 Daniel Satgé
Daniel Satgé
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 24 May 2018
Sec. Primary Immunodeficiencies
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01149
This article is part of the Research Topic The Relationship between Cancer Predisposition and Primary Immunodeficiency View all 18 articles
Under the concept of cancer immune surveillance, individuals with primary immune deficiencies would be expected to develop many more malignancies and show an excess of all types of cancers, compared to people with a normal immune system. A review of the nine most frequent and best-documented human conditions with primary immune deficiency reveals a 1.6- to 2.3-fold global increase of cancer in the largest epidemiological studies. However, the spectrum of cancer types with higher frequencies is narrow, limited mainly to lymphoma, digestive tract cancers, and virus-induced cancers. Increased lymphoma is also reported in animal models of immune deficiency. Overstimulation of leukocytes, chronic inflammation, and viruses explain this tumor profile. This raises the question of cancers being foreign organisms or tissues. Organisms, such as bacteria, viruses, and parasites as well as non-compatible grafts are seen as foreign (non-self) and identified and destroyed or rejected by the body (self). As cancer cells rarely show strong (and unique) surface antibodies, their recognition and elimination by the immune system is theoretically questionable, challenging the immune surveillance concept. In the neonatal period, the immune system is weak, but spontaneous regression and good outcomes occur for some cancers, suggesting that non-immune factors are effective in controlling cancer. The idea of cancer as a group of cells that must be destroyed and eliminated appears instead as a legacy of methods and paradigms in microbiological medicine. As an alternative approach, cancer cells could be considered part of the body and could be controlled by an embryonic and neonatal environment.
According to the concept of immune surveillance proposed by Burnet (1), the immune system identifies and destroys nascent cancers (2, 3). This model is based on the idea that cancers, which are produced by an organism’s cells, present specific antigens recognized by the immune system. Accordingly, compared to normal individuals, individuals with weakened or deficient immune systems should develop an excess of all types of cancers, since white blood cells in immune-compromised patients are less able to recognize and destroy malignant cells.
In this study, the concept of immune surveillance was examined in nine of the most well-known and well-characterized primary immune deficiencies (PIDs) chosen from 300 conditions each with a simple gene defect in the context of immune impairment (4). The immune surveillance concept predicts a global increase in cancer in these PIDs, but instead these conditions associated with a narrow spectrum of particular malignancies. This suggests that several immune processes, such as overstimulation of immune cells, viral infections, reactions to impaired bone marrow tissue, and chronic inflammation, influence cancer in PIDs. The particular distribution of cancers in primary immune-deficient humans and in murine models challenges the concept of immune surveillance as the major cause of increased cancer frequency in these diseases. This observation raises the question of the strategic place of immune mechanisms in diseases that do not arise from intrinsically foreign tissue.
Nine PIDs were selected from 300 immune deficiency diseases with single gene defects (4), on the basis of their frequency and sufficiently documented cancer occurrences. Less frequent conditions were included if significantly associated with cancers. The diseases were chosen from the most frequently cited conditions in recent cancer and PID epidemiological studies (4–7) and from the most frequently cited conditions in PID and cancer reviews (8–11).
We excluded diseases deemed not well defined for a sufficiently long period (preventing assessment of a number of people with precisely the same condition) and diseases with a chromosome breakage-DNA repair defect (to prevent bias due to their effects on oncogenesis). The following conditions were retained, selective IgA deficiency, common variable immune deficiency (CVID), X-linked agammaglobulinemia, X-linked hyper-IgM syndrome, cartilage-hair hypoplasia, Warts,-hypogammaglobulinemia,-infections, and myelokathexis (WHIM) syndrome, severe congenital neutropenia, and natural killer (NK) cell deficiency. For each condition, a search in PubMed was conducted, cross-searching the name of the disease with the words “cancer” and “malignancies.” Articles were selected if their title indicated information on health problems, cancer occurrence, and cancer frequency, or the functional or structural impact of the disease on tissues. Articles reporting in vitro experimental data were excluded.
From the 1,112 identified articles, 223 abstracts were selected for reading, 152 articles were read in full, and 80 articles and book chapters were included in the bibliography.
Table 1 displays cancer distributions in the nine chosen PID conditions (4–6, 9, 10, 12–44).
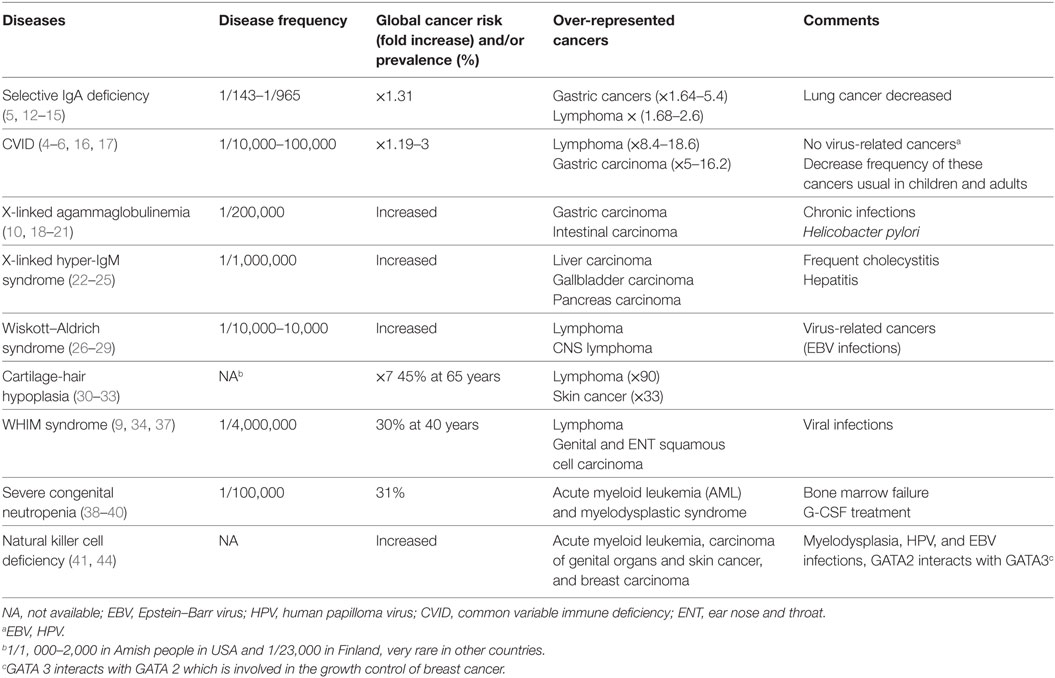
Table 1. Frequency of nine inherited diseases with primary immune deficiency and their cancer risk.
Based on the concept of immune surveillance, people with PIDs who are less able to eliminate cancer at its early stages should develop an excess of malignancies, particularly an excess of the most frequent cancers, leukemia, brain tumors, lymphomas, embryonic tumors, and germ cell tumors in childhood and colon, breast, lung, and prostate cancers in adults. The nine PIDs chosen for this review, based on their frequency and documented cancer occurrence, account for a large proportion of all PID patients. For instance, seven of the nine diseases per the United States Immune Deficiency NETwork accounted for 56% (2,047/3,658) of enrolled conditions (4). These diseases are also the most frequent conditions cited in reviews on PID (8, 10, 11), if we exclude DNA repair defects. Thus, the chosen conditions are truly representative for analysis of PID-associated cancer frequency. A recently published series of studies of patients with primary immune deficiency, which included large cohorts and a comparison group, from either Australia (6), the Netherlands (7), or the USA (4), indicate that PID causes a significant increased risk of all cancers of 1.6-, 2.3-, or 1.42-fold, respectively, compared to the general population. This is much lower than the approximately 10,000-fold (45) increase estimated in past reports.
Despite this global increased risk, the PID-cancer profile observed in the two largest studies, together including 4,790 patients, contained strikingly few cancer types. The Australian patients presented an excess of non-Hodgkin lymphoma (SIR 8.82), leukemia (SIR 5.36), and stomach cancer (SIR 6.10). Tumors were predominantly associated with antibody deficiencies (6). In the US study, the authors observed a 10-fold or 8.340-fold excess of lymphoma for men and women, respectively, but no increase for the most frequent common solid malignancies: prostate, lung, breast, and colon cancer (4).
A closer look at cancers observed in the nine selected conditions using epidemiological studies as well as institutional experience, series, and case reports allows for more precise tumor profiling. There is a global tumor profile for the nine conditions, and concurrently, a tumor profile for each condition. An excess of lymphoma occurred in the most diverse set of conditions, associating with five PIDs; however, liver and biliary tract cancer only associated with one. The majority of over-represented cancers were associated with either two or three conditions. Each condition in turn was associated with a narrow, sometimes very narrow, spectrum of cancers. For example, severe congenital neutropenia associated only with leukemia (40), and X-linked agammaglobulinemia associated only with gastric and intestinal carcinoma (19).
On the other hand, some tumors were rare in some of these diseases. In selective IgA deficiency, deaths from lung cancer were more than twofold less frequent than in the general population (14). In the whole group of PIDs, the most frequent cancers in the USA—breast, lung, and colon cancer—had a lower incidence of 0.41, 0.48, and 0.19, respectively, for women; for men, colon and lung cancer incidences were 1 and 0.62, respectively, compared to the general population. Interestingly, these are the most frequent epithelial malignancies, suggesting an underrepresentation of these types of cancers in the largest available study conducted (4). In CVID, lung, breast, and colon cancer had a lower incidence compared to that of the general population (4). Thus, the incidence of most solid tumors in PIDs is not significantly higher than in the general population. This is well shown by these epidemiological studies which took into account the reduced life expectancy (due to infection-associated sequelae or autoimmunity), for instance in CIVD, Wiskott–Aldrich syndrome, X-linked agammaglobulinemia, and X-linked hyper IgM syndrome, which calculated age-adjusted cancer incidence (4) and adjusted evaluation for 5-year age group (6). Some PIDs, outside of the nine selected, are not associated with malignancies. In contrast, they appear to have global decreased cancer incidences. This is the case for X-linked chronic granulomatous disease where no cancer was registered among 326 patients (4). Globally, cancer distribution in PID does not seem to result from a unique general mechanism applying to every disease. This does not fit with a defective immune surveillance model. The cancer distribution rather suggests that different mechanisms are involved, leading to special and sometimes unique tumor profiles.
The largest and most recent study of patients with PID-associated cancers points to a restricted role of the immune system in protection from cancer (4).
Lymphoma is unquestionably the most over-represented malignancy in PID, with an approximate 8- to 13-fold increased risk compared to that of the general population (4, 6, 7). These are mainly B cell lymphoma occurring in extranodal sites, often in patients with preceding polyclonal lymphocyte infiltrations (11). As emphasized by Hauck et al., in PIDs, cancer (lymphoma) affects cells altered by the deficiency itself (lymphocytes) (46). This increase in lymphoma has been linked to recurrent and persistent viral and bacterial infections, which characterize a great majority of PIDs sometimes in an antigen-specific manner (32, 47, 48). The overstimulated defective lymphoid tissue leads to lymphoid hyperplasia observed in 50% of patients with CVID. They show lympho-adenopathies and splenomegaly (17), a state favoring lymphoma transformation.
An increased incidence of leukemia, presenting as abnormal bone marrow, is observed in some PIDs. In severe congenital neutropenia, patients experience bone marrow failure (40). The bone marrow of NK cell-deficient patients is characterized by multilineage dysplasia (44). In these conditions, hematopoietic lineages could be the target of chronic overstimulation to counterbalance the impaired production of white cells. Elevated intrinsic G-CSF observed in severe congenital neutropenia also increases the risk for acute myeloid leukemia and myelodysplastic syndrome (MDS), in addition to the treatment with G-CSF (38). Reviewing chronic myelocytic leukemia (CML) in immune deficiency, Gale and Opelz found no global excess of CML in patients with immune deficiency. They suggested that failure of immune suppression per se does not explain most cases of CML and conclude that immune surveillance does no contribute significantly toward preventing new cases of CML (49). The role of immune surveillance could as well be questioned for other PID-associated leukemias.
Stomach cancer is over-represented in epidemiological studies on malignancies in PID, especially in humoral defects such as CVID and selective IgA deficiency (4–6). In conditions with humoral defects, the impaired immune system permits recurring Helicobacter pylori gastric colonization, which leads to gastritis, and, for some patients, to severe atrophy and intestinal metaplasia which are two major risk factors for gastric adenocarcinoma (16, 17). An increased incidence of intestinal cancer has been found in patients with X-linked agammaglobulinemia. In this disease, a team observed inflammatory bowel diseases and infectious enteritis, which are risk factors for cancer, for 11.3% of patients (21). Patients with X-linked hyper-IgM syndrome who present frequent protracted diarrhea also develop more colon cancer (23). Additionally, patients with X-linked hyper-IgM syndrome are much more prone to hepatitis and cholangitis, mainly linked to persistent infection of Cryptosporidium Parvum, and are anticipated to develop cancer in organs altered by chronic inflammation and cirrhosis (22).
Cancers related to oncogenic viral infections account for 12% of cancers in humans and an even higher percentage in patients with primary or acquired immune deficiencies (9, 50). Cancers of the genital, anal, and oropharyngeal regions and of the skin (50, 51) are more frequent in those PIDs that are vulnerable to human papilloma virus (HPV) infections, particularly patients with NK cell deficiency who develop an excess of cervix-uterine HPV-related dysplasia and cancer as well as oropharyngeal squamous cell carcinomas and skin carcinomas (44). Genital and oral HPV-related squamous cell carcinomas are also observed in patients with WHIM syndrome (35, 36). Epstein–Barr virus (EBV) infections cause lymphoproliferative diseases and lymphoma, which are also observed in WHIM syndrome (34, 37), and in cartilage–hair hypoplasia (32). Additionally, EBV causes EBV-smooth muscle tumors, which have been reported in liver, adrenals, and smooth muscle of patients with NK cell deficiencies (42, 44). Significantly, in PIDs there is an excess of malignancies stemming from organs targeted by viruses but no excess of cancers in other tissues.
The molecular pathogenesis of PIDs associated with malignancy cannot be developed for each clinical condition included in Table 1; this is beyond the scope of the current article. NK cell deficiency, for example, has been associated with mutations in five genes, GATA2, MCM4, RTLLE1, IRF8, and FCGR3A (51). At the same time, GATA2 deficiency has been linked to four clinical syndromes: NK cell deficiency, but also monocytopenia and mycobacterial infection syndrome, familial MDS and Emberger syndrome (44). GATA2 is a transcription factor highly expressed in immature hematopoietic cells. The gene is necessary for survival and renewal of hematopoietic cells. It is critical for genesis and function of hematopoietic stem cells and thus blood cell lineage (44). Myeloid malignancy in GATA2 deficiency is related to differentiation arrest and in part to a novel function of the mutated gene. Currently, it remains unclear how germline GATA2 loss-of-function mutations result in myeloid neoplasms (52). In this context the hypothesis of an overstimulation of myeloid cells could be suggested.
As animal models are considered to support the concept of immune surveillance, it is interesting to compare the spontaneous tumor occurrence in immune deficient mice to that in PID patients. An early study showed no difference in the incidence of spontaneous lung adenoma between athymic-nude mice, which are deficient for T cells, and immunocompetent mice (53). More recent works using immunodeficient mouse strains with defects in performin, interferon gamma, recombination activating gene (Rag2), signal transducer, and activator of transcription 1 (Stat1), and other genes reported an increased cancer incidence; however, the distribution of cancer resembled that observed in humans with PID. In a review of 11 strains, eight showed an excess of lymphoma and one an excess of plasmacytoma, either alone (6) or associated with other malignancies (3). Only two strains showed an excess of carcinomas and no excess of lymphoma (2). One team who studied spontaneous tumors in mice lacking the tumor necrosis factor (TNF)-apoptosis-inducing ligand (TRAIL) concluded that TRAIL-R did not protect against mammary cancers or against colon cancers, but did protect against lymphoid malignancies, which affected more than 25% of the deficient mice (54). An ongoing process of immune activation through IL6 upregulation has been proposed to explain plasma cell hyperplasia followed by plasmacytoma in mice lacking the interleukin-12 receptor Beta2 (55).
The literature describing mouse models of immune deficiency report increased incidence of lymphoma, while carcinomas are rare, as seen in human PID (see above). Notably, for the two mouse strains with only carcinomas, the tumor distribution is quite narrow. Mice lacking Rag2 developed mainly intestinal adenomas and colon carcinomas (56), which usually occur following intestinal infection (57). Mice lacking both Rag2 and Stat1 developed an excess of colon cancer and breast carcinomas (56). As STAT1 is involved in breast cancer pathways (58), the increase in breast cancer is likely due to STAT1’s role in tumorigenesis instead of immune surveillance. Similarly, a mouse model with a deficiency of granulocyte-macrophage colony stimulating factor exhibited an excess of both lymphoma and solid tumors. Interestingly, mice on antimicrobial therapy developed neither lymphoma nor solid malignancies compared to non-treated mice (p < 0.001). As the treated mice showed a marked reduction of both chronic infection and cancers, the authors proposed a role for chronic infection in the onset of malignancies (59). Thus, carcinomas observed in immune-deficient mouse strains are likely the consequence of specific processes affected by the deficient gene, for example, tissue damage secondary to unresolved inflammation, instead of a general decrease in immune surveillance, which should cause an excess of many types of carcinomas.
Other clinical observations indicate that immune defects increase the incidence of lymphoma instead of all cancers and cast doubt on the protective role of the immune system in cancer. First, the excess of lymphoma (but not other cancers) observed in Nijmegen breakage syndrome is associated with chronic stimulation of defective lymphocytes (60). In fact, only conditions with a chromosome breakage-DNA repair defect presenting with an immune deficiency, i.e., ataxia telangiectasia, Nijmegen breakage syndrome, and Bloom syndrome, develop an excess of lymphoma. In contrast, conditions with a chromosome breakage-DNA repair defect without immune deficiency, such as xeroderma pigmentosum, Fanconi anemia, Werner syndrome, and Rothmund–Thomson syndrome, develop other types of cancers. Second, although malignant melanoma, renal cell carcinoma, and non-small cell lung carcinoma have historically been considered as proof of immune therapy principles, these two cancers are not found more frequently in PIDs than in the general population (46). This is particularly clear in the nine conditions presented in Table 1. Third, although the immune system is weak and immature in neonates and young infants (61), there is no increased incidence of cancer at this period (62). In fact, a notable number of various neonatal cancers, some very aggressive, e.g., neuroblastoma, leukemia, and pontine glioma, may undergo spontaneous regression (62–64). Furthermore, in neonates, a cure for sarcoma is possible despite incomplete tumor resection (65). For this reason, the neonatal period was dubbed, three decades ago, the “oncogenic grace period” (66). Fourth, although people with Down syndrome have mildly to moderately weakened immune systems favoring pneumonias and opportunistic infections (67), adults with Down syndrome experience half the burden of solid tumors observed in the general population, notably showing a reduced frequency in the most common cancers: breast, prostate, and lung (68, 69).
As the immune system’s role is to identify and destroy foreign organisms, bacteria, viruses, and parasites, the concept of cancer immune surveillance presupposes that cancer tissues are foreign (non-self) to the body of the patient (self). In this theoretical framework, cancer would be a foreign biological process characterized by genetic modifications. However, it is a genetic modification (gene or chromosome mutation) sufficient to consider a tissue foreign to the body? We should then consider that a person who has a constitutional genetic mosaicism as partly composed of “non-self” tissues. For instance, a woman with mosaic Turner syndrome with 20% 46, XX cells and 80% 45, X0 could be considered to be composed of 80% foreign tissue. A genetic point of view is really debatable for considering that a mutated tissue is foreign to the body. Even if they are genetically modified and even if they become uncontrolled by the integrating and coordinating system of the body, cancer tissues remain composed of body cells.
If cancers are recognized as foreign structures, the body would naturally reject them as it does for bacteria, viruses, parasites, and non-compatible grafts. Foreign tissues induce antibody production by the immune system, but tumor-specific antigens are not common in human tumors. Nonetheless, mouse models show that antigen expression is important for the process of recognizing and eliminating cancer cells (70). However, tumor reactive antibodies mirror tissue damage and reflect a response to tissue necrosis, rather than targeting a specific cell type (71). The observation that cancer cells, excepting those of virus-induced malignancies (70, 71), do not usually harbor strong specific antigens which suggests that cancers are not considered foreign by the body. Cancer could as well be seen as a diseased tissue. Some researchers propose that cancers can be considered as abnormal organs that develop in a manner similar to that of normal organs (72). Moreover, specific immune factors, such as a given major cytokine-like interferon gamma, or major immune effector cells, such as CD4 T lymphocytes, have both pro- and anti-tumoral effects (73, 74). These and other puzzling observations (75) suggest that the immune system has a complex reaction to tumor cells and does not always and systematically protect against malignancies.
Approaching cancer as a foreign organism implies that the main tumor and any tumor cells must be destroyed to cure the patient, similar to treatments aimed at killing bacteria, viruses, and parasites. This point of view seems a legacy of concepts and paradigms developed during the nineteenth and twentieth centuries with the progress of microbial medicine (76). We should not remain constrained by a framework that has been very effective for microbiology but narrows our understanding of oncogenesis. It is time to open our minds to other currently neglected approaches. We may question how best to fight against a disease composed of our own cells, which have escaped bodily control and gone awry. We may also question current cancer treatments that destroy normal cells likely because cancer is not so different from normal tissue.
Cancer could also be considered as a developmental disease, leading us to search for the processes that protect against malignancy during the embryonic and neonatal periods. The observation that embryonic carcinoma cells can incorporate into a normal blastocyst and produce a normal mouse composed of a mixture of normal cells (the host) and normalized cancer cells (the incorporated teratocarcinoma) (77) and that cancer cells in the presence of normal tissue may reverse to a normal phenotype (78) enlarges our perspective on research for cancer control and treatment. While the present study does not discuss the effectiveness of immune therapy and the progresses in treatment (79), we suggest that immune surveillance is probably not, in natural conditions, as effective as expected.
This review has some limitations, since only nine conditions were included. However, these conditions account for more than half of cancers reported in the largest epidemiological study on cancer in PIDs (4). Epidemiological studies are high-quality works, where the biological diagnosis is difficult to challenge. Isolated case reports and series are well documented, published in high-quality journals. Thus, repetitive errors on tumor diagnosis are unlikely.
From the presented data we may draw the following conclusions: (1) The increased risk in frequency of cancer in PIDs is moderate, nearly twofold, much less than formally estimated. (2) The spectrum of cancer in PIDs is narrow. (3) Over-represented malignancies are mainly lymphoma, digestive tract tumors, and cutaneous-mucus carcinoma, which can be explained by an overstimulation of lymphocytes, chronic digestive infection, and cutaneous-mucus viral infections. (4) Animal models of immune deficiency develop a similar spectrum of malignancies as observed in human. Thus, since a defect of immune surveillance should theoretically favor all types of cancers, the data raise the hypothesis that immune surveillance does not play a major role in the increase of cancer in PIDs. While analyzing proposed mechanisms of oncogenesis and unmasking discrepancies with the immune surveillance model, three aspects of oncogenesis were emphasized, the overstimulation of a tissue leading to cancer, the role of tissue inflammation and fibrosis, and the possibility of cancer control by the body as it is observed during the neonatal period.
From an immediate point of view, it is disappointing to find that the role of the immune system is not, in a typical condition (i.e., non therapeutic conditions, particularly when excluding CAR T-cell therapy and immune checkpoint therapy), as important as expected. From another point of view, it is very exciting news implying that each PID with its specific excess or decrease of cancers has something to tell us about precise aspects of carcinogenesis. It is thus necessary to precisely identify the cancer types that occur and to articulate these data to functional and tissue alterations linked to the disease. These all provide many pathways for understanding cancer under precise conditions and possibly to fight against neoplasia.
The author confirms being the sole contributor of this work and approved it for publication.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
A grant from the Fondation Jérôme Lejeune supports the study. Christiane Satgé is acknowledged for the preparation of the manuscript.
1. Burnet MF. Cancer. A biological approach. The processes of control. Br Med J (1957) 6:779–86. doi:10.1136/bmj.1.5022.779
2. Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest (2007) 117(5):1137–46. doi:10.1172/JCI31405
3. Finn OJ. Immuno-oncology: understanding the function and dysfunction of the immune system in cancer. Ann Oncol (2012) 23(Suppl 8):viii6–9. doi:10.1093/annonc/mds256
4. Mayor PC, Eng KH, Singel KL, Abrams SI, Odunsi K, Moysich KB, et al. Cancer in primary immunodeficiency diseases: cancer incidence in the United States immune deficiency network registry. J Allergy Clin Immunol (2018) 141(3):1028–35. doi:10.1016/j.jaci.2017.05.024
5. Mellemkjaer L, Hammarstrom L, Andersen V, Yuen J, Heilmann C, Barington T, et al. Cancer risk among patients with IgA deficiency or common variable immunodeficiency and their relatives: a combined Danish and Swedish study. Clin Exp Immunol (2002) 130(3):495–500. doi:10.1046/j.1365-2249.2002.02004.x
6. Vajdic CM, Mao L, van Leeuwen MT, Kirkpatrick P, Grulich AE, Riminton S. Are antibody deficiency disorders associated with a narrower range of cancers than other forms of immunodeficiency? Blood (2010) 116(8):1228–34. doi:10.1182/blood-2010-03-272351
7. Jonkman-Berk BM, van den Berg JM, Ten Berge IJ, Bredius RG, Driessen GJ, Dalm VA, et al. Primary immunodeficiencies in the Netherlands: national patient data demonstrate the increased risk of malignancy. Clin Immunol (2015) 156(2):154–62. doi:10.1016/j.clim.2014.10.003
8. Salavoura K, Kolialexi A, Tsangaris G, Mavrou A. Development of cancer in patients with primary immunodeficiencies. Anticancer Res (2008) 28(2B):1263–9.
9. Rezaei N, Hedayat M, Aghamohammadi A, Nichols KE. Primary immunodeficiency diseases associated with increased susceptibility to viral infections and malignancies. J Allergy Clin Immunol (2011) 127(6):1329.e–41.e. doi:10.1016/j.jaci.2011.02.047
10. van der Werff Ten Bosch J, van den Akker M. Genetic predisposition and hematopoietic malignancies in children: primary immunodeficiency. Eur J Med Genet (2016) 59(12):647–53. doi:10.1016/j.ejmg.2016.03.001
11. Mortaz E, Tabarsi P, Mansouri D, Khosravi A, Garssen J, Velayati A, et al. Cancers related to immunodeficiencies: update and perspectives. Front Immunol (2016) 7:365. doi:10.3389/fimmu.2016.00365
12. Quiding-Järbrink M, Sundström P, Lundgren A, Hansson M, Bäckström M, Johansson C, et al. Decreased IgA antibody production in the stomach of gastric adenocarcinoma patients. Clin Immunol (2009) 131(3):463–71. doi:10.1016/j.clim.2009.01.010
13. Wobser M, Kerstan A, Kneitz H, Goebeler M, Kunzmann V, Rosenwald A, et al. Primary cutaneous marginal zone lymphoma with sequential development of nodal marginal zone lymphoma in a patient with selective immunoglobulin A deficiency. J Cutan Pathol (2013) 40(12):1035–41. doi:10.1111/cup.12230
14. Ludvigsson JF, Neovius M, Hammarström L. IgA deficiency and mortality: a population-based cohort study. J Clin Immunol (2013) 33(8):1317–24. doi:10.1007/s10875-013-9948-4
15. Ludvigsson JF, Neovius M, Ye W, Hammarström L. IgA deficiency and risk of cancer: a population-based matched cohort study. J Clin Immunol (2015) 35(2):182–8. doi:10.1007/s10875-014-0124-2
16. Quinti I, Soresina A, Spadaro G, Martino S, Donnanno S, Agostini C, et al. Italian primary immunodeficiency network. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol (2007) 27(3):308–16. doi:10.1007/s10875-007-9075-1
17. Gangemi S, Allegra A, Musolino C. Lymphoproliferative disease and cancer among patients with common variable immunodeficiency. Leuk Res (2015) 39(4):389–96. doi:10.1016/j.leukres.2015.02.002
18. Smith CIE, Conley ME. X-linked ammaglobulinemia and autosomal recessive ammaglobulinemia. 3rd ed. In: Ochs HD, Smith CIE, Puck JM, editors. Primary Immunodeficiency Diseases. A Molecular and Genetic Approach. New York: Oxford University press (2014). p. 299–323.
19. Staines Boone AT, Torres Martínez MG, López Herrera G, de Leija Portilla JO, Espinosa Padilla SE, Espinosa Rosales FJ, et al. Gastric adenocarcinoma in the context of X-linked agammaglobulinemia: case report and review of the literature. J Clin Immunol (2014) 34(2):134–7. doi:10.1007/s10875-013-9971-5
20. Conley ME. Are patients with X-linked agammaglobulinemia at increased risk of developing acute lymphoblastic leukemia? J Clin Immunol (2015) 35(2):98–9. doi:10.1007/s10875-015-0132-x
21. Barmettler S, Otani IM, Minhas J, Abraham RS, Chang Y, Dorsey MJ, et al. Gastrointestinal manifestations in X-linked agammaglobulinemia. J Clin Immunol (2017) 37(3):287–94. doi:10.1007/s10875-017-0374-x
22. Hayward AR, Levy J, Facchetti F, Notarangelo L, Ochs HD, Etzioni A, et al. Cholangiopathy and tumors of the pancreas, liver, and biliary tree in boys with X-linked immunodeficiency with hyper-IgM. J Immunol (1997) 158(2):977–83.
23. Winkelstein JA, Marino MC, Ochs H, Fuleihan R, Scholl PR, Geha R, et al. The X-linked hyper-IgM syndrome: clinical and immunologic features of 79 patients. Medicine (2003) 82(6):373–84. doi:10.1097/01.md.0000100046.06009.b0
24. Mitsui-Sekinaka K, Imai K, Sato H, Tomizawa D, Kajiwara M, Nagasawa M, et al. Clinical features and hematopoietic stem cell transplantations for CD40 ligand deficiency in Japan. J Allergy Clin Immunol (2015) 136(4):1018–24. doi:10.1016/j.jaci.2015.02.020
25. De la Morena MT, Leonard D, Torgerson TR, Cabral-Marques O, Slatter M, Aghamohammadi A, et al. Long-term outcomes of 176 patients with X-linked hyper-IgM syndrome treated with or without hematopoietic cell transplantation. J Allergy Clin Immunol (2017) 139(4):1282–92. doi:10.1016/j.jaci.2016.07.039
26. Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr (1994) 125(6 Pt 1):876–85. doi:10.1016/S0022-3476(05)82002-5
27. Palenzuela G, Bernard F, Gardiner Q, Mondain M. Malignant B cell non-Hodgkin’s lymphoma of the larynx in children with Wiskott Aldrich syndrome. Int J Pediatr Otorhinolaryngol (2003) 67(9):989–93. doi:10.1016/S0165-5876(03)00155-1
28. Imai K, Morio T, Zhu Y, Jin Y, Itoh S, Kajiwara M, et al. Clinical course of patients with WASP gene mutations. Blood (2004) 103(2):456–64. doi:10.1182/blood-2003-05-1480
29. Coccia P, Mastrangelo S, Ruggiero A, Scalzone M, Rosolen A, Maurizi P, et al. Treatment of pharyngeal non-Hodgkin lymphoma in a patient with Wiskott-Aldrich syndrome. Pediatr Blood Cancer (2012) 59(2):318–9. doi:10.1002/pbc.23393
30. Mäkitie O, Pukkala E, Teppo L, Kaitila I. Increased incidence of cancer in patients with cartilage-hair hypoplasia. J Pediatr (1999) 134(3):315–8. doi:10.1016/S0022-3476(99)70456-7
31. Eisner JM, Russell M. Cartilage hair hypoplasia and multiple basal cell carcinomas. J Am Acad Dermatol (2006) 54(2 Suppl):S8–10. doi:10.1016/j.jaad.2005.03.055
32. Taskinen M, Ranki A, Pukkala E, Jeskanen L, Kaitila I, Mäkitie O. Extended follow-up of the Finnish cartilage-hair hypoplasia cohort confirms high incidence of non-Hodgkin lymphoma and basal cell carcinoma. Am J Med Genet A (2008) 146A(18):2370–5. doi:10.1002/ajmg.a.32478
33. Kainulainen L, Lassila O, Ruuskanen O. Cartilage-hair hypoplasia: follow-up of immunodeficiency in two patients. J Clin Immunol (2014) 34(2):256–9. doi:10.1007/s10875-013-9981-3
34. Imashuku S, Miyagawa A, Chiyonobu T, Ishida H, Yoshihara T, Teramura T, et al. Epstein-Barr virus-associated T-lymphoproliferative disease with hemophagocytic syndrome, followed by fatal intestinal B lymphoma in a young adult female with WHIM syndrome. Warts, hypogammaglobulinemia, infections, and myelokathexis. Ann Hematol (2002) 81(8):470–3. doi:10.1007/s00277-002-0489-9
35. Cipriani NA, Blair E, Taxy JB. WHIM syndrome and oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod (2010) 109(1):105–8. doi:10.1016/j.tripleo.2009.08.011
36. Beaussant Cohen S, Fenneteau O, Plouvier E, Rohrlich PS, Daltroff G, Plantier I, et al. Description and outcome of a cohort of 8 patients with WHIM syndrome from the French Severe Chronic Neutropenia Registry. Orphanet J Rare Dis (2012) 7:71. doi:10.1186/1750-1172-7-71
37. Yoshii Y, Kato T, Ono K, Takahashi E, Fujimoto N, Kobayashi S, et al. Primary cutaneous follicle center lymphoma in a patient with WHIM syndrome. J Eur Acad Dermatol Venereol (2016) 30(3):529–30. doi:10.1111/jdv.12927
38. Beekman R, Valkhof MG, Sanders MA, van Strien PM, Haanstra JR, Broeders L, et al. Sequential gain of mutations in severe congenital neutropenia progressing to acute myeloid leukemia. Blood (2012) 119(22):5071–7. doi:10.1182/blood-2012-01-406116
39. Carlsson G, Fasth A, Berglöf E, Lagerstedt-Robinson K, Nordenskjöld M, Palmblad J, et al. Incidence of severe congenital neutropenia in Sweden and risk of evolution to myelodysplastic syndrome/leukaemia. Br J Haematol (2012) 158(3):363–9. doi:10.1111/j.1365-2141.2012.09171.x
40. Skokowa J, Steinemann D, Katsman-Kuipers JE, Zeidler C, Klimenkova O, Klimiankou M, et al. Cooperativity of RUNX1 and CSF3R mutations in severe congenital neutropenia: a unique pathway in myeloid leukemogenesis. Blood (2014) 123(14):2229–37. doi:10.1182/blood-2013-11-538025
41. Ballas ZK, Turner JM, Turner DA, Goetzman EA, Kemp JD. A patient with simultaneous absence of "classical" natural killer cells (CD3-, CD16+, and NKH1+) and expansion of CD3+, CD4-, CD8-, NKH1+ subset. J Allergy Clin Immunol (1990) 85(2):453–9. doi:10.1016/0091-6749(90)90155-W
42. Shaw RK, Issekutz AC, Fraser R, Schmit P, Morash B, Monaco-Shawver L, et al. Bilateral adrenal EBV-associated smooth muscle tumors in a child with a natural killer cell deficiency. Blood (2012) 119(17):4009–12. doi:10.1182/blood-2011-10-385377
43. Orange JS. Natural killer cell deficiency. J Allergy Clin Immunol (2013) 132(3):515–25. doi:10.1016/j.jaci.2013.07.020
44. Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood (2014) 123(6):809–21. doi:10.1182/blood-2013-07-515528
45. Mueller BU, Pizzo PA. Cancer in children with primary or secondary immunodeficiencies. J Pediatr (1995) 126(1):1–10. doi:10.1016/S0022-3476(95)70491-4
46. Hauck F, Voss R, Urban C, Seidel MG. Intrinsic and extrinsic causes of malignancies in patients with primary immunodeficiency disorders. J Allergy Clin Immunol (2018) 141(1):59–68. doi:10.1016/j.jaci.2017.06.009
47. Suarez F, Lortholary O, Hermine O, Lecuit M. Infection-associated lymphomas derived from marginal zone B cells: a model of antigen-driven lymphoproliferation. Blood (2006) 107(8):3034–44. doi:10.1182/blood-2005-09-3679
48. Chua I, Quinti I, Grimbacher B. Lymphoma in common variable immunodeficiency: interplay between immune dysregulation, infection and genetics. Curr Opin Hematol (2008) 15(4):368–74. doi:10.1097/MOH.0b013e328302c7b6
49. Gale RP, Opelz G. Is there immune surveillance against chronic myeloid leukaemia? Possibly, but not much. Leuk Res (2017) 57:109–11. doi:10.1016/j.leukres.2017.03.003
50. Pierangeli A, Antonelli G, Gentile G. Immunodeficiency-associated viral oncogenesis. Clin Microbiol Infect (2015) 21(11):975–83. doi:10.1016/j.cmi.2015.07.009
51. Mace EM, Orange JS. Genetic causes of human NK cell deficiency and their effect on NK cell subsets. Front Immunol (2016) 7:545. doi:10.3389/fimmu.2016.00545
52. Wlodarski MW, Collin M, Horwitz MS. GATA2 deficiency and related myeloid neoplasms. Semin Hematol (2017) 54(2):81–6. doi:10.1053/j.seminhematol.2017.05.002
53. Stutman O. Tumor development after 3-methylcholanthrene in immunologically deficient athymic-nude mice. Science (1974) 183(4124):534–6. doi:10.1126/science.183.4124.534
54. Zerafa N, Westwood JA, Cretney E, Mitchell S, Waring P, Iezzi M, et al. Cutting edge: TRAIL deficiency accelerates hematological malignancies. J Immunol (2005) 175(9):5586–90. doi:10.4049/jimmunol.175.9.5586
55. Airoldi I, Di Carlo E, Cocco C, Sorrentino C, Fais F, Cilli M, et al. Lack of Il12rb2 signaling predisposes to spontaneous autoimmunity and malignancy. Blood (2005) 106(12):3846–53. doi:10.1182/blood-2005-05-2034
56. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature (2001) 410(6832):1107–11. doi:10.1038/35074122
57. Wang C, Gong G, Sheh A, Muthupalani S, Bryant EM, Puglisi DA, et al. Interleukin-22 drives nitric oxide-dependent DNA damage and dysplasia in a murine model of colitis-associated cancer. Mucosal Immunol (2017) 10(6):1504–17. doi:10.1038/mi.2017.9
58. Jeon M, You D, Bae SY, Kim SW, Nam SJ, Kim HH, et al. Dimerization of EGFR and HER2 induces breast cancer cell motility through STAT1-dependent ACTA2 induction. Oncotarget (2016) 8(31):50570–81. doi:10.18632/oncotarget.10843
59. Enzler T, Gillessen S, Manis JP, Ferguson D, Fleming J, Alt FW, et al. Deficiencies of GM-CSF and interferon gamma link inflammation and cancer. J Exp Med (2003) 197(9):1213–9. doi:10.1084/jem.20021258
60. Gregorek H, Chrzanowska KH, Dzierzanowska-Fangrat K, Wakulińska A, Pietrucha B, Zapaśnik A, et al. Nijmegen breakage syndrome: long-term monitoring of viral and immunological biomarkers in peripheral blood before development of malignancy. Clin Immunol (2010) 135(3):440–7. doi:10.1016/j.clim.2010.01.008
61. Levy O, Wynn JL. A prime time for trained immunity: innate immune memory in newborns and infants. Neonatology (2014) 105(2):136–41. doi:10.1159/000356035
62. Raciborska A, Bilska K, Węcławek-Tompol J, Ussowicz M, Pogorzała M, Janowska J, et al. Solid cancers in the premature and the newborn: report of three national referral centers. Pediatr Neonatol (2016) 57:295–301. doi:10.1016/j.pedneo.2015.08.007
63. van den Berg H, Hopman AH, Kraakman KC, de Jong D. Spontaneous remission in congenital leukemia is not related to (mosaic) trisomy 21: case presentation and literature review. Pediatr Hematol Oncol (2004) 21(2):135–44. doi:10.1080/08880010490273000
64. Schomerus L, Merkenschlager A, Kahn T, Hirsch W. Spontaneous remission of a diffuse brainstem lesion in a neonate. Pediatr Radiol (2007) 37(4):399–402. doi:10.1007/s00247-007-0424-2
65. Miura K, Han G, Sano M, Tsutsui Y. Regression of congenital fibrosarcoma to hemangiomatous remnant with histological and genetic findings. Pathol Int (2002) 52(9):612–8. doi:10.1046/j.1440-1827.2002.01394.x
66. Bolande RP. Spontaneous regression and cytodifferentiation of cancer in early life: the oncogenic grace period. Surv Synth Pathol Res (1985) 4(4):296–311.
67. Kusters MA, Verstegen RH, Gemen EF, de Vries E. Intrinsic defect of the immune system in children with Down syndrome: a review. Clin Exp Immunol (2009) 156(2):189–93. doi:10.1111/j.1365-2249.2009.03890.x
68. Satgé D, Sommelet D, Geneix A, Nishi M, Malet P, Vekemans MJ. A tumor profile in Down syndrome. Am J Med Genet (1998) 78:207–16. doi:10.1002/(SICI)1096-8628(19980707)78:3<207::AID-AJMG1>3.0.CO;2-M
69. Hasle H, Friedman JM, Olsen JH, Rasmussen SA. Low risk of solid tumors in persons with Down syndrome. Genet Med (2016) 18(11):1151–7. doi:10.1038/gim.2016.23
70. DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature (2012) 482(7385):405–9. doi:10.1038/nature10803
71. Preiss S, Kammertoens T, Lampert C, Willimsky G, Blankenstein T. Tumor-induced antibodies resemble the response to tissue damage. Int J Cancer (2005) 115(3):456–62. doi:10.1002/ijc.20914
72. Egeblad M, Nakasone ES, Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell (2010) 18(6):884–901. doi:10.1016/j.devcel.2010.05.012
73. DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell (2009) 16(2):91–102. doi:10.1016/j.ccr.2009.06.018
74. Zaidi R, Merlino G. The two faces of interferon-gamma in cancer. Clin Cancer Res (2011) 17:6118–24. doi:10.1158/1078-0432.CCR-11-0482
75. de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer (2006) 6(1):24–37. doi:10.1038/nrc1782
76. Wion D, Appaix F, Burruss M, Berger F, van der Sanden B. Cancer research in need of a scientific revolution: using "paradigm shift" as a method of investigation. J Biosci (2015) 40(3):657–66. doi:10.1007/s12038-015-9543-3
77. Mintz B, Illmensee K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc Natl Acad Sci U S A (1975) 9:3585–9. doi:10.1073/pnas.72.9.3585
78. Bizzarri M, Cucina A, Proietti S. Tumor reversion: mesenchymal-epithelial transition as a critical step in managing the tumor-microenvironment cross-talk. Curr Pharm Des (2017) 23(32):4705–15. doi:10.2174/1381612823666170609082757
Keywords: primary immune deficiencies, immune surveillance, cancer incidence, tumor profile, cancer, lymphoma, gastrointestinal cancer, virus-induced cancer
Citation: Satgé D (2018) A Tumor Profile in Primary Immune Deficiencies Challenges the Cancer Immune Surveillance Concept. Front. Immunol. 9:1149. doi: 10.3389/fimmu.2018.01149
Received: 11 January 2018; Accepted: 07 May 2018;
Published: 24 May 2018
Edited by:
Andrew R. Gennery, Newcastle University, United KingdomReviewed by:
Shigeaki Nonoyama, National Defense Medical College, JapanCopyright: © 2018 Satgé. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel Satgé, ZGFuaWVsc2F0Z2VAb3JhbmdlLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.