sh/W-sh mice, beside their MC deficiency, probably also contribute to the exacerbation of severe CHS responses in this strain, as suggested by Dudeck et al. (24).
The influence of the dose of haptens used for sensitization and/or challenge was studied in detail in KitW/W-v mice (60). The authors reported that the ear swelling and the recruitment of leukocytes was reduced in KitW/W-v mice as compared to Kit+/+ mice in a CHS model consisting of sensitization with a low dose of oxazolone, and challenge with the same hapten. These values were restored to levels observed in Kit+/+ mice after intradermal engraftment of bone marrow-derived cultured MCs (i.e., BMCMCs) into the ear pinnae of KitW/W-v mice (60). However, when using a higher dose of oxazolone for the sensitization, the authors found opposite results, i.e., significantly increased ear swelling in MC-deficient KitW/W-v mice as compared to both Kit+/+ mice and KitW/W-v mice engrafted with BMCMCs (60). These results suggest that, depending on the severity of the CHS model used, MCs can have either a positive or a negative immunomodulatory role. We recently confirmed this using a different hapten (DNFB) and Kit-independent Cpa3-cre+; Mcl1fl/fl MC-deficient mice, indicating that the plasticity of MC responses in CHS models is probably not due solely to KIT-related phenotypic abnormalities and does not depend on the type of hapten used (39).
Potential Mechanisms of MC Activation in CHS
Multiple lines of evidence indicate that skin MCs are activated both during the sensitization and challenge phases in CHS models (Figure 2). Skin exposure to haptens induces innate immune responses which could directly or indirectly activate MCs. Indeed, Dudeck et al. reported MC degranulation in skin sections obtained 4 h after sensitization with DNFB (59). A recent report also shows that DNFB, but not oxazolone, can induce direct degranulation of rat PMCs in vitro (74). MC activation has been best visualized in the effector phase using two-photon microscopy. First, Dudeck et al. used Mcpt5-EYFP reporter mice to demonstrate that perivascular MCs exhibit important changes in their morphology upon hapten challenge (24). More recently, we precisely monitored MC degranulation in a severe DNFB-induced CHS models using fluorescently labeled avidin to stain MC intracellular granules (39). Before DNFB challenge, only ~10% of dermal MCs were marginally degranulated, exhibiting just a few fluorescent avidin+ granules outside of the cell. However, as early as 1 day after DNFB challenge, ~60% of dermal MCs were extensively degranulated and this number reached ~90% at day 2 (39).
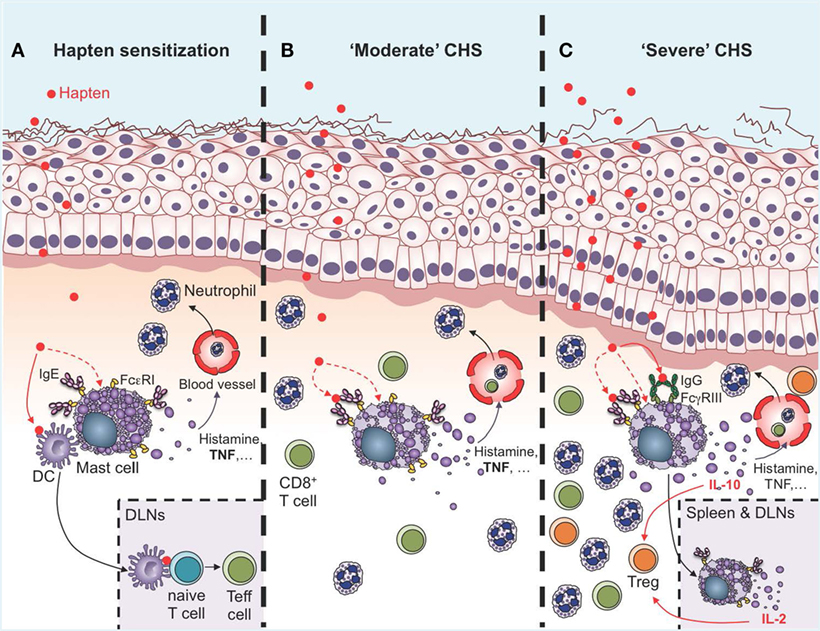
Figure 2. Roles of mast cells (MCs) in the sensitization and effector phases of contact hypersensitivity (CHS). (A) During the sensitization phase, MCs are activated directly or indirectly by haptens to release of a diverse spectrum of mediators, including histamine and TNF, which induce vasodilatation and recruitment of leukocytes, mainly neutrophils. Multiple lines of evidence suggest that IgE can amplify MC activation in the sensitization phase though antigen-independent cytokinergic effects. Direct cell-to-cell contacts between MCs and dendritic cells (DCs), as well as MC-derived TNF, can amplify DC migration to the draining lymph nodes (DLNs), where these cells prime naïve T cells to become effector cells (Teff cells) via antigen presentation. (B) During “moderate” CHS responses, MCs and MC-derived TNF amplify ear swelling, leukocyte recruitment (mainly neutrophils and CD8+ T cells), and epidermal hyperplasia. Evidence suggests that IgE and FcεRI can amplify the pro-inflammatory functions of MCs in moderate CHS (C). During more severe (and chronic) CHS responses, MCs represent an early source of IL-10 in the skin, which amplifies recruitment of regulatory T cells (Treg) and limits ear swelling and epidermal hyperplasia. MC activation in the skin is amplified by the engagement of FcγRIII by hapten-IgG immune complexes. Additionally, MCs can migrate to the DLNs and the spleen in an IgE-dependent manner, where they produce IL-2 which helps in maintaining the Teff:Treg ratio at the site of inflammation, and thereby contributes to limiting the severity of CHS responses.
How MCs are activated upon challenge in CHS models is not fully understood. One possibility could be that, similarly to what is observed during immediate hypersensitivity reactions in the skin, activation of MCs in CHS could require crosslinking of antigen-specific IgE bound to FcεRI on the surface of MCs. Indeed, mice deficient for IgE have reduced ear swelling and leukocyte infiltration in models of CHS to oxazolone or DNFB (58). Surprisingly however, contact sensitivity to oxazolone in IgE−/− mice could be restored by administration of hapten-irrelevant IgE before sensitization (58). The authors obtained similar results after transfer of four different IgE clones directed against dinitrophenyl or trinitrophenyl. Interestingly, two of these IgE clones (SPE-7 and C38-2) were previously shown to be cytokinergic, that is, to induce the release of cytokines in MCs in an antigen-independent manner (75, 76). Bryce and colleagues found that exposure to oxazolone upregulates mRNA levels for the MC-specific gene Mcpt6, and for genes encoding several cytokines and chemokines required for efficient sensitization. mRNA levels for most of these genes, including Mcpt6, were reduced in oxazolone-treated IgE−/− mice, and restored by prior administration of IgE of irrelevant specificity (58). Taken together, these results suggest that IgE can control sensitization though antigen-independent cytokinergic effects on MCs in this CHS model (58) (Figure 2A). More recently, Kobayashi and colleagues also reported reduced ear swelling and leukocyte infiltration in mice deficient for the β chain of FcεRI in a CHS model to oxazolone (77). In the same model, administration of a recombinant soluble form of human FcεRI, which binds both human and mouse IgE, after oxazolone challenge also significantly reduced CHS responses, suggesting that IgE could amplify MC activation in the effector phase of this CHS model (77).
Mast cells express the activating IgG receptor FcγRIII and could thus also be activated by IgG immune complexes during CHS responses. Evidence of this has been reported by Grimbaldeston et al. in a model of severe CHS, and will be discussed below in the section Pro-inflammatory functions of MCs in CHS in more detail (61). However, it should be noted that whether, and in specifically what circumstances, antibodies (IgE or IgG) play a significant role in CHS is still a matter of debate. First, CHS is largely considered to be mediated by CD8+ T cells, which are primed in lymphoid organs during the sensitization phase and recruited in the skin upon re-exposure with the hapten (3). Second, in 2006, O’Leary et al. reported normal CHS responses to DNFB and oxazolone in two types of T cell- and B cell-deficient mice, i.e., Rag2−/− and SCID mice (5). CHS reactions in these T and B cell-deficient mice still required a sensitization phase, and the authors discovered that such antibody-independent adaptive immune response to haptens was mediated by NK cells (5).
Pro-Inflammatory Functions of MCs in CHS
Data obtained by independent groups using both Kit mutant and Kit-independent MC-deficient mice have confirmed that MCs can have important pro-inflammatory functions in CHS models of moderate severity (24, 27, 39, 56–60) (Figure 2B). MCs can amplify neutrophil recruitment at the site of hapten exposure both during the sensitization phase (4), and after challenge (24, 57). Interestingly, recruitment of neutrophils seems to be a key step for hapten sensitization, and neutrophil-depleted mice exhibit decreased migration of DCs to the local draining lymph nodes (DLNs) and reduced contact allergen-induced T cell priming (4). Recent evidence indicates that MCs might also interact directly with DCs during CHS responses. Dudeck et al. used Mcpt5-RFP; Cd11c-EGFP triple transgenic mice to track simultaneously RFP+ MCs and EGFP+ DCs in the skin of living mice during CHS responses using intravital two-photon microscopy (43). Using this approach, they observed targeted cell-to-cell interactions between MCs and DCs. Skin DCs dynamically “scanned” MCs at early time-points of CHS responses, while at a later stage, long-lasting MC–DC interactions occurred, with evidence of DC-to-MC molecule transfers, including major histocompatibility complex class II (MHCII) proteins. The authors also provided evidence that such transfer of MHCII-conferred antigen-presenting capability to MCs enabled MCs to prime allogenic T cells in this model of moderate CHS (43).
While direct cell-to-cell contacts likely are involved in the pro-inflammatory functions of MCs in CHS, as suggested in this study (43), soluble factors released by MCs might also potentiate the inflammatory response and leukocyte recruitment in CHS. Among these, TNF is a key mediator in hapten-induced CHS responses (78). MCs represent a potential source of both pre-formed and de novo synthesized TNF (79–82). Biedermann and colleagues first demonstrated the importance of MC-derived TNF in CHS by showing that MC-deficient KitW/W-v mice engrafted locally with TNF−/− BMCMCs have reduced ear swelling and neutrophil recruitment in the skin following challenge with the hapten TNCB as compared to KitW/W-v mice engrafted with WT BMCMCs (57). Using a similar approach in KitW-sh/W-sh mice, it was latter demonstrated that MC-derived TNF is also required for optimal expression of oxazolone- and FITC-induced CHS (83, 84). In these studies, the authors reported that MCs and MC-derived TNF can promote nerve fiber elongation in the epidermis and dermis (83) as well as DC migration to the DLNs (84). More recently, the importance of MC-derived TNF in CHS was ascertained using Mcpt5-Cre+; TNFfl/fl, in which TNF is deleted specifically in connective tissue MCs (59). In this elegant study, the authors first used single-cell PCR in FACS-sorted peritoneal cells to demonstrate that the Tnf gene was efficiently deleted in MCs, but not in B cells and macrophages from Mcpt5-Cre+; TNFfl/fl mice (59). They then confirmed the role of MC-derived TNF in CHS by showing that Mcpt5-Cre+; TNFfl/fl mice have reduced ear swelling, as well as neutrophil and CD8+ T cell infiltration in the skin, following hapten challenge in a model of moderate DNFB-induced CHS (59). Confirming previous findings by Suto et al. (84), Mcpt5-Cre+; TNFfl/fl mice also had reduced migration of DCs from inflamed skin to skin-DLNs as compared to their littermate controls in this CHS model (84).
Besides TNF, MCs can produce and release a variety of pro-inflammatory factors which might contribute to CHS responses. Among these, immunohistochemistry data indicate that skin MCs produce CXCL2 (MIP-2) during CHS responses, and it was further reported that CXCL2 (MIP-2) blockade markedly reduces ear swelling and inflammation in this model (57). MC-deficient KitW/W-v mice had reduced levels of CXCL2 in the skin as compared to Kit+/+ littermates in this CHS model, and intradermal engraftment of BMCMCs into KitW/W-v mice restored CXCL2 levels to those observed in Kit+/+ mice (57). Altogether, these data suggest that MC-derived CXCL2 can play an important role in CHS, but formal demonstration of this will require more direct evidence using mice in which only MCs are deficient for this chemokine.
Skin MCs might also contribute to CHS through their ability to release histamine. However, acute CHS responses to the hapten TNCB were identical between WT mice and mice deficient for histidine decarboxylase (HDC) (85, 86), the enzyme responsible for synthesis of endogenous histamine from histidine in mammals. By contrast, the authors observed slightly reduced epidermal hyperplasia and leukocyte infiltration in HDC−/− mice as compared to WT controls upon repeated challenges with TNCB, suggesting that histamine might contribute to these features in more chronic models of CHS (86). However, it should be noted that: (1) HDC-deficient mice have multiple phenotypic abnormalities, including decreased MC numbers and altered storage of various proteases in MC granules (87, 88) and (2) while MCs likely represent the major source of histamine in the skin, histamine can also be produced by other cell types, including basophils (89) and neutrophils (90, 91).
Protective Functions of MCs in Severe and Chronic CHS Responses
Two studies performed in Kit mutant mice indicated that MCs can reduce the ear swelling, leukocyte recruitment, and tissue damage in models of severe acute CHS responses induced by either DNFB or oxazolone (60, 61) (Figure 2C). The mechanism of such protective effects was studied by Grimbaldeston et al., who showed that adoptive transfer of WT, but not IL-10−/−, BMCMCs into MC-deficient Kit-mutant mice reversed the exaggerated ear swelling and skin pathology (61). These results suggested that, in the model and experimental conditions tested, the enhanced CHS responses are due to the absence of MCs and MC-derived IL-10 in the Kit-mutant mice. However, these experiments did not prove that the same phenotypic abnormalities would necessarily be produced if MC-derived IL-10 was removed from the WT mice.
We, therefore, recently decided to use a different approach to: (1) investigate whether MCs indeed represent a significant source of IL-10 during severe CHS responses and (2) determine whether deletion of IL-10 from endogenous MCs in non-Kit mutant mice could limit skin pathology during severe CHS responses. We used fluorochrome-labeled avidin in IL-10-GFP reporter mice, in which the GFP is placed under the control of the il10 gene promotor, to simultaneously monitor MC granule release and MC il10 gene activation in severe versus moderate models of DNFB-induced CHS (39). In the severe model of CHS, we found that dermal MCs released intracellular granule content into the surrounding microenvironment a few hours after induction of the pathology and that MCs represented one of the first immune cells to exhibit Il10 gene activation at such sites of inflammation (Figure 3). Finally, using Mcpt5-Cre+; Il10fl/fl mice, in which the il10 gene is specifically ablated in MCs, we confirmed that MC-derived IL-10 can significantly limit both the inflammation and the tissue pathology observed in severe CHS reactions (39) (Figure 2C).
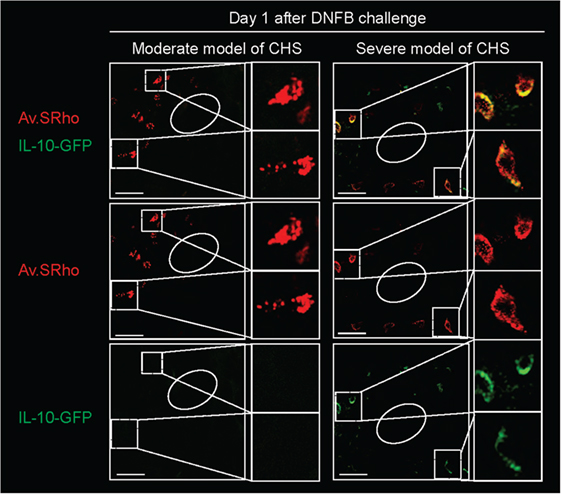
Figure 3. Skin mast cells (MCs) represent an early source of IL-10 during severe contact hypersensitivity (CHS) responses. We used Av.SRho in IL-10-GFP reporter mice to preform a longitudinal monitoring of both the release of dermal MC granules and activation of Il10 gene transcription (IL-10-GFP, as detected by emission of GFP fluorescent signal) at the site of severe CHS responses using intravital 2-photon microscopy. Images show representative 3D photographs of the ear pinna one at day 1 upon DNFB challenge. Upper panel: merged fluorescence of Av.SRho (red) and IL-10-GFP (green). Middle panel: Av.SRho (red) fluorescence. Lower panel: IL-10-GFP (green) fluorescence. White dashed lines identify the magnified areas and white circles identify hair follicles (Scale bars: 50 µm). Adapted from Reber et al. (39).
However, we found that the enhancement of both the tissue swelling and the epidermal thickness associated with the reactions observed in Mcpt5-Cre+; Il10fl/fl mice was less pronounced than those observed in the Kit-independent MC-deficient mice. This suggested that MCs might help to limit these features of this acute model of severe CHS by both IL-10-dependent and IL-10-independent mechanisms. In contrast to the data obtained in the severe CHS model, in a more moderate CHS model we did not find evidence of il10 gene activation in dermal MCs (39). These data align with previous results obtained by Dudeck et al. reporting statistically undistinguishable ear swelling in controls and Mcpt5-Cre+; Il10fl/fl mice in a similar moderate model of CHS (24).
The mechanism by which MCs can produce IL-10 during severe CHS responses was further assessed by Grimbaldeston and colleagues (61). It was reported that, when transferred intravenously into naïve recipient mice before sensitization, purified fractions of IgG obtained from mice immunized with urushiol can suppress CHS responses to the same hapten (92). The authors hypothesized that IgG immune complexes could suppress CHS responses, at least in part, by promoting production of IL-10 in MCs. Indeed, mouse MCs express the activating IgG receptor FcγRIII, as well as the inhibitory receptor FcγRIIB (93). Grimbaldeston et al. showed that IgG immune complexes can induce release of IL-10 in mouse BMCMCs in vitro. They also demonstrated that administration of an antigen-specific IgG1 antibody can suppress many features of the hapten-induced cutaneous responses in a model of passive transfer of CHS reactivity to Rag2−/− mice (61). Finally, they reported that CHS responses in MC-deficient KitW/W-v mice could be reduced to levels observed in Kit+/+ mice after engraftment with WT BMCMCs, but not after engraftment with BMCMCs derived from FcγRIII−/− or FcRγ−/− mice (FcRγ is the common γ-chain shared by the high-affinity IgE receptor FcεRI and all IgG activating Fc receptors) (61). Altogether, these results suggest that IgG-induced aggregation of FcγRIII in MCs induces IL-10 production and is important for MC anti-inflammatory function during severe acute CHS responses (Figure 2C).
Most of the data on the role of MCs in CHS were obtained using acute models. However, chronic allergic contact dermatitis (CACD) is a chronic disease resulting from recurrent allergic skin inflammation triggered by episodic skin contact with relevant allergens (63). CACD might thus be better mimicked using more chronic models of CHS, in which mice are repeatedly challenged with haptens over the course of several days or weeks. Hershko et al. developed a model of chronic CHS consisting of sensitization with oxazolone on the ear followed in 1 week by the first of repeated challenges of the ear three times per week for up to a total of ten challenges (62). In this model, ear swelling was markedly increased in MC-deficient KitW-sh/W-sh mice compared to their WT counterparts. Adoptive transfer of WT but not IL-2−/− BMCMCs into KitW-sh/W-sh mice dampened the inflammatory response, suggesting that MC production of IL-2 could suppress chronic CHS responses (62).
Interestingly, the authors observed similar results using KitW-sh/W-sh mice engrafted with BMCMCs intravenously, a protocol which does not efficiently restore the MC population into the ear pinna (18, 62). These findings argue that IL-2 production by MCs located at sites distal from the ear could help reduce CHS responses in this chronic model. Using local intradermal engraftment with BMCMCs expressing the fluorescent GFP protein, the authors found that MCs may migrate from the injection site in the ear to the DLNs and then to the spleen (62). Increased MC numbers were also observed in this model in the spleen of WT mice, suggesting that endogenous MCs could also potentially migrate to the spleen in this chronic CHS model. Interestingly, such accumulation of MCs in the spleen was not observed in Igh-7−/− mice, which lack the heavy chain of IgE antibodies (62). Moreover, and similar to the results obtained with MC-deficient mice, oxazolone challenge of Igh-7−/− mice resulted in more severe eczematous dermatitis than was seen in Igh-7+/+ mice (62). Finally, the authors reported that in the absence of MC IL-2 production, the ratio of activated to regulatory T cells (Treg) at the site of inflammation was increased. They thus suggested that in this chronic CHS model the spleen could serve as a site were MCs are recruited in an IgE-dependent manner and then produce IL-2, which maintains the effector T cells:Treg ratio at the site of inflammation and controls the severity of the disease (62) (Figure 2C). The immunosuppressive functions of Treg in CHS reactions have been extensively reviewed (94, 95). Interestingly, it was reported that Treg can directly suppress MC degranulation through cell–cell contact involving the OX40–OX40L interaction (96). Although this mechanism was not studied directly in the context of CHS responses, it is possible that such direct interactions might also regulate MC functions during CHS responses.
More recently, Gimenez-Rivera et al. developed a chronic CHS model consisting of sensitization with oxazolone followed by once-monthly challenges with the hapten for 3 or 4 months (63). Using this model, they observed more pronounced ear swelling in both KitW-sh/W-sh mice and DT-treated Mcpt5-Cre+; iDTR MC-deficient mice as compared to their respective controls. Moreover, adoptive transfer of BMCMCs into KitW-sh/W-sh mice restored responses to levels observed in WT mice (63). This study did not directly assess whether, similar to the findings of Hershko and colleagues (62), MCs in the spleen can also suppress CHS responses in this chronic model. However, the authors did report that MC-deficient mice have CHS responses comparable to those of the WT control mice at skin sites that had not been previously challenged with oxazolone, but that the MC-deficient mice exhibited exacerbated CHS responses in repeatedly challenged skin sites. They, therefore, concluded that local, and not systemic, effects are critical for the immunosuppressive effects of MCs in this chronic model (62, 63). Levels of Th1 cytokines and accumulation of antigen-specific IFN-γ-producing CD8+ TRM cells were increased at skin sites chronically exposed to oxazolone in MC-deficient KitW-sh/W-sh mice, as compared to Kit+/+ mice. In addition, depletion of CD8+ T cells using an anti-CD8 depleting antibody reduced ear swelling in KitW-sh/W-sh mice to levels observed in Kit+/+ mice. Altogether, the authors concluded from these data that MCs can interfere with the exacerbated allergic skin inflammation in this chronic CHS model, at least in part, via effects on CD8+ TRM cells (63).
Conclusion and Future Perspectives
In this article, we have reviewed evidence that MCs can either limit or contribute to the inflammation and tissue pathology of CHS depending on the severity of the model used. It is tempting to speculate that, depending on their activation threshold, MCs could either become pro-inflammatory, i.e., “type 1” or inflammatory MCs, or anti-inflammatory, i.e., “type 2” or immunoregulatory MCs, as previously reported for other immune cells, such as macrophages (49). Whether these different functions of MCs can occur in settings other than CHS responses and the extent to which they reflect distinct MC phenotypes within the same population or different MC sub-populations expressing specific phenotypes and transcriptional programs remains to be investigated. Although care should be taken in extrapolating results obtained in mice to humans, our findings raise the possibility that immunoregulatory MCs might also help to dampen tissue inflammation associated with human contact dermatitis.
Inflammatory reactions, such as observed in models of CHS, are usually associated with complex and rapidly evolving biological processes. We have reviewed herein a broad panel of innovative genetic and imaging tools to investigate, and to directly visualize, the roles played by MCs in different mouse models mimicking the human responses (Table 1). The use of the Cre/LoxP approach is a particularly powerful tool to either deplete MC populations or delete selected genes specifically in MCs. This permits investigation of the roles of the MC, and MC expression of defined molecular targets, during disease development. Moreover, we have introduced the use of fluorochrome-labeled avidin as a non-genetic approach to probe and monitor skin MC granules. This offers a new alternative to the more time-consuming and expensive double transgenic approach and permits the unambiguous investigation of both MC gene activation and degranulation by intravital imaging in living mice in a genetically untouched environment.
We have focused our review on available data obtained using mouse models, but it goes without saying that one should always be cautious when extrapolating to humans the results obtained in mice. Clearly, there are likely to be multiple differences in both immune responses and disease pathogenesis in the two species. However, we propose that studies using newly described mouse models, in which MCs and their products can be monitored and manipulated, promise to reveal previously unsuspected roles of MCs in health and disease.
Author Contributions
All authors were involved in drafting this review, and all authors approved the final version of the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer HS and handling Editor declared their shared affiliation.
Funding
NG was supported by the French “Fondation pour la Recherche Médicale FRM” award number SPE20130326582, the Société Française de Dermatologie (SFD), the Société Française l’Allergologie (SFA), the European Commission (Marie Skłodowska-Curie Individual Fellowship H2020-MSCA-IF-2016 749629), and the French “Institut National de la Santé et de la Recherche Médicale” (INSERM); TM is a Research Associate of the F.R.S.-FNRS and is supported by an “Incentive Grant for Scientific Research” of the F.R.S.-FNRS (F.4508.18), by the FRFS-WELBIO under grant CR-2017s-04 and by the Acteria Foundation; SJG is supported by NIH Grants U19 AI104209, R01 AR067145, and R01 AI32494; the United States-Israel Binational Science Foundation (Grant 2013263); the University of California, Tobacco-Related Disease Research Program; and the Sean N. Parker Center for Allergy and Asthma Research, Stanford University; LLR acknowledges support from the European Commission (Marie Skłodowska-Curie Individual Fellowship H2020-MSCA-IF-2014 656086) and the INSERM.
References
1. Belsito DV. A sherlockian approach to contact dermatitis. Dermatol Clin (1999) 17:705–13, x. doi:10.1016/S0733-8635(05)70117-0
2. Grabbe S, Schwarz T. Immunoregulatory mechanisms involved in elicitation of allergic contact hypersensitivity. Immunol Today (1998) 19:37–44. doi:10.1016/S0167-5699(97)01186-9
3. Vocanson M, Hennino A, Rozieres A, Poyet G, Nicolas JF. Effector and regulatory mechanisms in allergic contact dermatitis. Allergy (2009) 64:1699–714. doi:10.1111/j.1398-9995.2009.02082.x
4. Weber FC, Nemeth T, Csepregi JZ, Dudeck A, Roers A, Ozsvari B, et al. Neutrophils are required for both the sensitization and elicitation phase of contact hypersensitivity. J Exp Med (2015) 212:15–22. doi:10.1084/jem.20130062
5. O’Leary JG, Goodarzi M, Drayton DL, von Andrian UH. T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat Immunol (2006) 7:507–16. doi:10.1038/ni1332
6. Rafei-Shamsabadi DA, van de Poel S, Dorn B, Kunz S, Martin SF, Klose CSN, et al. Lack of type 2 innate lymphoid cells promote a type I driven enhanced immune response in contact hypersensitivity. J Invest Dermatol (2018). doi:10.1016/j.jid.2018.03.001
7. Abraham SN, St John AL. Mast cell-orchestrated immunity to pathogens. Nat Rev Immunol (2010) 10:440–52. doi:10.1038/nri2782
8. Reber LL, Frossard N. Targeting mast cells in inflammatory diseases. Pharmacol Ther (2014) 142:416–35. doi:10.1016/j.pharmthera.2014.01.004
9. Galli SJ, Grimbaldeston M, Tsai M. Immunomodulatory mast cells: negative, as well as positive, regulators of immunity. Nat Rev Immunol (2008) 8:478–86. doi:10.1038/nri2327
10. Galli SJ, Nakae S, Tsai M. Mast cells in the development of adaptive immune responses. Nat Immunol (2005) 6:135–42. doi:10.1038/ni1158
11. Galli SJ, Kalesnikoff J, Grimbaldeston MA, Piliponsky AM, Williams CM, Tsai M. Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu Rev Immunol (2005) 23:749–86. doi:10.1146/annurev.immunol.21.120601.141025
12. Rodewald HR, Feyerabend TB. Widespread immunological functions of mast cells: fact or fiction? Immunity (2012) 37:13–24. doi:10.1016/j.immuni.2012.07.007
13. Reber LL, Marichal T, Galli SJ. New models for analyzing mast cell functions in vivo. Trends Immunol (2012) 33:613–25. doi:10.1016/j.it.2012.09.008
14. Kitamura Y, Go S, Hatanaka K. Decrease of mast cells in W/Wv mice and their increase by bone marrow transplantation. Blood (1978) 52:447–52.
15. Oliveira SH, Lukacs NW. Stem cell factor: a hemopoietic cytokine with important targets in asthma. Curr Drug Targets Inflamm Allergy (2003) 2:313–8. doi:10.2174/1568010033483990
16. Reber L, Da Silva CA, Frossard N. Stem cell factor and its receptor c-Kit as targets for inflammatory diseases. Eur J Pharmacol (2006) 533:327–40. doi:10.1016/j.ejphar.2005.12.067
17. Dawicki W, Marshall JS. New and emerging roles for mast cells in host defence. Curr Opin Immunol (2007) 19:31–8. doi:10.1016/j.coi.2006.11.006
18. Grimbaldeston MA, Chen CC, Piliponsky AM, Tsai M, Tam SY, Galli SJ. Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am J Pathol (2005) 167:835–48. doi:10.1016/S0002-9440(10)62055-X
19. Kitamura Y. Heterogeneity of mast cells and phenotypic change between subpopulations. Annu Rev Immunol (1989) 7:59–76. doi:10.1146/annurev.iy.07.040189.000423
20. Piliponsky AM, Chen CC, Grimbaldeston MA, Burns-Guydish SM, Hardy J, Kalesnikoff J, et al. Mast cell-derived TNF can exacerbate mortality during severe bacterial infections in C57BL/6-KitW-sh/W-sh mice. Am J Pathol (2010) 176:926–38. doi:10.2353/ajpath.2010.090342
21. Grimbaldeston MA, Metz M, Yu M, Tsai M, Galli SJ. Effector and potential immunoregulatory roles of mast cells in IgE-associated acquired immune responses. Curr Opin Immunol (2006) 18:751–60. doi:10.1016/j.coi.2006.09.011
22. Nakano T, Sonoda T, Hayashi C, Yamatodani A, Kanayama Y, Yamamura T, et al. Fate of bone marrow-derived cultured mast cells after intracutaneous, intraperitoneal, and intravenous transfer into genetically mast cell-deficient W/Wv mice. Evidence that cultured mast cells can give rise to both connective tissue type and mucosal mast cells. J Exp Med (1985) 162:1025–43.
23. Galli SJ, Tsai M, Marichal T, Tchougounova E, Reber LL, Pejler G. Approaches for analyzing the roles of mast cells and their proteases in vivo. Adv Immunol (2015) 126:45–127. doi:10.1016/bs.ai.2014.11.002
24. Dudeck A, Dudeck J, Scholten J, Petzold A, Surianarayanan S, Kohler A, et al. Mast cells are key promoters of contact allergy that mediate the adjuvant effects of haptens. Immunity (2011) 34:973–84. doi:10.1016/j.immuni.2011.03.028
25. Feyerabend TB, Weiser A, Tietz A, Stassen M, Harris N, Kopf M, et al. Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity (2011) 35:832–44. doi:10.1016/j.immuni.2011.09.015
26. Lilla JN, Chen CC, Mukai K, BenBarak MJ, Franco CB, Kalesnikoff J, et al. Reduced mast cell and basophil numbers and function in Cpa3-Cre; Mcl-1fl/fl mice. Blood (2011) 118:6930–8. doi:10.1182/blood-2011-03-343962
27. Otsuka A, Kubo M, Honda T, Egawa G, Nakajima S, Tanizaki H, et al. Requirement of interaction between mast cells and skin dendritic cells to establish contact hypersensitivity. PLoS One (2011) 6:e25538. doi:10.1371/journal.pone.0025538
28. Sawaguchi M, Tanaka S, Nakatani Y, Harada Y, Mukai K, Matsunaga Y, et al. Role of mast cells and basophils in IgE responses and in allergic airway hyperresponsiveness. J Immunol (2012) 188:1809–18. doi:10.4049/jimmunol.1101746
29. Dahdah A, Gautier G, Attout T, Fiore F, Lebourdais E, Msallam R, et al. Mast cells aggravate sepsis by inhibiting peritoneal macrophage phagocytosis. J Clin Invest (2014) 124:4577–89. doi:10.1172/JCI75212
30. Feyerabend TB, Terszowski G, Tietz A, Blum C, Luche H, Gossler A, et al. Deletion of Notch1 converts pro-T cells to dendritic cells and promotes thymic B cells by cell-extrinsic and cell-intrinsic mechanisms. Immunity (2009) 30:67–79. doi:10.1016/j.immuni.2008.10.016
31. Furumoto Y, Charles N, Olivera A, Leung WH, Dillahunt S, Sargent JL, et al. PTEN deficiency in mast cells causes a mastocytosis-like proliferative disease that heightens allergic responses and vascular permeability. Blood (2011) 118:5466–75. doi:10.1182/blood-2010-09-309955
32. Musch W, Wege AK, Mannel DN, Hehlgans T. Generation and characterization of alpha-chymase-Cre transgenic mice. Genesis (2008) 46:163–6. doi:10.1002/dvg.20378
33. Scholten J, Hartmann K, Gerbaulet A, Krieg T, Muller W, Testa G, et al. Mast cell-specific Cre/loxP-mediated recombination in vivo. Transgenic Res (2008) 17:307–15. doi:10.1007/s11248-007-9153-4
34. Li Y, Liu B, Harmacek L, Long Z, Liang J, Lukin K, et al. The transcription factors GATA2 and MITF regulate Hdc gene expression in mast cells and are required for IgE/mast cell-mediated anaphylaxis. J Allergy Clin Immunol (2017). doi:10.1016/j.jaci.2017.10.043
35. Pejler G, Ronnberg E, Waern I, Wernersson S. Mast cell proteases: multifaceted regulators of inflammatory disease. Blood (2010) 115:4981–90. doi:10.1182/blood-2010-01-257287
36. Shimshek DR, Kim J, Hubner MR, Spergel DJ, Buchholz F, Casanova E, et al. Codon-improved Cre recombinase (iCre) expression in the mouse. Genesis (2002) 32:19–26. doi:10.1002/gene.10023
37. Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol (2001) 1:4. doi:10.1186/1471-213X-1-4
38. Gaudenzio N, Sibilano R, Marichal T, Starkl P, Reber LL, Cenac N, et al. Different activation signals induce distinct mast cell degranulation strategies. J Clin Invest (2016) 126:3981–98. doi:10.1172/JCI85538
39. Reber LL, Sibilano R, Starkl P, Roers A, Grimbaldeston MA, Tsai M, et al. Imaging protective mast cells in living mice during severe contact hypersensitivity. JCI Insight (2017) 2:e92900. doi:10.1172/jci.insight.92900
40. Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci (2010) 13:133–40. doi:10.1038/nn.2467
41. Cheng LE, Hartmann K, Roers A, Krummel MF, Locksley RM. Perivascular mast cells dynamically probe cutaneous blood vessels to capture immunoglobulin E. Immunity (2013) 38:166–75. doi:10.1016/j.immuni.2012.09.022
42. Luche H, Weber O, Nageswara Rao T, Blum C, Fehling HJ. Faithful activation of an extra-bright red fluorescent protein in “knock-in” Cre-reporter mice ideally suited for lineage tracing studies. Eur J Immunol (2007) 37:43–53. doi:10.1002/eji.200636745
43. Dudeck J, Medyukhina A, Frobel J, Svensson CM, Kotrba J, Gerlach M, et al. Mast cells acquire MHCII from dendritic cells during skin inflammation. J Exp Med (2017) 214:3791–811. doi:10.1084/jem.20160783
44. Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity (2002) 17:211–20. doi:10.1016/S1074-7613(02)00365-5
45. Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol (2004) 22:1567–72. doi:10.1038/nbt1037
46. Drew E, Merzaban JS, Seo W, Ziltener HJ, McNagny KM. CD34 and CD43 inhibit mast cell adhesion and are required for optimal mast cell reconstitution. Immunity (2005) 22:43–57. doi:10.1016/j.immuni.2004.11.014
47. Kinet JP, Blank U, Ra C, White K, Metzger H, Kochan J. Isolation and characterization of cDNAs coding for the beta subunit of the high-affinity receptor for immunoglobulin E. Proc Natl Acad Sci U S A (1988) 85:6483–7. doi:10.1073/pnas.85.17.6483
48. Bulfone-Paus S, Nilsson G, Draber P, Blank U, Levi-Schaffer F. Positive and negative signals in mast cell activation. Trends Immunol (2017) 38(9):657–67. doi:10.1016/j.it.2017.01.008
49. Galli SJ, Borregaard N, Wynn TA. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol (2011) 12:1035–44. doi:10.1038/ni.2109
50. Joulia R, Gaudenzio N, Rodrigues M, Lopez J, Blanchard N, Valitutti S, et al. Mast cells form antibody-dependent degranulatory synapse for dedicated secretion and defence. Nat Commun (2015) 6:6174. doi:10.1038/ncomms7174
51. Sali A, Matsumoto R, McNeil HP, Karplus M, Stevens RL. Three-dimensional models of four mouse mast cell chymases. Identification of proteoglycan binding regions and protease-specific antigenic epitopes. J Biol Chem (1993) 268:9023–34.
52. Stevens RL, Fox CC, Lichtenstein LM, Austen KF. Identification of chondroitin sulfate E proteoglycans and heparin proteoglycans in the secretory granules of human lung mast cells. Proc Natl Acad Sci U S A (1988) 85:2284–7. doi:10.1073/pnas.85.7.2284
53. Tharp MD, Seelig LL, Tigelaar RE, Bergstresser PR. Conjugated avidin binds to mast-cell granules. J Histochem Cytochem (1985) 33:27–32. doi:10.1177/33.1.2578142
54. Joulia R, L’Faqihi FE, Valitutti S, Espinosa E. IL-33 fine tunes mast cell degranulation and chemokine production at the single-cell level. J Allergy Clin Immunol (2017) 140(2):497–509.e10. doi:10.1016/j.jaci.2016.09.049
55. McNeil BD, Pundir P, Meeker S, Han L, Undem BJ, Kulka M, et al. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature (2014) 519:237–41. doi:10.1038/nature14022
56. Askenase PW, Van Loveren H, Kraeuter-Kops S, Ron Y, Meade R, Theoharides TC, et al. Defective elicitation of delayed-type hypersensitivity in W/Wv and SI/SId mast cell-deficient mice. J Immunol (1983) 131:2687–94.
57. Biedermann T, Kneilling M, Mailhammer R, Maier K, Sander CA, Kollias G, et al. Mast cells control neutrophil recruitment during T cell-mediated delayed-type hypersensitivity reactions through tumor necrosis factor and macrophage inflammatory protein 2. J Exp Med (2000) 192:1441–52. doi:10.1084/jem.192.10.1441
58. Bryce PJ, Miller ML, Miyajima I, Tsai M, Galli SJ, Oettgen HC. Immune sensitization in the skin is enhanced by antigen-independent effects of IgE. Immunity (2004) 20:381–92. doi:10.1016/S1074-7613(04)00080-9
59. Dudeck J, Ghouse SM, Lehmann CH, Hoppe A, Schubert N, Nedospasov SA, et al. Mast-cell-derived TNF amplifies CD8(+) dendritic cell functionality and CD8(+) T cell priming. Cell Rep (2015) 13:399–411. doi:10.1016/j.celrep.2015.08.078
60. Norman MU, Hwang J, Hulliger S, Bonder CS, Yamanouchi J, Santamaria P, et al. Mast cells regulate the magnitude and the cytokine microenvironment of the contact hypersensitivity response. Am J Pathol (2008) 172:1638–49. doi:10.2353/ajpath.2008.070559
61. Grimbaldeston MA, Nakae S, Kalesnikoff J, Tsai M, Galli SJ. Mast cell-derived interleukin 10 limits skin pathology in contact dermatitis and chronic irradiation with ultraviolet B. Nat Immunol (2007) 8:1095–104. doi:10.1038/ni1503
62. Hershko AY, Suzuki R, Charles N, Alvarez-Errico D, Sargent JL, Laurence A, et al. Mast cell interleukin-2 production contributes to suppression of chronic allergic dermatitis. Immunity (2011) 35:562–71. doi:10.1016/j.immuni.2011.07.013
63. Gimenez-Rivera VA, Siebenhaar F, Zimmermann C, Siiskonen H, Metz M, Maurer M. Mast cells limit the exacerbation of chronic allergic contact dermatitis in response to repeated allergen exposure. J Immunol (2016) 197:4240–6. doi:10.4049/jimmunol.1600236
64. Galli SJ, Hammel I. Unequivocal delayed hypersensitivity in mast cell-deficient and beige mice. Science (1984) 226:710–3. doi:10.1126/science.6494907
65. Mekori YA, Galli SJ. Undiminished immunologic tolerance to contact sensitivity in mast cell-deficient W/Wv and Sl/Sld mice. J Immunol (1985) 135:879–85.
66. Mekori YA, Chang JC, Wershil BK, Galli SJ. Studies of the role of mast cells in contact sensitivity responses. Passive transfer of the reaction into mast cell-deficient mice locally reconstituted with cultured mast cells: effect of reserpine on transfer of the reaction with DNP-specific cloned T cells. Cell Immunol (1987) 109:39–52.
67. Abt MC, Artis D. The intestinal microbiota in health and disease: the influence of microbial products on immune cell homeostasis. Curr Opin Gastroenterol (2009) 25:496–502. doi:10.1097/MOG.0b013e328331b6b4
68. Hill DA, Siracusa MC, Abt MC, Kim BS, Kobuley D, Kubo M, et al. Commensal bacteria-derived signals regulate basophil hematopoiesis and allergic inflammation. Nat Med (2012) 18:538–46. doi:10.1038/nm.2657
69. Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity (2012) 37(1):158–70. doi:10.1016/j.immuni.2012.04.011
70. Chervenick PA, Boggs DR. Decreased neutrophils and megakaryocytes in anemic mice of genotype W/Wv. J Cell Physiol (1969) 73:25–30. doi:10.1002/jcp.1040730104
71. Tsai M, Grimbaldeston MA, Yu M, Tam SY, Galli SJ. Using mast cell knock-in mice to analyze the roles of mast cells in allergic responses in vivo. Chem Immunol Allergy (2005) 87:179–97. doi:10.1159/000087644
72. Zhou JS, Xing W, Friend DS, Austen KF, Katz HR. Mast cell deficiency in Kit(W-sh)mice does not impair antibody-mediated arthritis. J Exp Med (2007) 204:2797–802. doi:10.1084/jem.20071391
73. Nigrovic PA, Gray DH, Jones T, Hallgren J, Kuo FC, Chaletzky B, et al. Genetic inversion in mast cell-deficient (Wsh) mice interrupts corin and manifests as hematopoietic and cardiac aberrancy. Am J Pathol (2008) 173:1693–701. doi:10.2353/ajpath.2008.080407
74. Manabe Y, Yoshimura M, Sakamaki K, Inoue A, Kakinoki A, Hokari S, et al. 1-Fluoro-2,4-dinitrobenzene and its derivatives act as secretagogues on rodent mast cells. Eur J Immunol (2017) 47:60–7. doi:10.1002/eji.201646536
75. Kalesnikoff J, Huber M, Lam V, Damen JE, Zhang J, Siraganian RP, et al. Monomeric IgE stimulates signaling pathways in mast cells that lead to cytokine production and cell survival. Immunity (2001) 14:801–11. doi:10.1016/S1074-7613(01)00159-5
76. Kitaura J, Song J, Tsai M, Asai K, Maeda-Yamamoto M, Mocsai A, et al. Evidence that IgE molecules mediate a spectrum of effects on mast cell survival and activation via aggregation of the FcepsilonRI. Proc Natl Acad Sci U S A (2003) 100:12911–6. doi:10.1073/pnas.1735525100
77. Kobayashi M, Nunomura S, Gon Y, Endo D, Kishiro S, Fukunaga M, et al. Abrogation of high-affinity IgE receptor-mediated mast cell activation at the effector phase prevents contact hypersensitivity to oxazolone. J Invest Dermatol (2010) 130:725–31. doi:10.1038/jid.2009.289
78. Piguet PF, Grau GE, Hauser C, Vassalli P. Tumor necrosis factor is a critical mediator in hapten induced irritant and contact hypersensitivity reactions. J Exp Med (1991) 173:673–9. doi:10.1084/jem.173.3.673
79. Gordon JR, Galli SJ. Mast cells as a source of both preformed and immunologically inducible TNF-alpha/cachectin. Nature (1990) 346:274–6. doi:10.1038/346274a0
80. Kunder CA, St John AL, Li G, Leong KW, Berwin B, Staats HF, et al. Mast cell-derived particles deliver peripheral signals to remote lymph nodes. J Exp Med (2009) 206:2455–67. doi:10.1084/jem.20090805
81. Malaviya R, Ikeda T, Ross E, Abraham SN. Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-alpha. Nature (1996) 381:77–80. doi:10.1038/381077a0
82. Gordon JR, Galli SJ. Promotion of mouse fibroblast collagen gene expression by mast cells stimulated via the Fc epsilon RI. Role for mast cell-derived transforming growth factor beta and tumor necrosis factor alpha. J Exp Med (1994) 180:2027–37. doi:10.1084/jem.180.6.2027
83. Kakurai M, Monteforte R, Suto H, Tsai M, Nakae S, Galli SJ. Mast cell-derived tumor necrosis factor can promote nerve fiber elongation in the skin during contact hypersensitivity in mice. Am J Pathol (2006) 169:1713–21. doi:10.2353/ajpath.2006.060602
84. Suto H, Nakae S, Kakurai M, Sedgwick JD, Tsai M, Galli SJ. Mast cell-associated TNF promotes dendritic cell migration. J Immunol (2006) 176:4102–12. doi:10.4049/jimmunol.176.7.4102
85. Ohtsu H, Kuramasu A, Tanaka S, Terui T, Hirasawa N, Hara M, et al. Plasma extravasation induced by dietary supplemented histamine in histamine-free mice. Eur J Immunol (2002) 32:1698–708. doi:10.1002/1521-4141(200206)32:6<1698::AID-IMMU1698>3.0.CO;2-7
86. Seike M, Furuya K, Omura M, Hamada-Watanabe K, Matsushita A, Ohtsu H. Histamine H(4) receptor antagonist ameliorates chronic allergic contact dermatitis induced by repeated challenge. Allergy (2010) 65:319–26. doi:10.1111/j.1398-9995.2009.02240.x
87. Ohtsu H, Tanaka S, Terui T, Hori Y, Makabe-Kobayashi Y, Pejler G, et al. Mice lacking histidine decarboxylase exhibit abnormal mast cells. FEBS Lett (2001) 502:53–6. doi:10.1016/S0014-5793(01)02663-1
88. Wiener Z, Buzas E, Kovacs P, Csaba G, Szabo D, Kittel A, et al. Highly reduced peritoneal mast cell number and decreased c-kit expression in histidine decarboxylase knock out mice. Inflamm Res (2001) 50(Suppl 2):S55–6. doi:10.1007/pl00022406
89. Windelborg Nielsen B, Engberg TM, Herlin T, Bjerke T, Schiotz PO. Histamine release from cord blood basophils. Int Arch Allergy Appl Immunol (1990) 93:314–22. doi:10.1159/000235260
90. Ghosh AK, Hirasawa N, Ohtsu H, Watanabe T, Ohuchi K. Defective angiogenesis in the inflammatory granulation tissue in histidine decarboxylase-deficient mice but not in mast cell-deficient mice. J Exp Med (2002) 195:973–82. doi:10.1084/jem.20011782
91. Xu X, Zhang D, Zhang H, Wolters PJ, Killeen NP, Sullivan BM, et al. Neutrophil histamine contributes to inflammation in mycoplasma pneumonia. J Exp Med (2006) 203:2907–17. doi:10.1084/jem.20061232
92. Dunn IS, Liberato DJ, Stampf JL, Castagnoli N Jr, Byers VS. Regulation of murine contact sensitivity to urushiol components by serum factors. J Invest Dermatol (1987) 89:296–8. doi:10.1111/1523-1747.ep12471534
93. Bruhns P, Jonsson F. Mouse and human FcR effector functions. Immunol Rev (2015) 268:25–51. doi:10.1111/imr.12350
94. Honda T, Miyachi Y, Kabashima K. Regulatory T cells in cutaneous immune responses. J Dermatol Sci (2011) 63:75–82. doi:10.1016/j.jdermsci.2011.06.004
95. Zielinski CE, Zuberbier T, Maurer M. Immunoregulation in cutaneous allergy: prevention and control. Curr Opin Allergy Clin Immunol (2012) 12:498–503. doi:10.1097/ACI.0b013e3283574ccb
Keywords: interleukin-10, tumor necrosis factor-alpha, avidin, two photon microscopy, mouse models, mast cells, contact hypersensitivity, IgE
Citation: Gaudenzio N, Marichal T, Galli SJ and Reber LL (2018) Genetic and Imaging Approaches Reveal Pro-Inflammatory and Immunoregulatory Roles of Mast Cells in Contact Hypersensitivity. Front. Immunol. 9:1275. doi: 10.3389/fimmu.2018.01275
Received: 12 February 2018; Accepted: 22 May 2018;
Published: 05 June 2018
Edited by:
Marcus Maurer, Charité Universitätsmedizin Berlin, GermanyReviewed by:
Carlo Pucillo, Università degli Studi di Udine, ItalyAxel Lorentz, University of Hohenheim, Germany
Silvia Bulfone-Paus, University of Manchester, United Kingdom
Hanna Siiskonen, Charité Universitätsmedizin Berlin, Germany
Copyright: © 2018 Gaudenzio, Marichal, Galli and Reber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicolas Gaudenzio, bmljb2xhcy5nYXVkZW56aW9AaW5zZXJtLmZy;
Laurent L. Reber, bGF1cmVudC5yZWJlckBpbnNlcm0uZnI=