 Morad-Remy Muhsin-Sharafaldine
Morad-Remy Muhsin-Sharafaldine Alexander D. McLellan
Alexander D. McLellan- Department of Microbiology and Immunology, University of Otago, Dunedin, New Zealand
Tumor cells release lipid particles known as extracellular vesicles (EV) that contribute to cancer metastasis, to the immune response, and to thrombosis. When tumors are exposed to radiation or chemotherapy, apoptotic vesicles (ApoVs) are released in abundance as the plasma membrane delaminates from the cytoskeleton. Recent studies have suggested that ApoVs are distinct from the EVs released from living cells, such as exosomes or microvesicles. Depending on their treatment conditions, tumor-released ApoV have been suggested to either enhance or suppress anti-cancer immunity. In addition, tumor-derived ApoV possess procoagulant activity that could increase the thrombotic state in cancer patients undergoing chemotherapy or radiotherapy. Since ApoVs are one of the least appreciated type of EVs, we focus in this review on the distinctive characterization of tumor ApoVs and their proposed mechanistic effects on cancer immunity, coagulation, and metastasis.
Introduction
The term extracellular vesicle (EV) describes membrane particles released from eukaryote cells, as well as prokaryote microorganisms (1). It is now evident that EV plays important roles in multiple biological systems involved in the control of homeostasis of the organism. For example, intercellular signaling is mediated by EV in processes, such as bone calcification, immune tolerance and activation, neuron-glia communication, wound repair, and hemostasis (2–7). Furthermore, EVs have been implicated in pathological conditions, such as viral and prion transfer, cardiovascular disease, thrombosis, autoimmune diseases (e.g., rheumatoid arthritis and multiple sclerosis), sickle cell anemia, and cancer (8–14).
Extracellular vesicle can be isolated via differential and/or density gradient centrifugation based on their relative size and density. Depending on their parental cell type and their cellular site of origin, EVs differ in terms of size, composition, density, and other biochemical and structural properties (15, 16). Exosomes, one of the smallest EV, are released from a large spectrum of living cells and range from 40–100 nm in diameter [isolated at sedimentation speeds of ≥100,000 × g; Ref. (17)]. While the differentiation or activation state of primary cells is critical for exosome release (18), Wolfers et al. have shown that murine and human tumor cell lines constitutively release exosomes (19). After their discovery by electron microscopy in 1981 (20), numerous studies have shown that exosomes may function as intercellular messengers in a diverse range of roles controlling cellular physiology and pathology (5, 19, 21–24).
Living cells also secrete larger, membrane-derived EV known as microvesicles (MV). These were first described by Chargaff and West in 1946 and were later characterized as a predominant procoagulant product of degranulating platelets (25, 26). MVs range in size (0.1–2 µm in diameter) and have been shown to be constitutively released by tumor cells potentially carrying oncogenes (27). In addition, “migratory benign cells-derived EV” structures, termed migrasomes (≤3 μm), that harbor internal EV, have also recently been described (28). Tumor-derived EV (100–500 nm) that transport epidermal growth factor receptor variant III, overexpressed in the parental human U373 astrocytoma tumor cell line have been referred to as “oncosomes” (29). Other studies that have used this term and verified the tumor origin of “oncosomes” due to other oncogenic cargo (30–33). However, it is still not clear from the original (29), or clarified definitions (34), if oncosomes contain “oncogenes” (i.e., transforming nucleic acids), “oncogenic receptors” (oncogene products; i.e., epidermal growth factor receptor variant III polypeptide), or whether this terminology merely reflect their functional transforming ability (transfer of oncogenic activity). Since the term oncosomes originates from a single cell line studied (29), the size range, cargo, or morphological features cannot yet be extrapolated to other tumor cells. Moreover, oncosomes have yet to be included in the ISEV guidelines (35, 36).
During apoptosis, cellular contents are packaged into apoptotic blebs (0.03–5 µm) for clearance with minimal perturbation/inflammation to the surrounding tissues (15, 37). The term “apoptotic bodies” usually refers to the larger (1–5 µm) bleb fraction (38, 39) that may contain a proportion of nuclear content and are released when the plasma membrane delaminates from the cytoskeleton (37). Due to their variable size range, apoptotic blebs are isolated at different sedimentation speeds [1,000–110,000 × g; Ref. (15, 40–43)]. An obvious problem with definition occurs when studies isolate smaller apoptotic blebs at high speeds (≥100,000 × g); resulting in likely contamination with exosomes due to the similar sedimentation forces used in the isolation of these two EV sub-types. However, contaminating exosomes will have distinguishable markers (discussed later) that could be utilized to enhance apoptotic vesicles (ApoVs) purity. In addition, there is no consensus regarding the nomenclature for the smaller fraction of apoptotic blebs (0.03–1 µm). For example, the terms apoptotic microparticles, small ApoVs, and even apoptotic bodies have been used to define the smaller apoptotic blebs (15, 40, 44, 45). For this reason, the term ApoVs will be used in this review to describe lipid encapsulated EV released from dying (apoptotic) cells.
Extracellular vesicles are becoming increasingly studied due to their release by cancer cells and their reported influence on the immune system, metastasis, angiogenesis, and coagulation (46–49). Although tumor-derived ApoVs are released in relative abundance following chemotherapeutic treatment, as compared to exosomes and MV (12, 47), limited research has been directed toward ApoV. Here, we will focus on the ApoV and their functional implications in cancer, the immune system, and coagulation.
Cell Death: A General Overview
At present, at least six cellular processes leading to cellular death have been described: mitotic catastrophe, senescence, necrosis, necroptosis, autophagy, and apoptosis (50, 51). However, it is unclear whether the six cellular death mechanisms are strictly independent, or if they all eventually overlap to some degree.
During eukaryotic cell division (mitosis), DNA-damaging agents cause cells to lose or gain a single chromosome (an aneuploid state) that, if left unchecked, could lead to severe genomic instability (52, 53). This may result in irreversible damage and death of the aberrant dividing cell in a manner known as mitotic catastrophe (51, 53). Prior to the discovery of mitotic catastrophe, Hayflick et al. showed that normal cells eventually cease to divide in vitro despite the availability of favorable conditions for cell growth (54). This inflammatory death mechanism, defined as senescence, is now known to be triggered by several signals such as DNA damage or shortened/dysfunctional telomeres (38, 50, 51, 55, 56). Necrosis is the uncontrolled breakdown of the cell membrane and consequent release of intracellular contents and proinflammatory molecules into the extracellular matrix (57, 58). Several pathological conditions, such as infection, ischemia, or inflammation can cause necrosis and that is generally characterized by cellular swelling and organelle degradation (57, 59). Necrosis can be triggered in a controlled manner, a process known as necroptosis, and driven by receptor-interacting protein kinase 1, 3, and pseudokinase mixed lineage kinase domain-like (60, 61). Autophagy is triggered when redundant or unwanted proteins are excessively targeted for degradation by the cell’s proteolytic mechanisms (50). One of the main mediators of autophagy is ubiquitin, often leading to degradation within proteasomes (50, 62, 63). Apart from apoptosis, autophagy and necrosis are the only other types of cell death that are characterized by membrane blebbing (50, 58, 64).
Apoptosis is a highly controlled process and is activated via two main pathways: the extrinsic (or receptor) pathway is characterized by the binding of a ligand to a death receptor of a cell (65). Activation of these death receptors by their ligands may lead to the assembly of the Fas-associated death domain and caspase-8 (66). Apoptosis is orchestrated via the activation of a (usually inactive) cytoplasmic family of proteins known as caspases (67–69). The activation of one may lead to the activation of another and thus initiate apoptosis in a cascade fashion. Hence, within the extrinsic pathway, recruited, activated caspase-8 cleaves caspase-3 which will cleave other caspases, eventually leading to apoptosis (69, 70). The other pathway, known as the intrinsic pathway, also converges at caspase-3 (67). However, the intrinsic (or mitochondrial) pathway is usually triggered via stress signals that may lead the mitochondrion to the leakage of proapoptotic factors, including cytochrome c, into the cytoplasm (67, 69, 70). This results in the formation/activation of several protein complexes including caspase-9 which then cleaves caspase-3 leading to downstream disassembly of cellular components (70). One of the main features of apoptosis is the formation and release of membrane blebs or ApoV (71). One advantage of the apoptotic process is that proteins and nucleic acids, that would otherwise trigger an immune response, are packaged within these apoptotic blebs for rapid clearance by the immune system (72, 73). On the other hand, materials that act as autoantigens may also be packaged into apoptotic blebs (45).
General Mechanisms of EV Formation
The mechanism of EV release is tightly regulated and differs between exosomes, MV, and ApoV (Figure 1). The exosomal machinery begins with the cell membrane invaginating inwards toward the intracellular matrix by endocytosis, forming an endosome (74). This early endosome is formed by the aid of proteins such as Ras-related in the brain GTPases and soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins (75). In later stages, further invagination of the endosome leads to the formation of intraluminal vesicles (ILV) and this late endosome is now referred to as a multivesicular body (MVB) (38, 76). Although their specific role remains unclear, tetraspanins, the endosomal sorting complexes required for transport (ESCRT) complexes, and ALG-2-interacting protein X, are involved in ILV formation (15). The excision of ILV into MVB requires ESCRT-III proteins, such as vacuolar protein sorting-associated protein 20, 24, and Snf7 (77). MVB can then either be targeted for degradation/recycling or may fuse with the cell membrane releasing ILV known as exosomes (15, 38, 74). Alternatively, lipid-metabolizing enzymes, such as neutral sphingomyelinase and phospholipase D2, can drive the formation of MVB and ILV in an ESCRT-independent manner (78, 79). However, it remains unknown how ESCRT and lipid-metabolizing enzymes cooperate to induce exosome formation (78).
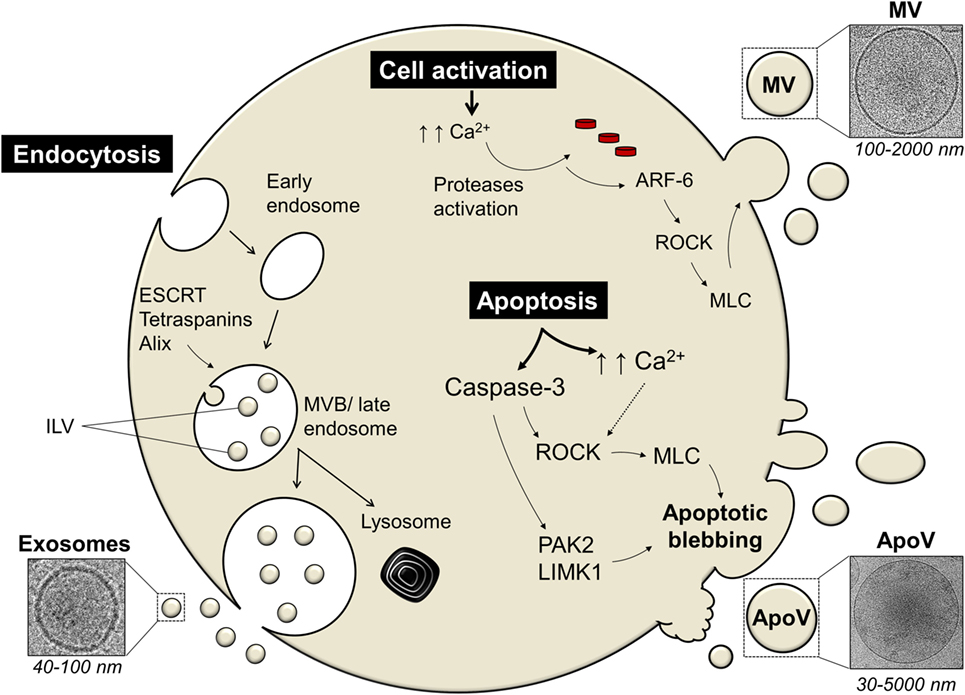
Figure 1. The mechanism of extracellular vesicles (EVs) release differs among different types of EV. Exosomes are secreted via the endosomal pathway, starting with the inward invagination of the cell membrane forming an early endosome. In later stages, further invagination of the endosome leads to the formation of intraluminal vesicles (ILV) and this late endosome is now referred to as a multivesicular body [MVB; Ref. (38, 76)]. The MVB can either be targeted for cellular destruction/recycling by lysosomes or it may fuse with the cell membrane thus releasing ILV as exosomes. Proteins involved in exosomal machinery include the endosomal sorting complexes required for transport (ESCRT), tetraspanins (such as CD9), ALG-2-interacting protein X (Alix), and tumor susceptibility gene 101 [TSG101; Ref. (74)]. Microvesicles (MV) and apoptotic vesicles (ApoV) are secreted via the direct outward budding of the cell membrane. Cellular activation or apoptosis results in an increased influx of calcium ions (Ca2+) which triggers proteases, such as calpain or gelsolin (80). These activated proteases lead to cytoskeletal disruption. ADP-ribosylation factor-6 (ARF-6) initiates a signaling cascade eventually activating the Rho-associated protein kinase (ROCK) signaling pathway which in turn activates myosin light-chain (MLC) kinases that activates and phosphorylates MLC that would initiate blebbing eventually leading to ApoV/MV release (81, 82). Apoptosis initiation also activates apoptotic enzymes known as caspases. Caspase-3 can cleave ROCK transforming it into a constitutive truncated form which then enhances the phosphorylation and activation of MLC that leads to ApoV shedding (83). Caspase-3-mediated blebbing can also act on other blebbing-mediated proteins, such as LIM domain kinase 1 (LIMK1) or p21-activated kinase (PAK2); reviewed in Ref. (71).
Cells also release EV through the direct outward membrane budding and depending on the state of the cell, the EV is termed MV or ApoV. Initially, the shedding mechanism of MV or ApoV is thought to begin with the influx of calcium into activated or dying cells, respectively, resulting in the activation of calcium-dependent proteases, such as calpain and gelsolin (38, 80, 84, 85). This leads to the disruption of the membrane cytoskeleton, exposure of phosphatidylserine (PS), and initial formation of membrane protrusions (81, 85). ADP-ribosylation factor-6 initiates a signaling cascade eventually activating the Rho-associated protein kinase (ROCK) signaling pathway which in turn activates myosin light-chain (MLC) kinases that activates and phosphorylates MLC (38, 82, 86). The cell membrane then begins to bleb due to increased hydrostatic pressure following MLC-driven actomyosin contractions (87). Eventually ApoV or MV is released (81, 82). The mechanism of MV/ApoV scission remains unclear. However, it has been shown that vacuolar protein sorting four participates in the scission of T cell-derived MV (88). No incision proteins have yet been identified in ApoV release. One could speculate that the budding off of ApoV may resemble the mechanism of ILV excision. It has been hypothesized; however, that ApoV may simply break-off due to shear force under flow conditions and/or interaction of a recipient phagocyte “pinching” off ApoV (71).
Despite the mechanistic similarities between ApoV and MV, the apoptotic cell passes through a three-step apoptotic disassembly process to finally release ApoV [reviewed in Ref. (71)]. One major pathway of apoptosis is orchestrated via the activation of cytoplasmic caspases (67–69). Sequential activation of caspases leads to dismantling and repackaging of cellular and nuclear content caspase-3 (69, 70). The blebbing of the apoptotic cell marks the first step of apoptotic disassembly and is linked to caspase-3 (83, 89, 90). There is evidence that caspase-3 can cleave ROCK-1 transforming it into a constitutive truncated form which then enhances the phosphorylation and activation of MLC (89–91). Caspase-3 can also act on other blebbing-mediated proteins, such as LIM domain kinase 1 or p21-activated kinase (92, 93). The blebbing cell then moves to stage two whereby membrane protrusions form via microtubule spikes and/or long string-like constructs known as apoptopodia (94, 95). Finally, the apoptotic cell fragments, and the formed ApoV, detached from the membrane protrusions and are released.
Molecular Profile of EV
The composition of EV largely depends on the type and differentiation state of the parental cell. Currently, differential ultracentrifugation is the most common technique to isolate EV. Exosomes are isolated using ≥100,000 × g along with pore filtration to remove larger EV fractions (21, 23). However, it has proved difficult to isolate the larger MV or ApoV, and to obtain a pure EV preparation as ApoV or MV purified from identical centrifugation speeds may cross-contaminate due to spontaneous cell death or MV release (47). Despite these limitations, various proteins, DNA, RNA, and lipid profiles have been identified to assist in the phenotyping of MV and ApoV (23, 47, 96–98). Current methodology for protein detection in EV includes western blotting, flow cytometry, and mass spectrometry (24, 47). It is now clear that many of the proteins enriched in EV are involved in EV formation or trafficking (3). Despite the different types of EV, there are well-documented markers that are commonly shared among EV fractions (24). Cluster of differentiation 147 (CD147), for example, is consistently observed to be enriched in tumor-derived EV (47, 99, 100). Acting as an Extracellular Matrix Metalloproteinase Inducer as a main role, CD147 is thus also referred to as “EMMPRIN.” Once matrix metalloproteinases (such as MMP1, MMP2, and MMP11) are induced by CD147, the enzymes help to break the extracellular matrix thus aiding in the proliferation of tumor cells (101–103). With a family of at least 30 proteins, tetraspanins levels are also elevated in EV (104–106). However, recent studies have shown the expression level of the tetraspanin CD9 is lower in ApoV compared to exosomes or MV (47, 96, 99, 107). Tetraspanins are glycoproteins suspected to be involved in cell motility, adhesion, and proliferation, and are known to complex with integrins (108, 109). It is, therefore, not surprising that adhesion molecules, such as integrins are also detected in EV (23, 47, 96, 99). Despite all the efforts, there has not been a stand-alone protein marker for ApoV that can distinguish them from other EV types (47, 96).
Interestingly, the DNA-associated proteins, histones, were assumed to be exclusive ApoV markers (46). Despite this, a proteomic study of dendritic cells (DC)-derived exosomes by Théry et al. detected histones in exosomes (15). They hypothesized that their presence may be due to the spontaneous DC apoptosis, contaminating the DC-derived exosome preparations with disintegrating nuclear material. However, it is now appreciated that histones localize within the cytoplasm, as well as the nucleus (110), and histones within exosomes and ApoV have been widely documented (45, 47, 111). Since EV contain nucleic acids, including RNA, it is likely that histones in EV act as chaperones for nucleic acids (46, 112). It has been proposed that the presence of RNA in exosomes may be due to the fact that MVB contain RNA-induced silencing complexes (113–115) where there is a direct interaction between histones and RNA (113). A study by Müller et al. suggests that the histone H3 modification is necessary for exosome release (116). Furthermore, glioma-derived EVs have been known to carry mRNA, micro RNA, and proteins that contribute to tumor growth (112, 117). The uptake of glioma-derived MV, containing oncogenic epidermal growth factor receptor, by endothelial cells have been known to greatly alter the nature of the endothelial cells in a manner that elevates tumor angiogenesis (117). However, a recent study revealed that there is a clear difference in the RNA profile between ApoV, MV, and exosomes, highlighting that ribosomal RNA and smaller RNA is highest in ApoV (118). Previous studies have identified that the RNA content in EV is reflective of the RNA content of the cell of origin (115, 119). Since DNA fragmentation is a feature of apoptosis, it was no surprise that DNA fragments have been detected in ApoV (120, 121). These DNA fragments can be transferred to recipient cells and reused when ApoVs are engulfed by phagocytes (121).
The exposure of PS on the outer leaflet of EV membranes is a common feature among EV (7, 21, 122). PS is a membrane aminophospholipid that is actively kept within the intracellular level (inner leaflet) of a living cell by the (predicted) ATP-dependent membrane lipid transporters known as flippases (123–125). Simultaneously, the other group of (predicted) lipid transporters, floppases, transports choline- and amino-phospholipids toward the outer membrane leaflet (126). The rate of floppase activity is usually 10 times slower compared to flippase (127). However, during apoptosis or cell activation, flippase is believed to be inhibited and different lipid transporters, scramblases, are activated (128–130). Unlike the other two lipid transporters, scramblases are calcium-dependent and ATP-independent (123, 131). The activity of scramblase transporters moves all major classes of phospholipids back and forth, thereby destroying the phospholipid asymmetry orchestrated by flippases and floppases (128). The presence of PS at the extracellular leaflet by scramblases is a key marker of apoptosis and is particularly enriched in ApoV (47, 96, 123).
Since EV originate from the cell membrane, they are expected to carry various cellular surface glycoproteins and glycolipids [commonly known as glycans; Ref. (132)]. Most studies have focused on exosomes for glycan profile, and demonstrate a complex and varying glycan content depending on the cell of origin (133–137). The use of plant-derived carbohydrate-binding proteins (lectins) and mass spectrometry has been an integral part of glycan profiling (136–139). Glycomic analysis remains challenging; however, due to the lack of specific glycan ligands and the fact that a large number of low molecular weight carbohydrate molecules are the building blocks of various molecular structures (140). Thus, the glycomic profiles of EV remain refractory to analysis and, therefore, a major hurdle to laboratories studying the molecular composition of EV. Nevertheless, studies have shown that the glycans detected in EV participate in protein trafficking, attachment, and EV uptake by recipient cells (134, 136, 141–144). The glycome of mammalian cells-derived EV shows consistency in terms of lectin interactions suggesting the presence of N-acetylglucosamine as an integral component of most EV glycans (140).
The surface of EV is negatively charged partially due to the heavy sialylation of surface glycans (47, 49, 138, 145–147). Increase in sialylation is also a common feature of tumor cells along with other proteins such as the superfamily tetraspanins (148, 149). It remains to be determined if this anionic-rich surface due to sialylation dictates the function of EV. Nevertheless, EVs have a sialic acid-rich surface and EV bind to the sialic-acid binding lectin CD169 (47, 49, 138). Interestingly, CD169 has a low (mM) affinity to sialic acids and thus only heavily sialylated structures (such as tumor-derived EV) are able to bind avidly to CD169+ macrophages (150). Moreover, CD169 binds to α2,6 and α2,3 sialic acids of EV but prefers the latter [discussed later in Ref. (47, 49)].
ApoV and the Immune System
Tumor antigens can be captured by antigen-presenting cells (APC) via direct cell-to-cell contact with living or apoptotic cells (151), heat-shock-associated and soluble proteins, or tumor-EV (19, 152–155). Tumor antigens captured by APC activate CD4 helper and cytotoxic lymphocyte-driven immune responses for tumor regression (156–159). Previous reports indicate that EVs display major histocompatibility complex 1 (MHC-I) and MHC-II on their surface and theoretically should be capable of antigen-presenting function (4, 81, 160). However, cross-presentation of apoptotic cell and EV-associated antigens are more often observed in vivo, rather than direct antigen presentation by the vesicles themselves (19, 161–163). Dendritic cells (DCs) appear to be the major APC subset able to efficiently present antigens derived from apoptotic cells to stimulate both MHC-II and MHC-I-restricted CD4 and CD8 T cell responses (162). Exploiting the ability of DC-derived exosomes to eradicate established tumors due to the expression of MHC-I and -II on the surface of the exosomes (164) can be useful for exosome-based vaccine therapy. Heat-shock proteins have been detected in exosomes (23, 99), and have been reported by Srivastava’s group to participate in immunogenic action via their interaction with APC (165).
Paradoxically, studies regarding apoptotic cell-associated antigens have either identified them as immunosuppressive (166–168) or immunostimulatory (169–171), depending on the experimental setting. Recent studies suggest that tumor-derived ApoV can downregulate the immunostimulatory effect of antigen-specific DC in vivo (47, 167). There is evidence that the immunosuppressive effect of apoptotic cells and ApoV is caused by the transforming growth factor β1 [TGF-β1; Ref. (167, 172, 173)]. PS participates in the immunosuppression by ApoV through the induction of TGF-β from tissue-resident macrophages (174). Interestingly, the same study showed that TGF-β was not released when apoptotic cells failed to express PS (174). A recent study also highlights that the culture content of chemotherapy-treated tumor cells (floating dead cells, supernatant, and potentially ApoV) promotes primary tumor growth (175). The study also showed that this tumorigenic effect was caused by macrophages releasing proinflammatory cytokines known to promote tumor growth, and was PS-dependent (175).
In contrast, exposure of DC to murine myeloid cell-derived ApoV resulted in DC maturation and the secretion of proinflammatory cytokines (176). Leukemic-derived ApoV can elicit DC-driven CD8 T cell activation (177) and the immunization of antigen-pulsed tumor-derived ApoV alone elicits a significant CD8-mediated and anti-cancer immune response in mice (47). In addition, compared to MV or exosomes, tumor-derived ApoV afforded the highest anti-tumor protection against a specific antigen, via unknown mechanisms (99). Intriguingly, ApoV elicited the highest protection despite containing the lowest level of tumor antigen (ovalbumin), as compared to MV and exosomes (99). Despite its documented role in immunosuppression (174, 175), PS may also act as an immune stimulant and, therefore, it is possible that PS enrichment on the surface of ApoV could be responsible for their superior immunogenic activity. Hoffman et al. identified that apoptotic cell clearance is enhanced by PS-receptor mediated micropinocytosis [a regulated form of endocytosis for solute molecules and antigens; Ref. (178, 179)]. The pathway is highly active among APC such as macrophages and DC (179). Moreover, it was shown that blocking PS on MV (by annexin V) disables their uptake by target cells (29).
For apoptotic cells and their ApoV to be efficiently cleared by phagocytes, their exposed PS functions as an “eat-me” signal to phagocytes (71, 178). First, PS binds to annexin V which in turn is recognized by phagocytes (123, 180). However, PS is also a known ligand for other receptors, such as β2-glycoprotein I (181), Mer receptor tyrosine kinase (182), lectin-like oxidized low density lipoprotein-receptor 1 (183), and PS-receptor (184), all of which are known to promote apoptotic cell clearance (182, 184–186). For example, PS-receptor-deficient mice died due to accumulation of uncleared apoptotic cells in lung alveoli (184). A sufficient threshold of PS exposure is necessary for phagocytic clearance. Borisenko et al. have measured phagocytosis of live, apoptotic, and live with the inclusion of exogenous PS (187). Their study concluded that phagocytosis is directly proportional to PS levels. Thus, PS exposure is a critical factor for the clearance of apoptotic materials. Although PS and its receptors are critical for phagocytic recognition, the Albert group have demonstrated that integrin subunits αVβ3 and αVβ5 on macrophages and DCs, respectively, participate in phagocytic clearance of apoptotic cells, but these studies did not investigate a possible involvement of PS in the phagocytic process (162, 188). Furthermore, oxidation levels on apoptotic cells and ApoV create a binding site for thrombospondin and the complement protein C3b, which are in turn recognized by phagocytes (38, 189, 190). CD44-deficiency has been shown to impair apoptotic cell clearance in the lungs (191). This coincides with our study that showed that ApoV express higher CD44 than MV and exosomes (99). The fact that apoptotic cells and their ApoVs are efficiently recognized by the immune system opens a potential use of ApoV for vaccination against cancer. Indeed, PS exposure is seen in all EV (38, 46, 99, 192), and PS participates in the uptake of exogenous antigens (29, 193). However, the slight increase of PS levels on ApoV (96, 99), above that observed for MV and exosomes could still be the threshold necessary to induce a superior immune response. There have been several clinical trials for cancer treatments using exosome-borne tumor antigens, but with poor outcome (194–197). Nevertheless, since immune cells efficiently recognize antigens on ApoV (47, 170, 177, 198), ApoVs remain of interest for cancer immunotherapy.
ApoV can act as immunostimulatory or immunosuppressive. This greatly highlights our lack of understanding of the complexity of ApoV interactions with the immune system. Adding to the complexity of our understanding, in vitro-generated ApoV is not immediately engulfed, as would be expected to happen in vivo. Therefore, the results of in vitro studies may not always reflect the true in vivo situation. Unfortunately, there are limited studies regarding tumor-derived ApoV and their implications for cancer and the immune system. Because tumor-derived ApoVs are strongly thrombotic (see below), this makes it difficult to conduct immune activation studies utilizing systemic application of ApoV regarding of tumor-derived ApoV (48, 99).
Could CD169 be the Gate for EV-Driven Lymph Node Metastasis?
One of the most important events of tumor metastasis is the migration of tumor cells from the primary site to the draining lymph node [LN; Ref. (199)]. Invading tumor cells eventually spread to other LNs in a sequential fashion starting and ending from the closest (draining) to most distal LN, respectively (200). In fact, there is convincing evidence that draining LNs are the best prognostic estimators for the status of the entire lymphatic nodal system (201). Within the subcapsular sinus (SCS) of LN, macrophages are the first to be exposed to antigens and have a role in presenting the captured antigens to APC, including B cells (202). Hood et al. have demonstrated that B16F10-derived exosomes accumulate in and prepare the draining lymph node for tumor invasion (203). This mechanism is often termed “seed and soil” in metastasis, where tumor-derived EVs are regarded as “seeds” preparing a particular site (the soil) for tumor cell invasion (204, 205). The studies suggest that tumor-derived EVs facilitate invasion by enhancing angiogenesis and immunosuppression in situ (22, 203, 206). Interestingly, a macrophage-restricted receptor known as sialoadhesin (CD169; Siglec-1) is abundantly expressed on the surface of macrophages within the SCS of LN, marginal zones of the spleen, and liver [Kupffer cells; Ref. (49)]. CD169 is a member of the sialic-acid binding Ig-like lectin family of proteins; this enriched level of CD169 expression allows these CD169+ macrophages to bind to glycoproteins bearing terminal sialic acids (207). Depending on the experimental setting, CD169+ macrophages mediate a tolerogenic or immunogenic response to self-antigens, infection, and tumor models (208–212).
Several studies that deplete the entire CD169+ macrophage population (using diphtheria toxin or clodronate liposomes) indicate that the function of CD169+ macrophages is immunogenic (213–216). In cancer, the Tanaka group’s CD169+ macrophage depletion model showed that the cells have a critical role in the anti-cancer effect (212). Interestingly, they show that upon immunization with dead tumor cells, CD169+ macrophages cross-present tumor antigens to CD8+ T cells thereby mediating a cytotoxic-mediated anti-cancer immune response. Consistent with these findings, Pucci et al. showed that tumor spread is significantly reduced when tumor-derived EV are captured by CD169 (209). The results showed that CD169 poses a physical barrier to block tumor-derived EV interactions with LN B cells preventing pro-metastatic humoral immunity (209).
The function of the high surface sialylated state of EV remains unclear, however, studies have identified that CD169+ macrophages exclusively capture EV in a CD169-sialic acid-dependent manner (47, 49). EV-immunized CD169-deficient mice display a significant elevated immunogenicity, suggesting that the function of the CD169 receptor itself may be immunoinhibitory [Figure 2; Ref. (47, 49)]. However, our recent study suggested that progression of primary tumor growth and LN metastasis was not significantly associated with CD169 expression in mice (217). Despite this, the direct implication of CD169 capture of EV (the “seed”) and CD169 (the “soil” receptor) in tumor metastasis has not been extensively investigated. Since cancer patients have an increased level of circulating tumor-derived EV bearing pro-metastatic factors highlights the urgency to further explore the role of the tumor EV receptor CD169 in cancer progression.
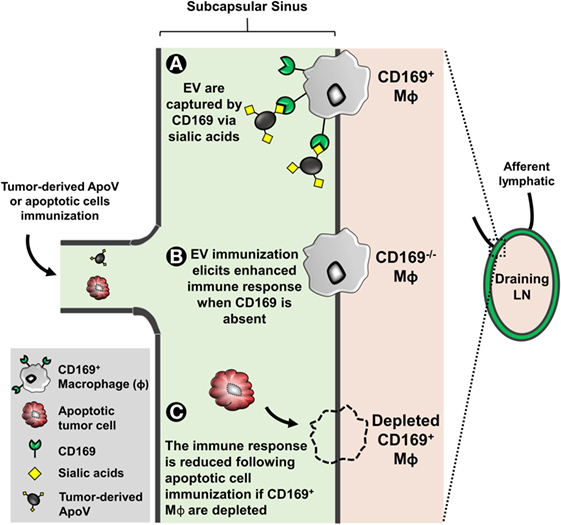
Figure 2. The subcapsular sinus of lymph nodes (LN) is rich in macrophages expressing the surface molecule CD169. EVs are enriched in surface sialylated ligands. (A) CD169+ macrophages can bind to these EV in a sialic acid-dependent manner. CD169 binds to α2,6 and α2,3 sialic acids but prefers the latter (47, 49). (B) EL4 (thymoma)-derived apoptotic vesicle (ApoV) immunization, CD169−/− mice elicit a significant enhanced cytotoxic response (47). The invasion of tumor cells from the primary site to the draining LN is unaffected by CD169 (217). However, the direct implication of EV and tumor metastasis with respect to CD169 remains unknown. (C) When depleted, CD169+ macrophages have a critical role in the anti-cancer effect (212). CD169+ macrophages cross-present tumor antigens from dead tumor cells to CD8+ T cells thereby mediating a cytotoxic-mediated anti-cancer immune response.
Tumor-Derived ApoV and Coagulation
Thrombosis is a pathophysiological condition characterized by localized clotting of the blood within a blood vessel leading to a blockage of blood flow (218, 219). In venous thromboembolism (VTE), the wall of the endothelium remains intact but may transform from an anticoagulant to a procoagulant surface (218, 219). Cancer patients have a fourfold increased risk of developing VTE (220, 221). Strikingly, this risk is increased to more than sixfold when the patients are receiving chemotherapy (221, 222). The risk of VTE depends on the cancer type and stage as well as the type of anti-cancer drug administered, which may alter the hemostatic state in patients (223, 224). Tamoxifen, gemcitabine, and platinum-based compounds, for example, are known to lower the levels of circulating anticoagulants (225–228). In contrast, thalidomide treatment for leukemia does not increase the risk of VTE unless combined with other drugs (229, 230). In general, solid tumors pose a greater risk of VTE and worsened prognosis as compared to lymphomas (231). It is now evident that the leading cause of death in cancer patients receiving chemotherapy is VTE (223, 224).
The shedding of procoagulant EV from human cancer cells was first reported in 1981 (232). In the later decades, tissue factor (TF; CD142) and PS exposure were identified as the main procoagulant components of EV (233). TF expression in tumor cells is linked to the mutations in p53 and phosphatase and tensin homolog PTEN (234), resulting in dysregulation of TF expression which may be upregulated 105-fold compared to non-malignant counterparts (235). TF is an integral membrane protein with a MW of approximately 45 kDa. TF is not only present on the surface of most non-hematopoietic tumors but is also present in EVs released from these tumor cells (47, 48, 99, 235–238). Although the majority of procoagulant TF is vesicle associated, an alternatively spliced soluble form of TF acts independently of FVII to stimulate angiogenesis (239, 240). Moreover, the cytoplasmic domain of TF is involved in signaling events that promote tumor metastasis, further demonstrating that TF displays pro-metastatic function independent of FVII (241).
The association of tumor-derived EV and thrombotic risk is now well-appreciated (193, 233, 242–244). Cancer patients have a significantly higher number of circulating EV compared to healthy controls (243–245). For example, pancreatic cancer patients undergoing chemotherapy are known to possess elevated levels of TF-bearing EV, thus increasing the risk of VTE (246). This phenomenon was observed by Zwicker et al. when they concluded that TF-bearing EVs were associated with VTE in cancer patients (244). Since VTE risk is increased by chemotherapy, this implicates tumor-derived ApoV released from dying tumor cells (Figure 3). Although the levels of detected EV in chemotherapy patients are increased, it remains difficult to determine their identity as either tumor, leukocyte, platelet, or endothelial-derived particles. In addition, there is a vast overlap between tumor-derived exosomes, MV, and ApoV with respect to their size, lipid compositions, surface markers, morphology, and functional behavior (21, 96, 99). In vitro assays indicate that tumor-derived ApoV are significantly procoagulant in a TF/PS-dependent manner (48, 99, 247). The predisposition to VTE observed in cancer patients may be due to the close association of tumor with the extensive networks of vasculature, allowing the direct interaction of tumor-derived EV with blood-borne coagulation factors. However, the depth of the contribution of local release of tumor EV to the systemic hypercoagulable state observed in cancer patients remains to be elucidated (220, 222, 248, 249).
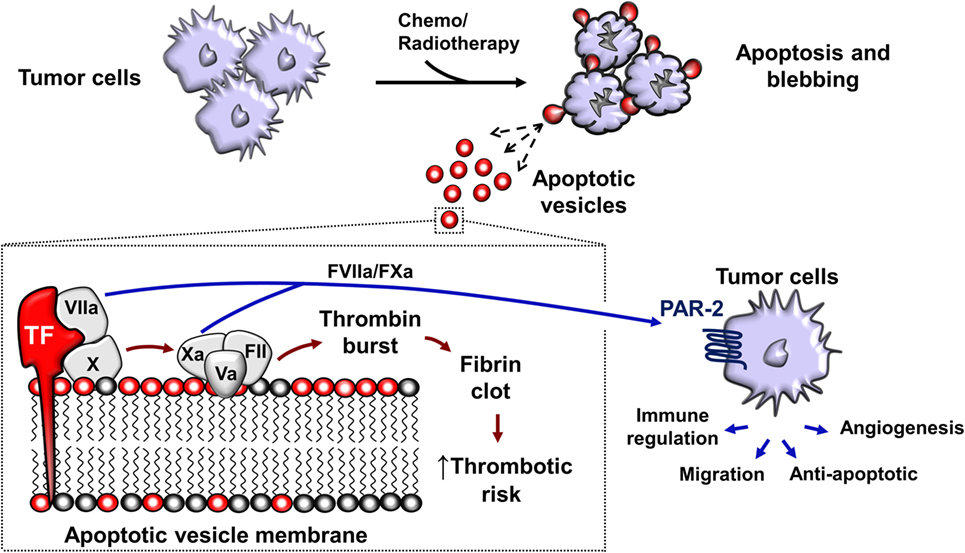
Figure 3. Chemotherapy or radiotherapy-exposed tumor cells may initiate apoptosis and trigger the release apoptotic vesicles (ApoV). Tumor-derived ApoVs express tissue factor (TF) and harbor an anionic-rich surface due to phosphatidylserine (PS) exposure. TF/PS procoagulant complex binds with activated factor VII (FVIIa) and activates the extrinsic coagulation cascade that eventually leads to a fibrin clot. In vitro, tumor-derived ApoV are significantly more procoagulant than MV, exosomes, or their cell of origin (47, 48, 99). FVIIa and FXa also cleaves protease-activated receptor-2 (PAR-2) present on the surface of tumor cells (250–252). This results in signal transduction events important in angiogenesis that enhances tumor blood supply and tumor cell growth (234). ApoVs are, therefore, a potential source of increasing the risk of thrombosis and metastasis in cancer patients undergoing chemotherapy.
The presence of TF on ApoV may contribute to metastasis, since TF-mediated coagulation aids cancer progression (241, 253). In 1995, Bromberg et al. were the first to demonstrate a coagulation-independent role for metastasis by TF through mutation of a TF region required for the initiation of coagulation (241). Interestingly, despite the dramatically lower ability of the extracellular mutant to initiate coagulation, metastasis still occurred. This coagulation-independent effect was likely due to the additional function of TF in the activation of the protease-activated receptor-2 (PAR-2), present on the surface of tumor cells (250–252). TF binds to and activates factor VII, activated factor VII (FVIIa) cleaves PAR-2, resulting in signal transduction events important in angiogenesis that enhances tumor blood supply and tumor cell growth (234). Later studies suggested that the formation of the TF:FVIIa complex was necessary to induce metastasis via the inhibition of apoptosis, promoting cell adhesion, and angiogenesis (254–256). TF expression on tumor cells has been widely implicated in triggering thrombotic events in cancer patients (236). With the possibility that metastatic tumor cells may upregulate TF up to ~1,000-fold compared to non-metastatic cells (235), TF can be considered as a potent pro-metastatic molecule present on solid and myeloid leukemia-derived ApoV (47, 48).
Conclusion
Despite the current advances in the field, much remains to be identified about ApoV and their composition, immune clearance, and their implications in cancer and coagulation. Furthermore, it remains unclear if tumor-derived ApoV generated by varying apoptotic agents possess distinct molecular, or morphological, or functional differences. There is greater need to improve on the isolation methods of EV to enhance their purity and decrease any co-contamination between different subtypes. Since ApoV suppress or stimulate an immune response, then they can potentially be used to treat autoimmunity or cancer, respectively. As for the latter, the immunogenic nature of ApoV opens the possibility to exploit their molecular composition for clinical utility as prophylactic and therapeutic cancer vaccines.
Author Contributions
MM-S wrote the bulk of the review and made the figures. AM contributed to planning, scientific input, and editing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer RX and handling Editor declared their shared affiliation.
Funding
This work cited from our laboratory was funded by a University of Otago Research Grant and the Otago Medical Research Foundation.
References
1. Jang SC, Kim SR, Yoon YJ, Park KS, Kim JH, Lee J, et al. In vivo kinetic biodistribution of nano-sized outer membrane vesicles derived from bacteria. Small (2015) 11(4):456–61. doi:10.1002/smll.201401803
2. Anderson HC. Vesicles associated with calcification in the matrix of epiphyseal cartilage. J Cell Biol (1969) 41(1):59–72. doi:10.1083/jcb.41.1.59
3. Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol (2014) 14(3):195–208. doi:10.1038/nri3622
4. Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, et al. B lymphocytes secrete antigen-presenting vesicles. J Exp Med (1996) 183(3):1161–72. doi:10.1084/jem.183.3.1161
5. Frühbeis C, Fröhlich D, Kuo WP, Krämer-Albers E-M. Extracellular vesicles as mediators of neuron-glia communication. Front Cell Neurosci (2013) 7:182. doi:10.3389/fncel.2013.00182
6. Sonnemann KJ, Bement WM. Wound repair: toward understanding and integration of single-cell and multicellular wound responses. Annu Rev Cell Dev Biol (2011) 27:237. doi:10.1146/annurev-cellbio-092910-154251
7. Heijnen HF, Schiel AE, Fijnheer R, Geuze HJ, Sixma JJ. Activated platelets release two types of membrane vesicles: microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood (1999) 94(11):3791–9.
8. Robertson C, Booth SA, Beniac DR, Coulthart MB, Booth TF, McNicol A. Cellular prion protein is released on exosomes from activated platelets. Blood (2006) 107(10):3907–11. doi:10.1182/blood-2005-02-0802
9. Gould SJ, Booth AM, Hildreth JE. The Trojan exosome hypothesis. Proc Natl Acad Sci U S A (2003) 100(19):10592–7. doi:10.1073/pnas.1831413100
10. VanWijk MJ, VanBavel E, Sturk A, Nieuwland R. Microparticles in cardiovascular diseases. Cardiovasc Res (2003) 59(2):277–87. doi:10.1016/S0008-6363(03)00367-5
11. Furie B, Furie BC. Role of platelet P-selectin and microparticle PSGL-1 in thrombus formation. Trends Mol Med (2004) 10(4):171–8. doi:10.1016/j.molmed.2004.02.008
12. Distler JH, Pisetsky DS, Huber LC, Kalden JR, Gay S, Distler O. Microparticles as regulators of inflammation: novel players of cellular crosstalk in the rheumatic diseases. Arthritis Rheum (2005) 52(11):3337–48. doi:10.1002/art.21350
13. Simak J, Gelderman MP, Yu H, Wright V, Baird AE. Circulating endothelial microparticles in acute ischemic stroke: a link to severity, lesion volume and outcome. J Thromb Haemost (2006) 4(6):1296–302. doi:10.1111/j.1538-7836.2006.01911.x
14. Lima LG, Chammas R, Monteiro RQ, Moreira ME, Barcinski MA. Tumor-derived microvesicles modulate the establishment of metastatic melanoma in a phosphatidylserine-dependent manner. Cancer Lett (2009) 283(2):168–75. doi:10.1016/j.canlet.2009.03.041
15. Théry C, Boussac M, Véron P, Ricciardi-Castagnoli P, Raposo G, Garin J, et al. Proteomic analysis of dendritic cell-derived exosomes: a secreted subcellular compartment distinct from apoptotic vesicles. J Immunol (2001) 166(12):7309–18. doi:10.4049/jimmunol.166.12.7309
16. Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol (2002) 2(8):569–79. doi:10.1038/nri855
17. Taylor DD, Akyol S, Gercel-Taylor C. Pregnancy-associated exosomes and their modulation of T cell signaling. J Immunol (2006) 176(3):1534–42. doi:10.4049/jimmunol.176.3.1534
18. McLellan AD. Exosome release by primary B cells. Crit Rev Immunol (2009) 29(3):203–17. doi:10.1615/CritRevImmunol.v29.i3.20
19. Wolfers J, Lozier A, Raposo G, Regnault A, Thery C, Masurier C, et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med (2001) 7(3):297–303. doi:10.1038/85438
20. Trams EG, Lauter CJ, Salem N Jr, Heine U. Exfoliation of membrane ecto-enzymes in the form of micro-vesicles. Biochim Biophys Acta (1981) 645(1):63–70. doi:10.1016/0005-2736(81)90512-5
21. Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol (2009) 9(8):581–93. doi:10.1038/nri2567
22. Taylor DD, Gercel-Taylor C. Tumour-derived exosomes and their role in cancer-associated T-cell signalling defects. Br J Cancer (2005) 92(2):305–11. doi:10.1038/sj.bjc.6602316
23. Théry C, Regnault A, Garin J, Wolfers J, Zitvogel L, Ricciardi-Castagnoli P, et al. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein Hsc73. J Cell Biol (1999) 147(3):599–610. doi:10.1083/jcb.147.3.599
24. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol (2013) 200(4):373–83. doi:10.1083/jcb.201211138
25. Wolf P. The nature and significance of platelet products in human plasma. Br J Haematol (1967) 13(3):269–88. doi:10.1111/j.1365-2141.1967.tb08741.x
26. Chargaff E, West R. The biological significance of the thromboplastic protein of blood. J Biol Chem (1946) 166(1):189–97.
27. Smalley DM, Sheman NE, Nelson K, Theodorescu D. Isolation and identification of potential urinary microparticle biomarkers of bladder cancer. J Proteome Res (2008) 7(5):2088–96. doi:10.1021/pr700775x
28. Ma L, Li Y, Peng J, Wu D, Zhao X, Cui Y, et al. Discovery of the migrasome, an organelle mediating release of cytoplasmic contents during cell migration. Cell Res (2015) 25(1):24–38. doi:10.1038/cr.2014.135
29. Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol (2008) 10(5):619–24. doi:10.1038/ncb1725
30. Di Vizio D, Kim J, Hager MH, Morello M, Yang W, Lafargue CJ, et al. Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res (2009) 69(13):5601–9. doi:10.1158/0008-5472.CAN-08-3860
31. Di Vizio D, Morello M, Dudley AC, Schow PW, Adam RM, Morley S, et al. Large oncosomes in human prostate cancer tissues and in the circulation of mice with metastatic disease. Am J Pathol (2012) 181(5):1573–84. doi:10.1016/j.ajpath.2012.07.030
32. Tatischeff I. Cell-derived extracellular vesicles open new perspectives for cancer research. Cancer Res Front (2015) 1(2):208–24. doi:10.17980/2015.208
33. Zappulli V, Friis KP, Fitzpatrick Z, Maguire CA, Breakefield XO. Extracellular vesicles and intercellular communication within the nervous system. J Clin Invest (2016) 126(4):1198–207. doi:10.1172/JCI81134
34. Meehan B, Rak J, Di Vizio D. Oncosomes – large and small: what are they, where they came from? J Extracell Vesicles (2016) 5:1–2. doi:10.3402/jev.v5.33109
35. Lötvall J, Hill AF, Hochberg F, Buzás EI, Di Vizio D, Gardiner C, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the international society for extracellular vesicles. J Extracell Vesicles (2014) 3:26913. doi:10.3402/jev.v3.26913
36. Coumans FAW, Brisson AR, Buzas EI, Dignat-George F, Drees EEE, El-Andaloussi S, et al. Methodological guidelines to study extracellular vesicles. Circ Res (2017) 120(10):1632–48. doi:10.1161/CIRCRESAHA.117.309417
37. Wickman G, Julian L, Olson MF. How apoptotic cells aid in the removal of their own cold dead bodies. Cell Death Differ (2012) 19(5):735–42. doi:10.1038/cdd.2012.25
38. Akers JC, Gonda D, Kim R, Carter BS, Chen CC. Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J Neurooncol (2013) 113(1):1–11. doi:10.1007/s11060-013-1084-8
39. Hristov M, Erl W, Linder S, Weber PC. Apoptotic bodies from endothelial cells enhance the number and initiate the differentiation of human endothelial progenitor cells in vitro. Blood (2004) 104(9):2761–6. doi:10.1182/blood-2003-10-3614
40. Bhatnagar S, Schorey JS. Exosomes released from infected macrophages contain Mycobacterium avium glycopeptidolipids and are proinflammatory. J Biol Chem (2007) 282(35):25779–89. doi:10.1074/jbc.M702277200
41. Orozco AF, Jorgez CJ, Horne C, Marquez-Do DA, Chapman MR, Rodgers JR, et al. Membrane protected apoptotic trophoblast microparticles contain nucleic acids: relevance to preeclampsia. Am J Pathol (2008) 173(6):1595–608. doi:10.2353/ajpath.2008.080414
42. Litvack ML, Post M, Palaniyar N. IgM promotes the clearance of small particles and apoptotic microparticles by macrophages. PLoS One (2011) 6(3):e17223. doi:10.1371/journal.pone.0017223
43. Atkin-Smith GK, Paone S, Zanker DJ, Duan M, Phan TK, Chen W, et al. Isolation of cell type-specific apoptotic bodies by fluorescence-activated cell sorting. Sci Rep (2017) 7:39846. doi:10.1038/srep39846
44. Reich CF III, Pisetsky DS. The content of DNA and RNA in microparticles released by Jurkat and HL-60 cells undergoing in vitro apoptosis. Exp Cell Res (2009) 315(5):760–8. doi:10.1016/j.yexcr.2008.12.014
45. Schiller M, Bekeredjian-Ding I, Heyder P, Blank N, Ho AD, Lorenz HM. Autoantigens are translocated into small apoptotic bodies during early stages of apoptosis. Cell Death Differ (2007) 15(1):183–91. doi:10.1038/sj.cdd.4402239
46. Gyorgy B, Szabo TG, Pasztoi M, Pal Z, Misjak P, Aradi B, et al. Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol Life Sci (2011) 68(16):2667–88. doi:10.1007/s00018-011-0689-3
47. Black LV, Saunderson SC, Coutinho FP, Muhsin-Sharafaldine MR, Damani TT, Dunn AC, et al. The CD169 sialoadhesin molecule mediates cytotoxic T-cell responses to tumour apoptotic vesicles. Immunol Cell Biol (2016) 94(5):430–8. doi:10.1038/icb.2015.111
48. Muhsin-Sharafaldine MR, Kennedy BR, Saunderson SC, Buchanan CR, Dunn AC, Faed JM, et al. Mechanistic insight into the procoagulant activity of tumor-derived apoptotic vesicles. Biochim Biophys Acta (2017) 1861(2):286–95. doi:10.1016/j.bbagen.2016.11.020
49. Saunderson SC, Dunn AC, Crocker PR, McLellan AD. CD169 mediates the capture of exosomes in spleen and lymph node. Blood (2014) 123(2):208–16. doi:10.1182/blood-2013-03-489732
50. Okada H, Mak TW. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat Rev Cancer (2004) 4(8):592–603. doi:10.1038/nrc1412
51. Ricci MS, Zong W-X. Chemotherapeutic approaches for targeting cell death pathways. Oncologist (2006) 11(4):342–57. doi:10.1634/theoncologist.11-4-342
52. Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol (2009) 10(7):478–87. doi:10.1038/nrm2718
53. Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol (2011) 12(6):385–92. doi:10.1038/nrm3115
54. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res (1965) 37(3):614–36. doi:10.1016/0014-4827(65)90211-9
55. Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol (2007) 8(9):729–40. doi:10.1038/nrm2233
56. Vallejo AN, Weyand CM, Goronzy JJ. T-cell senescence: a culprit of immune abnormalities in chronic inflammation and persistent infection. Trends Mol Med (2004) 10(3):119–24. doi:10.1016/j.molmed.2004.01.002
57. Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol (1995) 146(1):3–15.
58. Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol (2004) 16(6):663–9. doi:10.1016/j.ceb.2004.09.011
59. Proskuryakov SY, Gabai V, Konoplyannikov A. Necrosis is an active and controlled form of programmed cell death. Biochemistry (Mosc) (2002) 67(4):387–408. doi:10.1023/A:1015289521275
60. Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell (2008) 135(7):1311–23. doi:10.1016/j.cell.2008.10.044
61. Yoon S, Kovalenko A, Bogdanov K, Wallach D. MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity (2017) 47(1):51–65.e7. doi:10.1016/j.immuni.2017.06.001
62. Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell (2009) 34(3):259–69. doi:10.1016/j.molcel.2009.04.026
63. Bursch W, Hochegger K, Torok L, Marian B, Ellinger A, Hermann RS. Autophagic and apoptotic types of programmed cell death exhibit different fates of cytoskeletal filaments. J Cell Sci (2000) 113(7):1189–98.
64. Barros LF, Kanaseki T, Sabirov R, Morishima S, Castro J, Bittner CX, et al. Apoptotic and necrotic blebs in epithelial cells display similar neck diameters but different kinase dependency. Cell Death Differ (2003) 10:687. doi:10.1038/sj.cdd.4401236
65. Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene (2006) 25(34):4798–811. doi:10.1038/sj.onc.1209608
66. Rytömaa M, Martins LM, Downward J. Involvement of FADD and caspase-8 signalling in detachment-induced apoptosis. Curr Biol (1999) 9(18):1043–S2. doi:10.1016/S0960-9822(99)80454-0
67. Cohen GM. Caspases: the executioners of apoptosis. Biochem J (1997) 326(Pt 1):1–16. doi:10.1042/bj3260001
68. Thornberry NA, Lazebnik Y. Caspases: enemies within. Science (1998) 281(5381):1312–6. doi:10.1126/science.281.5381.1312
69. Grütter MG. Caspases: key players in programmed cell death. Curr Opin Struct Biol (2000) 10(6):649–55. doi:10.1016/S0959-440X(00)00146-9
70. Mehmet H. Apoptosis: caspases find a new place to hide. Nature (2000) 403(6765):29–30. doi:10.1038/47377
71. Atkin-Smith GK, Poon IKH. Disassembly of the dying: mechanisms and functions. Trends Cell Biol (2017) 27(2):151–62. doi:10.1016/j.tcb.2016.08.011
72. Savill J, Gregory C, Haslett C. Eat me or die. Science (2003) 302(5650):1516–7. doi:10.1126/science.1092533
73. Kerr JFR, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer (1972) 26(4):239–57. doi:10.1038/bjc.1972.33
74. Bobrie A, Colombo M, Raposo G, Théry C. Exosome secretion: molecular mechanisms and roles in immune responses. Traffic (2011) 12(12):1659–68. doi:10.1111/j.1600-0854.2011.01225.x
75. Bock JB, Matern HT, Peden AA, Scheller RH. A genomic perspective on membrane compartment organization. Nature (2001) 409:839. doi:10.1038/35057024
76. van Deurs B, Holm PK, Kayser L, Sandvig K, Hansen SH. Multivesicular bodies in HEp-2 cells are maturing endosomes. Eur J Cell Biol (1993) 61(2):208–24.
77. Wollert T, Wunder C, Lippincott-Schwartz J, Hurley JH. Membrane scission by the ESCRT-III complex. Nature (2009) 458:172. doi:10.1038/nature07836
78. Xu R, Greening DW, Zhu HJ, Takahashi N, Simpson RJ. Extracellular vesicle isolation and characterization: toward clinical application. J Clin Invest (2016) 126(4):1152–62. doi:10.1172/JCI81129
79. Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science (2008) 319(5867):1244–7. doi:10.1126/science.1153124
80. Miyoshi H, Umeshita K, Sakon M, Imajoh-Ohmi S, Fujitani K, Gotoh M, et al. Calpain activation in plasma membrane bleb formation during tert-butyl hydroperoxide-induced rat hepatocyte injury. Gastroenterology (1996) 110(6):1897–904. doi:10.1053/gast.1996.v110.pm8964416
81. Muralidharan-Chari V, Clancy J, Plou C, Romao M, Chavrier P, Raposo G, et al. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr Biol (2009) 19(22):1875–85. doi:10.1016/j.cub.2009.09.059
82. Schlienger S, Campbell S, Claing A. ARF1 regulates the Rho/MLC pathway to control EGF-dependent breast cancer cell invasion. Mol Biol Cell (2014) 25(1):17–29. doi:10.1091/mbc.E13-06-0335
83. Sebbagh M, Renvoizé C, Hamelin J, Riché N, Bertoglio J, Bréard J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat Cell Biol (2001) 3:346. doi:10.1038/35070019
84. Khorchid A, Ikura M. How calpain is activated by calcium. Nat Struct Mol Biol (2002) 9(4):239–41. doi:10.1038/nsb0402-239
85. Pasquet JM, Dachary-Prigent J, Nurden AT. Calcium influx is a determining factor of calpain activation and microparticle formation in platelets. Eur J Biochem (1996) 239(3):647–54. doi:10.1111/j.1432-1033.1996.0647u.x
86. Kawaguchi K, Saito K, Asami H, Ohta Y. ADP ribosylation factor 6 (Arf6) Acts through FilGAP protein to down-regulate Rac protein and regulates plasma membrane blebbing. J Biol Chem (2014) 289(14):9675–82. doi:10.1074/jbc.M113.546051
87. Charras GT, Yarrow JC, Horton MA, Mahadevan L, Mitchison T. Non-equilibration of hydrostatic pressure in blebbing cells. Nature (2005) 435(7040):365–9. doi:10.1038/nature03550
88. Choudhuri K, Llodrá J, Roth EW, Tsai J, Gordo S, Wucherpfennig KW, et al. Polarized release of T-cell-receptor-enriched microvesicles at the immunological synapse. Nature (2014) 507:118. doi:10.1038/nature12951
89. Mills JC, Stone NL, Erhardt J, Pittman RN. Apoptotic membrane blebbing is regulated by myosin light chain phosphorylation. J Cell Biol (1998) 140(3):627–36. doi:10.1083/jcb.140.3.627
90. Leverrier Y, Ridley AJ. Apoptosis: caspases orchestrate the ROCKn’ bleb. Nat Cell Biol (2001) 3:E91. doi:10.1038/35070151
91. Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol (2001) 3(4):339–45. doi:10.1038/35070009
92. Tomiyoshi G, Horita Y, Nishita M, Ohashi K, Mizuno K. Caspase-mediated cleavage and activation of LIM-kinase 1 and its role in apoptotic membrane blebbing. Genes Cells (2004) 9(6):591–600. doi:10.1111/j.1356-9597.2004.00745.x
93. Rudel T, Bokoch GM. Membrane and morphological changes in apoptotic cells regulated by caspase-mediated activation of PAK2. Science (1997) 276(5318):1571–4. doi:10.1126/science.276.5318.1571
94. Moss DK, Betin VM, Malesinski SD, Lane JD. A novel role for microtubules in apoptotic chromatin dynamics and cellular fragmentation. J Cell Sci (2006) 119(11):2362–74. doi:10.1242/jcs.02959
95. Poon IKH, Chiu Y-H, Armstrong AJ, Kinchen JM, Juncadella IJ, Bayliss DA, et al. Unexpected link between an antibiotic, pannexin channels and apoptosis. Nature (2014) 507:329. doi:10.1038/nature13147
96. Osteikoetxea X, Balogh A, Szabo-Taylor K, Nemeth A, Szabo TG, Paloczi K, et al. Improved characterization of EV preparations based on protein to lipid ratio and lipid properties. PLoS One (2015) 10(3):e0121184. doi:10.1371/journal.pone.0121184
97. Thakur BK, Zhang H, Becker A, Matei I, Huang Y, Costa-Silva B, et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res (2014) 24:766. doi:10.1038/cr.2014.44
98. Ratajczak MZ, Ratajczak J. Horizontal transfer of RNA and proteins between cells by extracellular microvesicles: 14 years later. Clin Transl Med (2016) 5(1):7. doi:10.1186/s40169-016-0087-4
99. Muhsin-Sharafaldine MR, Saunderson SC, Dunn AC, Faed JM, Kleffmann T, McLellan AD. Procoagulant and immunogenic properties of melanoma exosomes, microvesicles and apoptotic vesicles. Oncotarget (2016) 7(35):56279–94. doi:10.18632/oncotarget.10783
100. Millimaggi D, Mari M, D’Ascenzo S, Carosa E, Jannini EA, Zucker S, et al. Tumor vesicle-associated CD147 modulates the angiogenic capability of endothelial cells. Neoplasia (2007) 9(4):349–57. doi:10.1593/neo.07133
101. Zucker S, Hymowitz M, Conner C, Zarrabi HM, Hurewitz AN, Matrisian L, et al. Measurement of matrix metalloproteinases and tissue inhibitors of metalloproteinases in blood and tissues: clinical and experimental applications. Ann N Y Acad Sci (1999) 878(1):212–27. doi:10.1111/j.1749-6632.1999.tb07687.x
102. Zucker S, Hymowitz M, Rollo EE, Mann R, Conner CE, Cao J, et al. Tumorigenic potential of extracellular matrix metalloproteinase inducer. Am J Pathol (2001) 158(6):1921–8. doi:10.1016/S0002-9440(10)64660-3
103. Voigt H, Vetter-Kauczok CS, Schrama D, Hofmann UB, Becker JC, Houben R. CD147 impacts angiogenesis and metastasis formation. Cancer Invest (2009) 27(3):329–33. doi:10.1080/07357900802392675
104. Andreu Z, Yáñez-Mó M. Tetraspanins in extracellular vesicle formation and function. Front Immunol (2014) 5:442. doi:10.3389/fimmu.2014.00442
105. Hemler ME. Tetraspanin proteins mediate cellular penetration, invasion, and fusion events and define a novel type of membrane microdomain. Annu Rev Cell Dev Biol (2003) 19(1):397–422. doi:10.1146/annurev.cellbio.19.111301.153609
106. Zöller M. Tetraspanins: push and pull in suppressing and promoting metastasis. Nat Rev Cancer (2009) 9(1):40–55. doi:10.1038/nrc2543
107. Mathivanan S, Simpson RJ. ExoCarta: a compendium of exosomal proteins and RNA. Proteomics (2009) 9(21):4997–5000. doi:10.1002/pmic.200900351
108. Hemler ME. Tetraspanin proteins promote multiple cancer stages. Nat Rev Cancer (2014) 14(1):49–60. doi:10.1038/nrc3640
109. Berditchevski F. Complexes of tetraspanins with integrins: more than meets the eye. J Cell Sci (2001) 114(23):4143–51.
110. Burgess RJ, Zhang Z. Histone chaperones in nucleosome assembly and human disease. Nat Struct Mol Biol (2013) 20(1):14–22. doi:10.1038/nsmb.2461
111. Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A (2016) 113(8):E968–77. doi:10.1073/pnas.1521230113
112. Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Curry WT, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol (2008) 10(12):1470–6. doi:10.1038/ncb1800
113. Palma J, Yaddanapudi SC, Pigati L, Havens MA, Jeong S, Weiner GA, et al. MicroRNAs are exported from malignant cells in customized particles. Nucleic Acids Res (2012) 40(18):9125–38. doi:10.1093/nar/gks656
114. Gibbings DJ, Ciaudo C, Erhardt M, Voinnet O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat Cell Biol (2009) 11(9):1143–9. doi:10.1038/ncb1929
115. Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol (2007) 9:654–9. doi:10.1038/ncb1596
116. Müller G, Schneider M, Gassenhuber J, Wied S. Release of exosomes and microvesicles harbouring specific RNAs and glycosylphosphatidylinositol-anchored proteins from rat and human adipocytes is controlled by histone methylation. Am J Mol Biol (2012) 2(3):187–209. doi:10.4236/ajmb.2012.23020
117. Al-Nedawi K, Meehan B, Rak J. Microvesicles: messengers and mediators of tumor progression. Cell Cycle (2009) 8(13):2014–8. doi:10.4161/cc.8.13.8988
118. Crescitelli R, Lässer C, Szabó TG, Kittel A, Eldh M, Dianzani I, et al. Distinct RNA profiles in subpopulations of extracellular vesicles: apoptotic bodies, microvesicles and exosomes. J Extracell Vesicles (2013) 2(1):20677. doi:10.3402/jev.v2i0.20677
119. Nolte-’t Hoen EN, Buermans HP, Waasdorp M, Stoorvogel W, Wauben MH, ’t Hoen PA. Deep sequencing of RNA from immune cell-derived vesicles uncovers the selective incorporation of small non-coding RNA biotypes with potential regulatory functions. Nucleic Acids Res (2012) 40(18):9272–85. doi:10.1093/nar/gks658
120. Halicka HD, Bedner E, Darzynkiewicz Z. Segregation of RNA and separate packaging of DNA and RNA in apoptotic bodies during apoptosis. Exp Cell Res (2000) 260(2):248–56. doi:10.1006/excr.2000.5027
121. Holmgren L, Szeles A, Rajnavölgyi E, Folkman J, Klein G, Ernberg I, et al. Horizontal transfer of DNA by the uptake of apoptotic bodies. Blood (1999) 93(11):3956–63.
122. Freyssinet JM. Cellular microparticles: what are they bad or good for? J Thromb Haemost (2003) 1(7):1655–62. doi:10.1046/j.1538-7836.2003.00309.x
123. Balasubramanian K, Mirnikjoo B, Schroit AJ. Regulated externalization of phosphatidylserine at the cell surface: implications for apoptosis. J Biol Chem (2007) 282(25):18357–64. doi:10.1074/jbc.M700202200
125. Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science (2014) 344(6188):1164–8. doi:10.1126/science.1252809
126. Groen A, Romero MR, Kunne C, Hoosdally SJ, Dixon PH, Wooding C, et al. Complementary functions of the flippase ATP8B1 and the floppase ABCB4 in maintaining canalicular membrane integrity. Gastroenterology (2011) 141(5):1927–37.e1–4. doi:10.1053/j.gastro.2011.07.042
127. Bitbol M, Devaux PF. Measurement of outward translocation of phospholipids across human erythrocyte membrane. Proc Natl Acad Sci U S A (1988) 85(18):6783–7. doi:10.1073/pnas.85.18.6783
128. Hankins HM, Baldridge RD, Xu P, Graham TR. Role of flippases, scramblases and transfer proteins in phosphatidylserine subcellular distribution. Traffic (2015) 16(1):35–47. doi:10.1111/tra.12233
129. Zhou Q, Zhao J, Stout JG, Luhm RA, Wiedmer T, Sims PJ. Molecular cloning of human plasma membrane phospholipid scramblase a protein mediating transbilayer movement of plasma membrane phospholipids. J Biol Chem (1997) 272(29):18240–4. doi:10.1074/jbc.272.29.18240
130. Suzuki J, Umeda M, Sims PJ, Nagata S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature (2010) 468(7325):834–8. doi:10.1038/nature09583
131. Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science (2013) 341(6144):403–6. doi:10.1126/science.1236758
132. Varki A, Lowe JB. Chapter 6: Biological roles of glycans. In: Varki A, Cummings RD, Esko JD, editors. Essentials of Glycobiology. 2nd edition. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press (2009). Available from: https://www.ncbi.nlm.nih.gov/books/NBK1897/
133. Zonneveld MI, Brisson AR, van Herwijnen MJC, Tan S, van de Lest CHA, Redegeld FA, et al. Recovery of extracellular vesicles from human breast milk is influenced by sample collection and vesicle isolation procedures. J Extracell Vesicles (2014) 3(1):24215. doi:10.3402/jev.v3.24215
134. Echevarria J, Royo F, Pazos R, Salazar L, Falcon-Perez JM, Reichardt N-C. Microarray-based identification of lectins for the purification of human urinary extracellular vesicles directly from urine samples. Chembiochem (2014) 15(11):1621–6. doi:10.1002/cbic.201402058
135. da Silva RP, Heiss C, Black I, Azadi P, Gerlach JQ, Travassos LR, et al. Extracellular vesicles from Paracoccidioides pathogenic species transport polysaccharide and expose ligands for DC-SIGN receptors. Sci Rep (2015) 5:14213. doi:10.1038/srep14213
136. Gerlach JQ, Krüger A, Gallogly S, Hanley SA, Hogan MC, Ward CJ, et al. Surface glycosylation profiles of urine extracellular vesicles. PLoS One (2013) 8(9):e74801. doi:10.1371/journal.pone.0074801
137. Krishnamoorthy L, Bess JW Jr, Preston AB, Nagashima K, Mahal LK. HIV-1 and microvesicles from T cells share a common glycome, arguing for a common origin. Nat Chem Biol (2009) 5:244. doi:10.1038/nchembio.151
138. Batista BS, Eng WS, Pilobello KT, Hendricks-Muñoz KD, Mahal LK. Identification of a conserved glycan signature for microvesicles. J Proteome Res (2011) 10(10):4624–33. doi:10.1021/pr200434y
139. Gupta G, Surolia A, Sampathkumar S-G. Lectin microarrays for glycomic analysis. OMICS (2010) 14(4):419–36. doi:10.1089/omi.2009.0150
140. Gerlach JQ, Griffin MD. Getting to know the extracellular vesicle glycome. Mol Biosyst (2016) 12(4):1071–81. doi:10.1039/c5mb00835b
141. Wang S, Cesca F, Loers G, Schweizer M, Buck F, Benfenati F, et al. Synapsin I is an oligomannose-carrying glycoprotein, acts as an oligomannose-binding lectin, and promotes neurite outgrowth and neuronal survival when released via glia-derived exosomes. J Neurosci (2011) 31(20):7275–90. doi:10.1523/JNEUROSCI.6476-10.2011
142. Shimoda A, Tahara Y, Sawada S-I, Sasaki Y, Akiyoshi K. Glycan profiling analysis using evanescent-field fluorescence-assisted lectin array: importance of sugar recognition for cellular uptake of exosomes from mesenchymal stem cells. Biochem Biophys Res Commun (2017) 491(3):701–7. doi:10.1016/j.bbrc.2017.07.126
143. Barrès C, Blanc L, Bette-Bobillo P, André S, Mamoun R, Gabius H-J, et al. Galectin-5 is bound onto the surface of rat reticulocyte exosomes and modulates vesicle uptake by macrophages. Blood (2010) 115(3):696–705. doi:10.1182/blood-2009-07-231449
144. Hogan MC, Manganelli L, Woollard JR, Masyuk AI, Masyuk TV, Tammachote R, et al. Characterization of PKD protein-positive exosome-like vesicles. J Am Soc Nephrol (2009) 20(2):278–88. doi:10.1681/ASN.2008060564
145. Gomes J, Gomes-Alves P, Carvalho S, Peixoto C, Alves P, Altevogt P, et al. Extracellular vesicles from ovarian carcinoma cells display specific glycosignatures. Biomolecules (2015) 5(3):1741. doi:10.3390/biom5031741
146. Takanori A, Kei K, Nami H, Masashi K, Takanori I. Evaluation of desialylation effect on zeta potential of extracellular vesicles secreted from human prostate cancer cells by on-chip microcapillary electrophoresis. Jpn J App Phys (2014) 53(6S):06JL1. doi:10.7567/JJAP.53.06JL01
147. Escrevente C, Grammel N, Kandzia S, Zeiser J, Tranfield EM, Conradt HS, et al. Sialoglycoproteins and N-glycans from secreted exosomes of ovarian carcinoma cells. PLoS One (2013) 8(10):e78631. doi:10.1371/journal.pone.0078631
148. Wang P-H. Altered glycosylation in cancer: sialic acids and sialyltransferases. J Cancer Mol (2005) 1(2):73–81. doi:10.29685/JCM.200512.0001
149. Ashman LK, Zöller M. Tetraspanins in cancer. In: Berditchevski F, Rubinstein E, editor. Tetraspanins. Proteins and Cell Regulation, vol 9. Dordrecht: Springer (2013). p. 257–98.
150. Crocker PR, Vinson M, Kelm S, Drickamer K. Molecular analysis of sialoside binding to sialoadhesin by NMR and site-directed mutagenesis. Biochem J (1999) 341(Pt 2):355–61. doi:10.1042/bj3410355
151. Matheoud D, Baey C, Vimeux L, Tempez A, Valente M, Louche P, et al. Dendritic cells crosspresent antigens from live B16 cells more efficiently than from apoptotic cells and protect from melanoma in a therapeutic model. PLoS One (2011) 6(4):e19104. doi:10.1371/journal.pone.0019104
152. Harshyne LA, Watkins SC, Gambotto A, Barratt-Boyes SM. Dendritic cells acquire antigens from live cells for cross-presentation to CTL. J Immunol (2001) 166(6):3717–23. doi:10.4049/jimmunol.166.6.3717
153. Zeelenberg IS, van Maren WWC, Boissonnas A, Van Hout-Kuijer MA, Den Brok MHMGM, Wagenaars JAL, et al. Antigen localization controls T cell-mediated tumor immunity. J Immunol (2011) 187(3):1281–8. doi:10.4049/jimmunol.1003905
154. Basu S, Binder RJ, Ramalingam T, Srivastava PK. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity (2001) 14(3):303–13. doi:10.1016/S1074-7613(01)00111-X
155. Valenti R, Huber V, Iero M, Filipazzi P, Parmiani G, Rivoltini L. Tumor-released microvesicles as vehicles of immunosuppression. Cancer Res (2007) 67(7):2912–5. doi:10.1158/0008-5472.CAN-07-0520
156. Mitchell DA, Nair SK, Gilboa E. Dendritic cell/macrophage precursors capture exogenous antigen for MHC class I presentation by dendritic cells. Eur J Immunol (1998) 28(6):1923–33. doi:10.1002/(SICI)1521-4141(199806)28:06<1923::AID-IMMU1923>3.0.CO;2-9
157. Knuth A, Wölfel T, Klehmann E, Boon T, Meyer zum Büschenfelde KH. Cytolytic T-cell clones against an autologous human melanoma: specificity study and definition of three antigens by immunoselection. Proc Natl Acad Sci U S A (1989) 86(8):2804–8. doi:10.1073/pnas.86.8.2804
158. Heiser A, Coleman D, Dannull J, Yancey D, Maurice MA, Lallas CD, et al. Autologous dendritic cells transfected with prostate-specific antigen RNA stimulate CTL responses against metastatic prostate tumors. J Clin Invest (2002) 109(3):409–17. doi:10.1172/JCI0214364
159. Ludewig B, Ochsenbein AF, Odermatt B, Paulin D, Hengartner H, Zinkernagel RM. Immunotherapy with dendritic cells directed against tumor antigens shared with normal host cells results in severe autoimmune disease. J Exp Med (2000) 191(5):795–804. doi:10.1084/jem.191.5.795
160. Winau F, Weber S, Sad S, de Diego J, Hoops SL, Breiden B, et al. Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity (2006) 24(1):105–17. doi:10.1016/j.immuni.2005.12.001
161. Saunderson SC, McLellan AD. Role of lymphocyte subsets in the immune response to primary B cell–derived exosomes. J Immunol (2017) 199(7):2225–35. doi:10.4049/jimmunol.1601537
162. Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature (1998) 392(6671):86–9. doi:10.1038/32183
163. Heusermann W, Hean J, Trojer D, Steib E, von Bueren S, Graff-Meyer A, et al. Exosomes surf on filopodia to enter cells at endocytic hot spots, traffic within endosomes, and are targeted to the ER. J Cell Biol (2016) 213(2):173–84. doi:10.1083/jcb.201506084
164. Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza D, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell derived exosomes. Nat Med (1998) 4(5):594–600. doi:10.1038/nm0598-594
165. Srivastava P. Interaction of heat shock proteins with peptides and antigen presenting cells: chaperoning of the innate and adaptive immune responses. Annu Rev Immunol (2002) 20:395–425. doi:10.1146/annurev.immunol.20.100301.064801
166. Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature (1997) 390(6658):350–1. doi:10.1038/37022
167. Xie Y, Bai O, Yuan J, Chibbar R, Slattery K, Wei Y, et al. Tumor apoptotic bodies inhibit CTL responses and antitumor immunity via membrane-bound transforming growth factor-β1 inducing CD8+ T-cell anergy and CD4+ Tr1 cell responses. Cancer Res (2009) 69(19):7756–66. doi:10.1158/0008-5472.CAN-09-0496
168. Wermeling F, Chen Y, Pikkarainen T, Scheynius A, Winqvist O, Izui S, et al. Class A scavenger receptors regulate tolerance against apoptotic cells, and autoantibodies against these receptors are predictive of systemic lupus. J Exp Med (2007) 204(10):2259–65. doi:10.1084/jem.20070600
169. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini J-L, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med (2007) 13(1):54–61. doi:10.1038/nm1523
170. Bellone M, Iezzi G, Rovere P, Galati G, Ronchetti A, Protti MP, et al. Processing of engulfed apoptotic bodies yields T cell epitopes. J Immunol (1997) 159(11):5391–9.
171. Apetoh L, Tesniere A, Ghiringhelli F, Kroemer G, Zitvogel L. Molecular interactions between dying tumor cells and the innate immune system determine the efficacy of conventional anticancer therapies. Cancer Res (2008) 68(11):4026–30. doi:10.1158/0008-5472.CAN-08-0427
172. Chen H, Tritton TR, Kenny N, Absher M, Chiu JF. Tamoxifen induces TGF-β1 activity and apoptosis of human MCF-7 breast cancer cells in vitro. J Cell Biochem (1996) 61(1):9–17. doi:10.1002/(SICI)1097-4644(19960401)61:1<9::AID-JCB2>3.0.CO;2-Z
173. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest (1998) 101(4):890. doi:10.1172/JCI1112
174. Huynh M-LN, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-β1 secretion and the resolution of inflammation. J Clin Invest (2002) 109(1):41. doi:10.1172/JCI0211638
175. Sulciner ML, Serhan CN, Gilligan MM, Mudge DK, Chang J, Gartung A, et al. Resolvins suppress tumor growth and enhance cancer therapy. J Exp Med (2017) 215(1):115–140. doi:10.1084/jem.20170681
176. Fransen JH, Hilbrands LB, Ruben J, Stoffels M, Adema GJ, van der Vlag J, et al. Mouse dendritic cells matured by ingestion of apoptotic blebs induce T cells to produce interleukin-17. Arthritis Rheum (2009) 60(8):2304–13. doi:10.1002/art.24719
177. Ruben JM, van den Ancker W, Bontkes HJ, Westers TM, Hooijberg E, Ossenkoppele GJ, et al. Apoptotic blebs from leukemic cells as a preferred source of tumor-associated antigen for dendritic cell-based vaccines. Cancer Immunol Immunother (2014) 63(4):335–45. doi:10.1007/s00262-013-1515-6
178. Hoffmann PR, deCathelineau AM, Ogden CA, Leverrier Y, Bratton DL, Daleke DL, et al. Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J Cell Biol (2001) 155(4):649–59. doi:10.1083/jcb.200108080
179. Lim JP, Gleeson PA. Macropinocytosis: an endocytic pathway for internalising large gulps. Immunol Cell Biol (2011) 89(8):836–43. doi:10.1038/icb.2011.20
180. Martínez MC, Freyssinet J-M. Deciphering the plasma membrane hallmarks of apoptotic cells: phosphatidylserine transverse redistribution and calcium entry. BMC Cell Biol (2001) 2(1):20. doi:10.1186/1471-2121-2-20
181. Balasubramanian K, Chandra J, Schroit AJ. Immune clearance of phosphatidylserine-expressing cells by phagocytes: the role of β2-glycoprotein i in macrophage recognition. J Biol Chem (1997) 272(49):31113–7. doi:10.1074/jbc.272.49.31113
182. Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature (2001) 411(6834):207–11. doi:10.1038/35079659
183. Sambrano GR, Steinberg D. Recognition of oxidatively damaged and apoptotic cells by an oxidized low density lipoprotein receptor on mouse peritoneal macrophages: role of membrane phosphatidylserine. Proc Natl Acad Sci U S A (1995) 92(5):1396–400. doi:10.1073/pnas.92.5.1396
184. Li MO, Sarkisian MR, Mehal WZ, Rakic P, Flavell RA. Phosphatidylserine receptor is required for clearance of apoptotic cells. Science (2003) 302(5650):1560–3. doi:10.1126/science.1087621
185. Oka K, Sawamura T, Kikuta K-I, Itokawa S, Kume N, Kita T, et al. Lectin-like oxidized low-density lipoprotein receptor 1 mediates phagocytosis of aged/apoptotic cells in endothelial cells. Proc Natl Acad Sci U S A (1998) 95(16):9535–40. doi:10.1073/pnas.95.16.9535
186. Maiti SN, Balasubramanian K, Ramoth JA, Schroit AJ. β-2-glycoprotein 1-dependent macrophage uptake of apoptotic cells binding to lipoprotein receptor-related protein receptor family members. J Biol Chem (2008) 283(7):3761–6. doi:10.1074/jbc.M704990200
187. Borisenko GG, Matsura T, Liu S-X, Tyurin VA, Jianfei J, Serinkan FB, et al. Macrophage recognition of externalized phosphatidylserine and phagocytosis of apoptotic Jurkat cells—existence of a threshold. Arch Biochem Biophys (2003) 413(1):41–52. doi:10.1016/S0003-9861(03)00083-3
188. Albert ML, Kim J-I, Birge RB. [alpha]v[beta]5 integrin recruits the CrkII-Dock180-Rac1 complex for phagocytosis of apoptotic cells. Nat Cell Biol (2000) 2:899. doi:10.1038/35046549
189. Takizawa F, Tsuji S, Nagasawa S. Enhancement of macrophage phagocytosis upon iC3b deposition on apoptotic cells. FEBS Lett (1996) 397(2–3):269–72. doi:10.1016/S0014-5793(96)01197-0
190. Friedl P, Vischer P, Freyberg M. The role of thrombospondin-1 in apoptosis. Cell Mol Life Sci (2002) 59(8):1347–57. doi:10.1007/s00018-002-8512-9
191. Teder P, Vandivier RW, Jiang D, Liang J, Cohn L, Puré E, et al. Resolution of lung inflammation by CD44. Science (2002) 296(5565):155–8. doi:10.1126/science.1069659
192. Segawa K, Nagata S. An apoptotic “eat me” signal: phosphatidylserine exposure. Trends Cell Biol (2015) 25(11):639–50. doi:10.1016/j.tcb.2015.08.003
193. Del Conde I, Shrimpton CN, Thiagarajan P, Lopez JA. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood (2005) 106(5):1604–11. doi:10.1182/blood-2004-03-1095
194. Escudier B, Dorval T, Chaput N, André F, Caby M-P, Novault S, et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of the first phase I clinical trial. J Transl Med (2005) 3(1):10. doi:10.1186/1479-5876-3-10
195. Viaud S, Terme M, Flament C, Taieb J, Andre F, Novault S, et al. Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: a role for NKG2D ligands and IL-15Ralpha. PLoS One (2009) 4(3):e4942. doi:10.1371/journal.pone.0004942
196. Dai S, Wei D, Wu Z, Zhou X, Wei X, Huang H, et al. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol Ther (2008) 16(4):782–90. doi:10.1038/mt.2008.1
197. Besse B, Charrier M, Lapierre V, Dansin E, Lantz O, Planchard D, et al. Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. Oncoimmunology (2016) 5(4):e1071008. doi:10.1080/2162402X.2015.1071008
198. Kokhaei P, Choudhury A, Mahdian R, Lundin J, Moshfegh A, Osterborg A, et al. Apoptotic tumor cells are superior to tumor cell lysate, and tumor cell RNA in induction of autologous T cell response in B-CLL. Leukemia (2004) 18(11):1810–5. doi:10.1038/sj.leu.2403517
199. Carter BA, Jensen RA, Simpson JF, Page DL. Benign transport of breast epithelium into axillary lymph nodes after biopsy. Am J Clin Pathol (2000) 113(2):259–65. doi:10.1309/7EF8-F1W7-YVNT-H8H5
200. Van Trappen PO, Pepper MS. Lymphatic dissemination of tumour cells and the formation of micrometastases. Lancet Oncol (2002) 3(1):44–52. doi:10.1016/S1470-2045(01)00621-0
201. Murray CA, Leong WL, McCready DR, Ghazarian DM. Histopathological patterns of melanoma metastases in sentinel lymph nodes. J Clin Pathol (2004) 57(1):64–7. doi:10.1136/jcp.57.1.64
202. Gray EE, Friend S, Suzuki K, Phan TG, Cyster JG. Subcapsular sinus macrophage fragmentation and CD169+ bleb acquisition by closely associated IL-17-committed innate-like lymphocytes. PLoS One (2012) 7(6):e38258. doi:10.1371/journal.pone.0038258
203. Hood JL, San RS, Wickline SA. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res (2011) 71(11):3792–801. doi:10.1158/0008-5472.CAN-10-4455
204. Kaplan RN, Rafii S, Lyden D. Preparing the "soil": the premetastatic niche. Cancer Res (2006) 66(23):11089–93. doi:10.1158/0008-5472.CAN-06-2407
205. Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer (2009) 9(4):285–93. doi:10.1038/nrc2621
206. Hood JL, Pan H, Lanza GM, Wickline SA. Consortium for translational research in advanced I, nanomedicine. Paracrine induction of endothelium by tumor exosomes. Lab Invest (2009) 89(11):1317–28. doi:10.1038/labinvest.2009.94
207. O’Neill ASG, van den Berg TK, Mullen GED. Sialoadhesin – a macrophage-restricted marker of immunoregulation and inflammation. Immunology (2013) 138(3):198–207. doi:10.1111/imm.12042
208. Bernhard CA, Ried C, Kochanek S, Brocker T. CD169+ macrophages are sufficient for priming of CTLs with specificities left out by cross-priming dendritic cells. Proc Natl Acad Sci U S A (2015) 112(17):5461–6. doi:10.1073/pnas.1423356112
209. Pucci F, Garris C, Lai CP, Newton A, Pfirschke C, Engblom C, et al. SCS macrophages suppress melanoma by restricting tumor-derived vesicle-B cell interactions. Science (2016) 352(6282):242–6. doi:10.1126/science.aaf1328
210. Iannacone M, Moseman EA, Tonti E, Bosurgi L, Junt T, Henrickson SE, et al. Subcapsular sinus macrophages prevent CNS invasion on peripheral infection with a neurotropic virus. Nature (2010) 465(7301):1079–83. doi:10.1038/nature09118
211. Ravishankar B, Shinde R, Liu H, Chaudhary K, Bradley J, Lemos HP, et al. Marginal zone CD169+ macrophages coordinate apoptotic cell-driven cellular recruitment and tolerance. Proc Natl Acad Sci U S A (2014) 111(11):4215–20. doi:10.1073/pnas.1320924111
212. Asano K, Nabeyama A, Miyake Y, Qiu C-H, Kurita A, Tomura M, et al. CD169-Positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity (2011) 34(1):85–95. doi:10.1016/j.immuni.2010.12.011
213. Chow A, Huggins M, Ahmed J, Hashimoto D, Lucas D, Kunisaki Y, et al. CD169(+) macrophages provide a niche promoting erythropoiesis under homeostasis, myeloablation and in JAK2V617F-induced polycythemia vera. Nat Med (2013) 19(4):429–36. doi:10.1038/nm.3057
214. Barral P, Polzella P, Bruckbauer A, van Rooijen N, Besra GS, Cerundolo V, et al. CD169(+) macrophages present lipid antigens to mediate early activation of invariant nkt cells in lymph nodes. Nat Immunol (2010) 11(4):303–12. doi:10.1038/ni.1853
215. Junt T, Moseman EA, Iannacone M, Massberg S, Lang PA, Boes M, et al. Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature (2007) 450(7166):110–4. doi:10.1038/nature06287
216. Kawasaki N, Vela JL, Nycholat CM, Rademacher C, Khurana A, van Rooijen N, et al. Targeted delivery of lipid antigen to macrophages via the CD169/sialoadhesin endocytic pathway induces robust invariant natural killer T cell activation. Proc Natl Acad Sci U S A (2013) 110(19):7826–31. doi:10.1073/pnas.1219888110
217. Muhsin-Sharafaldine MR, Saunderson SC, Dunn AC, McLellan AD. Melanoma growth and lymph node metastasis is independent of host CD169 expression. Biochem Biophys Res Commun (2017) 486(4):965–70. doi:10.1016/j.bbrc.2017.03.138
218. Mackman N. Triggers, targets and treatments for thrombosis. Nature (2008) 451(7181):914–8. doi:10.1038/nature06797
219. Rosendaal FR. Venous thrombosis: a multicausal disease. Lancet (1999) 353(9159):1167–73. doi:10.1016/S0140-6736(98)10266-0
220. Khorana AA, Francis CW, Culakova E, Kuderer NM, Lyman GH. Thromboembolism is a leading cause of death in cancer patients receiving outpatient chemotherapy. J Thromb Haemost (2007) 5(3):632–4. doi:10.1111/j.1538-7836.2007.02374.x
221. Ornstein DL, Zacharski LR. Cancer, thrombosis, and anticoagulants. Curr Opin Pulm Med (2000) 6(4):301–8. doi:10.1097/00063198-200007000-00009
222. Heit JA, Silverstein MD, Mohr DN, Petterson TM, O’Fallon WM, Melton LJ III. Risk factors for deep vein thrombosis and pulmonary embolism: a population-based case-control study. Arch Intern Med (2000) 160(6):809–15. doi:10.1001/archinte.160.6.809
223. Ashrani AA, Rajkumar SV. Chemotherapy-associated thrombosis. Cancer Treat Res (2009) 148:181–206. doi:10.1007/978-0-387-79962-9_11
224. Haddad TC, Greeno EW. Chemotherapy-induced thrombosis. Thromb Res (2006) 118(5):555–68. doi:10.1016/j.thromres.2005.10.015
225. Mannucci PM, Bettega DD, Chantarangkul VV, Tripodi AA, Sacchini VV, Veronesi UU. Effect of tamoxifen on measurements of hemostasis in healthy women. Arch Intern Med (1996) 156(16):1806–10. doi:10.1001/archinte.1996.00440150056006
226. Pemberton K, Melissari E, Kakkar V. The influence of tamoxifen in vivo on the main natural anticoagulants and fibrinolysis. Blood Coagul Fibrinolysis (1993) 4(6):935–42. doi:10.1097/00001721-199312000-00011
227. Verso M, Agnelli G, Barni S, Gasparini G, LaBianca R. A modified Khorana risk assessment score for venous thromboembolism in cancer patients receiving chemotherapy: the Protecht score. Intern Emerg Med (2012) 7(3):291–2. doi:10.1007/s11739-012-0784-y
228. Roselli M, Ferroni P, Riondino S, Mariotti S, Laudisi A, Vergati M, et al. Impact of chemotherapy on activated protein C-dependent thrombin generation–association with VTE occurrence. Int J Cancer (2013) 133(5):1253–8. doi:10.1002/ijc.28104
229. Rajkumar SV, Blood E, Vesole D, Fonseca R, Greipp PR; Eastern Cooperative Oncology Group. Phase III clinical trial of thalidomide plus dexamethasone compared with dexamethasone alone in newly diagnosed multiple myeloma: a clinical trial coordinated by the eastern cooperative oncology group. J Clin Oncol (2006) 24(3):431–6. doi:10.1200/JCO.2005.03.0221
230. Rajkumar SV, Hayman S, Gertz MA, Dispenzieri A, Lacy MQ, Greipp PR, et al. Combination therapy with thalidomide plus dexamethasone for newly diagnosed myeloma. J Clin Oncol (2002) 20(21):4319–23. doi:10.1200/JCO.2002.02.116
231. Sørensen HT, Mellemkjaer L, Olsen JH, Baron JA. Prognosis of cancers associated with venous thromboembolism. N Engl J Med (2000) 343(25):1846–50. doi:10.1056/NEJM200012213432504
232. Dvorak HF, Quay SC, Orenstein NS, Dvorak AM, Hahn P, Bitzer AM, et al. Tumor shedding and coagulation. Science (1981) 212(4497):923–4. doi:10.1126/science.7195067
233. Date K, Hall J, Greenman J, Maraveyas A, Madden LA. Tumour and microparticle tissue factor expression and cancer thrombosis. Thromb Res (2013) 131(2):109–15. doi:10.1016/j.thromres.2012.11.013
234. van den Berg YW, Osanto S, Reitsma PH, Versteeg HH. The relationship between tissue factor and cancer progression: insights from bench and bedside. Blood (2012) 119(4):924–32. doi:10.1182/blood-2011-06-317685
235. Mueller BM, Reisfeld RA, Edgington TS, Ruf W. Expression of tissue factor by melanoma cells promotes efficient hematogenous metastasis. Proc Natl Acad Sci U S A (1992) 89(24):11832–6. doi:10.1073/pnas.89.24.11832
236. Geddings JE, Mackman N. Tumor-derived tissue factor-positive microparticles and venous thrombosis in cancer patients. Blood (2013) 122(11):1873–80. doi:10.1182/blood-2013-04-460139
237. Rickles FR, Patierno S, Fernandez PM. Tissue factor, thrombin, and cancer. Chest (2003) 124(3 Suppl):58S–68S. doi:10.1378/chest.124.3_suppl.58S
238. Ueno T, Toi M, Koike M, Nakamura S, Tominaga T. Tissue factor expression in breast cancer tissues: its correlation with prognosis and plasma concentration. Br J Cancer (2000) 83:164. doi:10.1054/bjoc.2000.1272
239. van den Berg YW, van den Hengel LG, Myers HR, Ayachi O, Jordanova E, Ruf W, et al. Alternatively spliced tissue factor induces angiogenesis through integrin ligation. Proc Natl Acad Sci U S A (2009) 106(46):19497–502. doi:10.1073/pnas.0905325106
240. Hobbs JE, Zakarija A, Cundiff DL, Doll JA, Hymen E, Cornwell M, et al. Alternatively spliced human tissue factor promotes tumor growth and angiogenesis in a pancreatic cancer tumor model. Thromb Res (2007) 120(Suppl 2):S13–21. doi:10.1016/S0049-3848(07)70126-3
241. Bromberg ME, Konigsberg WH, Madison JF, Pawashe A, Garen A. Tissue factor promotes melanoma metastasis by a pathway independent of blood coagulation. Proc Natl Acad Sci U S A (1995) 92(18):8205–9. doi:10.1073/pnas.92.18.8205
242. Lechner D, Weltermann A. Chemotherapy-induced thrombosis: a role for microparticles and tissue factor? Semin Thromb Hemost (2008) 34(2):199–203. doi:10.1055/s-2008-1079261
243. Tesselaar MET, Romijn FPHTM, Van Der Linden IK, Bertina RM, Osanto S. Microparticle-associated tissue factor activity in cancer patients with and without thrombosis. J Thromb Haemost (2009) 7(8):1421–3. doi:10.1111/j.1538-7836.2009.03504.x
244. Zwicker JI, Liebman HA, Neuberg D, Lacroix R, Bauer KA, Furie BC, et al. Tumor-derived tissue factor–bearing microparticles are associated with venous thromboembolic events in malignancy. Clin Cancer Res (2009) 15(22):6830–40. doi:10.1158/1078-0432.CCR-09-0371
245. Thaler J, Ay C, Weinstabl H, Dunkler D, Simanek R, Vormittag R, et al. Circulating procoagulant microparticles in cancer patients. Ann Hematol (2011) 90(4):447–53. doi:10.1007/s00277-010-1111-1
246. Echrish H, Madden L, Greenman J, Maraveyas A. PO-84 expression of tissue factor (TF) and growth factor receptors on pancreatic cell lines: correlation with TF activity and cell invasion. Thromb Res (2010) 125:S188–9. doi:10.1016/S0049-3848(10)70134-1
247. Davila M, Amirkhosravi A, Coll E, Desai H, Robles L, Colon J, et al. Tissue factor-bearing microparticles derived from tumor cells: impact on coagulation activation. J Thromb Haemost (2008) 6(9):1517–24. doi:10.1111/j.1538-7836.2008.02987.x
248. Blom JW, Vanderschoot JP, Oostindier MJ, Osanto S, van der Meer FJ, Rosendaal FR. Incidence of venous thrombosis in a large cohort of 66,329 cancer patients: results of a record linkage study. J Thromb Haemost (2006) 4(3):529–35. doi:10.1111/j.1538-7836.2006.01804.x
249. Huang H, Korn JR, Mallick R, Friedman M, Nichols C, Menzin J. Incidence of venous thromboembolism among chemotherapy-treated patients with lung cancer and its association with mortality: a retrospective database study. J Thromb Thrombolysis (2012) 34(4):446–56. doi:10.1007/s11239-012-0741-7
250. Darmoul D, Marie J, Devaud H, Gratio V, Laburthe M. Initiation of human colon cancer cell proliferation by trypsin acting at protease-activated receptor-2. Br J Cancer (2001) 85(5):772–9. doi:10.1054/bjoc.2001.1976
251. Massi D, Naldini A, Ardinghi C, Carraro F, Franchi A, Paglierani M, et al. Expression of protease-activated receptors 1 and 2 in melanocytic nevi and malignant melanoma. Hum Pathol (2005) 36(6):676–85. doi:10.1016/j.humpath.2005.04.008
252. Ge L, Shenoy SK, Lefkowitz RJ, DeFea K. Constitutive protease-activated receptor-2-mediated migration of MDA MB-231 breast cancer cells requires both β-arrestin-1 and-2. J Biol Chem (2004) 279(53):55419–24. doi:10.1074/jbc.M410312200
253. Bromberg ME, Sundaram R, Homer RJ, Garen A, Konigsberg WH. Role of tissue factor in metastasis: functions of the cytoplasmic and extracellular domains of the molecule. Thromb Haemost (1999) 82(1):88–92. doi:10.1055/s-0037-1614634
254. Hembrough TA, Swartz GM, Papathanassiu A, Vlasuk GP, Rote WE, Green SJ, et al. Tissue factor/factor VIIa inhibitors block angiogenesis and tumor growth through a nonhemostatic mechanism. Cancer Res (2003) 63(11):2997–3000.
255. Sorensen BB, Rao LV, Tornehave D, Gammeltoft S, Petersen LC. Antiapoptotic effect of coagulation factor VIIa. Blood (2003) 102(5):1708–15. doi:10.1182/blood-2003-01-0157
Keywords: extracellular vesicles, apoptotic vesicles, membrane blebs, chemotherapy, thrombosis
Citation: Muhsin-Sharafaldine M-R and McLellan AD (2018) Tumor-Derived Apoptotic Vesicles: With Death They Do Part. Front. Immunol. 9:957. doi: 10.3389/fimmu.2018.00957
Received: 30 January 2018; Accepted: 17 April 2018;
Published: 07 May 2018
Edited by:
Ivan Poon, La Trobe University, AustraliaReviewed by:
Rong Xu, La Trobe University, AustraliaMuriel Moser, Free University of Brussels, Belgium
Copyright: © 2018 Muhsin-Sharafaldine and McLellan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander D. McLellan, YWxleC5tY2xlbGxhbkBvdGFnby5hYy5ueg==