 Jessica C. Santos
Jessica C. Santos Leah M. Pyter
Leah M. Pyter- 1Department of Basic and Applied Immunology, School of Medicine of Ribeirao Preto, University of Sao Paulo, Sao Paulo, Brazil
- 2Departments of Psychiatry and Behavioral Health and Neuroscience, The Institute for Behavioral Medicine Research, Ohio State University Wexner Medical Center, Columbus, OH, United States
Behavioral comorbidities (depression, anxiety, fatigue, cognitive disturbances, and neuropathic pain) are prevalent in cancer patients and survivors. These mental and neurological health issues reduce quality-of-life, which is a significant societal concern given the increasing rates of long-term survival after various cancers. Hypothesized causes of behavioral comorbidities with cancer include tumor biology, stress associated with the cancer experience, and cancer treatments. A relatively recent leading mechanism by which these causes contribute to changes in neurobiology that underlie behavior is inflammation. Indeed, both basic and clinical research indicates that peripheral inflammation leads to central inflammation and behavioral changes in other illness contexts. Given the limitations of assessing neuroimmunology in clinical populations, this review primarily synthesizes evidence of neuroimmune and neuroinflammatory changes due to two components of cancer (tumor biology and cancer treatments) that are associated with altered affective-like or cognitive behaviors in rodents. Specifically, alterations in microglia, neuroinflammation, and immune trafficking to the brain are compiled in models of tumors, chemotherapy, and/or radiation. Evidence-based neuronal mechanisms by which these neuroimmune changes may lead to changes in behavior are proposed. Finally, converging evidence in clinical cancer populations is discussed.
Introduction
Over the past decade, advances in cancer diagnosis and therapy have increased the number of cancer survivors, substantially improving the relative percentages of 5-year survivors for the most common types of cancer in the United States (1). Regardless, cognitive impairments, fatigue, psychiatric comorbidities, and peripheral neuropathy, attributed largely to neurotoxic effects of cancer therapy, remain highly prevalent among cancer patients and survivors (2, 3). The cancer-related cognitive impairments are well-recognized and commonly referred to as “chemofog” or “chemobrain” [reviewed in Ref. (4)]. Indeed, intensity or duration of chemotherapy relates to the severity of chemobrain (5, 6), whereas psychological factors (e.g., depression) and surgery are largely independent (7). The cognitive domains most commonly implicated include learning and memory, concentration, executive function, and processing speed (8, 9), while the common psychiatric comorbidities include anxiety and depression (10). Cancer-related fatigue is characterized by persistent physical and mental tiredness that are not explained by recent activity and interfere with functional abilities [reviewed by Bower (11)]. Chemotherapy-induced peripheral neuropathy (CIPN) is another maladaptive and debilitating side effect of cancer treatment consisting of allodynia, hyperalgesia, and neuropathic pain, observed in 30–68% of patients and persisting even after completion of chemotherapy (12). Of note, fatigue strongly correlates more with pain than with cognitive impairments or mood in patients with cancer (13–15). Together, these behavioral symptoms are debilitating and reduce quality-of-life by limiting functional independence, reducing adherence to cancer treatment, undermining social and professional life, and generating a high psychosocial stress burden (16–18). They can manifest acutely or chronically, persisting in 35–75% of cancer patients for months or even years after they are cancer-free (19–21). Such large discrepancies in prevalence are likely related to differences in cancer types and treatments or methodological assessments across studies. However, the biological mechanisms underlying these comorbidities remain unclear. Therefore, preventative approaches for behavioral changes have not been standardized and effective treatment remains a serious clinical problem (22). Substantial evidence has associated cancer treatment, especially chemotherapy, with brain damage and these behavioral comorbidities. The mechanisms by which chemotherapy induces brain neurotoxicity are hypothesized to involve neuroinflammation, damage associated molecular patterns (DAMPs) (23), impaired neurogenesis (24–32), oxidative stress, myelin degradation, and blood–brain barrier (BBB) degradation (33). Similarly, CIPN involves peripheral neuron damage and axonal degeneration (34, 35) via neuroinflammatory mechanisms in the spinal cord (34, 36–42).
Radiation therapy directed at the brain also has obvious effects on behavior and neuroimmunology (43, 44), whereas radiation directed outside of the brain was long considered localized and, therefore, without consequences on the brain. However, recent evidence in various cancer populations indicates that radiation directed away from the brain still induces fatigue, as well as executive function and memory problems that persist for years after therapy (45–47), potentially through the actions of radiation-induced bystander effects (e.g., inflammation).
In addition to cancer treatments, numerous studies demonstrate that tumor biology by itself is able to influence neurocognitive function and affect. For example, behavioral impairments are observed in treatment-free, tumor-bearing mice (30, 48–56) and in cancer patients before they start chemotherapy (57–69). Tumorigenesis is a complex and multistep process, consisting of tumor initiation, progression, and dissemination. The solid tumor microenvironment contains various non-tumor cell populations such as endothelial, stromal, and innate inflammatory immune cells that support tumor progression (70). Thus, peripheral-to-central inflammation has been implicated as a key pathway underlying these tumor-induced changes in behavior. In addition, tumors can affect endocrine stress pathways, thereby indirectly modulating neuroimmunology and behavior [reviewed by Pyter (71)]. This review will focus on the recent and expanding primary literature supporting a role for innate immunity and inflammation in tumor- and cancer treatment-induced behavioral symptoms.
Indeed, innate immune cell activation within the central nervous system (CNS) is a key factor driving neuroinflammation, with resident microglial cells as the primary cellular venue (72). Pattern recognition receptors, such as toll-like receptors (TLRs) and NOD-like receptors (NLRs), are constitutively expressed by microglia, astrocytes, and oligodendrocytes in the brain. These receptors recognize pathogen associated molecular patterns and DAMPs, which are “sterile” inflammatory signals released by dying cells in the periphery or brain (73). TLR activation elicits canonical NF-κB signaling, whereas NLR activation induces the assembly and activation of inflammasomes (multiprotein cytosolic complexes), each of which trigger pro-inflammatory caspases to cleave the pro-inflammatory cytokines, interleukin (IL)-1β, IL-18, and IL-33, into their active forms (74). Mounting evidence implicates microglial activation and its associated neuroinflammation in the pathogenesis of multiple neurological and psychiatric disorders, such as depression, Alzheimer’s disease, multiple sclerosis, cognitive impairments, and normal aging (75–81). In terms of these chronic peripheral inflammatory conditions, basic science data indicate that cytokines can stimulate peripheral nerves (e.g., vagus) and/or humorally transduce inflammatory signals into the CNS and drive behavioral changes (82). In addition, recent studies indicate that TBI, stroke, and experimental autoimmune encephalomyelitis (multiple sclerosis model) increase BBB permeability (83–85), allowing inflammatory mediators and peripheral immune cells to directly enter the brain. Thus, it is possible that tumors or cancer treatments may also influence brain function by altering innate immune cell trafficking directly to the brain.
The pathway between cancer and the CNS is hypothesized to be bidirectional. Indeed, the concept that depression or stress may precipitate chronic inflammatory diseases, including cancer (86), has existed for centuries and has been reviewed elsewhere (87). Here, we focus on one direction of this bidirectional relationship: the tumor- and tumor treatment-induced neuroinflammation contributing to affective-like, pain, and cognitive behaviors. Cancer-related fatigue and its underlying immune mechanisms are thoroughly reviewed elsewhere (11). Understanding how tumor biology and cancer treatments can interact to lead to changes in the brain will allow for improved targeting by therapeutic interventions focused on behavioral issues and thereby increase quality-of-life and survival for cancer patients. Although behavioral comorbidities are relevant for both brain and peripheral tumor patients, brain tumors and their treatments impact the brain much more directly than peripheral tumors. Indeed, brain tumor effects on behavior are confounded by the fact that they physically disrupt the brain/brain immune system and their treatments directly target brain tissue; therefore, only tumors outside of the brain will be discussed here. It is important to note that despite relatively consistent behavioral issues reported among some cancer patients, their tumors, and cancer treatments are heterogeneous and complex. Finally, we focus on the most common cancer treatments of chemotherapy and radiation, however, cancer patients are also treated with other anticancer (e.g., immunotherapy), anti-nausea, anti-infection drugs, which likely further contribute to mental health issues.
Rodent Models of Cancer, Neuroimmunology and Behavior
Current basic research using rodent cancer models implicates several putative mechanisms underlying behavioral changes. These non-human models allow for a more neurobiological understanding of the effects of tumors and tumor treatments on behavior compared to clinical research. They can also elucidate the effects of specific cancer therapies by themselves by using tumor-free mice, thereby simplifying the complex interactions between tumors and multiple tumor treatments inherent to clinical populations.
The methods for identifying current reports in the English language on how cancer and cancer treatments drive behavioral and/or neuroimmune changes in rodent models consisted of PubMed searches through April 2018 using combinations of the MeSH search terms: (“rodent”; “cancer” or “neoplasms, experimental,” or “tumor”; “inflammation” or “cytokine” or “microglia” or “neuroinflammation”; “behavior” or “cognition” or “learning” or “affect” or “depression” or “anxiety”; “chemotherapy” or “chemobrain” or “radiation” or “neuropathy” or “neuropathic pain”). Notably, only tumor models consisting of tumors located outside of the brain were considered. Here, we present tumor-bearing models with and without cancer treatments, followed by tumor-free models with cancer treatments.
Neuroimmunology in Tumor-Bearing Rodent Models
In solid peripheral neoplasms, tumor and non-tumor cells in the tumor microenvironment (e.g., leukocytes, fibroblasts, endothelial cells) secrete inflammatory mediators that attract additional immune cells, and promote tumor growth, development, and metastasis (70, 88, 89). Among the most common inflammatory mediators increased by tumors are cytokines and chemokines, including IL-1, TNF-α, IL-6, IL-8, IFN-α, IL-10, IL-12, TGF-β, and CXCR4 (90, 91). These inflammatory mediators are released into circulation and can be transduced into the brain potentially via neural and humoral pathways (92) leading to neuroinflammation, which in turn influences behavior (89) (Figure 1). Of note, increases in circulating cytokines are detectable only in some tumor models and during specific stages of the tumor development (89), although these humoral elevations are not mandatory to induce neuroinflammation and behavioral alterations (82).
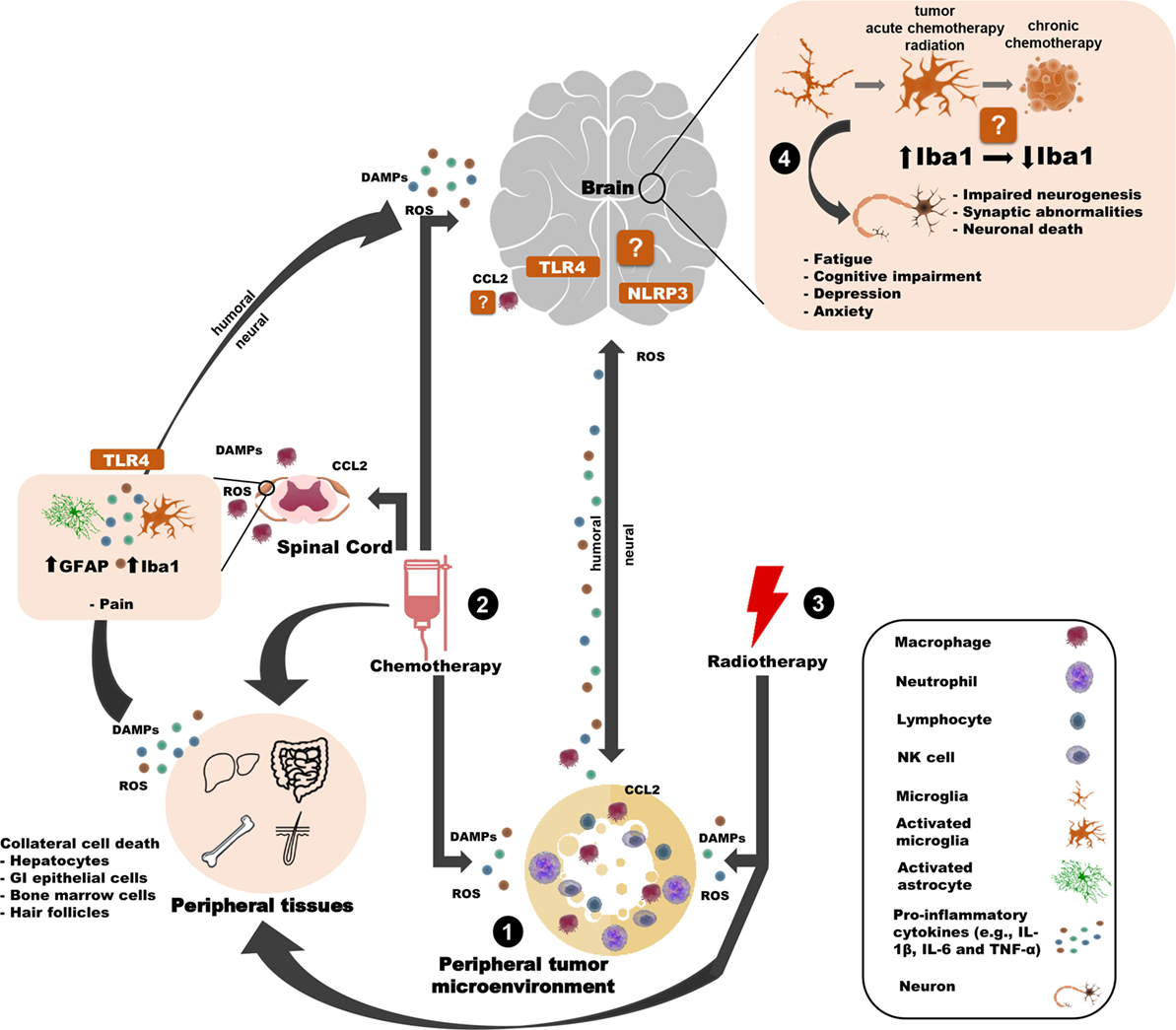
Figure 1. Potential innate immune mechanisms by which peripheral cancer and cancer treatments can induce behavioral changes. (1) The tumor microenvironment releases pro-inflammatory mediators (e.g, cytokines) that can influence the brain and behavior through humoral or neural routes. (2) Chemotherapy induces cell death of tumor cells and healthy cells (in the brain and the periphery), thereby causing the release of DAMPs, ROS, cytokines, and chemokines and contributing to many side effects. For example, chemotherapy-induced peripheral neuropathy is associated with astroglial and microglial activation in the spinal cord and TLR4 activation in DRG neurons. Similar inflammasome activity may occur in the brain. Chemotherapy may also weaken the blood–brain barrier, allowing peripheral immune cells to traffic into/closer to the brain. (3) Peripheral radiotherapy induces cell death of tumor cells and healthy “bystander” cells and (indirectly) contributes to microglial activation and behavioral deficits. (4) Together, the tumor and cancer treatments influence microglia. Tumors and radiotherapy (indirectly) activate microglia, whereas chemotherapy may affect microglia differently over time. Microglia interface with neurons to affect behavior, potentially through. Certain elements of this work were taken and then adapted from somersault18:24 (Library of Science & Medical Illustrations). To view their site, visit http://www.somersault1824.com/. They are licensed under the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/ or send a letter to Creative Commons, PO Box 1866, Mountain View, CA 94042, USA.
Our previous review focuses on the behavioral consequences of tumors in rodents without cancer treatments (89). Several of these behavioral studies also report concomitant tumor-induced immune changes in the brain and/or in the periphery. For example, brain pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α), as well as inflammatory enzymes and signaling factors [nitric oxide synthase (iNOS), indolamine 2,3-deoxygenase, cyclooxygenase-2 (COX-2)], increase along with affective-like behavior, fatigue, or cognitive impairments, when various solid tumors are generated in the periphery [(30, 48, 50, 52, 54–56), but see Ref. (93)]. Likewise, circulating pro-inflammatory cytokine increases are frequently observed in solid tumor models [(30, 51, 53–55, 93, 94), but see Ref. (48, 51)]. These inflammatory changes are hypothesized to drive the accompanying behavioral changes, however, rarely are statistical relationships between the two assessed. Of the reports statistically linking behavior and inflammation, our lab and others demonstrate that circulating cytokines (51), tumor mass, and/or tumor-derived cytokine gene expression are positively associated with neuroinflammation, fatigue, or anxiety-like behavior in female mice with peripheral tumors (94). Indeed, the consistent increase in brain IL-1β levels in tumor-bearing mice (50, 55, 56, 95) suggests a putative role for inflammasomes not only during chemotherapy, but also in cancer-induced depressive-like behavior. However, a different mammary tumor model reports neuroinflammation, but in the absence of affective-like behavior, cognitive deficits, or changes in neurogenesis (93). These discrepancies in behavioral and neurobiological outcomes might be due to differing methodological approaches. While microglial activation is well-established in brain tumor models (96), recent evidence indicates that brain microglia may be the cellular source of this neuroinflammation in various peripheral tumor models, as is observed through increased ionized calcium-binding adaptor molecule 1 (Iba1) immunoreactivity or Cd11b gene expression in the cortex and hippocampus (brain regions that regulate affect, energy, and cognition) (55, 56, 94, 95) (Table 1). Similarly, microglial activation in the spinal cord is associated with bone pain in bone cancer models (34).
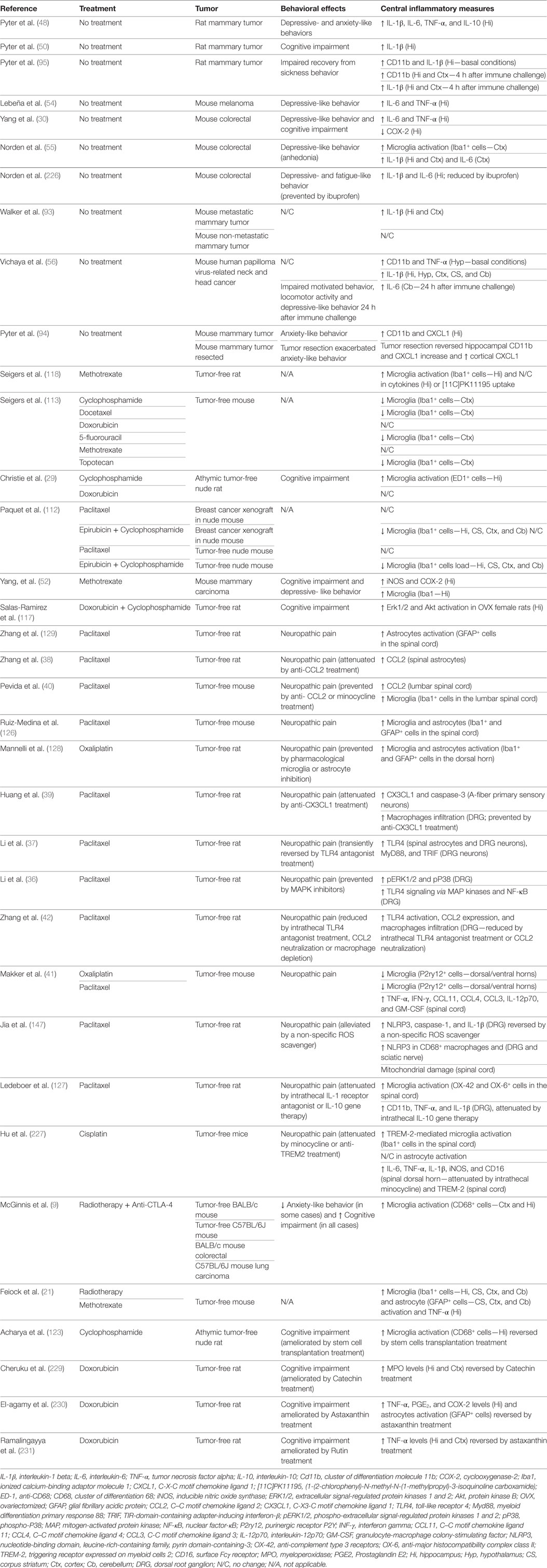
Table 1. Summary of cancer- and cancer treatment-induced neuroinflammatory changes in rodents.
Furthermore, elevations in Cd11b and other neuroinflammatory mediators, as well depressive-like and sickness behaviors, are attenuated by minocycline anti-inflammatory treatment in murine models of colon cancer and human papilloma virus (HPV)-related neck and head cancer (55, 56). As further evidence that tumors appear to be causal, and perhaps have long-lasting consequences on microglial-related changes, complete surgical resection of non-metastatic mammary tumors partially reverses tumor-induced neuroinflammation and circulating cytokines, but amplifies anxiety-like behavior (94).
Although hippocampal microglial activation (Cd11b expression) at rest is consistent among the majority of these tumor models, the results concerning regional expression of Cd11b mRNA in the brain to a subsequent peripheral immune challenge are mixed. One such challenge, lipopolysaccharide (LPS) injection (i.p.) increases cortical and hippocampal Cd11b expression, but decreases its expression in the hypothalamus of mammary tumor-bearing rats (95). Moreover, in HPV-related neck and head tumor-bearing mice, LPS does not change hippocampal and cortical Cd11b expression (56). Such discrepancies in the neuroinflammatory response are likely related to differences in cancer types and LPS doses. Taken together, these results indicate that baseline inflammation and neuroinflammatory responses to secondary immune challenges are influenced by tumors, although the identification of specific underlying mechanisms requires further investigation.
While microglial cells are the primary drivers of neuroinflammation within the CNS, increasing evidence suggests that other neuroimmune mechanisms are associated with behavioral changes. For example, psychological stress induces myeloid-derived cell trafficking to the brain, which in turn, induces affective-like behavior (81). While this review focuses on cancer and cancer treatments, it is important to note that stress associated with a cancer diagnosis may exacerbate tumor inflammatory activation, increasing tumor burden and development of metastases (97–100), and leading to more severe behavioral symptoms (51, 101, 102) through similar neuroimmune mechanisms. Furthermore, bone marrow-derived monocytes, including perivascular cells, meningeal macrophages, dendritic cells, and monocytes, have been implicated in the brain innate response in several neurologic and psychiatric diseases, as well as peripheral acute infections with sickness behavior (103–105). Indeed, in addition to potential humoral and neural routes by which peripheral inflammation is transduced into neuroinflammation, tumors affect immune trafficking to various areas of the body (94). Thus, immune trafficking of circulating monocytes to the brain may also play a role in tumor-induced changes in neurobiology and behavior (106–108) and warrants investigation.
Neuroimmunology and Cancer Treatments in Tumor-Bearing Rodents
Chemotherapy is a common adjuvant cancer treatment (109). While most basic science reports focus separately on either tumors or chemotherapy, a few combine the two for a more clinically-relevant (albeit complex) model. The combination of tumors and chemotherapy could additively increase peripheral inflammation or potential BBB disruption, thereby allowing peripheral inflammatory mediators to reach the brain, induce neurotoxicity and neuroinflammation, and contribute to cognitive and affective symptoms (8, 25, 52).
Both human and non-human research suggests that cancer treatment is causally related to the development of mood and anxiety disorders, although the potential underlying mechanisms remain broad. For example, antimetabolite chemotherapy (methotrexate) induces significant depressive-like behavior and cognitive impairments associated with an upregulation of pro-inflammatory enzymes (iNOS and COX-2) and activation of microglia in the brains of mammary tumor-bearing mice (52). In contrast, methotrexate suppresses peripheral cytokine levels in other studies (110, 111). Combined epirubicin and cyclophosphamide chemotherapies reduce stereological hippocampal microglial Iba-1 expression in the hippocampus, cortex, striatum, and cerebellum of both tumor-free and nude mice xenografted with patients’ tumor samples compared to xenografted mice treated with saline (112). These Iba1 reductions may represent chemotherapy-induced microglial cell death (113). Behavioral changes were not assessed in this study. Taken together, the mixed inflammatory results from tumor-bearing models treated with chemotherapy indicate that tumor-free models are still necessary to clarify the individual roles of chemotherapeutic agents and tumors in associated behavioral impairments.
Emerging basic and clinical research indicates that stress and cancer treatments interact to influence tumor-associated immune and behavioral symptoms (101, 114). For example, physical restraint stress in tumor-bearing mice treated with cyclophosphamide impairs the antitumoral immune response, thereby reducing the therapeutic effects of the chemotherapy treatment (115). The potential synergistic effects of stress on cancer treatment-induced neuroinflammation remain to be determined. Finally, limited data are available on radiation as another potential cancer treatment contributor to neuroinflammation and/or behavioral consequences in tumor-bearing rodent models. One recent study evaluated these changes in tumor-free and tumor-bearing mice that received peripheral radiotherapy or immunotherapy (anti-CTLA-4 antibody) or both. Of note, the mice received precise peripheral irradiation to the tumor site in the right flank. Immunotherapy alone or in combination with radiotherapy induces cognitive impairments, increases in CD68+ microglial immunostaining and central cytokine production (9). Thus, the current literature concerning neuroinflammatory-dependent behavioral changes in rodent cancer models indicates that a variety of cancer treatments are likely relevant, despite their different mechanisms of action.
Neuroimmunology and Cancer Treatments in Tumor-Free Rodent Models
The most extensive investigation regarding the potential mechanisms by which cancer treatments alter behavior is derived from studies using chemotherapeutic agents in tumor-free rodent models. Within this literature, some reports focus on behavioral consequences, neuroimmune consequences, or both. Notably, the reported behavioral effects vary based upon the particular agents and administration paradigms used, as well as the specific behavioral tests employed. The short- and long-term behavioral changes following chemotherapy treatment predominantly consist of impaired performance in learning and memory tasks including reference and working spatial performance, novel object recognition, and object placement without affecting general motor function (113, 116, 117). The majority of these studies report generalized hippocampal and cortical cellular or myelin (protective sheath of neuronal axons) damage in the brain, with some evidence of microglial cell death (25, 29, 118–120). Biochemical testing of some chemotherapeutic agents indicates that they should not be able to cross the BBB (121), alternatively suggesting that chemotherapy metabolites or other indirect mechanisms, such as peripheral immune cell infiltration, may be driving these neurobiological consequences (25). How neuroinflammation can alter neuronal function to cause behavioral changes is discussed in section “Link Between Neuroimmunology and the Neuroscience of Behavior.”
Several reports indicate a role for neuroimmune activation in chemotherapy-induced behavioral deficits (Table 1). For example, methotrexate induces microglia activation (Iba-1+ staining) in the hippocampus 1 and 3 weeks after treatment in tumor-free rats, in addition to reducing hippocampal blood vessel density (122). However, the peripheral cytokine levels and positron emission tomography scans for the uptake of [11C]PK11195 (a marker that has been associated with neuroinflammation and increased microglia activation) do not support neuroinflammatory changes underlying to immunohistochemistry results. Similarly, clinically-relevant dosing of chronic cyclophosphamide and doxorubicin treatments in tumor-free rats induces impairments in hippocampal-based memory and reduces neurogenesis (29). Coincident with the behavioral and neurogenesis changes, cyclophosphamide, but not doxorubicin, induces microglia activation (ED-1+ staining). In another report, impairments in different cognitive domains were observed in tumor-free mice after cyclophosphamide, docetaxel, doxorubicin, 5-fluorouracil, methotrexate, or topotecan treatment (116), while a reduced number of microglial cells (Iba-1+ cells) were observed in the prefrontal cortex for all these treatments (three weeks after treatment) compared with control mice, except methotrexate and doxorubicin treatment (113). In addition, chronic cyclophosphamide treatment in athymic nude rats induces microglial activation (increased CD68+ cells) in the hippocampus, as well as cognitive impairments in hippocampal and cortical-dependent tasks (123). These mixed microglial results indicate that multiple chemotherapeutic mechanisms of action may converge to trigger neuroinflammation and behavioral changes and that neuroinflammation may be due to microglial activation, microglial cell (or other cell) death or even disruptions in microglial homeostasis. Interestingly, some studies have reported a correlation between chemotherapy-induced circulating and central pro-inflammatory cytokine (IL-6, TNF-α, and IL-1β) concentration and behavioral changes acutely, but not chronically, suggesting that different mechanisms might be driving the initiation and the persistence of these comorbidities (124, 125).
Several chemotherapeutic drugs, such as platinum-based drugs (cisplatin, oxaliplatin), vinca alkaloids (vincristine), and taxanes (paclitaxel and docetaxel), trigger CIPN [as reviewed by Starobova and Vetter (35)] that is associated with neuroinflammation, although the literature is rather conflicting. Some studies indicate a key role for microglial activation [increased Iba-1, OX-42 (complement type 3 receptors), OX-6 (major histocompatibility complex class II) immunoreactivity, and Cd11b gene expression] in the spinal cord, or specifically, the dorsal root ganglion (DRG) of sensory neurons within the spinal cord (40, 126, 127). This microglial activation can be reversed by minocycline antibiotic treatment (40, 128) or intrathecal anti-inflammatory IL-10 gene therapy (127). However, the majority of CIPN studies implicate astrocyte activation (increased GFAP immunoreactivity and astrocyte hypertrophy) (36, 37, 41, 126, 128, 129). With CIPN, paracrine activation of CCL2/CCR2 signaling occurs and/or increased levels of CX3CL1 drive immune trafficking of activated macrophages to DRG, inducing nerve damage (39), which can be inhibited by anti-CCL2 antibody treatments or macrophage depletion (38–40, 42). Moreover, recent studies reported an increase in TLR4 signaling in spinal cord astrocytes and neurons in the DRG (36, 37). Potential pathways by which paclitaxel chemotherapy contributes to CIPN via TLR4 activation are the downstream canonical (myeloid differentiation primary response gene 88) and non-canonical pathways (TIR-domain-containing adapter-inducing interferon-β), culminating in NF-κB activation and upregulation of pro-inflammatory cytokines and chemokines including TNF-α, IFN-γ, IL-6, and CCL2 (36, 37, 41). TLR4 activation has been also associated with the sensitization of the ionic channel transient receptor potential vanilloid subtype 1 (TRPV1) (36, 130), found in nociceptors. Oxaliplatin chemotherapy also induces CIPN by upregulating pro-inflammatory cytokines and chemokines (IL1-β, TNF-α, IL-6, IL-8, and CCL2), which sensitizes nociceptors. The adaptive immune system is also likely involved in these responses as both paclitaxel and oxaliplatin increase the circulating levels of CD4+ and CD8+ T-cells (41). In addition, one study (131) indicates that the NLRP3 inflammasome pathway may also be activated in vitro in primed murine bone marrow-derived macrophage during anthracycline-induced IL-1β release. This suggests that some of the associated side effects, including behavioral changes, may be attenuated by IL-1β suppression. Indeed, intrathecal injection of IL-1ra transiently reversed paclitaxel-induced allodynia (127). NLRP3 inflammasome activation is driven by mitochondrial damage and reactive oxygen species (ROS) production in infiltrated macrophages of DRG and peripheral nerves and is also thought to play a role in paclitaxel-induced CIPN. Of note, some studies implicate neurotoxic effects of antineoplastic agents, which impair axonal trafficking leading to myelin and axon damage in CIPN, suggesting that the cellular damage may precede the neuroinflammation in the DRG [reviewed by Nicolini et al. (132)].
Finally, the induction of biological consequences on cells that are not directly transected by radiation treatment due to the signaling of those cells that are, is termed radiation-induced bystander effects. Both in vitro and in vivo models demonstrate off-target consequences of radiation on epigenetics, DNA health, apoptosis, cell proliferation, tumorigenesis, and inflammation (133). Indeed, peripheral radiation treatment to the right hind limb in tumor-free mice increases microglial Iba1+ cell numbers and TNF-α gene expression in the brain, comparable to the neuroinflammation observed following chemotherapy treatment (21). Whole-body radiation-induced neuroinflammation is associated with pro-inflammatory gene expression and reduced locomotion (134), although the direct brain radiation may be responsible for these effects. Radiation in rodents is also associated with general increases in circulating inflammation (135, 136), which coincide with fatigue (i.e., reduced locomotion) (137, 138). Despite the modest behavioral data currently available after radiation treatment, the reported peripheral and neuroinflammatory responses suggest that radiation can contribute to behavioral changes.
Other major gaps in understanding neurobiobehavioral changes in the context of cancer and cancer treatments pertain to the role of peripheral myeloid cells (monocytes, macrophages, and dendritic cells) and their potential localization to brain areas that interface with the peripheral circulation, such as the choroid plexus, perivascular spaces, and meninges. The chemokine CCL2 regulates myeloid cell infiltration (and potential inflammation) to different tissues, including these brain areas. For example, while CCL2 ablation in mice increases the peripheral pro-inflammatory cytokine response to LPS, it decreases the neuroinflammatory response in the entorhinal and frontal cortices and the hippocampus (139). Furthermore, CCL2 released by brain glioma tumors plays a key role in recruiting myeloid cells to the brain (140). However, the extent to which chemokine release by tumors in the periphery may influence immune cell trafficking to the brain and behavior remains unclear. On the other hand, CCL2 or peripheral immune cell trafficking to the brain may play a role in the context of chemotherapy-induced behavioral changes, as CCL2 ablation improves 5-FU chemotherapy-induced fatigue in tumor-free mice (141). A similar trafficking mechanism in spinal cord and DRG is also hypothesized to influence the development and persistence of CIPN (39, 42). The common neutropenia and lymphopenia side effects of chemotherapy may appear to conflict with the potential for increased innate immune cell trafficking to the brain and inflammation at first glance (142). When in fact, this immunogenic cell death results in production of DAMPs (proteins, nucleic acids, purines, and ROS), priming of CD4+ and CD8+ lymphocytes and a robust antigen-specific immune response against dead cell-associated antigens (143–145). Thus, cancer treatments overall consistently increase inflammation. These inflammatory mediators activate inflammasomes as well as TLRs to induce immunosurveillance or tumor progression (146), but also contribute to neuroinflammation, depression and neuropathic pain (147).
In summary, converging lines of evidence suggest that cancer and cancer treatments induce neuroinflammatory and behavioral changes in rodent models (Figure 1). Nevertheless, expansion of these initial basic science findings is required (Table 1). Specifically, the moderate variability in current microglial-related results from brain samples of models of tumors and cancer treatments necessitates a thorough temporal screening of brain microglial functioning and neuroinflammatory responses throughout tumor development and chemotherapy/radiotherapy treatments. This type of investigation would help to identify the cellular source/s of inflammation in the brain and elucidate the causal role of microglia in associated behavioral changes. Finally, several alternative pathways, including the sympathetic nervous system and the hypothalamic–pituitary–adrenal axis, modulate immune functions and, therefore, may be involved in the interaction among peripheral cancer, inflammation, and the brain.
Link Between Neuroimmunology and the Neuroscience of Behavior
Support for neuroimmune signaling that is associated with changes in behavior is extensively reviewed elsewhere in the context of peripheral tumors alone (89) or chemotherapy (20, 23, 148). As previously discussed, peripheral inflammation due to cancer or chemotherapy can trigger microglial activation and associated neuroinflammation. In some reports, this neuroinflammation is associated with changes in neurons. For example, chronic cyclophosphamide chemotherapy treatment induces cognitive impairment, microglial activation, and impaired neuronal architecture (123). Attenuation of this neuroinflammation reverses the neural and behavioral changes, suggesting that the neuroinflammation preceded the structural changes to the neurons. Alternatively, chemotherapy may damage brain tissue directly, heralding in the inevitable local neuroinflammatory response. Indeed, chemotherapy induces brain cell death (e.g., via ROS production), synaptic damage, DAMP production, disruption of the BBB, mitochondrial dysfunction, white matter damage, and alterations in neurotransmitter availability (149, 150). For example, reports from multiple labs indicate that antimetabolite chemotherapy (5-fluorouracil; 5-FU) crosses the BBB thereby directly reducing myelination and neurogenesis, as well as disrupting learning and memory (27, 151, 152). In addition, intrahippocampal human neural stem cell treatment reverses the hippocampal microglial activation and impaired neuronal architecture, as well as cognitive impairments induced by chronic cyclophosphamide treatment in athymic nude rats (123), which suggests that the neuronal damage caused microglial activation. Regardless of the order of the events, converging evidence indicates that different inflammatory microenvironments can drive various microglia phenotypes that interact with CD4+ CD45+ cells to induce neuroprotection, neurodestruction, or unchanged neurobiology (153). While these direct microglial–neuron interactions have not yet been demonstrated in the context of cancer, other examples are available. Chronic stress-induced depressive-like behavior is mediated by an initial phase of microglial activation and proliferation followed by microglial apoptosis and suppressed hippocampal neurogenesis (154). Indeed, this dynamic microglial pattern may be similar to that over early and late periods of time after chemotherapy treatment. Furthermore, although microglia are most well-recognized for innate immune functions, increasing data indicate that non-pathological microglial functions are essential for normal brain development, as well as structural and functional processes in the adult CNS (155). In the healthy brain, microglia regulate the development and plasticity of neuronal circuit architecture, modulate synapse development, activity, and elimination, as well as modulate neurogenesis (156, 157). Thus, microglial activation under inflammatory conditions (potentially cancer or cancer treatments) also likely interferes with these basic neurobiological functions and thereby alters behavior.
Alternatively, microglia may indirectly affect neuronal function via astrocytes. Microglial activation has been shown to induce ATP release, which in turn stimulates purinergic receptors on astrocytes to modulate nearby neuronal electrophysiology (158). Although this work is in vitro, the interaction between the various glial cells and neurons constitute another putative mechanism for cancer-associated behavioral changes and an interesting area for future studies. Furthermore, a recent study indicates that the serotonergic pathways downstream of the serotonin (5-HT)2B receptor in microglial cells contribute to neuronal synaptic refinement and brain maturation (159). These same 5-HT receptors can inhibit TLRs, thereby counteracting inflammation (160), and may, therefore, be a potential target to prevent cancer-associated behavioral changes. Indeed, an increasing number of studies suggest an involvement of the serotonergic system in the modulation of innate and adaptive immune functions (160–162).
Cancer Patients
Neuroimmunology and Cancer Treatments
For this section, the approach for finding original reports in the English language that considered neuroinflammatory factors and/or psychological and behavioral symptoms in cancer patients with tumors outside of the CNS before and after cancer treatment consisted of PubMed searches through April 2018 using combinations of the MeSH search terms: “depression,” or “anxiety,” or “cognition,” or “neuropsychological test”; “cancer,” or “tumor” or “chemotherapy” or “radiation”; “inflammation” or “imaging” or “microglia” or “brain.”
In the field of cancer research, it is well-accepted that affective disorders and cognitive impairments are more highly prevalent in cancer patients before, during and even years after cancer treatment relative to non-cancer controls (10, 163, 164). Although, cancer and cancer treatments share some potentially confounding physical symptoms (cachexia, fatigue, sleep disturbances) with major depressive disorder (MDD), meta-analyses and systematic reviews indicate that affective (MDD and “depressive”) and cognitive impairments are independent of these physical symptoms in cancer patients (165–168).
Although it has often been proposed that neuroinflammation may underlie the affective and cognitive deficits observed in cancer patients (23, 89, 169), scant neuroscientific data are attainable in patients. The most relevant clinical approach for understanding the relationship between neuroscience and cancer-associated behavioral comorbidities is neuroimaging. Of this neuroimaging work in cancer patients, studies focused on the effects of chemotherapy are most abundant, reviewed in Ref. (170). In the dominant breast cancer literature, cross-sectional neuroimaging approaches have yielded mostly consistent chemotherapy-induced deteriorations in neurostructure (using diffusion tensor imaging), some of which have been correlated with poor cognitive performance (171, 172). Specifically, cancer treatments reduce brain white and/or gray matter in the corpus callosum and cortex (173) or reduce hippocampal volume (171). These structural impairments are detectable over 20 years post-chemotherapy (174), and in fact, may be progressive (175, 176). Neuroinflammation is a top potential mechanism by which this occurs (177). Functional magnetic resonance imaging results (e.g., hippocampal activation during a cognitive task or at rest) are more mixed for chemotherapy-treated survivors (4, 178–181), perhaps due to the increased complexity of these assessments during active behavior. Neuroimaging cannot yet directly address the neuroinflammatory hypothesis; however, alterations in neuroimaging have been recently associated with peripheral inflammation in cancer patients treated with chemotherapy or radiation (182) and are associated with peripheral immune activation in other populations (183–186). Altered neuroimaging is also demonstrated in cancer patients prior to treatment, indicating that tumors outside of the brain influence brain network dynamics on their own (187, 188), possibly through immune signaling. For example, in breast cancer survivors at least 6 months after cancer treatment completion, peripheral inflammation is more strongly associated with amygdala reactivity to socially threatening images than in cancer-free controls (182).
Although less direct than neuroimaging, the positive association between cancer behavioral comorbidities and circulating inflammatory markers corroborates the neuroinflammatory theory and is well-supported (11, 58). In addition to baseline peripheral inflammatory markers, in vitro reactivity of peripheral immune cells is elevated in cancer patients with negative behavioral symptoms (189, 190), as are allelic profiles characterized by cytokine deregulation (191), and genetic polymorphisms of the inflammatory pathway [(192–194), but see Ref. (195)]. Furthermore, cytokine-based immunotherapy (IFN-α, IL-2 infusions) causes depression and cognitive impairments in cancer patients and other medically ill patients (196–198). Finally, there is a single neurobiological record of four adult (non-brain) cancer patients after high-dose chemotherapy treatment (199). Neuropathology is similar among these cancer patients and includes the loss of myelin and axons, as well as fluid and macrophage infiltration in various CNS regions. Similar brain pathology has been reported in autopsies of children with leukemia who were treated with chemotherapy (200). Taken together, these studies are consistent with the hypothesis that neuroimmune activation may be a key underlying mechanism of chemotherapy-induced cognitive impairment in cancer patients.
Many clinical studies that focus on chemotherapy effects on brain and behavior include cancer patients that also receive ionizing radiation therapy, although the individual role of radiation is rarely delineated. Thus, the effects of radiation therapy on neurobiology are poorly understood compared with chemotherapy. This oversight is relevant to many cancer patients; for example, radiation is used to treat approximately 56% of breast cancer patients (201). Abscopal effects, by which radiation used to treat a proximal tumor also reduces distal tumors, are thought to be immune-mediated (202). Specifically, dendritic cells and macrophages phagocytose cells damaged by radiation and then present tumor debris to adaptive immune cells to trigger widespread anti-tumor actions (203). As a result, circulating cytokines are elevated during radiation therapy in some cancer studies (204–206), but not others (207, 208). Breast cancer patients with higher baseline circulating inflammatory markers (C-reactive protein, myeloid-derived cells, IL-6) are also predisposed to fatigue after radiation (209). Thus, the potential for radiation-induced peripheral inflammation to potentiate neuroinflammation remains a viable hypothesis in need of further testing.
Anti-Inflammatory Interventions (Pharmacological and Non-Pharmacological) in Rodents and Humans
To date, there are no standard clinical interventions for cancer behavioral comorbidities. Interventions used to reduce these behavioral issues by targeting inflammatory mechanisms include exercise, psychosocial interventions (210), and pharmacological anti-inflammatory treatments. Of note, pharmacological anti-inflammatory treatments have potential hematologic toxicity and cardiovascular side effects and may interact with other cancer treatments (211); therefore, greater emphasis has been placed on non-pharmacological interventions.
In breast cancer patients after chemotherapy, 12 weeks of hatha yoga improves self-reported cognitive function while reducing circulating inflammatory markers (212). Similar results were observed after 6-weeks of aerobic walking and resistance training (213) with the addition of increases in circulating anti-inflammatory markers. In the latter study, reductions in inflammation correlate with cognitive improvements. The same duration of lyengar yoga also reduces fatigue (214) while decreasing pro-inflammatory NF-κB activity (215). Furthermore, Qigong intervention (Chinese coordinated body posturing and movement) reduces circulating C-reactive protein as well as improves self-reported cognitive functioning (216).
In a subset of depressed cancer patients, 4 months of psychosocial intervention (relaxation and stress reduction exercises and education) improves mood while reducing inflammatory markers (217). Furthermore, cognitive–behavioral stress management intervention reduces pro-inflammatory gene expression of circulating immune cells from breast cancer patients, while decreasing negative affect and increasing positive affect relative to standard-of-care controls (218). However, other cognitive-based training that reduces depression and anxiety, increases inflammatory cytokine production in stimulated immune cells in vitro in breast cancer patients (219, 220). Finally, resistance-based exercise reduces radiation-induced increases in circulating pro-inflammatory cytokines, which mediates slight improvements in fatigue and pain (205). In another breast cancer subpopulation, characterized by mild to moderate depression and pain, a nonsteroidal anti-inflammatory drug that specifically inhibits COX-2 (celecoxib) improves depressive symptoms better than a non-selective COX inhibitor (211). Drugs that interfere with TNF-α signaling also improve fatigue in chemotherapy-treated cancer patients (221, 222).
In rodent models, similar interventions to reduce cancer treatment side effects include exercise and pharmacological anti-inflammatory treatments. Several studies indicate that voluntary (223) or forced (224, 225) aerobic exercise prevent cognitive impairments in chemotherapy-treated or brain-irradiated, tumor-free mice compared to sedentary control groups, while increasing hippocampal neurogenesis. Ibuprofen treatment reduces fatigue and depressive-like behaviors in tumor-bearing mice, while reducing IL-1β and IL-6 mRNA expression in the hippocampus, compared to healthy control mice (226). Furthermore, minocycline administration reduces central levels of pro-inflammatory cytokines and microglial activation, attenuating depressive-like behavior in tumor-bearing mice (55). Similarly, minocycline administration or functional blockade of a receptor expressed on myeloid cells attenuates cisplatin-induced CIPN (227) by suppressing the microglial pro-inflammatory response.
Alternative interventions include plant-derived adjuvant therapy drugs, such as those used in traditional Ayurvedic medicine (228). For example, pretreatment with rutin, astaxantin, or catechin significantly prevents the behavioral and neurobiological impairments induced by doxorubicin treatment in rodents (229–231). These bioceuticals also decrease TNF-α, prostaglandin E2, and COX-2 levels in hippocampus (230, 231). Furthermore, tetrahydrocurcumin exerts neuroprotective effects for vincristine-induced CIPN by decreasing oxidative stress, calcium and TNF-α levels in rats (232). These studies demonstrate the immunomodulatory, anti-inflammatory, and neuroprotective properties of these plant-based drugs in the context of chemotherapy.
Conclusion
The current review organizes and evaluates the evidence supporting how cancer and cancer treatments can influence neuroimmune pathways, leading to behavioral and neurobiological changes. Notable progress has been made in cancer diagnoses and treatment, prioritizing the need for understanding and intervention that addresses the mental welfare of cancer survivors. Additional basic science research using various modeling approaches is required to untangle and to understand the interactions among various cancer treatments and their paradigms, tumor biology, and stress. These models will be essential to determining the role of neuroimmune pathways in neuronal and behavioral consequences of cancer. Complementary neuroimmune-focused information is warranted in clinical research, potentially via postmortem brain autopsies, magnetic resonance spectroscopy, and further studies of anti-inflammatory interventions. Finally, the extent to which cancer-induced behavioral changes differ from the same changes in other disease contexts can contribute to the understanding of factors that influence onset versus persistence of these comorbidities in cancer patients and survivors. In summary, increasing recent basic and clinical science evidence points to potentially additive neuroimmune mechanisms due to various components of the cancer experience in cancer-associated behavioral comorbidities (depression, anxiety, fatigue, cognitive disturbances, and neuropathic pain).
Author Contributions
The authors contributed equally for conceptualization, data synthesis, and manuscript preparation of this review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a shared affiliation, though no other collaboration, with one of the authors LP.
Acknowledgments
The authors thank Ashley Lahoud and Browning Haynes for their help with the literature search. The authors are supported by grants, CA201681 and CA216290, from the National Cancer Institute and Ohio State Medical Center (LP) and CAPES—Brazil for the Graduate Research Scholar Award (88881.133418/2016-01 – JS).
References
1. Jemal A, Ward EM, Johnson CJ, Cronin KA, Ma J, Ryerson AB, et al. Annual report to the nation on the status of cancer, 1975-2014, featuring survival. J Natl Cancer Inst (2017) 109:1–22. doi:10.1093/jnci/djx030
2. Carmack CL, Basen-Engquist K, Yuan Y, Greisinger A, Rodriguez-Bigas M, Wolff RA, et al. Feasibility of an expressive-disclosure group intervention for post-treatment colorectal cancer patients: results of the healthy expressions study. Cancer (2011) 117:4993–5002. doi:10.1002/cncr.26110
3. Cleeland CS, Workshop PD, Bennett GJ, Ph D, Dantzer R, Ph D, et al. Are the symptoms of cancer and cancer treatment due to a shared biologic mechanism? A cytokine-immunologic model of cancer symptoms. Cancer (2003) 97:2919–25. doi:10.1002/cncr.11382
4. Kesler SR, Blayney DW. Neurotoxic effects of anthracycline- vs nonanthracycline-based chemotherapy on cognition in breast cancer survivors. JAMA Oncol (2016) 2:185–92. doi:10.1001/jamaoncol.2015.4333
5. Collins B, Mackenzie J, Tasca GA, Scherling C, Smith A. Cognitive effects of chemotherapy in breast cancer patients: a dose-response study. Psychooncology (2013) 22:1517–27. doi:10.1002/pon.3163
6. Hodgson KD, Hutchinson AD, Wilson CJ, Nettelbeck T. A meta-analysis of the effects of chemotherapy on cognition in patients with cancer. Cancer Treat Rev (2013) 39:297–304. doi:10.1016/j.ctrv.2012.11.001
7. Ahles TA, Root JC, Ryan EL. Cancer- and cancer treatment-associated cognitive change: an update on the state of the science. J Clin Oncol (2012) 30:3675–86. doi:10.1200/JCO.2012.43.0116
8. Wardill HR, Mander KA, Van Sebille YZA, Gibson RJ, Logan RM, Bowen JM, et al. Cytokine-mediated blood brain barrier disruption as a conduit for cancer/chemotherapy-associated neurotoxicity and cognitive dysfunction. Int J Cancer (2016) 139:2635–45. doi:10.1002/ijc.30252
9. McGinnis GJ, Friedman D, Young KH, Torres ERS, Thomas CR Jr, Gough MJ, et al. Neuroinflammatory and cognitive consequences of combined radiation and immunotherapy in a novel preclinical model. Oncotarget (2016) 5:9155–73. doi:10.18632/oncotarget.13551
10. Evans DL, Charney DS, Lewis L, Golden RN, Gorman JM, Krishnan KRR, et al. Mood disorders in the medically ill: scientific review and recommendations. Biol Psychiatry (2005) 58:175–89. doi:10.1016/j.biopsych.2005.05.001
11. Bower JE. Cancer-related fatigue – mechanisms, risk factors, and treatments. Nat Rev Clin Oncol (2014) 11:597–609. doi:10.1038/nrclinonc.2014.127
12. Seretny M, Currie GL, Sena ES, Ramnarine S, Grant R, Macleod MR, et al. Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: a systematic review and meta-analysis. Pain (2014) 155:2461–70. doi:10.1016/j.pain.2014.09.020
13. Paulsen Ø, Laird B, Aass N, Lea T, Fayers P, Kaasa S, et al. The relationship between pro-inflammatory cytokines and pain, appetite and fatigue in patients with advanced cancer. PLoS One (2017) 12:e0177620. doi:10.1371/journal.pone.0177620
14. Matzka M, Köck-Hódi S, Jahn P, Mayer H. Relationship among symptom clusters, quality of life, and treatment-specific optimism in patients with cancer. Support Care Cancer (2018). doi:10.1007/s00520-018-4102-8
15. Nguyen LT, Alexander K, Yates P. Psychoeducational intervention for symptom management of fatigue, pain, and sleep disturbance cluster among cancer patients: a pilot quasi-experimental study. J Pain Symptom Manage (2018) 55(6):1459–72. doi:10.1016/j.jpainsymman.2018.02.019
16. Hess LM, Insel KC. Chemotherapy-related change in cognitive function: a conceptual model. Oncol Nurs Forum (2007) 34:981–94. doi:10.1188/07.ONF.981-994
17. Boykoff N, Moieni M, Subramanian SK. Confronting chemobrain: an in-depth look at survivors’ reports of impact on work, social networks, and health care response. J Cancer Surviv (2009) 3:223–32. doi:10.1007/s11764-009-0098-x
18. Stilley C, Bender C. The impact of cognitive function on medication management: three studies. Health Psychol (2010) 29:50–5. doi:10.1037/a0016940
19. Janelsins MC, Kohli S, Mohile SG, Usuki K, Ahles TA, Morrow GR. An update on cancer- and chemotherapy-related cognitive dysfunction: current status. Semin Oncol (2011) 38:431–8. doi:10.1053/j.seminoncol.2011.03.014
20. Janelsins MC, Kesler SR, Ahles TA, Morrow GR. Prevalence, mechanisms, and management of cancer-related cognitive impairment. Int Rev Psychiatry (2014) 26:102–13. doi:10.3109/09540261.2013.864260
21. Feiock C, Yagi M, Maidman A, Rendahl A, Hui S, Seelig D. Central nervous system injury – a newly observed bystander effect of radiation. PLoS One (2016) 11:e0163233. doi:10.1371/journal.pone.0163233
22. Wefel JS, Kesler SR, Noll KR, Schagen SB. Clinical characteristics, pathophysiology, and management of noncentral nervous system cancer-related cognitive impairment in adults. CA Cancer J Clin (2015) 65:123–38. doi:10.3322/caac.21258
23. Vichaya EG, Chiu GS, Krukowski K, Lacourt TE, Kavelaars A, Dantzer R, et al. Mechanisms of chemotherapy-induced behavioral toxicities. Front Neurosci (2015) 9:131. doi:10.3389/fnins.2015.00131
24. McAfoose J, Baune BT. Exploring visual-spatial working memory: a critical review of concepts and models. Neuropsychol Rev (2009) 19:130–42. doi:10.1007/s11065-008-9063-0
25. Yang M, Kim JS, Song MS, Kim SH, Kang SS, Bae CS, et al. Cyclophosphamide impairs hippocampus-dependent learning and memory in adult mice: possible involvement of hippocampal neurogenesis in chemotherapy-induced memory deficits. Neurobiol Learn Mem (2010) 93:487–94. doi:10.1016/j.nlm.2010.01.006
26. Mondie CM, Vandergrift KA, Wilson CL, Gulinello ME, Weber ET. The chemotherapy agent, thioTEPA, yields long-term impairment of hippocampal cell proliferation and memory deficits but not depression-related behaviors in mice. Behav Brain Res (2010) 209:66–72. doi:10.1016/j.bbr.2010.01.016
27. Nokia MS, Anderson ML, Shors TJ. Chemotherapy disrupts learning, neurogenesis and theta activity in the adult brain. Eur J Neurosci (2012) 36:3521–30. doi:10.1111/ejn.12007
28. Lyons L, ELBeltagy M, Bennett G, Wigmore P. Fluoxetine counteracts the cognitive and cellular effects of 5-Fluorouracil in the rat hippocampus by a mechanism of prevention rather than recovery. PLoS One (2012) 7:e30010. doi:10.1371/journal.pone.0030010
29. Christie LA, Acharya MM, Parihar VK, Nguyen A, Martirosian V, Limoli CL. Impaired cognitive function and hippocampal neurogenesis following cancer chemotherapy. Clin Cancer Res (2012) 18:1954–65. doi:10.1158/1078-0432.CCR-11-2000
30. Yang M, Kim J, Kim JS, Kim SH, Kim JC, Kang MJ, et al. Hippocampal dysfunctions in tumor-bearing mice. Brain Behav Immun (2014) 36:147–55. doi:10.1016/j.bbi.2013.10.022
31. Winocur G, Wojtowicz JM, Tannock IF. Memory loss in chemotherapy-treated rats is exacerbated in high-interference conditions and related to suppression of hippocampal neurogenesis. Behav Brain Res (2015) 281:239–44. doi:10.1016/j.bbr.2014.12.028
32. Rendeiro C, Sheriff A, Bhattacharya TK, Gogola JV, Baxter JH, Chen H, et al. Long-lasting impairments in adult neurogenesis, spatial learning and memory from a standard chemotherapy regimen used to treat breast cancer. Behav Brain Res (2016) 315:10–22. doi:10.1016/j.bbr.2016.07.043
33. Dietrich J, Prust M, Kaiser J. Chemotherapy, cognitive impairment and hippocampal toxicity. Neuroscience (2015) 309:224–32. doi:10.1016/j.neuroscience.2015.06.016
34. Zhao H, Alam A, Chen Q, Eusman MA, Pal A, Eguchi S, et al. The role of microglia in the pathobiology of neuropathic pain development: what do we know? Br J Anaesth (2017) 118:504–16. doi:10.1093/bja/aex006
35. Starobova H, Vetter I. Pathophysiology of chemotherapy-induced peripheral neuropathy. Front Mol Neurosci (2017) 10:174. doi:10.3389/fnmol.2017.00174
36. Li Y, Zhang H, Kosturakis AK, Cassidy RM, Zhang H, Kennamer-chapman RM, et al. MAPK signaling downstream to TLR4 contributes to paclitaxel-induced peripheral neuropathy. Brain Behav Immun (2015) 49:255–66. doi:10.1016/j.bbi.2015.06.003
37. Li Y, Zhang H, Zhang H, Kosturakis AK, Jawad AB, Dougherty PM. Toll-like receptor 4 signaling contributes to paclitaxel-induced peripheral neuropathy. J Pain (2014) 15:712–25. doi:10.1016/j.jpain.2014.04.001
38. Zhang H, Boyette-davis JA, Kosturakis AK, Li Y, Yoon S, Walters ET, et al. Induction of monocyte chemoattractant protein-1 (MCP-1) and its receptor CCR2 in primary sensory neurons contributes to paclitaxel-induced peripheral neuropathy. J Pain (2013) 14:1031–44. doi:10.1016/j.jpain.2013.03.012
39. Huang Z, Li D, Liu C, Cui Y, Zhu H, Zhang W, et al. CX3CL1-mediated macrophage activation contributed to paclitaxel-induced DRG neuronal apoptosis and painful peripheral neuropathy. Brain Behav Immun (2014) 40:155–65. doi:10.1016/j.bbi.2014.03.014
40. Pevida M, Lastra A, Hidalgo A, Baamonde A, Menéndez L. Spinal CCL2 and microglial activation are involved in paclitaxel-evoked cold hyperalgesia. Brain Res Bull (2013) 95:21–7. doi:10.1016/j.brainresbull.2013.03.005
41. Makker PGS, Duffy SS, Lees JG, Perera CJ, Tonkin S, Butovsky O, et al. Characterisation of immune and neuroinflammatory changes associated with chemotherapy-induced peripheral neuropathy. PLoS One (2017) 12(1):e0170814. doi:10.1371/journal.pone.0170814
42. Zhang H, Li Y, De Carvalho-Barbosa M, Kavelaars A, Heijnen CJ, Albrecht PJ, et al. Dorsal root ganglion infiltration by macrophages contributes to paclitaxel chemotherapy-induced peripheral neuropathy. J Pain (2016) 17:775–86. doi:10.1016/j.jpain.2016.02.011
43. Makale MT, McDonald CR, Hattangadi-Gluth JA, Kesari S. Mechanisms of radiotherapy-associated cognitive disability in patients with brain tumours. Nat Rev Neurol (2016) 13:52–64. doi:10.1038/nrneurol.2016.185
44. Ramanan S, Kooshki M, Zhao W, Hsu FC, Robbins ME. PPARα ligands inhibit radiation-induced microglial inflammatory responses by negatively regulating NF-κB and AP-1 pathways. Free Radic Biol Med (2008) 45:1695–704. doi:10.1016/j.freeradbiomed.2008.09.002
45. Noal S, Levy C, Hardouin A, Rieux C, Heutte N, Ségura C, et al. One-year longitudinal study of fatigue, cognitive functions, and quality of life after adjuvant radiotherapy for breast cancer. Int J Radiat Oncol Biol Phys (2011) 81:795–803. doi:10.1016/j.ijrobp.2010.06.037
46. Shibayama O, Yoshiuchi K, Inagaki M, Matsuoka Y, Yoshikawa E, Sugawara Y, et al. Association between adjuvant regional radiotherapy and cognitive function in breast cancer patients treated with conservation therapy. Cancer Med (2014) 3:702–9. doi:10.1002/cam4.174
47. Phillips KM, Jim HS, Small BJ, Laronga C, Andrykowski MAJP. Cognitive functioning after cancer treatment: a three-year longitudinal comparison of breast cancer survivors treated with chemotherapy or radiation and non-cancer controls. Cancer (2012) 118:1925–32. doi:10.1002/cncr.26432
48. Pyter LM, Pineros V, Galang JA, McClintock MK, Prendergast BJ. Peripheral tumors induce depressive-like behaviors and cytokine production and alter hypothalamic-pituitary-adrenal axis regulation. Proc Natl Acad Sci U S A (2009) 106:9069–74. doi:10.1073/pnas.0811949106
49. Qi H, Ma J, Liu YM, Yang L, Peng L, Wang H, et al. Allostatic tumor-burden induces depression-associated changes in hepatoma-bearing mice. J Neurooncol (2009) 94:367–72. doi:10.1007/s11060-009-9887-3
50. Pyter LM, Cochrane SF, Ouwenga RL, Patel PN, Pineros V, Prendergast BJ. Mammary tumors induce select cognitive impairments. Brain Behav Immun (2010) 24:903–7. doi:10.1016/j.bbi.2010.02.004
51. Lamkin DM, Lutgendorf SK, Lubaroff D, Sood AK, Beltz TG, Johnson AK. Cancer induces inflammation and depressive-like behavior in the mouse: modulation by social housing. Brain Behav Immun (2011) 25:555–64. doi:10.1016/j.bbi.2010.12.010
52. Yang M, Kim J, Kim J, Jang S, Kim S, Kim J, et al. Acute treatment with methotrexate induces hippocampal dysfunction in a mouse model of breast cancer. Brain Res Bull (2012) 89:50–6. doi:10.1016/j.brainresbull.2012.07.003
53. Fang CK, Chen HW, Chiang IT, Chen CC, Liao JF, Su TP, et al. Mirtazapine inhibits tumor growth via immune response and serotonergic system. PLoS One (2012) 7:e38886. doi:10.1371/journal.pone.0038886
54. Lebeña A, Vegas O, ómez-Lázaro EG, Arregi A, Garmendia L, Beitia G, et al. Melanoma tumors alter proinflammatory cytokine production and monoamine brain function, and induce depressive-like behavior in male mice. Behav Brain Res (2014) 272:83–92. doi:10.1016/j.bbr.2014.06.045
55. Norden DM, Bicer S, Clark Y, Jing R, Henry CJ, Wold LE, et al. Tumor growth increases neuroinflammation, fatigue and depressive-like behavior prior to alterations in muscle function. Brain Behav Immun (2015) 43:76–85. doi:10.1016/j.bbi.2014.07.013
56. Vichaya EG, Vermeer DW, Christian DL, Molkentine JM, Mason KA, Lee JH, et al. Neuroimmune mechanisms of behavioral alterations in a syngeneic murine model of human papilloma virus-related head and neck cancer. Psychoneuroendocrinology (2017) 79:59–66. doi:10.1016/j.psyneuen.2017.02.006
57. Wefel JS, Lenzi R, Theriault RL, Davis RN, Meyers CA. The cognitive sequelae of standard-dose adjuvant chemotherapy in women with breast carcinoma: results of a prospective, randomized, longitudinal trial. Cancer (2004) 100:2292–9. doi:10.1002/cncr.20272
58. Meyers CA, Albitar M, Estey E. Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer (2005) 104:788–93. doi:10.1002/cncr.21234
59. Hermelink K, Untch M, Lux MP, Kreienberg R, Beck T, Bauerfeind I, et al. Cognitive function during neoadjuvant chemotherapy for breast cancer: results of a prospective, multicenter, longitudinal study. Cancer (2007) 109:1905–13. doi:10.1002/cncr.22610
60. Ahles TA, Saykin AJ, McDonald BC, Furstenberg CT, Cole BF, Hanscom BS, et al. Cognitive function in breast cancer patients prior to adjuvant treatment. Breast Cancer Res Treat (2008) 110:143–52. doi:10.1007/s10549-007-9686-5
61. Cimprich B, Reuter-Lorenz P, Nelson J, Clark PM, Therrien B, Normolle D, et al. Prechemotherapy alterations in brain function in women with breast cancer. J Clin Exp Neuropsychol (2010) 32:324–31. doi:10.1080/13803390903032537
62. Wefel JS, Vidrine DJ, Veramonti TL, Meyers CA, Marani SK, Hoekstra HJ, et al. Cognitive impairment in men with testicular cancer prior to adjuvant therapy. Cancer (2011) 117:190–6. doi:10.1002/cncr.25298
63. Vin-Raviv N, Hillyer GC, Hershman DL, Galea S, Leoce N, Bovbjerg DH, et al. Racial disparities in posttraumatic stress after diagnosis of localized breast cancer: the BQUAL study. J Natl Cancer Inst (2013) 105:563–72. doi:10.1093/jnci/djt024
64. Oliveira Miranda D, Soares De Lima TA, Ribeiro Azevedo L, Feres O, Ribeiro Da Rocha JJ, Pereira-Da-Silva G. Proinflammatory cytokines correlate with depression and anxiety in colorectal cancer patients. Biomed Res Int (2014) 2014:1–5. doi:10.1155/2014/739650
65. Patel SK, Wong AL, Wong FL, Breen EC, Hurria A, Smith M, et al. Inflammatory biomarkers, comorbidity, and neurocognition in women with newly diagnosed breast cancer. J Natl Cancer Inst (2015) 107:1–7. doi:10.1093/jnci/djv131
66. Hermelink K, Voigt V, Kaste J, Neufeld F, Wuerstlein R, Bühner M, et al. Elucidating pretreatment cognitive impairment in breast cancer patients: the impact of cancer-related post-traumatic stress. J Natl Cancer Inst (2015) 107:djv099. doi:10.1093/jnci/djv099
67. Kesler SR, Adams M, Packer M, Rao V, Henneghan AM, Blayney DW, et al. Disrupted brain network functional dynamics and hyper-correlation of structural and functional connectome topology in patients with breast cancer prior to treatment. Brain Behav (2017) 7:1–10. doi:10.1002/brb3.643
68. Hermelink K, Bühner M, Sckopke P, Neufeld F, Kaste J, Voigt V, et al. Chemotherapy and post-traumatic stress in the causation of cognitive dysfunction in breast cancer patients. J Natl Cancer Inst (2017) 109:1–15. doi:10.1093/jnci/djx057
69. Miranda DO, Anatriello E, Azevedo LR, Cordeiro JFC, Peria FM, Flória-Santos M, et al. Elevated serum levels of proinflammatory cytokines potentially correlate with depression and anxiety in colorectal cancer patients in different stages of the antitumor therapy. Cytokine (2017) 104:72–7. doi:10.1016/j.cyto.2017.09.030
70. Sheu B-C, Chang W-C, Cheng C-Y, Lin H-H, Chang D-Y, Huang S-C. Cytokine regulation networks in the cancer microenvironment. Front Biosci (2008) 13:6255–68. doi:10.2741/3152
71. Pyter LM. The influence of cancer on endocrine, immune, and behavioral stress responses. Physiol Behav (2016) 166:4–13. doi:10.1016/j.physbeh.2015.09.031
72. Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol (2009) 27:119–45. doi:10.1146/annurev.immunol.021908.132528
73. Hanke ML, Kielian T. Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin Sci (2011) 121:367–87. doi:10.1042/CS20110164
74. Arend WP, Palmer G, Gabay C. IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev (2008) 223:20–38. doi:10.1111/j.1600-065X.2008.00624.x
75. Walker AK, Kavelaars A, Heijnen CJ, Dantzer R. Neuroinflammation and comorbidity of pain and depression. Pharmacol Rev (2013) 66:80–101. doi:10.1124/pr.113.008144
76. Pimplikar SW. Neuroinflammation in Alzheimer’s disease: from pathogenesis to a therapeutic target. J Clin Immunol (2014) 34:64–9. doi:10.1007/s10875-014-0032-5
77. Giannetti P, Politis M, Su P, Turkheimer F, Malik O, Keihaninejad S, et al. Microglia activation in multiple sclerosis black holes predicts outcome in progressive patients: an in vivo [(11)C](R)-PK11195-PET pilot study. Neurobiol Dis (2014) 65:203–10. doi:10.1016/j.nbd.2014.01.018
78. Ownby RL. Neuroinflammation and cognitive aging. Curr Psychiatry Rep (2010) 12:39–45. doi:10.1007/s11920-009-0082-1
79. Hein AM, O’Banion MK. Neuroinflammation and memory: the role of prostaglandins. Mol Neurobiol (2011) 40:15–32. doi:10.1007/s12035-009-8066-z
80. Singhal G, Jaehne EJ, Corrigan F, Toben C, Baune BT. Inflammasomes in neuroinflammation and changes in brain function: a focused review. Front Neurosci (2014) 8:315. doi:10.3389/fnins.2014.00315
81. Niraula A, Sheridan JF, Godbout JP. Microglia priming with aging and stress. Neuropsychopharmacology (2017) 42:318–33. doi:10.1038/npp.2016.185
82. Quan N. In-depth conversation: spectrum and kinetics of neuroimmune afferent pathways. Brain Behav Immun (2014) 40:1–8. doi:10.1016/j.bbi.2014.02.006
83. Clausen F, Lorant T, Lewén A, Hillered L. T lymphocyte trafficking: a novel target for neuroprotection in traumatic brain injury. J Neurotrauma (2007) 24:1295–307. doi:10.1089/neu.2006.0258
84. Ritzel RM, Patel AR, Grenier JM, Crapser J, Verma R, Jellison ER, et al. Functional differences between microglia and monocytes after ischemic stroke. J Neuroinflammation (2015) 12:106. doi:10.1186/s12974-015-0329-1
85. Vainchtein ID, Vinet J, Brouwer N, Brendecke S, Biagini G, Biber K, et al. In acute experimental autoimmune encephalomyelitis, infiltrating macrophages are immune activated, whereas microglia remain immune suppressed. Glia (2014) 62:1724–35. doi:10.1002/glia.22711
86. Balon R. Mood, anxiety, and physical illness: body and mind, or mind and body? Depress Anxiety (2006) 23:377–87. doi:10.1002/da.20217
87. McKenna MC, Zevon MA, Corn B, Rounds J. Psychosocial factors and the development of breast cancer: a meta-analysis. Health Psychol (1999) 18:520–31. doi:10.1037/0278-6133.18.5.520
88. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9:162–74. doi:10.1038/nri2506
89. Schrepf A, Lutgendorf SK, Pyter LM. Pre-treatment effects of peripheral tumors on brain and behavior: neuroinflammatory mechanisms in humans and rodents. Brain Behav Immun (2015) 49:1–17. doi:10.1016/j.bbi.2015.04.010
90. Esquivel-Velázquez M, Ostoa-Saloma P, Palacios-Arreola MI, Nava-Castro KE, Castro JI, Morales-Montor J. The role of cytokines in breast cancer development and progression. J Interferon Cytokine Res (2015) 35:1–16. doi:10.1089/jir.2014.0026
91. Mumm JB, Oft M. Cytokine-based transformation of immune surveillance into tumor-promoting inflammation. Oncogene (2008) 27:5913–9. doi:10.1038/onc.2008.275
92. DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem (2016) 139:136–53. doi:10.1111/jnc.13607
93. Walker WH, Borniger JC, Surbhi T, Zalenski AA, Muscarella SL, Fitzgerald JA, et al. Mammary tumors induce central pro-inflammatory cytokine expression, but not behavioral deficits in Balb/C mice. Sci Rep (2017) 7:1–13. doi:10.1038/s41598-017-07596-9
94. Pyter LM, Suarez-Kelly LP, Carson WE, Kaur J, Bellisario J, Bever SR. Novel rodent model of breast cancer survival with persistent anxiety-like behavior and inflammation. Behav Brain Res (2017) 330:108–17. doi:10.1016/j.bbr.2017.05.011
95. Pyter LM, El Mouatassim Bih S, Sattar H, Prendergast BJ. Peripheral tumors alter neuroinflammatory responses to lipopolysaccharide in female rats. Brain Res (2014) 1552:55–63. doi:10.1016/j.brainres.2014.01.012
96. Wu S-Y, Watabe K. The roles of microglia/macrophages in tumor progression of brain cancer and metastatic disease. Front Biosci (Landmark Ed) (2017) 22:1805–29. doi:10.2741/4573
97. Hermes GL, Delgado B, Tretiakova M, Cavigelli SA, Krausz T, Conzen SD, et al. Social isolation dysregulates endocrine and behavioral stress while increasing malignant burden of spontaneous mammary tumors. Proc Natl Acad Sci U S A (2009) 106:22393–8. doi:10.1073/pnas.0910753106
98. Vegas O, Beitia G, Sánchez-Martin JR, Arregi A, Azpiroz A. Behavioral and neurochemical responses in mice bearing tumors submitted to social stress. Behav Brain Res (2004) 155:125–34. doi:10.1016/j.bbr.2004.04.006
99. Vegas O, Garmendia L, Arregi A, Beitia G, Azpiroz A. Effects of antalarmin and nadolol on the relationship between social stress and pulmonary metastasis development in male OF1 mice. Behav Brain Res (2009) 205:200–6. doi:10.1016/j.bbr.2009.06.033
100. Azpiroz A, De Miguel Z, Fano E, Vegas O. Relations between different coping strategies for social stress, tumor development and neuroendocrine and immune activity in male mice. Brain Behav Immun (2008) 22:690–8. doi:10.1016/j.bbi.2007.10.007
101. Volden PA, Conzen SD. The influence of glucocorticoid signaling on tumor progression. Brain Behav Immun (2013) 30:S26–31. doi:10.1016/j.bbi.2012.10.022
102. Lamkin DM, Sloan EK, Patel AJ, Chiang BS, Pimentel MA, Ma JCY, et al. Chronic stress enhances progression of acute lymphoblastic leukemia via β-adrenergic signaling. Brain Behav Immun (2012) 26:635–41. doi:10.1016/j.bbi.2012.07.040
103. Bossù P, Spalletta G, Caltagirone C, Ciaramella A. Myeloid dendritic cells are potential players in human neurodegenerative diseases. Front Immunol (2015) 6:632. doi:10.3389/fimmu.2015.00632
104. Hesske L, Vincenzetti C, Heikenwalder M, Prinz M, Reith W, Fontana A, et al. Induction of inhibitory central nervous system-derived and stimulatory blood-derived dendritic cells suggests a dual role for granulocyte-macrophage colony-stimulating factor in central nervous system inflammation. Brain (2018) 133(Pt 6):1637–54. doi:10.1093/brain/awq081
105. Hirako IC, Ataide MA, Faustino L, Assis PA, Sorensen EW, Menezes GB, et al. Splenic differentiation and emergence of CCR5+CXCL9+CXCL10+ monocyte-derived dendritic cells in the brain during cerebral malaria. Nat Commun (2016) 7:13277. doi:10.1038/ncomms13277
106. Wohleb ES, McKim DB, Shea DT, Powell ND, Tarr AJ, Sheridan JF, et al. Re-establishment of anxiety in stress-sensitized mice is caused by monocyte trafficking from the spleen to the brain. Biol Psychiatry (2014) 75:970–81. doi:10.1016/j.biopsych.2013.11.029
107. Wohleb ES, McKim DB, Sheridan JF, Godbout JP. Monocyte trafficking to the brain with stress and inflammation: a novel axis of immune-to-brain communication that influences mood and behavior. Front Neurosci (2015) 8:447. doi:10.3389/fnins.2014.00447
108. Weber MD, Godbout JP, Sheridan JF. Repeated social defeat, neuroinflammation, and behavior: monocytes carry the signal. Neuropsychopharmacology (2017) 42:46–61. doi:10.1038/npp.2016.102
109. DeSantis CE, Chieh Lin C, Mariotto AB, Siegel RL, Stein KD, Kramer JL, et al. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin (2014) 64:252–71. doi:10.3322/caac.21235
110. Cutolo M. Anti-inflammatory mechanisms of methotrexate in rheumatoid arthritis. Ann Rheum Dis (2001) 60:729–35. doi:10.1136/ard.60.8.729
111. Chan ESL, Cronstein BN. Molecular action of methotrexate in inflammatory diseases. Arthritis Res (2002) 4:266–73. doi:10.1186/ar419
112. Paquet C, Boche D, El Bouchtaoui M, Gourmaud S, Janin A, Bousquet G. Effect of anti-cancer drugs on microglia in patient-derived breast cancer xenografted mouse models. Neuropathology (2017) 37:91–3. doi:10.1111/neup.12323
113. Seigers R, Loos M, Van Tellingen O, Boogerd W, Smit AB, Schagen SB. Neurobiological changes by cytotoxic agents in mice. Behav Brain Res (2016) 299:19–26. doi:10.1016/j.bbr.2015.10.057
114. Armaiz-pena GN, Cole SW, Lutgendorf SK, Sood AK. Neuroendocrine influences on cancer progression. Brain Behav Immun (2013) 30:S19–25. doi:10.1016/j.bbi.2012.06.005
115. Zorzet S, Perissin L, Rapozzi V, Giraldi T. Restraint stress reduces the antitumor efficacy of cyclophosphamide in tumor-bearing mice. Brain Behav Immun (1998) 33:23–33. doi:10.1006/brbi.1997.0504
116. Seigers R, Loos M, Van Tellingen O, Boogerd W, Smit AB, Schagen SB. Cognitive impact of cytotoxic agents in mice. Psychopharmacology (Berl) (2015) 232:17–37. doi:10.1007/s00213-014-3636-9
117. Salas-Ramirez KY, Bagnall C, Frias L, Abdali SA, Ahles TA, Hubbard K. Doxorubicin and cyclophosphamide induce cognitive dysfunction and activate the ERK and AKT signaling pathways. Behav Brain Res (2015) 292:133–41. doi:10.1016/j.bbr.2015.06.028
118. Seigers R, Schagen SB, Coppens CM, van der Most PJ, van Dam FSAM, Koolhaas JM, et al. Methotrexate decreases hippocampal cell proliferation and induces memory deficits in rats. Behav Brain Res (2009) 201:279–84. doi:10.1016/j.bbr.2009.02.025
119. Seigers R, Pourtau L, Schagen SB, van Dam FS, Koolhaas JM, Konsman JP, et al. Inhibition of hippocampal cell proliferation by methotrexate in rats is not potentiated by the presence of a tumor. Brain Res Bull (2010) 81:472–6. doi:10.1016/j.brainresbull.2009.10.006
120. Yang M, Kim J-S, Kim J, Kim S-H, Kim J-C, Kim J, et al. Neurotoxicity of methotrexate to hippocampal cells in vivo and in vitro. Biochem Pharmacol (2011) 82:72–80. doi:10.1016/j.bcp.2011.03.020
121. Eiseman JL, Eddington ND, Leslie J, MacAuley C, Sentz DL, Zuhowski M, et al. Plasma pharmacokinetics and tissue distribution of paclitaxel in CD2F1 mice. Cancer Chemother Pharmacol (1994) 34:465–71. doi:10.1007/BF00685656
122. Seigers R, Timmermans J, van der Horn HJ, de Vries EF, Dierckx RA, Visser L, et al. Methotrexate reduces hippocampal blood vessel density and activates microglia in rats but does not elevate central cytokine release. Behav Brain Res (2010) 207:265–72. doi:10.1016/j.bbr.2009.10.009
123. Acharya MM, Martirosian V, Chmielewski NN, Hanna N, Tran KK, Liao AC, et al. Stem cell transplantation reverses chemotherapy-induced cognitive dysfunction. Cancer Res (2015) 75:676–86. doi:10.1158/0008-5472.CAN-14-2237
124. Smith LB, Leo MC, Anderson C, Wright TJ, Weymann KB, Wood LJ. The role of IL-1β and TNF-α signaling in the genesis of cancer treatment related symptoms (CTRS); a study using cytokine receptor-deficient mice. Brain Behav Immun (2014) 38:66–76. doi:10.1016/j.bbi.2013.12.022
125. Elsea CR, Kneiss JA, Wood LJ. Induction of IL-6 by cytotoxic chemotherapy is associated with loss of lean body and fat mass in tumor-free female mice. Biol Res Nurs (2015) 17:549–57. doi:10.1177/1099800414558087
126. Ruiz-Medina J, Baulies A, Bura SA, Valverde O. Paclitaxel-induced neuropathic pain is age dependent and devolves on glial response. Eur J Pain (2013) 17:75–85. doi:10.1002/j.1532-2149.2012.00172.x
127. Ledeboer A, Jekich BM, Sloane EM, Mahoney JH, Langer SJ, Milligan ED, et al. Intrathecal interleukin-10 gene therapy attenuates paclitaxel-induced mechanical allodynia and proinflammatory cytokine expression in dorsal root ganglia in rats. Brain Behav Immun (2007) 21:686–98. doi:10.1016/j.bbi.2006.10.012
128. Mannelli LDC, Pacini A, Micheli L, Tani A, Zanardelli M, Ghelardini C. Glial role in oxaliplatin-induced neuropathic pain. Exp Neurol (2014) 261:22–33. doi:10.1016/j.expneurol.2014.06.016
129. Zhang H, Yoon SY, Zhang H, Dougherty PM. Evidence that spinal astrocytes but not microglia contribute to the pathogenesis of paclitaxel-induced painful neuropathy. J Pain (2012) 13:293–303. doi:10.1016/j.jpain.2011.12.002
130. Hara T, Chiba T, Abe K, Makabe A, Ikeno S, Kawakami K, et al. Effect of paclitaxel on transient receptor potential vanilloid 1 in rat dorsal root ganglion. Pain (2013) 154:882–9. doi:10.1016/j.pain.2013.02.023
131. Sauter KAD, Wood LJ, Wong J, Iordanov M, Magun BE. Doxorubicin and daunorubicin induce processing and release of interleukin-1β through activation of the NLRP3 inflammasome. Cancer Biol Ther (2011) 11:1008–16. doi:10.4161/cbt.11.12.15540
132. Nicolini G, Monfrini M, Scuteri A. Axonal transport impairment in chemotherapy-induced peripheral neuropathy. Toxics (2015) 3:322–41. doi:10.3390/toxics3030322
134. York JM, Blevins NA, Meling DD, Peterlin MB, Gridley DS, Cengel KA, et al. The biobehavioral and neuroimmune impact of low-dose ionizing radiation. Brain Behav Immun (2012) 26:218–27. doi:10.1016/j.bbi.2011.09.006
135. Khayyal MT, El-Ghazaly MA, El-Hazek RM, Nada AS. The effects of celecoxib, a COX-2 selective inhibitor, on acute inflammation induced in irradiated rats. Inflammopharmacology (2009) 17:255–66. doi:10.1007/s10787-009-0014-z
136. Haveman J, Geerdink AG, Rodermond HM. TNF, IL-1 and IL-6 in circulating blood after total-body and localized irradiation in rats. Oncol Rep (1998) 5:679–83.
137. Van der Meeren A, Monti P, Lebaron-Jacobs L, Marquette C, Gourmelon P. Characterization of the acute inflammatory response after irradiation in mice and its regulation by interleukin 4 (Il4). Radiat Res (2001) 155:858–65. doi:10.1667/0033-7587(2001)155[0858:COTAIR]2.0.CO;2
138. Renner M, Feng R, Springer D, Chen MK, Ntamack A, Espina A, et al. A murine model of peripheral irradiation-induced fatigue. Behav Brain Res (2016) 307:218–26. doi:10.1016/j.bbr.2016.03.035
139. Thompson WL, Karpus WJ, Van Eldik LJ. MCP-1-deficient mice show reduced neuroinflammatory responses and increased peripheral inflammatory responses to peripheral endotoxin insult. J Neuroinflammation (2008) 5:1–13. doi:10.1186/1742-2094-5-35
140. Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res (2016) 76:5671–82. doi:10.1158/0008-5472.CAN-16-0144
141. Mahoney SE, Davis JM, Murphy EA, McClellan JL, Gordon B, Pena MM. Effects of 5-fluorouracil chemotherapy on fatigue: role of MCP-1. Brain Behav Immun (2013) 27:155–61. doi:10.1016/j.bbi.2012.10.012
142. Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol (2011) 13:324–33. doi:10.1093/neuonc/noq157
143. Fucikova J, Moserova I, Urbanova L, Bezu L, Kepp O, Cremer I, et al. Prognostic and predictive value of DAMPs and DAMP-associated processes in cancer. Front Immunol (2015) 6:402. doi:10.3389/fimmu.2015.00402
144. Pol J, Vacchelli E, Aranda F, Castoldi F, Eggermont A, Cremer I, et al. Trial watch: immunogenic cell death inducers for anticancer chemotherapy. Oncoimmunology (2015) 4(4):e1008866. doi:10.1080/2162402X.2015.1008866
145. Bracci L, Schiavoni G, Sistigu A, Belardelli F. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ (2014) 21:15–25. doi:10.1038/cdd.2013.67
146. Lin C, Zhang J. Inflammasomes in inflammation-induced cancer. Front Immunol (2017) 8:271. doi:10.3389/fimmu.2017.00271
147. Jia M, Wu C, Gao F, Xiang H, Sun N, Peng P, et al. Activation of NLRP3 inflammasome in peripheral nerve contributes to paclitaxel-induced neuropathic pain. Mol Pain (2017) 13:1–11. doi:10.1177/1744806917719804
148. Kaiser J, Bledowski C, Dietrich J. Neural correlates of chemotherapy-related cognitive impairment. Cortex (2014) 54:33–50. doi:10.1016/j.cortex.2014.01.010
149. Dietrich J, Han R, Yang Y, Mayer-Pröschel M, Noble M. CNS progenitor cells and oligodendrocytes are targets of chemotherapeutic agents in vitro and in vivo. J Biol (2006) 5:22. doi:10.1186/jbiol50
150. Wick A, Wick W, Hirrlinger J, Gerhardt E, Dringen R, Dichgans J, et al. Chemotherapy-induced cell death in primary cerebellar granule neurons but not in astrocytes: in vitro paradigm of differential neurotoxicity. J Neurochem (2004) 91:1067–74. doi:10.1111/j.1471-4159.2004.02774.x
151. Mustafa S, Walker A, Bennett G, Wigmore PM. 5-Fluorouracil chemotherapy affects spatial working memory and newborn neurons in the adult rat hippocampus. Eur J Neurosci (2008) 28:323–30. doi:10.1111/j.1460-9568.2008.06325.x
152. Han R, Yang YM, Dietrich J, Luebke A, Mayer-Pröschel M, Noble M. Systemic 5-fluorouracil treatment causes a syndrome of delayed myelin destruction in the central nervous system. J Biol (2008) 7:12. doi:10.1186/jbiol69
153. Kipnis J, Avidan H, Caspi RR, Schwartz M. Dual effect of CD4+CD25+ regulatory T cells in neurodegeneration: a dialogue with microglia. Proc Natl Acad Sci U S A (2004) 101:14663–9. doi:10.1073/pnas.0404842101
154. Kreisel T, Frank MG, Licht T, Reshef R, Baratta MV, Maier SF, et al. Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol Psychiatry (2013) 19:699–709. doi:10.1038/mp.2013.155
155. Yirmiya R, Rimmerman N, Reshef R. Depression as a microglial disease. Trends Neurosci (2015) 38:637–58. doi:10.1016/j.tins.2015.08.001
156. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science (2011) 333:1456–8. doi:10.1126/science.1202529
157. Wake H, Moorhouse AJ, Miyamoto A, Nabekura J. Microglia: actively surveying and shaping neuronal circuit structure and function. Trends Neurosci (2013) 36:209–17. doi:10.1016/j.tins.2012.11.007
158. Pascual O, Ben Achour S, Rostaing P, Triller A, Bessis A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci U S A (2012) 109:E197–205. doi:10.1073/pnas.1111098109
159. Kolodziejczak M, Béchade C, Gervasi N, Irinopoulou T, Banas SM, Cordier C, et al. Serotonin modulates developmental microglia via 5-HT2B receptors: potential implication during synaptic refinement of retinogeniculate projections. ACS Chem Neurosci (2015) 6:1219–30. doi:10.1021/cn5003489
160. Szabo A, Gogolak P, Koncz G, Foldvari Z, Pazmandi K, Miltner N, et al. Immunomodulatory capacity of the serotonin receptor 5-HT2B in a subset of human dendritic cells. Sci Rep (2018) 8:1765. doi:10.1038/s41598-018-20173-y
161. Dürk T, Panther E, Müller T, Sorichter S, Ferrari D, Pizzirani C, et al. 5-Hydroxytryptamine modulates cytokine and chemokine production in LPS-primed human monocytes via stimulation of different 5-HTR subtypes. Int Immunol (2005) 17:599–606. doi:10.1093/intimm/dxh242
162. Herr N, Bode C, Duerschmied D. The effects of serotonin in immune cells. Front Cardiovasc Med (2017) 4:48. doi:10.3389/fcvm.2017.00048
163. Wang XM, Walitt B, Saligan L, Tiwari AFY, Cheung CW, Zhang ZJ. Chemobrain: a critical review and causal hypothesis of link between cytokines and epigenetic reprogramming associated with chemotherapy. Cytokine (2015) 72:86–96. doi:10.1016/j.cyto.2014.12.006
164. Saligan LN, Kim HS. A systematic review of the association between immunogenomic markers and cancer-related fatigue. Brain Behav Immun (2012) 26:830–48. doi:10.1016/j.bbi.2012.05.004
165. Wang J, Wu X, Lai W, Long E, Zhang X, Li W, et al. Prevalence of depression and depressive symptoms among outpatients: a systematic review and meta-analysis. BMJ Open (2017) 7:e017173. doi:10.1136/bmjopen-2017-017173
166. Laoutidis ZG, Mathiak K. Antidepressants in the treatment of depression/depressive symptoms in cancer patients: a systematic review and meta-analysis. BMC Psychiatry (2013) 13:140. doi:10.1186/1471-244X-13-140
167. Krebber AMH, Buffart LM, Kleijn G, Riepma IC, De Bree R, Leemans CR, et al. Prevalence of depression in cancer patients: a meta-analysis of diagnostic interviews and self-report instruments. Psychooncology (2014) 23:121–30. doi:10.1002/pon.3409
168. Caruso R, GiuliaNanni M, Riba MB, Sabato S, Grassi L. Depressive spectrum disorders in cancer: diagnostic issues and intervention. A critical review. Curr Psychiatry Rep (2017) 19:33. doi:10.1007/s11920-017-0785-7
169. Miller AH, Ancoli-Israel S, Bower JE, Capuron L, Irwin MR. Neuroendocrine-immune mechanisms of behavioral comorbidities in patients with cancer. J Clin Oncol (2008) 26:971–82. doi:10.1200/JCO.2007.10.7805
170. Andryszak P, Wiłkość M, Izdebski P, Żurawski B. A systemic literature review of neuroimaging studies in women with breast cancer treated with adjuvant chemotherapy. Contemp Oncol (2017) 21:6–15. doi:10.5114/wo.2017.66652
171. Bergouignan L, Lefranc JP, Chupin M, Morel N, Spano JP, Fossati P. Breast cancer affects both the hippocampus volume and the episodic autobiographical memory retrieval. PLoS One (2011) 6:e25349. doi:10.1371/journal.pone.0025349
172. Kesler SR, Wefel JS, Hosseini SMH, Cheung M, Watson CL, Hoeft F. Default mode network connectivity distinguishes chemotherapy-treated breast cancer survivors from controls. Proc Natl Acad Sci U S A (2013) 110:11600–5. doi:10.1073/pnas.1214551110
173. Deprez S, Amant F, Smeets A, Peeters R, Leemans A, Van Hecke W, et al. Longitudinal assessment of chemotherapy-induced structural changes in cerebral white matter and its correlation with impaired cognitive functioning. J Clin Oncol (2012) 30:274–81. doi:10.1200/JCO.2011.36.8571
174. Koppelmans V, de Groot M, de Ruiter MB, Boogerd W, Seynaeve C, Vernooij MW, et al. Global and focal white matter integrity in breast cancer survivors 20 years after adjuvant chemotherapy. Hum Brain Mapp (2014) 35:889–99. doi:10.1002/hbm.22221
175. Brown RT, Madan-Swain A, Walco GA, Cherrick I, Ievers CE, Conte PM, et al. Cognitive and academic late effects among children previously treated for acute lymphocytic leukemia receiving chemotherapy as CNS prophylaxis. J Pediatr Psychol (1998) 23:333–40. doi:10.1093/jpepsy/23.5.333
176. Stemmer SM, Stears JC, Burton BS, Jones RB, Simon JH. White matter changes in patients with breast cancer treated with high-dose chemotherapy and autologous bone marrow support. AJNR Am J Neuroradiol (1994) 15:1267–73.
177. Ahles TA, Saykin AJ. Candidate mechanisms for chemotherapy-induced cognitive changes. Nat Rev Cancer (2007) 7:192–201. doi:10.1038/nrc2073
178. de Ruiter MB, Reneman L, Boogerd W, Veltman DJ, van Dam FSAM, Nederveen AJ, et al. Cerebral hyporesponsiveness and cognitive impairment 10 years after chemotherapy for breast cancer. Hum Brain Mapp (2011) 32:1206–19. doi:10.1002/hbm.21102
179. Kesler SR, Bennett FC, Mahaffey ML, Spiegel D. Regional brain activation during verbal declarative memory in metastatic breast cancer. Clin Cancer Res (2009) 15:6665–73. doi:10.1158/1078-0432.CCR-09-1227
180. López Zunini RA, Scherling C, Wallis N, Collins B, MacKenzie J, Bielajew C, et al. Differences in verbal memory retrieval in breast cancer chemotherapy patients compared to healthy controls: a prospective fMRI study. Brain Imaging Behav (2013) 7:460–77. doi:10.1007/s11682-012-9213-0
181. Wang L, Apple AC, Schroeder MP, Ryals AJ, Voss JL, Gitelman D, et al. Reduced prefrontal activation during working and long-term memory tasks and impaired patient-reported cognition among cancer survivors postchemotherapy compared with healthy controls. Cancer (2016) 122:258–68. doi:10.1002/cncr.29737
182. Muscatell KA, Eisenberger NI, Dutcher JM, Cole SW, Bower JE. Links between inflammation, amygdala reactivity, and social support in breast cancer survivors. Brain Behav Immun (2016) 53:34–8. doi:10.1016/j.bbi.2015.09.008
183. Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Critchley HD. Inflammation causes mood changes through alterations in subgenual cingulate activity and mesolimbic connectivity. Biol Psychiatry (2009) 66:407–14. doi:10.1016/j.biopsych.2009.03.015
184. O’Connor MF, Irwin MR, Wellisch DK. When grief heats up: pro-inflammatory cytokines predict regional brain activation. Neuroimage (2009) 47:891–6. doi:10.1016/j.neuroimage.2009.05.049
185. Satizabal CL, Zhu YC, Mazoyer B, Dufouil C, Tzourio C. Circulating IL-6 and CRP are associated with MRI findings in the elderly: the 3C-Dijon Study. Neurology (2012) 78:720–7. doi:10.1212/WNL.0b013e318248e50f
186. Haroon E, Woolwine BJ, Chen X, Pace TW, Parekh S, Spivey JR, et al. IFN-alpha-induced cortical and subcortical glutamate changes assessed by magnetic resonance spectroscopy. Neuropsychopharmacology (2014) 39:1777–85. doi:10.1038/npp.2014.25
187. Scherling C, Collins B, MacKenzie J, Bielajew C, Smith A. Prechemotherapy differences in response inhibition in breast cancer patients compared to controls: a functional magnetic resonance imaging study. J Clin Exp Neuropsychol (2012) 34:543–60. doi:10.1080/13803395.2012.666227
188. Kesler SR, Gugel M, Huston-Warren E, Watson C. Atypical structural connectome organization and cognitive impairment in young survivors of acute lymphoblastic leukemia. Brain Connect (2016) 6:273–82. doi:10.1089/brain.2015.0409
189. Bower JE, Ganz PA, Aziz N, Olmstead R, Irwin MR, Cole SW. Inflammatory responses to psychological stress in fatigued breast cancer survivors: relationship to glucocorticoids. Brain Behav Immun (2007) 21:251–8. doi:10.1016/j.bbi.2006.08.001
190. Collado-Hidalgo A, Bower JE, Ganz PA, Cole SW, Irwin MR. Inflammatory biomarkers for persistent fatigue in breast cancer survivors. Clin Cancer Res (2006) 12:2759–66. doi:10.1158/1078-0432.CCR-05-2398
191. de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer (2006) 6:24–37. doi:10.1038/nrc1782
192. Collado-Hidalgo A, Bower JE, Ganz PA, Irwin MR, Cole SW. Cytokine gene polymorphisms and fatigue in breast cancer survivors: early findings. Brain Behav Immun (2008) 22:1197–200. doi:10.1016/j.bbi.2008.05.009
193. Dhruva A, Aouizerat BE, Cooper B, Paul SM, Dodd M, West C, et al. Cytokine gene associations with self-report ratings of morning and evening fatigue in oncology patients and their family caregivers. Biol Res Nurs (2015) 17:175–84. doi:10.1177/1099800414534313
194. Kober KM, Smoot B, Paul SM, Cooper BA, Levine JD, Miaskowski C. Polymorphisms in cytokine genes are associated with higher levels of fatigue and lower levels of energy in women after breast cancer surgery. J Pain Symptom Manage (2016) 52:695–708. doi:10.1016/j.jpainsymman.2016.04.014
195. Reinertsen KV, Grenaker Alnaes GI, Landmark-Høyvik H, Loge JH, Wist E, Kristensen VN, et al. Fatigued breast cancer survivors and gene polymorphisms in the inflammatory pathway. Brain Behav Immun (2011) 25:1376–83. doi:10.1016/j.bbi.2011.04.001
196. Walker LG, Walker MB, Heys SD, Lolley J, Wesnes K, Eremin O. The psychological and psychiatric effects of rIL-2-therapy: a controlled clinical trial. Psychooncology (1997) 6:290–301. doi:10.1002/(SICI)1099-1611(199712)6:4<290::AID-PON283>3.0.CO;2-G
197. Capuron L, Ravaud A, Gualde N, Bosmans E, Dantzer R, Maes M, et al. Association between immune activation and early depressive symptoms in cancer patients treated with interleukin-2-based therapy. Psychoneuroendocrinology (2001) 26:797–808. doi:10.1016/S0306-4530(01)00030-0
198. Trask PC, Esper P, Riba M, Redman B. Psychiatric side effects of interferon therapy: prevalence, proposed mechanisms, and future directions. J Clin Oncol (2000) 18:2316–26. doi:10.1200/JCO.2000.18.11.2316
199. Moore-Maxwell CA, Datto MB, Hulette CM. Chemotherapy-induced toxic leukoencephalopathy causes a wide range of symptoms: a series of four autopsies. Mod Pathol (2004) 17:241–7. doi:10.1038/modpathol.3800049
200. Kay HEM, Knapton PJ, O’Sullivan JP, Wells DG, Harris RF, Innes EM, et al. Encephalopathy in acute leukaemia associated with methotrexate therapy. Arch Dis Child (1972) 47:344–54. doi:10.1136/adc.47.253.344
201. McGale P, Taylor C, Correa C, Cutter D, Duane F, Ewertz M, et al. Effect of radiotherapy after mastectomy and axillary surgery on 10-year recurrence and 20-year breast cancer mortality: meta-analysis of individual patient data for 8135 women in 22 randomised trials. Lancet (2014) 383:2127–35. doi:10.1016/S0140-6736(14)60488-8
202. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol (2013) 31:51–72. doi:10.1146/annurev-immunol-032712-100008
203. Lauber K, Ernst A, Orth M, Herrmann M, Belka C. Dying cell clearance and its impact on the outcome of tumor radiotherapy. Front Oncol (2012) 2:116. doi:10.3389/fonc.2012.00116
204. Bower JE, Ganz PA, Tao ML, Hu W, Thomas R, Sepah S, et al. Inflammatory biomarkers and fatigue during radiation therapy for breast and prostate cancer. Clin Cancer Res (2010) 15:5534–40. doi:10.1158/1078-0432.CCR-08-2584
205. Schmidt ME, Meynköhn A, Habermann N, Wiskemann J, Oelmann J, Hof H, et al. Resistance exercise and inflammation in breast cancer patients undergoing adjuvant radiation therapy: mediation analysis from a randomized, controlled intervention trial. Int J Radiat Oncol Biol Phys (2016) 94:329–37. doi:10.1016/j.ijrobp.2015.10.058
206. Ohba K, Omagari K, Nakamura T, Ikuno N, Saeki S, Matsuo I, et al. Abscopal regression of hepatocellular carcinoma after radiotherapy for bone metastasis. Gut (1998) 43:575–7. doi:10.1136/gut.43.4.575
207. Ahlberg K, Ekman T, Gaston-Johansson F. Levels of fatigue compared to levels of cytokines and hemoglobin during pelvic radiotherapy: a pilot study. Biol Res Nurs (2004) 5:203–10. doi:10.1177/1099800403259500
208. Geinitz H, Zimmermann FB, Stoll P, Thamm R, Kaffenberger W, Ansorg K, et al. Fatigue, serum cytokine levels, and blood cell counts during radiotherapy of patients with breast cancer. Int J Radiat Oncol Biol Phys (2001) 51:691–8. doi:10.1016/S0360-3016(01)01657-1
209. Wratten C, Kilmurray J, Nash S, Seldon M, Hamilton CS, O’Brien PC, et al. Fatigue during breast radiotherapy and its relationship to biological factors. Int J Radiat Oncol Biol Phys (2004) 59:160–7. doi:10.1016/j.ijrobp.2003.10.008
210. Antoni MH. Psychosocial intervention effects on adaptation, disease course and biobehavioral processes in cancer. Brain Behav Immun (2013) 30:S88–98. doi:10.1016/j.bbi.2012.05.009
211. Mohammadinejad P, Arya P, Esfandbod M, Kaviani A, Najafi M, Kashani L, et al. Celecoxib versus diclofenac in mild to moderate depression management among breast cancer patients: a double-blind, placebo-controlled, randomized trial. Ann Pharmacother (2015) 49:953–61. doi:10.1177/1060028015592215
212. Derry HM, Jaremka LM, Bennett JM, Peng J, Andridge R, Shapiro C, et al. Yoga and self-reported cognitive problems in breast cancer survivors: a randomized controlled trial. Psychooncology (2015) 24:958–66. doi:10.1002/pon.3707
213. Mustian KM, Janelsins MC, Peppone LJ, Kamen CS, Guido JJ, Heckler CE. EXCAP exercise effects on cognitive impairment and inflammation: a URCC NCORP RCT in 479 cancer patients. J Clin Oncol (2015) 33:9504. doi:10.1200/jco.2015.33.15_suppl.9504
214. Bower JE, Garet D, Sternlieb B, Ganz PA, Irwin MR, Olmstead R, et al. Yoga for persistent fatigue in breast cancer survivors: a randomized controlled trial. Cancer (2012) 118:3766–75. doi:10.1002/cncr.26702
215. Bower JE, Greendale G, Crosswell AD, Garet D, Sternlieb B, Ganz PA, et al. Yoga reduces inflammatory signaling in fatigued breast cancer survivors: a randomized controlled trial. Psychoneuroendocrinology (2014) 43:20–9. doi:10.1016/j.psyneuen.2014.01.019
216. Oh B, Butow PN, Mullan BA, Clarke SJ, Beale PJ, Pavlakis N, et al. Effect of medical Qigong on cognitive function, quality of life, and a biomarker of inflammation in cancer patients: a randomized controlled trial. Support Care Cancer (2012) 20:1235–42. doi:10.1007/s00520-011-1209-6
217. Andersen BL, Thornton LM, Shapiro CL, Farrar WB, Mundy BL, Yang HC, et al. Biobehavioral, immune, and health benefits following recurrence for psychological intervention participants. Clin Cancer Res (2010) 16:3270–8. doi:10.1158/1078-0432.CCR-10-0278
218. Antoni MH, Lutgendorf SK, Blomberg B, Carver CS, Lechner S, Diaz A, et al. Cognitive-behavioral stress management reverses anxiety-related leukocyte transcriptional dynamics. Biol Psychiatry (2012) 71:366–72. doi:10.1016/j.biopsych.2011.10.007
219. Savard J, Simard S, Ivers H, Morin CM. Randomized study on the efficacy of cognitive-behavioral therapy for insomnia secondary to breast cancer, part I: sleep and psychological effects. J Clin Oncol (2005) 23:6083–96. doi:10.1200/JCO.2005.09.548
220. Savard J, Simard S, Ivers H, Morin CM. Randomized study on the efficacy of cognitive-behavioral therapy for insomnia secondary to breast cancer, part II: immunologic effects. J Clin Oncol (2005) 23:6097–106. doi:10.1200/JCO.2005.12.513
221. Monk JP, Phillips G, Waite R, Kuhn J, Schaaf LJ, Otterson GA, et al. Assessment of tumor necrosis factor alpha blockade as an intervention to improve tolerability of dose-intensive chemotherapy in cancer patients. J Clin Oncol (2006) 24:1852–9. doi:10.1200/JCO.2005.04.2838
222. Bower JE, Lamkin DM. Inflammation and cancer-related fatigue: mechanisms, contributing factors, and treatment implications. Brain Behav Immun (2013) 30:S48–57. doi:10.1016/j.bbi.2012.06.011
223. Winocur G, Wojtowicz JM, Huang J, Tannock IF. Physical exercise prevents suppression of hippocampal neurogenesis and reduces cognitive impairment in chemotherapy-treated rats. Psychopharmacology (Berl) (2014) 231:2311–20. doi:10.1007/s00213-013-3394-0
224. Ji JF, Ji SJ, Sun R, Li K, Zhang Y, Zhang LY, et al. Forced running exercise attenuates hippocampal neurogenesis impairment and the neurocognitive deficits induced by whole-brain irradiation via the BDNF-mediated pathway. Biochem Biophys Res Commun (2014) 443:646–51. doi:10.1016/j.bbrc.2013.12.031
225. Fardell JE, Vardy J, Shah JD, Johnston IN. Cognitive impairments caused by oxaliplatin and 5-fluorouracil chemotherapy are ameliorated by physical activity. Psychopharmacology (Berl) (2012) 220:183–93. doi:10.1007/s00213-011-2466-2
226. Norden DM, Mccarthy DO, Bicer S, Devine RD, Reiser PJ, Godbout JP, et al. Ibuprofen ameliorates fatigue- and depressive-like behavior in tumor-bearing mice. Life Sci (2015) 143:65–70. doi:10.1016/j.lfs.2015.10.020
227. Hu LY, Zhou Y, Cui WQ, Hu XM, Du LX, Mi WL, et al. Triggering receptor expressed on myeloid cells 2 (TREM2) dependent microglial activation promotes cisplatin-induced peripheral neuropathy in mice. Brain Behav Immun (2018) 68:132–45. doi:10.1016/j.bbi.2017.10.011
228. Aggarwal BB, Shishodia S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochem Pharmacol (2006) 71:1397–421. doi:10.1016/j.bcp.2006.02.009
229. Cheruku SP, Ramalingayya GV, Chamallamudi MR, Biswas S, Nandakumar K, Nampoothiri M, et al. Catechin ameliorates doxorubicin-induced neuronal cytotoxicity in in vitro and episodic memory deficit in in vivo in Wistar rats. Cytotechnology (2018) 70(1):245–59. doi:10.1007/s10616-017-0138-8
230. El-agamy SE, Abdel-aziz AK, Wahdan S, Esmat A, Azab SS. Astaxanthin ameliorates doxorubicin-induced cognitive impairment (chemobrain) in experimental rat model: impact on oxidative, inflammatory, and apoptotic machineries. Mol Neurobiol (2017) 1–14. doi:10.1007/s12035-017-0797-7
231. Ramalingayya GV, Cheruku SP, Nayak PG, Kishore A, Shenoy R, Rao CM, et al. Rutin protects against neuronal damage in vitro and ameliorates doxorubicin-induced memory deficits in vivo in Wistar rats. Drug Des Dev Ther (2017) 11:1011–26. doi:10.2147/DDDT.S103511
Keywords: depression, cognition, cytokines, neuroinflammation, neuropathic pain
Citation: Santos JC and Pyter LM (2018) Neuroimmunology of Behavioral Comorbidities Associated With Cancer and Cancer Treatments. Front. Immunol. 9:1195. doi: 10.3389/fimmu.2018.01195
Received: 05 December 2017; Accepted: 14 May 2018;
Published: 07 June 2018
Edited by:
Mireia Guerau-de-Arellano, The Ohio State University, United StatesReviewed by:
Elisabeth G. Vichaya, University of Texas MD Anderson Cancer Center, United StatesAttila Szabo, University of Oslo, Norway
Copyright: © 2018 Santos and Pyter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leah M. Pyter, bGVhaC5weXRlckBvc3VtYy5lZHU=