 Miguel G. Toscano
Miguel G. Toscano Peter de Haan
Peter de Haan- 1Amarna Therapeutics SL, Sevilla, Spain
- 2Amarna Therapeutics BV, Leiden, Netherlands
Viruses efficiently transfer and express their genes in host cells and evolve to evade the host’s defense responses. These properties render them highly attractive for use as gene delivery vectors in vaccines, gene, and immunotherapies. Among the viruses used as gene delivery vectors, the macaque polyomavirus Simian Virus 40 (SV40) is unique in its capacity to evade intracellular antiviral defense responses upon cell entry. We here describe the unique way by which SV40 particles deliver their genomes in the nucleus of permissive cells and how they prevent presentation of viral antigens to the host’s immune system. The non-immunogenicity in its natural host is not only of benefit to the virus but also to us in developing effective SV40 vector-based treatments for today’s major human diseases.
Introduction
As intracellular parasites, viruses hijack the host cell machinery to replicate, spread and survive. Host cells use membrane-bound and cytoplasmic receptors to sense pathogen-associated molecular patterns (PAMPs). After receptor-binding, viral structural proteins may serve as PAMPs and bind toll-like receptors (TLRs) that are located on the cell surface or on endosomal membranes. After replication, virus-specific RNAs serve as PAMPs and bind cytoplasmic RIG-I-like receptors (RLRs). Activation of TLRs or RLRs leads to the assembly of inflammasomes that induce an inflammatory response (1, 2). Inflammation is a highly orchestrated cascade of processes aimed at confining the infection and ultimately in inducing an adaptive immune response directed to peptides (antigens) derived from viral proteins that are presented on major histocompatibility (MHC) molecules on the surface of cells of the immune system.
Simian Virus 40 (SV40), the type member of the Polyomaviridae family, was discovered in the fifties of the previous century as a contaminating virus in the polio vaccines that in those days were produced in primary cells from macaques (3, 4). Since then, SV40’s DNA genome was the first animal virus genome to be characterized (5, 6). SV40 served as the model virus to study molecular and biochemical processes in eukaryote organisms (7). The first mammalian viral gene delivery vector was derived from SV40 (8) and pioneering gene transfer studies using replication-defective SV40 vectors ultimately resulted in the recent approval of the first viral vector-based gene therapies to the market (9), albeit that the currently used vectors are derived from adeno-associated virus (AAV) or the human immunodeficiency virus type 1 (HIV-1).
SV40 is a macaque polyomavirus consisting of icosahedral particles of 45 nm in diameter (10, 11). The virus particle consists of 72 pentamers of the major viral protein VP1. On the inside of the capsid, each pentamer forms a hydrophobic pocket that is bound to one monomer of the viral proteins VP2 or VP3 (12). Each particle contains a single copy of the viral genome, a circular 5.2 kilobase pairs long double-stranded DNA molecule packaged with histones to form a mini-chromosome. The SV40 genomic DNA has two genes. The early gene encodes two non-structural replication-associated proteins: small T antigen and large T antigen. The late gene codes for the structural viral proteins VP1, VP2, and VP3, respectively (13, 14).
In macaques, SV40 causes chronic asymptomatic infections (15). Children who received the SV40-contaminated poliovirus vaccine did not develop an adaptive immune response to the virus particles and excreted SV40 in their stools within 5 weeks after vaccination (16). This indicates that SV40 capsids do not serve as PAMPs and that the virus does not replicate in human cells. Studies in animals administered with replication-defective SV40 vector particles in the absence of adjuvants (PAMPs) do not result in the induction of an adaptive immune response to SV40, demonstrating that SV40 particles are non-immunogenic in vivo (17, 18). This implies that SV40 after its entrance into permissive cells (Figure 1) is able to efficiently evade TLR binding and prevents presentation of viral antigens on MHC molecules to cells of the host’s immune system.
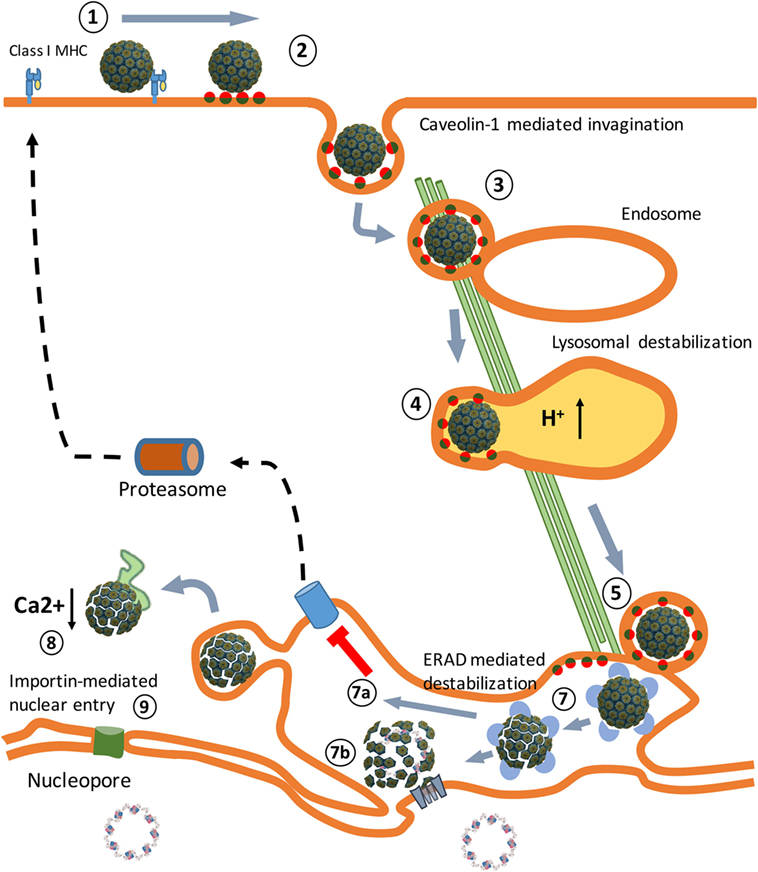
Figure 1. An overview of the Simian Virus 40 (SV40) cell entry process. (1) Binding of the virus particles to major histocompatibility class I (MHC-I) molecules that target them to lipid rafts, enriched in GM1 molecules. (2) Release from MHC-I molecules, binding to GM1 molecules, and caveolae vesicle formation. (3) Endosome internalization. (4) Endosome maturation and particle destabilization. (5) Release from endolysosomes and endoplasmic reticulum (ER) trafficking. (6) ER-associated degradation (ERAD)-mediated particle destabilization. (7a) ERAD-mediated cytosol transport. (7b) Viroporin-mediated nuclear entrance. (8) Cytosol destabilization. (9) Nucleopore-mediated nuclear entrance. The SV40 image was created using VMD software (19) PDB ID: 1SVA (20) and is a courtesy of Dr. J.-Y. Sgro, UW-Madison, USA (http://www.virology.wisc.edu/virusworld/).
The non-immunogenicity of SV40 combined with the absence of an immune memory for this macaque polyomavirus in the human population is of benefit to us, since it renders SV40 highly attractive for use as a gene delivery vector in gene and immunotherapies.
Endocytosis
SV40 binds MHC class I (MHC-I) molecules present on the surface of all body cells (21–23). Once bound, the SV40–MHC-I complexes migrate to caveolin-enriched membrane domains named caveolin pits also known as lipid rafts (Figure 2). Caveolin pits are cell surface membrane domains enriched in cholesterol, gangliosides, glycosphingolipids, and protein receptors including MHC-I molecules that are involved in endocytosis and transcytosis (24–27).
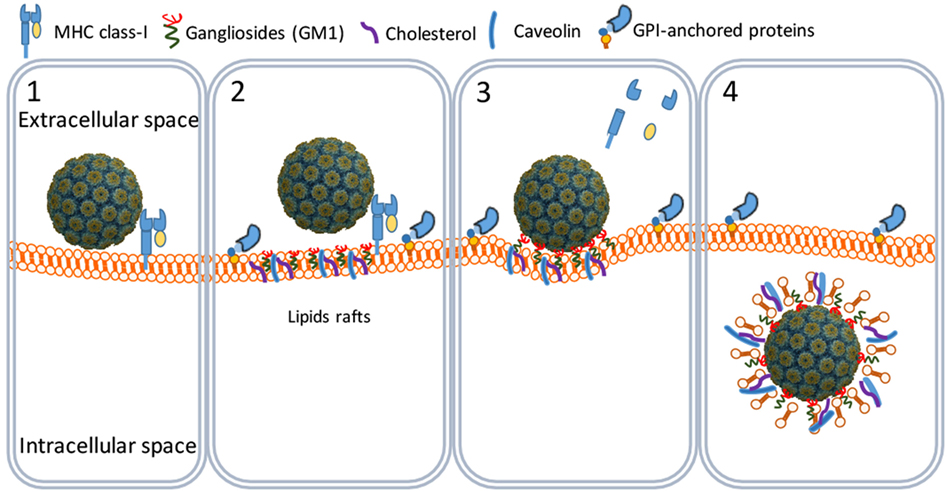
Figure 2. SV40 cell attachment and internalization. SV40 particles attach to the cell surface by binding to major histocompatibility class I molecules (1) that shuttle the virus particles to GM1-rich lipid rafts (2). The particles bind GM1 and induce the formation of caveolar endocytic vesicles (3) and get into the cytosol through an active translocation mediated by glycosylphosphatidylinositol (GPI)-anchored proteins (4).
At the caveolin pits, the MHC-I molecules are degraded by metalloproteinases whereby the SV40 particles bind to membrane ganglioside GM1 molecules which are considered their endocytic receptors (28, 29). Binding of SV40 particles to GM1 induces a curvature of the cell membrane that results in the formation of endocytic vesicles known as caveolae (20, 24, 26, 30). Caveolae are circular or tubular vesicles of 70–100 nm in diameter and usually contain one SV40 particle. Next to GM1, cholesterol and tyrosine kinases are needed for the formation of caveolae, since nystatin (a cholesterol sequestering agent) and genistein (a tyrosine kinase inhibitor) efficiently block the translocation of SV40 particles into the caveolae (Figure 2) (29, 31–33).
The caveolar tyrosine kinases promote the recruitment of the cytoskeleton proteins actin and dynamin II (30, 34, 35) and assisted by Rab5 (a GTP-binding protein) and Arf1 (a GTP-ase) the caveolae traffic along the cytoskeleton to early endosomes (Figure 3) (36, 37). The SV40 particles remain bound to the membrane-associated GM1 molecules in endosomes during their maturation to late endosomes and endolysosomes. At this stage, the cell entry process of polyomaviruses differs from that of other viruses. Most viruses directly move from the endolysosome to the nucleus (38, 39). However, before they traffic to the nucleus, major part of their structural proteins is degraded by the lysosomal proteases yielding viral peptides which are loaded as antigens on MHC class II (MHC-II) molecules (40). MHC-II molecules are expressed in antigen-presenting cells (APCs) that are involved in the induction of adaptive immune responses.
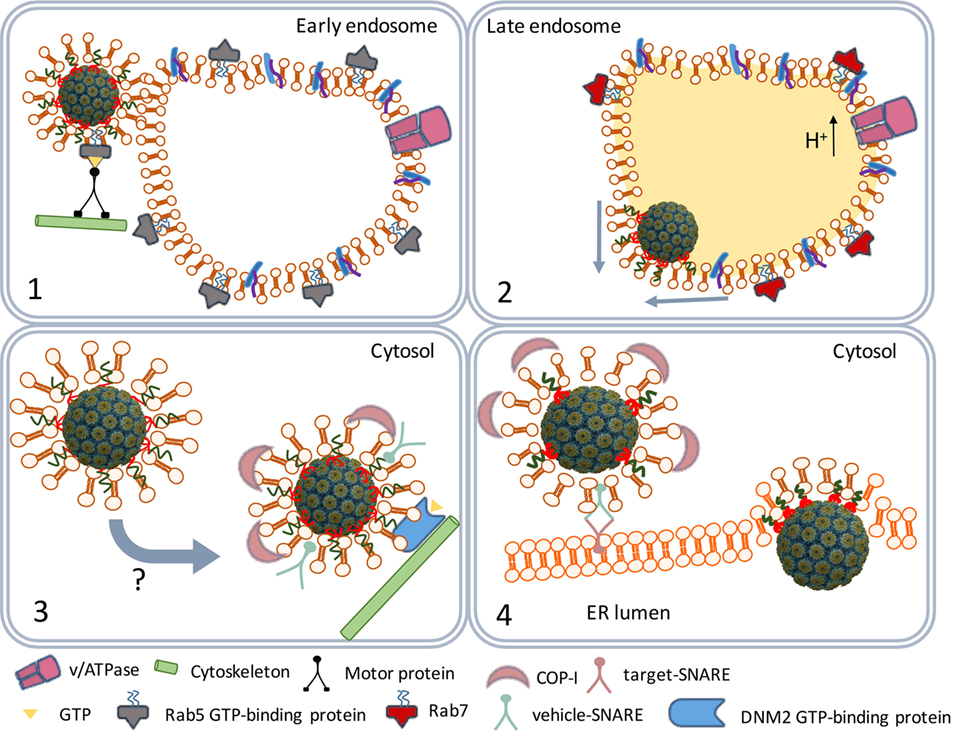
Figure 3. SV40 endocytosis and transport to the endoplasmic reticulum (ER). Scheme depicting the traffic of SV40-containing caveolar vesicles to early endosomes (1). The maturation of endosomes and fusion with lysosomes to endolysosomes (2). The traffic of coat protein I (COPI)-decorated vesicles to the ER (3). ER internalization (4).
Polyomaviruses, on the contrary, traffic from the endolysosome to the endoplasmic reticulum (ER) (36). The acidic environment in the endolysosome renders SV40 particles susceptible to successive disassembly steps later in the ER. However, before the virus particles are degraded by lysosomal enzymes they leave the endolysosome. The GM1 molecules that remained bound to the SV40 particles mediate the budding from the endolysosome membranes, it has remained unknown which factors are responsible for the timing of the budding process (41). The early exit from the endolysosomes prevents degradation of the SV40 structural proteins by lysosomal enzymes. As a result, SV40 antigens are not loaded on MHC-II molecules and presented by APCs to lymphocytes.
ER Processing and Nuclear Entry
The virus-containing vesicles traffic from the endolysosome to the ER using the trans-Golgi network, a bidirectional vesicle trafficking route between ER and Golgi apparatus (Figure 3) (42). Trans-Golgi network vesicles are coated with coat protein I (COPI) complex proteins originating from the lysosomal membranes (43, 44). The COPI-coated vesicles containing the SV40 particles fuse with the ER membranes (45) releasing the virus particles into the ER lumen.
The ER-associated degradation (ERAD) system is a protein quality control mechanism that recognizes nascent polypeptides and assists them in their correct folding or degradation by cytoplasmic proteasomes (46). The SV40 particles are recognized by the ERAD system as misfolded proteins. Peptide disulfide isomerase and ER protein 57 bind to and reduce the disulfide bonds that stabilize the VP1 pentamers (Figure 4). The pentamers become less tightly associated with each other and the VP2 and VP3 become exposed to the exterior (47). Indeed, in vitro studies confirmed that the SV40 particles in the ER are larger than those in the cytosol (48). The hydrophobicity of VP2 and VP3 renders the virus particles prone to aggregation. Aggregation is prevented by binding to the molecular chaperone BiP. Usually, proteins to be degraded bind a membrane-bound protein complex containing Hrd1 that targets them for degradation by cytoplasmic proteasomes. In this degradation process, specific peptides derived from the proteins are loaded as antigens on MHC-I molecules to be presented at the cell surface to cells of the host’s immune system. MHC-I molecules are involved in the induction of cellular immune responses. Polyomaviruses, however, do not bind Hrd1-containing complexes and are not loaded to proteasomes. SV40 thus has developed an effective mechanism to prevent being targeted for proteasome degradation (47, 49). This implies that SV40 is also capable of avoiding presentation on MHC-I molecules, thereby preventing the induction of cellular antiviral immune responses upon infection.
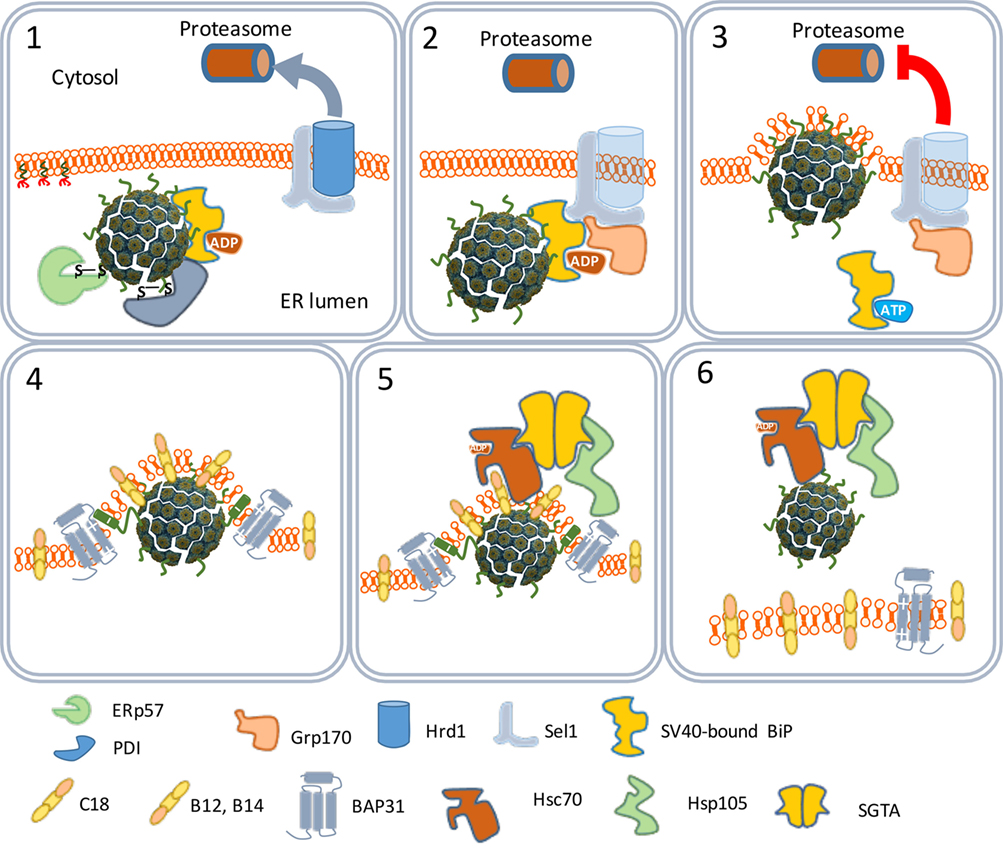
Figure 4. The traffic of SV40 particles from the endoplasmic reticulum (ER) to the cytosol. SV40 particles are destabilized by the ER-associated degradation machinery (1) and remain associated with the ER membrane (2). But instead of leaving the ER by binding Hrd1 and to be loaded to cytoplasmic proteasomes (3), the destabilized particles interact with ER membrane-resident proteins (4) and associate with cytosolic chaperones (5) to move to the cytosol (6).
One scenario to explain this phenomenon is that the virus particles use an extra step via the cytoplasm to evade proteasome degradation and reach the nucleus. In this scenario, the exit of the virus–BiP complexes from the ER to the cytosol is facilitated by proteins of the ERAD system in combination with cytosolic chaperones and takes place at particular domains on the ER membrane named foci (Figure 4) (50–52). At the foci, the destabilized virus particles are pulled-out from the ER and released into the cytoplasm (53). In the cytoplasm, VP1 is removed from the SV40 particles due to the action of chaperones (53) and the local physiological conditions (54). The nuclear localization signals present on VP2 and VP3 bind α/β importins (55–58) that mediate the transport of the SV40 genetic material into the nucleus through the nucleopores (Figure 5) (59–61).
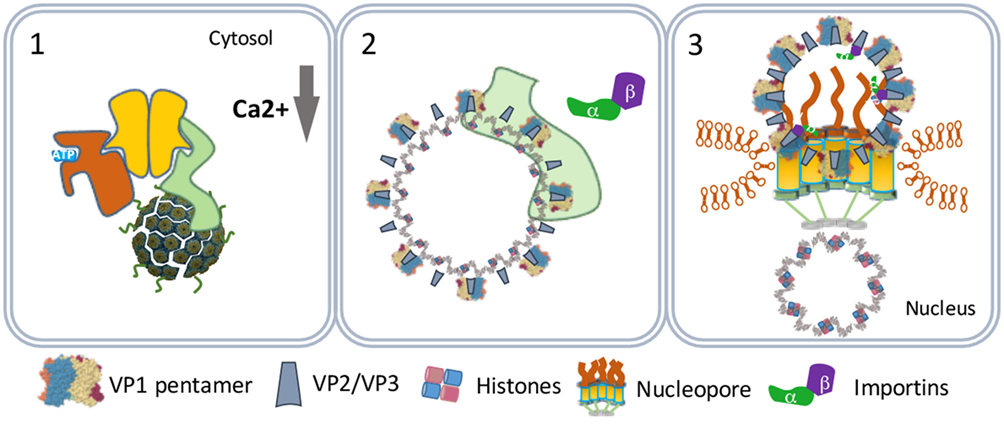
Figure 5. SV40 nuclear entry. SV40 particles further destabilize by low calcium in the cytosol (1) and lose VP1 by cytosolic chaperones (2). The particles bind nucleopores and viral genomes enter the nucleus (3).
In another scenario, the virus particles directly move from the ER lumen to the nucleus. This scenario relies on the capacity of purified VP2 and VP3 monomers to insert in membranes forming pore-like structures named viroporins (62, 63). The ERAD-mediated destabilized SV40 particles allow the formation of VP2/VP3 viroporins on the inner nuclear membranes. The viroporins subsequently pull the SV40 genomes into the nucleus (Figure 6) (64).
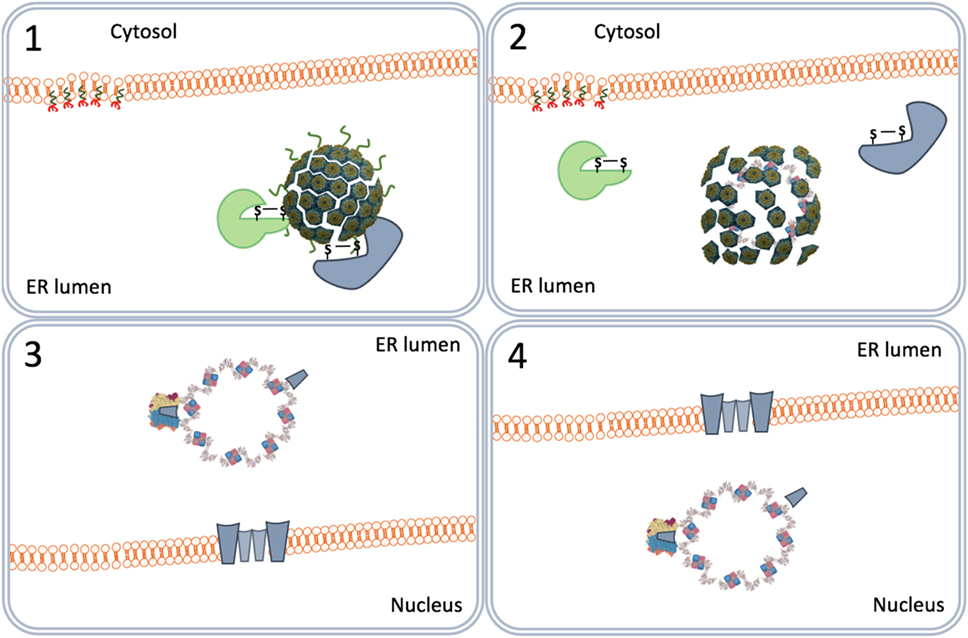
Figure 6. Nuclear entry by viroporins. The virus particles destabilized by the endoplasmic reticulum-associated degradation system (1) form VP2 and VP3 viroporins (2) on the inner nuclear membrane (3) that pull the viral genomes into the nucleus (4).
The Presence of SV40 in the Human Population
Polyomaviruses cause chronic symptomless infections in their hosts and since they are replication-competent sustained adaptive immune responses to the virus are induced in infected hosts (65–69). In humans, polyomaviral particles can be found in the kidney and urine of immunocompetent individuals and in kidney, brain, lung, or peripheral blood mononuclear cells in immunocompromised individuals.
Since the massive administration of children with SV40 in the fifties and sixties of the previous century, many studies have been performed to determine the consequences of this undesired vaccination. However, the epidemiological studies to identify SV40 seropositive individuals were inconclusive. The assays used in macaques to detect SV40 are not reliable in humans (70, 71). Only a small number of individuals vaccinated with SV40-contaminated polio vaccine developed antibodies to SV40 (16, 72) and the very low antibody titers suggest that SV40 does not replicate in human cells (73, 74). Finally, the presumed SV40 antibodies disappeared with time, indicating that the seropositive individuals were not chronically infected with SV40 (71, 75, 76). From a study with zoo workers that were in close contact with macaques for a long time, it was concluded that SV40 does not replicate in humans (70, 71, 74, 77). Overall, from all epidemiologic studies, the Institute of Medicine from the National Institutes of Health in the USA concluded that humans are not a host of SV40 and that SV40 is not a human pathogen (78).
Concluding Remarks
The successful entry into a host cell is a crucial step in the virus replication cycle. Among all viruses, polyomaviruses including SV40 have developed a unique way of entering a permissive cell and expressing its genetic information in the nucleus of an infected cell. The viral particles prevent activation of TLRs, escape from the proteasome, and thus evade antigen presentation to cells of the host’s immune system during this initial stage of infection. The serological analysis of hosts naturally infected with polyomaviruses shows long-lasting adaptive immune responses, indicating that replicating polyomaviruses activate RLRs (79).
Epidemiological studies revealed that humans are not a host for SV40 and that this macaque polyomavirus does not replicate in humans. Therefore, the human population is considered to be immunologically naïve for SV40. On the basis of these findings, it is expected that replication-defective SV40 gene delivery vectors are completely non-immunogenic in humans.
In a number of reports, it has been shown that replication-defective viral gene delivery vectors such as vectors derived from AAV and HIV-1 induce immune tolerance to the transgene proteins when administered to hosts that are naïve to the cognate virus (80–83). These studies indicate that replication-defective SV40 vectors are ideally suited for inducing immune tolerance to the transgene proteins in humans (17, 18). This is crucial for designing effective gene replacement therapies where long-term transgene expression in the target tissue is required to cure patients from inherited diseases. In addition, the capacity of SV40 vectors to induce immune tolerance opens the way to treat autoimmune diseases by restoring the immune tolerance to primary self-antigens involved in the autoimmune tissue destruction.Restoration of immune tolerance to self-antigens using viral gene delivery vectors is named reverse viral vector vaccination and has been a longstanding goal in autoimmunity research. To date, diabetes mellitus type 1, multiple sclerosis, and arthritis are the best studied autoimmune diseases. In rodent models of these diseases, it has been shown that replication-defective AAV or HIV-1 vectors encoding the primary self-antigens of the disease highly efficiently protect and cure the treated animals from the autoimmune disease (84–89). With our rapidly increasing knowledge on immunology, the list of autoimmune diseases is growing and includes the major degenerative diseases of our time such as cardiovascular diseases (90), neurodegenerative and psychiatric diseases (91), obesity, diabetes mellitus type 2 (92, 93), arthritis and pulmonary diseases (94). Moreover, the induction of immune tolerance in recipients to MHC-I molecules of donor cells will improve the success rate of tissue transplantations.
HIV-1 derived vector particles are instable, rapidly degraded when administered in vivo, and for these reasons only used for ex vivo gene therapy to treat blood-related genetic disorders or cancer. To date, AAV vectors are the most popular for use in in vivo gene therapy. However, the majority of the human population encountered wild-type AAV together with its helper virus (adenovirus, causing the common cold) and developed an immune memory against the AAV capsid proteins. Clinical studies revealed that because of the immune memory in humans, the in vivo efficacy of AAV vectors is very low. SV40 vectors are the only gene delivery vectors that can be used for inducing immune tolerance to transgene proteins in humans and for this reason the oldest viral gene delivery vector will be key to the successful development of effective interventions for today’s major diseases (95).
Author Contributions
MGT and PH wrote the manuscript and MGT designed the figures.
Conflict of Interest Statement
Both authors are employed by Amarna Therapeutics. The company holds patents on the production and use of polyomaviral gene delivery vectors.
Acknowledgments
MGT was co-funded by the Torres Quevedo program PTQ-13-06051 from the Spanish Ministry of Economy, Industry and Competitiveness. We thank Dr. Daniel C. DiMaio (Yale Cancer Center) for his comments to the manuscript.
References
1. Kumar H, Kawai T, Akira S. Pathogen recognition in the innate immune response. Biochem J (2009) 420:1–16. doi:10.1042/BJ20090272
2. Thompson AJV, Locarnini SA. Toll-like receptors, RIG-I-like RNA helicases and the antiviral innate immune response. Immunol Cell Biol (2007) 85:435–45. doi:10.1038/sj.icb.7100100
3. Eddy BE, Rowe WP, Hartley JW, Stewart SE, Huebner RJ. Hemagglutination with the SE polyoma virus. Virology (1958) 6:290–1. doi:10.1016/0042-6822(58)90078-3
4. Sweet BH, Hilleman MR. The vacuolating virus, SV 40. Proc Soc Exp Biol Med (1960) 105:420–7. doi:10.3181/00379727-105-26128
5. Danna KJ, Sack GH, Nathans D. Studies of simian virus 40 DNA. VII. A cleavage map of the SV40 genome. J Mol Biol (1973) 78:363–76. doi:10.1016/0022-2836(73)90122-8
6. Fiers W, Contreras R, Haegemann G, Rogiers R, Van de Voorde A, Van Heuverswyn H, et al. Complete nucleotide sequence of SV40 DNA. Nature (1978) 273:113–20. doi:10.1038/273113a0
7. Howley PM, Livingston DM. Small DNA tumor viruses: large contributors to biomedical sciences. Virology (2009) 384:256–9. doi:10.1016/j.virol.2008.12.006
8. Hamer DH, Leder P. Expression of the chromosomal mouse beta maj-globin gene cloned in SV40. Nature (1979) 281:35–40. doi:10.1038/281035a0
9. Touchot N, Flume M. Early insights from commercialization of gene therapies in Europe. Genes (Basel) (2017) 8:E78. doi:10.3390/genes8020078
10. Liddington RC, Yan Y, Moulai J, Sahli R, Benjamin TL, Harrison SC. Structure of simian virus 40 at 3.8-A resolution. Nature (1991) 354:278–84. doi:10.1038/354278a0
11. Modis Y, Trus BL, Harrison SC. Atomic model of the papillomavirus capsid. EMBO J (2002) 21:4754–62. doi:10.1093/emboj/cdf494
12. Chen XS, Stehle T, Harrison SC. Interaction of polyomavirus internal protein VP2 with the major capsid protein VP1 and implications for participation of VP2 in viral entry. EMBO J (1998) 17:3233–40. doi:10.1093/emboj/17.12.3233
13. Cole CN. Polyomaviridae: the viruses and their replication. Fields virology (Vol. 2). Philadelphia: Lippincott-Raven (1996). p. 1997–2025.
14. Hassell J, Brinton B. SV40 and Polyomavirus DNA Replication. In: DePamphilis ML, editor. DNA replication in eukaryotic cells. New York: Cold Spring Harbor Laboratory Press (1996). p. 639–677.
15. Shah K, Nathanson N. Human exposure to SV40: review and comment. Am J Epidemiol (1976) 103:1–12. doi:10.1093/oxfordjournals.aje.a112197
16. Melnick JL, Stinebaugh S. Excretion of vacuolating SV-40 virus (papova virus group) after ingestion as a contaminant of oral poliovaccine. Proc Soc Exp Biol Med (1962) 109:965–8. doi:10.3181/00379727-109-27392
17. Eid L, Bromberg Z, El-Latif MA, Zeira E, Oppenheim A, Weiss YG. Simian virus 40 vectors for pulmonary gene therapy. Respir Res (2007) 8:74. doi:10.1186/1465-9921-8-74
18. Kondo R, Feitelson MA, Strayer DS. Use of SV40 to immunize against hepatitis B surface antigen: implications for the use of SV40 for gene transduction and its use as an immunizing agent. Gene Ther (1998) 5:575–82. doi:10.1038/sj.gt.3300623
19. Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph (1996) 14:33–8. doi:10.1016/0263-7855(96)00018-5
20. Stehle T, Gamblin SJ, Yan Y, Harrison SC. The structure of simian virus 40 refined at 3.1 A resolution. Structure (1996) 4:165–82. doi:10.1016/S0969-2126(96)00020-2
21. Atwood WJ, Norkin LC. Class I major histocompatibility proteins as cell surface receptors for simian virus 40. J Virol (1989) 63:4474–7.
22. Breau WC, Atwood WJ, Norkin LC. Class I major histocompatibility proteins are an essential component of the simian virus 40 receptor. J Virol (1992) 66:2037–45.
23. Stang E, Kartenbeck J, Parton RG. Major histocompatibility complex class I molecules mediate association of SV40 with caveolae. Mol Biol Cell (1997) 8:47–57. doi:10.1091/mbc.8.1.47
24. Laude AJ, Prior IA. Plasma membrane microdomains: organization, function and trafficking (review). Mol Membr Biol (2009) 21:193–205. doi:10.1080/09687680410001700517
25. Palade GE. Fine structure of blood capillaries. J Appl Phys (1953) 24:1424. doi:10.1161/01.RES.27.3.482
26. Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR, Anderson RG. Caveolin, a protein component of caveolae membrane coats. Cell (1992) 68:673–82. doi:10.1016/0092-8674(92)90143-Z
27. Yamada E. The fine structure of the gall bladder epithelium of the mouse. J Biophys Biochem Cytol (1955) 1:445–58. doi:10.1083/jcb.1.5.445
28. Anderson HA, Chen Y, Norkin LC. MHC class I molecules are enriched in caveolae but do not enter with simian virus 40. J Gen Virol (1998) 79:1469–77. doi:10.1099/0022-1317-79-6-1469
29. Tsai B, Gilbert JM, Stehle T, Lencer W, Benjamin TL, Rapoport TA. Gangliosides are receptors for murine polyoma virus and SV40. EMBO J (2003) 22:4346–55. doi:10.1093/emboj/cdg439
30. Ewers H, Römer W, Smith AE, Bacia K, Dmitrieff S, Chai W, et al. GM1 structure determines SV40-induced membrane invagination and infection. Nat Cell Biol (2009) 12:11–8. doi:10.1038/ncb1999
31. Chen Y, Norkin LC. Extracellular simian virus 40 transmits a signal that promotes virus enclosure within caveolae. Exp Cell Res (1999) 246:83–90. doi:10.1006/excr.1998.4301
32. Dangoria NS, Breau WC, Anderson HA, Cishek DM, Norkin LC. Extracellular simian virus 40 induces an ERK/MAP kinase-independent signalling pathway that activates primary response genes and promotes virus entry. J Gen Virol (1996) 77(Pt 9):2173–82. doi:10.1099/0022-1317-77-9-2173
33. Pelkmans L, Kartenbeck J, Helenius A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat Cell Biol (2001) 3:473–83. doi:10.1038/35074539
34. Pelkmans L, Püntener D, Helenius A. Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science (2002) 296:535–9. doi:10.1126/science.1069784
35. Tagawa A, Mezzacasa A, Hayer A, Longatti A, Pelkmans L, Helenius A. Assembly and trafficking of caveolar domains in the cell. J Cell Biol (2005) 170:769–79. doi:10.1083/jcb.200506103
36. Engel S, Heger T, Mancini R, Herzog F, Kartenbeck J, Hayer A, et al. Role of endosomes in simian virus 40 entry and infection. J Virol (2011) 85:4198–211. doi:10.1128/JVI.02179-10
37. Mercer J, Schelhaas M, Helenius A. Virus entry by endocytosis. Annu Rev Biochem (2010) 79:803–33. doi:10.1146/annurev-biochem-060208-104626
38. Bartlett JS, Wilcher R, Samulski RJ. Infectious entry pathway of adeno-associated virus and adeno-associated virus vectors. J Virol (2000) 74:2777–85. doi:10.1128/JVI.74.6.2777-2785.2000
39. Kobiler O, Drayman N, Butin-Israeli V, Oppenheim A. Virus strategies for passing the nuclear envelope barrier. Nucleus (2014) 3:526–39. doi:10.4161/nucl.21979
40. Mantegazza AR, Magalhaes JG, Amigorena S, Marks MS. Presentation of phagocytosed antigens by MHC class I and II. Traffic (2012) 14:135–52. doi:10.1111/tra.12026
41. Qian M, Cai D, Verhey KJ, Tsai B. A lipid receptor sorts polyomavirus from the endolysosome to the endoplasmic reticulum to cause infection. PLoS Pathog (2009) 5:e1000465. doi:10.1371/journal.ppat.1000465
42. Orci L, Glick BS, Rothman JE. A new type of coated vesicular carrier that appears not to contain clathrin: its possible role in protein transport within the Golgi stack. Cell (1986) 46:171–84. doi:10.1016/0092-8674(86)90734-8
43. Norkin LC, Kuksin D. The caveolae-mediated sv40 entry pathway bypasses the Golgi complex en route to the endoplasmic reticulum. Virol J (2005) 2:38. doi:10.1186/1743-422X-2-38
44. Norkin LC, Anderson HA, Wolfrom SA, Oppenheim A. Caveolar endocytosis of simian virus 40 is followed by brefeldin A-sensitive transport to the endoplasmic reticulum, where the virus disassembles. J Virol (2002) 76:5156–66. doi:10.1128/JVI.76.10.5156-5166.2002
45. Brandizzi F, Barlowe C. Organization of the ER-Golgi interface for membrane traffic control. Nat Rev Mol Cell Biol (2013) 14:382–92. doi:10.1038/nrm3588
46. Byun H, Gou Y, Zook A, Lozano MM, Dudley JP. ERAD and how viruses exploit it. Front Microbiol (2014) 5:330. doi:10.3389/fmicb.2014.00330
47. Schelhaas M, Malmström J, Pelkmans L, Haugstetter J, Ellgaard L, Grünewald K, et al. Simian virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell (2007) 131:516–29. doi:10.1016/j.cell.2007.09.038
48. Inoue T, Tsai B. A large and intact viral particle penetrates the endoplasmic reticulum membrane to reach the cytosol. PLoS Pathog (2011) 7:e1002037. doi:10.1371/journal.ppat.1002037
49. Geiger R, Andritschke D, Friebe S, Herzog F, Luisoni S, Heger T, et al. BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat Cell Biol (2011) 13:1305–14. doi:10.1038/ncb2339
50. Bagchi P, Walczak CP, Tsai B. The endoplasmic reticulum membrane J protein C18 executes a distinct role in promoting simian virus 40 membrane penetration. J Virol (2015) 89:4058–68. doi:10.1128/JVI.03574-14
51. Goodwin EC, Lipovsky A, Inoue T, Magaldi TG, Edwards APB, Van Goor KEY, et al. BiP and multiple DNAJ molecular chaperones in the endoplasmic reticulum are required for efficient simian virus 40 infection. MBio (2011) 2:e101–11. doi:10.1128/mBio.00101-11
52. Walczak CP, Ravindran MS, Inoue T, Tsai B. A cytosolic chaperone complexes with dynamic membrane J-proteins and mobilizes a nonenveloped virus out of the endoplasmic reticulum. PLoS Pathog (2014) 10:e1004007. doi:10.1371/journal.ppat.1004007
53. Kuksin D, Norkin LC. Disassociation of the SV40 genome from capsid proteins prior to nuclear entry. Virol J (2011) 9:158–158. doi:10.1186/1743-422X-9-158
54. Li PP, Naknanishi A, Tran MA, Ishizu KI, Kawano M, Phillips M, et al. Importance of Vp1 calcium-binding residues in assembly, cell entry, and nuclear entry of simian virus 40. J Virol (2003) 77:7527–38. doi:10.1128/JVI.77.13.7527-7538.2003
55. Clever J, Dean DA, Kasamatsu H. Identification of a DNA binding domain in simian virus 40 capsid proteins Vp2 and Vp3. J Biol Chem (1993) 268:20877–83.
56. Gordon-Shaag A, Ben-nun-Shaul O, Roitman V, Yosef Y, Oppenheim A. Cellular transcription factor Sp1 recruits simian virus 40 capsid proteins to the viral packaging signal, ses. J Virol (2002) 76:5915–24. doi:10.1128/JVI.76.12.5915-5924.2002
57. Li PP, Nakanishi A, Shum D, Sun PC, Salazar AM, Fernandez CF, et al. Simian virus 40 Vp1 DNA-binding domain is functionally separable from the overlapping nuclear localization signal and is required for effective virion formation and full viability. J Virol (2001) 75:7321–9. doi:10.1128/JVI.75.16.7321-7329.2001
58. Soussi T. DNA-binding properties of the major structural protein of simian virus 40. J Virol (1986) 59:740–2.
59. Nakanishi A, Shum D, Morioka H, Otsuka E, Kasamatsu H. Interaction of the Vp3 nuclear localization signal with the importin 2/heterodimer directs nuclear entry of infecting simian virus 40. J Virol (2002) 76:9368–77. doi:10.1128/JVI.76.18.9368-9377.2002
60. Nakanishi A, Li PP, Qu Q, Jafri QH, Kasamatsu H. Molecular dissection of nuclear entry-competent SV40 during infection. Virus Res (2007) 124:226–30. doi:10.1016/j.virusres.2006.10.001
61. Yamada M, Kasamatsu H. Role of nuclear pore complex in simian virus 40 nuclear targeting. J Virol (1993) 67:119–30.
62. Daniels R, Rusan NM, Wilbuer AK, Norkin LC, Wadsworth P, Hebert DN. Simian virus 40 late proteins possess lytic properties that render them capable of permeabilizing cellular membranes. J Virol (2006) 80:6575–87. doi:10.1128/JVI.00347-06
63. Giorda KM, Raghava S, Zhang MW, Hebert DN. The viroporin activity of the minor structural proteins VP2 and VP3 is required for SV40 propagation. J Biol Chem (2013) 288:2510–20. doi:10.1074/jbc.M112.428425
64. Daniels R, Rusan NM, Wadsworth P, Hebert DN. SV40 VP2 and VP3 insertion into ER membranes is controlled by the capsid protein VP1: implications for DNA translocation out of the ER. Mol Cell (2006) 24:955–66. doi:10.1016/j.molcel.2006.11.001
65. de Sanjose S, Shah K, Engels EA, Viscidi RP. Lack of serological evidence for an association between simian virus 40 and lymphoma. Int J Cancer (2003) 107:507–8. doi:10.1002/ijc.11326
66. Eash S, Manley K, Gasparovic M, Querbes W, Atwood WJ. The human polyomaviruses. Cell Mol Life Sci (2006) 63:865–76. doi:10.1007/s00018-005-5454-z
67. Elsner C, Dörries K. Evidence of human polyomavirus BK and JC infection in normal brain tissue. Virology (1992) 191:72–80. doi:10.1016/0042-6822(92)90167-N
68. Ilyinskii PO, Daniel MD, Horvath CJ, Desrosiers RC. Genetic analysis of simian virus 40 from brains and kidneys of macaque monkeys. J Virol (1992) 66:6353–60.
69. Newman JS, Baskin GB, Frisque RJ. Identification of SV40 in brain, kidney and urine of healthy and SIV-infected rhesus monkeys. J Neurovirol (1998) 4:394–406. doi:10.3109/13550289809114538
70. Engels EA, Switzer WM, Heneine W, Viscidi RP. Serologic evidence for exposure to simian virus 40 in North American zoo workers. J Infect Dis (2004) 190:2065–9. doi:10.1086/425997
71. Vilchez RA, Butel JS. Re: lack of serologic evidence for prevalent simian virus 40 infection in humans. J Natl Cancer Inst (2004) 96:633–633. doi:10.1093/jnci/djh098
72. Carter JJ, Madeleine MM, Wipf GC, Garcea RL, Pipkin PA, Minor PD, et al. Lack of serologic evidence for prevalent simian virus 40 infection in humans. J Natl Cancer Inst (2003) 95:1522–30. doi:10.1093/jnci/djg074
73. Shah KV. Neutralizing antibodies to simian virus 40 (SV40) in human sera from India. Proc Soc Exp Biol Med (1966) 121:303–7. doi:10.3181/00379727-121-30765
74. Viscidi RP, Rollison DEM, Viscidi E, Clayman B, Rubalcaba E, Daniel R, et al. Serological cross-reactivities between antibodies to simian virus 40, BK virus, and JC virus assessed by virus-like-particle-based enzyme immunoassays. Clin Diagn Lab Immunol (2003) 10:278–85. doi:10.1128/CDLI.10.2.278-285.2003
75. Garcea RL, Imperiale MJ. Simian virus 40 infection of humans. J Virol (2003) 77:5039–45. doi:10.1128/JVI.77.9.5039-5045.2003
76. Lundstig A, Dillner J. Serological diagnosis of human polyomavirus infection. In: Ahsan N, editor. Polyomaviruses and Human Diseases Advances in Experimental Medicine and Biology. New York, NY: Springer (2006). p. 96–101.
77. Simon MA. Polyomaviruses of nonhuman primates: implications for research. Comp Med (2008) 58:51–6.
78. Institute of Medicine Immunization Safety Review Committee, Stratton K, Almario DA, McCormick MC. Immunization Safety Review: SV40 Contamination of Polio Vaccine and Cancer. Washington, DC: National Academies Press (US) (2002).
79. Horvath CJ, Simon MA, Bergsagel DJ, Pauley DR, King NW, Garcea RL, et al. Simian virus 40-induced disease in rhesus monkeys with simian acquired immunodeficiency syndrome. Am J Pathol (1992) 140:1431–40.
80. Boisgerault F, Mingozzi F. The skeletal muscle environment and its role in immunity and tolerance to AAV vector-mediated gene transfer. Curr Gene Ther (2015) 15:381–94. doi:10.2174/1566523215666150630121750
81. Dobrzynski E, Herzog RW. Tolerance induction by viral in vivo gene transfer. Clin Med Res (2005) 3:234–40. doi:10.3121/cmr.3.4.234
82. Sack BK, Herzog RW, Terhorst C, Markusic DM. Development of gene transfer for induction of antigen-specific tolerance. Mol Ther Methods Clin Dev (2014) 1:14013–9. doi:10.1038/mtm.2014.13
83. Zhang P, Sun B, Osada T, Rodriguiz R, Yang XY, Luo X, et al. Immunodominant liver-specific expression suppresses transgene-directed immune responses in murine Pompe disease. Hum Gene Ther (2012) 23:460–72. doi:10.1089/hum.2011.063
84. Elsner M, Terbish T, Jörns A, Naujok O, Wedekind D, Hedrich H-J, et al. Reversal of diabetes through gene therapy of diabetic rats by hepatic insulin expression via lentiviral transduction. Mol Ther (2009) 20:918–26. doi:10.1038/mt.2012.8
85. Eneljung T, Tengvall S, Jirholt P, Henningsson L, Holmdahl R, Gustafsson K, et al. Antigen-specific gene therapy after immunisation reduces the severity of collagen-induced arthritis. Clin Dev Immunol (2013) 2013:345092. doi:10.1155/2013/345092
86. Keeler GD, Kumar S, Palaschak B, Silverberg EL, Markusic DM, Jones NT, et al. Gene therapy-induced antigen-specific Tregs inhibit neuro-inflammation and reverse disease in a mouse model of multiple sclerosis. Mol Ther (2018) 26:173–83. doi:10.1016/j.ymthe.2017.09.001
87. Lüth S, Huber S, Schramm C, Buch T, Zander S, Stadelmann C, et al. Ectopic expression of neural autoantigen in mouse liver suppresses experimental autoimmune neuroinflammation by inducing antigen-specific Tregs. J Clin Invest (2008) 118:3403–10. doi:10.1172/JCI32132
88. Ren B, O’Brien BA, Swan MA, Koina ME, Nassif N, Wei MQ, et al. Long-term correction of diabetes in rats after lentiviral hepatic insulin gene therapy. Diabetologia (2007) 50:1910–20. doi:10.1007/s00125-007-0722-0
89. Tengvall S, Eneljung T, Jirholt P, Turesson O, Wing K, Holmdahl R, et al. Gene therapy induces antigen-specific tolerance in experimental collagen-induced arthritis. PLoS One (2016) 11:e0154630. doi:10.1371/journal.pone.0154630
90. Mundkur L, Ponnusamy T, Philip S, Rao L, Biradar S, Deshpande V, et al. Oral dosing with multi-antigenic construct induces atheroprotective immune tolerance to individual peptides in mice. Int J Cardiol (2014) 175(2):340–51. doi:10.1016/j.ijcard.2014.06.001
91. de Haan P, Klein HC, ’t Hart BA. Autoimmune aspects of neurodegenerative and psychiatric diseases: a template for innovative therapy. Front Psychiatry (2017) 8:46. doi:10.3389/fpsyt.2017.00046
92. Esser N, Legrand-Poels S, Piette J, Scheen AJ, Paquot N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res Clin Pract (2014) 105:141–50. doi:10.1016/j.diabres.2014.04.006
93. Tsai S, Clemente-Casares X, Revelo XS, Winer S, Winer DA. Are obesity-related insulin resistance and type 2 diabetes autoimmune diseases? Diabetes (2015) 64:1886–97. doi:10.2337/db14-1488
94. Eppert BL, Wortham BW, Flury JL, Borchers MT. Functional characterization of T cell populations in a mouse model of chronic obstructive pulmonary disease. J Immunol (2013) 190:1331–40. doi:10.4049/jimmunol.1202442
Keywords: Simian Virus 40, polyomavirus, immune evasion, viral vector, non-immunogenicity, immune tolerance
Citation: Toscano MG and de Haan P (2018) How Simian Virus 40 Hijacks the Intracellular Protein Trafficking Pathway to Its Own Benefit … and Ours. Front. Immunol. 9:1160. doi: 10.3389/fimmu.2018.01160
Received: 21 February 2018; Accepted: 09 May 2018;
Published: 28 May 2018
Edited by:
Shokrollah Elahi, University of Alberta, CanadaReviewed by:
Jianzhong Zhu, Yangzhou University, ChinaFrançois J. M. A. Meurens, INRA UMR703 Ecole Nationale Vétérinaire, Agroalimentaire et de l’alimentation de Nantes-Atlantique, France
Copyright: © 2018 Toscano and de Haan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miguel G. Toscano, bWlndWVsLmdhcmNpYUBhbWFybmF0aGVyYXBldXRpY3MuY29t