 Miles C. Andrews
Miles C. Andrews Alexandre Reuben
Alexandre Reuben Vancheswaran Gopalakrishnan
Vancheswaran Gopalakrishnan Jennifer A. Wargo1,4*
Jennifer A. Wargo1,4*

95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 04 May 2018
Sec. Cancer Immunity and Immunotherapy
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.00946
This article is part of the Research Topic Cancer Plasticity and The Microenvironment: Implications for Immunity and Therapy Response View all 16 articles
Cancer research has seen unprecedented advances over the past several years, with tremendous insights gained into mechanisms of response and resistance to cancer therapy. Central to this has been our understanding of crosstalk between the tumor and the microenvironment, with the recognition that complex interactions exist between tumor cells, stromal cells, overall host immunity, and the environment surrounding the host. This is perhaps best exemplified in cancer immunotherapy, where numerous studies across cancer types have illuminated our understanding of the genomic and immune factors that shape responses to therapy. In addition to their individual contributions, it is now clear that there is a complex interplay between genomic/epigenomic alterations and tumor immune responses that impact cellular plasticity and therapeutic responses. In addition to this, it is also now apparent that significant heterogeneity exists within tumors–both at the level of genomic mutations as well as tumor immune responses–thus contributing to heterogeneous clinical responses. Beyond the tumor microenvironment, overall host immunity plays a major role in mediating clinical responses. The gut microbiome plays a central role, with recent evidence revealing that the gut microbiome influences the overall immune set-point, through diverse effects on local and systemic inflammatory processes. Indeed, quantifiable differences in the gut microbiome have been associated with disease and treatment outcomes in patients and pre-clinical models, though precise mechanisms of microbiome-immune interactions are yet to be elucidated. Complexities are discussed herein, with a discussion of each of these variables as they relate to treatment response.
Interest in defining factors that influence the outcome of cancer therapy has existed for as long as the therapies themselves. Traditionally, a highly tumor-centric focus has dominated, resulting in a now well-characterized yet still incomplete view of the complex molecular and cellular tumoral dynamics relevant to cancer progression and to treatment response.
Several of these factors, particularly the overall somatic mutational burden of the tumor, have gained traction and even potential clinical utility in the prediction of response to immunotherapy, notwithstanding ongoing concerns about their limited accuracy. Qualitative genomic characterization of tumors may also be very informative, however, information derived from such analyses is subject to the limitations imposed by sampling error and heterogeneous composition of synchronous tumors in patients with multiple metastases. Despite initial enthusiasm, an appreciation of the limitations of genomic characterization alone is emerging, and a more comprehensive analysis of the multitude of factors influencing therapeutic responses is critically needed.
In this mini review, we provide an overview of genomic factors implicated in the response to cancer immunotherapy, utilizing melanoma as a model “immunogenic” tumor from which the majority of empirical evidence derives. We will also discuss immune determinants of response and resistance, highlighting recent data regarding tumor immune cell co-evolution influenced by immunogenic factors arising from tumor cells, and the immunoediting effects of the responding immune infiltrate, both of which are impacted by intra- and inter-tumoral heterogeneity. In addition to this, we will complement studies of the tumor microenvironment to better delineate the crosstalk between the tumor microenvironment and overall host immunity with the microbiome, as this has been shown to influence outcomes ranging from tumor growth and immunity to treatment-related response and toxicity. Though discussion of the gut microbiome will predominate, we will also describe the potential impact of the intra-tumoral microbiome on resistance to cancer therapy, thus providing a full discussion of the intersection of tumor genomics, immunity, and the microbiome in shaping therapeutic responses, as summarized in (Table 1).
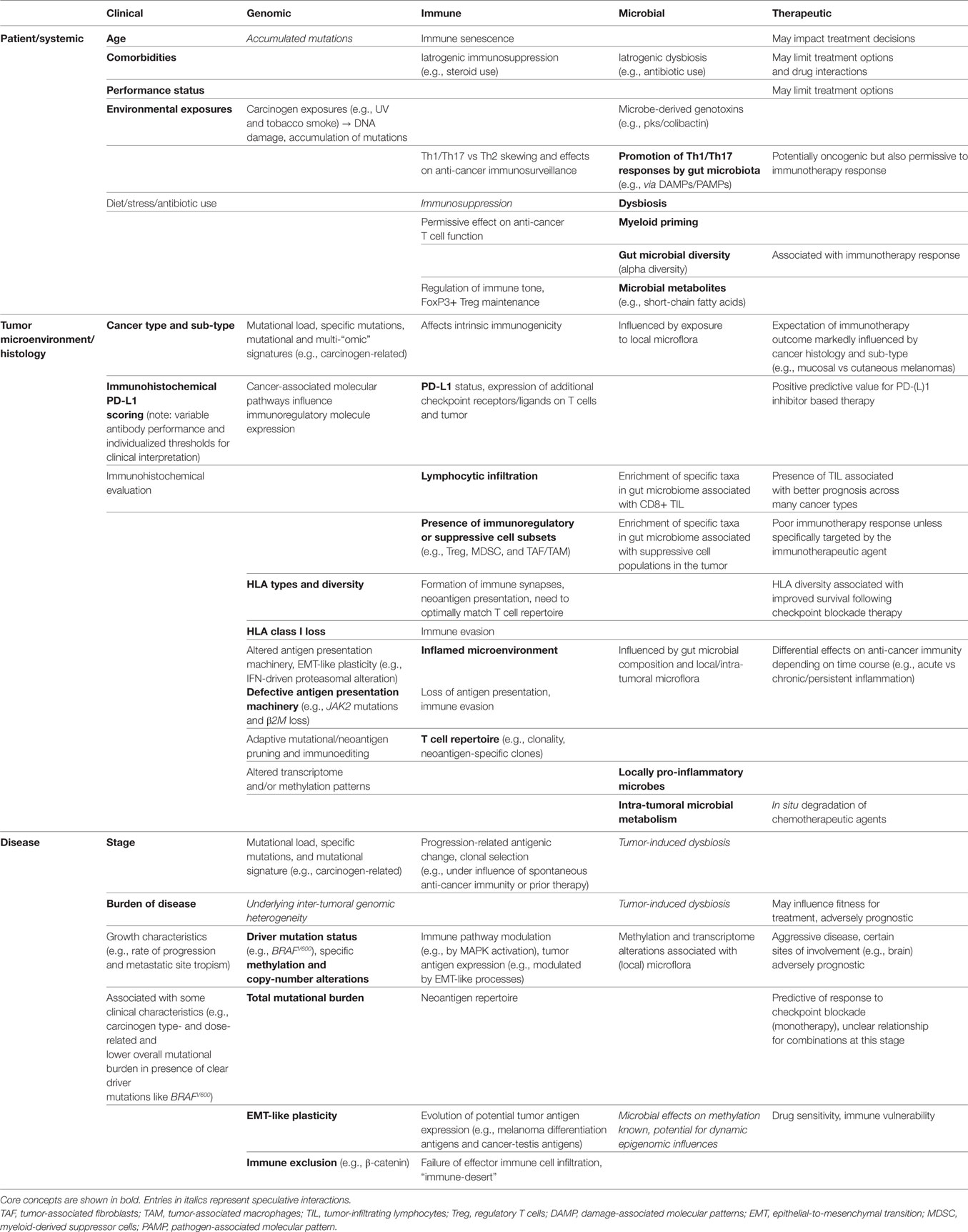
Table 1. Inter-relationships between clinical, genomic, immune, and microbial factors drawn from the patient (systemic), tumor microenvironment (histology), and disease-level domains, with associated influence on immunotherapeutic outcomes.
Long before the advent of modern genomic technologies, histologic sub-types of cancer were described, with differences in response to therapeutic intervention noted across these sub-types. This is well illustrated in melanoma, for which several clinicopathologic sub-types exist, including superficial spreading, acral, desmoplastic, and mucosal melanomas. With the advent of next generation sequencing, we have gained tremendous insight into the molecular underpinnings of these clinicopathologic observations and into the mechanisms driving differences between tumors themselves. Distinct genomic aberrations frequently define histologic sub-types and can confer notable differences in therapeutic sensitivity that have major clinical relevance (1, 2). Beyond this, other components of the tumor microenvironment have been noted to play a major role in therapeutic response and resistance, impacting upon tumor visibility and susceptibility (Figure 1), as discussed below.
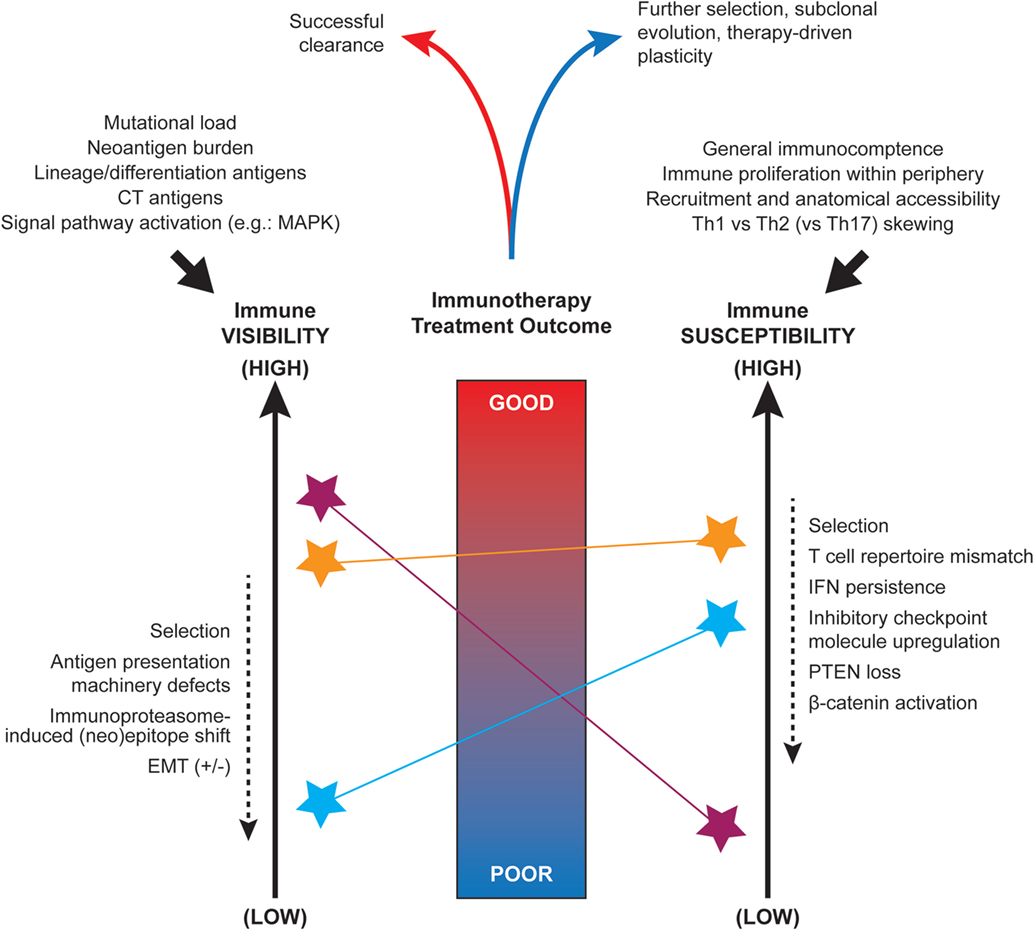
Figure 1. Factors influencing the immune visibility and susceptibility of tumors. Nomogram-conceptualization of the competing influences of tumor immune visibility (at left) and the susceptibility of tumor cells to immune attack (at right). Due to underlying intra- and inter-tumoral heterogeneity, distinct tumor cell sub-clones or microenvironments (denoted by colored stars) may display a range of visibility and susceptibility characteristics that must be integrated when predicting the overall outcome of spontaneous or immunotherapy treatment-induced anti-tumor responses. The initial set-point of immunogenicity is influenced by several factors including somatic mutations, antigen expression, and signal pathway activity (top left). The anti-tumor immune set-point is similarly influenced by a number of systemic factors such as availability of immune cell populations for recruitment and Th1 skewing (top right). Multiple factors have been implicated as dynamic modulators of these visibility and susceptibility states (dashed arrows at sides).
In addition to their influence on oncogenic signaling and proliferative potential, genomic mutations present in melanoma and other cancers may have a profound impact on anti-tumor immunity and can contribute to the “visibility” of a tumor to the immune system (3, 4). This is largely shaped by the antigenic characteristics of the tumor cells allowing their recognition by the immune system, but may be shaped by other influences of these oncogenic mutations on the tumor cells themselves as well as the microenvironment. This is important, as therapeutic targeting of oncogenically activated signaling pathways may alter anti-tumor immunity. A key example of how genomic alterations may impact tumor visibility is illustrated in the case of BRAF-mutant melanoma. Early observations demonstrated a link between MAPK signaling and the expression of melanoma-associated antigens (5, 6), with subsequent data revealing brisk infiltration of tumors with T lymphocytes in the setting of treatment with BRAF inhibitor-based therapy (3, 7). Interestingly, inhibition of oncoproteins such as BRAF may also be associated with increased expression of HLA molecules and heat shock proteins, which can further contribute to a tumor’s visibility (8, 9).
More generally, tumor cell immune visibility is fundamentally dependent on the presence (or absence) of molecular moieties that can be recognized by components of the host immune system. Tumor cell self-antigens represent the basis of cognate interactions with cellular elements of the adaptive immune system, but have varying degrees of tumor cell specificity. Such antigens include differentiation or lineage-specific antigens, aberrantly expressed antigens either absent or found at only low levels in adult tissues (3), or may be truly tumor cell-specific neoantigens derived from the protein products of somatically mutated genes. Tumor neoantigens are felt to predominantly mediate effective anti-tumor immune responses because neoantigen-reactive T cells escape deletion mechanisms during T cell ontogeny, and respond to these antigens as “foreign” rather than “self” (10, 11). In addition, epithelial-to-mesenchymal-like (EMT-like) plasticity in melanoma is thought to contribute to functional and antigenic variation that has the potential to influence the efficacy of immune-based therapies (12). Given the prominent role of the lymphocyte response to MAPK blockade in BRAF-mutated melanoma, these EMT-like shifts in melanoma cell state may well also contribute to BRAF inhibitor resistance at least in part by altering melanoma cell visibility via this antigenic shift (13).
Tumor genomics gains specific relevance to immune visibility in light of the significance of tumor-specific neoantigens in shaping immune responses. The mutational landscape varies across tumor types (14), and is shaped by factors influencing carcinogenesis such as UV irradiation and smoking (14). Interestingly, responses to immunotherapy are positively associated with the mutational burden of each particular tumor type, evidenced by higher response rates and clinical benefit in tumor types with an overall high mutational burden, such as melanoma, non-small cell lung cancer (NSCLC), clear cell renal cell cancer, and genitourinary cancers (14, 15). A recent study of 151 patients with predominantly melanoma (34%) or NSCLC (24%), mostly treated with anti-CTLA-4, anti-PD-1, or anti-PD-L1 blockade therapy, assessed the relationship between tumor mutational burden measured by hybrid capture next generation sequencing and clinical outcomes. Using pre-defined cut-offs, patients with higher tumor mutational burden experienced higher response rates and longer progression-free and overall survival than those with low to intermediate tumor mutational burden (16). These results were broadly applicable to the sub-group of patients (42% of the overall cohort) with non-melanoma/non-NSCLC histologies. Interestingly, the relationship between higher mutational load and better treatment outcomes was not evident for patients who received combined anti-CTLA-4 and anti-PD-1 therapy. Further supporting the tumor mutational burden-response relationship are tumors with microsatellite instability and mismatch-repair deficiency, which demonstrate an increased sensitivity to checkpoint blockade likely to be related to an associated increase in mutational load and neoantigen burden (17, 18). Indeed, demonstration of microsatellite instability-high or mismatch-repair deficient tumors upon biomarker testing forms the basis for the first site-agnostic drug approval made by the FDA, for anti-PD-1 therapy. The practical limitations of measuring tumor mutational burden for use as a predictive biomarker before therapy have been significantly met by robust estimation of overall mutational load using data obtained from targeted next generation sequencing technologies that are now relatively widely accessible in the clinic (19, 20). In addition, cancer-gene panel mutational profiling by liquid biopsy represents a promising alternative mutational burden-related methodology for predicting immunotherapy response, as reported in an analysis of NSCLC patients enrolled in clinical trials of the anti-PD-L1 agent atezolizumab (21).
However, the relevance of a tumor cell’s mutational repertoire to immune dynamics is moderated by additional factors that affect expression, processing, and intrinsic immunogenicity of any putative neoantigen. The complex processes involved in cleaving a peptide, loading it onto an MHC molecule, transporting it to the cell surface, and ensuring its stability are essential to induce the antigenic T cell responses required for tumor clearance. Epitope production is influenced by the molecular chaperones and proteasomal machinery involved in protein processing; not all epitopes produced may be immunogenic, and a form of stochastic competition between display of immunogenic and non-immunogenic epitopes may ensue. The initially beneficial IFN-rich microenvironment of a T cell-inflamed tumor ultimately promotes mismatch between the neoepitope and T cell repertoires due to a shift from utilization of the constitutive proteasome to the immunoproteasome, thereby influencing tumor visibility (22). Furthermore, defects in β2-microglobulin expression can further impair antigen processing and display, affecting stable expression of MHC I molecules for their adequate surface expression and subsequent T cell recognition (23, 24). MHC class I haplotype loss or overall downregulation has been associated not only with altered tumor cell growth characteristics, but also facilitates evasion of immune surveillance (25). In melanoma, MHC class I internalization induced by BRAF V600E has also been described, suggesting another potential mechanism underlying the enhanced tumor visibility resulting from BRAF inhibitor therapy (9). Specific MHC class I loss has also been demonstrated in the evasion of T cell therapy for colorectal cancer (26). In addition, not all neoantigens bind MHC with high affinity, and the combinatorial match between neoantigen and MHC molecules expressed in the same cell determine how intrinsically immunogenic a neoantigen can be.
Studies of patient samples and ex vivo evidence strongly support the dominance of mutational neoantigens as targets for lymphocyte recognition of tumor, even in cancer types with lower overall mutational burden (10, 27–29). The importance of considering the available HLA sub-types and the T cell repertoire also present in the tumor, and their importance as major determinants of the tumor sub-clonal pruning that results from ongoing cycles of immune recognition, attack, and clearance, is now being appreciated (30). Computational methods exist to infer neoantigen expression and HLA binding characteristics from genomic and transcriptomic data (31, 32), and have been shown to act as a surrogate for treatment response in the context of checkpoint blockade immunotherapy in melanoma (10, 29). In fact, the sole presence of a more diverse array of HLA molecules (i.e., HLA heterozygosity), presumably linked to the ability to present a wider breadth of neoantigens, has recently been associated with increased survival in melanoma and lung cancer patients treated with immune checkpoint blockade (33). Knowledge of the mutational landscape of a tumor is thus of great importance to estimating the outcome of both targeted and immune therapies, however, measures of mutational and neoantigen burden alone do not predict immunotherapeutic outcome perfectly and results have been conflicting in separate cohorts (29, 30, 34). Similarly, though predictive approaches have been utilized to identify neoantigen candidates based on somatic mutations, these algorithms remain suboptimal, likely due to the numerous moderating factors described above (35). Accordingly, predictive approaches are now being paired with additional filters provided by proteasomal cleavage algorithms, as well as expression data to evaluate somatic mutations which are adequately expressed. A smaller number of neoantigen candidates can then be tested with autologous lymphocytes through molecular cloning of tandem minigenes comprising numerous expressed neoantigens (11).
A tumor’s visibility to the immune system does not automatically imply its clearance, and numerous distinct factors can also influence its susceptibility to immune attack, which may be related to or completely independent of visibility.
In recent work, Chen and Mellman described the different immune infiltration profiles associated with response, which were classified as “inflamed,” “immune-excluded,” and “immune-desert” (36). Tumor immune susceptibility is inherently greater in patients of the “inflamed” type, where immune cells are present and capable of exerting their anti-tumor effects. Although immune visibility is critical to the establishment of an inflamed tumor microenvironment, the outcome of tumor inflammation can be influenced by a series of factors which build on a tumor’s visibility, such as chemokines, pro-inflammatory cytokines, and effector T cell density and function. Conversely, immunosuppressive cytokines and the presence of pro-tumor immune inhibitory cell types, such as tumor-associated (M2) macrophages, regulatory T cells (Treg), and myeloid-derived suppressor cells (MDSCs) can lead to development of an immune-desert tumor microenvironment, clearly detrimental to response.
Recently, an extensive genome-scale in vitro CRISPR/Cas9 screen revealed genes involved in antigen presentation and IFN-signaling to be most relevant to the ability of CD8 T cells to kill melanoma cells (37). IFN-γ signaling defects have been repeatedly implicated in cancer immunotherapy failure, including copy-number losses of IFN-γ pathway genes (principally IFNGR1/2, IRF1, and JAK2) in patients failing to respond to anti-CTLA-4 therapy (38). Loss-of-function mutations in JAK1 and JAK2 have also been described in the tumors of melanoma patients with either primary (39) or secondary (24) resistance to anti-PD-1 therapy. It must be noted that while tumoral inflammation appears a common if not necessary component of the anti-cancer immune response (regardless of therapeutic agent used), persistent activation of IFN-driven inflammatory signals adaptively leads to upregulation of inhibitory checkpoint molecules on lymphocytes and generation of an immunosuppressive microenvironment (40, 41). Thus, optimal immunotherapeutic outcomes may require more complex sequencing and/or intermittent dosing strategies than have yet been studied in patients.
In keeping with the concept of immune-inflamed and immune-excluded or immune-desert phenotypes described by Chen and Mellman, microenvironmental characteristics affecting lymphocyte entry and trafficking are critical to the efficacy of immunotherapy. Baseline lymphocytic infiltrate, particularly CD8 T cell density, is predictive of response to checkpoint inhibitor monotherapy (42), with early on treatment biopsies being more highly predictive of response than at baseline (43). Such “snapshots” of the immune infiltrate represent the combination over time of factors affecting T cell recruitment and T cell exclusion, such as a tumor cell-intrinsic activation of β-catenin (44). In fact, in work by Spranger and colleagues, it was demonstrated that the absence of tumor-derived β-catenin signaling allows production of CCL4, a chemokine which aids dendritic cell recruitment and thereby promotes T cell priming and anti-tumor responses (44). Furthermore, loss of expression of genes such as PTEN may influence the immune response through increased expression of immunosuppressive cytokines, such as VEGF and CCL2 (4). In fact, PTEN loss in melanoma patients was associated with progression on PD-1 blockade, possibly due to this mechanism, with CD8 T cell exclusion shown in regions of the tumor devoid of PTEN expression. Angiopoietic factors such as VEGF are frequently secreted by tumors and contribute to treatment failure (45). Pre-clinical models and translational studies of combined immune checkpoint blockade and anti-angiogenic agents suggest a potentially complex effect on tumor immunity, including beneficial effects on DC function and suppressive capacity of intra-tumoral MDSCs (46), enhanced anti-tumor humoral immunity (47), and increased lymphocyte trafficking and recruitment (47, 48).
Failure of spontaneous anti-tumor activity may largely be due to a dysfunctional “exhausted” T cell state associated with high expression of negative regulatory checkpoint molecules that are nonetheless amenable to treatment with modern checkpoint blockade immunotherapy (49). A more comprehensively inhibited T cell phenotype, typically with expression of numerous inhibitory checkpoint molecules including TIM-3, LAG-3, and others, may contribute to resistance to checkpoint inhibitor therapies in current clinical use (50). The presence of Treg as a key inhibitory factor on the anti-tumor response is, conversely, associated with poor response to checkpoint blockade.
That the majority of treatments for advanced cancers fail to produce curative outcomes is testament to the sheer diversity of cancer cell sub-populations present, limited in number only by the ability of our technologies to unravel their complexities at a molecular level. Heterogeneity of tumor cells, infiltrating immune cells, local vasculature, chemokine/cytokine gradients, and the underlying genetic basis for these variations are thus highly relevant to multiple aspects of immunotherapy efficacy. Tumoral heterogeneity influences both visibility and susceptibility of a tumor to immune attack, and has been described across cancer types (51–53).
Heterogeneity may arise from stochastic variation between cell sub-populations as cancer cells divide and accumulate mutations, or as a consequence of more plastic processes which shape cell state, gene expression, cellular function, and phenotype in response to prevailing selective processes over time or in different microenvironments (54–56). Heterogeneity may also arise as a direct consequence of sub-clonal immunoediting that occurs during both spontaneous and treatment-related anti-cancer immune responses, leading to non-uniform expression of target antigen (57) or essential antigen presentation machinery (25) across tumors.
The impact of tumor heterogeneity was recently highlighted in localized lung adenocarcinoma, demonstrating that a substantial proportion of tumor mutations are sub-clonal, i.e., restricted to regions of a tumor (58). This pattern extended to the neoantigens derived from these mutations, and patients with the highest proportion of sub-clonal neoantigens experienced shortened disease-free survival. Similar findings were seen when studying the T cell repertoire, where patients with the most heterogeneity in their T cell repertoire fared worst, highlighting the direct implications of genomic and immune heterogeneity on patient outcome (58).
Future treatment strategies will need to consider the effects of pre-existing tumoral heterogeneity as well as the adaptive treatment-induced changes that contribute to treatment failure. Furthermore, treatment strategies may also exert unique effects on tumor heterogeneity. In a recent melanoma study, prior therapy did not affect genomic inter-tumor heterogeneity whereas immune heterogeneity was more limited in patients previously treated with checkpoint blockade (52). As late stage patients become increasingly heavily pre-treated, the effects of these prior therapies on tumor heterogeneity will also need to be taken into consideration.
Although undeniably important, the metabolic, vascular, and immune dynamics active in the tumor microenvironment are only some of the contributing factors. It is now quite clear that overall host immunity as well as environmental influences (Figure 2) can shape therapeutic responses (59), and these factors will be discussed herein.
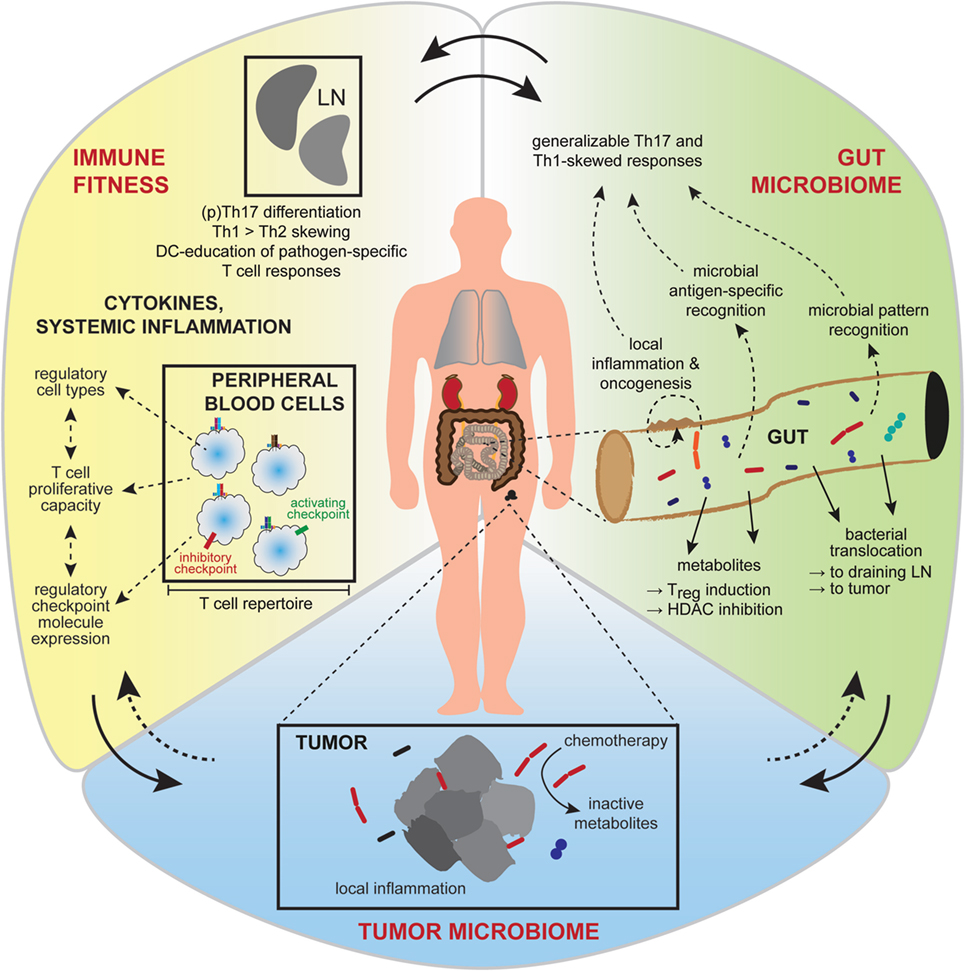
Figure 2. Interactions between the gut and intra-tumoral microbiota and the systemic immunity affect treatment outcome. Broader influences on tumor growth and responsiveness to immunotherapy are now realized, including contributions from the gut microbiome, the tumor microbiome, and systemic factors affecting general immune fitness. Interactions between these conceptual compartments are complex and incompletely understood. Gut microbiota (green) have diverse metabolic and antigen or pattern-molecule immunogenic effects on local gut inflammation, the effects of which contribute to local carcinogenesis or can become generalized to affect cancer growth and immunity at distant body sites. Favorable immune fitness (yellow), characterized by overall skewing toward cellular immune responses, a permissive cytokine milieu and activation-biased representation of anti-tumor, and regulatory immune cell types, is important for anti-tumor responses. The intra-tumoral microbiome (blue) is of emerging significance, having local inflammatory and metabolic effects that influence therapeutic sensitivity.
Effective anti-tumor immune responses require exposure of the tumor microenvironment to a wide network of innate and adaptive immune effector populations recruited from the systemic circulation. These cells must recognize and target tumor cells for elimination, based on the visibility factors described previously, in a critical process termed “immunosurveillance” (60). Three core phases of immunosurveillance have been described, spanning elimination of susceptible tumor cells through equilibrium (in which visibility-susceptibility mismatch or selective pruning of the most immune-susceptible tumor sub-clones leads to an anti-cancer stalemate), to escape (in which selection of low-visibility low-susceptibility tumor cells facilitates renewed tumor progression) (61). These dynamic phases occur spontaneously, but are undoubtedly influenced by exposure to immunotherapies, as shown in a parallel genomic and immune analysis of tumors from patients with advanced melanoma who received treatment with the anti-PD-1 agent nivolumab (30). In this study, clear patterns of mutational contraction and T cell clonal expansion occurred in what appeared to be a refocusing of the immune-cancer interaction and elimination of neoantigen-expressing sub-clones in patients who responded to therapy. While the specific cellular interactions that characterize each phase of immunosurveillance occur within the tumor microenvironment, the immune cells involved are sensitively dependent on adequate supply from the systemic compartment, and a prevailing immune phenotype conducive to anti-tumor activity (which may be therapeutically modifiable).
Recent work by Spitzer and colleagues provides key evidence supporting the direct relevance of systemic immune function to cancer immunotherapy (62). In this study, extensive high dimensional immune profiling using mass cytometry was performed in a MMTV-PyMT breast cancer mouse model to explore the dynamics of multiple immune cell populations in response to either effective, or ineffective, anti-cancer immunotherapy, in multiple body compartments. Many immune subsets were found to proliferate within the tumor microenvironment during the initiation of immune responses. Importantly, however, the proliferation of multiple immune cell populations, including B cells, NK cells, dendritic cells, and effector/memory T cells, during active tumor rejection was primarily sustained in secondary lymphoid organs and not the intra-tumoral compartment, indicating the significance of the systemic compartment to maintenance of effective anti-cancer immune responses (62).
Myeloid-derived suppressor cells have garnered much interest in recent years for their immune-suppressive capabilities across various cancer types. MDSCs are a phenotypically heterogeneous group of cells comprised of immature myeloid cells, and broadly divided into monocytic and granulocytic sub-types (63). MDSCs have a potent ability to suppress T cell responses through numerous specific mechanisms in lymphoid organs, such as production of indoleamine 2,3 dioxygenase or arginase-1, which locally deplete crucial amino acids such as tryptophan and arginine, thereby rendering T cells functionally anergic. MDSCs may also inhibit T cell responses through production of immunosuppressive cytokines including TGF-β and IL-10, or generation of reactive oxygen species (ROS) (64). Because of this ability to inhibit T cell responses and promote tumor development, MDSCs have been suggested to be a key therapeutic target in cancer. Though MDSCs are present in the tumor microenvironment and tend to increase with cancer development, their characterization in the periphery has become an area of intense investigation, including studies of their relationship to clinicopathologic attributes and patient outcome. Overall, MDSCs are generally more abundant in the peripheral blood of cancer patients compared with healthy subjects. However, their frequencies also increase from early stage to late stage disease and with higher histological grade. These trends have been observed in numerous histologies such as renal cell cancer (65), colorectal carcinoma (66), melanoma (67), as well as gastric cancer (68). Interestingly, higher frequencies of MDSCs in the periphery have also been predictive of patient relapse in breast cancer (69), melanoma (67, 70), differentiated thyroid cancer (71), glioblastoma (72), head and neck squamous cell carcinoma (73), pancreatic cancer (74), prostate cancer (75), and renal cell carcinoma (76). Their increased frequencies in the circulation have also been tied to the development of metastases in melanoma and colorectal cancer (66). Together, the somewhat graded relationship between MDSC abundance and cancer progression, including metastasis, suggests that circulating MDSCs at least reflect the immunosuppressive status of the tumor microenvironment, and likely fulfill a more direct role in the development of a systemically suppressed immune response. Unfortunately, inconsistency in the classification and functional characterization of MDSCs has limited our ability to accurately enumerate and isolate them, highlighting some of the challenges in translating the therapeutic targeting of MDSCs to the clinic.
Despite the importance of immune fitness in therapeutic response, our ability to assess anti-tumor immune responses in the peripheral circulation remains somewhat limited to date and uncertainties remain regarding the contributions made by immune populations at different times, and at different sites. Insofar as upregulation of inhibitory checkpoint molecules is associated with prior antigen exposure and activation, PD-1 expression on circulating T cells was found to enrich for neoantigen-specific T cells in the peripheral blood of melanoma patients (77). However, data showing that higher clonality of the T cell repertoire resident within tumors before therapy was associated with better outcome to PD-1 blockade (34, 42) suggests that much of the relevant effector immune population is already intra-tumoral even before therapy. Thus, the proportion of tumor-specific T cells present in the general circulation at any instant may be relatively low, limiting what can be concluded about therapeutic response from examination of the periphery in isolation, with currently available tools. Looking even more generally, the broad functional status of the adaptive immune system, defined as Th1-, Th17-, or Th2-skewing of circulating immune cells, may be more reliably associated with the prevalent immunophenotype of tumor-infiltrating lymphocytes (TIL), reflecting the likelihood of effective cytotoxic rather than tolerogenic cellular immune outcomes.
Systemic immunity is shaped by interactions with our environment, and there is now clear evidence that the gut microbiome contributes to the establishment and maintenance of systemic immune tone (78, 79). As the largest commensal microbial community (80, 81), the gut microflora has been extensively studied as a trigger for local inflammation in non-malignant conditions such as inflammatory bowel disease (82, 83). Overall, these microbes present a significant challenge to the host’s immune defenses, which must regulate tolerance to beneficial microbes while guarding against harmful pathogens. Moreover, recent evidence suggests that the gut microbiome plays a significant role in cancer development and response to cancer therapy as discussed in the following sections.
Gut microbiota represent a double-edged sword that can promote or inhibit cancer development, with both individual bacterial taxa and overall bacterial dysbiosis implicated in oncogenic initiation and progression. Helicobacter pylori–particularly those containing the virulence factor cagA–have been extensively characterized as an oncogenic initiator, particularly in gastric adenocarcinoma (84). In a murine model, a potentially pathogenic enterotoxigenic Bacteroides fragilis-induced a STAT3-dependent, Th17-mediated colitis associated with colonic tumor formation in pre-disposed mice; colitis and tumor formation were prevented by administration of blocking antibodies to IL-17 and IL-23 (85). Escherichia coli strains may harbor the polyketide synthase genomic island (pks), which encodes a genotoxin called colibactin that induces DNA damage in murine colonocytes. Furthermore, E. coli that are pks+ are more frequently identified in colon cancer patients compared with healthy controls (86, 87). On the other hand, bacteria can also be tumor suppressive as demonstrated by Butyrivibrio fibrisolvens, which resulted in colorectal tumor attenuation by producing copious amounts of butyrate in the presence of a high-fiber diet in a rat-azoxymethane model. Importantly, population-based metagenomic analyses in colon cancer patients have also revealed differential enrichment of bacterial taxa (especially Fusobacterium) in colorectal tumors when compared with controls (88–91).
As opposed to microbial taxa that drive oncogenesis in a highly penetrant manner, the relationship between dysbiosis and cancer is more complex and multifactorial. Dysbiosis is influenced by several extraneous factors including diet, antibiotic use, and smoking (92). In pre-clinical models of colon carcinoma, both germ-free status and antibiotic treatment have been found to be associated with reduced incidence of tumors (93–96). A dysbiotic microbiota can also influence cancer development at distant sites such as the liver (97) and pancreas through pro-inflammatory microbe-associated molecular patterns and metabolites released into the systemic circulation (98).
Several murine studies have established a clear requirement for a diverse and intact intestinal microbiota to achieve optimal response to distinct cancer treatment modalities through effects on both the innate and adaptive arms of the immune system. The gut microbiome is now implicated in modulating responses across a wide range of cancer therapies, including intra-tumoral therapy (99), chemotherapy (99, 100), and immune checkpoint blockade (101–103). Attempts to define the mechanisms underlying microbial associations with the efficacy of cancer therapies have revealed both microbe-specific and microbe-agnostic influences (Figure 2). For instance, administration of LPS alone, in the presence of its cognate pattern recognition receptor, largely restored the efficacy of intralesional immunotherapy administered to either germ-free or antibiotic-ablated mice implanted with melanoma or colon cancer (99). In these models, systemic microbial priming of myeloid lineage cells appeared essential to their subsequent intra-tumoral accumulation and production of pro-inflammatory and chemotactic cytokines or ROS, without which an effective secondary T cell infiltrate could not be recruited. Interestingly, responses to chemotherapy may be facilitated by disruption of the integrity of the gut epithelial barrier with subsequent bacterial translocation of Enterococcus hirae and Lactobacillus johnsonii and priming of immune responses demonstrated in a murine model (100). Bacteria such as E. hirae and Barnesiella intestinihominis can also affect immune responses directly at the tumor site, with depletion of intra-tumoral Treg and accumulation of γδT cells, respectively (104).
The initial demonstration that several bacterial taxa were associated with response to immune checkpoint blockade came from murine studies, which implicated Bacteroides thetaiotaomicron and B. fragilis in the case of CTLA-4 blockade (101) and Bifidobacterium, in the case of PD-L1 blockade (103). These studies were followed by several analyses of patient cohorts that confirm a clear role for the gut microbiome in modulating responses to immune checkpoint blockade across cancer types (102, 105). A reciprocal relationship between the mid-level taxa Clostridiales (favorable) and Bacteroidales (unfavorable) and likelihood of response were recently reported in a large cohort of melanoma patients (102). Importantly, fecal microbial transplantation into germ-free mice using stool from responding patients resulted in enhanced tumor control when compared with donor stool from non-responding patients. Other population-level studies have also reported similar findings with regards to over-representation of the Faecalibacterium genus of the Clostridiales order in pre-treatment samples of responders to anti-CTLA-4 and combination anti-CTLA-4 plus anti-PD-1 immunotherapies (106, 107). In another study, Bifidobacterium was also found to be enriched in melanoma patients who were responding positively to anti-PD-1 therapy, analogous to earlier results implicating this taxon in a murine model of PD-L1 (105).
Interestingly, the bacterial taxa identified in these human studies differ somewhat from those identified in murine experiments and even across the patient cohorts, suggesting the need for additional studies to address the significance of geographical and other variables influencing microbiome composition and response, and to confirm unifying taxa or functionalities to take forward to clinical development. These studies also imply that administration of single bacterial species may not be reliably effective in modulating responses to immunotherapy.
Our understanding of the influence of the gut microbiome on immunity and therapeutic responses is evolving, and it is evident that micro-organisms may share functionalities that convey immunomodulatory properties that are not immediately evident from taxonomic discovery. Indeed, several investigators have demonstrated a key integrative role of microbial metabolites such as short chain fatty acids (SCFAs) produced by microbial fermentation of undigested complex carbohydrates in mediating the effect of commensal bacteria on immune tone (108, 109). In fact, a significant proportion of metabolites found in human plasma are microbiome-derived and can affect immune cells by influencing their metabolic circuits or engaging with metabolite-specific receptors. SCFAs act as signaling molecules, by inhibiting histone deacetylases (HDAC). Their action on lymphocytes and neutrophils is mediated via the blockade of NF-κB and the subsequent downstream production of pro-inflammatory TNF (110). Importantly, SCFAs promote homeostasis by regulating the size and function of the colonic FoxP3+ Treg pool in an HDAC-dependent manner (108, 109). SCFAs can also exert a regulatory effect by signaling through G protein-coupled receptors, resulting in the limitation of neutrophil chemotaxis (111) and expansion of Treg function (112). Therefore, it is also not surprising that these metabolites can directly affect cancer cells by impacting immunosurveillance (113).
At the population-level, numerous studies have identified high diversity of the gut microbiota as being associated with improved survival outcomes in allogeneic hematopoietic stem cell transplantation patients, together with relatively lower rates of graft-vs-host disease (114, 115). Consistent with these findings, a beneficial effect was also reported in the context of anti-PD-1 therapy in melanoma patients, wherein high diversity of the gut microbiome at baseline was found to be associated with significantly improved progression-free survival rates (102). Importantly, a similar provocative observation was also made in the context of lung, renal, and bladder cancer patients where disruption of microbial diversity of the gut was found to have a detrimental effect on the efficacy of immune checkpoint blockade (116). In these studies, the authors demonstrated that antibiotic usage shortly before, during or shortly after the initiation of treatment with immune checkpoint blockade was associated with significantly reduced progression-free and overall survival. In addition, metagenomic sequencing implicated the species Akkermansia muciniphila to be abundant in responders to anti-PD-1, and capable of restoring its efficacy in germ-free mice transplanted with non-responder patient stool (116).
In addition to the gut microbiome, bacteria within tumors themselves may influence cancer development as well as therapeutic responses. This has been studied most extensively in colorectal cancer, where certain bacterial taxa such as Fusobacterium nucleatum and Streptococcus bovis have been associated with primary tumors (88, 117, 118) as well as in metastatic sites (119). Fusobacterium, specifically F. nucleatum, is enriched in colorectal carcinomas relative to normal colonic tissues (88, 89). The abundance of F. nucleatum within colon cancer tissues inversely correlates with recurrence-free survival, and appears adversely prognostic, comparable to increasing AJCC stage (120). Profiling the fecal microbiota or screening for known microbial markers associated with colon carcinogenesis such as the genotoxin colibactin and its encoding genotoxin cluster pks found in oncogenic E. coli, may provide a novel strategy for population screening, and may additionally provide clues as to the underlying mechanisms driving such microbial associations with the accumulated genetic damage that characterizes malignancy (121, 122). In this regard, detailed study of the molecular products of the pks island confirm that colibactin directly damages DNA (122) and mediates pks+ E. coli promotion of tumor formation in a murine ApcMin/+; Il10−/− model (123). While chronic DNA damage is undesirable, it is clear that a high mutational burden may in fact be beneficial in patients receiving checkpoint blockade immunotherapy, thus the relevance of such microbial genotoxicity on contemporary treatment outcomes warrants particular study. Furthermore, certain pathogenic taxa are linked to enrichment of inflammatory and DNA damage-response pathways in tumor transcriptomes, together with a distinct methylation and microsatellite instability profile (124). In light of these data, it is probable that the genomic and immune characteristics of intestinal tumors may be sensitively linked to the geographic microbial niches in which they arise.
Importantly, therefore, bacterial-associated molecular alterations in tumors span genomic, epigenetic and immune domains with the immunomodulatory effects of tumor-associated microbes appearing equally as diverse as the observed genomic and biochemical effects. In the context of Fusobacterium, direct molecular interaction between the bacterial Fap2 protein and TIGIT present on human NK cells contributes to tumor immune evasion; this interaction was shown to inhibit TIGIT-mediated activation of NK cell killing of colon adenocarcinoma cells, and was more generally suppressive of TIL (NK and T cells) killing using patient-derived matched TIL and tumor cells from melanoma patients (125). Furthermore, F. nucleatum appears to promote colorectal cancer cell chemoresistance to select agents in a complex multi-step sequence of molecular changes involving TLR4 and MYD88 activation and culminating in activation of autophagy (120). Colonic Th17-responses represent a common and partially unifying feature of many microbiota-associated local and systemic inflammatory states and have been associated with poor-responses to anti-cancer therapies (100). As previously noted, colon cancer formation associated with enterotoxigenic B. fragilis has been shown to involve Stat3-driven colitis and induction of a Th17 response that was prevented by IL-17 and IL-23 blockade in mouse models (85). B. fragilis-induced Th17-driven tumorigenesis involves the promotion of a suppressive myeloid environment characterized by monocytic MDSCs and consequently suppressed T cell proliferation (126). While Th17 skewing may thus form a major contribution to carcinogenesis and influence systemic chemotherapeutic responses in non-intestinal tumor models, these same mechanisms, and the specific effect of B. fragilis, were required for the efficacy of anti-CTLA-4 checkpoint blockade immunotherapy (101). This emphasizes the new complexity that has arisen with the advent of checkpoint molecule-targeted immunotherapies, for which the distinction between “favorable” and “unfavorable” microbiota is potentially reversed depending on whether the context at hand is one of cancer development, or immunotherapy-based treatment of an already-established cancer.
In addition to their roles in carcinogenesis and immunomodulation, intra-tumoral bacteria may also modulate responses to cytotoxic cancer therapy. Mycoplasma hyorhinis, which was associated with fibroblasts in the tumor microenvironment in pancreatic ductal adenocarcinoma tumors, was found to be able to direct drug metabolism and diminish the efficacy of gemcitabine. Further analyses of bacterial genes implicated the enzyme cytidine deaminase contained in the Gammaproteobacteria class to be necessary and sufficient to mediate conversion of gemcitabine to its inactive form, by expression of the long isoform of the enzyme cytidine deaminase, in a colon cancer murine model (127). Depletion of bacteria within the tumor and a robust anti-cancer response to gemcitabine were noted in tumor-bearing mice treated with the combination of gemcitabine and ciprofloxacin delivered directly to the tumor site. A high prevalence of Gammaproteobacteria was subsequently identified in pancreatic adenocarcinoma samples from patients, and retained the ability to confer gemcitabine resistance after ex vivo co-culture with colon cancer cell lines (127). How these intra-tumoral bacteria interact with infiltrating immune cells has not been completely elucidated.
Direct spatial microbe-tumor interactions are not only relevant to gastrointestinal cancers. Recent analyses of the microbiota present in breast cancer tissue compared with normal breast tissue revealed distinct microbial communities, driven by a lower abundance of Methylobacterium in cancerous tissues (128). The authors performed a parallel comparison of microbiota present at distant sites, with the provocative finding that urinary microbiota were also distinct between cancer patients and controls, even after correction for menopausal status. The mechanisms through which bacteria induce carcinogenesis may include induction of inflammation (85), altered cell signaling (129) and inhibition of T cell and natural killer cell responses (125), however the precise role of intra-tumoral bacteria in carcinogenesis across tumor types remains incompletely elucidated.
Our future conceptualization of what matters for good outcomes to cancer immunotherapy requires a thoroughly integrated understanding of what contributes to cancer formation and immune evasion in the first place. Tumor mutational load provides an instructive example; while a high tumor mutational load is clearly important for response to checkpoint blockade immunotherapy, it still lacks adequate negative predictive value to be trusted in the clinic, and performs poorly for combination checkpoint blockade. If highly mutated tumors were truly (simplistically) vulnerable to immune clearance, it should be considered remarkable that they are observed at all. More likely, the snapshot measurement of mutational load is limited by numerous other factors, such as the neoantigen characteristics of the available pool of mutations, genomic methylation and transcriptome patterns, the intrinsic immunomodulatory effects of the tumor over time, and the extrinsic immunomodulatory effects of the patient’s environment and microbiota. Recent research highlights the critical need to model these interactions systematically and dynamically, taking adaptive evolution into consideration rather than relying on static measurements at single moments in time.
The importance of integrated biomarker models in cancer is highlighted well by the rapidly expanding interest in the study of the commensal microbiota in the context of cancer development and progression. Microbes influence the response to traditional cytotoxic agents through a diverse combination of effects on cellular metabolism, local pharmacokinetics, and could plausibly affect bioavailability of orally administered agents which are increasingly common in the era of targeted therapy, although this remains to be studied. Perhaps of greater significance, local interactions between specific bacteria and host tissues contribute to locoregional inflammation and carcinogenesis. Molecular interactions with pattern receptors (e.g., TLR4 and downstream MYD88), and immunosuppressive signals mediated by engagement with cell surface inhibitory molecules (e.g., TIGIT) or elaboration of suppressive cytokines (e.g., VEGF and CCL2) result in immune evasion that likely contributes to ineffective immunosurveillance of nascent, developing and established neoplasia.
It is not yet fully know to what extent the immunomodulatory effects of cancer-associated microbes may influence cancer immunotherapies, however, it is highly likely that these effects will not be consistent. For instance, immunosuppressive mechanisms such as MDSC-induction by B. fragilis suggest a negative impact on anti-cancer immunotherapy treatment response, however, the often simultaneous induction of Th17- and Th1-biased systemic immunity by the same organism(s) appears beneficial to checkpoint blockade immunotherapy response. This apparent contradiction will likely prove to be a complex and difficult issue to resolve as the field progresses, particularly as it relates to how best–or how safely–to manipulate the gut microbial composition to optimize treatment outcomes, toxicity, and long-term general health.
As more mechanisms are identified by which microbes directly influence tumor genomics, it will be important to evaluate whether the commonly observed EMT-like processes that accompany tumor progression involve a feed-forward loop precipitated by tumor-induced dysbiosis and subsequent microbe-directed epigenetic reprogramming of tumor cells. Another important issue that urgently requires attention is the relative significance of individual microbial taxa as opposed to unifying functional or metabolic characteristics of multiple taxa, in cancer development and treatment response. Metabolomic and whole genome shotgun sequencing studies are underway to address this, and will be highly relevant to the identification of the most readily targeted predisposing, permissive and perpetuating factors in cancer microbiology; it may be that critical intermediary metabolites or activated metabolic pathways prove to be most amenable to therapeutic modification. Further study is also required to integrate the significance of the microbiota with what is already known about the influence of lifestyle factors on cancer outcomes. Consideration should be made to the impact of duration, mode and type of microbiota “exposure” when integrating the microbiome into the new cancer-immunity model. As we continue to develop a deeper understanding of the myriad factors impacting cancer immunotherapy response, we will undoubtedly develop and refine new therapeutic strategies for maximal patient benefit.
All authors conceived, wrote, reviewed, and approved the final manuscript.
MA and AR report no relevant conflict of interest or financial disclosures. VG and JW are inventors on a US patent application (PCT/US17/53717) submitted by The University of Texas MD Anderson Cancer Center that covers methods to enhance checkpoint blockade therapy by the microbiome. JW has honoraria from speakers’ bureau of Dava Oncology, Bristol-Myers Squibb, and Illumina, and is an advisory board member for GlaxoSmithKline, Novartis, and Roche/Genentech.
MA is supported by a National Health and Medical Research Council of Australia CJ Martin Early Career Fellowship (#1148680). AR is supported by the Kimberley Clark Foundation Award for Scientific Achievement provided by MD Anderson’s Odyssey Fellowship Program.
1. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol (2006) 24(26):4340–6. doi:10.1200/JCO.2006.06.2984
2. Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, et al. The catalogue of somatic mutations in cancer (COSMIC). Curr Protoc Hum Genet (2008) Chapter 10:Unit 10 1. doi:10.1002/0471142905.hg1011s57
3. Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res (2013) 19(5):1225–31. doi:10.1158/1078-0432.CCR-12-1630
4. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov (2016) 6(2):202–16. doi:10.1158/2159-8290.CD-15-0283
5. Kono M, Dunn IS, Durda PJ, Butera D, Rose LB, Haggerty TJ, et al. Role of the mitogen-activated protein kinase signaling pathway in the regulation of human melanocytic antigen expression. Mol Cancer Res (2006) 4(10):779–92. doi:10.1158/1541-7786.MCR-06-0077
6. Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res (2010) 70(13):5213–9. doi:10.1158/0008-5472.CAN-10-0118
7. Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res (2012) 18(5):1386–94. doi:10.1158/1078-0432.CCR-11-2479
8. Donia M, Fagone P, Nicoletti F, Andersen RS, Hogdall E, Straten PT, et al. BRAF inhibition improves tumor recognition by the immune system: potential implications for combinatorial therapies against melanoma involving adoptive T-cell transfer. Oncoimmunology (2012) 1(9):1476–83. doi:10.4161/onci.21940
9. Bradley SD, Chen Z, Melendez B, Talukder A, Khalili JS, Rodriguez-Cruz T, et al. BRAFV600E Co-opts a conserved MHC class I internalization pathway to diminish antigen presentation and CD8+ T-cell recognition of melanoma. Cancer Immunol Res (2015) 3(6):602–9. doi:10.1158/2326-6066.CIR-15-0030
10. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med (2014) 371(23):2189–99. doi:10.1056/NEJMoa1406498
11. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science (2014) 344(6184):641–5. doi:10.1126/science.1251102
12. Woods K, Pasam A, Jayachandran A, Andrews MC, Cebon J. Effects of epithelial to mesenchymal transition on T cell targeting of melanoma cells. Front Oncol (2014) 4:367. doi:10.3389/fonc.2014.00367
13. Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A, et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov (2014) 4(7):816–27. doi:10.1158/2159-8290.CD-13-0424
14. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature (2013) 500(7463):415–21. doi:10.1038/nature12477
15. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature (2013) 499(7457):214–8. doi:10.1038/nature12213
16. Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther (2017) 16(11):2598–608. doi:10.1158/1535-7163.MCT-17-0386
17. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (2015) 348(6230):124–8. doi:10.1126/science.aaa1348
18. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med (2015) 372(26):2509–20. doi:10.1056/NEJMoa1500596
19. Johnson DB, Frampton GM, Rioth MJ, Yusko E, Xu Y, Guo X, et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res (2016) 4(11):959–67. doi:10.1158/2326-6066.CIR-16-0143
20. Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol (2018) 36(7):633–41. doi:10.1200/JCO.2017.75.3384
21. Gandara DR, Kowanetz M, Mok TSK, Rittmeyer A, Fehrenbacher L, Fabrizio D, et al. editors. Abstract 1295O: blood-based biomarkers for cancer immunotherapy: tumor mutational burden in blood (bTMB) is associated with improved atezolizumab (atezo) efficacy in 2L+ NSCLC (POPLAR and OAK). ESMO congress 2017. Ann Oncol (2017) 28(suppl 5):460. doi:10.1093/annonc/mdx380
22. Woods K, Knights AJ, Anaka M, Schittenhelm RB, Purcell AW, Behren A, et al. Mismatch in epitope specificities between IFNgamma inflamed and uninflamed conditions leads to escape from T lymphocyte killing in melanoma. J Immunother Cancer (2016) 4:10. doi:10.1186/s40425-016-0111-7
23. D’Urso CM, Wang ZG, Cao Y, Tatake R, Zeff RA, Ferrone S. Lack of HLA class I antigen expression by cultured melanoma cells FO-1 due to a defect in B2m gene expression. J Clin Invest (1991) 87(1):284–92. doi:10.1172/JCI114984
24. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med (2016) 375(9):819–29. doi:10.1056/NEJMoa1604958
25. Garrido C, Paco L, Romero I, Berruguilla E, Stefansky J, Collado A, et al. MHC class I molecules act as tumor suppressor genes regulating the cell cycle gene expression, invasion and intrinsic tumorigenicity of melanoma cells. Carcinogenesis (2012) 33(3):687–93. doi:10.1093/carcin/bgr318
26. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med (2016) 375(23):2255–62. doi:10.1056/NEJMoa1609279
27. Lennerz V, Fatho M, Gentilini C, Frye RA, Lifke A, Ferel D, et al. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc Natl Acad Sci U S A (2005) 102(44):16013–8. doi:10.1073/pnas.0500090102
28. Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science (2015) 350(6266):1387–90. doi:10.1126/science.aad1253
29. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science (2015) 350(6257):207–11. doi:10.1126/science.aad0095
30. Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell (2017) 171(4):934–49 e15. doi:10.1016/j.cell.2017.09.028
31. Liu C, Yang X, Duffy B, Mohanakumar T, Mitra RD, Zody MC, et al. ATHLATES: accurate typing of human leukocyte antigen through exome sequencing. Nucleic Acids Res (2013) 41(14):e142. doi:10.1093/nar/gkt481
32. Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, et al. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics (2009) 61(1):1–13. doi:10.1007/s00251-008-0341-z
33. Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science (2018) 359(6375):582–7. doi:10.1126/science.aao4572
34. Roh W, Chen PL, Reuben A, Spencer CN, Prieto PA, Miller JP, et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci Transl Med (2017) 9(379):eaah3560. doi:10.1126/scitranslmed.aah3560
35. Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, Justesen S, et al. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS One (2007) 2(8):e796. doi:10.1371/journal.pone.0000796
36. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature (2017) 541(7637):321–30. doi:10.1038/nature21349
37. Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, et al. Identification of essential genes for cancer immunotherapy. Nature (2017) 548(7669):537–42. doi:10.1038/nature23477
38. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-gamma pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell (2016) 167(2):397–404 e9. doi:10.1016/j.cell.2016.08.069
39. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov (2017) 7(2):188–201. doi:10.1158/2159-8290.CD-16-1223
40. Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell (2016) 167(6):1540–54.e12. doi:10.1016/j.cell.2016.11.022
41. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell (2017) 168(4):707–23. doi:10.1016/j.cell.2017.01.017
42. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature (2014) 515(7528):568–71. doi:10.1038/nature13954
43. Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov (2016) 6(8):827–37. doi:10.1158/2159-8290.CD-15-1545
44. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature (2015) 523(7559):231–5. doi:10.1038/nature14404
45. Johnson BF, Clay TM, Hobeika AC, Lyerly HK, Morse MA. Vascular endothelial growth factor and immunosuppression in cancer: current knowledge and potential for new therapy. Expert Opin Biol Ther (2007) 7(4):449–60. doi:10.1517/14712598.7.4.449
46. Du Four S, Maenhout SK, Niclou SP, Thielemans K, Neyns B, Aerts JL. Combined VEGFR and CTLA-4 blockade increases the antigen-presenting function of intratumoral DCs and reduces the suppressive capacity of intratumoral MDSCs. Am J Cancer Res (2016) 6(11):2514–31.
47. Wu X, Giobbie-Hurder A, Liao X, Lawrence D, McDermott D, Zhou J, et al. VEGF neutralization plus CTLA-4 blockade alters soluble and cellular factors associated with enhancing lymphocyte infiltration and humoral recognition in melanoma. Cancer Immunol Res (2016) 4(10):858–68. doi:10.1158/2326-6066.CIR-16-0084
48. Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res (2010) 70(15):6171–80. doi:10.1158/0008-5472.CAN-10-0153
49. Daud AI, Loo K, Pauli ML, Sanchez-Rodriguez R, Sandoval PM, Taravati K, et al. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J Clin Invest (2016) 126(9):3447–52. doi:10.1172/JCI87324
50. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun (2016) 7:10501. doi:10.1038/ncomms10501
51. Albino AP, Lloyd KO, Houghton AN, Oettgen HF, Old LJ. Heterogeneity in surface antigen and glycoprotein expression of cell lines derived from different melanoma metastases of the same patient. Implications for the study of tumor antigens. J Exp Med (1981) 154(6):1764–78. doi:10.1084/jem.154.6.1764
52. Reuben A, Spencer CN, Prieto PA, Gopalakrishnan V, Reddy SM, Miller JP, et al. Genomic and immune heterogeneity are associated with differential responses to therapy in melanoma. NPJ Genom Med (2017) 2:10. doi:10.1038/s41525-017-0013-8
53. Morris LG, Riaz N, Desrichard A, Senbabaoglu Y, Hakimi AA, Makarov V, et al. Pan-cancer analysis of intratumor heterogeneity as a prognostic determinant of survival. Oncotarget (2016) 7(9):10051–63. doi:10.18632/oncotarget.7067
54. Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer (2006) 6(12):924–35. doi:10.1038/nrc2013
55. Tirosh I, Izar B, Prakadan SM, Wadsworth MH II, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science (2016) 352(6282):189–96. doi:10.1126/science.aad0501
56. Marusyk A, Tabassum DP, Altrock PM, Almendro V, Michor F, Polyak K. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature (2014) 514(7520):54–8. doi:10.1038/nature13556
57. Velazquez EF, Jungbluth AA, Yancovitz M, Gnjatic S, Adams S, O’Neill D, et al. Expression of the cancer/testis antigen NY-ESO-1 in primary and metastatic malignant melanoma (MM) – correlation with prognostic factors. Cancer Immun (2007) 7:11.
58. Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang J, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science (2014) 346(6206):256–9. doi:10.1126/science.1256930
59. Cogdill AP, Andrews MC, Wargo JA. Hallmarks of response to immune checkpoint blockade. Br J Cancer (2017) 117(1):1–7. doi:10.1038/bjc.2017.136
60. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol (2002) 3(11):991–8. doi:10.1038/ni1102-991
61. Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest (2007) 117(5):1137–46. doi:10.1172/JCI31405
62. Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, et al. Systemic immunity is required for effective cancer immunotherapy. Cell (2017) 168(3):487–502.e15. doi:10.1016/j.cell.2016.12.022
63. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi:10.1038/nri2506
64. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol (2016) 37(3):208–20. doi:10.1016/j.it.2016.01.004
65. Florcken A, Takvorian A, Singh A, Gerhardt A, Ostendorf BN, Dorken B, et al. Myeloid-derived suppressor cells in human peripheral blood: optimized quantification in healthy donors and patients with metastatic renal cell carcinoma. Immunol Lett (2015) 168(2):260–7. doi:10.1016/j.imlet.2015.10.001
66. Zhang B, Wang Z, Wu L, Zhang M, Li W, Ding J, et al. Circulating and tumor-infiltrating myeloid-derived suppressor cells in patients with colorectal carcinoma. PLoS One (2013) 8(2):e57114. doi:10.1371/journal.pone.0057114
67. Stanojevic I, Miller K, Kandolf-Sekulovic L, Mijuskovic Z, Zolotarevski L, Jovic M, et al. A subpopulation that may correspond to granulocytic myeloid-derived suppressor cells reflects the clinical stage and progression of cutaneous melanoma. Int Immunol (2016) 28(2):87–97. doi:10.1093/intimm/dxv053
68. Wang L, Chang EW, Wong SC, Ong SM, Chong DQ, Ling KL. Increased myeloid-derived suppressor cells in gastric cancer correlate with cancer stage and plasma S100A8/A9 proinflammatory proteins. J Immunol (2013) 190(2):794–804. doi:10.4049/jimmunol.1202088
69. Gonda K, Shibata M, Ohtake T, Matsumoto Y, Tachibana K, Abe N, et al. Myeloid-derived suppressor cells are increased and correlated with type 2 immune responses, malnutrition, inflammation, and poor prognosis in patients with breast cancer. Oncol Lett (2017) 14(2):1766–74. doi:10.3892/ol.2017.6305
70. Tanaka T, Fujita M, Hasegawa H, Arimoto A, Nishi M, Fukuoka E, et al. Frequency of myeloid-derived suppressor cells in the peripheral blood reflects the status of tumor recurrence. Anticancer Res (2017) 37(7):3863–9. doi:10.21873/anticanres.11766
71. Angell TE, Lechner MG, Smith AM, Martin SE, Groshen SG, Maceri DR, et al. Circulating myeloid-derived suppressor cells predict differentiated thyroid cancer diagnosis and extent. Thyroid (2016) 26(3):381–9. doi:10.1089/thy.2015.0289
72. Raychaudhuri B, Rayman P, Ireland J, Ko J, Rini B, Borden EC, et al. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro Oncol (2011) 13(6):591–9. doi:10.1093/neuonc/nor042
73. Vasquez-Dunddel D, Pan F, Zeng Q, Gorbounov M, Albesiano E, Fu J, et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J Clin Invest (2013) 123(4):1580–9. doi:10.1172/JCI60083
74. Porembka MR, Mitchem JB, Belt BA, Hsieh CS, Lee HM, Herndon J, et al. Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth. Cancer Immunol Immunother (2012) 61(9):1373–85. doi:10.1007/s00262-011-1178-0
75. Brusa D, Simone M, Gontero P, Spadi R, Racca P, Micari J, et al. Circulating immunosuppressive cells of prostate cancer patients before and after radical prostatectomy: profile comparison. Int J Urol (2013) 20(10):971–8. doi:10.1111/iju.12086
76. Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res (2009) 69(4):1553–60. doi:10.1158/0008-5472.CAN-08-1921
77. Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med (2016) 22(4):433–8. doi:10.1038/nm.4051
78. Lee YK, Mazmanian SK. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science (2010) 330(6012):1768–73. doi:10.1126/science.1195568
79. Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science (2012) 336(6086):1268–73. doi:10.1126/science.1223490
80. Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science (2005) 307(5717):1915–20. doi:10.1126/science.1104816
81. Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, et al. Metagenomic analysis of the human distal gut microbiome. Science (2006) 312(5778):1355–9. doi:10.1126/science.1124234
82. Kim D, Zeng MY, Nunez G. The interplay between host immune cells and gut microbiota in chronic inflammatory diseases. Exp Mol Med (2017) 49(5):e339. doi:10.1038/emm.2017.24
83. Neish AS. Microbes in gastrointestinal health and disease. Gastroenterology (2009) 136(1):65–80. doi:10.1053/j.gastro.2008.10.080
84. Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet (1984) 1(8390):1311–5. doi:10.1016/S0140-6736(84)91816-6
85. Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med (2009) 15(9):1016–22. doi:10.1038/nm.2015
86. Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science (2012) 338(6103):120–3. doi:10.1126/science.1224820
87. Buc E, Dubois D, Sauvanet P, Raisch J, Delmas J, Darfeuille-Michaud A, et al. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS One (2013) 8(2):e56964. doi:10.1371/journal.pone.0056964
88. Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res (2012) 22(2):292–8. doi:10.1101/gr.126573.111
89. Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res (2012) 22(2):299–306. doi:10.1101/gr.126516.111
90. McCoy AN, Araujo-Perez F, Azcarate-Peril A, Yeh JJ, Sandler RS, Keku TO. Fusobacterium is associated with colorectal adenomas. PLoS One (2013) 8(1):e53653. doi:10.1371/journal.pone.0053653
91. Sanapareddy N, Legge RM, Jovov B, McCoy A, Burcal L, Araujo-Perez F, et al. Increased rectal microbial richness is associated with the presence of colorectal adenomas in humans. ISME J (2012) 6(10):1858–68. doi:10.1038/ismej.2012.43
92. Bhatt AP, Redinbo MR, Bultman SJ. The role of the microbiome in cancer development and therapy. CA Cancer J Clin (2017) 67(4):326–44. doi:10.3322/caac.21398
93. Reddy BS, Narisawa T, Wright P, Vukusich D, Weisburger JH, Wynder EL. Colon carcinogenesis with azoxymethane and dimethylhydrazine in germ-free rats. Cancer Res (1975) 35(2):287–90.
94. Vannucci L, Stepankova R, Kozakova H, Fiserova A, Rossmann P, Tlaskalova-Hogenova H. Colorectal carcinogenesis in germ-free and conventionally reared rats: different intestinal environments affect the systemic immunity. Int J Oncol (2008) 32(3):609–17. doi:10.3892/ijo.32.3.609
95. Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature (2012) 491(7423):254–8. doi:10.1038/nature11465
96. Klimesova K, Kverka M, Zakostelska Z, Hudcovic T, Hrncir T, Stepankova R, et al. Altered gut microbiota promotes colitis-associated cancer in IL-1 receptor-associated kinase M-deficient mice. Inflamm Bowel Dis (2013) 19(6):1266–77. doi:10.1097/MIB.0b013e318281330a
97. Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell (2012) 21(4):504–16. doi:10.1016/j.ccr.2012.02.007
98. Ochi A, Nguyen AH, Bedrosian AS, Mushlin HM, Zarbakhsh S, Barilla R, et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J Exp Med (2012) 209(9):1671–87. doi:10.1084/jem.20111706
99. Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science (2013) 342(6161):967–70. doi:10.1126/science.1240527
100. Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, Hannani D, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science (2013) 342(6161):971–6. doi:10.1126/science.1240537
101. Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science (2015) 350(6264):1079–84. doi:10.1126/science.aad1329
102. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science (2018) 359(6371):97–103. doi:10.1126/science.aan4236
103. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science (2015) 350(6264):1084–9. doi:10.1126/science.aac4255
104. Daillere R, Vetizou M, Waldschmitt N, Yamazaki T, Isnard C, Poirier-Colame V, et al. Enterococcus hirae and Barnesiella intestinihominis facilitate cyclophosphamide-induced therapeutic immunomodulatory effects. Immunity (2016) 45(4):931–43. doi:10.1016/j.immuni.2016.09.009
105. Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science (2018) 359(6371):104–8. doi:10.1126/science.aao3290
106. Frankel AE, Coughlin LA, Kim J, Froehlich TW, Xie Y, Frenkel EP, et al. Metagenomic shotgun sequencing and unbiased metabolomic profiling identify specific human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia (2017) 19(10):848–55. doi:10.1016/j.neo.2017.08.004
107. Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol (2017) 28(6):1368–79. doi:10.1093/annonc/mdx108
108. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504(7480):451–5. doi:10.1038/nature12726
109. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature (2013) 504(7480):446–50. doi:10.1038/nature12721
110. Usami M, Kishimoto K, Ohata A, Miyoshi M, Aoyama M, Fueda Y, et al. Butyrate and trichostatin A attenuate nuclear factor kappaB activation and tumor necrosis factor alpha secretion and increase prostaglandin E2 secretion in human peripheral blood mononuclear cells. Nutr Res (2008) 28(5):321–8. doi:10.1016/j.nutres.2008.02.012
111. Vinolo MA, Rodrigues HG, Hatanaka E, Sato FT, Sampaio SC, Curi R. Suppressive effect of short-chain fatty acids on production of proinflammatory mediators by neutrophils. J Nutr Biochem (2011) 22(9):849–55. doi:10.1016/j.jnutbio.2010.07.009
112. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science (2013) 341(6145):569–73. doi:10.1126/science.1241165
113. Zitvogel L, Ayyoub M, Routy B, Kroemer G. Microbiome and anticancer immunosurveillance. Cell (2016) 165(2):276–87. doi:10.1016/j.cell.2016.03.001
114. Taur Y, Jenq RR, Perales MA, Littmann ER, Morjaria S, Ling L, et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood (2014) 124(7):1174–82. doi:10.1182/blood-2014-02-554725
115. Jenq RR, Taur Y, Devlin SM, Ponce DM, Goldberg JD, Ahr KF, et al. Intestinal Blautia is associated with reduced death from graft-versus-host disease. Biol Blood Marrow Transplant (2015) 21(8):1373–83. doi:10.1016/j.bbmt.2015.04.016
116. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science (2018) 359(6371):91–7. doi:10.1126/science.aan3706
117. Krishnan S, Eslick GD. Streptococcus bovis infection and colorectal neoplasia: a meta-analysis. Colorectal Dis (2014) 16(9):672–80. doi:10.1111/codi.12662
118. Mima K, Nishihara R, Qian ZR, Cao Y, Sukawa Y, Nowak JA, et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut (2016) 65(12):1973–80. doi:10.1136/gutjnl-2015-310101
119. Bullman S, Pedamallu CS, Sicinska E, Clancy TE, Zhang X, Cai D, et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science (2017) 358(6369):1443–8. doi:10.1126/science.aal5240
120. Yu T, Guo F, Yu Y, Sun T, Ma D, Han J, et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell (2017) 170(3):548–63 e16. doi:10.1016/j.cell.2017.07.008
121. Eklof V, Lofgren-Burstrom A, Zingmark C, Edin S, Larsson P, Karling P, et al. Cancer-associated fecal microbial markers in colorectal cancer detection. Int J Cancer (2017) 141(12):2528–36. doi:10.1002/ijc.31011
122. Vizcaino MI, Crawford JM. The colibactin warhead crosslinks DNA. Nat Chem (2015) 7(5):411–7. doi:10.1038/nchem.2221
123. Tomkovich S, Yang Y, Winglee K, Gauthier J, Muhlbauer M, Sun X, et al. Locoregional effects of microbiota in a preclinical model of colon carcinogenesis. Cancer Res (2017) 77(10):2620–32. doi:10.1158/0008-5472.CAN-16-3472
124. Lennard KS, Goosen RW, Blackburn JM. Bacterially-associated transcriptional remodelling in a distinct genomic subtype of colorectal cancer provides a plausible molecular basis for disease development. PLoS One (2016) 11(11):e0166282. doi:10.1371/journal.pone.0166282
125. Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity (2015) 42(2):344–55. doi:10.1016/j.immuni.2015.01.010
126. Thiele Orberg E, Fan H, Tam AJ, Dejea CM, Destefano Shields CE, Wu S, et al. The myeloid immune signature of enterotoxigenic Bacteroides fragilis-induced murine colon tumorigenesis. Mucosal Immunol (2017) 10(2):421–33. doi:10.1038/mi.2016.53
127. Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science (2017) 357(6356):1156–60. doi:10.1126/science.aah5043
128. Wang H, Altemus J, Niazi F, Green H, Calhoun BC, Sturgis C, et al. Breast tissue, oral and urinary microbiomes in breast cancer. Oncotarget (2017) 8(50):88122–38. doi:10.18632/oncotarget.21490
Keywords: cancer immunotherapy, biomarkers, heterogeneity, microbiome, cancer genomics, tumor microenvironment, systemic immunity
Citation: Andrews MC, Reuben A, Gopalakrishnan V and Wargo JA (2018) Concepts Collide: Genomic, Immune, and Microbial Influences on the Tumor Microenvironment and Response to Cancer Therapy. Front. Immunol. 9:946. doi: 10.3389/fimmu.2018.00946
Received: 25 January 2018; Accepted: 16 April 2018;
Published: 04 May 2018
Edited by:
Petranel Theresa Ferrao, University of Melbourne, AustraliaReviewed by:
Arthur Edward Frankel, University of South Alabama, United StatesCopyright: © 2018 Andrews, Reuben, Gopalakrishnan and Wargo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jennifer A. Wargo, andhcmdvQG1kYW5kZXJzb24ub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.