 Manuel D. Díaz-Muñoz
Manuel D. Díaz-Muñoz
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 23 May 2018
Sec. T Cell Biology
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01094
Fighting external pathogens requires an ever-changing immune system that relies on tight regulation of gene expression. Transcriptional control is the first step to build efficient responses while preventing immunodeficiencies and autoimmunity. Post-transcriptional regulation of RNA editing, location, stability, and translation are the other key steps for final gene expression, and they are all controlled by RNA-binding proteins (RBPs). Nowadays we have a deep understanding of how transcription factors control the immune system but recent evidences suggest that post-transcriptional regulation by RBPs is equally important for both development and activation of immune responses. Here, we review current knowledge about how post-transcriptional control by RBPs shapes our immune system and discuss the perspective of RBPs being the key players of a hidden immune cell epitranscriptome.
Immune cell development and function are not only regulated by gene networks controlled by transcription factors but also by post-transcriptional regulatory mechanisms, controlled by RNA-binding proteins (RBPs) and non-coding RNAs, that are essential for immune cell lineage commitment, maintenance, and modulation of immune responses (1, 2). Here, we review the state of the art, paying special attention to those mechanisms controlled by RBPs that shape gene expression in our immune system.
Pioneering studies discovered the presence of nucleotide regulatory sequences in the 3′ untranslated region (3′UTRs) of dozens of messenger (m) RNAs encoding cytokines, such as Tnf, Il1, Ifng, and Csf2 (3–6), that were responsible for the discrepancies in mRNA abundance and protein expression observed due to differential regulation of mRNA stability and translation. Some of these foundational studies also highlighted the key role of mRNA splicing in defining the qualitative nature of cellular transcripts. How differential splicing controls membrane association, cell signaling, and immunoglobulin (Ig) secretion of the mRNAs encoding the B-cell receptor (BCR) and the T-cell receptor (TCR) are exemplars of the importance of mRNA splicing in immunity (7–9). With the advent of transcriptome-wide datasets, provided initially by microarray and more recently by next-generation sequencing (NGS) technologies, the novel concept of “RNA regulons” has emerged (10, 11). RNA regulons are defined as networks of RNA molecules that are similarly modulated in order to trigger a given response. Coordinating such RNA regulons is often the responsibility of regulatory RBPs that have key roles in immunity like the polypyrimide track protein 1 (PTBP1), embryonic lethal abnormal vision like protein 1 (ELAVL1, also known as HuR), or the members of the zinc finger protein 36 family (ZFP36, ZFP36L1 and ZFP36L2; also known as TTP or Tis11-family of proteins).
In recent years, the number of genes identified as encoding RBPs has increased substantially with the development of new techniques that enable the mapping of protein:RNA interactions to a single nucleotide resolution (e.g., RNA interactome capture, SONAR, and Cross-Linking ImmunoPrecipitation; CLIP) (12–15). In most cases, proteins with well-known enzymatic activity were classified as novel RBPs (13). This raises the possibility that RNAs are not only transient messengers of genetic information but also they might act as facilitators or repressors of protein function. For example, long non-coding RNAs (lncRNAs), such as NeST, Xist, Air, and Hotair, modulate transcription by binding to proteins in histone-modifying complexes and targeting them to selected genes including Ifng (16–19). Other lncRNAs, such as lnc-EGFR and Flicr, modulate Treg differentiation and function and have been implicated in peripheral immune tolerance (20, 21). Circular RNAs (cRNAs) enable the recruitment of the activation-induced cytidine deaminase AID to actively transcribed switch (S) regions for Ig gene mutagenesis and class-switch antibody recombination (CSR) (22). cRNAs also mediate formation of AID complexes with distinct heterogeneous nuclear ribonucleoproteins (hnRNPs) and SERBP1, which are themselves required for CSR (23, 24). Although it remains unclear how cRNAs and RBPs recruit AID selectively to S regions, formation of cRNA:DNA hybrid G-quadruplexes may explain selectivity while preventing off-target AID-mediated mutagenesis and chromosomal translocations that can lead toward B-cell malignant transformation.
Recent progress in RNA biology has brought us to the realization that chemical modification of the RNA (called “epi-transcriptomics” by analogy to DNA methylation) can exert key roles in cell maintenance, development, and differentiation in the immune system. Methylation, hydroxylation, and uridinylation of ribonucleotides were discovered over 50 years ago (25). But, it has not been until recently that such modifications have been linked with RBP function and with the control of mRNA stability and translation (26–29). The impact of RNA epigenetic modification is highlighted by its role in suppressing antiviral responses. Incorporation of modified nucleosides (e.g., m5C, m6A, m5U, s2U, or pseudouridine) reduces RNA recognition by Toll-like receptors (TLRs) (30). Methylation of adenosine at the N6 position (m6A) has been linked to nuclear retention of antiviral RNAs, inhibition of interferon production (31), and HIV viral replication in T cells (32). m6A mRNA methylation is also important in T-cell homeostasis and differentiation (33). Conditional deletion of the methyltransferase METTL3, which catalyzes m6A, disrupts IL-7 mediated signaling and reduces decay of SOCS mRNAs, affecting naïve T-cell priming for proliferation and differentiation (33). Over-expression of METTL3 blocks HSP myeloid differentiation whereas its inhibition induces apoptosis. METTL3-dependent methylation of N6-adenosine increases AKT phosphorylation and MYC, BCL2 and PTEN translation in myeloid leukemia cells (34). These studies illustrate the importance of annotating the epi-transcriptome of immune cells. Furthermore, it is to be expected that epigenetic modification of RNA will be dynamically regulated. Scores of RBPs have the potential to influence these modifications, acting as “writers,” “readers,” or “erasers” of these chemical changes. This highlights the immense complexity of post-transcriptional regulation of gene expression by RBPs.
RNA-binding proteins control all aspects of mRNA biology (Figure 1A). In the nucleus, RBPs bind to nascent RNA and recruit the spliceosome, a multimeric ribonucleoprotein complex that edits the nucleotide sequence of nascent RNAs by joining selected exons while removing intronic regions (35–37). Selective recognition of splicing sites for exon inclusion is carried out by splicing factors (e.g., SC35, hnRNPLL, PTBP1, and ELAVL1) and enables the expansion of the proteome by generating alternative coding mRNA transcripts. In lymphocytes, splicing factors commonly act as on/off switches to allow alternative splicing and RNA transcript expression (38). Altered or loss of function of splicing factors such as U2AF1, hnRNPA1, SF3B1, and SRSF2 results in profound deficiencies in hematopoiesis and myelodysplastic syndromes (39–42). RNA splicing is tightly linked with 5′ RNA capping (5′-m7Gppp) and 3′ RNA polyadenylation (PolyA). Alternative usage of polyA sites is associated with 3′UTR shortening, increased mRNA stability, and enhanced protein production upon macrophage and lymphocyte activation (43–45). Alternative polyadenylation plays an important role in antiviral innate immunity (46). Differential expression of polyadenylation factors (e.g., CSTF2, CPEB1, PAB1, PAB2, and U1-snRNP) regulates the usage of weak upstream polyA sites and removes destabilizing miRNA and RBP binding sites (43, 47–51). mRNA processing and maturation in the nucleus are closely associated with mRNA export to the cytoplasm through the nuclear pores. mRNA can be exported upon assembly of the ribonucleoprotein complex TREX or interaction of PAB1 with the CRM1/XPO1 complex. Deregulation of CRM1/XPO1 is often found in chronic lymphocytic leukemia and other cancers (52).
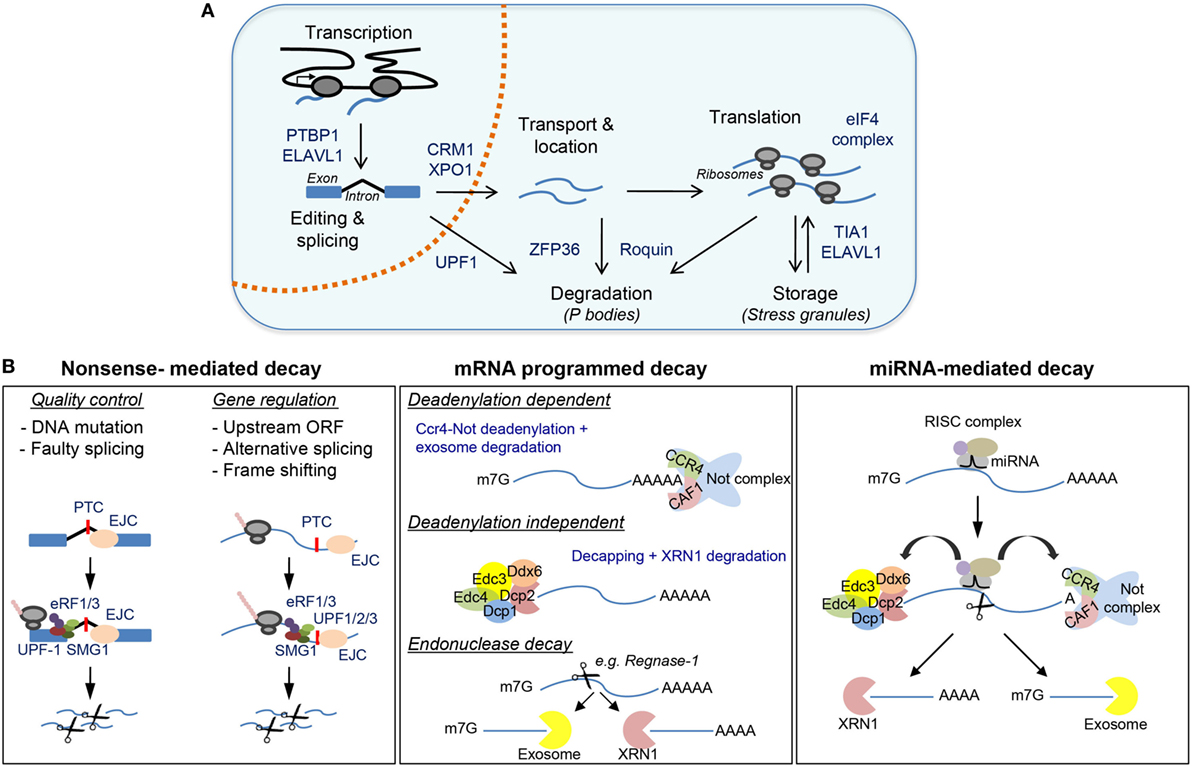
Figure 1. Post-transcriptional regulatory mechanisms controlled by RNA-binding proteins (RBPs) with a detailed view of messenger RNA (mRNA) decay. (A) Major post-transcriptional mechanisms regulated by RBPs include transcription, mRNA editing and splicing, mRNA transport and subcellular localization, mRNA translation, mRNA storage, and mRNA decay. (B) Major mechanisms for mRNA degradation include nonsense-mediated decay (NMD), mRNA programmed decay, and miRNA-mediated mRNA decay. Representative RBPs involved in these mechanisms are shown in (A,B).
In the cytoplasm, RBPs control the dynamic location and assembly of mRNAs into polyribosomes for protein synthesis. Recognition of the 5′-CAP and RNA regulatory motifs in the 5′UTR (e.g., Kozak and IRES sequences) by eukaryotic initiation factors enables formation of the 43S pre-initiation complex, ribosome assembly, and translation. mTOR-dependent regulation of the 4E-BP/eIF4E axis is essential for lymphocyte growth, proliferation, activation, and differentiation (53–56). Other RBPs, such as PTBP1, ELAVL1 and TIA1, can promote or repress translation after binding to specific RNA motifs present in the 5′UTR or 3′UTR (57, 58). Importantly, they can control the recognition and usage of alternative first-codon start sites. This allows translation of several open reading frames (ORFs) and expansion of the cell proteome (59). RNA accumulation within cytoplasmic RNA granules and regulation of translation are important mechanisms to halt viral replication during the antiviral immune response (60). RBP-dependent regulation of transcript stability and translation controls type I interferon (IFN) antiviral response. Dead-box helicases, including DDX9 and DDX58 (also known as RIG-I), recognize viral RNAs and activate the mitochondrial antiviral signaling protein (MAVS), NFkB and IRF3/7 to trigger the type-I (IFN) response (61–63). The extension of this antiviral response is controlled by RBPs such as OASL1, KSRP, and Elavl1. OASL1 expression modules IRF7 mRNA translation (64), whereas KSRP and Elavl1 binding to 3′UTR regulatory elements controls mRNA stability and translation of Ifnα and Ifnβ (65, 66). Interferon-induced, double-stranded RNA-activated protein kinase PKR promotes phosphorylation of eIF2a and silencing of CAP-dependent translation and stress granule (SG) formation (67–71). This global mechanism affects to hundreds of interferon-induced genes (72), and it is one of the 10 strategists used by viruses to escape from Ifn-mediated innate immunity (73, 74).
Mesanger RNA translation is coupled with mRNA stabilization and decay by RBP and miRNAs (Figure 1B). The 5′ CAP and the polyA tail not only enable efficient mRNA translation but also protect mRNA from degradation by the exosome. RNA decapping by DCP2 and mRNA deadenylation by polyA ribonuclease, including PAN2–PAN3, CCR4–NOT, and PARN, are central mechanisms widely used for mRNA translational silencing and decay. Several activators, including EDC4, enhance the catalytic activity of DCP2 and form the decapping complex. Decapped mRNAs are then susceptible to 5′- to 3′-exonuclease degradation, a process carried out by the XRN1 family of proteins. The abundance and activity of DCP2 are tightly modulated (75, 76). Recurrent mutations and/or chromosome translocations of the helicase DDX3X and the decapping activator protein NUDT16 are associated with different malignancies, including T-cell acute lymphoblastic leukemia, chronic lymphoblastic leukemia, natural killer/T-cell lymphomas, carcinomas, and medulloblastomas (77–83). PolyA tail shortening and 3′- to 5′-exonuclease degradation are promoted by destabilizing RBPs such as ZFP36, Roquin-1, and Roquin-2 (encoded by Rc3h1 and Rc3h2). Stable and actively translated mRNAs are bound by the RBP PABP, which also interacts with eIF4E to assemble the 43S pre-initiation complex and the ribosome. Recognition of constitutive decay AU- and GU-rich elements by mRNA decay activators such as Roquin1/2, ZFP36, KSRP, and CUGBP/CELF recruit polyA ribonucleases (84–86). This displaces PABP, shortens polyA tails and triggers exosome-mediated 3′–5′ decay (87). The activity of mRNA decay activators is highly regulated during immune cell activation. mRNA decay activators can compete with mRNA stabilizers such as ELAVL1 (88). Post-translational modification of RBPs controls the activity of mRNA regulons (89). For example, phosphorylation of ZFP36 by the p38-MK2 signaling pathway is not only essential for TNF production upon macrophage activation but also controls feedback regulatory networks that shape the inflammatory response (90). Deficiencies in MK2-p38 MAPK signaling or in ZFP36, Roquin-1, and Roquin-2 are associated with severe inflammatory and autoimmune pathologies linked to global changes in cytokine profile expression (91, 92).
Point mutations in RNA coding sequences, errors in mRNA splicing or abortive translation can trigger nonsense-mediated RNA decay (NMD) (Figure 1B). NMD is an RNA quality control mechanism that censors the synthesis of truncated proteins. During the first (or pioneer) round of translation, the cell tests whether mRNAs are correctly edited and spliced, and whether translation can take place from the start to the stop codon (93). The presence of components of the exon junction complex (marking unsuccessful splicing in newly transcribed mRNAs), the improper recognition of the 5′CAP by the cap-binding protein (CBP) complex (CBC) CBP80–CBP20 or the presence of premature termination codons triggers translation initiation silencing and NMD. The up-frameshift proteins 1, 2, and 3 (UPF1, UPF2, and UPF3) play a central role in NMD and enable XRN1- or SMG5/SMG7-mediated exonuclease decay or SMG6-mediated endonuclease decay (94). NMD is essential for embryo and neuronal development, hematopoiesis, and T-cell development and differentiation (95). The absence of UPF1 and UPF2 is associated with the accumulation of peptide by-products and cell death (96). UPF1 cooperates with siRNAs and miRNAs to control mRNA stability, myeloid cell differentiation, and inflammation (97, 98).
The importance of RBP in miRNA biogenesis and function has been reviewed extensively (99–102). Briefly, miRNAs are key agents of immune cell differentiation, homeostasis, and function. For example, conditional deletion in lymphocytes of the RBP DICER (an RBP required for the biogenesis of most miRNAs) blocks lymphoid development and differentiation (103, 104). Mechanistically, miRNA-mediated mRNA decay is linked to mRNA translational blockade, mRNA decapping and deadenylation (100). miRNAs act in close partnership with RBPs involved in mRNA stability control. RBPs can dampen miRNA-mediated decay by binding directly to miRNA precursors or, indirectly, by competing for binding motifs present in the 3′UTR of the mRNA targets. The RBP Lin28 binds directly to let-7 miRNA precursors, blocking their maturation which is required for reprograming of somatic cells and embryonic cell renewal (105–107). By contrast, ELAVL1 competes with miRNAs for binding to RNA regulatory motifs present in the 3′UTR of target mRNAs, enhancing their stability in macrophages (108). ELAVL1 also promotes miRISC complex dissociation from target mRNAs, thus increasing mRNA stability (109). On the contrary, physical interaction of ZFP36 with AGO2 (part of the miRISC complex) enhances miRNA-dependent mRNA degradation (110–112). Finally, the RBPs Pumilio 1 and 2 can reshape mRNA secondary structures allowing miRNA-21 and miRNA-22 recruitment and degradation of p27 mRNA (113). Recent evidence indicates that the expression of Pumilio 1 and 2 is closely linked to FOXP1/p21/p27 expression, thus having a key role in hematopoietic stem/progenitor cell (HSPC) proliferation and leukemic cell growth (114).
RNA location and storage are important mechanisms for timely protein synthesis in immune cells. Cytoplasmic RNA granules, including processing (P-) bodies and SGs, are assembled in both T and B cells upon activation with mitogens (58, 115). P-bodies are aggregates of ribonucleoprotein complexes containing RNAs targeted for degradation (116, 117). By contrast, SGs are associated with RNA translational silencing and storage (118–120). Why, when, and how P-bodies and SGs are assembled in activated lymphocytes remain poorly understood. However, it is plausible that they regulate the abundance and translation of key modulators of cell activation, proliferation, and selection (121). Roquin-dependent suppression of ICOS is linked to P-body assembly in CD4 T cells activated with anti-CD3 and anti-CD28 antibodies (115). In B cells, analysis of the mRNA targets bound by TIA1, a translation silencer found in SGs, showed that the translation of hundreds of transcripts might potentially be regulated by temporal location in RNA granules. TIA1 regulates p53 mRNA translation during B-cell activation and in response to genotoxic stress (58). TIA1 binding to U-rich motifs in the p53 3′UTR silences translation in activated B cells without affecting overall mRNA abundance. DNA damage triggers TIA1:p53 mRNA dissociation, p53 mRNA release from SGs and p53 protein synthesis. Thus, TIA1 has the potential to coordinate cell cycle arrest, DNA repair, and selection of B cells by controlling p53 expression. It is also possible that regulation of mRNA location and translation is part of a larger genetic program that enables to rapidly proliferating lymphocytes to switch quickly their transcriptome and translatome to cope with endogenous genotoxic stress during TCR/BCR expansion.
Genetically altered mouse models have revealed the importance of RBPs in myeloid and lymphocyte development and function (Figure 2). One of the most studied RBP in the immune system is Elavl1 (also known as HuR). Conditional deletion of Elavl1 from the pro-B-cell stage onward results in reduced B-cell numbers. B-cells populations are consistently reduced between 1.5- and 5-fold in the bone marrow and the periphery. This correlates with a global reduction in serum Igs (122, 123). By contrast, Elavl1 deficiency in myeloid-cells does not affect bone marrow progenitors or their capacity to differentiate in vivo and in vitro (124). Conditional deletion of ELAVL1 in thymocytes affects T-cell development and egress from the thymus, thus resulting in severe lymphopenia in the periphery. ELAVL1 is required for TCR-mediated signaling, activation and progression through positive selection. In the absence of ELAVL1, an array of cell cycle regulators, TCR, and death-receptor signaling components are deregulated, leading to an accumulation of CD4 and CD8 single positive thymocytes (125).
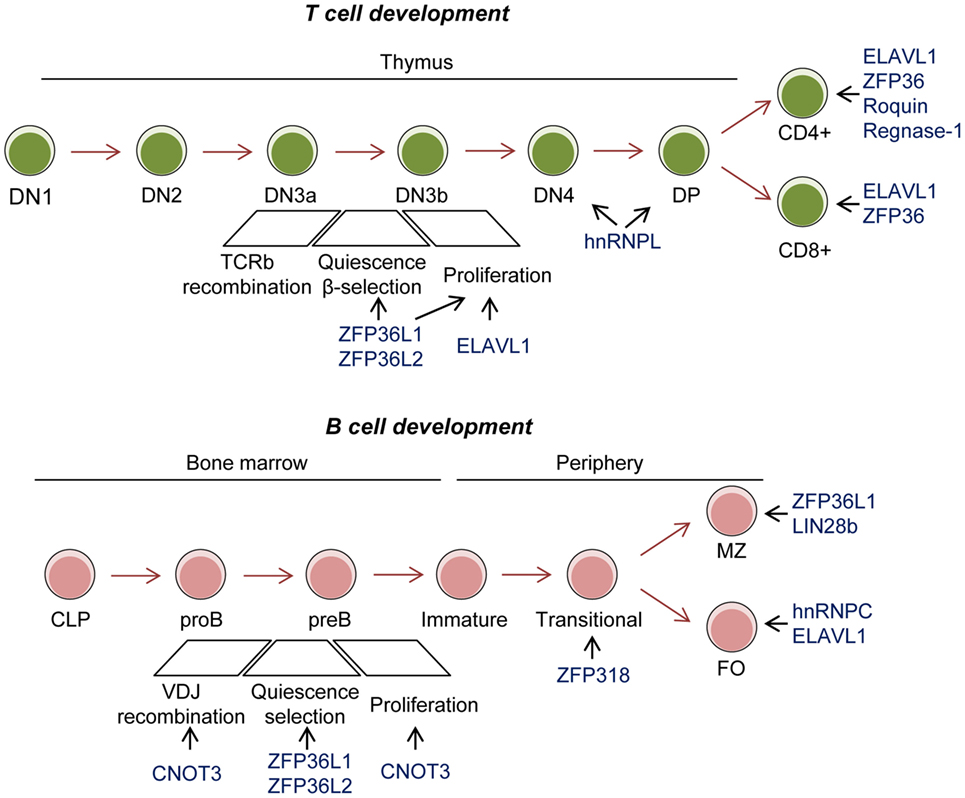
Figure 2. Role of RNA-binding proteins (RBPs) in T- and B-cell development. Conditional mouse models have revealed the importance of the RBP ZFP36L1 and ZFP36L2 in cell quiescence upon VDJ recombination to test and positively select those cells that have successfully recombined the BCR and TCR. C-NOT3 is related to successful VDJ recombination. These three RBPs and ELAVL1 are implicated in later expansion of double negative (DN) 3–4 T cells. ELAVL1, ZFP36, Roquin, and Regnase-1 are implicated in activation and differentiation of single positive T cells. ZFP318 is involved in IgD expression in transitional T2 B cells. ZFP36L1 and LIN28b have been involved in marginal zone (MZ) B-cell differentiation. hnRNPC and ELAVL1 are implicated in follicular (FO) B-cell maintenance.
Qualitative and quantitative control of the cell transcriptome by mRNA splicing is a key for lymphocyte development. For example, the B-cell development block arising from PTEN knockout in pro-B cells has been attributed, in part, to the defective splicing of Ikzf1 mRNA. In pro-B cells, PI3K signaling supresses the function of FOXO-1 that is required for the correct splicing of Ikzf1, a transcription factors which enables VDJ recombination in cooperation with PAX5 (126). Forward genetic screens in zebrafish have extended the list of RBPs involved in pre-mRNA-processing in T cells in thymus and they support the idea that global splicing pathways control lymphoid development (127). Expression of the splicing factors FUS, SC35, hnRPNPL, and hnRPNPLL is important for B- and T-cell development and for T-cell activation (128, 129). This was initially attributed to the role of SC35, hnRPNPL, and hnRNPLL in regulating alternative mRNA splicing of the receptor tyrosine phosphatase CD45, a key negative-feedback regulator of TCR signaling (130, 131). However, recent transcriptome analyses suggest that they might also regulate global splicing programs in T and B cells (132, 133). Analysis of hnRPNPL knockout mice shows decreased thymic cellularity, a partial block at double negative 4 and double-positive T-cell stages, and the reduced egress of mature T cells from the thymus to the periphery. Thymocytes deficient in hnRPNPL have an aberrant splicing program that reduces CD45 levels and, possibly, the expression of GTPase and cytoskeleton regulators, that are key for T-cell migration in response to CCL21 and CXCL2. hnRPNPL also modulates pre-TCR signaling as its deletion increases Lck activity, linked to increased proliferation of DN4 cells (134). Future studies are required to characterize the splicing profiles of the different immune cell populations during development and to identify the splicing regulators and their mechanisms of action.
Splicing has also an important role in myeloid cell development. Splicing factors are commonly mutated in hematopoietic malignancies. As a result, these diseases are associated with extensive alterations in mRNA splicing. Mouse genetics has revealed that mutagenesis of SRSF2 and SF3B1 drives myelodysplastic syndromes characterized by leukopenia, macrocytic anemia, myeloid, and erythroid dysplasia (135–137). B-cell lineage-specific expression of the mutant SF3B1-K700E reduces the number of mature B cells. Sub-clonal mutations of splicing factors, such as SF3B1, are often found after treatment of chronic lymphocytic leukemia, and they are predictive of poor outcomes (138, 139). Recently, chronic stimulation of TLR leading to HSPC dysfunction and myelodysplasia has been linked to altered RNA splicing due to abnormal ubiquitination of the splicing factor hnRNPA1 (42). Using gain and loss of function models, it has been shown that the E3 ubiquitin ligase TRAF6 can modulate the function of multiple RBPs, including hnRNPA1. This RBP controls mRNA splicing during hematopoiesis and myeloid development. hnRNPA1 regulates the alternative splicing of Arhgap1, an inhibitor of the small G-protein CDC42 that regulates LT-HSC self-renewal and differentiation. hnRNPA1 shuttling activity is required for the formation and survival of myeloid precursors. Disruption of hnRNPA1 shuttling can lead to tumorigenesis (140). In summary, RBPs play major roles in the development of the immune system and they preserve cells from malignant transformation by quantitatively and qualitatively affecting the transcriptome.
Regulation of mRNA stability and decay is essential in lymphocyte development. The RBPs ZFP36L1 and ZFP36L2 are redundant in lymphocyte development. However, conditional deletion of both RBPs early in lymphopoiesis results in a severe reduction of lymphocyte precursors in the thymus and bone marrow, lymphopenia and, eventually, malignant transformation of immature CD8 + thymocytes. Mechanistically, ZFP36L1 and ZFP36L2 regulate the expression of proliferative cell cycle regulators in developing thymocytes by controlling mRNA stability and translation (141, 142). In the absence of these RBPs, up-regulation of NOTCH1contributes to the bypass the β-selection checkpoint. In B cells, ZFP36L1 and ZFP36L2 are essential for cell quiescence necessary for VDJ recombination. In both cell types, they regulate the mRNA stability of an RNA regulon involved in transition into the S phase of the cell cycle (143, 144). Thus, it is possible they enforce quiescence in other developmental systems.
B-cell specific deletion of CNOT3, a subunit of the CCR4–NOT deadenylase complex, results in a developmental block at the pro- to pre-B-cell transition. CNOT3 has a dual function. It controls the efficient VH to DH-JH rearrangement in the distal region of IgH as well as it maintains in check P53-EBF1 expression. In its absence, p53 mRNA is abnormally stable and expressed. This switches on the expression of the p53-target genes p21, Bax, and Puma, which in turn induce cell growth arrest and death (145, 146). The interplay of RBPs and miRNAs in mRNA stability and decay is also essential for lymphocyte development. LIN28b is expressed in fetal BM and thymus. It inhibits miRNA let-7 in order to promote fetal immune cell development. Enforced expression of Lin28b in adult BM results in the anomalous expansion of B-1a, marginal zone (MZ) B cells, gamma/delta T cells, and natural killer (NK) T cells (147). An emerging picture from these studies suggests that the regulation of mRNA stability and decay is essential for immune cell development. RBPs coordinate fundamental cellular processes such as quiescence, cell cycle re-entry and proliferation coupled to TCR and BCR rearrangement and cell selection at given cell stages of development. If RBP function is faulty, this can lead to pathology.
Recent studies have highlighted the importance of RBPs in immune cell maintenance and differentiation in the periphery. For example, the RBP hnRNPC (aka AUF1) is required for BCL2, A1, and BCL-XL expression and maintenance of follicular (FO) B cells (148). hnRNPC regulates Bcl2 mRNA decay through the binding to AU-rich regulatory elements (AREs) present in the Bcl2 3’UTR. Gene targeting deletion of these Bcl2 AREs diminishes Bcl2 mRNA stability and protein levels in primary B cells, decreasing life- span of transitional (T) and FO B cells (149). Stringent regulation of mRNA abundance is also essential for the maintenance of marginal zone precursors (MZP) and mature marginal zone (MZ) B cells. Expression of the RBP ZFP36L1 enforces the identity of MZ B cells by limiting the expression of genes that promotes the FO B-cell phenotype. This novel epigenetic mechanism involves the repression by ZFP36L1 of the transcription factors KLF2 and IRF8. In the absence of ZFP36L1, MZ B-cell identity is lost as well as cell location in the splenic MZ (150).
The role of RBPs such as ZFP36, Roquin, Regnase-1, hnRNPC and ELAVL1 in myeloid cell activation has been studied extensively. Mice lacking the expression of ZFP36, Roquin, Regnase-1, and AUF1 share pro-inflammatory syndromes arising from a failure to limit cytokine production. Timely expression of cytokines and growth factors is regulated by RBPs that bind to RNA regulatory elements present in their 3′UTR. Constitutive decay elements (CDEs), AU-rich elements (AREs), and miRNA recognition elements control cytokine mRNA stability and decay. CDE and ARE show very low sequence complexity and can be bound by multiple RBPs sometimes mediating opposing functional outcomes. Widespread 3′UTR shortening and removal of decay elements may be a general mechanism used by T cells and macrophages to increase the expression of cytokines upon activation (43, 44), although their impact in protein expression might be limited (45) and subject to further regulation.
Post-translational control of RBP expression, subcellular location, and binding affinity is a reversible regulatory mechanism that affects RNA operons during macrophage activation (151, 152). Competitive binding of RBPs and miRNA to their RNA regulatory elements is coupled to cellular signaling pathways. Phosphorylation of ZFP36 at two conserved serine residues (S52 and S178 in mouse, S60 and S186 in human) by the p38-MK2 axis turns on the expression of essential immunomodulators such as TNF and COX-2. RAS-MEK signaling upstream of p38-MAPK mediates tumor cell intrinsic expression of PD-L1, partly by supressing ZFP36-dependent CD274 mRNA decay, and promotes tumor-immune evasion (153). Mechanistically, phosphorylated ZFP36 is sequestered by 14-3-3 proteins reducing its affinity for RNA. This prevents deadenylase recruitment and mRNA degradation (88, 154, 155). Selective targeting of mRNAs by ZFP36 not only regulates the expression of cytokines but also controls the magnitude of the pro-inflammatory response upon macrophage activation with LPS (90, 156). ZFP36 regulation of negative feedback regulators such as DUSP1, IER3, and TNFAIP3 (or A20) may also prevent TNF overexpression, apoptosis, and chronic systemic inflammatory syndrome (157–159). Recently, analysis of non-phosphorylatable Zfp36aa/aa knockin mice showed protection in models of bacterial infection and inflammatory arthritis (160, 161), corroborating the essential role of post-translational regulation of ZFP36 function in macrophage activation.
Similarly, functional activity of the RBP ZFP36L1 is regulated by the mTOR-p38-MK2 signaling pathway. ZFP36L1 controls a senescence-associated secretory phenotype that can either activate immune surveillance responses or promote tumor development and aging (162). Inhibition of mTOR impairs the senescence phenotype, partly by blocking 4EBP translation initiation of MK2. In turn, MK2 fails to phosphorylate and inactivate ZFP36L1, decreasing the mRNA expression of senescence-related mRNAs such as Cdkn1a, IL8, and IL1. Selective mutation of ZFP36L1 at Ser54, Ser92, and Ser203 impairs the senescence-associated secretory phenotype and blocks the pro-tumorigenic effects of senescence. Interestingly, mutation of MK2 phosphosites of ZFP36 and ZFP36L2 also reduces senescence to some extent, pointing out to the conservation of redundant functional mechanisms that are yet to be defined.
hnRNPC and Roquin have been also involved in cytokine mRNA destabilization and in the inflammatory response. Similar to ZFP36, deletion of these two RBPs aggravates endotoxemia and chronic inflammation (163–165). This suggests that, although each of these three proteins induces mRNA translational silencing and degradation of similar targets, they may have non-redundant functions. Roquin has been mostly studied in T cells. However, it is co-expressed with ZFP36 and hnRNPC in macrophages. Whether these RBPs cooperate in the control of the inflammatory response and by which mechanisms are questions that remain unanswered. It might be possible that they control different thresholds of macrophage activation at different given times. Sustained activation of the p38-MK2 and NFkB pathways by hnRNPC and Roquin seems to play a central role in regulating the function of ZFP36 (166, 167). These RBPs, additionally, regulate the stability and translation of their own mRNAs (168). Thus, future studies should integrate the knowledge about these three RBPs to identify the molecular mechanisms and activation thresholds that control cytokine mRNA decay and expression in macrophages during the inflammatory response.
It is widely believed that ELAVL1 function opposes the destabilizing activities of ZFP36 and hnRNPC, and the translational silencing function of TIA1 (88, 169–172). However, recent studies using conditional knockout mice suggest a more complex picture. ELAVL1 controls the expression of both pro-inflammatory and anti-inflammatory cytokines in a cell-dependent manner. Myeloid cell-specific deletion of ELAVL1 results in an exacerbated inflammatory phenotype with enhanced expression of chemotaxis-related and inflammatory mRNAs (including Tnf, Tgfb, Il10, Ccr2, and Ccl2) (124). ELAVL1 synergizes with the translational inhibitor TIA1 to suppress pro-inflammatory cytokine expression (170). These results contrast with other findings that linked ELAVL1 expression with increased Tnf mRNA stability and TNF synthesis in macrophages (88, 173). Similarly, results obtained after T cell-specific deletion of ELAVL1 suggest a more complex role than the opposition of ZFP36. Th2-polarized cells from heterozygous Elavl1 mice have decreased mRNA steady levels of Gata3, Il4, and Il13 without affecting protein abundance. However, Th2 cells from Elavl1 KO homozygous mice have an increased expression of Il2, Il4, and Il13 mRNA and protein (174). This could be explained if ELAVL1-dependent stabilization of cytokine mRNAs is out-weighed by the altered expression of other RNA regulons controlling key cellular functions. Indeed, ELAVL1 expression in T cells and macrophages is associated with the stabilization of RNA regulons involved in cell activation, signaling, proliferation, and differentiation (125, 175). In B cells, ELAVL1 preserves mRNA splicing of key metabolic genes during B-cell activation, allowing metabolic switch and cell growth. In the absence of ELAVL1, B cells fail to control the oxidative response induced upon mitogen activation and die by apoptosis (122). Regulation of ELAVL1 function is highly dynamic and depends on post-translational modification of the protein. Phosphorylation, methylation, and ubiquitination alter ELAVL1 subcellular location and/or function (176–179). Cellular stress and DNA damage responses are drivers of ELAVL1 function. ATM/ATR, CHK1/CHK2, PKC, AMPK, and MK2-p38 MAPK have been all involved in ELAVL1 phosphorylation and regulation (151, 152). Thus, ELAVL1 is a major post-transcriptional regulator of gene expression whose function is stringently regulated during immune cell activation.
RNA-binding proteins coordinate global changes in mRNA expression and splicing upon TCR- and CD28-mediated T-cell activation (180–183) (Figure 3A). It has been estimated that over 2,000 genes are subjected to alternative splicing in T cells upon activation, including genes involved T cell-specific and chemokine activation pathways and in MAPK, NFKB, and JAK/STAT signaling (184). Intron retention is globally reduced in activated T cells whereas alternative polyA site usage and 3′UTR shortening is increased. These effects correlate with a global increase in mRNA stability and translation (43, 185). Mapping of functional 3′ splice sites has been attempted by U2AF2 RNA immunoprecipitation (184). U2AF1 and U2AF2 form the heterodimer U2AF, which recruits the spliceosome to the 3′ splice site. Loss of function experiments suggest that U2AF1 is required for mRNA splicing and surface expression of CD25 and CD62L and the secretion of cytokines such as IL4, IL5, IL10, IL13, IL17, IFNg, RANTES, and TNF. The expression of SYNCRIP and ILF2, two splicing factors that bind to the U2AF complex, is also important for the secretion of a limited number of cytokines such as IL-21, suggesting that they might regulate T follicular helper (Tfh) cell differentiation (184).
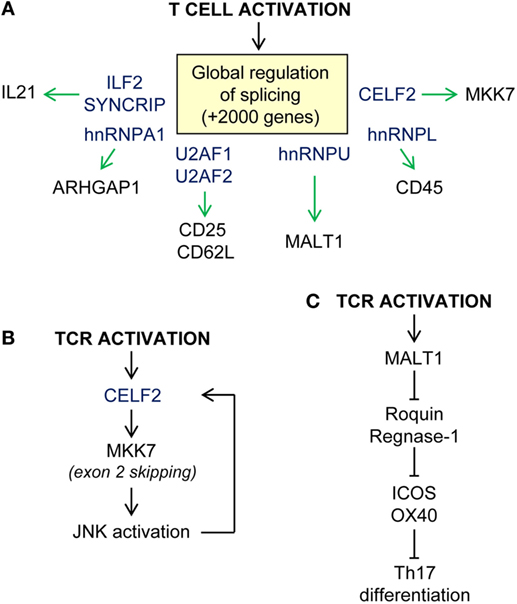
Figure 3. Examples of post-transcriptional regulation upon T-cell activation. (A) Global regulation of RNA splicing upon T-cell activation. Splicing regulators (in blue) control alternative splicing and/or expression of T-cell receptor (CD25, CD62L, CD45), kinases (MKK7), paracaspases (MALT1), and cytokines (IL21) among other proteins. (B) CELF2-MKK7-JNK axis of enhanced TCR-mediated activation. (C) T-cell differentiation in modulated by the extension of MALT1 paracaspase activation, cleavage of Roquin and Regnase-1 RBP, and release repression of their mRNA targets.
Other splicing factors such as hnRNPU, CELF2, and hnRNPL play essential roles in modulating TCR-dependent signaling and activation. hnRNPU is a negative regulator of the multifunctional protein MALT1, one of the components of the CARMA1–BCL10–MALT1 signaling complex. As part of this complex, MALT1 acts as a scaffold between antigen-engaged TCR/CD28 and downstream NF-kB-signaling pathways. TCR-mediated activation promotes Malt1 exon7 inclusion. Malt1 exon 7 encodes for a TRAF6-binding domain that allows TRAF6 recruitment and ubiquitination of IKK and activation of classical p65/p50 NFkB. This enables production of IL-2 and T-cell proliferation. Expression of hnRNPU limits Malt1 exon 7 inclusion acting as a negative feedback regulator (186). In this study, the authors identified other splicing factors that potentially compete to modulate Malt1 alternative splicing. hnRNPR, hnRNPLL, and SRSF3 might promote exon inclusion whereas hnRNPH1, hnRNPA1, U2AF1, and hnRNPU promote exon skipping. As noted above, a recent report shows that TRAF6-mediated ubiquitination of hnRNPA1 leads to global splicing changes in HSC (42). Thus, it is possible that coordinated regulation of different splicing factors shapes the T-cell transcriptome during development, activation, and differentiation.
Regulatory feed-forward loops allow activated T cells to respond to a rapidly changing environment. Evidence suggests that the splicing factor CELF2 and JNK are involved in a positive loop that allows the full activation of T cells (Figure 3B). CELF2 expression is required for exon 2 skipping of MKK7. This enables MKK7-protein interaction with JNK, enhancing signaling. In turn, JNK activation induces CELF2 stabilization and further exon skipping. hnRNPL is also a splicing repressor that has been involved in CD45 alternative splicing. Expression of hnRNPL leads to the expression of the smallest isoform of CD45 which is required for T-cell homeostasis (187, 188). hnRNPL-deficient mice have profound defects in thymic development and migration to the periphery that cannot be only subscribed to a defect in CD45 alternative splicing (134). Indeed, a recent study suggests that hnRNPL regulates the splicing of hundreds of genes upon PMA activation of Jurkat cells (189).
T-cell activation also induces the accumulation of RBPs in the cytoplasm (190). This directly correlates with the increase in RNA metabolism that supports T-cell growth, proliferation, cytokine production, and differentiation into the different T-cell subtypes. For example, TCR-mediated ERK activation phosphorylates hnRNPK at Ser284 and Ser353. This allows hnRNPK cytoplasmic location and translation inhibition of selected mRNA targets. hnRNPK also limits Vav-1-mediated proteolysis and enables IL-2 production (191, 192). Other RBPs such as ELAVL1, PTBP1, and ZFP36 have also been involved in IL2 mRNA translational regulation and protein synthesis upon TCR engagement (174, 193, 194).
The role of RBPs in cytokine expression upon antigen-mediated T-cell activation has been, and remains, an intensive area of research. T-cell differentiation and effector/memory functions are associated with distinctive cytokine profiles. Recent evidence suggests that mRNA translation silencing by RBPs is responsible for uncoupling cytokine mRNA abundance from protein synthesis in anergic self-reactive T cells (195). RBPs, such as Roquin-1 and Roquin-2, have a relevant role in controlling TCR signal strength, activation, and differentiation of mature T cells (Figure 3C). The number of CD4+T cells is normal in Roquin-1 knockout mice, although the CD8+T-cell population is expanded (196). Expression of Roquin-1 and Roquin-2 is reduced upon TCR engagement. Both gene transcription and activation of the paracaspase MALT1 contribute to decrease Roquin expression (197, 198). This promotes strong T-cell activation and effector functions regulating the expression of ICOS and OX-40 (165). Production of IL-10 also decreases Roquin-1 which limits further the expression of these co-inhibitors and promotes Tfh cell differentiation (198, 199). The increased number of Tfh cells in Roquin-1/2 double knockout and Sanroque mice causes a breach in self-tolerance and promote autoimmunity. These mice have elevated levels of IL-17 and IFNg, which correlates with an increased differentiation of Th17 and Tfh cells (197, 200). Roquin-1 function favors Th1 over Th2 T cell and increases serum levels of IFNg, IL6, IL17, and TNF. This is linked with the development of hepatitis and strong collagen-induced arthritis (201, 202). In addition, the RBP ARID5a promotes the development of autoimmune diseases such as experimental autoimmune encephalomyelitis and arthritis (203). Mechanistically, ARID5a stabilizes Il6 mRNA, likely by countering Regnase-1 endonuclease activity. In the absence of ARID5a, Il6 production is significantly reduced in vivo and Th1 T cells are favored over Th17 T cells. In summary, post-transcriptional regulation of cytokine expression by RBPs controls the magnitude of T-cell effector functions as well as balances antigen-dependent T-cell differentiation.
The role of alternative splicing and polyadenylation in the generation of different Ig isotypes and the production of membrane-bound or secreted Ig has been long appreciated (8, 9, 204). Alternative splicing of an mRNA transcript encoding for the Ig mu heavy chain (Igh) produces both IgD and IgM. Recent evidence suggests that the candidate RBP ZFP318 controls differential splicing in transitional T2 B cells enabling IgD expression (205, 206). Splicing also raises the production of rare Ig by-products upon induction of somatic hypermutation (SHM) by Plasmodium falciparum and other pathogens (207). These rare Ig by-products can be often found in B-cell non-Hodgkin’s lymphomas (B-NHL), B-cell chronic lymphocytic leukemia (B-CLL), and other lymphomas, marking their origin as the germinal center (GC) reaction (208). The mechanisms controlling the alternative splicing of these Ig transcripts remain incompletely understood, but might be linked to the observation that AID and some of its cofactors are bonafide splicing regulators. Activation-induced cytosine deaminase (AID) is expressed upon B-cell activation and is necessary for affinity maturation in GCs. Affinity maturation is the process by which SHM of the Ig locus enables the expansion of the antibody repertoire. AID is an RNA/DNA binding protein that binds to G-quadruplex structures formed upon active transcription of the Ig locus (209). RNA mediates the interaction of AID with hnRNPK and hnRNPL, which are required for DNA cleavage and end-joining essential to both class CSR and SHM (23). hnRNPI, hnRNPU, hnRNPC, PABP1, and SERBP1 have been also described as components of these AID-RNP complexes. Knockdown of any of these proteins reduces CSR to some extend (24).
RNA-binding proteins control many other aspects of the antibody response (Figure 4). T-cell independent and T-cell-dependent antibody responses are impaired in the absence of PTBP1 or ELAVL1. GCs are not formed in Elavl1fl/fl Mb1Cre mice, and affinity maturation is severely impaired (122, 123). ELAVL1 expression guarantees the correct mRNA splicing and expression of key enzymes of the glucose metabolism such as DLST. This is the E2 subunit of the 2-oxoglutarate dehydrogenase complex that controls the amount of reducing equivalents (NADH) that will be subsequently used for oxidative phosphorylation, ATP production, and ROS scavenging. Thus, after B-cell activation, ELAVL1 enables a B-cell metabolic switch that fuels B-cell growth and proliferation while preserving the cells from a detrimental oxidative stress response (122). Additionally, PTBP1 is essential for high-affinity antibody production (210). PTBP1 controls the expression of a substantial fraction of MYC-dependent genes to enable cell cycle progression and GC B-cell positive selection. Among these, PTBP1 regulates the splicing of key metabolic genes such as Pkm1 and Tyms but also controls other MYC target genes by an, as yet, unknown mechanism. Whether additional properties of PTBP1, such as its ability to regulate mRNA stability or translation, play a role in the selection of GC B cells remains uncertain. The RBP hnRNPLL might also have an important role during later B-cell to plasma cell differentiation (211). In the absence of hnRNPLL, plasma cells and antibody production are reduced. This is likely because hnRNPLL regulates both mRNA splicing and stability of key drivers of plasma cell differentiation such as Irf4 and Oct1. Finally, it is suggested that hnRNPLL might regulate mRNA translation of IgG1, although this needs further investigation.
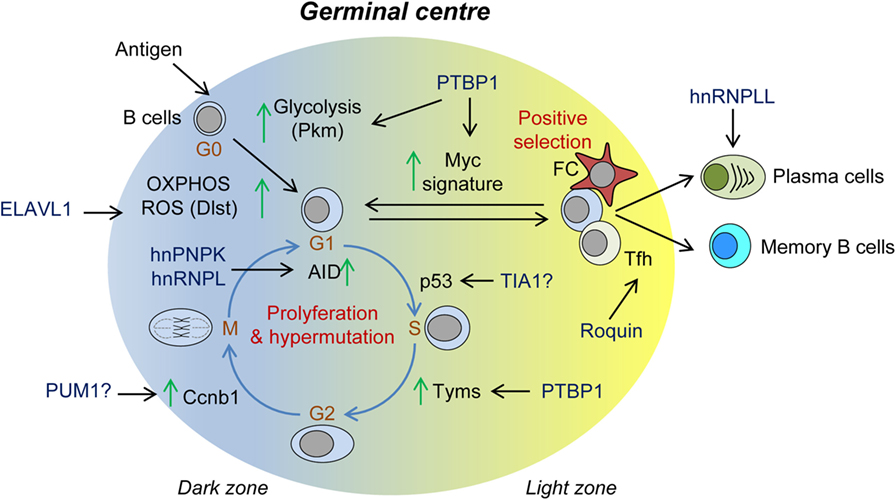
Figure 4. Role of RNA-binding proteins (RBPs) in the germinal center (GC) reaction. Summary of RBPs associated with: (1) B-cell metabolic switch upon B-cell activation; (2) cell cycle progression, proliferation, and somatic Ig hypermutation in the dark zone; (3) positive GC B-cell selection and Myc-mediated re-entry into further rounds of proliferation and Ig hypermutation; and (4) terminal differentiation into plasma B cells and memory B cells.
RNA-binding proteins control T-cell-mediated B-cell selection in GCs (Figure 4). Analysis of the Sanroque mouse model revealed the importance of Roquin-1 and Roquin-2 expression in T-follicular helper cells to prevent autoimmunity. Sanroque mice bare a point mutation in the ROQ domain of Roquin-1 and develop spontaneous GCs, plasmacytosis, polyclonal hypergammaglobulinemia, and production of autoantibodies and high-levels of IFNg. As a result, they develop hepatitis, nephritis, and anemia (165, 200). Roquin-1 and Roquin-2 are mRNA destabilizers that limit the extension of the GC response. They control the number and activation of Tfh cells as well as they restrain activation and differentiation of helper T cells and conversion of Treg to Tfr cells (212). Roquin-1 and Roquin-2 bind to CDEs present in the 3′UTR of several mRNA targets, including Icos, Ox40, Il6, Ifng, and Tnf, and recruit the CCR4-NOT deadenylation complex (115, 213–215). It has been proposed that Roquin not only can cooperate with miRNAs such as miR-146a but also can interfere with miR-17-92, causing PTEN up-regulation and controlling excessive PI3K-mTOR signaling and autoimmunity (212). Roquin can interact with other RBPs such as NUFIP2 to recognize 3′UTR RNA regulatory elements (216) and with the RNA nuclease Regnase-1 (encoded by Zc3h12a) (197) to regulate the expression of mRNA targets like Icos. Regnase-1 targets mostly mRNA undergoing active translation in the cytoplasm or in the endoplasmic reticulum (217, 218). By contrast, it is believed that Roquin targets for degradation silenced mRNAs that accumulate in P-bodies. Nevertheless, Regnase-1 knockout mice have a similar phenotype to the Sanroque mice (219).
Post-transcriptional control by RBPs is an essential extra layer of gene regulation that is fundamental for the development, homeostasis, and function of the immune system. The involvement of RBP at all stages of the biology of RNA can be conceptualized by considering RBPs as writers, editors, readers, and erasers of the transcriptome; an analogy that has been used previously in the context of epigenetic regulation at the level of DNA and histones. RBP writers (e.g., splicing factors) affect transcription and RNA processing. RBP editors (e.g., RNA methyltransferases and deaminases) modify the sequence content of the transcriptome. RBP readers bind RNA and define subcellular location (e.g., RNA granule components), and translation (e.g., eukaryotic translation factors). Finally, RBP erasers (e.g., destabilizing factors and nucleases) induce RNA decay. Acting in concert with transcription factors, epigenetic regulators, and signal transduction networks, they comprise a global regulatory network that we are just starting to appreciate.
A reductionist approach to study the role of RBPs at a single gene level is likely to be insufficient to uncover the broad implications of post-transcriptional RNA control in immune cell development, differentiation, and function. An integrative analysis of the protein:RNA interactome, transcriptome, and translatome is required to understand the global mechanisms of RBP-mediated control. Gaining knowledge of how individual RBPs target single RNA molecules and their function is a first step to further define these global cellular mechanisms. Collection of the protein:RNA interactome from hundreds of RBPs is extremely useful in order to infer possible interactions of different RBPs for single or global gene expression. Capturing the dynamic changes of cellular RBP-content in concert with the quantitative and qualitative changes in the transcriptome is a key to understand immune cell development and activation. For example, it has been reported that m6A modification of the RNA might differ between different RNA species and during T-cell activation (33, 220). Cellular responses to intrinsic and extrinsic signals induce post-translational modifications that alter both the expression and function of RBPs. Thus, careful selection of model systems is essential when studying the role of post-transcriptional regulation by RBP in the immune system. Finally, validation of the post-transcriptional regulatory mechanism by selecting model genes is essential. Gene-wide identification of the protein:RNA interactome annotates tens of thousands interactions with hundreds of transcripts (12). However, RNA binding by RBPs may not always have consequences for the qualitative or quantitative transcriptome. Thus, genetic modification and biochemical analysis of single protein:RNA interactions will be important to validate RBP action on genes that drive important immune phenotypes.
MDD-M, manuscript conception, literature review, and writing. MT, conception and writing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
MDD-M is supported by ATIP-Avenir-Plan Cancer (C18003BS) and Inserm-CNRS-University Paul Sabatier (U104318B). MT is supported by The Biotechnology and Biological Sciences Research Council (BB/J004472/1 and BB/J00152X/1), the Wellcome Trust (200823/Z/16/Z), and Bloodwise (14022).
1. Turner M. Is transcription the dominant force during dynamic changes in gene expression? Adv Exp Med Biol (2011) 780:1–13. doi:10.1007/978-1-4419-5632-3_1
2. Turner M, Galloway A, Vigorito E. Noncoding RNA and its associated proteins as regulatory elements of the immune system. Nat Immunol (2014) 15:484–91. doi:10.1038/ni.2887
3. Shaw G, Kamen R. A conserved AU sequence from the 3’ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell (1986) 46:659–67. doi:10.1016/0092-8674(86)90341-7
4. Caput D, Beutler B, Hartog K, Thayer R, Brown-Shimer S, Cerami A. Identification of a common nucleotide sequence in the 3’-untranslated region of mRNA molecules specifying inflammatory mediators. Proc Natl Acad Sci U S A (1986) 83:1670–4. doi:10.1073/pnas.83.6.1670
5. Koeffler HP, Gasson J, Tobler A. Transcriptional and posttranscriptional modulation of myeloid colony-stimulating factor expression by tumor necrosis factor and other agents. Mol Cell Biol (1988) 8:3432–8. doi:10.1128/MCB.8.8.3432
6. Turner M, Katsikis PD. A new mechanism of gene regulation mediated by noncoding RNA. J Immunol (2012) 189:3–4. doi:10.4049/jimmunol.1201339
7. Behlke MA, Loh DY. Alternative splicing of murine T-cell receptor beta-chain transcripts. Nature (1986) 322:379–82. doi:10.1038/322379a0
8. Rogers J, Choi E, Souza L, Carter C, Word C, Kuehl M, et al. Gene segments encoding transmembrane carboxyl termini of immunoglobulin gamma chains. Cell (1981) 26:19–27. doi:10.1016/0092-8674(81)90029-5
9. Danner D, Leder P. Role of an RNA cleavage/poly(A) addition site in the production of membrane-bound and secreted IgM mRNA. Proc Natl Acad Sci U S A (1985) 82:8658–62. doi:10.1073/pnas.82.24.8658
10. Keene JD. RNA regulons: coordination of post-transcriptional events. Nat Rev Genet (2007) 8:533–43. doi:10.1038/nrg2111
11. Bisogno LS, Keene JD. RNA regulons in cancer and inflammation. Curr Opin Genet Dev (2017) 48:97–103. doi:10.1016/j.gde.2017.11.004
12. Lee FCY, Ule J. Advances in CLIP technologies for studies of protein-RNA interactions. Mol Cell (2018) 69:354–69. doi:10.1016/j.molcel.2018.01.005
13. Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell (2012) 149:1393–406. doi:10.1016/j.cell.2012.04.031
14. Brannan KW, Jin W, Huelga SC, Banks CA, Gilmore JM, Florens L, et al. SONAR discovers RNA-binding proteins from analysis of large-scale protein-protein interactomes. Mol Cell (2016) 64:282–93. doi:10.1016/j.molcel.2016.09.003
15. Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB. CLIP identifies Nova-regulated RNA networks in the brain. Science (2003) 302:1212–5. doi:10.1126/science.1090095
16. Gomez JA, Wapinski OL, Yang YW, Bureau JF, Gopinath S, Monack DM, et al. The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-gamma locus. Cell (2013) 152:743–54. doi:10.1016/j.cell.2013.01.015
17. Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, et al. The air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science (2008) 322:1717–20. doi:10.1126/science.1163802
18. Jeon Y, Lee JT. YY1 tethers Xist RNA to the inactive X nucleation center. Cell (2011) 146:119–33. doi:10.1016/j.cell.2011.06.026
19. Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature (2011) 472:120–4. doi:10.1038/nature09819
20. Jiang R, Tang J, Chen Y, Deng L, Ji J, Xie Y, et al. The long noncoding RNA lnc-EGFR stimulates T-regulatory cells differentiation thus promoting hepatocellular carcinoma immune evasion. Nat Commun (2017) 8:15129. doi:10.1038/ncomms15129
21. Zemmour D, Pratama A, Loughhead SM, Mathis D, Benoist C. Flicr, a long noncoding RNA, modulates Foxp3 expression and autoimmunity. Proc Natl Acad Sci U S A (2017) 114:E3472–80. doi:10.1073/pnas.1700946114
22. Zheng S, Vuong BQ, Vaidyanathan B, Lin JY, Huang FT, Chaudhuri J. Non-coding RNA generated following lariat debranching mediates targeting of AID to DNA. Cell (2015) 161:762–73. doi:10.1016/j.cell.2015.03.020
23. Hu W, Begum NA, Mondal S, Stanlie A, Honjo T. Identification of DNA cleavage- and recombination-specific hnRNP cofactors for activation-induced cytidine deaminase. Proc Natl Acad Sci U S A (2015) 112:5791–6. doi:10.1073/pnas.1506167112
24. Mondal S, Begum NA, Hu W, Honjo T. Functional requirements of AID’s higher order structures and their interaction with RNA-binding proteins. Proc Natl Acad Sci U S A (2016) 113:E1545–54. doi:10.1073/pnas.1601678113
25. Harcourt EM, Kietrys AM, Kool ET. Chemical and structural effects of base modifications in messenger RNA. Nature (2017) 541:339–46. doi:10.1038/nature21351
26. Lewis CJ, Pan T, Kalsotra A. RNA modifications and structures cooperate to guide RNA-protein interactions. Nat Rev Mol Cell Biol (2017) 18:202–10. doi:10.1038/nrm.2016.163
27. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature (2014) 505:117–20. doi:10.1038/nature12730
28. Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, et al. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun (2016) 7:12626. doi:10.1038/ncomms12626
29. Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, et al. 5’ UTR m(6)A promotes cap-independent translation. Cell (2015) 163:999–1010. doi:10.1016/j.cell.2015.10.012
30. Kariko K, Buckstein M, Ni HP, Weissman D. Suppression of RNA recognition by toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity (2005) 23:165–75. doi:10.1016/j.immuni.2005.06.008
31. Zheng Q, Hou J, Zhou Y, Li Z, Cao X. The RNA helicase DDX46 inhibits innate immunity by entrapping m(6)A-demethylated antiviral transcripts in the nucleus. Nat Immunol (2017) 18:1094–103. doi:10.1038/ni1217-1361a
32. Lichinchi G, Gao S, Saletore Y, Gonzalez GM, Bansal V, Wang Y, et al. Dynamics of the human and viral m(6)A RNA methylomes during HIV-1 infection of T cells. Nat Microbiol (2016) 1:16011. doi:10.1038/nmicrobiol.2016.11
33. Li HB, Tong J, Zhu S, Batista PJ, Duffy EE, Zhao J, et al. m(6)A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature (2017) 548:338–42. doi:10.1038/nature23450
34. Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med (2017) 23:1369–76. doi:10.1038/nm.4416
35. Matera AG, Wang Z. A day in the life of the spliceosome. Nat Rev Mol Cell Biol (2014) 15:108–21. doi:10.1038/nrm3742
36. Hang J, Wan R, Yan C, Shi Y. Structural basis of pre-mRNA splicing. Science (2015) 349:1191–8. doi:10.1126/science.aac8159
37. Yan C, Hang J, Wan R, Huang M, Wong CC, Shi Y. Structure of a yeast spliceosome at 3.6-angstrom resolution. Science (2015) 349:1182–91. doi:10.1126/science.aac7629
38. Ergun A, Doran G, Costello JC, Paik HH, Collins JJ, Mathis D, et al. Differential splicing across immune system lineages. Proc Natl Acad Sci U S A (2013) 110:14324–9. doi:10.1073/pnas.1311839110
39. Yip BH, Steeples V, Repapi E, Armstrong RN, Llorian M, Roy S, et al. The U2AF1S34F mutation induces lineage-specific splicing alterations in myelodysplastic syndromes. J Clin Invest (2017) 127:2206–21. doi:10.1172/JCI91363
40. Komeno Y, Huang YJ, Qiu J, Lin L, Xu Y, Zhou Y, et al. SRSF2 is essential for hematopoiesis, and its myelodysplastic syndrome-related mutations dysregulate alternative pre-mRNA splicing. Mol Cell Biol (2015) 35:3071–82. doi:10.1128/MCB.00202-15
41. Mian SA, Rouault-Pierre K, Smith AE, Seidl T, Pizzitola I, Kizilors A, et al. SF3B1 mutant MDS-initiating cells may arise from the haematopoietic stem cell compartment. Nat Commun (2015) 6:10004. doi:10.1038/ncomms10004
42. Fang J, Bolanos LC, Choi K, Liu X, Christie S, Akunuru S, et al. Ubiquitination of hnRNPA1 by TRAF6 links chronic innate immune signaling with myelodysplasia. Nat Immunol (2017) 18:236–45. doi:10.1038/ni.3654
43. Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. Proliferating cells express mRNAs with shortened 3’ untranslated regions and fewer microRNA target sites. Science (2008) 320:1643–7. doi:10.1126/science.1155390
44. Pai AA, Baharian G, Page Sabourin A, Brinkworth JF, Nedelec Y, Foley JW, et al. Widespread shortening of 3’ untranslated regions and increased exon inclusion are evolutionarily conserved features of innate immune responses to infection. PLoS Genet (2016) 12:e1006338. doi:10.1371/journal.pgen.1006338
45. Gruber AR, Martin G, Muller P, Schmidt A, Gruber AJ, Gumienny R, et al. Global 3’ UTR shortening has a limited effect on protein abundance in proliferating T cells. Nat Commun (2014) 5:5465. doi:10.1038/ncomms6465
46. Jia X, Yuan S, Wang Y, Fu Y, Ge Y, Ge Y, et al. The role of alternative polyadenylation in the antiviral innate immune response. Nat Commun (2017) 8:14605. doi:10.1038/ncomms14605
47. Beisang D, Rattenbacher B, Vlasova-St Louis IA, Bohjanen PR. Regulation of CUG-binding protein 1 (CUGBP1) binding to target transcripts upon T cell activation. J Biol Chem (2012) 287:950–60. doi:10.1074/jbc.M111.291658
48. Batra R, Stark TJ, Clark E, Belzile JP, Wheeler EC, Yee BA, et al. RNA-binding protein CPEB1 remodels host and viral RNA landscapes. Nat Struct Mol Biol (2016) 23:1101–10. doi:10.1038/nsmb.3310
49. Kaida D, Berg MG, Younis I, Kasim M, Singh LN, Wan L, et al. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature (2010) 468:664–8. doi:10.1038/nature09479
50. Berg MG, Singh LN, Younis I, Liu Q, Pinto AM, Kaida D, et al. U1 snRNP determines mRNA length and regulates isoform expression. Cell (2012) 150:53–64. doi:10.1016/j.cell.2012.05.029
51. Jenal M, Elkon R, Loayza-Puch F, van Haaften G, Kuhn U, Menzies FM, et al. The poly(A)-binding protein nuclear 1 suppresses alternative cleavage and polyadenylation sites. Cell (2012) 149:538–53. doi:10.1016/j.cell.2012.03.022
52. Mancini A, Niemann-Seyde SC, Pankow R, El Bounkari O, Klebba-Farber S, Koch A, et al. THOC5/FMIP, an mRNA export TREX complex protein, is essential for hematopoietic primitive cell survival in vivo. BMC Biol (2010) 8:1. doi:10.1186/1741-7007-8-1
53. So L, Lee J, Palafox M, Mallya S, Woxland CG, Arguello M, et al. The 4E-BP-eIF4E axis promotes rapamycin-sensitive growth and proliferation in lymphocytes. Sci Signal (2016) 9:ra57. doi:10.1126/scisignal.aad8463
54. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity (2009) 30:832–44. doi:10.1016/j.immuni.2009.04.014
55. Yang K, Shrestha S, Zeng H, Karmaus PW, Neale G, Vogel P, et al. T cell exit from quiescence and differentiation into Th2 cells depend on raptor-mTORC1-mediated metabolic reprogramming. Immunity (2013) 39:1043–56. doi:10.1016/j.immuni.2013.09.015
56. Zhang S, Pruitt M, Tran D, Du Bois W, Zhang K, Patel R, et al. B cell-specific deficiencies in mTOR limit humoral immune responses. J Immunol (2013) 191:1692–703. doi:10.4049/jimmunol.1201767
57. Galban S, Kuwano Y, Pullmann R, Martindale JL, Kim HH, Lal A, et al. RNA-binding proteins HuR and PTB promote the translation of hypoxia-inducible factor 1 alpha. Mol Cell Biol (2008) 28:93–107. doi:10.1128/MCB.00973-07
58. Diaz-Munoz MD, Kiselev VY, Le Novere N, Curk T, Ule J, Turner M. Tia1 dependent regulation of mRNA subcellular location and translation controls p53 expression in B cells. Nat Commun (2017) 8:530. doi:10.1038/s41467-017-00454-2
59. Ingolia NT. Ribosome profiling: new views of translation, from single codons to genome scale. Nat Rev Genet (2014) 15:205–13. doi:10.1038/nrg3645
60. McCormick C, Khaperskyy DA. Translation inhibition and stress granules in the antiviral immune response. Nat Rev Immunol (2017) 17:647–60. doi:10.1038/nri.2017.63
61. Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, et al. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell (2010) 141:315–30. doi:10.1016/j.cell.2010.03.029
62. Reikine S, Nguyen JB, Modis Y. Pattern recognition and signaling mechanisms of RIG-I and MDA5. Front Immunol (2014) 5:342. doi:10.3389/fimmu.2014.00342
63. Zhang Z, Yuan B, Lu N, Facchinetti V, Liu YJ. DHX9 pairs with IPS-1 to sense double-stranded RNA in myeloid dendritic cells. J Immunol (2011) 187:4501–8. doi:10.4049/jimmunol.1101307
64. Lee MS, Kim B, Oh GT, Kim YJ. OASL1 inhibits translation of the type I interferon-regulating transcription factor IRF7. Nat Immunol (2013) 14:346–55. doi:10.1038/ni.2535
65. Lin WJ, Zheng X, Lin CC, Tsao J, Zhu X, Cody JJ, et al. Posttranscriptional control of type I interferon genes by KSRP in the innate immune response against viral infection. Mol Cell Biol (2011) 31:3196–207. doi:10.1128/MCB.05073-11
66. Herdy B, Karonitsch T, Vladimer GI, Tan CS, Stukalov A, Trefzer C, et al. The RNA-binding protein HuR/ELAVL1 regulates IFN-beta mRNA abundance and the type I IFN response. Eur J Immunol (2015) 45:1500–11. doi:10.1002/eji.201444979
67. White JP, Cardenas AM, Marissen WE, Lloyd RE. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe (2007) 2:295–305. doi:10.1016/j.chom.2007.08.006
68. Ruggieri A, Dazert E, Metz P, Hofmann S, Bergeest JP, Mazur J, et al. Dynamic oscillation of translation and stress granule formation mark the cellular response to virus infection. Cell Host Microbe (2012) 12:71–85. doi:10.1016/j.chom.2012.05.013
69. Clavarino G, Claudio N, Couderc T, Dalet A, Judith D, Camosseto V, et al. Induction of GADD34 is necessary for dsRNA-dependent interferon-beta production and participates in the control of chikungunya virus infection. PLoS Pathog (2012) 8:e1002708. doi:10.1371/journal.ppat.1002708
70. Yoo JS, Takahasi K, Ng CS, Ouda R, Onomoto K, Yoneyama M, et al. DHX36 enhances RIG-I signaling by facilitating PKR-mediated antiviral stress granule formation. PLoS Pathog (2014) 10:e1004012. doi:10.1371/journal.ppat.1004012
71. Reineke LC, Lloyd RE. The stress granule protein G3BP1 recruits protein kinase R to promote multiple innate immune antiviral responses. J Virol (2015) 89:2575–89. doi:10.1128/JVI.02791-14
72. Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature (2011) 472:481–5. doi:10.1038/nature09907
73. Walsh D, Mathews MB, Mohr I. Tinkering with translation: protein synthesis in virus-infected cells. Cold Spring Harb Perspect Biol (2013) 5:a012351. doi:10.1101/cshperspect.a012351
74. Garcia-Sastre A. Ten strategies of interferon evasion by viruses. Cell Host Microbe (2017) 22:176–84. doi:10.1016/j.chom.2017.07.012
75. Erickson SL, Corpuz EO, Maloy JP, Fillman C, Webb K, Bennett EJ, et al. Competition between decapping complex formation and ubiquitin-mediated proteasomal degradation controls human Dcp2 decapping activity. Mol Cell Biol (2015) 35:2144–53. doi:10.1128/MCB.01517-14
76. D’Lima NG, Ma J, Winkler L, Chu Q, Loh KH, Corpuz EO, et al. A human microprotein that interacts with the mRNA decapping complex. Nat Chem Biol (2017) 13:174–80. doi:10.1038/nchembio.2249
77. Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations driving CLL and their evolution in progression and relapse. Nature (2015) 526:525–30. doi:10.1038/nature15395
78. Jiang L, Gu ZH, Yan ZX, Zhao X, Xie YY, Zhang ZG, et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat Genet (2015) 47:1061–6. doi:10.1038/ng.3358
79. Brandimarte L, Pierini V, Di Giacomo D, Borga C, Nozza F, Gorello P, et al. New MLLT10 gene recombinations in pediatric T-acute lymphoblastic leukemia. Blood (2013) 121:5064–7. doi:10.1182/blood-2013-02-487256
80. Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science (2011) 333:1157–60. doi:10.1126/science.1208130
81. Northcott PA, Jones DT, Kool M, Robinson GW, Gilbertson RJ, Cho YJ, et al. Medulloblastomics: the end of the beginning. Nat Rev Cancer (2012) 12:818–34. doi:10.1038/nrc3410
82. Anadon C, van Tetering G, Ferreira HJ, Moutinho C, Martinez-Cardus A, Villanueva A, et al. Epigenetic loss of the RNA decapping enzyme NUDT16 mediates C-MYC activation in T-cell acute lymphoblastic leukemia. Leukemia (2017) 31:1622–5. doi:10.1038/leu.2017.99
83. Valentin-Vega YA, Wang YD, Parker M, Patmore DM, Kanagaraj A, Moore J, et al. Cancer-associated DDX3X mutations drive stress granule assembly and impair global translation. Sci Rep (2016) 6:25996. doi:10.1038/srep25996
84. Lai WS, Carballo E, Strum JR, Kennington EA, Phillips RS, Blackshear PJ. Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol Cell Biol (1999) 19:4311–23. doi:10.1128/MCB.19.6.4311
85. Leppek K, Schott J, Reitter S, Poetz F, Hammond MC, Stoecklin G. Roquin promotes constitutive mRNA decay via a conserved class of stem-loop recognition motifs. Cell (2013) 153:869–81. doi:10.1016/j.cell.2013.04.016
86. Sgromo A, Raisch T, Bawankar P, Bhandari D, Chen Y, Kuzuoglu-Ozturk D, et al. A CAF40-binding motif facilitates recruitment of the CCR4-NOT complex to mRNAs targeted by Drosophila Roquin. Nat Commun (2017) 8:14307. doi:10.1038/ncomms14307
87. Schoenberg DR, Maquat LE. Regulation of cytoplasmic mRNA decay. Nat Rev Genet (2012) 13:246–59. doi:10.1038/nrg3160
88. Tiedje C, Ronkina N, Tehrani M, Dhamija S, Laass K, Holtmann H, et al. The p38/MK2-driven exchange between tristetraprolin and HuR regulates AU-rich element-dependent translation. PLoS Genet (2012) 8:e1002977. doi:10.1371/journal.pgen.1002977
89. Venigalla RK, Turner M. RNA-binding proteins as a point of convergence of the PI3K and p38 MAPK pathways. Front Immunol (2012) 3:398. doi:10.3389/fimmu.2012.00398
90. Tiedje C, Diaz-Munoz MD, Trulley P, Ahlfors H, Laass K, Blackshear PJ, et al. The RNA-binding protein TTP is a global post-transcriptional regulator of feedback control in inflammation. Nucleic Acids Res (2016) 44:7418–40. doi:10.1093/nar/gkw474
91. Fu M, Blackshear PJ. RNA-binding proteins in immune regulation: a focus on CCCH zinc finger proteins. Nat Rev Immunol (2017) 17:130–43. doi:10.1038/nri.2016.129
92. Jeltsch KM, Heissmeyer V. Regulation of T cell signaling and autoimmunity by RNA-binding proteins. Curr Opin Immunol (2016) 39:127–35. doi:10.1016/j.coi.2016.01.011
93. Popp MW, Maquat LE. Leveraging rules of nonsense-mediated mRNA decay for genome engineering and personalized medicine. Cell (2016) 165:1319–22. doi:10.1016/j.cell.2016.05.053
94. Serdar LD, Whiteside DL, Baker KE. ATP hydrolysis by UPF1 is required for efficient translation termination at premature stop codons. Nat Commun (2016) 7:14021. doi:10.1038/ncomms14021
95. Wittkopp N, Huntzinger E, Weiler C, Sauliere J, Schmidt S, Sonawane M, et al. Nonsense-mediated mRNA decay effectors are essential for zebrafish embryonic development and survival. Mol Cell Biol (2009) 29:3517–28. doi:10.1128/MCB.00177-09
96. Weischenfeldt J, Damgaard I, Bryder D, Theilgaard-Monch K, Thoren LA, Nielsen FC, et al. NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev (2008) 22:1381–96. doi:10.1101/gad.468808
97. Saul MJ, Stein S, Grez M, Jakobsson PJ, Steinhilber D, Suess B. UPF1 regulates myeloid cell functions and S100A9 expression by the hnRNP E2/miRNA-328 balance. Sci Rep (2016) 6:31995. doi:10.1038/srep31995
98. Jin H, Suh MR, Han J, Yeom KH, Lee Y, Heo I, et al. Human UPF1 participates in small RNA-induced mRNA downregulation. Mol Cell Biol (2009) 29:5789–99. doi:10.1128/MCB.00653-09
99. Forero A, So L, Savan R. Re-evaluating strategies to define the immunoregulatory roles of miRNAs. Trends Immunol (2017) 38:558–66. doi:10.1016/j.it.2017.06.001
100. Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet (2015) 16:421–33. doi:10.1038/nrg3965
101. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol (2014) 15:509–24. doi:10.1038/nrm3838
102. Mehta A, Baltimore D. MicroRNAs as regulatory elements in immune system logic. Nat Rev Immunol (2016) 16:279–94. doi:10.1038/nri.2016.40
103. Muljo SA, Ansel KM, Kanellopoulou C, Livingston DM, Rao A, Rajewsky K. Aberrant T cell differentiation in the absence of Dicer. J Exp Med (2005) 202:261–9. doi:10.1084/jem.20050678
104. Koralov SB, Muljo SA, Galler GR, Krek A, Chakraborty T, Kanellopoulou C, et al. Dicer ablation affects antibody diversity and cell survival in the B lymphocyte lineage. Cell (2008) 132:860–74. doi:10.1016/j.cell.2008.02.020
105. Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of microRNA processing by Lin28. Science (2008) 320:97–100. doi:10.1126/science.1154040
106. Piskounova E, Polytarchou C, Thornton JE, LaPierre RJ, Pothoulakis C, Hagan JP, et al. Lin28A and Lin28B inhibit let-7 microRNA biogenesis by distinct mechanisms. Cell (2011) 147:1066–79. doi:10.1016/j.cell.2011.10.039
107. Bin G, Jiarong Z, Shihao W, Xiuli S, Cheng X, Liangbiao C, et al. Aire promotes the self-renewal of embryonic stem cells through Lin28. Stem Cells Dev (2012) 21:2878–90. doi:10.1089/scd.2012.0097
108. Lu YC, Chang SH, Hafner M, Li X, Tuschl T, Elemento O, et al. ELAVL1 modulates transcriptome-wide miRNA binding in murine macrophages. Cell Rep (2014) 9:2330–43. doi:10.1016/j.celrep.2014.11.030
109. Kundu P, Fabian MR, Sonenberg N, Bhattacharyya SN, Filipowicz W. HuR protein attenuates miRNA-mediated repression by promoting miRISC dissociation from the target RNA. Nucleic Acids Res (2012) 40:5088–100. doi:10.1093/nar/gks148
110. Qi MY, Wang ZZ, Zhang Z, Shao Q, Zeng A, Li XQ, et al. AU-rich-element-dependent translation repression requires the cooperation of tristetraprolin and RCK/P54. Mol Cell Biol (2012) 32:913–28. doi:10.1128/MCB.05340-11
111. Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen JM, et al. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell (2005) 120:623–34. doi:10.1016/j.cell.2004.12.038
112. El Gazzar M, McCall CE. MicroRNAs distinguish translational from transcriptional silencing during endotoxin tolerance. J Biol Chem (2010) 285:20940–51. doi:10.1074/jbc.M110.115063
113. Kedde M, van Kouwenhove M, Zwart W, Oude Vrielink JA, Elkon R, Agami R. A pumilio-induced RNA structure switch in p27-3’ UTR controls miR-221 and miR-222 accessibility. Nat Cell Biol (2010) 12:1014–20. doi:10.1038/ncb2105
114. Naudin C, Hattabi A, Michelet F, Miri-Nezhad A, Benyoucef A, Pflumio F, et al. PUMILIO/FOXP1 signaling drives expansion of hematopoietic stem/progenitor and leukemia cells. Blood (2017) 129:2493–506. doi:10.1182/blood-2016-10-747436
115. Glasmacher E, Hoefig KP, Vogel KU, Rath N, Du L, Wolf C, et al. Roquin binds inducible costimulator mRNA and effectors of mRNA decay to induce microRNA-independent post-transcriptional repression. Nat Immunol (2010) 11:725–33. doi:10.1038/ni.1902
116. Hubstenberger A, Courel M, Benard M, Souquere S, Ernoult-Lange M, Chouaib R, et al. P-body purification reveals the condensation of repressed mRNA regulons. Mol Cell (2017) 68:144–57.e5. doi:10.1016/j.molcel.2017.09.003
117. Youn JY, Dunham WH, Hong SJ, Knight JDR, Bashkurov M, Chen GI, et al. High-density proximity mapping reveals the subcellular organization of mRNA-associated granules and bodies. Mol Cell (2018) 69:517–32.e11. doi:10.1016/j.molcel.2017.12.020
118. Decker CJ, Parker R. P-bodies and stress granules: possible roles in the control of translation and mRNA degradation. Cold Spring Harb Perspect Biol (2012) 4:a012286. doi:10.1101/cshperspect.a012286
119. Khong A, Matheny T, Jain S, Mitchell SF, Wheeler JR, Parker R. The stress granule transcriptome reveals principles of mRNA accumulation in stress granules. Mol Cell (2017) 68:808–20.e5. doi:10.1016/j.molcel.2017.10.015
120. Protter DS, Parker R. Principles and properties of stress granules. Trends Cell Biol (2016) 26:668–79. doi:10.1016/j.tcb.2016.05.004
121. Kotani T, Yasuda K, Ota R, Yamashita M. Cyclin B1 mRNA translation is temporally controlled through formation and disassembly of RNA granules. J Cell Biol (2013) 202:1041–55. doi:10.1083/jcb.201302139
122. Diaz-Munoz MD, Bell SE, Fairfax K, Monzon-Casanova E, Cunningham AF, Gonzalez-Porta M, et al. The RNA-binding protein HuR is essential for the B cell antibody response. Nat Immunol (2015) 16:415–25. doi:10.1038/ni.3115
123. DeMicco A, Naradikian MS, Sindhava VJ, Yoon JH, Gorospe M, Wertheim GB, et al. B cell-intrinsic expression of the HuR RNA-binding protein is required for the T cell-dependent immune response in vivo. J Immunol (2015) 195:3449–62. doi:10.4049/jimmunol.1500512
124. Yiakouvaki A, Dimitriou M, Karakasiliotis I, Eftychi C, Theocharis S, Kontoyiannis DL. Myeloid cell expression of the RNA-binding protein HuR protects mice from pathologic inflammation and colorectal carcinogenesis. J Clin Invest (2012) 122:48–61. doi:10.1172/JCI45021
125. Papadaki O, Milatos S, Grammenoudi S, Mukherjee N, Keene JD, Kontoyiannis DL. Control of thymic T cell maturation, deletion and egress by the RNA-binding protein HuR. J Immunol (2009) 182:6779–88. doi:10.4049/jimmunol.0900377
126. Alkhatib A, Werner M, Hug E, Herzog S, Eschbach C, Faraidun H, et al. FoxO1 induces Ikaros splicing to promote immunoglobulin gene recombination. J Exp Med (2012) 209:395–406. doi:10.1084/jem.20110216
127. Iwanami N, Sikora K, Richter AS, Monnich M, Guerri L, Soza-Ried C, et al. Forward genetic screens in zebrafish identify pre-mRNA-processing pathways regulating early T cell development. Cell Rep (2016) 17:2259–70. doi:10.1016/j.celrep.2016.11.003
128. Hicks GG, Singh N, Nashabi A, Mai S, Bozek G, Klewes L, et al. Fus deficiency in mice results in defective B-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat Genet (2000) 24:175–9. doi:10.1038/72842
129. Wang HY, Xu X, Ding JH, Bermingham JR Jr, Fu XD. SC35 plays a role in T cell development and alternative splicing of CD45. Mol Cell (2001) 7:331–42. doi:10.1016/S1097-2765(01)00181-2
130. Oberdoerffer S, Moita LF, Neems D, Freitas RP, Hacohen N, Rao A. Regulation of CD45 alternative splicing by heterogeneous ribonucleoprotein, hnRNPLL. Science (2008) 321:686–91. doi:10.1126/science.1157610
131. Wu Z, Yates AL, Hoyne GF, Goodnow CC. Consequences of increased CD45RA and RC isoforms for TCR signaling and peripheral T cell deficiency resulting from heterogeneous nuclear ribonucleoprotein L-like mutation. J Immunol (2010) 185:231–8. doi:10.4049/jimmunol.0903625
132. Wu Z, Jia X, de la Cruz L, Su XC, Marzolf B, Troisch P, et al. Memory T cell RNA rearrangement programmed by heterogeneous nuclear ribonucleoprotein hnRNPLL. Immunity (2008) 29:863–75. doi:10.1016/j.immuni.2008.11.004
133. Cho V, Mei Y, Sanny A, Chan S, Enders A, Bertram EM, et al. The RNA-binding protein hnRNPLL induces a T cell alternative splicing program delineated by differential intron retention in polyadenylated RNA. Genome Biol (2014) 15:R26. doi:10.1186/gb-2014-15-1-r26
134. Gaudreau MC, Heyd F, Bastien R, Wilhelm B, Moroy T. Alternative splicing controlled by heterogeneous nuclear ribonucleoprotein L regulates development, proliferation, and migration of thymic pre-T cells. J Immunol (2012) 188:5377–88. doi:10.4049/jimmunol.1103142
135. Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, et al. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell (2015) 27:617–30. doi:10.1016/j.ccell.2015.04.006
136. Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, et al. Physiologic expression of Sf3b1(K700E) causes impaired erythropoiesis, aberrant splicing, and sensitivity to therapeutic spliceosome modulation. Cancer Cell (2016) 30:404–17. doi:10.1016/j.ccell.2016.08.006
137. Mupo A, Seiler M, Sathiaseelan V, Pance A, Yang Y, Agrawal AA, et al. Hemopoietic-specific Sf3b1-K700E knock-in mice display the splicing defect seen in human MDS but develop anemia without ring sideroblasts. Leukemia (2017) 31:720–7. doi:10.1038/leu.2016.251
138. Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell (2013) 152:714–26. doi:10.1016/j.cell.2013.01.019
139. Wang L, Brooks AN, Fan J, Wan Y, Gambe R, Li S, et al. Transcriptomic characterization of SF3B1 mutation reveals its pleiotropic effects in chronic lymphocytic leukemia. Cancer Cell (2016) 30:750–63. doi:10.1016/j.ccell.2016.10.005
140. Iervolino A, Santilli G, Trotta R, Guerzoni C, Cesi V, Bergamaschi A, et al. hnRNP A1 nucleocytoplasmic shuttling activity is required for normal myelopoiesis and BCR/ABL leukemogenesis. Mol Cell Biol (2002) 22:2255–66. doi:10.1128/MCB.22.7.2255-2266.2002
141. Hodson DJ, Janas ML, Galloway A, Bell SE, Andrews S, Li CM, et al. Deletion of the RNA-binding proteins ZFP36L1 and ZFP36L2 leads to perturbed thymic development and T lymphoblastic leukemia. Nat Immunol (2010) 11:717–24. doi:10.1038/ni.1901
142. Vogel KU, Bell LS, Galloway A, Ahlfors H, Turner M. The RNA-binding proteins Zfp36l1 and Zfp36l2 enforce the thymic beta-selection checkpoint by limiting DNA damage response signaling and cell cycle progression. J Immunol (2016) 197:2673–85. doi:10.4049/jimmunol.1600854
143. Galloway A, Saveliev A, Lukasiak S, Hodson DJ, Bolland D, Balmanno K, et al. RNA-binding proteins ZFP36L1 and ZFP36L2 promote cell quiescence. Science (2016) 352:453–9. doi:10.1126/science.aad5978
144. Zhang L, Reynolds TL, Shan X, Desiderio S. Coupling of V(D)J recombination to the cell cycle suppresses genomic instability and lymphoid tumorigenesis. Immunity (2011) 34:163–74. doi:10.1016/j.immuni.2011.02.003
145. Inoue T, Morita M, Hijikata A, Fukuda-Yuzawa Y, Adachi S, Isono K, et al. CNOT3 contributes to early B cell development by controlling Igh rearrangement and p53 mRNA stability. J Exp Med (2015) 212:1465–79. doi:10.1084/jem.20150384
146. Yang CY, Ramamoorthy S, Boller S, Rosenbaum M, Rodriguez Gil A, Mittler G, et al. Interaction of CCR4-NOT with EBF1 regulates gene-specific transcription and mRNA stability in B lymphopoiesis. Genes Dev (2016) 30:2310–24. doi:10.1101/gad.285452.116
147. Yuan J, Nguyen CK, Liu X, Kanellopoulou C, Muljo SA. Lin28b reprograms adult bone marrow hematopoietic progenitors to mediate fetal-like lymphopoiesis. Science (2012) 335:1195–200. doi:10.1126/science.1216557
148. Sadri N, Lu JY, Badura ML, Schneider RJ. AUF1 is involved in splenic follicular B cell maintenance. BMC Immunol (2010) 11:1. doi:10.1186/1471-2172-11-1
149. Diaz-Munoz MD, Bell SE, Turner M. Deletion of AU-rich elements within the Bcl2 3’UTR reduces protein expression and B cell survival in vivo. PLoS One (2015) 10:e0116899. doi:10.1371/journal.pone.0116899
150. Newman R, Ahlfors H, Saveliev A, Galloway A, Hodson DJ, Williams R, et al. Maintenance of the marginal-zone B cell compartment specifically requires the RNA-binding protein ZFP36L1. Nat Immunol (2017) 18:683–93. doi:10.1038/ni.3724
151. Vlasova-St Louis I, Bohjanen PR. Feedback regulation of kinase signaling pathways by AREs and GREs. Cells (2016) 5:E4. doi:10.3390/cells5010004
152. Grammatikakis I, Abdelmohsen K, Gorospe M. Posttranslational control of HuR function. Wiley Interdiscip Rev RNA (2017) 8:e1372. doi:10.1002/wrna.1372
153. Glodde N, Holzel M. RAS and PD-L1: a masters’ liaison in cancer immune evasion. Immunity (2017) 47:1007–9. doi:10.1016/j.immuni.2017.12.001
154. Stoecklin G, Stubbs T, Kedersha N, Wax S, Rigby WFC, Blackwell TK, et al. MK2-induced tristetraprolin: 14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J (2004) 23:1313–24. doi:10.1038/sj.emboj.7600163
155. Clement SL, Scheckel C, Stoecklin G, Lykke-Andersen J. Phosphorylation of tristetraprolin by MK2 impairs AU-rich element mRNA decay by preventing deadenylase recruitment. Mol Cell Biol (2011) 31:256–66. doi:10.1128/MCB.00717-10
156. Schott J, Reitter S, Philipp J, Haneke K, Schafer H, Stoecklin G. Translational regulation of specific mRNAs controls feedback inhibition and survival during macrophage activation. PLoS Genet (2014) 10:e1004368. doi:10.1371/journal.pgen.1004368
157. Hammer M, Mages J, Dietrich H, Servatius A, Howells N, Cato ACB, et al. Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. J Exp Med (2006) 203:15–20. doi:10.1084/jem.20051753
158. Frazier WJ, Wang X, Wancket LM, Li XA, Meng X, Nelin LD, et al. Increased inflammation, impaired bacterial clearance, and metabolic disruption after gram-negative sepsis in Mkp-1-deficient mice. J Immunol (2009) 183:7411–9. doi:10.4049/jimmunol.0804343
159. Akilov OE, Ustyugova IV, Zhi L, Hasan T, Wu MX. Enhanced susceptibility to Leishmania infection in resistant mice in the absence of immediate early response gene X-1. J Immunol (2009) 183:7994–8003. doi:10.4049/jimmunol.0900866
160. Ross EA, Smallie T, Ding Q, O’Neil JD, Cunliffe HE, Tang T, et al. Dominant suppression of inflammation via targeted mutation of the mRNA destabilizing protein tristetraprolin. J Immunol (2015) 195:265–76. doi:10.4049/jimmunol.1402826
161. Ross EA, Naylor AJ, O’Neil JD, Crowley T, Ridley ML, Crowe J, et al. Treatment of inflammatory arthritis via targeting of tristetraprolin, a master regulator of pro-inflammatory gene expression. Ann Rheum Dis (2017) 76:612–9. doi:10.1136/annrheumdis-2016-209424
162. Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol (2015) 17:1205–17. doi:10.1038/ncb3225
163. Lu JY, Sadri N, Schneider RJ. Endotoxic shock in AUF1 knockout mice mediated by failure to degrade proinflammatory cytokine mRNAs. Genes Dev (2006) 20:3174–84. doi:10.1101/gad.1467606
164. Montufar-Solis D, Vigneswaran N, Nakra N, Schaefer JS, Klein JR. Hematopoietic not systemic impairment of Roquin expression accounts for intestinal inflammation in Roquin-deficient mice. Sci Rep (2014) 4:4920. doi:10.1038/srep04920
165. Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui LX, Hill KM, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature (2005) 435:452–8. doi:10.1038/nature03555
166. Sarkar S, Han J, Sinsimer KS, Liao B, Foster RL, Brewer G, et al. RNA-binding protein AUF1 regulates lipopolysaccharide-induced IL10 expression by activating IkappaB kinase complex in monocytes. Mol Cell Biol (2011) 31:602–15. doi:10.1128/MCB.00835-10
167. Murakawa Y, Hinz M, Mothes J, Schuetz A, Uhl M, Wyler E, et al. RC3H1 post-transcriptionally regulates A20 mRNA and modulates the activity of the IKK/NF-kappaB pathway. Nat Commun (2015) 6:7367. doi:10.1038/ncomms8367
168. Patial S, Curtis AD II, Lai WS, Stumpo DJ, Hill GD, Flake GP, et al. Enhanced stability of tristetraprolin mRNA protects mice against immune-mediated inflammatory pathologies. Proc Natl Acad Sci U S A (2016) 113:1865–70. doi:10.1073/pnas.1519906113
169. Mukherjee N, Jacobs NC, Hafner M, Kennington EA, Nusbaum JD, Tuschl T, et al. Global target mRNA specification and regulation by the RNA-binding protein ZFP36. Genome Biol (2014) 15:R12. doi:10.1186/gb-2014-15-1-r12
170. Katsanou V, Papadaki O, Milatos S, Blackshear PJ, Anderson P, Kollias G, et al. HuR as a negative posttranscriptional modulator in inflammation. Mol Cell (2005) 19:777–89. doi:10.1016/j.molcel.2005.08.007
171. Barker A, Epis MR, Porter CJ, Hopkins BR, Wilce MC, Wilce JA, et al. Sequence requirements for RNA binding by HuR and AUF1. J Biochem (2012) 151:423–37. doi:10.1093/jb/mvs010
172. Raghavan A, Robison RL, McNabb J, Miller CR, Williams DA, Bohjanen PR. HuA and tristetraprolin are induced following T cell activation and display distinct but overlapping RNA binding specificities. J Biol Chem (2001) 276:47958–65. doi:10.1074/jbc.M109511200
173. Dean JL, Wait R, Mahtani KR, Sully G, Clark AR, Saklatvala J. The 3’ untranslated region of tumor necrosis factor alpha mRNA is a target of the mRNA-stabilizing factor HuR. Mol Cell Biol (2001) 21:721–30. doi:10.1128/MCB.21.3.721-730.2001
174. Gubin MM, Techasintana P, Magee JD, Dahm GM, Calaluce R, Martindale JL, et al. Conditional knockout of the RNA-binding protein HuR in CD4(+) T cells reveals a gene dosage effect on cytokine production. Mol Med (2014) 20:93–108. doi:10.2119/molmed.2013.00127
175. Blackinton JG, Keene JD. Functional coordination and HuR-mediated regulation of mRNA stability during T cell activation. Nucleic Acids Res (2016) 44:426–36. doi:10.1093/nar/gkv1066
176. Kim HH, Abdelmohsen K, Gorospe M. Regulation of HuR by DNA damage response kinases. J Nucleic Acids (2010) 2010:981487. doi:10.4061/2010/981487
177. Li H, Park S, Kilburn B, Jelinek MA, Henschen-Edman A, Aswad DW, et al. Lipopolysaccharide-induced methylation of HuR, an mRNA-stabilizing protein, by CARM1. Coactivator-associated arginine methyltransferase. J Biol Chem (2002) 277:44623–30. doi:10.1074/jbc.M206187200
178. Abdelmohsen K, Srikantan S, Yang X, Lal A, Kim HH, Kuwano Y, et al. Ubiquitin-mediated proteolysis of HuR by heat shock. EMBO J (2009) 28:1271–82. doi:10.1038/emboj.2009.67
179. Zhou HL, Geng C, Luo G, Lou H. The p97-UBXD8 complex destabilizes mRNA by promoting release of ubiquitinated HuR from mRNP. Genes Dev (2013) 27:1046–58. doi:10.1101/gad.215681.113
180. Ansel KM. RNA regulation of the immune system. Immunol Rev (2013) 253:5–11. doi:10.1111/imr.12062
181. Butte MJ, Lee SJ, Jesneck J, Keir ME, Haining WN, Sharpe AH. CD28 costimulation regulates genome-wide effects on alternative splicing. PLoS One (2012) 7:e40032. doi:10.1371/journal.pone.0040032
182. Grigoryev YA, Kurian SM, Nakorchevskiy AA, Burke JP, Campbell D, Head SR, et al. Genome-wide analysis of immune activation in human T and B cells reveals distinct classes of alternatively spliced genes. PLoS One (2009) 4:e7906. doi:10.1371/journal.pone.0007906
183. Martinez NM, Pan Q, Cole BS, Yarosh CA, Babcock GA, Heyd F, et al. Alternative splicing networks regulated by signaling in human T cells. RNA (2012) 18:1029–40. doi:10.1261/rna.032243.112
184. Whisenant TC, Peralta ER, Aarreberg LD, Gao NJ, Head SR, Ordoukhanian P, et al. The activation-induced assembly of an RNA/protein interactome centered on the splicing factor U2AF2 regulates gene expression in human CD4 T cells. PLoS One (2015) 10:e0144409. doi:10.1371/journal.pone.0144409
185. Ni T, Yang W, Han M, Zhang Y, Shen T, Nie H, et al. Global intron retention mediated gene regulation during CD4+ T cell activation. Nucleic Acids Res (2016) 44:6817–29. doi:10.1093/nar/gkw591
186. Meininger I, Griesbach RA, Hu D, Gehring T, Seeholzer T, Bertossi A, et al. Alternative splicing of MALT1 controls signalling and activation of CD4(+) T cells. Nat Commun (2016) 7:11292. doi:10.1038/ncomms11292
187. Motta-Mena LB, Heyd F, Lynch KW. Context-dependent regulatory mechanism of the splicing factor hnRNP L. Mol Cell (2010) 37:223–34. doi:10.1016/j.molcel.2009.12.027
188. Preussner M, Schreiner S, Hung LH, Porstner M, Jack HM, Benes V, et al. HnRNP L and L-like cooperate in multiple-exon regulation of CD45 alternative splicing. Nucleic Acids Res (2012) 40:5666–78. doi:10.1093/nar/gks221
189. Cole BS, Tapescu I, Allon SJ, Mallory MJ, Qiu J, Lake RJ, et al. Global analysis of physical and functional RNA targets of hnRNP L reveals distinct sequence and epigenetic features of repressed and enhanced exons. RNA (2015) 21:2053–66. doi:10.1261/rna.052969.115
190. Mondino A, Jenkins MK. Accumulation of sequence-specific RNA-binding proteins in the cytosol of activated T cells undergoing RNA degradation and apoptosis. J Biol Chem (1995) 270:26593–601. doi:10.1074/jbc.270.44.26593
191. Chang JW, Koike T, Iwashima M. hnRNP-K is a nuclear target of TCR-activated ERK and required for T-cell late activation. Int Immunol (2009) 21:1351–61. doi:10.1093/intimm/dxp106
192. Habelhah H, Shah K, Huang L, Ostareck-Lederer A, Burlingame AL, Shokat KM, et al. ERK phosphorylation drives cytoplasmic accumulation of hnRNP-K and inhibition of mRNA translation. Nat Cell Biol (2001) 3:325–30. doi:10.1038/35060131
193. Ogilvie RL, Abelson M, Hau HH, Vlasova I, Blackshear PJ, Bohjanen PR. Tristetraprolin down-regulates IL-2 gene expression through AU-rich element-mediated mRNA decay. J Immunol (2005) 174:953–61. doi:10.4049/jimmunol.174.2.953
194. La Porta J, Matus-Nicodemos R, Valentin-Acevedo A, Covey LR. The RNA-binding protein, polypyrimidine tract-binding protein 1 (PTBP1) is a key regulator of CD4 T cell activation. PLoS One (2016) 11:e0158708. doi:10.1371/journal.pone.0158708
195. Villarino AV, Katzman SD, Gallo E, Miller O, Jiang S, McManus MT, et al. Posttranscriptional silencing of effector cytokine mRNA underlies the anergic phenotype of self-reactive T cells. Immunity (2011) 34:50–60. doi:10.1016/j.immuni.2010.12.014
196. Bertossi A, Aichinger M, Sansonetti P, Lech M, Neff F, Pal M, et al. Loss of Roquin induces early death and immune deregulation but not autoimmunity. J Exp Med (2011) 208:1749–56. doi:10.1084/jem.20110578
197. Jeltsch KM, Hu D, Brenner S, Zoller J, Heinz GA, Nagel D, et al. Cleavage of roquin and regnase-1 by the paracaspase MALT1 releases their cooperatively repressed targets to promote T(H)17 differentiation. Nat Immunol (2014) 15:1079–89. doi:10.1038/ni.3008
198. Schaefer JS, Montufar-Solis D, Klein JR. A role for IL-10 in the transcriptional regulation of Roquin-1. Gene (2014) 549:134–40. doi:10.1016/j.gene.2014.07.056
199. Cai G, Nie X, Zhang W, Wu B, Lin J, Wang H, et al. A regulatory role for IL-10 receptor signaling in development and B cell help of T follicular helper cells in mice. J Immunol (2012) 189:1294–302. doi:10.4049/jimmunol.1102948
200. Lee SK, Silva DG, Martin JL, Pratama A, Hu X, Chang PP, et al. Interferon-gamma excess leads to pathogenic accumulation of follicular helper T cells and germinal centers. Immunity (2012) 37:880–92. doi:10.1016/j.immuni.2012.10.010
201. Ji YR, Kim HJ, Yu DH, Bae KB, Park SJ, Yi JK, et al. Enforced expression of roquin protein in T cells exacerbates the incidence and severity of experimental arthritis. J Biol Chem (2012) 287:42269–77. doi:10.1074/jbc.M112.374835
202. Kim HJ, Ji YR, Kim MO, Yu DH, Shin MJ, Yuh HS, et al. The role of Roquin overexpression in the modulation of signaling during in vitro and ex vivo T-cell activation. Biochem Biophys Res Commun (2012) 417:280–6. doi:10.1016/j.bbrc.2011.11.101
203. Masuda K, Ripley B, Nishimura R, Mino T, Takeuchi O, Shioi G, et al. Arid5a controls IL-6 mRNA stability, which contributes to elevation of IL-6 level in vivo. Proc Natl Acad Sci U S A (2013) 110:9409–14. doi:10.1073/pnas.1307419110
204. Moore KW, Rogers J, Hunkapiller T, Early P, Nottenburg C, Weissman I, et al. Expression of IgD may use both DNA rearrangement and RNA splicing mechanisms. Proc Natl Acad Sci U S A (1981) 78:1800–4. doi:10.1073/pnas.78.3.1800
205. Enders A, Short A, Miosge LA, Bergmann H, Sontani Y, Bertram EM, et al. Zinc-finger protein ZFP318 is essential for expression of IgD, the alternatively spliced Igh product made by mature B lymphocytes. Proc Natl Acad Sci U S A (2014) 111:4513–8. doi:10.1073/pnas.1402739111
206. Pioli PD, Debnath I, Weis JJ, Weis JH. Zfp318 regulates IgD expression by abrogating transcription termination within the Ighm/Ighd locus. J Immunol (2014) 193:2546–53. doi:10.4049/jimmunol.1401275
207. Pieper K, Tan J, Piccoli L, Foglierini M, Barbieri S, Chen Y, et al. Public antibodies to malaria antigens generated by two LAIR1 insertion modalities. Nature (2017) 548:597–601. doi:10.1038/nature23670
208. Bende RJ, Aarts WM, Pals ST, van Noesel CJ. Immunoglobulin diversification in B cell malignancies: internal splicing of heavy chain variable region as a by-product of somatic hypermutation. Leukemia (2002) 16:636–44. doi:10.1038/sj.leu.2402405
209. Qiao Q, Wang L, Meng FL, Hwang JK, Alt FW, Wu H. AID recognizes structured DNA for class switch recombination. Mol Cell (2017) 67:361–73.e4. doi:10.1016/j.molcel.2017.06.034
210. Monzon-Casanova E, Screen M, Diaz-Munoz MD, Coulson RMR, Bell SE, Lamers G, et al. The RNA-binding protein PTBP1 is necessary for B cell selection in germinal centers. Nat Immunol (2018) 19(3):267–78. doi:10.1038/s41590-017-0035-5
211. Chang X, Li B, Rao A. RNA-binding protein hnRNPLL regulates mRNA splicing and stability during B-cell to plasma-cell differentiation. Proc Natl Acad Sci U S A (2015) 112:E1888–97. doi:10.1073/pnas.1422490112
212. Essig K, Hu D, Guimaraes JC, Alterauge D, Edelmann S, Raj T, et al. Roquin suppresses the PI3K-mTOR signaling pathway to inhibit T helper cell differentiation and conversion of Treg to Tfr cells. Immunity (2017) 47:1067–82.e12. doi:10.1016/j.immuni.2017.11.008
213. Vogel KU, Edelmann SL, Jeltsch KM, Bertossi A, Heger K, Heinz GA, et al. Roquin paralogs 1 and 2 redundantly repress the Icos and Ox40 costimulator mRNAs and control follicular helper T cell differentiation. Immunity (2013) 38:655–68. doi:10.1016/j.immuni.2012.12.004
214. Pratama A, Ramiscal RR, Silva DG, Das SK, Athanasopoulos V, Fitch J, et al. Roquin-2 shares functions with its paralog Roquin-1 in the repression of mRNAs controlling T follicular helper cells and systemic inflammation. Immunity (2013) 38:669–80. doi:10.1016/j.immuni.2013.01.011
215. Schlundt A, Heinz GA, Janowski R, Geerlof A, Stehle R, Heissmeyer V, et al. Structural basis for RNA recognition in roquin-mediated post-transcriptional gene regulation. Nat Struct Mol Biol (2014) 21:671–8. doi:10.1038/nsmb.2855
216. Rehage N, Davydova E, Conrad C, Behrens G, Maiser A, Stehklein JE, et al. Binding of NUFIP2 to Roquin promotes recognition and regulation of ICOS mRNA. Nat Commun (2018) 9:299. doi:10.1038/s41467-017-02582-1
217. Srivastava M, Duan G, Kershaw NJ, Athanasopoulos V, Yeo JH, Ose T, et al. Roquin binds microRNA-146a and Argonaute2 to regulate microRNA homeostasis. Nat Commun (2015) 6:6253. doi:10.1038/ncomms7253
218. Mino T, Murakawa Y, Fukao A, Vandenbon A, Wessels HH, Ori D, et al. Regnase-1 and Roquin regulate a common element in inflammatory mRNAs by spatiotemporally distinct mechanisms. Cell (2015) 161:1058–73. doi:10.1016/j.cell.2015.04.029
219. Matsushita K, Takeuchi O, Standley DM, Kumagai Y, Kawagoe T, Miyake T, et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature (2009) 458:1185–90. doi:10.1038/nature07924
Keywords: post-transcriptional RNA regulation, RNA binding proteins, immune cell development, immune cell homeostasis, immune cell activation, T-cell mediated immunity, humoral response
Citation: Díaz-Muñoz MD and Turner M (2018) Uncovering the Role of RNA-Binding Proteins in Gene Expression in the Immune System. Front. Immunol. 9:1094. doi: 10.3389/fimmu.2018.01094
Received: 09 March 2018; Accepted: 02 May 2018;
Published: 23 May 2018
Edited by:
Loretta Tuosto, Sapienza Università di Roma, ItalyReviewed by:
Zhaocai Zhou, Shanghai Institutes for Biological Sciences (CAS), ChinaCopyright: © 2018 Díaz-Muñoz and Turner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manuel D. Díaz-Muñoz, manuel.diaz-munoz@inserm.fr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.