 Adriana Lima Vallochi*
Adriana Lima Vallochi* Livia Teixeira
Livia Teixeira Karina da Silva Oliveira
Karina da Silva Oliveira Clarissa Menezes Maya-Monteiro
Clarissa Menezes Maya-Monteiro Patricia T. Bozza*
Patricia T. Bozza*
- Laboratório de Imunofarmacologia, Instituto Oswaldo Cruz, Fundação Oswaldo Cruz (FIOCRUZ), Rio de Janeiro, Brazil
Lipid droplets (lipid bodies, LDs) are dynamic organelles that have important roles in regulating lipid metabolism, energy homeostasis, cell signaling, membrane trafficking, and inflammation. LD biogenesis, composition, and functions are highly regulated and may vary according to the stimuli, cell type, activation state, and inflammatory environment. Increased cytoplasmic LDs are frequently observed in leukocytes and other cells in a number of infectious diseases. Accumulating evidence reveals LDs participation in fundamental mechanisms of host-pathogen interactions, including cell signaling and immunity. LDs are sources of eicosanoid production, and may participate in different aspects of innate signaling and antigen presentation. In addition, intracellular pathogens evolved mechanisms to subvert host metabolism and may use host LDs, as ways of immune evasion and nutrients source. Here, we review mechanisms of LDs biogenesis and their contributions to the infection progress, and discuss the latest discoveries on mechanisms and pathways involving LDs roles as regulators of the immune response to protozoan infection.
Introduction
Lipid droplets (LDs) are dynamic and complex organelles, composed of a hydrophobic core enriched in neutral lipids such as triglyceride, cholesteryl ester, and retinyl ester, a surrounding monolayer of phospholipid, cholesterol, and a varied array of associated proteins with diverse functions in cell homeostasis, metabolism, and signaling (1, 2). The accumulation of lipids within the cytoplasmic LDs is a highly conserved feature, due to its role in energy homeostasis, membrane synthesis and cell signaling, conferring an evolutionary advantage to the organism. Indeed, LDs are present in virtually all cell types and organisms, including in unicellular protozoan parasites (3, 4), and in leukocytes and other cells involved in immune response in mammalians (5, 6).
Interest in LDs biogenesis and functions has dramatically increased in recent years due to their association with metabolic and inflammatory diseases, including diabetes (7), atherosclerosis (8, 9), obesity, cancer (10, 11), allergy (12), neurodegenerative diseases (13, 14), and numerous infectious diseases [reviewed in Bozza et al. (15), van der Meer-Janssen et al. (16), and Saka and Valdivia (17)]. Increased numbers of LDs in leukocytes and other cells are observed in infectious diseases caused by bacteria, virus, fungus, and protozoan parasites (17–26). LD accumulation in the cytoplasm of distinct host cell types has been reported after infection with Plasmodium berguei (27, 28), Trypanosoma cruzi (20, 29, 30), Toxoplasma gondii (31–34), and Leishmania sp. (35–37) and their contributions in disease pathogenesis are starting to be unveilled.
Different signaling pathways have been implicated in LD biogenesis in leukocytes and other cells involved in inflammatory and/or infectious reactions. Although molecular mechanisms that govern LD biogenesis are not fully characterized, the observations gathered so far indicate the involvement of both pathogen and host factors. Moreover, acumulating evidence indicates that infection-driven LD formation involves specific and well-regulated mechanisms that involve innate immune receptors, scavenger receptors, and nuclear receptors dependent pathways (18, 21, 26, 30, 38–42). Newly formed host LDs play key roles in host-pathogen interactions. Of note host LDs triggered during infection are major intracellular domains involved in eicosanoid synthesis with multiple functions in inflammation and immune signaling (18, 20, 21, 26, 30, 43, 44). More recently, parasite LDs were also demonstrated to compartmentalize eicosanoid synthesis with roles in parasite physiology and escape mechanisms (45, 46). In addition to functioning as signaling platforms, host LDs properties in energy storage and metabolic homeostasia may be explored by intracellular pathogens for their survival and growth. Accordingly, different authors showed a close association of LDs with parasite vacuoles (PVs) (20, 21, 33, 34, 47–49). This association may enable the nutrients’ flow between the host and the parasites, which is of crucial importance for parasite metabolism and may contribute to avoid the host response. Besides the acquisition of nutrients and imbalance of host metabolic pathways, the parasites explore many additional biological host responses with impacts to disease pathogenesis.
Here, we will address the mechanisms of formation and functions of LDs in the interactions between protozoan parasites and mammalian host. We will highlight the LDs role in host-pathogen interactions, discussing the crosstalk between pathogen and host cell on the immune response and survival mechanisms during infection.
LD: A Dynamic Cell Organelle
The Organelle Complexity
Lipid droplets are roughly spherical structures consisting mostly of triacylglycerols and sterol esters, lacking a delimiting classical bilayer membrane but surrounding by a monolayer of phospholipids and cholesterol and a variety of associated proteins (50, 51). This unique organization defies understanding how occur transport pathways to receive or deliver protein and lipid, once that arrangement does not fit in classical traffic vesicular mechanisms. On the other hand, favors its distinction from other organelles under electron microscope. More than 160 lipid species have been identified in LDs, with important variations in contents according to the cell type. For instance, triglycerides are largely dominant in white adipocytes, whereas sterol esters are more abundant in macrophages and retinyl esters in stellate cells (52–56). LDs associated proteins vary according to the cell and stimulatory condition and contains perilipin family proteins, lipid biosynthetic enzymes, lipolytic enzymes; enzymes involved in signaling and inflammatory mediator synthesis, and membrane-trafficking proteins (55, 57–65). Proteins associate with LDs surface through amphipathic and/or hydrophobic molecular moieties (66–69). Also, some proteins were detected within the hydrophobic core of the LD (59, 60, 70, 71). However, the most intriguing fact about protein content of LDs certainly is the presence of proteins with predicted membrane insertion domains within these organelles (59, 60, 65, 72). Among the proteins associated with LDs are the well-characterized perilipin familys’ proteins that share sequence similarities, previously termed as the PAT family, often used as molecular markers of LDs. It includes perilipin 1, perilipin 2 [PLIN2/adipose differentiation-related protein (ADRP)], perilipin 3 [tail-interacting protein of 47 kDa (TIP-47)], perilipin 4 (S3-12), and perilipin 5 (LSDA5, LSDP5, MLDP, and OXPAT) (73). Perilipins are major structural proteins present at the surface of LDs involved in regulating LD formation, incorporation, and accumulation of lipid in different cells types (74–79). The enzymes Acyl-CoA: diacylglycerol acyltransferase 2 (DGAT2) and acyl-CoA: cholesterol acyltransferase (ACAT) are responsible for the synthesis of the main LD core lipids (TAGs and CE). DGAT2 is inserted exclusively into the external leaflet of the endoplasmic reticulum (ER) membrane and, therefore, can diffuse onto the LD surface during LD formation, promoting TAGs synthesis and the continuation of LD growth (80). LDs also compartmentalize the lipolysis enzyme, adipose tissue triacylglycerol lipase (ATGL), as well as perilipins, that modulate both hormone-sensitive lipase and ATGL activities (80–82).
Lipid droplets are organelles that are intimately related to the ER (1, 83). The first and still accepted model of LDs formation suggests that these organelles are derived from the accumulation of newly synthesized lipids within the double layer of the ER membrane, with subsequent budding off from the ER to the cytoplasm after reaching a critical size (50, 83). According to this model, newly formed LDs detaches are surrounded by a phospholipid monolayer derived from the cytoplasmic leaflet of the ER coated with proteins that lack transmembrane spawning domains (3, 70, 83–85). However, accumulating evidence, based on imaging and composition of LDs, reported the presence of membrane-associated and transmembrane spanning proteins, as well as ribosomal structures and ribosomal associated proteins, giving rise to new hypothetical models for LD biogenesis (2, 65, 70, 86). These different groups suggested that invaginations of ER inside the LDs could be the membranous structure and hydrophilic ambient that allow proteins in the core of LDs (65, 72, 86), and/or that LDs may remain associated to ER through physical bridges (2, 87). Together, these biogenesis models contemplate the existence of subcompartments inside these organelles made possible through the incorporation of ER-derived membranes enabling sequential enzymatic reactions to occur within LDs as in the case of eicosanoid synthesis (5, 86, 88, 89).
Lipid droplets have gained importance as specialized and inducible cytoplasmic organelle due to their various proteic and lipidic components, and their participation in fundamental cellular processes. LD functions extend beyond the regulation of lipid metabolism in cell signaling, immunological activation, membrane trafficking participation, and control of the synthesis and secretion of inflammatory mediators. The LD in infectious diseases also suggests functions beyond the mere interaction with different viral proteins or bacteria; they can participate in important celular pathways of immune system.
One of the best characterized functions of LDs in leukocytes and other cells of the immune response are their capacity to act as platforms for heithened eicosanoid synthesis (90, 91). Eicosanoids—including prostaglandins, leukotrienes, and lipoxins—are non-storable but promptly newly synthesized signaling molecules, derived from the enzymatic oxygenation of arachidonic acid (AA), that control key cellular processes, including cell activation, metabolism, migration, proliferation, and death (92). Intracellular compartmentalization of eicosanoid synthesis within leukocytes and other cells has emerged as a key feature that regulates the amount and may also regulate the eicosanoid produced, and LDs has been established as main locales involved in the increased eicosanoid synthesis in inflammatory conditions (90). The substrate for eicosanoid synthesis— AA—is present in LDs, esterified in phospholipids and neutral lipids, where it can be converted into eicosanoids by specialized enzymes (6, 55, 93–95). cPLA2 and ATGL were shown to co-localize with LDs in different cells where it participates in the release of AA-associated LD from phospholipids and triglyceride pool, respectively (55, 96–98). Notably, P. aeruginosa toxin ExoU also mobilizes AA from host LDs due to their phospholipase activity as part of their pathogenic activity (94). The major eicosanoid-forming enzymes—cyclooxygenase, 5-lipoxygenase (5-LO), 15-LO, 5-LO-activating protein, and the terminal enzymes including PGE2 synthase and LTC4 synthase—were described within LDs of stimulated cells obtained under inflammatory and infectious conditions (18, 21, 26, 30, 43, 44, 59, 60, 99). Increased LD biogenesis correlates with an enhanced capacity of the cells to produce eicosanoids and suggests that the compartmentalization of the eicosanoid-synthetic machinery within LD have roles in the cellular ability to generate eicosanoids (18, 21, 59, 100). Direct proof of eicosanoid synthesis at LDs came only after the development of a lipid immunolabeling technique termed EicosaCell (88, 101). Since eicosanoids are not stored but are synthesized and released immediately, the EicosaCell method enables to immobilize and label eicosanoids at the exact locale of their synthesis, and by the use of this method the synthesis of leukotrienes and prostaglandins occuring at LDs have been confirmed (21, 30, 43, 101). Accordingly, it has been established that LDs are specialized, inducible cytoplasmic domains that have central roles in the control of synthesis and secretion of inflammatory mediators under different pathophysiological conditions, including in infections [reviewed in Bozza et al. (90)].
Mechanisms of LD Formation in Infectious Diseases Involves Specific and Regulated Mechanisms
Lipid droplet biogenesis in host cells during infections is a highly regulated cellular event. Although still incomplete, great advances on the understanding of the molecular events that govern LD formation in cells during host-pathogen interactions have been achieved in recent years. As detailed below, accumulation of LDs can be observed rapidly after infection with a variety of pathogens, and results from a balance between new lipid synthesis, increased lipid uptake, and regulated lypolisis involving both transcriptional dependent and independent mechanisms. Of note, during infection, not only infected cells and/or cells in direct contact with pathogens exhibit increased accumulation of LDs in their cytoplasm, but also non-infected neighboring cells at the inflammatory microenvironment may also present increased numbers of LDs indicating that paracrine mechanisms of bystander amplification through host and pathogen-derived signals also take place.
Pattern recognition receptors have been found to be involved in the generation of signals that induce LD formation. The main family of pattern recognition receptors with large range capacity of sensing diverse pathogens is the toll-like receptors (TLR). Cells from TLR4-null mutant mice and cells from signaling inactive mice fail to form LD after stimulation by LPS or during experimental sepsis, establishing the dependency of TLR4 for macrophages LD formation (18, 43). TLR4 signaling triggers the production of inflammatory mediators like platelet activator factor (PAF) and MCP-1/CCL2 that amplify the LD-induced response (18, 21, 43). Other proteins found to be required for productive signaling in response to LPS, include LPS-binding protein, CD14, CD11b/CD18, and myeloid differentiation factor-2 because their neutralization inhibits LPS-induced LD formation (18, 43). Indeed, different pathogens may be specifically recognized through innate receptors to trigger LDs in host cells. TLR2-dependent mechanisms have been implicated in LD formation triggered by Mycobacterium bovis (BCG), Mycobacterium leprae, T. cruzi, Chlamydia pneumonia, Histoplasma capsulatum, and schistosomal-lipids (21, 26, 38, 44, 102, 103). The studies of M. bovis (BCG) infection and stimulation by the purified cell wall component lipoarabinomannan highlighted the importance of TLR2 in LD formation, but not essential roles of TLR4 or TLR6 (21, 38, 104). However, classic TLR2 ligands as Pam3Cys and Zymozan and non-pathogenic mycobacteria Mycobacterium smegmatis failed to induce LDs formation or PGE2 production, while still inducing TLR2-dependent TNF-α production (38). This finding suggests that TLR2 activation, although essential is not sufficient to trigger pathways of LDs formation and other cofactors may be involved. Indeed, CD36, a multi-ligand scavenger receptor, has been demonstrated to cooperate with TLR2 in BCG signaling and LDs biogenesis in a mechanism dependent of compartmentalization within lipid rafts (39). Similarly, in M. leprae infection, TLR2 and TLR6 were implicated in LD formation and bystander amplification in macrophages with no internalized bacteria. This suggests an important role for these receptors in LD biogenesis, not only by direct recognition of microbial components, but also by leading to secondary autocrine/paracrine signaling (21, 44). Accordingly, conditioned medium from M. leprae-infected wild-type cells induced LD in wild-type and TLR2-deficient cells (40). Notably, many cytokines and chemokines have been described as being able to lead LDs formation (12, 18, 43, 99, 105–107). Indeed, MCP-1/CCL2 has been implicated in sepsis triggered LD formation, whereas IL-10 and CCL3 were shown to participate in mechanisms of mycobacterial LD formation (41, 43). Lipid signaling is also involved in LD formation and may participate in mechanisms of bystandar LD formation. PAF and PAF-like lipids are involved in LPS and oxidized low-density lipoprotein (LDL)-induced LDs formation, since pretreatment with PAF receptor antagonist significantly inhibits the number of these organelles during experimental sepsis (18, 43, 107). In addition, cis-unsaturated fatty acids including AA and oleic acid are potent inducers of LD formation in leukocytes and other cells (106, 108). TLR2 also mediates LD formation during protozoan parasite infection. T. cruzi infections induced LD formation in macrophages in a TLR2-dependent manner. Apoptotic cell uptake by these macrophages potentiated the LD formation and PGE2 production via TGF-β signaling. The inhibition of LD formation by the fatty acid synthase inhibitor C75 reverted the potentiating effects of the apoptotic cells on LD biogenesis, eicosanoid synthesis, and parasite replication, suggesting a role for LD in lipid mediator production and parasite escape mechanisms. Curiously, the effect was not restricted to parasitized cells, suggesting the involvement of paracrine mediators in LD biogenesis (30). Other classes of pattern recognition receptors have also been implicated in LD biogenesis, including dectin-1 (26).
Downstream mechanisms involved in infectious triggered LD formation are starting to be described. In this sense, signaling pathways involved in expression of genes related to influx/efflux of lipids and de novo synthesis of lipids during infectious process have been evaluated. Recently, it was reported that bacterial components may modulate expression and function of PPARγ. PPARγ is a member of lipid-activated nuclear receptor family that directly regulates several genes participating in fatty acids uptake, lipids storage, and inflammatory response. PPARγ heterodimerize with retinoid X receptor that binds directly to DNA response elements present in target gene. In macrophages and dendritic cells, PPARγ have proven to be a key regulator of lipid metabolism and inflammatory genes (109). In addition, PPARγ signaling pathway has been identified as central to foam cells formation in atherosclerotic lesions (110–113). During M. bovis BCG infection, TLR2 regulates the expression and activation of PPARγ activation as well as LDs formation. PPARγ inhibition by its antagonist significantly reduces number of LDs in BCG-infected macrophages, indicating its involvement in LDs biogenesis during mycobacterial infection (38). PPARγ involvement in LDs biogenesis may occur at different levels. PPARγ regulates expression of genes related to de novo lipid synthesis such as fatty acid synthase enzyme and PLIN2/ADRP, increasing TAG and cholesterol storage and reducing cholesterol efflux (114–116). PPARγ can also increase the influx of exogenous lipids due to increased expression of scavenger receptor CD36 (39). By contrast, liver X receptor, whose activation increases cholesterol efflux, has its transcriptional activity inhibited when viral and bacterial antigens are recognize via TLR3 and TLR4 receptors (117). Together these data show that LDs formation depends on specific signaling pathways activation, which regulate influx and efflux balance of lipids in cells. C. pneumoniae express a variety of potential TLR ligands and induces foam cell formation, facilitating atherogenesis. C. pneumoniae-induced LD formation also involves MAPK phosphorylation, including JNK and ERK, participates in macrophage LD formation. C. pneumoniae infection induced the down-regulation of PPARα and PPARγ expression (118), in contrary to the upregulation observed in BCG-infected macrophages where upregulation of PPARγ and PPARγ-dependent LD formation was described (39). Surprisingly, JNK and ERK inhibitors blocked LD formation and suppressed the down-regulation of PPARα and PPARγ. PPARα or PPARγ agonists reverted the infection-induced JNK and ERK phosphorylation while PPARα and PPARγ antagonist enhanced these phosphorylations. The increased C. pneumoniae-induced LD formation in LDL-treated THP-1 macrophages was interpreted to be the result of crosstalk between MAPK and PPARα/γ signaling pathways (118). Changes in the intracellular concentration of PPAR ligands can influence PPAR-dependent gene regulation (119).
Increasing amounts of data regarding the molecular mechanisms that govern lipogenesis, as well as regulation of the influx and efflux of lipids, suggest that these mechanisms operate synergistically for LD formation during infection (34, 39, 40).
LDs in Parasite Infections
Parasite Lipid Homeostasis and Their Own LDs
Throughout the entire infection process beginning with the host cell invasion, there is a huge involvement of lipids from both members’ interactions (120–122). Some parasites build and survive inside their PV; others leave them to live in host cytoplasm. The vacuole protects parasites but complicates access to some essential factors for parasite proliferation as, e.g., lipids. Intracellular parasites scavenge cholesterol from LDL or host LDs. LDs are a vital source of energy, and an essential element for the homeostasis (detoxification) of blood-stage parasites, as Plasmodium spp. and Schistosoma spp. Intracellular parasites have their own LDs—as an energy storage source, for metabolic homeostasis, and to modulate and escape the host response. The roles of parasites’ LDs and their interplay with host cells will be discussed in the following section.
Protozoa Are Unable to Synthesize Cholesterol De Novo and May Retrieve It From Host LDs
Despite possessing complex molecular machinery to interact in a variety of ways with their host cell, protozoa are unable to synthesize cholesterol de novo. T. cruzi recruits host lysosomes for fusion with the PV to initiate its egress from the PV and enter into the host cell cytoplasm, where replication begins (123). Parasites of the genus Leishmania evolved mechanisms to survive and multiply within acidified vesicles enriched in lysosomal enzymes (124). Otherwise, T. gondii and Plasmodium spp. have a so-called “active invasion,” and both built a nonfusogenic PV. The T. gondii PV interacts with many host structures such as LDs, microtubules, endosomes, the ER, mitochondria, and Golgi vesicles, which closely associated with the vacuole for nutrient acquisition (34, 125). T. gondii produces a network of tubule-like structures called the intravacuolar tubulovesicular network, a membranous interface derived from multilamellar vesicles secreted by the parasite (126, 127). However, protozoan parasites rely on their ability to steal cholesterol from different sources of the host (128–130).
As in mammals, T. cruzi captures cholesterol from LDLs via LDL receptor (LDLr). Experiments using LDLr knockout cells showed a 62% decrease in the total parasite burden compared with wild-type T. cruzi-infected mice. LDLr signaling is also essential in the invasion and fusion of PV with host lysosomes (129). T. brucei also needs LDL or high-density lipoproteins (HDL) for its multiplication (131).
Leishmania amazonensis has specific binding sites to HDL and LDL in its membranes. The parasite can capture cholesterol from LDL, and this process involves detergent-resistant membrane lipid microdomains. In all L. amazonensis extensions, host cholesterol is found; the intracellular compartments engaged in parasite lipid traffic and the presence of the LDLr remain to be discovered (130). Recently, Carvalho and colleagues found that some components of lipid metabolism had an association with lipoprotein lipase (LPL), apolipoprotein E (ApoE), and PPARα gene polymorphisms and presented the risk markers of visceral leishmaniasis (VL). Individuals with clinical manifestation of VL filed high TAG and very-low-density lipoprotein cholesterol (VLDL-C) levels, and low high-density lipoprotein cholesterol (HDL-C) levels. Furthermore, the LPL mutation (H+/H+ genotype and the H+ allele) was associated with elevated VLDL-C and TAG levels, reduced HDL-C levels and revealed an odds ratio of 21.3. The L162V polymorphism in the PPARα gene showed an odds ratio of 8.77 for Leu/Val when compared with Leu/Leu genotype. Thus, high TAG and VLDL-C levels were associated with susceptibility to VL, whereas low HDL levels with resistance to L. infantum infection. The mutated LPL and the PPARα Leu/Val genotypes may be considered risk markers for the development of VL (132).
Trypanosoma cruzi infection is capable of modulating cholesterol metabolism in the host tissue in both acute and chronic patients (133) as well as L. major in mice macrophages (48). Quantitative PCR analysis shows an 8,000-fold increase of LDLr levels in heart tissue of T. cruzi-infected mice compared with the control. Both, the accumulation of LDL and its receptor were observed around amastigote nests within infected cardiomyocytes (129). Like T. cruzi, T. brucei, and L. amazonensis, T. gondii also scavenges cholesterol from plasma (128). Experiments using LDLr knockout cells showed a 45% reduction in parasite load in T. gondii-infected LDLr knockout mice compared with control mice (134). However, Milovanović and colleagues showed a decrease in HDL and cholesterol in T. gondii-infected outbred mice, but LDL was unaltered until day 42 of infection. Moreover, they only observed an increase in LDL and consistent triglyceride levels at this end-point. The parasitemia only correlated with HDL levels and with LDL when the LDL levels were above 300 (135).
Host LDs are sources of lipids for many intracellular parasites. Intracellular parasites have evolved ways to attract host LDs to their phagosomal compartment and engulf them intact by unknown mechanisms (20, 33, 34, 48, 49). Although the Toxoplasma PV shares no characteristics with a phagosome, Toxoplasma also internalizes host LDs into the PV lumen. In Toxoplasma-infected cells, this process is independent of host Golgi or endoplasmatic reticulum, and the parasite has cholesterol equally distributed, but the PV membrane is low in cholesterol. When in excess, parasites release cholesterol and phosphatidylcholine using membrane-associated ABC-transport proteins (136). Overexpressing TgRab51, a Rab5 homolog, parasite mutants have increased numbers of LD by enhanced uptake of cholesterol and had their growth accelerated (137).
Toxoplasma gondii scavenges host LDs’ cholesterol and their lipolytic enzymatic activities to grow for its infectibility. It intercepts and engulfs host Rab7-associated LDs to the PV lumen, and the parasite lipase releases lipids from host LD making them accessible to the parasite. These LDs can be captured by endocytosis (34) and the parasite can modify the host-derived lipids (138), largely exploring the host lipidome (34). Conversely, Plasmodium cannot store cholesterol, because they do not express an ACAT enzyme, indicating that the parasite needs to scavenge lipids from the host constantly (139). The blood-stage form of Plasmodium scavenges cholesterol from HDL particles or directly from the erythrocyte membrane; how cholesterol is then transported to the parasite is still unknown. It is important to note that Plasmodium expresses sterol translocators and transporter genes and they can collect membrane components in their plasma membrane (140, 141). Furthermore, the Plasmodium PV has physical contact with the host plasma membrane, and the intracellular parasites can access cholesterol (142). Plasmodium scavenges cholesterol from LDL and HDL particles and LDs from hepatocytes. Plasmodium-infected hepatocytes have genes encoding sterol biosynthesis and metabolism components upregulated (143). Both LDL-derived and host synthesized cholesterol are co-transported to the parasite’ PV, but sterol biosynthetic pathway, or LDL defective cell lines, do not hamper parasite growth, suggesting compensatory ways to provide cholesterol to the parasite (144). Interestingly, the liver form’s PV is enriched in cholesterol and tightly associated only with the host ER. It is permeable to small solutes through channels stably maintained by host-derived cholesterol accumulated at the PV membrane, suggesting that sterols might contribute to parasite nutrient acquisition (144, 145). Then, intracellular parasites have many strategies to scavenge cholesterol, and host LDs are the essential source (Figure 1).
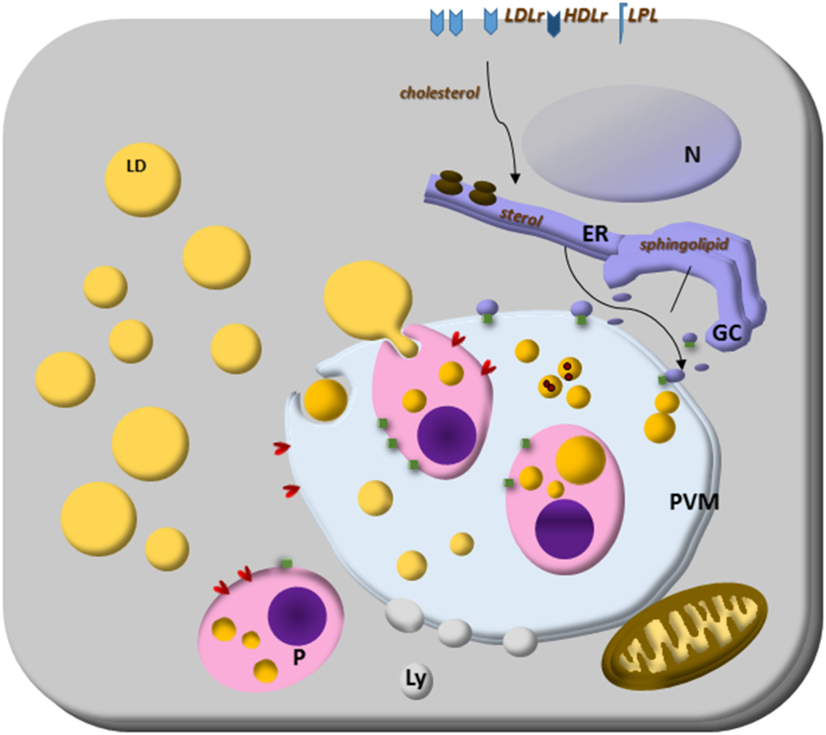
Figure 1. Lipid droplets (LDs) on parasites lipid homeostasis. Trypanosoma spp., Leishmania spp., Plasmodium spp., and Toxoplasma gondii cannot synthesize cholesterol, and the energy and lipid source for them is the host lipid synthesis and host LDs, as well as low-density lipoproteins, eventually high-density lipoproteins particles and lipoprotein lipase. The close relationship with other organelles through Rab proteins can be lipid sources, such as endoplasmic reticulum (ER) for sterol and Golgi complex (GC) for sphingolipid sources. Although the mechanisms are still unknown, some parasites can store lipids on their LD. Lipoprotein- and ABC-proteins are involved in the mammalian lipid transport, as well as important enzymes to lipid synthesis like ACAT and DGAT proteins. These proteins are also present in T. gondii and ACAT-like proteins are present in Trypanosoma cruzi. The parasite transfer lipids to their own LD to be stored, but in Plasmodium spp., or they metabolize them as in Plasmodium and T. gondii. The parasite and host LDs are involved in essential cell functions that can favor the survival of the parasite, such as energy and lipid source for intracellular parasites proliferation. They also can isolate lipids or toxic metabolites from the cytoplasm for detoxification, such as hemozoin from heme on Plasmodium-infected cell LD.
The two parasite ACAT enzymes, which esterified cholesterol (146, 147), are known and orchestrate the formation of LDs into the parasites (31, 34, 128, 138, 148). The parasite also hijacks host Golgi-derived vesicles by Rab14, Rab30, and Rab43 transport, small GTPases family members, within the PV and acquires their sphingolipid content (125).
In Figure 1, we summarize the essential and general ways discussed above by which parasites capture cholesterol from membrane receptors and host LDs. The organelle interactions highlight their importance on the traffic of lipids and metabolites. Moreover, the LD formation on both host and parasite cytoplasm, as well as the LD’s phagocytosis, discussed below are also illustrated as general characteristics of intracellular parasites.
Intraerythrocytic Plasmodium falciparum and T. gondii tachyzoites can synthesize neutral lipids, such as triacylglycerol, and store them in LDs present in the parasite cytoplasm (34, 148). The three forms of T. cruzi’s life also have intracellular LD formation. Epimastigotes cultured in media with higher concentrations of serum showed an increase in both cytoplasmic LD and LD-associated to reservosomes. In LD fraction present in purified reservosomes, the major neutral lipid was cholesterol and cholesteryl ester. No sterol synthesized by parasite pathways, such as ergosterol or ergosterol esther was found, which reinforces the idea that the lipid content of the LD-associated with reservosomes comes from the uptake of exogenous lipids (149). Once endocytosed, these lipids may be used in biosynthetic pathways of the T. cruzi. Carriers ABCA1-like, closely related to the mammal’s cholesterol and phospholipid carriers (150), were detected associated with the plasma membrane, intracellular vesicles and flagellar pocket in T. cruzi (151) and may participate in the cholesterol transport captured into the cytoplasm. Furthermore, the presence of Rab 18 in reservosomes could contribute from the traffic of lipids to other organelles (152).
Interestingly, the LDs formation in T. gondii cytoplasm was observed only when the host cells are in a higher concentration of lipids (34). Then, the presence of LDs on P. falciparum and T. gondii was associated with parasite energy store and homeostasis; since they are in a higher concentration of lipids, they could be toxic to these parasites (34, 148).
LDs in a Mechanism of Heme Detoxification
Considering that the cholesterol source for an extracellular parasite residing within the mesenteric veins and capillaries of their hosts, would not be a problem, Schistosoma mansoni indeed induce a reduction in serum total cholesterol of their host (153). Curiously, this capability of hijack cholesterol from its host is correlated with decrease LD formation in the host. The S. mansoni experimental model also shows the decreased levels of serum cholesterol, and of LDs in the liver cells of ApoE deficient mice fed a high-fat diet. Importantly, it also induced a systemic reduction of lipid since it was observed in adipose tissue around liver, heart, and blood vessels (153). The mechanisms responsible for this decrease remain unknown, but it may be dependent on the presence of parasite eggs and are related to the immune granulomas formed around them (153, 154). It was reported that eggs of Schistosoma japonicum require cholesteryl ester for their maturation. The uptake of these lipids came from HDL particles and CD36-related protein-mediated internalization (155).
Noteworthy, S. mansoni may also secrete parasite LDs. Numerous LD in the gut of parasites had hemozoin associated, and the authors suggested the role of LD in a mechanism of heme detoxification during parasite blood feeding (156, 157). In agreement with this view, other authors observed a prominent production of LD in Plasmodium berghei-infected hepatocytes and inside the parasitophorous vacuole and the food vacuole of P. falciparum-infected erythrocytes (27, 158, 159). These data suggest that the LDs might play roles in the detoxification of heme in the Plasmodium parasites (148). As summarized in Figure 1, the data discussed in the Sections “Protozoa Are Unable to Synthesize Cholesterol De Novo and May Retrieve It From Host LDs” and “LDs in a Mechanism of Heme Detoxification”indicate that LD may exert a parasite energy store and a functional role in parasite homeostasis including detoxification functions.
LDs in Parasite May Modulate Host Response
Intracellular parasites, such as T. cruzi, Leishmania infantum chagasi, P. falciparum, and T. gondii, also have cytoplasmic LDs. Notably, experiments with T. cruzi and L. i. chagasi are shedding light into LD functions in parasites. They went beyond sites for energy storage and suggested their involvement in the modulation of the host immune response, and participation in mechanisms of virulence (46, 160). These recent findings are illustrated in Figure 2 and discussed below.
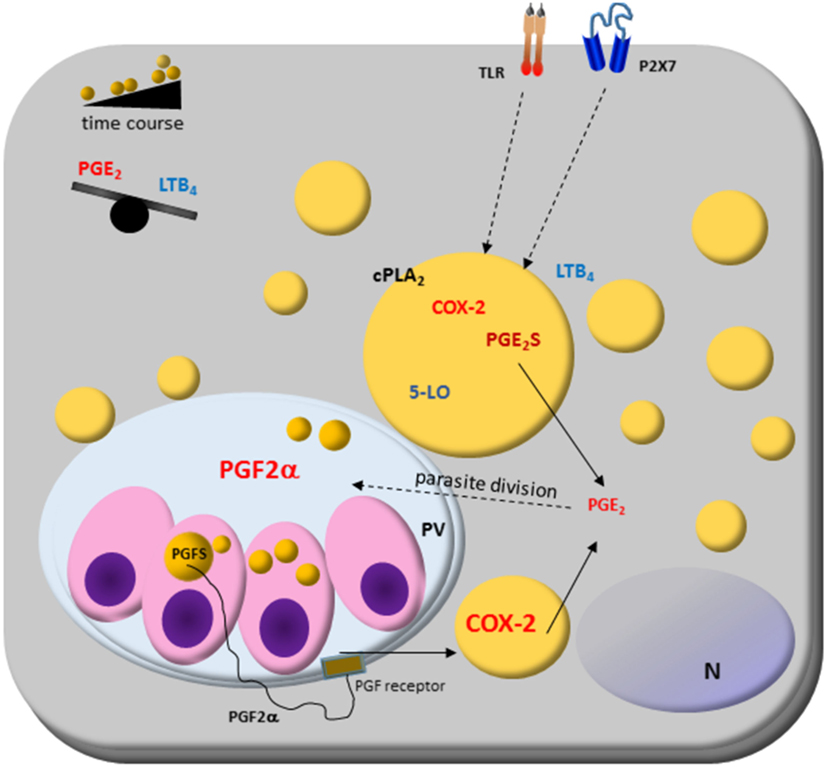
Figure 2. Host cell lipid droplet (LD) biogenesis in response to interaction with protozoan parasites. Increased numbers of LDs occur in the cytoplasm of many cell types after infection with different parasite-containing vesicles as well as on cytoplasm of these parasites, such as Trypanosoma spp., Leishmania spp., Plasmodium spp., and Toxoplasma gondii. The signaling from toll-like receptors (TLRs) by pathogens and pathogen-derived molecules triggers signaling pathways, such as PPARs and MAPKs, which are involved in the formation of LD. LDs produce lipid mediators; they compartmentalize both the substrate and the enzymatic machinery necessary for eicosanoid syntheses, such as cPLA2, cyclooxygenases (COX)-2, and prostaglandin synthases. Prostaglandin E2 (PGE2) production benefits parasite survival, as shown in Trypanosoma, Leishmania, Plasmodium, and Toxoplasma infections. On the other hand, leukotriene B4 (LTB4) production by host cells via P2 × 7 receptor is related to parasite killing as seen in Leishmania infection, where LDs biogenesis and an anti-inflammatory balance in the PGE2/LTB4 axis could facilitate the Leishmania transmissibility and infection in vivo. Cytoplasmic LD in Leishmania express prostaglandin F2α synthase (PGFS) responsible for PGF2α production. The PGF2α receptor (FP) is present on parasite vacuole (PV) surface, suggesting these LDs could act as a virulence factor. Then, LD could be involved in inflammation and immune evasion.
Novel findings show that L. i. chagasi cytoplasmatic LDs are intracellular sites of prostaglandin synthesis, mostly PGF2a production (45). L. i. chagasi has increased LD numbers and amplified expression of prostaglandin F2α synthase according to metacyclogenesis stage: metacyclic and amastigotes LD formation raised, and is the PGF2a source. Accordingly, it was demonstrated that the FP receptor is localized in the early phagocytic vacuoles and on surface parasitophorous vacuoles during macrophages infection with metacyclic forms of L. i. chagasi. Furthermore, the inhibition of the FP receptor in the macrophages diminished the L. i. chagasi parasite load 72 h after infection showing that the parasites may mobilize the eicosanoid machinery in the most infective stage of the parasite and suggest that the Leishmania’s LD could act as a virulence factor (45).
Trypanosoma cruzi LDs have also been associated with eicosanoid metabolism. The LDs of amastigotes growing in vivo have increased the size and electron-density than those from amastigotes cultured in vitro (46). Trypomastigotes forms also increased LD after both host interaction and exogenous AA stimulation. The AA content in LDs purified from AA-stimulated parasites increased and these parasites released high amounts of PGE2 and showed PGE2 synthase expression. Importantly, a PGE2 synthesis occurred within LDs from AA-stimulated trypomastigotes, indicating that LDs are essential sources of PGE2, a potent lipid mediator that inhibits many aspects of immune responses, and can be implicated in the pathogen survival (46). These data demonstrate novel functions for parasite-derived LDs in eicosanoid metabolism and evasion of the host immune response (45, 46).
Parasites Induce LDs on Host Cells: Friend or Foe?
Increasing data about the modulation of host lipid metabolism through targeting LD formation and accumulation by intracellular pathogens emerge as a typical phenotype to viral, bacterial, and parasitic infections and highlights the involving of LD in different aspects of inflammation [reviewed by Bozza et al. (5) and Saka and Valdivia (17)]. However, the understanding of LD formation and their dynamics in the interplay between hosts and parasites is still limited. Here, we will discuss the mechanisms known that parasites induce LD formation on host cells. Figures 2 and 3 summarize the data knew until now.
LDs in Eicosanoids Production and the Regulation of Host-Parasite Interactions
As a major source of eicosanoids production, LDs have been implicated in the regulation of parasite-induced inflammation [reviewed in Bozza et al. (5, 90)]. Eicosanoids are bioactive molecules produced by the oxidation of AA by specific enzymes, such as cyclooxygenases (COX), lipoxygenases, and terminal eicosanoid synthases. Prostaglandins have a wide variety of activities, including down-regulation of macrophage functions, and regulatory roles in immune responses. Notably, COX-2 and PGE-synthase compartmentalize within LDs (10, 18, 21), and LDs were the main site for heightened PGE2 production during T. cruzi infection (30).
The crucial role of PGE2 in the modulation of the host immune response can be illustrated by its higher plasma levels in patients with localized or diffuse cutaneous leishmaniasis than patient control (161). Also, the PGE2 secretion through PLA2 induced in the progression of cutaneous disease in L. amazonensis infection (162). More than that, PGE2 production benefits parasite survival, as showed in Leishmania spp. (35, 163–165), T. cruzi (20, 30, 166), and T. gondii infections (32, 33, 37).
This association between LD formation and PGE2 production was observed during in vivo infection with T. cruzi (20). Infected macrophages exhibit positive immunostaining for COX-2 and PGES, and both co-localize with PLIN2/ADRP staining, a LD structural protein, which confirms the presence of these enzymes inside LD in T. cruzi-infected macrophages. The association of PGE2 production and parasite division highlighted the LD participation on evasion mechanisms of T. cruzi (30, 167).
In Toxoplasma-infected macrophages and muscle cells, the host LD formation is associated with reduced host microbicides properties, with an increased PGE2 production, a potent inhibitor of Th1 response, and with a decreased nitric oxide production (32, 33). In muscle cells, the number of LD increased in a time course manner earlier as 6 h, until 48 h post-infection. The LD formation correlated with higher COX-2 and PGE2 expression, which could be contributing to IL-12 and interferon (IFN)-γ secretion from muscle cells (33). The production of the PGE2 with IL-12 and IFN-γ seems contradictory. However, IFN-γ is essential to control T. gondii infection—and the infections with others parasites [reviewed in Ref. (168–170)].
The secretory serine protease of Leishmania donovani is involved in down-regulation of macrophage microbicidal activity by inducing COX-2-mediated PGE2 release (171). The cytokines, such as IL-12 and IFN-γ, could favor the cyst formation in muscle cells and the establishment and maintenance of the chronic disease and transmission source via consumption of raw or undercooked meat containing T. gondii (33). The mechanisms of these processes await further investigation. In dendritic cells, L. amazonensis infection likewise induced LD formation and the modulation of cholesterol uptake pathways (172). Differently, L. i. chagasi-infected macrophages did not show LD formation, only the parasites showed LD formation (45).
The pathogenesis of severe malarial anemia is also due to the release of soluble mediators of inflammation as part of host immune response driven by phagocytosis of malarial pigment (hemozoin) present in parasitized RBC or free release on lysis of these cells (173). Although the suppression of COX-2-derived PGE2 is also associated with enhanced severity of malaria, how it alters the erythropoietic cascade remains to be determined (174, 175). Recently, increased LDs were demonstrated during malaria infection in different mice strains, however, there are not an association of number of LDs and disease severity (28).
Importantly, the stimulation of PLA2-COX-2-PGE2 pathway that can suppress macrophage’s activation is observed only in macrophages infected with live L. major parasites (37), highlighting the active role of the parasite with its cell host. Nevertheless, the protein level of COX-2 is unchanged in L. donovani (176) and L. major infection, suggesting that LD formation may not correlate with PGE2 production in L. donovani and L. major infections (48). However, recent data showed the essential role of PGE2 in suppressing L. major-infected macrophages, and this suppression was mediated by B1 cell IL-10 production (177).
A key observation is the host LDs in close apposition—and even inside—parasite-containing phagosomes, such as in T. cruzi (20, 49), L. major (48), L. amazonensis (172), and T. gondii infections (33, 34). Nolan et al. (34) showed that the host LDs are necessary for Toxoplasma infectivity in human foreskin fibroblasts (HFFs) and mouse embryonic fibroblasts (MEFs). The host LDs progressively increased with a peak of 8 h post-invasion (p.i.) that coincided with PLIN2/ADRP and GDAT2 upregulation transcription, suggesting stimulation of TAG synthesis in LD, and the host LDs cluster around PV. After that, their number decline and abruptly dropped until parasite egress at 24 h in HFF. In MEF, the host LD and GDAT2 decrease but the LDs remain around the PV. The authors suggested that the LD biogenesis and breakdown for lipolysis, the distribution of LD, and the DGAT2 expression levels reflect the highly dynamic status of host neutral lipid metabolism during infection. T. gondii may drive it, but it is also be part of a host cell defense mechanism in response to parasite-mediated lipid imbalances (34).
Lipid droplet formation in L. major-infected macrophages was very quick, time-dependent, and independent of parasite viability. It suggests that the trigger for LD formation by L. major also has a cellular origin, as T. cruzi and T. gondii infections do (30, 33, 37). Microarray data and transcriptomic analysis from L. major-infected macrophages support this formation. They revealed an enhanced cholesterol-uptake coupled with a decreased cholesterol efflux (48) and cholesterol uptake combined with activation of de novo TAG synthesis (37). Furthermore, LD formation was independent of cytoskeleton movement and L. major phagocytosis (37). As suggested for mycobacterial infections (41, 178), the paracrine induction of LD formation is also observed in the parasites T. cruzi (30), Toxoplasma (33) andL. major (37) infection. Then, soluble factors from infected cells or parasites may act in a paracrine manner to induce LD formation in uninfected bystander cells suggesting that receptors are triggering and downstream signaling pathways are sufficient to cause LD formation (30, 37, 178). It seems to have more and fundamental participants to achieve a homeostasis pattern. In L. amazonensis infection, Afonso e collaborators suggested that PGE2 and TGF-β1 pathways are involved with the enhanced parasite burden in L. amazonensis (179).
The roles of LDs in leukotriene synthesis during infection by intracellular parasites have not yet been established. This would be an important point to address since there is solid evidence that leukotriene B4 (LTB4) production by host cells is related to parasite killing in Leishmania infection by nitric oxide and reactive oxygen species production (180–183). Although there is evidence that 5-LO enzyme co-localize with LDs in hepatic stellate cells from schistosomal granulomas, where it is involved in the release of Cys-LTs in a TGF-β-regulated manner, potentially influencing pathogenesis and liver fibrosis in schistosomiasis (184).
The Leishmania vector (sand fly, Lutzomia longipalpis) saliva components have a central role in parasite infection. They can modulate eicosanoids metabolism and LDs formation in the host cells (164, 185, 186). The Lutzomia longipalpis saliva activates macrophages to produce PGE2 but not LTB4 (186) and the sand flies salivary gland sonicate (SGS) increases PGE2 production by neutrophils during L. infantum infection (164). The SGS during L. infantum inoculation increased the parasite viability inside peritoneal cells, and COX-2 inhibition blocked this action. The LD biogenesis on host or parasite and an anti-inflammatory balance in the PGE2/LTB4 axis could facilitate the Leishmania transmissibility and infection in vivo (160, 186).
The lipid derived pro-resolving mediators called lipoxins, resolvins, and protectins are potent anti-inflammatory mediators involved in the resolution of inflammation. As reviewed by Russell and Schwarze, lipoxins are protective to host control of disease caused by T. gondii, T. cruzi, and P. berghei but not by Mycobacterium tuberculosis bacteria. It could be related to the balance between pathogen-control and excessive immune response (187). However, the role of LDs in the synthesis of lipid mediators involved in the resolution of inflammation has not yet been characterized and it would be of interest to investigate whether LD is also involved in lipoxin- or resolvin-mediated mechanisms of pathogen evasion or destruction.
Lipid Droplets in Vesicular Interactions and the Resistance of Host to Parasite Infections
Interferons are effector key molecules involved in protective responses to viruses and intracellular pathogens [reviewed in Schroder et al. (168), Schneider et al. (169), and Pilla-Moffett et al. (170)]. They have their antimicrobial effects by a considerable remodeling transcriptional expression profile of target cells bearing the IFN receptors (188, 189). One of these IFN-stimulated genes encoding viperin (for virus inhibitory protein, ER-associated, and IFN-inducible), a canonical protein evolutionary conserved and found to be highly upregulated in response to LPS, dsDNA, and RNA analogs, and in infection with various viruses. Interestingly, viperin localizes to the cytoplasmic leaflet of the ER and LD, and LDs are sites of viral assembly (190, 191), and has antiviral activity against various human viruses (192).
Saka and Valdivia (17) already draw the attention for the exciting data showing that two signaling pathway (JNK and IFN) are involved in the cPLA2 activation required for LD biogenesis (193), as well as in immunity to Chlamydia trachomatis (194) and C. pneumoniae (118). In Figure 3, we highlighted the receptors described to induce LD formation and the key molecules and pathways involved in the modulation of parasite response. Others possible partners we also discuss in the following text.
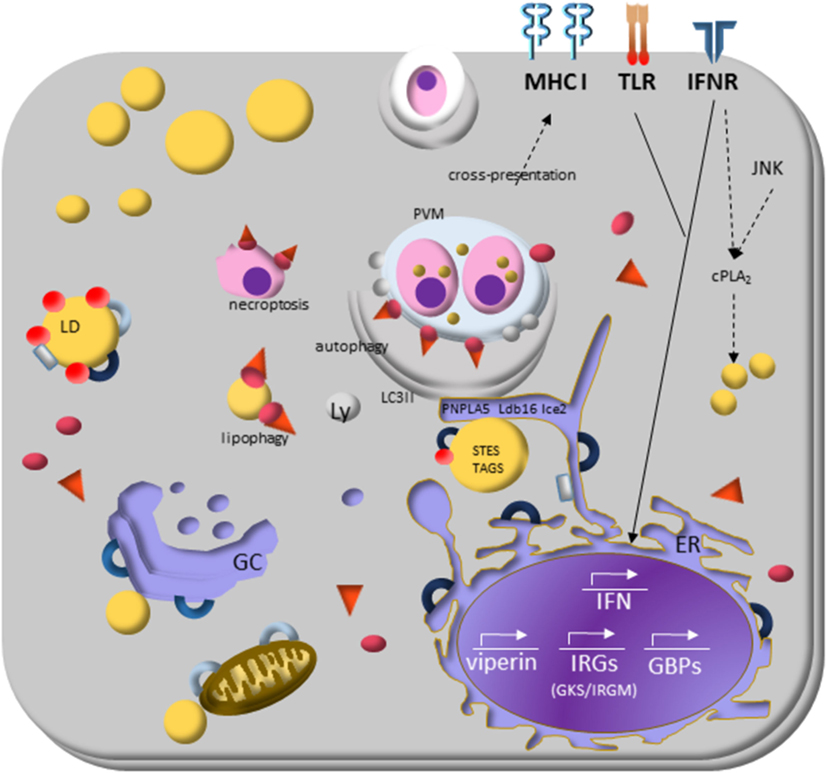
Figure 3. Lipid droplets (LDs) and the molecular mechanisms regulating immune response during protozoan infection. Interferon (IFN)-γ receptor signaling induces transduction of GTPases family molecules—immunity-related GTPases (GKS e IRGM proteins) and GPBs subfamilies—that play essential roles in membrane vesicular trafficking, autophagy, and antimicrobial and anti-inflammatory responses. IRGM are abundant proteins on LDs and “protects self-vesicles” from GKS and guanylate binding proteins (GBPs) destruction by autophagy machinery. The inactive GKS and GBP proteins built a pool of molecules available on cytoplasm and could have a stable association with the IRGM-deficient membrane, as parasitophorous vacuole membrane and eventually as LDs, to target them to autophagy, or lipophagy, respectively. LDs provide fatty acids, and lipids employed the phagophore membrane, implying their enzymes [steryl esters (STEs) and TAGs] on autophagosome biogenesis. IFN-γ signaling induces upregulation of LDs, and it appears to involve perilipin 2 (PLIN2)/ADRP that physically associates with IRGM. LD is also related to cross-presentation pathway since depletion of LDs by pharmacological inhibition of DGAT and by PLIN2/ADRP deficiency led to decreased LD biogenesis and cross-presentation since MHC-I surface expression, and the rate of antigen-presenting cell conjugated with T CD8+ lymphocyte are reduced.
Interferon-induced resistance genes include members of two GTPase families named immunity-related GTPases (IRGs) and guanylate binding proteins (GBPs). The IRGs proteins are subdivided into GKS and GMS IRGs class. The current model of IRGs interaction describes that, in IFN-γ-activated cells, the effector GKS proteins are cytosolic and stay in the inactive GDP-bound state by GMS regulatory proteins (195, 196). The specific membrane targeting of the GMS proteins prevent accumulation of activated GKS proteins and enable GKS proteins to distinguish organelles membranes from that of pathogen vacuoles. The GMS proteins are tightly associated with endomembrane of cellular organelles and restricted to specific organelles: Irgm1 localizes mainly to the Golgi complex membrane, endolysosomal compartment, mitochondria, peroxisomes, and LDs in uninfected cells; Irgm2 localizes to the Golgi and Irgm3 to the ER and LD (197).
Several members of both GTPases families can recognize specific host lipid molecules, to translocate and to adhere precisely to parasitophorous vacuole membranes (PVMs), labeling it for disruption or delivery to lysosomes to inhibit intracellular pathogen growth [reviewed in MacMicking (189)]. The specificity of these intracellular targeting events is well documented (168, 197–199) and the underlying mechanism is beginning to be deciphered (200–202). When GMS proteins are absent in the cell, GKS proteins activate spontaneously and form aggregate-like structures, preferentially on PVMs. The consequence is the disruption of the vacuole and the release of the pathogen into the cytosol or both vacuole and parasite disruption, resulting in either necroptosis or autophagy (203, 204).
Bougnères et al. (205) showed the Irgm3 role in LD formation, as Irgm3-deficient DCs do not accumulate LDs (205). DCs isolated from Irgm3-deficient mice showed the impaired capacity to stimulate specific CD8+ T cells owing to antigen phagocytosis and did not increase LD biogenesis upon IFN-γ stimulation when compared with wild-type mice. The increased antigen cross-presentation competence correlated with LD biogenesis and the chemically inhibited LD biogenesis blocked it. The data show that DCs from PLIN2 (ADRP) deficient mice had similar defects in antigen cross-presentation that Irgm3-deficient mice had and that Irgm3 interacts with PLIN2 suggest that LDs play a significant role in regulating cross-presentation (205). The absence of Irgm3 protein can explain the lack of LD biogenesis, where unprotected by IRGM protein, LD suffer an attack of GKS-GBP proteins bounding on their membrane delivering them to autophagy pathway (201).
The essential functions of GBPs family proteins seem to be their ability to target membranes and to oligomerize in them. Several GBPs harbor C-terminal CaaX motifs for isoprenylation that enable them to associate with intracellular endomembranes (206). They bind and hydrolyze guanosine phosphated, allowing GBPs to form homo- and hetero-oligomers. Once docked on PV membrane, GKS and GBPs proteins can recruit antimicrobial protein complexes, including autophagic cascade and inflammasome pathway to destroy the parasite vacuolar compartment [reviewed by Schroder et al. (168)]. Interestingly, GBP proteins stimulate caspase-11-dependent pyroptosis in macrophages infected with Gram-negative vacuole resident bacteria leading the activation of the non-canonical inflammasome (207, 208).
As inactive proteins, GKS and GBP have lipid binding substrates on LD and T. gondii PVMs. Haldar et al. (201) proposed that the high expression of GMS proteins on LDs make them regulatory proteins by increasing the availability of GKS and GBP inactive proteins allowing them to bind on PVMs and deliver them to autophagosomes for parasite degradation. It was recently shown that murine GBP2 positively regulates the recruitment of Irga6 (a GKS protein) to T. gondii PVMs (209) and directly targets the membrane of T. gondii after disruption of PVM (202). Then, the vacuolar contents released into the cytosol may be removed through autophagy (198, 203). The vacuolar contents released may be ubiquitinated, resulting in the recruitment of p62. ATG3 and p62 also promote the cross-presentation of vacuolar antigens, derived from lysed vacuoles, on MHC class I (210).
Haldar and colleagues showed that Irgm1 and Irgm3 co-localize on LD of IFN-γ-treated MEFs and that Gbp1 effector protein p62 co-localizes with IRGM-deficient LDs MEFs. Ichimura and Komatsu showed that p62Gbp1 binds directly to the LC3 to deliver its cargo to autophagosomes (211). As LDs from IRGM-deficient mice were inside autophagosomes upon IFN-γ activation, it could suggest that IRGM was not a role in LD formation. Instead, Irgm1 and Irgm3 proteins could protect LDs from degradation in MEFs and p62Gbp1 could start LDs lipophagy (201).
Lipophagy occurs when lysosomes degrade LD after its sequestration by autophagosomes. The recognition of LD by autophagic machinery is unknown [reviewed by Martinez-Lopez and Singh (212)]. Lipophagy could be related to lipid homeostasis, where control size and the number of LDs are making a significant balance of energy source and survival mechanisms, such as cell stress. Accumulating evidence suggest the participation of LD in the regulation of the autophagic process and their destruction by autophagy via lipophagy (212–214). LDs also act in concert with the proteasomal and autophagic pathways of protein degradation, proposing their role on isolation stress products inside LD, such as apo B in hepatocytes and a-synuclein in Parkinson’s disease (215, 216). On atherosclerosis, however, it seems that IRGM has a pathogenic role. IRGM proteins participate in the positive feedback where ox-LDL up-regulates IRGM in macrophage, which, in turn, modulates the CD36 to promote the ox-LDL uptake (217). Studies on other systems are necessary to investigate the IRGM pathogenic role.
Curiously, IRGM proteins physically interact with the central autophagy regulators and make molecular complexes with the pathogen sensor NOD2. In response to PAMPs, the RIG-1, and TLR3 signaling boost the NOD2-IRGM association. NOD2-IRGM complex promotes the assembly of autophagy regulators highlighting the critical role of IRGM proteins as organizer of the core autophagy machinery and as a vital component of antimicrobial and anti-inflammatory responses (200, 218, 219).
Recently, Shpilka and collaborators suggested that LDs provide fatty acids and lipids for the formation and elongation of the phagophore membrane (220). This work also showed the involvement of the enzymes responsible for the TAG and steryl esters synthesis with autophagosome biogenesis in yeasts strains. Moreover, they showed the essential participation of ER-LD contact-site proteins, Ldb16 and Ice2, in autophagosome formation. They suggested that these two organelles act together, and the lipid transfer from LD to the ER is necessary for autophagosome formation (220). Previously, the contribution of TAG mobilization from the LDs, by the lipase PNPLA5, to autophagosome biogenesis was shown (213).
As the autophagic machinery is also implicated in the formation and growth of LDs (221), feedback between these two processes could drive LD growth, autophagosome biogenesis, and cellular homeostasis.
Our current understanding of IFN-inducible GTPases role on host resistance to the intracellular protozoa and on the LD role to self-organelle recognition appoint to LD as important cell function modulator, implying the participation of LDs on parasite immune response.
Final Remarks and Future Perspectives
A number of key findings have evidenced a protagonist role for LDs in host parasite biology. LDs were shown to have major roles in metabolism, signaling and detoxification, and actively participates in innate immunity response to infection. Parasite-host interactions modulate the lipid metabolism of both organisms, and these studies provide valuable knowledge on cell biology, immune response, and drugs development. In leukocytes and other cells involved in infectious conditions, LD biogenesis is highly induced through regulated mechanisms that involve innate immune receptors and transcriptional dependent and independent pathways. Notably, it is now well established that LDs are main sites for the generation of lipid mediators involved in the host immune responses. The evolving understanding of the interplay of LDs and critical pathways on immune response, as IFN-induced resistance genes, autophagy, antigen cross-presentation and areas that still need to have roles of LDs investigated like resolution of inflammation, could illuminate our knowledge of the signaling ways that control parasite infections or, better to say, establish homeostasis with host and parasite living together. Furthermore, it is not a surprise that the intracellular parasites have developed strategies to evade or use LDs for surviving. Parasites have adapted to exploit LDs as a source of lipids for membrane building, a crucial step for parasite growth, and as a negative regulator of immune response.
Although great advances in the understanding of the mechanisms of LD biogenesis and its roles in lipid metabolism and inflammatory mediator production have been achieved, critical questions remain about the formation and the functions that LDs play in infectious diseases. In conclusion, recent studies have identified LDs as multifunctional organelles with key functions in lipid storage and cell signaling in inflammation, and as such, they are emerging as attractive target candidates for therapeutic intervention. Future studies will be necessary to characterize the role of LDs as targets for therapeutic intervention in infectious diseases that progress with increased LD accumulation. These will need to include the development of selective LD inhibitors. Moreover, the safety characterization of LD inhibition is required, as lipid accumulation within LDs may act as a protective mechanism in lipid homeostasis against cellular lipotoxicity.
Author Contributions
AV and PB contributed conception and design of the review. AV, KO, and LT organized the database. AV wrote the first draft of the manuscript. AV, PB, CM-M, KO, and LT contributed to writing a subset discussion and to manuscript revision, read, and approved the submitted version.
Conflict of Interest Statement
The authors have no other affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject discussed in the manuscript.
Acknowledgments
We would like to recognize present and past members of the Laboratory of Immunopharmacology for their valuable contributions. We acknowledge the comments and input of Dr. Jens Rietdorf (CDTS, FIOCRUZ). We apologize to investigators whose relevant work has not been cited because of space constraints. The work of the authors is supported by Fundação de Amparo à Pesquisa do Rio de Janeiro (FAPERJ, Brasil); Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brasil); PAPES/Fiocruz; Oswaldo Cruz Institute (IOC), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Capes).
References
1. Walther TC, Chung J, Farese RV. Lipid droplet biogenesis. Annu Rev Cell Dev Biol (2017) 33:491–510. doi:10.1146/annurev-cellbio-100616-060608
2. Pol A, Gross SP, Parton RG. Biogenesis of the multifunctional lipid droplet: lipids, proteins, and sites. J Cell Biol (2014) 204:635–46. doi:10.1083/jcb.201311051
3. Murphy DJ. The biogenesis and functions of lipid bodies in animals, plants and microorganisms. Prog Lipid Res (2001) 40:325–438. doi:10.1016/S0163-7827(01)00013-3
4. Ding Y, Yang L, Zhang S, Wang Y, Du Y, Pu J, et al. Identification of the major functional proteins of prokaryotic lipid droplets. J Lipid Res (2012) 53:399–411. doi:10.1194/jlr.M021899
5. Bozza PT, Magalhães KG, Weller PF. Leukocyte lipid bodies – Biogenesis functions in inflammation. Biochim Biophys Acta (2009) 1791:540–51. doi:10.1016/j.bbalip.2009.01.005
6. Melo RCN, D’Avila H, Wan H-C, Bozza PT, Dvorak AM, Weller PF. Lipid bodies in inflammatory cells: structure, function, and current imaging techniques. J Histochem Cytochem (2011) 59:540–56. doi:10.1369/0022155411404073
7. Borén J, Taskinen MR, Olofsson SO, Levin M. Ectopic lipid storage and insulin resistance: a harmful relationship. J Intern Med (2013) 274:25–40. doi:10.1111/joim.12071
8. Paul A, Chang BH, Li L, Yechoor VK, Chan L. Deficiency of adipose differentiation-related protein impairs foam cell formation and protects against atherosclerosis. Circ Res (2008) 102:1492–501. doi:10.1161/CIRCRESAHA.107.168070
9. Yuan Y, Li P, Ye J. Lipid homeostasis and the formation of macrophage-derived foam cells in atherosclerosis. Protein Cell (2012) 3:173–81. doi:10.1007/s13238-012-2025-6
10. Accioly MT, Pacheco P, Maya-Monteiro CM, Carrossini N, Robbs BK, Oliveira SS, et al. Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res (2008) 68:1732–40. doi:10.1158/0008-5472.CAN-07-1999
11. Bozza PT, Viola JPB. Lipid droplets in inflammation and cancer. Prostaglandins Leukot Essent Fatty Acids (2010) 82:243–50. doi:10.1016/j.plefa.2010.02.005
12. Vieira-de-Abreu A, Assis EF, Gomes GS, Castro-Faria-Neto HC, Weller PF, Bandeira-Melo C, et al. Allergic challenge-elicited lipid bodies compartmentalize in vivo leukotriene C 4 synthesis within eosinophils. Am J Respir Cell Mol Biol (2005) 33:254–61. doi:10.1165/rcmb.2005-0145OC
13. Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell (2015) 160:177–90. doi:10.1016/j.cell.2014.12.019
14. Cabirol-Pol M-J, Khalil B, Rival T, Faivre-Sarrailh C, Besson MT. Glial lipid droplets and neurodegeneration in a Drosophila model of complex I deficiency. Glia (2018) 66:874–88. doi:10.1002/glia.23290
15. Bozza PT, Melo RCN, Bandeira-Melo C. Leukocyte lipid bodies regulation and function: contribution to allergy and host defense. Pharmacol Ther (2007) 113:30–49. doi:10.1016/j.pharmthera.2006.06.006
16. van der Meer-Janssen YPM, van Galen J, Batenburg JJ, Helms JB. Lipids in host-pathogen interactions: pathogens exploit the complexity of the host cell lipidome. Prog Lipid Res (2010) 49:1–26. doi:10.1016/j.plipres.2009.07.003
17. Saka HA, Valdivia R. Emerging roles for lipid droplets in immunity and host-pathogen interactions. Annu Rev Cell Dev Biol (2012) 28:411–37. doi:10.1146/annurev-cellbio-092910-153958
18. Pacheco P, Bozza FA, Gomes RN, Bozza M, Weller PF, Castro-Faria-Neto HC, et al. Lipopolysaccharide-induced leukocyte lipid body formation in vivo: innate immunity elicited intracellular loci involved in eicosanoid metabolism. J Immunol (2002) 169:6498–506. doi:10.4049/jimmunol.169.11.6498
19. Cardona PJ, Llatjós R, Gordillo S, Díaz J, Ojanguren I, Ariza A, et al. Evolution of granulomas in lungs of mice infected aerogenically with Mycobacterium tuberculosis. Scand J Immunol (2000) 52:156–63. doi:10.1046/j.1365-3083.2000.00763.x
20. Melo RC, D’Avila H, Fabrino DL, Almeida PE, Bozza PT. Macrophage lipid body induction by Chagas disease in vivo: putative intracellular domains for eicosanoid formation during infection. Tissue Cell (2003) 35:59–67. doi:10.1016/S0040-8166(02)00105-2
21. D’Avila H, Melo RCN, Parreira GG, Werneck-Barroso E, Castro-Faria-Neto HC, Bozza PT. Mycobacterium bovis bacillus Calmette-Guerin induces TLR2-mediated formation of lipid bodies: intracellular domains for eicosanoid synthesis in vivo. J Immunol (2006) 176:3087–97. doi:10.4049/jimmunol.176.5.3087
22. Kumar Y, Cocchiaro J, Valdivia RH. The obligate intracellular pathogen Chlamydia trachomatis targets host lipid droplets. Curr Biol (2006) 16:1646–51. doi:10.1016/j.cub.2006.06.060
23. Samsa MM, Mondotte JA, Iglesias NG, Assunção-Miranda I, Barbosa-Lima G, Da Poian AT, et al. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog (2009) 5:e1000632. doi:10.1371/journal.ppat.1000632
24. Barba G, Harper F, Harada T, Kohara M, Goulinet S, Matsuura Y, et al. Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc Natl Acad Sci U S A (1997) 94:1200–5. doi:10.1073/pnas.94.4.1200
25. Hope RG, Murphy DJ, McLauchlan J. The domains required to direct core proteins of hepatitis C virus and GB virus-B to lipid droplets share common features with plant oleosin proteins. J Biol Chem (2002) 277:4261–70. doi:10.1074/jbc.M108798200
26. Sorgi CA, Secatto A, Fontanari C, Turato WM, Belanger C, de Medeiros AI, et al. Histoplasma capsulatum cell wall-glucan induces lipid body formation through CD18, TLR2, and dectin-1 receptors: correlation with leukotriene B4 generation and role in HIV-1 infection. J Immunol (2009) 182:4025–35. doi:10.4049/jimmunol.0801795
27. Rodríguez-Acosta A, Finol HJ, Pulido-Méndez M, Márquez A, Andrade G, González N, et al. Liver ultrastructural pathology in mice infected with Plasmodium berghei. J Submicrosc Cytol Pathol (1998) 30:299–307.
28. Borges TKS, Alves ÉAR, Vasconcelos HAR, Carneiro FP, Nicola AM, Magalhães KG, et al. Differences in the modulation of reactive species, lipid bodies, cyclooxygenase-2, 5-lipoxygenase and PPAR-γ in cerebral malaria-susceptible and resistant mice. Immunobiology (2017) 222:604–19. doi:10.1016/j.imbio.2016.11.010
29. Melo RCN, Fabrino DL, Dias FF, Parreira GG. Lipid bodies: structural markers of inflammatory macrophages in innate immunity. Inflamm Res (2006) 55:342–8. doi:10.1007/s00011-006-5205-0
30. D’Avila H, Freire-de-Lima CG, Roque NR, Teixeira L, Barja-Fidalgo C, Silva AR, et al. Host cell lipid bodies triggered by Trypanosoma cruzi infection and enhanced by the uptake of apoptotic cells are associated with prostaglandin E2 generation and increased parasite growth. J Infect Dis (2011) 204:951–61. doi:10.1093/infdis/jir432
31. Coppens I. Contribution of host lipids to toxoplasma pathogenesis. Cell Microbiol (2006) 8:1–9. doi:10.1111/j.1462-5822.2005.00647.x
32. Mota LA, Roberto Neto J, Monteiro VG, Lobato CS, Oliveira MA, Cunha Md, et al. Culture of mouse peritoneal macrophages with mouse serum induces lipid bodies that associate with the parasitophorous vacuole and decrease their microbicidal capacity against Toxoplasma gondii. Mem Inst Oswaldo Cruz (2014) 109:767–74. doi:10.1590/0074-0276140119
33. Gomes AF, Magalhães KG, Rodrigues RM, de Carvalho L, Molinaro R, Bozza PT, et al. Toxoplasma gondii-skeletal muscle cells interaction increases lipid droplet biogenesis and positively modulates the production of IL-12, IFN-g and PGE2. Parasit Vectors (2014) 7:47. doi:10.1186/1756-3305-7-47
34. Nolan SJ, Romano JD, Coppens I. Host lipid droplets: an important source of lipids salvaged by the intracellular parasite Toxoplasma gondii. PLoS Pathog (2017) 13:e1006362. doi:10.1371/journal.ppat.1006362
35. Pinheiro RO, Nunes MP, Pinheiro CS, D’Avila H, Bozza PT, Takiya CM, et al. Induction of autophagy correlates with increased parasite load of Leishmania amazonensis in BALB/c but not C57BL/6 macrophages. Microbes Infect (2009) 11:181–90. doi:10.1016/j.micinf.2008.11.006
36. Rodríguez NE, Lockard RD, Turcotte EA, Araújo-Santos T, Bozza PT, Borges VM, et al. Lipid bodies accumulation in Leishmania infantum -infected C57BL/6 macrophages. Parasite Immunol (2017) 39:e12443. doi:10.1111/pim.12443
37. Rabhi S, Rabhi I, Trentin B, Piquemal D, Regnault B, Goyard S, et al. Lipid droplet formation, their localization and dynamics during Leishmania major macrophage infection. PLoS One (2016) 11:1–19. doi:10.1371/journal.pone.0148640
38. Almeida PE, Silva AR, Maya-Monteiro CM, Torocsik D, D’Avila H, Dezso B, et al. Mycobacterium bovis bacillus Calmette-Guerin infection induces TLR2-dependent peroxisome proliferator-activated receptor expression and activation: functions in inflammation, lipid metabolism, and pathogenesis. J Immunol (2009) 183:1337–45. doi:10.4049/jimmunol.0900365
39. Almeida PE, Roque NR, Magalhães KG, Mattos KA, Teixeira L, Maya-Monteiro C, et al. Differential TLR2 downstream signaling regulates lipid metabolism and cytokine production triggered by Mycobacterium bovis BCG infection. Biochim Biophys Acta (2014) 1841:97–107. doi:10.1016/j.bbalip.2013.10.008
40. Mattos KA, Oliveira VC, Berrêdo-Pinho M, Amaral JJ, Antunes LC, Melo RC, et al. Mycobacterium leprae intracellular survival relies on cholesterol accumulation in infected macrophages: a potential target for new drugs for leprosy treatment. Cell Microbiol (2014) 16:797–815. doi:10.1111/cmi.12279
41. Mattos KA, Lara FA, Oliveira VGC, Rodrigues LS, D’Avila H, Melo RCN, et al. Modulation of lipid droplets by Mycobacterium leprae in Schwann cells: a putative mechanism for host lipid acquisition and bacterial survival in phagosomes. Cell Microbiol (2011) 13:259–73. doi:10.1111/j.1462-5822.2010.01533.x
42. Rajaram MVS, Brooks MN, Morris JD, Torrelles JB, Azad AK, Schlesinger LS. Mycobacterium tuberculosis activates human macrophage peroxisome proliferator-activated receptor linking mannose receptor recognition to regulation of immune responses. J Immunol (2010) 185:929–42. doi:10.4049/jimmunol.1000866
43. Pacheco P, Vieira-de-Abreu A, Gomes RN, Barbosa-Lima G, Wermelinger LB, Maya-Monteiro CM, et al. Monocyte chemoattractant protein-1/CC chemokine ligand 2 controls microtubule-driven biogenesis and leukotriene b4-synthesizing function of macrophage lipid bodies elicited by innate immune response. J Immunol (2007) 179:8500–8. doi:10.4049/jimmunol.179.12.8500
44. Mattos KA, D’Avila H, Rodrigues LS, Oliveira VGC, Sarno EN, Atella GC, et al. Lipid droplet formation in leprosy: toll-like receptor-regulated organelles involved in eicosanoid formation and Mycobacterium leprae pathogenesis. J Leukoc Biol (2010) 87:371–84. doi:10.1189/jlb.0609433
45. Araújo-Santos T, Rodríguez NE, Moura-Pontes S, Dixt UG, Abánades DR, Bozza PT, et al. Role of prostaglandin F2α production in lipid bodies from Leishmania infantum chagasi: insights on virulence. J Infect Dis (2014) 210:1951–61. doi:10.1093/infdis/jiu299
46. Toledo DA, D’Avila H, Melo RC. Host lipid bodies as platforms for intracellular survival of protozoan parasites. Front Immunol (2016) 7:174. doi:10.3389/fimmu.2016.00174
47. Cocchiaro J, Kumar Y, Fischer ER, Hackstadt T, Valdivia RH. Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proc Natl Acad Sci U S A (2008) 105:9379–84. doi:10.1073/pnas.0712241105
48. Rabhi I, Rabhi S, Ben-Othman R, Rasche A, Consortium S, Daskalaki A, et al. Transcriptomic signature of Leishmania infected mice macrophages: a metabolic point of view. PLoS Negl Trop Dis (2012) 6:e1763. doi:10.1371/journal.pntd.0001763
49. Melo RC, Dvorak AM. Lipid body–phagosome interaction in macrophages during infectious diseases: host defense or pathogen survival strategy? PLoS Pathog (2012) 8:e1002729. doi:10.1371/journal.ppat.1002729
50. Tauchi-Sato K, Ozeki S, Houjou T, Taguchi R, Fujimoto T. The surface of lipid droplets is a phospholipid monolayer with a unique fatty acid composition. J Biol Chem (2002) 277:44507–12. doi:10.1074/jbc.M207712200
51. Murphy DJ. The dynamic roles of intracellular lipid droplets: from archaea to mammals. Protoplasma (2012) 249(3):541–85. doi:10.1007/s00709-011-0329-7
52. Bartz R, Li W-H, Venables B, Zehmer JK, Roth MR, Welti R, et al. Lipidomics reveals that adiposomes store ether lipids and mediate phospholipid traffic. J Lipid Res (2007) 48:837–47. doi:10.1194/jlr.M600413-JLR200
53. Grillitsch K, Connerth M, Köfeler H, Arrey TN, Rietschel B, Wagner B, et al. Lipid particles/droplets of the yeast Saccharomyces cerevisiae revisited: lipidome meets proteome. Biochim Biophys Acta (2011) 1811:1165–75. doi:10.1016/j.bbalip.2011.07.015
54. Leber R, Zinser E, Paltauf F, Daum G, Zellnig G. Characterization of lipid particles of the yeast, Saccharomyces cerevisiae. Yeast (1994) 10:1421–8. doi:10.1002/yea.320101105
55. Yu W, Bozza PT, Tzizik DM, Gray JP, Cassara J, Dvorak AM, et al. Co-compartmentalization of MAP kinases and cytosolic phospholipase A2 at cytoplasmic arachidonate-rich lipid bodies. Am J Pathol (1998) 152:759–69.
56. Blaner WS, O’Byrne SM, Wongsiriroj N, Kluwe J, D’Ambrosio DM, Jiang H, et al. Hepatic stellate cell lipid droplets: a specialized lipid droplet for retinoid storage. Biochim Biophys Acta (2009) 1791(6):467–73. doi:10.1016/j.bbalip.2008.11.001
57. McGookey DJ, Anderson RG. Morphological characterization of the cholesteryl ester cycle in cultured mouse macrophage foam cells. J Cell Biol (1983) 97:1156–68. doi:10.1083/jcb.97.4.1156
58. Yu W, Cassara J, Weller PF. Phosphatidylinositide 3-kinase localizes to cytoplasmic lipid bodies in human polymorphonuclear leukocytes and other myeloid-derived cells. Blood (2000) 95:1078–85.
59. Bozza PT, Yu W, Penrose JF, Morgan ES, Dvorak AM, Weller PF. Eosinophil lipid bodies: specific, inducible intracellular sites for enhanced eicosanoid formation. J Exp Med (1997) 186:909–20. doi:10.1084/jem.186.6.909
60. Dvorak AM, Morgan E, Schleimer RP, Ryeom SW, Lichtenstein LM, Weller PF. Ultrastructural immunogold localization of prostaglandin endoperoxide synthase (cyclooxygenase) to non-membrane-bound cytoplasmic lipid bodies in human lung mast cells, alveolar macrophages, type II pneumocytes, and neutrophils. J Histochem Cytochem (1992) 40:759–69. doi:10.1177/40.6.1316915
61. Umlauf E, Csaszar E, Moertelmaier M, Schuetz GJ, Parton RG, Prohaska R. Association of stomatin with lipid bodies. J Biol Chem (2004) 279:23699–709. doi:10.1074/jbc.M310546200
62. Fujimoto T, Yamazaki S, Eto-Kimura A, Takeshige K, Muta T. The amino-terminal region of toll-like receptor 4 is essential for binding to MD-2 and receptor translocation to the cell surface. J Biol Chem (2004) 279:47431–7. doi:10.1074/jbc.M408724200
63. Liu P, Ying Y, Zhao Y, Mundy DI, Zhu M, Anderson RG. Chinese hamster ovary K2 cell lipid droplets appear to be metabolic organelles involved in membrane traffic. J Biol Chem (2004) 279:3787–92. doi:10.1074/jbc.M311945200
64. Brasaemle DL, Dolios G, Shapiro L, Wang R. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. J Biol Chem (2004) 279:46835–42. doi:10.1074/jbc.M409340200
65. Wan HC, Melo RCN, Jin Z, Dvorak AM, Weller PF. Roles and origins of leukocyte lipid bodies: proteomic and ultrastructural studies. FASEB J (2007) 21:167–78. doi:10.1096/fj.06-6711com
66. Boulant S, Montserret R, Hope RG, Ratinier M, Targett-Adams P, Lavergne J-P, et al. Structural determinants that target the hepatitis C virus core protein to lipid droplets. J Biol Chem (2006) 281:22236–47. doi:10.1074/jbc.M601031200
67. Bussell R, Eliezer D. A structural and functional role for 11-mer repeats in α-synuclein and other exchangeable lipid binding proteins. J Mol Biol (2003) 329:763–78. doi:10.1016/S0022-2836(03)00520-5
68. Ostermeyer AG, Ramcharan LT, Zeng Y, Lublin DM, Brown DA. Role of the hydrophobic domain in targeting caveolin-1 to lipid droplets. J Cell Biol (2004) 164:69–78. doi:10.1083/jcb.200303037
69. Subramanian V, Garcia A, Sekowski A, Brasaemle DL. Hydrophobic sequences target and anchor perilipin A to lipid droplets. J Lipid Res (2004) 45:1983–91. doi:10.1194/jlr.M400291-JLR200
70. Robenek H, Buers I, Hofnagel O, Robenek MJ, Troyer D, Severs NJ. Compartmentalization of proteins in lipid droplet biogenesis. Biochim Biophys Acta (2009) 1791:408–18. doi:10.1016/j.bbalip.2008.12.001
71. Robenek H, Robenek MJ, Troyer D. PAT family proteins pervade lipid droplet cores. J Lipid Res (2005) 46:1331–8. doi:10.1194/jlr.M400323-JLR200
72. Robenek MJ, Severs NJ, Schlattmann K, Plenz G, Zimmer K-P, Troyer D, et al. Lipids partition caveolin-1 from ER membranes into lipid droplets: updating the model of lipid droplet biogenesis. FASEB J (2004) 18:866–8. doi:10.1096/fj.03-0782fje
73. Itabe H, Yamaguchi T, Nimura S, Sasabe N. Perilipins: a diversity of intracellular lipid droplet proteins. Lipids Health Dis (2017) 16:1–11. doi:10.1186/s12944-017-0473-y
74. Brasaemle DL, Barber T, Wolins NE, Serrero G, Blanchette-Mackie EJ, Londos C. Adipose differentiation-related protein is an ubiquitously expressed lipid storage droplet-associated protein. J Lipid Res (1997) 38:2249–63.
75. Wolins NE, Rubin B, Brasaemle DL. TIP47 associates with lipid droplets. J Biol Chem (2001) 276:5101–8. doi:10.1074/jbc.M006775200
76. Yamaguchi T, Matsushita S, Motojima K, Hirose F, Osumi T. MLDP, a novel PAT family protein localized to lipid droplets and enriched in the heart, is regulated by peroxisome proliferator-activated receptor α. J Biol Chem (2006) 281:14232–40. doi:10.1074/jbc.M601682200
77. Tansey JT, Sztalryd C, Hlavin EM, Kimmel AR, Londos C. The central role of perilipin A in lipid metabolism and adipocyte lipolysis. IUBMB Life (2004) 56:379–85. doi:10.1080/15216540400009968
78. Sztalryd C, Xu G, Dorward H, Tansey JT, Contreras JA, Kimmel AR, et al. Perilipin A is essential for the translocation of hormone-sensitive lipase during lipolytic activation. J Cell Biol (2003) 161:1093–103. doi:10.1083/jcb.200210169
79. Greenberg AS, Egan JJ, Wek SA, Garty NB, Blanchette-Mackie EJ, Londos C. Perilipin, a major hormonally regulated adipocyte-specific phosphoprotein associated with the periphery of lipid storage droplets. J Biol Chem (1991) 266:11341–6.
80. Meyers A, Weiskittel TM, Dalhaimer P. Lipid droplets: formation to breakdown. Lipids (2017) 52:465–75. doi:10.1007/s11745-017-4263-0
81. Lass A, Zimmermann R, Oberer M, Zechner R. Lipolysis – a highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog Lipid Res (2011) 50:14–27. doi:10.1016/j.plipres.2010.10.004
82. Greenberg AS, Kraemer FB, Soni KG, Jedrychowski MP, Yan Q-W, Graham CE, et al. Lipid droplet meets a mitochondrial protein to regulate adipocyte lipolysis. EMBO J (2011) 30:4337–9. doi:10.1038/emboj.2011.371
83. Martin S, Parton RG. Lipid droplets: a unified view of a dynamic organelle. Nat Rev Mol Cell Biol (2006) 7:373–8. doi:10.1038/nrm1912
84. Brown DA. Lipid droplets: proteins floating on a pool of fat. Curr Biol (2001) 11:446–9. doi:10.1016/S0960-9822(01)00257-3
85. Brasaemle DL, Wolins NE. Packaging of fat: an evolving model of lipid droplet assembly and expansion. J Biol Chem (2012) 287:2273–9. doi:10.1074/jbc.R111.309088
86. Melo RCN, Paganoti GF, Dvorak AM, Weller PF. The internal architecture of leukocyte lipid body organelles captured by three-dimensional electron microscopy tomography. PLoS One (2013) 8:e59578. doi:10.1371/journal.pone.0059578
87. Kassan A, Herms A, Fernández-Vidal A, Bosch M, Schieber NL, Reddy BJN, et al. Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in ER microdomains. J Cell Biol (2013) 203:985–1001. doi:10.1083/jcb.201305142
88. Bandeira-Melo C, Paiva LA, Amorim NRT, Weller PF, Bozza PT. EicosaCell: an imaging-based assay to identify spatiotemporal eicosanoid synthesis. Methods Mol Biol (2017) 1554:127–41. doi:10.1007/978-1-4939-6759-9_6
89. Melo RC, Weller PF. Lipid droplets in leukocytes: organelles linked to inflammatory responses. Exp Cell Res (2016) 340:193–7. doi:10.1016/j.yexcr.2015.10.028
90. Bozza PT, Bakker-Abreu I, Navarro-Xavier RA, Bandeira-Melo C. Lipid body function in eicosanoid synthesis: an update. Prostaglandins Leukot Essent Fatty Acids (2011) 85:205–13. doi:10.1016/j.plefa.2011.04.020
91. Weller PF, Bozza PT, Yu W, Dvorak AM. Cytoplasmic lipid bodies in eosinophils: central roles in eicosanoid generation. Int Arch Allergy Immunol (1999) 118:450–2. doi:10.1159/000024161
92. Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol (2008) 9:162–76. doi:10.1038/nrm2335
93. Weller PF, Longworth DL, Jaffe JJ. Leukotriene C4 synthesis catalyzed by Dirofilaria immitis gluthathione S-transferase. Am J Trop Med Hyg (1989) 40:171–5.
94. Plotkowski M-C, Brandão BA, de Assis M-C, Feliciano L-FP, Raymond B, Freitas C, et al. Lipid body mobilization in the ExoU-induced release of inflammatory mediators by airway epithelial cells. Microb Pathog (2008) 45:30–7. doi:10.1016/j.micpath.2008.01.008
95. Triggiani M, Oriente A, Seeds MC, Bass DA, Marone G, Chilton FH. Migration of human inflammatory cells into the lung results in the remodeling of arachidonic acid into a triglyceride pool. J Exp Med (1995) 182:1181–90. doi:10.1084/jem.182.5.1181
96. Moreira LS, Piva B, Gentile LB, Mesquita-Santos FP, D’Avila H, Maya-Monteiro CM, et al. Cytosolic phospholipase A2-driven PGE2 synthesis within unsaturated fatty acids-induced lipid bodies of epithelial cells. Biochim Biophys Acta (2009) 1791:156–65. doi:10.1016/j.bbalip.2009.01.003
97. Dichlberger A, Schlager S, Maaninka K, Schneider WJ, Kovanen PT. Adipose triglyceride lipase regulates eicosanoid production in activated human mast cells. J Lipid Res (2014) 55:2471–8. doi:10.1194/jlr.M048553
98. Schlager S, Goeritzer M, Jandl K, Frei R, Vujic N, Kolb D, et al. Adipose triglyceride lipase acts on neutrophil lipid droplets to regulate substrate availability for lipid mediator synthesis. J Leukoc Biol (2015) 98:837–50. doi:10.1189/jlb.3A0515-206R
99. Bozza PT, Yu W, Cassara J, Weller PF. Pathways for eosinophil lipid body induction: differing signal transduction in cells from normal and hypereosinophilic subjects. J Leukoc Biol (1998) 64:563–9. doi:10.1002/jlb.64.4.563
100. Bozza PT. Mechanisms of platelet-activating factor-induced lipid body formation: requisite roles for 5-lipoxygenase and de novo protein synthesis in the compartmentalization of neutrophil lipids. J Exp Med (1996) 183:1515–25. doi:10.1084/jem.183.4.1515
101. Bandeira-Melo C, Gillard G, Ghiran I, Weller PF. EliCell: a gel-phase dual antibody capture and detection assay to measure cytokine release from eosinophils. J Immunol Methods (2000) 244:105–15. doi:10.1016/S0022-1759(00)00264-7
102. Magalhaes KG, Almeida PE, Atella GC, Maya-Monteiro CM, Castro-Faria-Neto HC, Pelajo-Machado M, et al. Schistosomal-derived lysophosphatidylcholine are involved in eosinophil activation and recruitment through Toll-like receptor-2-dependent mechanisms. J Infect Dis (2010) 202:1369–79. doi:10.1086/656477
103. Cao F, Castrillo A, Tontonoz P, Re F, Byrne GI. Chlamydia pneumoniae-induced macrophage foam cell formation is mediated by toll-like receptor 2. Infect Immun (2007) 75:753–9. doi:10.1128/IAI.01386-06
104. D’Avila H, Almeida PE, Roque NR, Catro-Faria-Neto HC, Bozza PT. Toll-Like receptor-2-mediated C-C chemokine receptor 3 and eotaxin-driven eosinophil influx induced by Mycobacterium bovis BCG pleurisy. Infect Immun (2007) 75:1507–11. doi:10.1128/IAI.01326-06
105. Bandeira-Melo C, Phoofolo M, Weller PF. Extranuclear lipid bodies, elicited by CCR3-mediated signaling pathways, are the sites of chemokine-enhanced leukotriene C4 production in eosinophils and basophils. J Biol Chem (2001) 276:22779–87. doi:10.1074/jbc.M101436200
106. Bozza PT, Payne JL, Morham SG, Langenbach R, Smithies O, Weller PF. Leukocyte lipid body formation and eicosanoid generation: cyclooxygenase-independent inhibition by aspirin. Proc Natl Acad Sci U S A (1996) 93:11091–6. doi:10.1073/pnas.93.20.11091
107. Silva AR, Pacheco P, Vieira-de-Abreu A, Maya-Monteiro CM, D’Alegria B, Magalhães KG, et al. Lipid bodies in oxidized LDL-induced foam cells are leukotriene-synthesizing organelles: a MCP-1/CCL2 regulated phenomenon. Biochim Biophys Acta (2009) 1791:1066–75. doi:10.1016/j.bbalip.2009.06.004
108. Weller PE, Ryeom W, Pieard ST, Aekerman SJ, Dvorak M. Cytoplasmic lipid bodies of neutrophils: formation induced by cis-unsaturated fatty acids and mediated by protein kinase C. J Cell Biol (1991) 113:137–46. doi:10.1083/jcb.113.1.137
109. Széles L, Torocsik D, Nagy L. PPARγ in immunity and inflammation: cell types and diseases. Biochim Biophys Acta (2007) 1771:1014–30. doi:10.1016/j.bbalip.2007.02.005
110. Chawla A, Boisvert WA, Lee C-H, Laffitte BA, Barak Y, Joseph SB, et al. A PPARγ-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell (2001) 7:161–71. doi:10.1016/S1097-2765(01)00164-2
111. Nagy L, Tontonoz P, Alvarez JGA, Chen H, Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARγ. Cell (1998) 93:229–40. doi:10.1016/S0092-8674(00)81574-3
112. Ricote M, Huang J, Fajas L, Li A, Welch J, Najib J, et al. Expression of the peroxisome proliferator-activated receptor (PPAR) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proc Natl Acad Sci U S A (1998) 95:7614–9. doi:10.1073/pnas.95.13.7614
113. Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPARγ promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell (1998) 93:241–52. doi:10.1016/S0092-8674(00)81575-5
114. Gao J, Serrero G. Adipose differentiation related protein (ADRP) expressed in transfected COS-7 cells selectively stimulates long chain fatty acid uptake. J Biol Chem (1999) 274:16825–30. doi:10.1074/jbc.274.24.16825
115. Imamura M, Inoguchi T, Ikuyama S, Taniguchi S, Kobayashi K, Nakashima N, et al. ADRP stimulates lipid accumulation and lipid droplet formation in murine fibroblasts. Am J Physiol Endocrinol Metab (2002) 283:E775–83. doi:10.1152/ajpendo.00040.2002
116. Larigauderie G, Furman C, Jaye M, Lasselin C, Copin C, Fruchart J-C, et al. Adipophilin enhances lipid accumulation and prevents lipid efflux from THP-1 macrophages: potential role in atherogenesis. Arterioscler Thromb Vasc Biol (2004) 24:504–10. doi:10.1161/01.ATV.0000115638.27381.97
117. Castrillo A, Joseph SB, Vaidya SA, Haberland M, Fogelman AM, Cheng G, et al. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol Cell (2003) 12:805–16. doi:10.1016/S1097-2765(03)00384-8
118. Cheng B, Wu X, Sun S, Wu Q, Mei C, Xu Q, et al. MAPK–PPARα/γ signal transduction pathways are involved in Chlamydia pneumoniae-induced macrophage-derived foam cell formation. Microb Pathog (2014) 69–70:1–8. doi:10.1016/j.micpath.2014.03.001
119. Georgiadi A, Kersten S. Mechanisms of gene regulation by fatty acids. Adv Nutr An Int Rev J (2012) 3:127–34. doi:10.3945/an.111.001602
120. Ramakrishnan S, Serricchio M, Striepen B, Bütikofer P. Lipid synthesis in protozoan parasites: a comparison between kinetoplastids and apicomplexans. Prog Lipid Res (2013) 52:488–512. doi:10.1016/j.plipres.2013.06.003
121. Rub A, Arish M, Husain SA, Ahmed N, Akhter Y. Host-lipidome as a potential target of protozoan parasites. Microbes Infect (2013) 15:649–60. doi:10.1016/j.micinf.2013.06.006
122. Coppens I. Targeting lipid biosynthesis and salvage in apicomplexan parasites for improved chemotherapies. Nat Rev Microbiol (2013) 11:823–35. doi:10.1038/nrmicro3139
123. Tardieux I, Webster P, Ravesloot J, Boron W, Lunn JA, Heuser JE, et al. Lysosome recruitment and fusion are early events required for trypanosome invasion of mammalian cells. Cell (1992) 71:1117–30. doi:10.1016/S0092-8674(05)80061-3
124. Antoine J-C, Prina E, Lang T, Courret N. The biogenesis and properties of the parasitophorous vacuoles that harbour Leishmania in murine macrophages. Trends Microbiol (1998) 6:392–401. doi:10.1016/S0966-842X(98)01324-9
125. Romano JD, Coppens I. Host organelle hijackers: a similar modus operandi for Toxoplasma gondii and Chlamydia trachomatis: co-infection model as a tool to investigate pathogenesis. Pathog Dis (2013) 69:72–86. doi:10.1111/2049-632X.12057
126. Sibley LD, Niesman IR, Parmley SF, Cesbron-Delauw MF. Regulated secretion of multi-lamellar vesicles leads to formation of a tubulo-vesicular network in host-cell vacuoles occupied by Toxoplasma gondii. J Cell Sci (1995) 108:1669–77.
127. Mercier C, Dubremetz JF, Rauscher B, Lecordier L, Sibley LD, Cesbron-Delauw MF. Biogenesis of nanotubular network in toxoplasma parasitophorous vacuole induced by parasite proteins. Mol Biol Cell (2002) 13:2397–409. doi:10.1091/mbc.E02-01-0021
128. Coppens I, Sinai AP, Joiner KA. Toxoplasma gondii exploits host low-density lipoprotein receptor-mediated endocytosis for cholesterol acquisition. J Cell Biol (2000) 149:167–80. doi:10.1083/jcb.149.1.167
129. Nagajyothi F, Weiss LM, Silver DL, Desruisseaux MS, Scherer PE, Herz J, et al. Trypanosoma cruzi utilizes the host low density lipoprotein receptor in invasion. PLoS Negl Trop Dis (2011) 5:e953. doi:10.1371/journal.pntd.0000953
130. De Cicco NNT, Pereira MG, Corrêa JR, Andrade-Neto VV, Saraiva FB, Chagas-Lima AC, et al. LDL uptake by Leishmania amazonensis: involvement of membrane lipid microdomains. Exp Parasitol (2012) 130:330–40. doi:10.1016/j.exppara.2012.02.014
131. Black S, Vandeweerd V. Serum lipoproteins are required for multiplication of Trypanosoma brucei brucei under axenic culture conditions. Mol Biochem Parasitol (1989) 37:65–72. doi:10.1016/0166-6851(89)90103-5
132. Carvalho MDT, Alonso DP, Vendrame CMV, Costa DL, Costa CHN, Werneck GL, et al. Lipoprotein lipase and PPAR alpha gene polymorphisms, increased very-low-density lipoprotein levels, and decreased high-density lipoprotein levels as risk markers for the development of visceral Leishmaniasis by Leishmania infantum. Mediators Inflamm (2014) 2014:230129. doi:10.1155/2014/230129
133. Johndrow C, Nelson R, Tanowitz H, Weiss LM, Nagajyothi F. Trypanosoma cruzi infection results in an increase in intracellular cholesterol. Microbes Infect (2014) 16:337–44. doi:10.1016/j.micinf.2014.01.001
134. Portugal LR, Fernandes LR, Pietra Pedroso VS, Santiago HC, Gazzinelli RT, Alvarez-Leite JI. Influence of low-density lipoprotein (LDL) receptor on lipid composition, inflammation and parasitism during Toxoplasma gondii infection. Microbes Infect (2008) 10:276–84. doi:10.1016/j.micinf.2007.12.001
135. Milovanović I, Trbovich AM, Vujanic M, Klun I, Bobic B, Nikolic A, et al. Toxoplasma gondii infection induces lipid metabolism alterations in the murine host. Int J Infect Dis (2008) 12:e172–3. doi:10.1016/j.ijid.2008.05.430
136. Ehrenman K, Sehgal A, Lige B, Stedman TT, Joiner KA, Coppens I. Novel roles for ATP-binding cassette G transporters in lipid redistribution in Toxoplasma. Mol Microbiol (2010) 76:1232–49. doi:10.1111/j.1365-2958.2010.07169.x
137. Robibaro B, Stedman TT, Coppens I, Ngô HM, Pypaert M, Bivona T, et al. Toxoplasma gondii Rab5 enhances cholesterol acquisition from host cells. Cell Microbiol (2002) 4:139–52. doi:10.1046/j.1462-5822.2002.00178.x
138. Charron AJ, Sibley LD. Host cells: mobilizable lipid resources for the intracellular parasite Toxoplasma gondii. J Cell Sci (2002) 115:3049–59.
139. Coppens I, Vielemeyer O. Insights into unique physiological features of neutral lipids in Apicomplexa: from storage to potential mediation in parasite metabolic activities. Int J Parasitol (2005) 35:597–615. doi:10.1016/j.ijpara.2005.01.009
140. Hiller NL, Akompong T, Morrow JS, Holder AA, Haldar K. Identification of a stomatin orthologue in vacuoles induced in human erythrocytes by malaria parasites. J Biol Chem (2003) 278:48413–21. doi:10.1074/jbc.M307266200
141. Di Girolamo F, Raggi C, Birago C, Pizzi E, Lalle M, Picci L, et al. Plasmodium lipid rafts contain proteins implicated in vesicular trafficking and signalling as well as members of the PIR superfamily, potentially implicated in host immune system interactions. Proteomics (2008) 8:2500–13. doi:10.1002/pmic.200700763
142. Lauer S, VanWye J, Harrison T, McManus H, Samuel BU, Hiller NL, et al. Vacuolar uptake of host components, and a role for cholesterol and sphingomyelin in malarial infection. EMBO J (2000) 19:3556–64. doi:10.1093/emboj/19.14.3556
143. Albuquerque SS, Carret C, Grosso AR, Tarun AS, Peng X, Kappe SHI, et al. Host cell transcriptional profiling during malaria liver stage infection reveals a coordinated and sequential set of biological events. BMC Genomics (2009) 10:270. doi:10.1186/1471-2164-10-270
144. Labaied M, Jayabalasingham B, Bano N, Cha S-J, Sandoval J, Guan G, et al. Plasmodium salvages cholesterol internalized by LDL and synthesized de novo in the liver. Cell Microbiol (2011) 13:569–86. doi:10.1111/j.1462-5822.2010.01555.x
145. Bano N, Romano JD, Jayabalasingham B, Coppens I. Cellular interactions of Plasmodium liver stage with its host mammalian cell. Int J Parasitol (2007) 37:1329–41. doi:10.1016/j.ijpara.2007.04.005
146. Nishikawa Y, Quittnat F, Stedman TT, Voelker DR, Choi J-Y, Zahn M, et al. Host cell lipids control cholesteryl ester synthesis and storage in intracellular Toxoplasma. Cell Microbiol (2005) 7:849–67. doi:10.1111/j.1462-5822.2005.00518.x
147. Lige B, Sampels V, Coppens I. Characterization of a second sterol-esterifying enzyme in Toxoplasma highlights the importance of cholesterol storage pathways for the parasite. Mol Microbiol (2013) 87:951–67. doi:10.1111/mmi.12142
148. Vielemeyer O, McIntosh MT, Joiner KA, Coppens I. Neutral lipid synthesis and storage in the intraerythrocytic stages of Plasmodium falciparum. Mol Biochem Parasitol (2004) 135:197–209. doi:10.1016/j.molbiopara.2003.08.017
149. Pereira MG, Nakayasu ES, Sant’Anna C, De Cicco NNT, Atella GC, de Souza W, et al. Trypanosoma cruzi epimastigotes are able to store and mobilize high amounts of cholesterol in reservosome lipid inclusions. PLoS One (2011) 6:e22359. doi:10.1371/journal.pone.0022359
150. Attie A. ABCA1: at the nexus of cholesterol, HDL and atherosclerosis. Trends Biochem Sci (2007) 32:172–9. doi:10.1016/j.tibs.2007.02.001
151. Torres C, Pérez-Victoria FJ, Parodi-Talice A, Castanys S, Gamarro F. Characterization of an ABCA-like transporter involved in vesicular trafficking in the protozoan parasite Trypanosoma cruzi. Mol Microbiol (2004) 54:632–46. doi:10.1111/j.1365-2958.2004.04304.x
152. Sant’Anna C, Nakayasu ES, Pereira MG, Lourenço D, De Souza W, Almeida IC, et al. Subcellular proteomics of Trypanosoma cruzi reservosomes. Proteomics (2009) 9:1782–94. doi:10.1002/pmic.200800730
153. Doenhoff MJ, Stanley RG, Griffiths K, Jackson CL. An anti-atherogenic effect of Schistosoma mansoni infections in mice associated with a parasite-induced lowering of blood total cholesterol. Parasitology (2002) 125:415–21. doi:10.1017/S0031182002002275
154. Stanley RG, Jackson CL, Griffiths K, Doenhoff MJ. Effects of Schistosoma mansoni worms and eggs on circulating cholesterol and liver lipids in mice. Atherosclerosis (2009) 207:131–8. doi:10.1016/j.atherosclerosis.2009.04.037
155. Okumura-Noji K, Miura Y, Lu R, Asai K, Ohta N, Brindley PJ, et al. CD36-related protein in Schistosoma japonicum: candidate mediator of selective cholesteryl ester uptake from high-density lipoprotein for egg maturation. FASEB J (2013) 27:1236–44. doi:10.1096/fj.12-219816
156. Lvova M, Zhukova M, Kiseleva E, Mayboroda O, Hensbergen P, Kizilova E, et al. Hemozoin is a product of heme detoxification in the gut of the most medically important species of the family Opisthorchiidae. Int J Parasitol (2016) 46:147–56. doi:10.1016/j.ijpara.2015.12.003
157. Corrêa Soares JB, Maya-Monteiro CM, Bittencourt-Cunha PR, Atella GC, Lara FA, D’Avila JC, et al. Extracellular lipid droplets promote hemozoin crystallization in the gut of the blood fluke Schistosoma mansoni. FEBS Lett (2007) 581:1742–50. doi:10.1016/j.febslet.2007.03.054
158. Jackson KE, Klonis N, Ferguson DJP, Adisa A, Dogovski C, Tilley L. Food vacuole-associated lipid bodies and heterogeneous lipid environments in the malaria parasite, Plasmodium falciparum. Mol Microbiol (2004) 54:109–22. doi:10.1111/j.1365-2958.2004.04284.x
159. Palacpac NM, Hiramine Y, Mi-ichi F, Torii M, Kita K, Hiramatsu R, et al. Developmental-stage-specific triacylglycerol biosynthesis, degradation and trafficking as lipid bodies in Plasmodium falciparum-infected erythrocytes. J Cell Sci (2004) 117:1469–80. doi:10.1242/jcs.00988
160. Araújo-Santos T, Prates DB, França-Costa J, Luz NF, Andrade BB, Miranda JC, et al. Prostaglandin E2/leukotriene B4 balance induced by Lutzomyia longipalpis saliva favors Leishmania infantum infection. Parasit Vectors (2014) 7:1–8. doi:10.1186/s13071-014-0601-8
161. França-Costa J, Van Weyenbergh J, Boaventura VS, Luz NF, Malta-Santos H, Oliveira MC, et al. Arginase I, polyamine, and prostaglandin E 2 pathways suppress the inflammatory response and contribute to diffuse cutaneous leishmaniasis. J Infect Dis (2015) 211:426–35. doi:10.1093/infdis/jiu455
162. Passero LF, Laurenti MD, Tomokane TY, Corbett CEP, Toyama MH. The effect of phospholipase A2 from Crotalus durissus collilineatus on Leishmania (Leishmania) amazonensis infection. Parasitol Res (2008) 102:1025–33. doi:10.1007/s00436-007-0871-6
163. Lonardoni MVC, Barbieri CL, Russo M, Jancar S. Modulation of Leishmania (L.) amazonensis growth in cultured mouse macrophages by prostaglandins and platelet activating factor. Mediators Inflamm (1994) 3:137–41. doi:10.1155/S0962935194000177
164. Prates DB, Araújo-Santos T, Luz NF, Andrade BB, França-Costa J, Afonso L, et al. Lutzomyia longipalpis saliva drives apoptosis and enhances parasite burden in neutrophils. J Leukoc Biol (2011) 90:575–82. doi:10.1189/jlb.0211105
165. Saha A, Biswas A, Srivastav S, Mukherjee M, Das PK, Ukil A. Prostaglandin E 2 negatively regulates the production of inflammatory cytokines/chemokines and IL-17 in visceral leishmaniasis. J Immunol (2014) 193:2330–9. doi:10.4049/jimmunol.1400399
166. Freire-de-Lima CG, Nascimento DO, Soares MBP, Bozza PT, Castro-Faria-Neto HC, de Mello FG, et al. Uptake of apoptotic cells drives the growth of a pathogenic trypanosome in macrophages. Nature (2000) 403:199–203. doi:10.1038/35003208
167. Almeida PE, Toledo DAM, Rodrigues GSC, D’Avila H. Lipid bodies as sites of prostaglandin E2 synthesis during chagas disease: impact in the parasite escape mechanism. Front Microbiol (2018) 9:499. doi:10.3389/fmicb.2018.00499
168. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-γ: an overview of signals, mechanisms and functions. J Leukoc Biol (2004) 75:163–89. doi:10.1189/jlb.0603252
169. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol (2014) 32:513–45. doi:10.1146/annurev-immunol-032713-120231
170. Pilla-Moffett D, Barber MF, Taylor GA, Coers J. Interferon-inducible GTPases in host resistance, inflammation and disease. J Mol Biol (2016) 428:3495–513. doi:10.1016/j.jmb.2016.04.032
171. Das P, De T, Chakraborti T. Leishmania donovani secretory serine protease alters macrophage inflammatory response via COX-2 mediated PGE-2 production. Indian J Biochem Biophys (2014) 51:542–51.
172. Lecoeur H, Giraud E, Prévost MC, Milon G, Lang T. Reprogramming neutral lipid metabolism in mouse dendritic leucocytes hosting live Leishmania amazonensis amastigotes. PLoS Negl Trop Dis (2013) 7(6):e2276. doi:10.1371/journal.pntd.0002276
173. Perkins DJ, Were T, Davenport GC, Kempaiah P, Hittner JB, Ong’echa JM. Severe malarial anemia: innate immunity and pathogenesis. Int J Biol Sci (2011) 7:1427–42. doi:10.7150/ijbs.7.1427
174. Anyona SB, Kempaiah P, Davenport GC, Vulule JM, Hittner JB, Ong’echa JM, et al. Suppressed circulating bicyclo-PGE2 levels and leukocyte COX-2 transcripts in children co-infected with P. falciparum malaria and HIV-1 or bacteremia. Biochem Biophys Res Commun (2013) 436:585–90. doi:10.1016/j.bbrc.2013.05.089
175. Anyona SB, Kempaiah P, Raballah E, Davenport GC, Were T, Konah SN, et al. Reduced systemic bicyclo-prostaglandin-E2 and cyclooxygenase-2 gene expression are associated with inefficient erythropoiesis and enhanced uptake of monocytic hemozoin in children with severe malarial anemia. Am J Hematol (2012) 87:782–9. doi:10.1002/ajh.23253
176. Matte C, Maion G, Mourad W, Olivier M. Leishmania donovani-induced macrophages cyclooxygenase-2 and prostaglandin E2 synthesis. Parasite Immunol (2001) 23:177–84. doi:10.1046/j.1365-3024.2001.00372.x
177. Arcanjo AF, Nunes MP, Silva-Junior EB, Leandro M, da Rocha JDB, Morrot A, et al. B-1 cells modulate the murine macrophage response to Leishmania major infection. World J Biol Chem (2017) 8:151. doi:10.4331/wjbc.v8.i2.151
178. D’Avila H, Roque NR, Cardoso RM, Castro-Faria-Neto HC, Melo RCN, Bozza PT. Neutrophils recruited to the site of Mycobacterium bovis BCG infection undergo apoptosis and modulate lipid body biogenesis and prostaglandin E2 production by macrophage. Cell Microbiol (2008) 10:2589–604. doi:10.1111/j.1462-5822.2008.01233.x
179. Afonso L, Borges VM, Cruz H, Ribeiro-Gomes FL, DosReis GA, Dutra AN, et al. Interactions with apoptotic but not with necrotic neutrophils increase parasite burden in human macrophages infected with Leishmania amazonensis. J Leukoc Biol (2008) 84:389–96. doi:10.1189/jlb.0108018
180. Peters-Golden M, Canetti C, Mancuso P, Coffey MJ. Leukotrienes: underappreciated mediators of innate immune responses. J Immunol (2005) 174:589–94. doi:10.4049/jimmunol.174.2.589
181. Serezani CH, Perrela JH, Russo M, Peters-Golden M, Jancar S. Leukotrienes are essential for the control of Leishmania amazonensis infection and contribute to strain variation in susceptibility. J Immunol (2006) 177:3201–8. doi:10.4049/jimmunol.177.5.3201
182. Tavares NM, Araújo-Santos T, Afonso L, Nogueira PM, Lopes UG, Soares RP, et al. Understanding the mechanisms controlling Leishmania amazonensis infection in vitro: the role of LTB4 derived from human neutrophils. J Infect Dis (2014) 210:656–66. doi:10.1093/infdis/jiu158
183. Chaves MM, Marques-da-Silva C, Monteiro APT, Canetti C, Coutinho-Silva R. Leukotriene B4 modulates P2X7 receptor-mediated Leishmania amazonensis elimination in murine macrophages. J Immunol (2014) 192:4765–73. doi:10.4049/jimmunol.1301058
184. Paiva LA, Maya-Monteiro CM, Bandeira-Melo C, Silva PMR, El-Cheikh MC, Teodoro AJ, et al. Interplay of cysteinyl leukotrienes and TGF-β in the activation of hepatic stellate cells from Schistosoma mansoni granulomas. Biochim Biophys Acta (2010) 1801:1341–8. doi:10.1016/j.bbalip.2010.08.014
185. Carregaro V, Valenzuela JG, Cunha TM, Verri WA, Grespan R, Matsumura G, et al. Phlebotomine salivas inhibit immune inflammation-induced neutrophil migration via an autocrine DC-derived PGE 2/IL-10 sequential pathway. J Leukoc Biol (2008) 84:104–14. doi:10.1189/jlb.1107797
186. Araújo-Santos T, Prates DB, Andrade BB, Nascimento DO, Clarêncio J, Entringer PF, et al. Lutzomyia longipalpis saliva triggers lipid body formation and prostaglandin E2 production in murine macrophages. PLoS Negl Trop Dis (2010) 4:e873. doi:10.1371/journal.pntd.0000873
187. Russell CD, Schwarze J. The role of pro-resolution lipid mediators in infectious disease. Immunology (2014) 141:166–73. doi:10.1111/imm.12206
188. Katze MG, He Y, Gale M. Viruses and interferon: a fight for supremacy. Nat Rev Immunol (2002) 2:675–87. doi:10.1038/nri888
189. MacMicking JD. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol (2012) 12:367–82. doi:10.1038/nri3210
190. Hinson ER, Cresswell P. The antiviral protein, viperin, localizes to lipid droplets via its N-terminal amphipathic alpha-helix. Proc Natl Acad Sci U S A (2009) 106:20452–7. doi:10.1073/pnas.0911679106
191. Hinson ER, Cresswell P. The N-terminal amphipathic α-helix of viperin mediates localization to the cytosolic face of the endoplasmic reticulum and inhibits protein secretion. J Biol Chem (2009) 284:4705–12. doi:10.1074/jbc.M807261200
192. Fitzgerald KA. The interferon inducible gene: viperin. J Interf Cytokine Res (2011) 31:131–5. doi:10.1089/jir.2010.0127
193. Gubern A, Barceló-Torns M, Barneda D, López JM, Masgrau R, Picatoste F, et al. JNK and ceramide kinase govern the biogenesis of lipid droplets through activation of group IVA phospholipase A2. J Biol Chem (2009) 284:32359–69. doi:10.1074/jbc.M109.061515
194. Vignola MJ, Kashatus DF, Taylor GA, Counter CM, Valdivia RH. cPLA 2 regulates the expression of type I interferons and intracellular immunity to Chlamydia trachomatis. J Biol Chem (2010) 285:21625–35. doi:10.1074/jbc.M110.103010
195. Martens S, Sabel K, Lange R, Uthaiah R, Wolf E, Howard JC. Mechanisms regulating the positioning of mouse p47 resistance GTPases LRG-47 and IIGP1 on cellular membranes: retargeting to plasma membrane induced by phagocytosis. J Immunol (2004) 173:2594–606. doi:10.4049/jimmunol.173.4.2594
196. Hunn JP, Koenen-Waisman S, Papic N, Schroeder N, Pawlowski N, Lange R, et al. Regulatory interactions between IRG resistance GTPases in the cellular response to Toxoplasma gondii. EMBO J (2008) 27:2495–509. doi:10.1038/emboj.2008.176
197. Maric-Biresev J, Hunn JP, Krut O, Helms JB, Martens S, Howard JC. Loss of the interferon-γ-inducible regulatory immunity-related GTPase (IRG), Irgm1, causes activation of effector IRG proteins on lysosomes, damaging lysosomal function and predicting the dramatic susceptibility of Irgm1-deficient mice to infection. BMC Biol (2016) 14:33. doi:10.1186/s12915-016-0255-4
198. Howard JC, Hunn JP, Steinfeldt T. The IRG protein-based resistance mechanism in mice and its relation to virulence in Toxoplasma gondii. Curr Opin Microbiol (2011) 14:414–21. doi:10.1016/j.mib.2011.07.002
199. Hunn JP, Feng CG, Sher A, Howard JC. The immunity-related GTPases in mammals: a fast-evolving cell-autonomous resistance system against intracellular pathogens. Mamm Genome (2011) 22:43–54. doi:10.1007/s00335-010-9293-3
200. Chauhan S, Mandell MA, Deretic V. IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol Cell (2015) 58:507–21. doi:10.1016/j.molcel.2015.03.020
201. Haldar AK, Saka HA, Piro AS, Dunn JD, Henry SC, Taylor GA, et al. IRG and GBP host resistance factors target aberrant, “non-self” vacuoles characterized by the missing of “self” IRGM proteins. PLoS Pathog (2013) 9:e1003414. doi:10.1371/journal.ppat.1003414
202. Kravets E, Degrandi D, Ma Q, Peulen T-O, Klümpers V, Felekyan S, et al. Guanylate binding proteins directly attack Toxoplasma gondii via supramolecular complexes. Elife (2016) 5:1–30. doi:10.7554/eLife.11479
203. Ling YM, Shaw MH, Ayala C, Coppens I, Taylor GA, Ferguson DJ, et al. Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J Exp Med (2006) 203:2063–71. doi:10.1084/jem.20061318
204. Zhao YO, Khaminets A, Hunn JP, Howard JC. Disruption of the Toxoplasma gondii parasitophorous vacuole by IFNγ-inducible immunity-related GTPases (IRG Proteins) triggers necrotic cell death. PLoS Pathog (2009) 5:e1000288. doi:10.1371/journal.ppat.1000288
205. Bougnères L, Helft J, Tiwari S, Vargas P, Chang BHJ, Chan L, et al. A role for lipid bodies in the cross-presentation of phagocytosed antigens by MHC class I in dendritic cells. Immunity (2009) 31:232–44. doi:10.1016/j.immuni.2009.06.022
206. Britzen-Laurent N, Bauer M, Berton V, Fischer N, Syguda A, Reipschläger S, et al. Intracellular trafficking of guanylate-binding proteins is regulated by heterodimerization in a hierarchical manner. PLoS One (2010) 5:e14246. doi:10.1371/journal.pone.0014246
207. Pilla DM, Hagar JA, Haldar AK, Mason AK, Degrandi D, Pfeffer K, et al. Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proc Natl Acad Sci U S A (2014) 111:6046–51. doi:10.1073/pnas.1321700111
208. Meunier E, Dick MS, Dreier RF, Schürmann N, Broz DK, Warming S, et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature (2014) 509:366–70. doi:10.1038/nature13157
209. Ohshima J, Sasai M, Liu J, Yamashita K, Ma JS, Lee Y, et al. RabGDIα is a negative regulator of interferon-γ–inducible GTPase-dependent cell-autonomous immunity to Toxoplasma gondii. Proc Natl Acad Sci U S A (2015) 112:E4581–90. doi:10.1073/pnas.1510031112
210. Haldar AK, Foltz C, Finethy R, Piro AS, Feeley EM, Pilla-Moffett DM, et al. Ubiquitin systems mark pathogen-containing vacuoles as targets for host defense by guanylate binding proteins. Proc Natl Acad Sci U S A (2015) 112:E5628–37. doi:10.1073/pnas.1515966112
211. Ichimura Y, Komatsu M. Selective degradation of p62 by autophagy. Semin Immunopathol (2010) 32:431–6. doi:10.1007/s00281-010-0220-1
212. Martinez-Lopez N, Singh R. Autophagy and lipid droplets in the liver. Annu Rev Nutr (2015) 35:215–37. doi:10.1146/annurev-nutr-071813-105336
213. Dupont N, Chauhan S, Arko-Mensah J, Castillo EF, Masedunskas A, Weigert R, et al. Neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Curr Biol (2014) 24:609–20. doi:10.1016/j.cub.2014.02.008
214. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature (2009) 458:1131–5. doi:10.1038/nature07976
215. Fujimoto T, Ohsaki Y. The proteasomal and autophagic pathways converge on lipid droplets. Autophagy (2006) 2:299–301. doi:10.4161/auto.2904
216. Ohsaki Y, Cheng J, Fujita A, Tokumoto T, Fujimoto T. Cytoplasmic lipid droplets are sites of convergence of proteasomal and autophagic degradation of apolipoprotein B. Mol Biol Cell (2006) 17:2674–83. doi:10.1091/mbc.E05-07-0659
217. Xia F, Li R, Wang C, Yang S, Tian L, Dong H, et al. IRGM1 regulates oxidized LDL uptake by macrophage via actin-dependent receptor internalization during atherosclerosis. Sci Rep (2013) 3:1867. doi:10.1038/srep01867
218. Singh SB, Ornatowski W, Vergne I, Naylor J, Delgado M, Roberts E, et al. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat Cell Biol (2010) 12:1154–65. doi:10.1038/ncb2119
219. Traver MK, Henry SC, Cantillana V, Oliver T, Hunn JP, Howard JC, et al. Immunity-related GTPase M (IRGM) proteins influence the localization of guanylate-binding protein 2 (GBP2) by modulating macroautophagy. J Biol Chem (2011) 286:30471–80. doi:10.1074/jbc.M111.251967
220. Shpilka T, Welter E, Borovsky N, Amar N, Mari M, Reggiori F, et al. Lipid droplets and their component triglycerides and steryl esters regulate autophagosome biogenesis. EMBO J (2015) 34:e201490315. doi:10.15252/embj.201490315
Keywords: lipid droplets, lipid bodies, parasite, intracellular pathogen, eicosanoids, inflammation, immune response
Citation: Vallochi AL, Teixeira L, Oliveira KdS, Maya-Monteiro CM and Bozza PT (2018) Lipid Droplet, a Key Player in Host-Parasite Interactions. Front. Immunol. 9:1022. doi: 10.3389/fimmu.2018.01022
Received: 31 January 2018; Accepted: 24 April 2018;
Published: 23 May 2018
Edited by:
Celio Geraldo Freire-de-Lima, Universidade Federal do Rio de Janeiro, BrazilReviewed by:
Joshua D. Nosanchuk, Albert Einstein College of Medicine, United StatesCarlos Arterio Sorgi, Universidade de São Paulo Ribeirão Preto, Brazil
Copyright: © 2018 Vallochi, Teixeira, Oliveira, Maya-Monteiro and Bozza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adriana Lima Vallochi, dmFsbG9jaGlAaW9jLmZpb2NydXouYnI=;
Patricia T. Bozza, cGJvenphQGlvYy5maW9jcnV6LmJy