 Richard B. Pouw1,2
Richard B. Pouw1,2 Irene Gómez Delgado3
Irene Gómez Delgado3 Alberto López Lera4
Alberto López Lera4 Santiago Rodríguez de Córdoba5
Santiago Rodríguez de Córdoba5 Diana Wouters1Taco W. Kuijpers2,6
Diana Wouters1Taco W. Kuijpers2,6 Pilar Sánchez-Corral3*
Pilar Sánchez-Corral3*
- 1Department of Immunopathology, Sanquin Research and Landsteiner Laboratory of the Academic Medical Center, University of Amsterdam, Amsterdam, Netherlands
- 2Department of Pediatric Hematology, Immunology and Infectious Diseases, Emma Children’s Hospital, Academic Medical Center, Amsterdam, Netherlands
- 3Complement Research Group, Hospital La Paz Institute for Health Research (IdiPAZ), La Paz University Hospital, Center for Biomedical Network Research on Rare Diseases (CIBERER), Madrid, Spain
- 4Immunology Unit, Hospital La Paz Institute for Health Research (IdiPAZ), La Paz University Hospital, Center for Biomedical Network Research on Rare Diseases (CIBERER), Madrid, Spain
- 5Biological Research Center (CIB)-CSIC, Center for Biomedical Network Research on Rare Diseases (CIBERER), Madrid, Spain
- 6Department of Blood Cell Research, Sanquin Research and Landsteiner Laboratory of the Academic Medical Center, University of Amsterdam, Amsterdam, Netherlands
Dysregulation of the complement alternative pathway (AP) is a major pathogenic mechanism in atypical hemolytic-uremic syndrome (aHUS). Genetic or acquired defects in factor H (FH), the main AP regulator, are major aHUS drivers that associate with a poor prognosis. FH activity has been suggested to be downregulated by homologous FH-related (FHR) proteins, including FHR-3 and FHR-1. Hence, their relative levels in plasma could be disease-relevant. The genes coding for FH, FHR-3, and FHR-1 (CFH, CFHR3, and CFHR1, respectively) are polymorphic and located adjacent to each other on human chromosome 1q31.3. We have previously shown that haplotype CFH(H3)–CFHR3*B–CFHR1*B associates with aHUS and reduced FH levels. In this study, we used a specific enzyme-linked immunosorbent assay to quantify FHR-3 in plasma samples from controls and patients with aHUS genotyped for the three known CFHR3 alleles (CFHR3*A, CFHR3*B, and CFHR3*Del). In the 218 patients carrying at least one copy of CFHR3, significant differences between CFHR3 genotype groups were found, with CFHR3*A/Del patients having the lowest FHR-3 concentration (0.684–1.032 µg/mL), CFHR3*B/Del and CFHR3*A/A patients presenting intermediate levels (1.437–2.201 µg/mL), and CFHR3*A/B and CFHR3*B/B patients showing the highest concentration (2.330–4.056 µg/mL) (p < 0.001). These data indicate that CFHR3*A is a low-expression allele, whereas CFHR3*B, associated with increased risk of aHUS, is a high-expression allele. Our study reveals that the aHUS-risk haplotype CFH(H3)–CFHR3*B–CFHR1*B generates twofold more FHR-3 than the non-risk CFH(H1)–CFHR3*A–CFHR1*A haplotype. In addition, FHR-3 levels were higher in patients with aHUS than in control individuals with the same CFHR3 genotype. These data suggest that increased plasma levels of FHR-3, altering the balance between FH and FHR-3, likely impact the FH regulatory functions and contribute to the development of aHUS.
Introduction
Atypical hemolytic-uremic syndrome (aHUS) is a thrombotic microangiopathy characterized by hematological and renal alterations, although neurological and cardiovascular damage is also frequent (1, 2). Genetic and/or acquired defects in the complement alternative pathway that disturb the activation-regulation balance are present in 40–60% of patients, potentiating the initial endothelial damage in the microvasculature (3–5). The prognosis of patients with aHUS who have mutations in complement factor H (FH) is particularly poor and is associated with terminal renal insufficiency at disease onset and disease recurrence in the transplanted kidney (6).
Factor H is the main complement regulator in the fluid phase, and it also binds to autologous cellular surfaces to protect them from complement-mediated damage (7). The FH gene, CFH, is located within a gene cluster that includes five additional genes (CFHR1 to CFHR5) coding for the homologous FH-related proteins (FHR-1 to FHR-5), which likely compete with FH for ligand binding and act as complement deregulators (8, 9). Several aHUS-predisposing genetic variants within the CFH/CFHR gene cluster have been found. A particular CFH haplotype named CFH(H3) increases aHUS penetrance in carriers and modulates the clinical phenotype (10–12), and the homozygous deletion of the CFHR3 and CFHR1 genes (ΔCFHR3–CFHR1) predisposes patients to an autoimmune form of aHUS characterized by the generation of anti-FH blocking antibodies (13–15). Additional aHUS-risk variants within the CFH/CFHR region are the CFHR1*B and the CFHR3*B alleles (16, 17).
The molecular basis for the contribution of the genetic variants CFH(H3), CFHR3*B, and CFHR1*B to the pathogenic mechanism of aHUS have not yet been determined. These variants include non-synonymous single-nucleotide polymorphisms (SNPs) with potential functional consequences, but changes in gene expression cannot be excluded, particularly in the case of CFH(H3) and CFHR3*B, which include SNPs within their 5′-untranslated region (UTR).
We have shown that the aHUS-risk haplotype CFH(H3) is nearly always associated with the CFHR3*B and CFHR1*B alleles, thus generating an extended CFH(H3)–CFHR3*B–CFHR1*B haplotype, which predisposes to aHUS and favors a poorer progression of renal function at disease onset (17); we also demonstrated that patients homozygous for this haplotype have lower FH levels. To check the hypothesis that the aHUS-risk CFHR3*B allele (tagged by rs385390A; rs446868C; rs138675433C; rs149352569A) gives rise to higher protein levels than the non-risk CFHR3*A allele (rs385390C; rs446868A; rs138675433T; rs149352569T), we determined FHR-3 levels in patients with aHUS and in control individuals genotyped for CFHR3*A, CFHR3*B, and ΔCFHR3–CFHR1 (referred to as CFHR3*Del). By using an FHR-3-specific enzyme-linked immunosorbent assay (ELISA) (18), we demonstrate that CFHR3*A is a low-expression allele and CFHR3*B is a high-expression allele and that increased FHR-3 levels in plasma are associated with aHUS.
Materials and Methods
Patients and Controls
A total of 230 patients from the Spanish aHUS registry with known CFH, CFHR3, and CFHR1 genotypes were selected for the study. Genotyping had been previously determined by direct sequencing and copy number variation analyzed by multiplex ligation-dependent probe amplification (MLPA) or by using an in-house comparative genomic hybridization microarray (19). Studies to identify mutations in complement genes had also been performed on most of these patients. Blood samples were drawn during an acute aHUS episode or during remission, centrifuged to obtain serum and ethylenediaminetetraacetic acid (EDTA) plasma, and stored at −80°C until use. Peripheral-blood leukocytes (PBLs) were used to prepare genomic DNA by standard procedures. Plasma and DNA samples from 49 healthy Spanish individuals were also obtained and used in the study. Patients and controls provided written informed consent, as approved by the ethical committees from La Paz University Hospital or the Biological Research Center.
CFHR3 Genotyping
Genotyping of the CFHR3*A and CFHR3*B alleles was performed by Sanger sequencing of CFHR3 exon 5, as described previously (17). The CFHR3–CFHR1 genomic deletion was analyzed by using the SALSA MLPA probemix P236-A3 ARMD mix-1 (MRC-Holland, Amsterdam, Netherlands); this deletion is referred to as the CFHR3*Del allele, and it does not generate FHR-3 and FHR-1.
FHR-3 Quantitation
FHR-3 levels in serum or EDTA plasma samples from the 230 patients with aHUS and the 49 controls were determined by using a specific sandwich FHR-3 ELISA as described previously (18).
Statistical Analyses
The statistical significance of FHR-3 levels in the various geno-type or age groups was analyzed with IBM SPSS Software.
Results
FHR-3 Levels in Spanish Controls Suggest Differential Expression of CFHR3*A and CFHR3*B
We had previously determined FHR-3 levels in serum samples from 100 Dutch controls and had shown that FHR-3 concentration correlated with the number of CFHR3 copies (18). We have now confirmed this observation in 47 Spanish control individuals containing 1 or 2 copies of CFHR3. FHR-3 levels ranged between 0.14 and 1.16 µg/mL (mean 0.61 ± 2.40; 95% CI 0.54–0.68), and FHR-3 concentration in individuals with 2 copies doubled the concentration observed in individuals with only one copy (0.68 vs. 0.36 µg/mL, p < 0.0001). Interestingly, genotyping of the Spanish controls for the CFHR3*A and CFHR3*B alleles provided additional data. FHR-3 levels were significantly lower in individuals with the CFHR3*A/A genotype (0.55 ± 0.15 µg/mL) than in individuals with the CFHR3*A/B (0.78 ± 0.18 µg/mL; p = 0.001) or CFHR3*B/B (0.82 ± 0.08 µg/mL; p = 0.033) genotypes, thus suggesting a lower expression of the CFHR3*A allele. A plausible explanation for this finding is that genetic variants located within the 5′-UTR of CFHR3 exon 1 (rs385390 and rs446868) determine a different expression of the CFHR3*A and CFHR3*B alleles (Figure 1A). No data on the effect of these SNPs on CFHR3 expression could be obtained from the Gene Expression Omnibus and Human Protein Atlas databases. However, data on gene expression correlations for the CFHR3 SNP rs385390 (c.1-90A>C) available at the GTExPortal showed lower mRNA levels of the A variant (tagging the CFHR3*A allele) than the C variant (tagging the CFHR3*B allele) in liver tissue (Figure 1B).
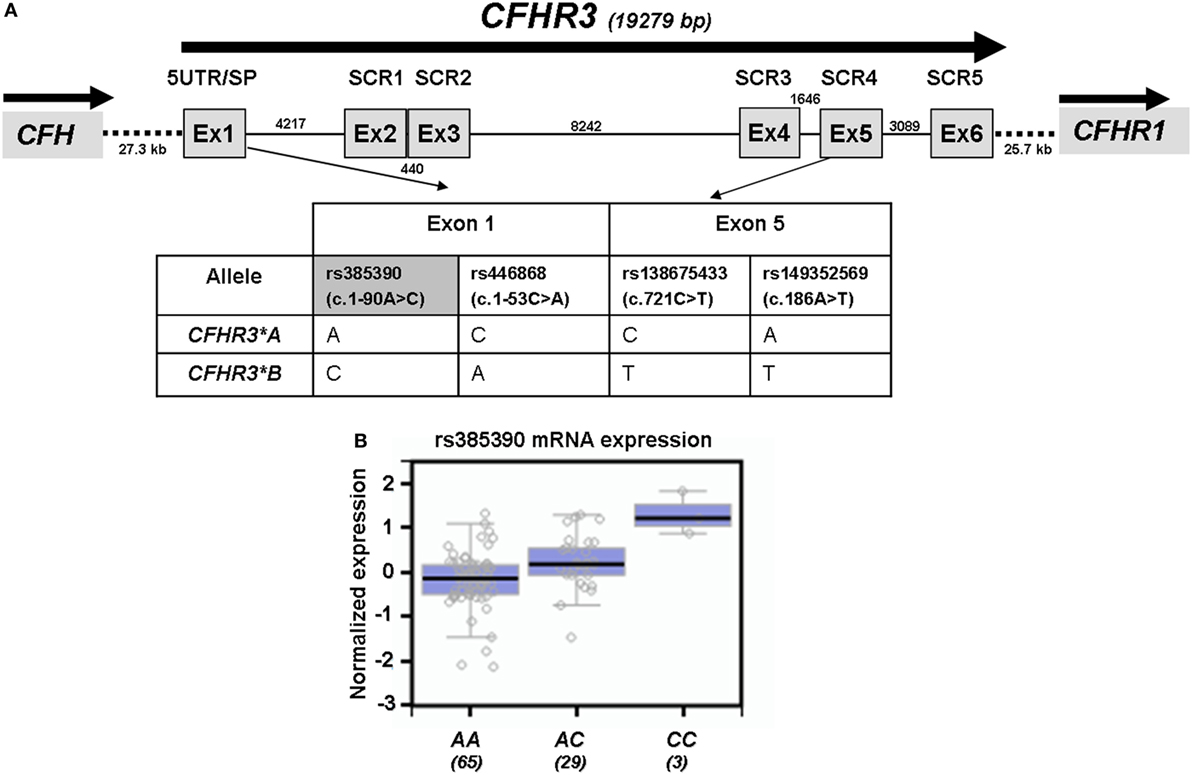
Figure 1. Genetic variants associated with the CFHR3*A and CFHR3*B alleles. (A) Exon–intron structure of the CFHR3 gene showing the location of the genetic variants within exon 1 (rs385390; rs446868) and exon 5 (rs138675433; rs149352569) that tag the CFHR3*A and CFHR3*B alleles. (B) In silico data of gene expression correlations from the GTExPortal show that individuals who are homozygous for the rs385390 A variant present lower mRNA expression in liver tissue than individuals homozygous for the rs385390 C variant, or than heterozygous individuals. Number of individuals with each genotype is shown between brackets. GTExPortal: Portal for the Genotype-Tissue Expression project (https://www.gtexportal.org). GTExPortal is supported by the Common Fund of the National Institutes of Health. The data in panel (B) were obtained on October 2017.
CFHR3*A Is a Low-Expression Allele, and CFHR3*B Is a High-Expression Allele
To confirm the association between FHR-3 levels and the CFHR3*A and CFHR3*B alleles, we determined FHR-3 levels in 230 patients with aHUS of known CFHR3 genotypes (17). The 22 patients with aHUS with the CFHR3*Del/Del genotype presented minimal FHR-3 levels corresponding to the lower limit of detection of the ELISA. The other 208 patients, presenting CFHR3 genotypes A/Del, B/Del, A/A, A/B, or B/B, showed a vast range of FHR-3 concentrations (0.124–13.450 µg/mL; mean 2.278 ± 1.723) (Table 1). Statistical analyses of these data were performed with nonparametric tests. The Kruskal–Wallis H test showed that there was a statistically significant difference in FHR-3 levels between the five CFHR3 genotypes [χ2(4) = 53.568; p < 0.001]. Next, a pairwise two-sided multiple comparison analysis was performed using the Dwass-Steel-Critchlow-Fligner method. Significant differences between the A/Del and B/Del genotypes (p = 0.0043), as well as between the A/A and B/B genotypes (p = 0.0008) were found, thus revealing that the CFHR3*A allele was associated with lower FHR-3 concentrations than the CFHR3*B allele (Table 2; Figure 2).
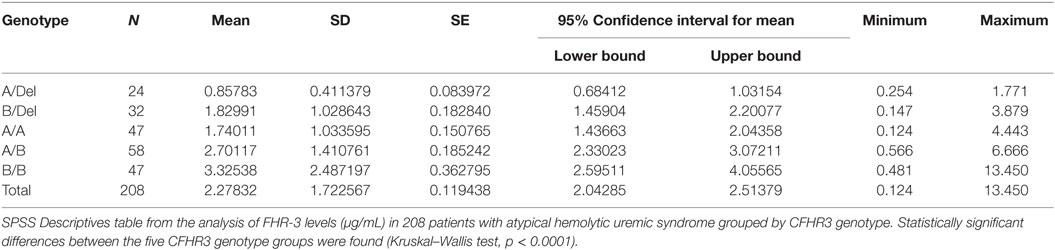
Table 1. FHR-3 levels differ between CFHR3 genotype groups.
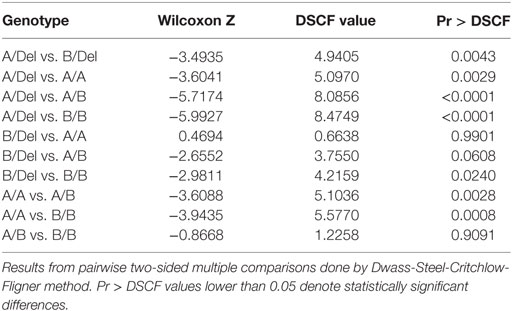
Table 2. The CFHR3*A allele generates lower FHR-3 levels than the CFHR3*B allele.
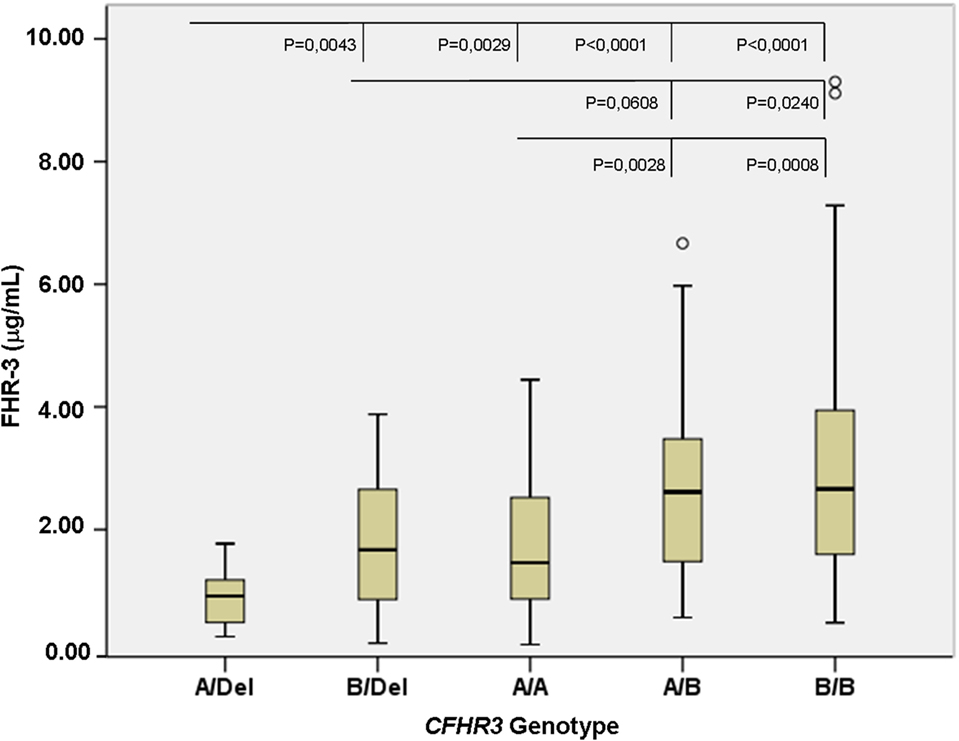
Figure 2. FHR-3 levels are associated with the CFHR3*A/B/Del genotypes. Boxplot of FHR-3 levels in 208 patients with atypical hemolytic uremic syndrome who were grouped according to their CFHR3 genotype; patients with the CFHR3 Del/Del genotype were not included because they have homozygous FHR-3 deficiency. The statistical significance obtained in pairwise two-sided multiple comparisons is also shown. Outliers in the CFHR3 A/B and B/B genotype groups are denoted by symbols (o).
FHR-3 Levels and Genetic Background
Statistical analyses showed that plasma FHR-3 levels were associated with the CFHR3*A/B/del genotypes; however, protein levels within each CFHR3 genotype group showed great variation. To explore the possibility that CFHR3 gene expression was also modulated by genetic factors in the adjacent CFH and CFHR1 genes, we analyzed the CFH–CFHR3–CFHR1 genotypes of the 230 patients with aHUS. Genotyping of CFH (H1/H2/H3/H4a/H4b/H5/H6/H7/H8), CFHR3 (A/B/Del), and CFHR1 (A/B/Del) in these patients had already been performed [(17); Table S1 in Supplementary Material]. As many as 63 different CFH–CFHR3–CFHR1 genotype combinations were observed (Table S2 in Supplementary Material), but only 23 of them were found in 3 or more patients (Table 3). The CFHR3*Del/Del genotype is associated with three CFH genotypic variants (H4a, H4b, or H2), and 90% of patients with CFHR3*A/Del or CFHR3*B/Del present 1 out of 4 different CFH–CFHR3–CFHR1 genotypes. Greater heterogeneity was found in the CFHR3*A/A and CFHR3*A/B groups, whereas 75% of the CFHR3*B/B patients were found to carry only two different CFH-CFHR3-CFHR1 genotypes. However, when comparing FHR-3 levels within each CFHR3 genotype group, no statistically significant differences between CFH–CFHR3–CFHR1 genotypes were found, suggesting that genetic variants in CFH and CFHR1 do not substantially contribute to the expression of the CFHR3*A and CFHR3*B alleles.
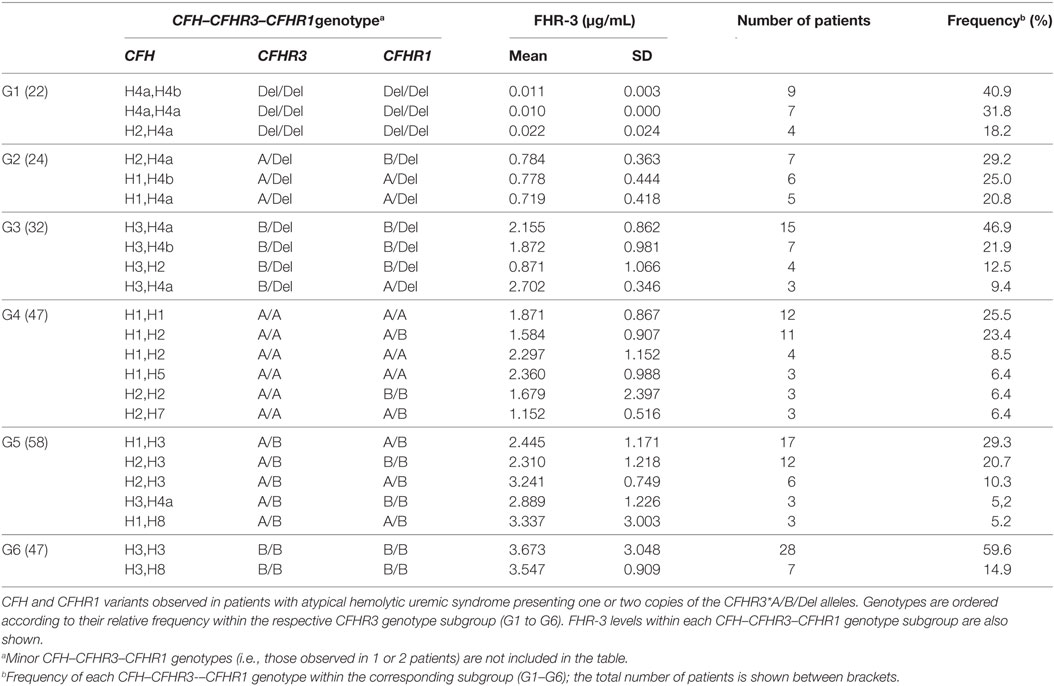
Table 3. CFH–CFHR3–CFHR1 genotypes and FHR-3 levels.
From the data in Table 3, we can also conclude that the CFHR3*Del allele mostly presents in the CFH(H4a) and CFH(H4b) haplotypes, the CFHR3*A allele in the CFH(H1) and CFH(H2) haplotypes and the CFHR3*B allele in the CFH(H3) and CFH(H8) haplotypes. These conclusions were further supported by the inferred CFH–CFHR3–CFHR1 haplotypes. Among the 208 patients expressing FHR-3 (groups G2–G6 in Table 3), 150 patients had CFH–CFHR3–CFHR1 genotypes homozygous for at least two genes, thus allowing segregation of the corresponding CFH–CFHR3–CFHR1 chromosomes. Some 25 different CFH–CFHR3–CFHR1 haplotypes were observed, the most frequent being CFH(H3)–CFHR3*B–CFHR1*B (31.7%), CFH(H4a)–CFHR3*Del–CFHR1*Del(20%) and CFH(H1)–CFHR3*A–CFHR3*A (16.3%). An analysis of FHR-3 levels in patients homozygous or hemizygous for these haplotypes rev-ealed higher expression in those patients with the CFH(H3)–CFHR3*B–CFHR1*B than CFH(H1)–CFHR3*A–CFHR3*A haplotype (1.87 ± 1.36 µg/mL vs. 0.9 ± 0.43 µg/mL; Kruskal–Wallis test, p < 0.001).
FHR-3 levels were also higher in patients with mutations in the complement genes CFH, CFI, MCP, C3, or CFB, than in patients without mutations (2.52 ± 1.62 vs. 2.06 ± 1.44 µg/mL, p < 0.05). This difference is probably explained by the different frequency of the CFHR3*B allele in patients with mutations (79%) and without mutations (59%).
FHR-3 Levels Are Higher in Patients With aHUS Than in Controls With the Same CFHR3 Genotype
Having observed that FHR-3 levels are associated with CFHR3*A/B/Del genotypes, we then compared FHR-3 levels in control individuals and in patients with aHUS with the same CFHR3 genotype. FHR-3 levels were significantly higher in the patients with aHUS than in the controls with the same CFHR3 genotypes A/Del, A/A or A/B, whereas in the CFHR3 genotypes B/Del and B/B the differences in FHR-3 levels were not significant due to the small sample size of the control cohort (Figure 3). These results suggest that factors other than the CFHR3*A/B/Del alleles contribute to FHR-3 levels. To determine whether FHR-3 levels change with age, all patients with aHUS and at least one copy of CFHR3 were included in one of three subgroups, according to their age at blood sampling. No significant differences in FHR-3 concentration were observed when comparing all the patients together, or when they were further subdivided according to their CFHR3 genotype (Figure 4).
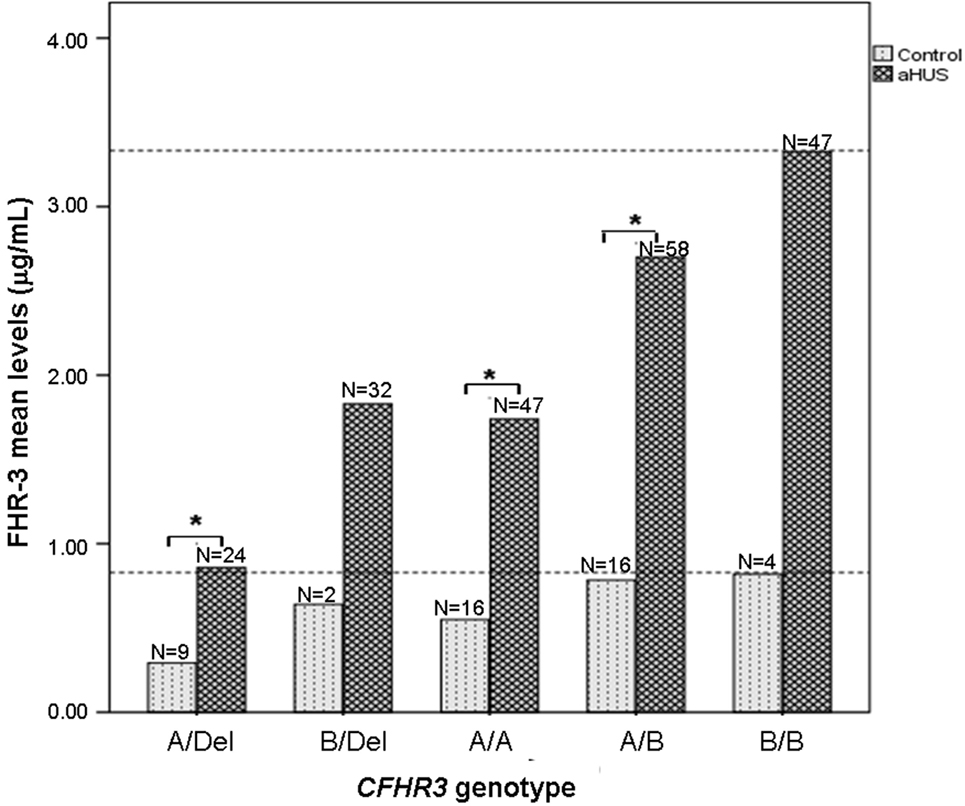
Figure 3. FHR-3 levels are higher in patients with atypical hemolytic uremic syndrome (aHUS) than in controls. Histogram showing the mean FHR-3 levels in controls and in patients with aHUS with different CFHR3 genotypes. Statistically significant differences (Kruskal–Wallis test, p < 0.05) are denoted by an asterisk. Dashed lines indicate the highest FHR-3 level observed in controls and patients.
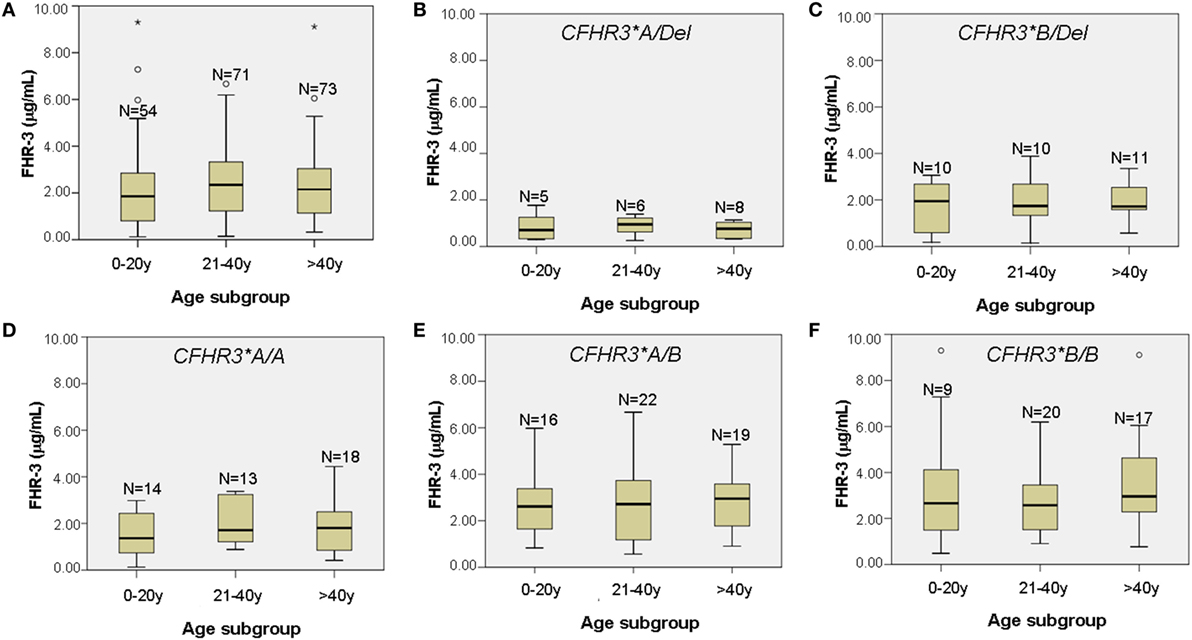
Figure 4. FHR-3 levels in patients with atypical hemolytic uremic syndrome (aHUS) do not change with age. (A) Boxplot of FHR-3 levels in 198 aHUS patients with the CFHR3 genotypes A/Del, B/Del, A/A, A/B, or B/B, subgrouped according to their age at blood sampling (in years). (B–F) Boxplots of FHR-3 levels in each of the five CFHR3 genotype groups. The number of patients within each age subgroup is also shown. Outliers are denoted by symbols (o).
Discussion
In this study, we show that the aHUS-risk CFHR3*B allele determines higher FHR-3 levels than the non-risk CFHR3*A allele. This observation was anticipated by the analysis of 49 healthy Spanish controls genotyped for the CFHR3*A/B/Del alleles and was further demonstrated in 230 patients with aHUS, most of Spanish origin.
FHR-3 levels in the 49 Spanish controls were 0.58 ± 0.26 µg/mL, which was very similar to the 0.69 µg/mL previously determined in 100 Dutch controls (18). In fact, these results support the evidence indicating that the actual concentration of FHR-3 in plasma is indeed much lower than the initial estimation of FHR-3 levels at 70–100 µg/mL (20). Moreover, we observed that the FHR-3 levels were significantly higher in the aHUS patients than in the control individuals (2.06 ± 1.77 vs. 0.58 ± 0.26 µg/mL, p < 0.0001). This result was not due to a significant difference in the allele frequency of CFHR3*Del between patients and controls, because when the 22 aHUS patients and the 2 controls with the CFHR3*Del/Del genotype and homozygous FHR-3 deficiency were excluded, a similar difference in FHR-3 levels was observed (2.28 ± 1.72 vs. 0.61 ± 0.24 µg/mL, p < 0.0001).
These results suggest that increased FHR-3 levels predispose to aHUS. This observation is in line with a previous study (21) that determined FHR-3 levels in 21 patients with aHUS (1.60 ± 0.57 µg/mL) and 21 controls (1.06 ± 0.53 µg/mL), although the difference in FHR-3 levels between their patient and control cohorts was smaller than in our study, which could well be related to the cohort size and the variation in allele frequency. In our study, 4% of the controls and 9% of the patients with aHUS carried the CFHR3*Del/Del genotype (i.e., homozygous CFHR3-CFHR1 deletion), frequencies comparable to the 2.9% and 12.4% observed in a French study comparing 70 controls and 117 patients with aHUS (22). However, in the study by Schäfer et al. (Table S2 in Supplementary Material), as many as 14% of controls and 38% of patients with aHUS presented homozygous deficiency of FHR-3, and these high and very different frequencies limit the relevance of their observations when comparing aHUS patients and controls. High frequencies of homozygous CFHR3–CFHR1 deletion have been reported in aHUS patients with anti-FH autoantibodies or with mutations in complement factor I (22), as well as in Middle Eastern and North African control populations (23, 24); the reason for the high frequency of the homozygous CFHR3-CFHR1 deletion in certain control populations could be related with its protective role against age-related macular degeneration (25) and IgA nephropathy (26). In conclusion, the comparison of FHR-3 levels could be biased by the ethnic origin of controls and patients, and by the frequency of anti-FH autoantibodies or factor I mutations in the patient cohorts. These facts have to be taken into account when trying to establish proper comparisons between control and patient cohorts.
To adequately analyze the contribution of the CFHR3*A and CFHR3*B alleles to FHR-3 levels, patients and controls with the CFHR3*Del/Del genotype were excluded. An analysis of the control cohort suggested a higher expression of the CFHR3*B allele that was clearly confirmed in the aHUS cohort. Patients with the CFHR3*A/Del genotype showed the lowest FHR-3 levels (0.684–1.032 µg/mL), patients with genotypes B/Del and A/A presented intermediate levels (1.437–2.201 µg/mL), and patients with genotypes A/B and B/B had the highest FHR-3 levels (2.330–4.056 µg/mL), explaining the wide individual range in FHR-3 concentrations between 0.684 and 4.056 µg/mL.
More importantly, statistically significant differences in FHR-3 levels were observed between the CFHR3 genotypes A/Del and B/Del (p = 0.0043), A/A and A/B (p = 0.0028), and A/A and B/B (p = 0.0008), demonstrating that the CFHR3*A allele is associated with lower FHR-3 concentrations than the aHUS-risk CFHR3*B allele. These data provide a genetic explanation for the increased FHR-3 levels observed in patients with aHUS, who present a higher frequency of the CFHR3*B allele than control individuals. However, whether higher FHR-3 levels increase susceptibility to aHUS is currently unknown, and the potential pathogenic mechanism will remain elusive until the actual physiological role of FHR-3 within the complement system is fully understood.
Recombinant FHR-3 binds C3b and C3d with similar affinity to FH (27); once bound, however, it cannot regulate complement activation because it lacks domains homologous to the N-terminal region of FH (28, 29). Therefore, increased competition between FHR-3 and FH for C3b binding will theoretically result in reduced complement regulation. Because the molar FHR-3 concentration in plasma is approximately 140 times lower than the molar FH concentration (18), competition between FHR-3 and FH for plasma-C3b binding is likely to be irrelevant. Nevertheless, the FHR-3/FH ratio could be more relevant for appropriate regulation of complement activation on cellular surfaces, such as the damaged endothelium of patients with aHUS. A recent study (30) shows a 1.3–1.9 increase in the FHR-1/FH molar ratio in patients with IgA nephropathy with disease progression. In our study, FHR-3 levels are 3.6 times higher in aHUS than in controls, while FH levels are only 1.2 times higher (not shown), but to demonstrate whether the increased FHR-3/FH ratio actually reduces complement regulation on the endothelial surface will require further investigation. Indirect evidence for competition between FH and FHR-3 on the cellular surface is provided by the functional characterization of a FH:FHR-3 hybrid protein identified in a familial case of aHUS (31). The authors suggest that the loss of FH regulatory activity of the hybrid FH:FHR-3 protein on cell-based assays could be explained because its C-terminal domains (belonging to FHR-3) prevent the correct orientation and function of the N-terminal domains (belonging to FH). A slightly different FH:FHR-3 hybrid protein, which also shows reduced FH regulatory activity on the cellular surface, has been reported in a sporadic case of aHUS (32). Based on these findings, we speculate that inefficient complement regulation on the cellular surface could also result from an altered FH/FHR-3 ratio. This would explain why the CFH(H3)–CFHR3*B–CFHR1*B haplotype, associated with reduced FH levels (17) and enhanced FHR-3 levels (this study) increases the risk of aHUS. The CFH(H3)-CFHR3*B–CFHR1*B haplotype also carries the CFH (rs1065489; p936D<E) and CFHR3 variants (rs385390, rs426736, and rs371075) that were shown to confer protection against meningococcal disease (33); quantitation of FHR-3 levels and CFHR3*A/B/Del genotyping in these patients will likely help establish the actual relationship between this haplotype and protection against N. meningitidis infection.
Another interesting conclusion from our study is that FHR-3 concentration is not only determined by the number of copies of the CFHR3*A and CFHR3*B alleles. Stratification by the CFHR3*A/B/Del genotype still showed higher FHR-3 levels in patients with aHUS than in controls (Figure 3), suggesting that additional genetic and/or acquired factors contribute to the increased FHR-3 concentration in patients with aHUS. We cannot rule out the possibility that genetic variants within the adjacent CFH and CFHR1 genes modulate CFHR3 expression, but our current results do not favor this hypothesis (Table 3). Because plasma levels of FH have a wide range of variation and were shown to increase with age (34), we explored whether this was also the case for FHR-3 levels by analyzing data from the patients with aHUS. FHR-3 levels remain unchanged in the three age subgroups, either when considering the whole patient cohort, or when each CFHR3 genotype was analyzed separately (Figure 4). A similar result was observed in the two age subgroups from the control cohort, but the small sample size and the absence of pediatric controls limit the relevance of this observation. Although FHR-3 does not appear to be an acute phase reactant (18), FHR-3 levels in patient samples could be associated with disease activity, in particular with the decreased renal function observed in aHUS. In this context, two recent studies in IgA nephropathy patients suggest that impaired renal function increases the FHR-1/FH ratio (30, 35). To fully understand the increased FHR-3 levels observed in patients with aHUS, it would be necessary to monitor FHR-3 concentration in serial samples from patients along their clinical evolution, and renal function/damage over time.
In conclusion, in this study we show that CFHR3*A is a low-expression allele, and CFHR3*B is a high-expression allele, and that, next to CFHR3 copies, other genetic factors determine the FHR-3 levels. We also show that the aHUS-risk CFH(H3)–CFHR3*B–CFHR1*B haplotype is associated with increased FHR-3 levels, and speculate that it leads to an imbalance between the local FH and FHR-3 concentration that predisposes patients to aHUS. These results uncover that genotyping for the CFHR3*A, CFHR3*B, and CFHR3*Del alleles is necessary for a proper interpretation of FHR-3 levels within a certain pathological context, and we propose to incorporate these analysis to the current genetic workflow in aHUS patients.
Ethics Statement
Patients and controls provided written informed consent, as approved by the ethical committees from La Paz University Hospital or the Biological Research Center.
Author Contributions
RP, DW, and TK were responsible for quantitation of FHR-3 in patients and controls. IG genotyped the control individuals, performed statistical analyses, and prepared figures and tables. AL searched databases for CFHR3 expression and prepared figures. SR was responsible for CFH–CFHR3–CFHR1 genotyping in the patients with aHUS. PS-C designed the study, analyzed the data, and wrote the first draft of the manuscript. All the authors revised the data and contributed to the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We appreciate the technical assistance of Lorena Risueño, César Vélez, and Álex Otero in the management of biological samples of patients and controls. We thank Rosario Madero (IdiPAZ Biostatistics) for statistical help, and Vega Mauleón and Hoi Tong (IdiPAZ UCICEC) for blood sampling of the healthy Spanish volunteers. This study was funded by the Spanish Instituto de Salud Carlos III and the European Program FEDER (grants PI12/00597 and PI16/00723 to PS-C, and grant SAF2015-66287-R to SR), and by the Complement II-CM network (B2017/BMD-3673). RP and TK received funding from the European Union's seventh Framework program under EC-GA no. 279185 (EUCLIDS; www.euclids-project.eu). AL is supported by the Spanish Center for Biomedical Network Research on Rare Diseases (CIBERER).
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.00848/full#supplementary-material.
References
1. Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med (2009) 361(17):1676–87. doi:10.1056/NEJMra0902814
2. Noris M, Remuzzi G. Cardiovascular complications in atypical haemolytic uraemic syndrome. Nat Rev Nephrol (2014) 10(3):174–80. doi:10.1038/nrneph.2013.280
3. Sánchez-Corral P, Melgosa M. Advances in understanding the aetiology of atypical Haemolytic Uraemic Syndrome. Br J Haematol (2010) 150(5):529–42. doi:10.1111/j.1365-2141.2010.08295.x
4. Kavanagh D, Goodship TH, Richards A. Atypical hemolytic uremic syndrome. Semin Nephrol (2013) 33(6):508–30. doi:10.1016/j.semnephrol.2013.08.003
5. Rodríguez de Córdoba S, Hidalgo MS, Pinto S, Tortajada A. Genetics of atypical hemolytic uremic syndrome (aHUS). Semin Thromb Hemost (2014) 40(4):422–30. doi:10.1055/s-0034-1375296
6. Le Quintrec M, Zuber J, Moulin B, Kamar N, Jablonski M, Lionet A, et al. Complement genes strongly predict recurrence and graft outcome in adult renal transplant recipients with atypical hemolytic and uremic syndrome. Am J Transplant (2013) 13(3):663–75. doi:10.1111/ajt.12077
7. Meri S. Self-nonself discrimination by the complement system. FEBS Lett (2016) 590(15):2418–34. doi:10.1002/1873-3468.12284
8. Skerka C, Chen Q, Fremeaux-Bacchi V, Romenina LT. Complement factor H related proteins (CFHRs). Mol Immunol (2013) 56:170–80. doi:10.1016/j.molimm.2013.06.001
9. Józsi M, Tortajada A, Uzonyi B, Goicoechea de Jorge E, Rodríguez de Córdoba S. Factor H-related proteins determine complement-activating surfaces. Trends Immunol (2015) 36(6):374–84. doi:10.1016/j.it.2015.04.008
10. Caprioli J, Castelletti F, Bucchioni S, Bettinaglio P, Bresin E, Pianetti G, et al. Complement factor H mutations and gene polymorphisms in haemolytic uraemic syndrome: the C-257T, the A2089G and the G2881T polymorphisms are strongly associated with the disease. Hum Mol Genet (2003) 12(24):3385–95. doi:10.1093/hmg/ddg363
11. Bresin E, Rurali E, Caprioli J, Sanchez-Corral P, Fremeaux-Bacchi V, Rodriguez de Cordoba S, et al. Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol (2013) 24(3):475–86. doi:10.1681/ASN.2012090884
12. Sansbury FH, Cordell HJ, Bingham C, Bromilow G, Nicholls A, Powell R, et al. Factors determining penetrance in familial atypical haemolytic uraemic syndrome. J Med Genet (2014) 51(11):756–64. doi:10.1136/jmedgenet-2014-102498
13. Dragon-Durey MA, Loirat C, Cloarec S, Macher MA, Blouin J, Nivet H, et al. Anti-factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol (2005) 16(2):555–63. doi:10.1681/ASN.2004050380
14. Zipfel PF, Edey M, Heinen S, Józsi M, Richter H, Misselwitz J, et al. Deletion of complement factor H-related genes CFHR1 and CFHR3 is associated with atypical hemolytic uremic syndrome. PLoS Genet (2007) 3(3):e41. doi:10.1371/journal.pgen.0030041
15. Józsi M, Licht C, Strobel S, Zipfel SL, Richter H, Heinen S, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood (2008) 111(3):1512–4. doi:10.1182/blood-2007-09-109876
16. Abarrategui-Garrido C, Martínez-Barricarte R, López-Trascasa M, de Córdoba SR, Sánchez-Corral P. Characterization of complement factor H-related (CFHR) proteins in plasma reveals novel genetic variations of CFHR1 associated with atypical hemolytic uremic syndrome. Blood (2009) 114(19):4261–71. doi:10.1182/blood-2009-05-223834
17. Bernabéu-Herrero ME, Jiménez-Alcázar M, Anter J, Pinto S, Sánchez Chinchilla D, Garrido S, et al. Complement factor H, FHR-3 and FHR-1 variants associate in an extended haplotype conferring increased risk of atypical hemolytic uremic syndrome. Mol Immunol (2015) 67(2 Pt B):276–86. doi:10.1016/j.molimm.2015.06.021
18. Pouw RB, Brouwer MC, Geissler J, van Herpen LV, Zeerleder SS, Wuillemin WA, et al. Complement factor H-related protein 3 serum levels are low compared to factor H and mainly determined by gene copy number variation in CFHR3. PLoS One (2016) 11(3):e0152164. doi:10.1371/journal.pone.0152164
19. Tortajada A, Yébenes H, Abarrategui-Garrido C, Anter J, García-Fernández JM, Martínez-Barricarte R, et al. C3 glomerulopathy-associated CFHR1 mutation alters FHR oligomerization and complement regulation. J Clin Invest (2013) 123(6):2434–46. doi:10.1172/JCI68280
20. Fritsche LG, Lauer N, Hartmann A, Stippa S, Keilhauer CN, Oppermann M, et al. An imbalance of human complement regulatory proteins CFHR1, CFHR3 and factor H influences risk for age-related macular degeneration (AMD). Hum Mol Genet (2010) 19(23):4694–704. doi:10.1093/hmg/ddq399
21. Schäfer N, Grosche A, Reinders J, Hauck SM, Pouw RB, Kuijpers TW, et al. Complement regulator FHR-3 is elevated either locally or systemically in a selection of autoimmune diseases. Front Immunol (2016) 7:542. doi:10.3389/fimmu.2016.00542
22. Dragon-Durey MA, Blanc C, Marliot F, Loirat C, Blouin J, Sautes-Fridman C, et al. The high frequency of complement factor H related CFHR1 gene deletion is restricted to specific subgroups of patients with atypical haemolytic uraemic syndrome. J Med Genet (2009) 46(7):447–50. doi:10.1136/jmg.2008.064766
23. Hageman GS, Hancox LS, Taiber AJ, Gehrs KM, Anderson DH, Johnson LV, et al. Extended haplotypes in the complement factor H (CFH) and CFH-related (CFHR) family of genes protect against age-related macular degeneration: characterization, ethnic distribution and evolutionary implications. Ann Med (2006) 38(8):592–604. doi:10.1080/07853890601097030
24. Leban N, Abarrategui-Garrido C, Fariza-Requejo E, Amiñoso-Carbonero C, Pinto S, Chibani JB, et al. Factor H and CFHR1 polymorphisms associated with atypical haemolytic uraemic syndrome (aHUS) are differently expressed in Tunisian and in Caucasian populations. Int J Immunogenet (2012) 39(2):110–3. doi:10.1111/j.1744-313X.2011.01071.x
25. Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet (2006) 38(10):1173–7. doi:10.1038/ng1890
26. Gharavi AG, Kiryluk K, Choi M, Li Y, Hou P, Xie J, et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet (2011) 43(4):321–7. doi:10.1038/ng.787
27. Hellwage J, Jokiranta TS, Koistinen V, Vaarala O, Meri S, Zipfel PF. Functional properties of complement factor H-related proteins FHR-3 and FHR-4: binding to the C3d region of C3b and differential regulation by heparin. FEBS Lett (1999) 462(3):345–52. doi:10.1016/S0014-5793(99)01554-9
28. Kühn S, Skerka C, Zipfel PF. Mapping of the complement regulatory domains in the human factor H-like protein 1 and in factor H. J Immunol (1995) 155:5663–70.
29. Sharma AK, Pangburn MK. Identification of three physically and functionally distinct binding sites for C3b in human complement factor H by deletion mutagenesis. Proc Natl Acad Sci U S A (1996) 93(20):10996–1001. doi:10.1073/pnas.93.20.10996
30. Tortajada A, Gutiérrez E, Goicoechea de Jorge E, Anter J, Segarra A, Espinosa M, et al. Elevated factor H-related protein 1 and factor H pathogenic variants decrease complement regulation in IgA nephropathy. Kidney Int (2017) 92(4):953–63. doi:10.1016/j.kint.2017.03.041
31. Francis NJ, McNicholas B, Awan A, Waldron M, Reddan D, Sadlier D, et al. A novel hybrid CFH/CFHR3 gene generated by a microhomology-mediated deletion in familial atypical hemolytic uremic syndrome. Blood (2012) 119(2):591–601. doi:10.1182/blood-2011-03-339903
32. Challis RC, Araujo GS, Wong EK, Anderson HE, Awan A, Dorman AM, et al. A de novo deletion in the regulators of complement activation cluster producing a hybrid complement factor H/complement factor H-related 3 gene in atypical hemolytic uremic syndrome. J Am Soc Nephrol (2016) 27(6):1617–24. doi:10.1681/ASN.2015010100
33. Davila S, Wright VJ, Khor CC, Sim KS, Binder A, Breunis WB, et al. Genome-wide association study identifies variants in the CFH region associated with host susceptibility to meningococcal disease. Nat Genet (2010) 42(9):772–6. doi:10.1038/ng.640
34. Esparza-Gordillo J, Soria JM, Buil A, Almasy L, Blangero J, Fontcuberta J, et al. Genetic and environmental factors influencing the human factor H plasma levels. Immunogenetics (2004) 56(2):77–82. doi:10.1007/s00251-004-0660-7
Keywords: complement, factor H, factor H-related protein 3, CFHR3 gene, atypical hemolytic-uremic syndrome
Citation: Pouw RB, Gómez Delgado I, López Lera A, Rodríguez de Córdoba S, Wouters D, Kuijpers TW and Sánchez-Corral P (2018) High Complement Factor H-Related (FHR)-3 Levels Are Associated With the Atypical Hemolytic-Uremic Syndrome-Risk Allele CFHR3*B. Front. Immunol. 9:848. doi: 10.3389/fimmu.2018.00848
Received: 16 January 2018; Accepted: 05 April 2018;
Published: 24 April 2018
Edited by:
Zvi Fishelson, Tel Aviv University, IsraelReviewed by:
Veronique Fremeaux-Bacchi, Assistance Publique Hopitaux De Paris (AP-HP), FranceChristine Skerka, Leibniz-Institut für Naturstoff-Forschung und Infektionsbiologie, Hans Knöll Institut, Germany
Copyright: © 2018 Pouw, Gómez Delgado, López Lera, Rodríguez de Córdoba, Wouters, Kuijpers and Sánchez-Corral. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pilar Sánchez-Corral, cGlsYXIuc2FuY2hlei1jb3JyYWxAaWRpcGF6LmVz