 Ana Stojanovic
Ana Stojanovic Margareta P. Correia
Margareta P. Correia Adelheid Cerwenka
Adelheid Cerwenka- 1Innate Immunity (D080), German Cancer Research Center (DKFZ), Heidelberg, Germany
- 2Department of Immunobiochemistry, Medical Faculty Mannheim, University of Heidelberg, Mannheim, Germany
Natural killer group 2, member D (NKG2D) receptor is a type II transmembrane protein expressed by both innate and adaptive immune cells, including natural killer (NK) cells, CD8+ T cells, invariant NKT cells, γδ T cells, and some CD4+ T cells under certain pathological conditions. NKG2D is an activating NK receptor that induces cytotoxicity and production of cytokines by effector cells and supports their proliferation and survival upon engagement with its ligands. In both innate and T cell populations, NKG2D can costimulate responses induced by other receptors, such as TCR in T cells or NKp46 in NK cells. NKG2D ligands (NKG2DLs) are remarkably diverse. Initially, NKG2DL expression was typically attributed to stressed, infected, or transformed cells, thus signaling “dysregulated-self.” However, many reports indicated their expression under homeostatic conditions, usually in the context of cell activation and/or proliferation. Myeloid cells, including macrophages and dendritic cells (DCs), are among the first cells sensing and responding to pathogens and tissue damage. By secreting a plethora of soluble mediators, by presenting antigens to T cells and by expressing costimulatory molecules, myeloid cells play vital roles in inducing and supporting responses of other immune cells in lymphoid organs and tissues. When activated, both macrophages and DCs upregulate NKG2DLs, thereby enabling them with additional mechanisms for regulating lymphocyte responses. In this review, we will focus on the expression of NKG2D by innate and adaptive lymphocytes, the regulation of NKG2DL expression on myeloid cells, and the contribution of the NKG2D/NKG2DL axis to the crosstalk of myeloid cells with NKG2D-expressing lymphocytes. In addition, we will highlight pathophysiological conditions associated with NKG2D/NKG2DL dysregulation and discuss the putative involvement of the NKG2D/NKG2DL axis in the lymphocyte/myeloid cell crosstalk in these diseases.
The Natural Killer Group 2, Member D (NKG2D) Receptor
Natural killer group 2, member D is a type II transmembrane protein with a C-type lectin-like extracellular domain, expressed on the cell surface as a disulfide-linked homodimer. The receptor possesses a short intracellular tail with charged amino acid residues that enable its association with the adaptor molecules DAP10 and/or DAP12, which is essential for NKG2D surface expression (1–3). In mice, alternative splicing of NKG2D results in two distinct isoforms: NKG2D-short and NKG2D-long. The NKG2D-short isoform can associate with both DAP10 and DAP12, whereas the NKG2D-long isoform associates only with DAP10 (2). Freshly isolated naïve natural killer (NK) cells express only the NKG2D-long isoform that forms complexes with DAP10. Activated NK cells express both NKG2D isoforms and can therefore pair with both adapters (2). However, human NK cells only express the NKG2D-long isoform, and consequently, NKG2D appears to only use DAP10 for signal transduction. NKG2D triggering induces cytotoxic responses, secretion of cytokines and chemokines, and supports proliferation and survival of responding effector cells (4).
The NKG2D receptor gene (KLRK1, killer cell lectin-like receptor K1) was first described in 1991 in human NK cells (5). Since then, several other lymphocytes have been shown to express NKG2D, namely, all αβCD8+ T cells in human, activated, but not naïve, αβCD8+ T cells in mice, γδ T cells, invariant NKT (iNKT) cells, and some CD4+ T cells under certain pathological conditions. More recently, NKG2D was detected on the surface of innate lymphoid cells (ILCs), including helper ILC1 and ILC3 (6–8). Importantly, those different NKG2D-expressing immune cell types reside in distinct morphological sites, such as lymphoid organs, skin, and epithelial and sub-epithelial tissues. Indeed, NKG2D-expressing cells and upregulation of NKG2D ligands (NKG2DLs) were shown to play important roles in inflammation, anti-tumor, and anti-viral responses in different organs (9, 10). In addition, the pathophysiology of several autoimmune conditions, as well as acute and chronic allograft rejection, involved dysregulated expression or/and activation of NKG2D and its ligands (11–17).
Expression of NKG2D on immune effector cells is regulated by cell activation and microenvironmental factors. In NK cells, which constitutively express NKG2D, the cytokines IL-2, IL-12, and IL-15 were shown to upregulate, while IL-21, IFN-γ, and TGF-β negatively regulated NKG2D surface expression (18). The triggering of the NKG2D receptor in mouse and human NK cells can induce effector functions, such as cytotoxicity and cytokine production. However, several studies revealed that pre-activation with cytokines, such as IL-2 or IL-15, was required to trigger NK cell responses upon NKG2D engagement (19, 20). This feature might have evolved to assure NKG2D activation only in cases when potential “danger signals” are present in the context of inflammation, characterized by the production of inflammatory cytokines. In addition, NKG2D can act as a costimulatory molecule, able to induce cytolytic activity in resting NK cells, when cotriggered with other activating receptors, such as NKp46 or 2B4 (9). Several mechanisms account for cytokine-mediated priming of NKG2D responsiveness, such as IL-2-mediated activation of mTORC1 and upregulation of amino acid transporters (20), or IL-15-induced phosphorylation of the adaptor molecule DAP10 (19) and activation of cytosolic phospholipase A2 accompanied by production of arachidonic acid (21). In human NK cells and resting mouse NK cells, which express the NKG2D-long isoform, the signal is transduced via DAP10 and propagates through Grb2/Vav and the PI3K pathway, similar to the costimulatory molecule CD28 (22), which might explain the need for additional signals for cell activation. In mouse, activation induces changes in Nkg2d mRNA alternative splicing, leading to the expression of the NKG2D-short isoform. Its coupling to the ITAM-bearing adaptor DAP12, which recruits and activates signaling via Syk and ZAP70 protein kinases, has been implicated in the triggering of the NKG2D-short isoform without the need of cytokine “priming” or coreceptor activation (22). These results signify that NKG2D function on NK cells depends on the NK cell activation status and tightly correlates with the presence of additional microenvironmental signals, such as cytokines or ligands of other receptors expressed on target or neighboring cells. Therefore, it is not surprising that NKG2D-deficient mice do not show a major phenotype until crossed to TRAMP or Eμ-Myc mice, which spontaneously develop prostate cancer and lymphoma, respectively (23).
NKG2DLs: Expression and Regulation
Although NKG2D is largely not polymorphic (only two alleles with a single aa difference exist in human) and shows strong evolutionary conservation with ~70% sequence identity between mouse and human, this receptor is able to bind a broad range of stress-induced ligands that, in contrast, show a high degree of polymorphism (24, 25). In the context of transplantation, polymorphic NKG2DLs can cause donor–recipient incompatibility and lead to allograft rejection by inducing the formation of antibodies directed against NKG2DL epitopes and complement-dependent cytotoxicity (26–29).
NKG2D ligands comprise several MHC class I-like molecules, which include murine UL16-binding protein-like transcript 1 (MULT1), retinoic acid early transcripts α-ε (RAE-1 α-ε) and H60 a-c in mice, and MHC class I-related genes A and B (MICA and MICB) and UL16-binding protein (ULBP) family in human. All NKG2DLs have α1α2 domains responsible for binding to the NKG2D receptor, however, only low sequence similarity can be observed between various ligands, indicating a significant level of variability. It was proposed that the variability of these ligands increased with coevolution with pathogens, allowing their differential expression patterns among cells and tissues, distinct intracellular trafficking, and differential affinity for the NKG2D receptor, which might influence the strength of the delivered signal (24).
NKG2D ligand expression is most frequently associated with infection, cell stress, and transformation, thus alerting for “stressed- and damaged-self.” Distinct forms of cell stress can induce cell surface expression of NKG2DLs, including DNA damage, oxidative stress, heat-shock, or the ER stress response (30–35). For example, the p53 pathway, involved in the DNA damage response, was shown to strongly upregulate ULBP1 and ULBP2 at both mRNA and protein level. Similarly, heat-shock-induced transcription factor HSF1 can drive MICA promoter activation (36). Other transcription factors, including E2F, NF-kB, ATF4, the Sp-family, and AP-1, were also shown to be involved in NKG2DL mRNA transcription (36–38). Sauer et al. showed that histone acetylation and binding of acetyltransferases CBP and p300 to NKG2DL promoter regions increased NKG2DL expression by tumor cells (39), suggesting the importance of an open chromatin state in the regulation of NKG2DL expression. In addition, NKG2DL expression has been associated with viral infections, including CMV, influenza, hepatitis B, Epstein–Barr, and adenovirus (40), as well with as some bacterial infections (e.g., E. coli, M. tuberculosis) (24). Accordingly, triggering of toll-like receptors (TLRs), that sense microbial products, also induced NKG2DL expression in macrophages and dendritic cells (DCs) (41, 42). Certain viruses, such as HIV, engage the DNA damage pathway, while other viruses, such as MCMV, induce NKG2DL expression via PI3K-mediated activation (35).
NKG2D ligand expression is regulated at several levels (transcriptional, post-transcriptional, and post-translational) and depends on the cell type, its activation and metabolic state, and the microenvironmental context (35, 36). Cytokines, such as IFN-γ, IFN-α, and TGF-β, were reported to regulate NKG2DL expression on mouse and human cell lines (10). NKG2DL surface expression is further controlled by miRNAs and mRNA-binding proteins that target NKG2DLs at the transcript level (43–45), by alternative splicing (37), and by Ub-mediated degradation of NKG2DL protein(s) (46, 47). In various cell types and tissues, NKG2DL RNA transcripts were detected in the absence of protein (40). In gut and bronchial epithelia, ligands can be stored intracellularly and transferred to the cell surface upon stimulation (40). In addition, NKG2DLs can be delivered to effector cells via exosome secretion, which represents a potent mechanism exploited by cancer cells to downregulate NKG2D expression on effector cells (48). NKG2DL surface expression can be downregulated by proteolytic shedding at the plasma membrane; however, this process depends on the type of ligand, and it seems to be regulated by the microenvironment (49). For example, elevated serum levels of soluble MIC proteins were detected in patients with different types of cancer and correlated with unfavorable prognosis. Soluble MIC was shown to induce NK and CD8+ T cell dysfunction by inducing loss of the CD3ζ signaling molecule, or by decreasing NKG2D surface expression through endocytosis and degradation upon its engagement (50). By contrast, soluble MULT1, a high affinity mouse NKG2DL, was shown to counteract NK cell desensitization and to cause NK cell activation and tumor rejection in vivo (51). Opposite to cancer, in autoimmune disease, although increased levels of MIC proteins in serum have been detected, NKG2D expression was not affected in the analyzed patients (50).
Although most frequently associated with infection, cellular stress, and transformation, NKG2DLs are also detected on certain cell types in the absence of pathophysiological conditions [reviewed in Ref. (40)]. These include subsets of uncommitted thymocytes, activated T and myeloid cells, class-switching B cells, regulatory T cells, myeloid-derived suppressor cells (MDSCs), bone marrow stromal cells, committed myelomonocytic progenitors, pluripotent mesenchymal cells, and epithelial cells of the respiratory and gut mucosa. Other cells, such as myoblasts, hepatocytes, neurons, and mouse embryonic cells, were also reported to express NKG2DLs, but their regulation and function on these cells is less clear. In the case of hematopoietic cells, it has been reported that NK cells can eliminate activated immune cells in an NKG2D-dependent manner, thereby restraining T cell responses or excessive inflammation mediated by myeloid cell activation. In the thymus, NKG2D might be involved in the regulation of the T cell repertoire (40), while at the epithelial barriers, NKG2D/NKG2DL expression can be linked to continuous presence of commensal microbiota and constitutive state of low-grade, the so-called physiological inflammation (52). Recently, Thompson and colleagues reported that endothelial cells in lymph nodes constitutively expressed Rae-1, whose engagement at steady state led to downmodulation of NKG2D expression and function in circulating NK cells (53). Human activated NK cells can themselves upregulate certain ULBP family members after culture with the cytokines IL-12/15/18 (54). In this case, intercellular NKG2D activation led to ADAM17-mediated release of TNF-α, thus promoting NK cell cytokine release. By contrast, NKG2DLs expressed on NK cells from diabetic NOD mice were postulated to negatively regulate expression of the NKG2D receptor (55). Moreover, NK cells could acquire NKG2DLs via trogocytosis upon interaction with target cells, which led to their fratricide, thereby resulting in downmodulation of the immune response (56).
Expression of NKG2DLs by Myeloid Cells
In both human and mice, various myeloid cell subsets were reported to express NKG2DLs (Figure 1). In many cases, NKG2DL induction on myeloid cells is a direct consequence of infection. For example, human macrophages upregulate NKG2DL expression upon influenza or Sendai virus infection (57). In infection settings, macrophages can directly sense pathogens using various innate immune receptors, some of which have been shown to directly regulate NKG2DL expression when triggered in vitro. Signaling via different TLRs can induce NKG2DLs in both monocytes and macrophages, but the nature of the induced ligand(s), the levels of their expression, and the magnitude of subsequent NKG2D-driven responses might vary. It is tempting to speculate that such differential responses were evolutionarily driven and tailored to fit the defense strategy against an invading pathogen. In primary human macrophages, TLR triggering induces MICA and MICB expression. Eissmann et al. showed that LPS not only induced MICA expression, but also decreased the levels of miRNAs involved in targeting MICA transcripts for degradation (58). In addition, while TLR4 signaling was required for MICA gene transcription, the ATM/ATR pathway, involved in the DNA damage response, controlled the expression of the MICA protein (58), highlighting the complexity of regulation of NKG2DL expression. The Davis lab also showed that TLR7/8, but not TLR3 ligation, induced both MICA and MICB, and that MICA expression correlated with macrophage activation, measured by the production of proinflammatory cytokines (58). Similar to macrophages, TLR-stimulated monocytes upregulated MICA, but not other NKG2DLs, along with CD80 and MHC class I and II (59). Besides MICA/B, monocytes and macrophages can express other NKG2DLs. TLR2-driven ULBP1 expression was reported on M. tuberculosis infected monocytes and alveolar macrophages, leading to their NKG2D-dependent lysis by NK cells (60). ULBP1 could also be induced on monocytes by growth factors, but not by the cytokines TNF-α, IL-1β, and IFN-α, or by the TLR4 ligand LPS (61).
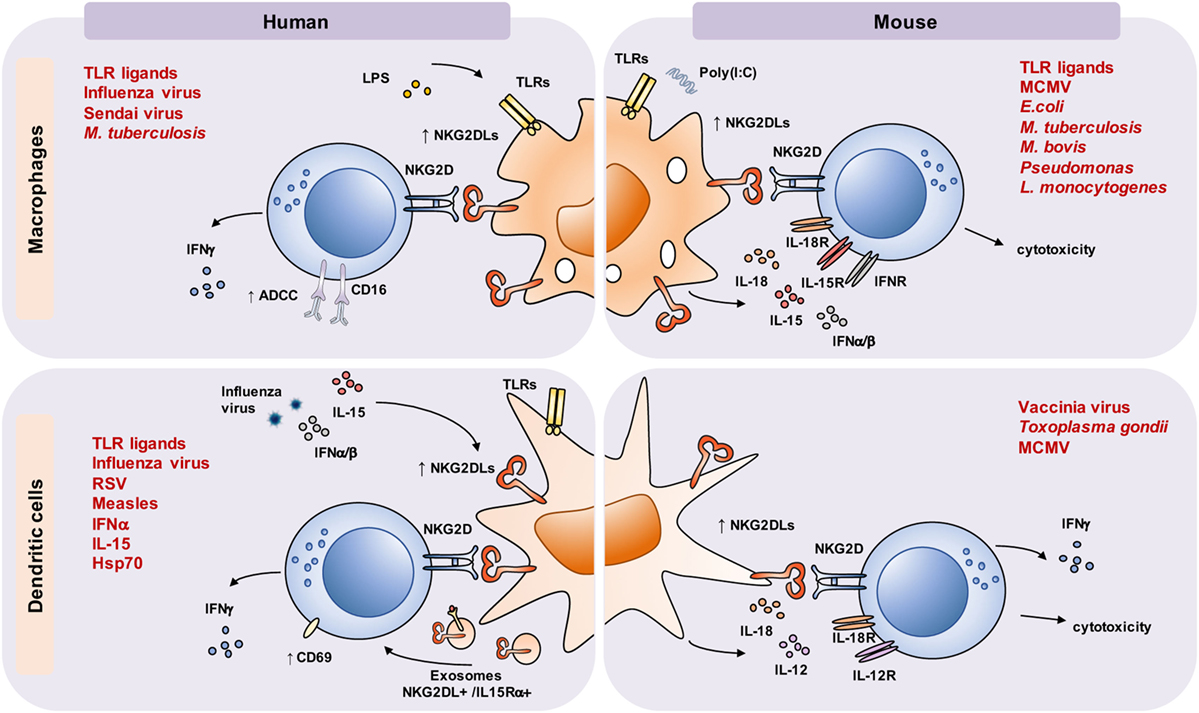
Figure 1. Macrophages and dendritic cells (DCs) activated by toll-like receptor (TLR) ligands, cytokines, viral or bacterial infection upregulate NKG2D ligands (NKG2DLs) and regulate natural killer (NK) cell effector responses. In both mouse and human, TLR activation and viral and bacterial infection were shown to upregulate NKG2DLs on macrophages and DCs. In addition, cytokines produced by myeloid cells upon infection, such as IL-15 or type I IFNs, can also induce NKG2DL expression on DCs and, importantly, increase natural killer group 2, member D (NKG2D) expression on interacting lymphocytes. Induced NKG2DLs interact with NKG2D expressed by NK cells and lead to their activation, resulting in secretion of IFN-γ, cytotoxicity, CD69 upregulation, and increased killing of antibody-coated cells by antibody-dependent cellular cytotoxicity (ADCC). NKG2DLs and IL-15Rα/IL-15 can also be delivered to NKG2D+ effector cells via exosomes. NK cell responses are further supported by soluble factors released by myeloid cells, including IL-12 and IL-18, which strongly synergize in IFN-γ induction. In turn, IFN-γ released by NK cells supports myeloid cell activation and release of soluble factors, creating a myeloid-lymphoid feedback activation loop. In some instances, activated NK cells can kill NKG2DL-expressing macrophages and DCs, thereby limiting their numbers, their responses, or improper stimulation. Abbreviations: RSV, respiratory syncytial virus; MCMV, mouse cytomegalovirus.
Similar to human monocytes and macrophages, mouse macrophages were reported to express NKG2DLs in the context of infection, such as M. tuberculosis (62) or P. aeruginosa (63). Murine peritoneal macrophages expressed Rae-1, but not other NKG2DLs, upon stimulation via TLRs, both in vitro and in vivo (42). Exposure to G− (E. coli) and G+ (S. aureus, L. monocytogenes) bacteria, Mycobacterium bovis BCG or infection with CMV, all induced Rae-1 expression (42, 64), reinforcing that macrophage activation and pathogen recognition is linked to NKG2DL upregulation. Cytokines produced upon response to pathogens, such as TNF-α, type I IFNs, or IFN-γ, were not required for Rae-1 expression in these settings.
Besides infection, evidence exists of NKG2DL association with myeloid cell differentiation and acquisition of an activated effector phenotype. This is not surprising, as induction of NKG2DLs was correlated with cell proliferation (65) and DNA damage (30, 31), which might occur during effector responses in tissues. It was shown that human CD34+ hematopoietic stem cells (HSC) expressed low levels of NKG2DLs, which were increased upon commitment to the myeloid lineage (61, 66). In mice, HSC transplantation in irradiated animals gave rise to Gr1+ and CD11b+ cells expressing Rae-1 and H60 in the bone marrow (29). Similarly, myeloid cells with immunosuppressive function that accumulated in tumor-bearing mice were reported to express Rae-1 (51, 67). So far, the reason for NKG2DL expression by immature myeloid cells is unclear. In tumor settings, engagement of NKG2D on NK cells led to NK cell activation and cytolysis of Rae-1-expressing myeloid cells (67). Whether NK cells can control myeloid lineage differentiation via regulating the numbers of developing progenitors is, so far, elusive.
Of note, besides expressing NKG2DLs, there are some indications that activated macrophages could also upregulate the NKG2D receptor, although these observations remain controversial. Thioglycolate-induction in vivo or stimuli, such as LPS, type I IFNs, and IFN-γ, were shown to drive mRNA and protein NKG2D expression on peritoneal and bone marrow-derived macrophages, respectively. When stimulated with immobilized ligands or ligand-expressing cells, NKG2D+ macrophages produced nitric oxide and TNF-α (2, 68, 69), indicating that this system might enhance macrophage-mediated elimination of pathogens, pathogen-infected or tumor cells. Indeed, it was also suggested that crosstalk with certain NKG2DL+ tumor cells might contribute to macrophage activation and lower the threshold for their responses (70). However, this effect might also be exploited in tumor settings and facilitate cancer immune evasion. For example, Qian et al. detected NKG2D expression on Gr1+ CD11b+ myeloid cells accumulating in blood, spleen, and bone marrow of tumor-bearing mice. Blockade of NKG2D in vitro impaired IL-4 and IL-10 production by these cells, while in vivo neutralization reduced their accumulation in mice bearing Rae-1+ tumors (71).
In DCs, NKG2DL upregulation was also associated with activation caused by infection and/or cellular stress (Figure 1). In mouse, vaccinia virus infection induced Rae-1 (72), while pulse with Toxoplasma gondii lysates led to both Rae-1 and MULT1 expression on the DC surface (73). In infected human monocyte-derived DCs, influenza virus induced ULBP proteins (74). Other RNA viruses, such as respiratory syncytial virus, led to ULBP1 upregulation, while measles virus, as well as exposure to poly(I:C), upregulated ULBP2 (41). These data indicate that different mechanisms can be employed by DCs to induce expression of distinct NKG2DLs in response to various virus types. IFN-α, which is primarily produced during viral infections, was reported to induce MIC ligand expression on human DCs. A similar effect was attributed to IL-15, while stimulation with LPS, poly(I:C), CD40L, TNF-α, IL-12, and IL-18 did not affect NKG2DL expression (75, 76). MICA expression was also induced by Hsp70 (77), although bacterial products or other molecules might as well have contributed to this effect (78). TLR ligands, such as LPS and poly(I:C), were able to upregulate ULBPs on human monocyte-derived DCs (41). ULBP1 was also detected on mature DCs in T cell areas of lymph nodes in situ, in close proximity to NKG2D-expressing CD8+ T cells (79), indicating its possible involvement in T cell priming.
The NKG2D/NKG2DL Axis in the Crosstalk of Myeloid Cells and NK Cells
Myeloid cells and innate lymphocytes form the first line of defense against invading pathogens. Their activation leads not only to the direct elimination of pathogens and their products, but also to the activation of proper adaptive immune responses. Moreover, it is becoming appreciated that these cells also respond to cues indicating tissue damage and, in addition to defense, orchestrate mechanisms of tissue repair (80). Since (i) both infection and damage were shown to upregulate NKG2DLs on myeloid cells, and (ii) factors produced in response to these events (by stroma, parenchymal, and immune cells) can induce or upregulate NKG2D on lymphocytes, an NKG2D-driven lymphocyte-myeloid cell crosstalk is expected to play an important role in these processes. Indeed, in many animal models, NKG2D genetic deletion or Ab-mediated blockade in vivo affected disease development and tissue repair, including tumor progression, autoimmunity, and wound healing (12, 13, 23, 65). However, the contribution of the individual cell populations to these observations remain not fully addressed. This particularly applies to cells expressing NKG2DLs, whose depletion or conditional targeting is experimentally challenging. In addition, the generation of NKG2DL-deficient mice is complicated by the existence of multiple ligands organized as gene families. However, recent success in creating Rae-1-deficient mice (51) opens the possibility to generate bone marrow chimeras and to address the contribution of Rae-1 expression, at least in the hematopoietic versus non-hematopoietic compartment, in disease settings in vivo.
In the case of myeloid cells, while their role in activating NKG2D-expressing cells in vivo via NKG2DLs remains largely unknown, in vitro data provide convincing evidence of an NK cell crosstalk with DCs and macrophages (Figure 1). For example, in the MCMV model, virus-infected mouse DCs play a crucial role in NK cell activation by both inducing IFN-γ release (through IL-12/18 production) and NK cell cytotoxic responses via IFNα and NKG2D engagement (81). In human in vitro system, it was shown that DCs infected with influenza virus supported CD69 upregulation and IFN-γ production by NK cells via NKG2D and NKp46 (74). In addition, it was shown that human DC-derived exosomes displayed ULBP1, together with IL-15Rα, on their surface and were able to promote NKG2D-dependent activation of NK cells (82). Also, MICA and MICB, induced on DCs upon IFN-α or IL-15 treatment, contributed to NK cell activation (75, 76). In these studies, the authors showed that type I IFNs and IL-15 induced MIC molecules on DCs, which was impaired in patients with chronic hepatitis C infection. Similarly, coculture of NK cells and DCs pulsed with T. gondii lysate increased DC IL-12 production and their ability to prime Ag-specific CD8+ T cell responses, which was impaired by NKG2D blockade (73). Besides mutual activation, the NK/DC crosstalk can also result in an NK cell cytolytic response, leading to DC elimination, which is considered essential in the regulation of the numbers and quality of the activated DCs and, consequently, the extent of the overall immune response. The outcome of the NK/DC crosstalk depends on the activation/maturation status of both interacting cell types and their relative abundance. Accordingly, human IL-2-activated NK cells increased mature DC responses, measured by the level of IL-12 release and ability to induce CD4+ T cell activation (83). In coculture with immature DCs (iDCs), activated NK cells were shown to support autologous DC maturation and activation at low NK/DC ratios, while increased numbers of NK cells resulted in iDC lysis (84). In these settings, cytotoxic NK cell responses were mainly mediated via the activating receptors NKp30 and DNAM-1 (85), although a partial contribution of NKG2D has also been observed (86). However, treatment of mature DCs with IL-10 induced their elimination by NK cells, which was mediated via NKG2D, while IL-10-treated iDCs resisted to NK-mediated cytotoxicity (87). As the authors of this study suggested, aberrant accumulation of iDCs in patients with chronic infections that are frequently associated with increased levels of IL-10 production, such as HIV, thus might be the consequence of IL-10-induced DC resistance to NK cell elimination (87). Remarkably, Schulz and colleagues showed that IL-10 also rendered autologous human macrophages susceptible to NK cell lysis, which involved NKG2D. In the presence of IL-10, NKG2D receptor expression on NK cells increased, while macrophages induced NKG2DL expression (88). Thus, IL-10 might exert its immunomodulatory pleotropic effect, not only via suppressing T cell responses, but also via inducing NK cell-mediated killing of activated myeloid cells, including antigen-presenting cells (APCs).
Similar to DCs, monocyte-derived macrophages were reported to express MICA and ULBP1-3 upon stimulation with LPS, rendering them susceptible to NK cell-mediated lysis (89). In alveolar macrophages, M. tuberculosis infection led to ULBP1 induction and their NKG2D-dependent lysis by NK cells (60). However, it was also reported that TLR-stimulated monocytes, which upregulate MICA, promoted IFN-γ release via interaction with NKG2D-expressing NK cells (59). In the presence of IL-12, NKG2DL-expressing monocytes were shown to not only stimulate IFN-γ release, but also to enhance antibody-dependent cellular cytotoxicity toward Ab-coated target cells (90). Results from the Davis lab indicated that NK cells and autologous human macrophages can engage two distinct types of interactions. On the one hand, low-dose LPS-stimulated macrophages can trigger NK cell proliferation, secretion of cytokines and increased killing of tumor targets via the 2B4/CD48 axis, while, on the other hand, macrophages activated with high doses of LPS expressed MICA and ULPBs, formed the so-called lytic synapse, and were lysed by NK cells via NKG2D (89). These data indicate that the activation status of macrophages can determine the outcome of their crosstalk with NK cells and that NKG2DL expression might be a signal for removal of activated macrophages to prevent exaggerated inflammation and tissue damage. In mice, activation of peritoneal macrophages with poly(I:C) induced Rae-1, H60, and MULT1 expression, along with IL-15, IL-18, and type I IFN production. These soluble factors could increase NKG2D expression on NK cells, leading to increased cytotoxicity in response to tumor cells expressing NKG2DLs. However, macrophages remained resistant to NK cell lysis due to the expression of Qa-1, a surface molecule engaging the inhibitory NK cell receptor NKG2A (91).
Besides a direct interaction that triggers immediate effector responses, such as cytokine production and cytotoxicity, myeloid cells expressing NKG2DLs can indirectly control the function of NKG2D expressed on effector lymphoid cells. It was shown that NKG2DL engagement can lead to NKG2D downregulation and that the constitutive presence of NKG2DLs can cause long-term desensitization of the NKG2D pathway (53, 92, 93). This phenomenon can be mediated by both membrane bound ligands and by ligands released in soluble form upon proteolytic shedding or via exosomes. While the effect of tumor-released soluble ligands/exosomes has been extensively studied, mainly as a mechanism of immune evasion, the contribution of myeloid cells to this phenomenon remains largely unrevealed. Only recently, a study showed that in vivo overexpression of Rae-1 on CD11chigh cells, comprising mainly DCs in mice, led to reduced NK cell-mediated cytotoxicity toward NKG2DL+ or MHC class I-deficient targets, compromising the ability of these animals to reject NKG2DL-expressing tumor cells, while the control of viral infection (MCMV) remained unaffected (94). Of note, continuous engagement of NKG2D was shown not only to affect NKG2D-dependent responses, but also to desensitize the signaling downstream of other activating receptors, such as NK1.1 and NKp46 (92, 95). Accordingly, Rae-1 expression on lymph node endothelial cells in steady state (53) or by myeloid cells in tumor-bearing animals (51) is responsible for NKG2D downregulaton and global desensitization of NK cells. Similarly, in cancer patients, the presence of MICA- and ULBP1-expressing myeloid cells in blood and tumor correlated with reduced NKG2D expression on NK cells (96). However, it will be important to further improve the mechanistic understanding of NKG2D downmodulation in disease conditions and to dissect whether NKG2D downregulation in patients truly results from the engagement with soluble and cell-expressing ligands or if other factors, such as TGF-β, might contribute in these settings.
In the past few years, ILCs, which mainly reside in tissues, have been identified as novel players in the regulation of tissue homeostasis, regeneration, and response to infection (80). Since helper ILC1 and ILC3 populations express NKG2D (6–8), investigating the importance of the NKG2D/NKG2DL axis in the putative crosstalk between ILCs and myeloid cells, particularly in tissues and/or pathologies where ILC populations play an important role, would be of high relevance.
NKG2D Expression on T Cell Subsets: When and Where?
As referred above, NKG2D is a widely expressed receptor detectable on NK cells and several subsets of T cells, including CD8+ T cells, subsets of γδ T cells and NKT cells in steady state, and CD4+ T cells under certain pathological conditions. This transversal expression from innate to adaptive immune lymphocytes makes NKG2D a remarkable NK receptor. Thus, the focus on the dynamics of a NKG2D/NKG2DL axis should go beyond the NK cell-myeloid cell crosstalk. Below, we discuss the so far described NKG2D-expressing T cell subsets (see Figure 2).
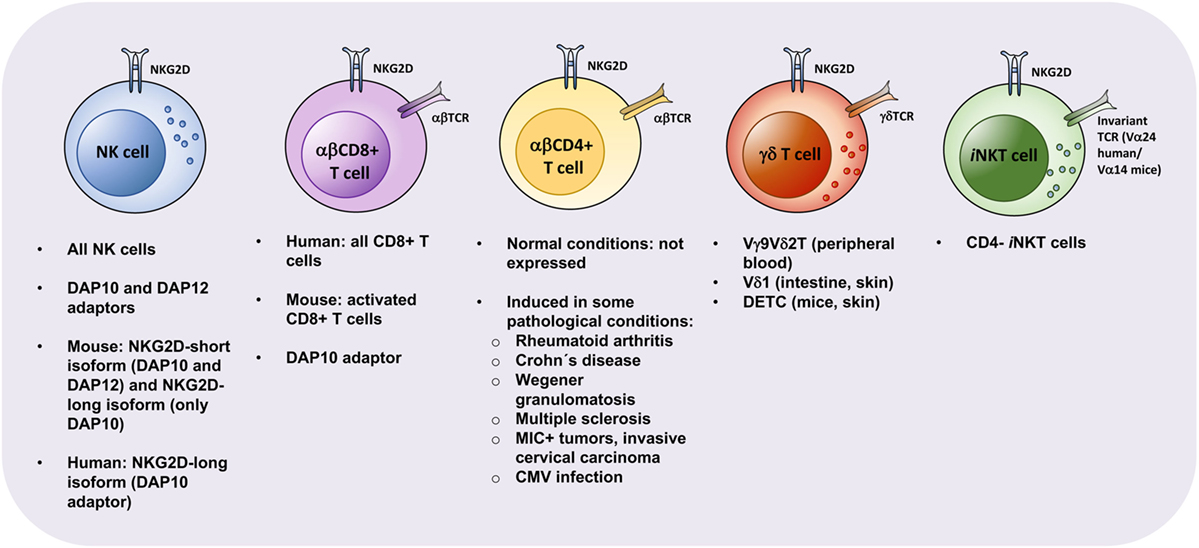
Figure 2. Properties of natural killer group 2, member D (NKG2D)-expressing lymphocytes. NKG2D can be expressed by natural killer (NK) cells, αβ CD8+ T cells, γδ T cells, invariant NKT (iNKT) cells, and αβ CD4+ T cells. However, its expression, alternative splicing, and coupling to adapter molecules vary between the indicated lymphoid subsets. Expression of NKG2D on certain lymphocytes correlates with activation (mouse αβ CD8+ T cells) or can be detected only in disease settings (αβ CD4+ T cells). While NK cells, iNKT cells and CD8+ T cells are circulatory and can be detected in peripheral blood, other lymphoid cells, such as helper innate lymphoid cells (data not shown) and certain γδ T cells, are tissue-resident and are involved in local tissue immune responses. Similarly, pathologic αβ CD4+ T cells are enriched in affected tissues in depicted autoimmune conditions. Abbreviations: DETCs, dendritic epidermal T cells; CMV, cytomegalovirus.
αβ CD8+ T Cells. CD8+ T cells are the most representative T cell subset expressing NKG2D. While in mice expression of NKG2D is restricted to activated CD8+ T cells, in humans, all CD8+ T cells express NKG2D constitutively (97). In human, as with NK cells, CD8+ T cells do not express the short NKG2D isoform. In mice, although activated CD8+ T cells express both NKG2D isoforms, they usually lack expression of DAP12, contrarily to NK cells. Thus, for both species, NKG2D in CD8+ T cells seems to primarily signal via DAP10 (2, 3). It was reported that DAP12 could also be expressed by T cells and that, unlike NK cells, human activated CD8+ T cells required simultaneous signaling mediated by both DAP10 and DAP12 pathways (98). However, several studies support that DAP10 appears to be the most important adaptor for NKG2D signaling in CD8+ T cells (3, 22, 99).
In conventional CD8+ T cells, NKG2D has been shown to mainly serve as a costimulatory receptor for TCR-induced signaling (22, 69, 99–101). Although the costimulatory function of NKG2D was more evident for activated CD8+ T cells that lack CD28 (99, 100), it was also observed in naïve CD8+ T cells (101). In fact, the cytoplasmic domain of DAP10 comprises a signaling motif similar to CD28, which activates PI3K and leads to similar, though not identical effects on T cell costimulation (102, 103). Besides its role in decreasing the TCR threshold in CD8+ T cells, as a costimulatory receptor, several studies have shown that NKG2D can also work as an activating receptor per se on CD8+ T cells under certain conditions, namely upon prolonged exposure to IL-15 (104–106). In fact, IL-15 appears to be a key factor in arming the NKG2D-mediated cytolysis of effector CD8+ T cells. Meresse et al. showed that, in celiac disease, NKG2D expressed on intraepithelial intestinal lymphocytes could mediate direct cytolysis, putatively due to overexpression of IL-15 in this disease (104). Other studies demonstrated that prolonged exposure of human peripheral blood CD8+ T cells to IL-15 in vitro led to a functional NKG2D receptor per se, without the need of TCR coengagement for activation. IL-15, besides being involved in NKG2D and DAP10 induction and upregulation (104–108), was demonstrated to synergize with the NKG2D downstream signaling pathway through activation of PI3K, JNK, ERK, and cPLA2 (21, 104, 109, 110), thereby enabling NKG2D to mediate direct activation and cytolysis in a TCR-independent manner. Also, it was shown that NKG2D could enhance IL-15-mediated PI3K signaling in activated CD8+ T cells, promoting CD8+ T cell survival and memory formation (111), showing a bi-directional importance of the IL-15–NKG2D downstream signaling interaction. Moreover, NKG2D on CD8+ T cells was shown to rescue CD4-unhelped CD8+ T cell memory recall responses, but not effector responses, by repressing the transcription factor T-bet (112). Accordingly, besides activating CD8+ T cells and driving cytotoxicity, NKG2D has also been implied in the survival and memory formation of CD8+ T cells.
αβ CD4+ T Cells. Under physiological conditions, expression of NKG2D is not detectable on conventional αβCD4+ T cells. However, NKG2D-expressing CD4+ T cells have been described in human under certain pathological conditions. NKG2D+ CD4+ T cells were initially found on peripheral blood and synovial fluid from rheumatoid arthritis (RA) patients, putatively as result of increased TNF-α and IL-15 levels in this disease. Groh et al. showed that those cells promoted the cytotoxic damage against synoviocytes with anomalous expression of NKG2DLs (14). Later on, NKG2D+ CD4+ T cells have been associated with several other autoimmune diseases, such as Crohn’s disease (108, 113, 114), Wegener granulomatosis (115, 116), type 2 diabetes (117), multiple sclerosis (118), and systemic lupus erythematosus (SLE) (119, 120). Moreover, NKG2D+ CD4+ T cells accumulated in patients with MIC+ tumors (121) and cervical carcinoma (122–124). Also, NKG2D+ CD4+ T cells were identified in patients suffering from a human T cell lymphotropic virus type I-associated neurological disease (125) and linked with human cytomegalovirus infection (126).
Of note, NKG2D+ CD4+ T cells appear to have similar features to a previously described CD4+ CD28− T cell phenotype, which prevails in several pathologies, namely autoimmune disorders (127–132). In fact, NKG2D+ CD4+ T cells have been detected mainly within the CD4+ CD28− T cell population (14, 126, 133). Thus, previous studies describing the CD4+ CD28− T cell population should be revisited by addressing a putative role of NKG2D in disease progression and severity.
Similar to CD8+ T cells, NKG2D plays a major role as a costimulatory receptor, enhancing TCR-mediated responses in NKG2D+ CD4+ T cells. Several studies showed the involvement of this population in the pathology of autoimmune diseases contributing to disease progression or severity in an NKG2D-dependent manner. However, increased frequencies of NKG2D+ CD4+ T cells inversely correlated with disease activity in juvenile-onset SLE, suggesting that these T cells may also have regulatory effects (134). Moreover, a recent study showed that NKG2D+ CD4+ T cells were involved in Treg killing in an NKG2D–NKG2DL-dependent manner in SLE (120).
Besides the existence of mounting evidence about the association of NKG2D+ CD4+ T cells and pathological conditions, shown to correlate with an increase in NKG2DLs, the functional crosstalk between myeloid cells expressing NKG2DL and NKG2D+ CD4+ T cells remains mainly unaddressed.
γδ T Cells. In both humans and mice, γδ T cells comprise a small population of peripheral blood cells (around 2–5%), but are abundant in tissues, particularly in the intestine, reproductive tract, and skin (135–137). Vγ9Vδ2 T cells (also known as Vγ2Vδ2), are the most abundant population in human peripheral blood (50–95%) (138), while Vδ1 γδ T cells are mainly enriched in the intestine (together with Vδ3 γδ T cells) and in the skin. Both Vγ9Vδ2 and Vδ1 γδ T cell subsets are described to express NKG2D on their cell surface, associating with DAP10 for signal triggering. In human, the IL-17+ Vγ9Vδ2 T population was reported to lack the expression of NKG2D, although the majority of circulating Vγ9Vδ2 T cells expressed NKG2D receptor (139).
In 1998, Groh and colleagues described that MICA and MICB could be recognized by intestinal Vδ1 γδ T cells through their γδ TCR (140). Shortly after, the same group showed that the cell surface NKG2D expressed on intestinal Vδ1 γδ T cells recognized MICA, and that NKG2D–MICA engagement resulted in target cell recognition and killing, suggesting that NKG2D might function as a costimulatory receptor on γδ T cells (141). MICA was later confirmed to be directly recognized by Vδ1 T cells through their γδ TCR, although weakly compared to recognition by NKG2D (142). Several studies showed that Vδ1 γδ T cells can recognize NKG2DLs expressed on cancer cells, triggering their cytotoxicity against targets (143, 144).
Das and colleagues showed that infection with M. tuberculosis induced MICA on the surface of dendritic and epithelial cells, both in vitro and in vivo (145). Moreover, MICA engagement by NKG2D expressed on Vγ9Vδ2 T cells resulted in a considerable increase of the TCR-dependent Vγ9Vδ2 T cells response (145). In the absence of antigen, NKG2D+ Vγ9Vδ2 T cells did not lyse MICA+ targets, indicating that the NKG2D–MICA interaction is not sufficient to trigger Vγ9Vδ2 T cell-mediated lysis (145). By contrast, a subsequent study showed that NKG2D expressed on Vγ9Vδ2 T cell could induce by itself Vγ9Vδ2 T cell activation and NKG2D-dependent cytolysis of target cells (146). NKG2D has been shown to be involved in Vγ9Vδ2 T cell recognition of leukemia and lymphoma (147), as well as of solid tumors (148, 149). Similar to Vδ1 γδ T cell recognition of MICA, ULBP4 was described to bind to both NKG2D and γδ TCR of Vγ9Vδ2 T cells, mediating their activation and cytotoxicity (150). Thus, altogether those studies indicate that tumor-expressed NKG2DLs can be specifically recognized by both TCR and NKG2D expressed on human γδ T cells.
Dendritic epidermal T cells (DETCs) are epithelial γδ T cells that reside in murine skin. While it is described that only 25% of splenic γδ T cells and 5% of thymic γδ T cells express NKG2D, basically all DETCs are NKG2D+ in mice (69). So far, human counterparts of mouse skin γδ T cells with the same dendritic-like characteristics have not been identified. Girardi and colleagues have shown that DETCs can kill carcinoma cells in an NKG2D-dependent manner (151), providing the first evidence of NKG2D-mediated DETC activation. Afterward, it was found that DETCs displayed impaired wound healing properties upon NKG2DL blocking (65, 152) and, as well, that the NKG2D/NKG2DL interaction was involved in allergen-induced activation of DETCs in contact hypersensitivity (153).
As with the other γδ T cell subsets, whether NKG2D expressed on DETCs works as a costimulatory receptor or as an activation receptor by itself is still controversial (153–156). Ibusuki et al. showed that NKG2D engagement alone was sufficient to trigger degranulation, but not cytokine production, in DETCs, which was mainly mediated via the DAP10–PI3K-dependent signaling pathway (157). However, in this study, DETCs were expanded in culture with IL-2, which may account for the observed surpass of the need for TCR cotriggering. In fact, NKG2D could not trigger cytotoxicity in freshly ex vivo isolated DETCs (157). Of note, since it is known that cytokines, such as IL-15, can synergize with the NKG2D downstream signaling pathway, it is important to consider that the majority of protocols using γδ T cells are preceded by an expansion phase including IL-2 or IL-15 cytokines. This fact might account for controversial views of NKG2D as costimulatory or stimulatory receptor by itself in NKG2D-expressing populations.
iNKT Cells. Invariant NKT cells represent a small population of blood cells (0.01–1% among peripheral blood T cells) in human that, however, can be found highly enriched in the liver, particularly in mice. Those cells express a semi-invariant TCR, characterized by Vα14-Jα18 in mice and Vα24-Jα18 in humans, which recognizes lipid-based antigens in the context of CD1d molecules. iNKT cells can be functionally distinguished by the expression of CD4 on the cell surface, both in human and mice. In general, CD4+ iNKT cells display a Th2-like profile, whereas CD4− iNKT are rather skewed toward Th1-like responses (158, 159). NKG2D surface expression is restricted mainly to the CD4 negative subset of iNKT cells (158, 160). Kuylenstierna and colleagues found that NKG2D stimulation in CD4− NKT cells could act as a costimulatory signal in response to suboptimal anti-CD3 triggering or CD1d-presented ligands. In the same study, NKG2D stimulation in CD4− NKT cells also mediated a direct NKG2D-dependent lysis of target cells, independent of invariant TCR engagement, thus demonstrating both a stimulatory and costimulatory role of NKG2D in those cells (160). Wang et al. showed that tumor-derived soluble MICs downregulated NKT cell NKG2D expression and consequently tumor cell killing in vitro (161), supporting an anti-tumor function of NKG2D+ NKT cells.
Besides playing a role in cancer, NKG2D+ NKT cells were shown to be enriched in certain pathologies, namely autoimmune diseases. Patients with type 2 diabetes showed increased NKG2D+ NKT cells in peripheral blood, when compared with healthy controls (162). Early onset SLE was associated with changes in the ratio of NKG2D/NKG2A expression in multiple cell types, including NKT cells (163). NKG2D+ NKT cells with a Th1-like profile were also increased in pre-eclampsia (164). Further studies focusing on the specific role of NKG2D expressed by iNKT cells, in both cancer and other pathologies, and on the potential crosstalk with myeloid cells via interaction with NKG2DLs, would be important.
Noteworthy, besides the existence of mounting evidence associating several subsets of NKG2D+ T cells and pathological conditions characterized by an increase in NKG2DLs, the functional crosstalk between myeloid cells expressing NKG2DL and NKG2D+ T cell subsets remains mainly unaddressed. In this regard, it would be important on the one hand, to determine the specific role of NKG2D in conditional knockout mice, where NKG2D could be selectively deleted on different cell populations; and on the other hand, to dissect in parallel the importance of the NKG2DL-expressing myeloid cells. Since myeloid cells are important APCs, the putative crosstalk between myeloid cells and T cells expressing NKG2D might be highly relevant. Particularly in pathological conditions, myeloid cells could not only trigger NKG2D expressed by T cells via NKG2DL but could also in parallel activate T cells via antigen presentation by MHC molecules, further enhancing or controlling specific T cell-mediated responses. As such, further studies focusing on the impact of NKG2DL expressed on myeloid cells in the crosstalk with T cells would be of major relevance.
Importantly, a connection between NKG2D, autoimmunity, and IL-15 is getting increasingly evident. First, several autoimmune diseases have been correlated with increased IL-15 cytokine levels (165, 166). Second, NKG2D induction, upregulation and its role in autoimmunity has been extensively demonstrated (167). Finally, a role of IL-15 in NKG2D upregulation and enhancement of downstream signaling has been shown. Interestingly, myeloid cells are widely regarded as main producers of IL-15, particularly under pathological conditions, trans-presenting IL-15 to responding cells. On the one hand, it is known that IL-15 is involved in NKG2DL upregulation (104, 110) and, on the other hand, it has been shown that this cytokine can upregulate or induce NKG2D expression (104–106, 110). In this regard, it is tempting to imagine a scenario where myeloid cells trans-present IL-15 to effector cells, leading to NKG2D induction and/or support of its signaling, while these cells at the same time trigger NKG2D via NKG2DL interaction.
NKG2DLs and Myeloid Cells in Disease Settings
Multiple diseases are associated with NKG2DL upregulation, where its expression might be either protective or detrimental. Dysregulated receptor/ligand expression was reported in various autoimmune diseases, such as RA, colitis, celiac disease, multiple sclerosis, type 1 diabetes, or atherosclerosis, where their involvement was postulated to mainly promote inflammation (11–17, 104). By contrast, in conditions such as wound healing, although NKG2DLs were induced by initial tissue damage and cellular stress, NKG2D was shown to support tissue remodeling and regeneration (65). However, in many of the studied diseases and respective animal models, it remains unclear which cells express NKG2DLs in the affected tissues and what is the myeloid cell contribution to the overall ligand expression and NKG2D-driven responses.
In tumor-bearing mice, Rae-1 was detected on a subset of MDSCs and contributed to NK cell activation (67). In glioblastoma multiforme (GBM) patients, MICA and ULBP1 were detected on microglia, tumor-infiltrating myeloid cells, and circulating monocytes (96). In this study, the authors showed that lactate dehydrogenase isoform 5 (LDH5), secreted by tumor cells, was elevated in GBM patients’ sera and responsible for NKG2DL upregulation on healthy monocytes. In vitro, IL-2-activated NK cells degranulated, produced IFN-γ, and induced apoptosis of autologous NKG2DL-bearing monocytes. However, in vivo, LDH5-mediated induction of NKG2DLs might serve as a cancer immune evasion mechanism, as NK cells from GBM patients displayed reduced surface NKG2D expression and impaired function (96). Similarly, NKG2DL-expressing monocytes were detected in the blood of breast, prostate, and virus-induced liver cancer patients (96).
In atherosclerotic plaques, both endothelial cells and macrophages have been reported to express MICA/B (15). In line with that, exposure of monocyte-derived macrophages to acetylated low-density lipoproteins in vitro led to MICA/B induction (168). In a mouse model of atherosclerosis, Rae-1 expression was detected on macrophages, not only in plaques but also in the liver, which is affected by metabolic changes associated with disease (15). The liver is enriched in NKG2D-expressing ILCs and NKT cells, whose crosstalk with the myeloid compartment might play a significant role in atherosclerosis. Consistent with that hypothesis, it was shown that NKG2D-deficient animals displayed smaller plaques in aortas, reduced liver damage, and reduced levels of proinflammatory cytokines, cholesterol, and triglycerides in serum (15).
La Scaleia et al. reported increased NKG2DL expression in the colon mucosa of pediatric patients with active inflammatory bowel disease (IBD). Their results indicated that the NKG2DL+ cells displayed a macrophage-like morphological phenotype (17). In addition, in active ulcerative colitis, MICA and MICB expression was significantly upregulated in peripheral blood monocytes (17). MICA/B+ macrophages were also detected in the duodenal tissue of patients with celiac disease, where those ligands were distributed intracellularly in the form of cytoplasmic aggregates (169). These data suggest that the crosstalk of myeloid cells with NKG2D+ innate and adaptive lymphocytes might play a significant role in IBD-associated inflammation. Moreover, intracellular NKG2DLs might have a specific, so far unappreciated function, not only in myeloid cells, but also in enterocytes, where peri- and supra-nuclear NKG2DL aggregates were also detected (169).
In experimental autoimmune encephalomyelitis, a mouse model of multiple sclerosis, Rae-1δ and Rae-1γ were induced at mRNA and protein level in spinal cord early upon disease onset. Djelloul et al. demonstrated that myeloid cells, including macrophages and microglia, expressed both Rae-1 and MULT1 (170). Expression of these ligands correlated with myeloid cell recruitment to affected tissue and their proliferation. Furthermore, this study identified M-CSF as factor driving NKG2DL expression on microglia.
As in the diseases discussed above NKG2D-expressing lymphocytes play significant roles, displaying either protective or detrimental properties, and myeloid cells exert potent regulatory roles in their activation, it would be of great importance to determine the contribution of NKG2D to their interaction. Moreover, NKG2D seems to play a costimulatory role in lymphocyte activation, acting often in the context of the proinflammatory environment. As myeloid cells contribute to an inflammatory environment via both soluble factors and NKG2DLs, it is tempting to speculate that their therapeutic targeting in combination with existing treatments might help to reduce tissue damage, especially in context of autoimmune diseases.
Conclusion and Future Perspectives
NKG2D is considered a major lymphocyte receptor detecting dysregulated cell homeostasis induced by infection or transformation. Distinct pathways activated by cellular stress can upregulate various NKG2DLs, thus conveying an alert-signal of potential cell dysfunction to the immune system. Importantly, NKG2DL upregulation is frequently accompanied by specific microenvironmental milieus that support NKG2D upregulation, NKG2D function, and NKG2DL induction. These milieus are characterized by the presence of proinflammatory cytokines, among which, IL-15 plays a central role. In tissues, tissue-resident and recruited NKG2D+ lymphocytes are crucial for detecting and eliminating infected and transformed cells. However, this function is greatly supported by the myeloid immune compartment. Similar mechanisms, leading to NKG2DL upregulation in infected and transformed cells, operate to induce their expression in myeloid cells as well. Thus, besides their classical functions, that include Ag presentation to T cells or cytokine-mediated activation of innate lymphocytes, myeloid cells use the NKG2D/NKG2DL axis to support a regulatory loop, leading to lymphocyte activation via NKG2D, which in turn can activate or eliminate myeloid cells. The NKG2D-mediated myeloid–lymphocyte interaction can have a dual effect on the lymphoid effector cell. On the one hand, it can lead to cell activation, which promotes cytotoxicity, survival, and/or cytokine production. On the other hand, it causes cell inactivation, as a consequence of ligand-induced receptor internalization and desensitization of activating pathways beyond NKG2D. Although some of the factors that regulate these processes are known, such as soluble ligands and chronic stimulation, it would be highly relevant to define molecular events that can shift NKG2D engagement toward activation, e.g., during anti-tumor responses, or inactivation in the case of autoimmunity.
In many experimental settings, especially in disease, the importance of the myeloid–lymphocyte activation/inactivation loop has not been fully addressed, namely due to the existence of a broad variety of NKG2DLs and due to their expression beyond the myeloid compartment, including stromal and parenchymal cells. Accordingly, it remains a major challenge to address the contribution of the myeloid compartment to NKG2D-dependent lymphocyte activation, especially in pathological settings. So far, conditional knockout animals lacking all NKG2DLs were not generated, but mice overexpressing Rae-1 ligands exist and their overexpression in specific cell types was obtained by the use of Cre-lox technology (94). However, the complexity of the NKG2DL system, concerning their differential expression, differential shedding susceptibility and the ability to induce NKG2D downregulation, might compromise the full understanding of complex pathologies. In addition, recent data indicating that the continuous engagement of the NKG2D receptor in steady state regulates its activity (53), suggest that the use of inducible systems of deletion or overexpression would be a better experimental choice.
Although the NKG2D-mediated crosstalk of myeloid cells and NK cells is relatively well understood, the outcome of the interaction of the different NKG2D-expressing T cell subsets with NKG2DL+ myeloid cells remain largely unknown. In many diseases, especially in autoimmune disorders, that are often associated with aberrant activation of myeloid cells, the presence of NKG2D-expressing T cells is well documented. It would be of great value to gain understanding of the relative contribution of NKG2D-expressing T cells in these settings and the involvement of NKG2D, especially for therapeutic targeting. For this purpose, mice carrying a conditional NKG2D deletion specifically in T cells would be valuable tools. In addition, novel populations of tissue-resident lymphocytes are emerging and their importance in regulating inflammation, tissue homeostasis, and regeneration is now eminent. Their topological position and fast responses to tissue damage, in cooperation with the myeloid compartment, along with the fact that NKG2DLs are the messengers of damage, impose them as attractive candidates that might utilize the NKG2D system to perform their functions. It is known that such scenarios operate in skin with tissue-resident γδ T cells being major players. Other barrier sites are awaiting further evaluation in the context of the NKG2DL/NKG2D crosstalk.
Author Contributions
AS and MPC wrote the manuscript; MPC generated the figures; AC read the manuscript and provided critical input.
Conflict of Interest Statement
AC is a member of the Scientific Advisory Board of Dragonfly Therapeutics.
Acknowledgments
We thank all the scientist who performed important work in the NKG2D field and inspired the writing of this review article, and we apologize to those ones we could not cite.
Funding
The authors of this review are supported by Deutsche Krebshilfe (German Cancer Aid) grants no. 70112233 and 110442 and DFG Priority Program 1937.
References
1. Wu J, Song Y, Bakker AB, Bauer S, Spies T, Lanier LL, et al. An activating immunoreceptor complex formed by NKG2D and DAP10. Science (1999) 285(5428):730–2. doi:10.1126/science.285.5428.730
2. Diefenbach A, Jamieson AM, Liu SD, Shastri N, Raulet DH. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat Immunol (2000) 1(2):119–26. doi:10.1038/77793
3. Gilfillan S, Ho EL, Cella M, Yokoyama WM, Colonna M. NKG2D recruits two distinct adapters to trigger NK cell activation and costimulation. Nat Immunol (2002) 3(12):1150–5. doi:10.1038/ni857
4. Jelencic V, Lenartic M, Wensveen FM, Polic B. NKG2D: a versatile player in the immune system. Immunol Lett (2017) 189:48–53. doi:10.1016/j.imlet.2017.04.006
5. Houchins JP, Yabe T, McSherry C, Bach FH. DNA sequence analysis of NKG2, a family of related cDNA clones encoding type II integral membrane proteins on human natural killer cells. J Exp Med (1991) 173(4):1017–20. doi:10.1084/jem.173.4.1017
6. Dyring-Andersen B, Geisler C, Agerbeck C, Lauritsen JP, Gudjonsdottir SD, Skov L, et al. Increased number and frequency of group 3 innate lymphoid cells in nonlesional psoriatic skin. Br J Dermatol (2014) 170(3):609–16. doi:10.1111/bjd.12658
7. Killig M, Glatzer T, Romagnani C. Recognition strategies of group 3 innate lymphoid cells. Front Immunol (2014) 5:142. doi:10.3389/fimmu.2014.00142
8. Klose CSN, Flach M, Mohle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell (2014) 157(2):340–56. doi:10.1016/j.cell.2014.03.030
9. Lanier LL. NKG2D receptor and its ligands in host defense. Cancer Immunol Res (2015) 3(6):575–82. doi:10.1158/2326-6066.CIR-15-0098
10. Nausch N, Cerwenka A. NKG2D ligands in tumor immunity. Oncogene (2008) 27(45):5944–58. doi:10.1038/onc.2008.272
11. Andersson AK, Sumariwalla PF, McCann FE, Amjadi P, Chang C, McNamee K, et al. Blockade of NKG2D ameliorates disease in mice with collagen-induced arthritis: a potential pathogenic role in chronic inflammatory arthritis. Arthritis Rheum (2011) 63(9):2617–29. doi:10.1002/art.30460
12. Ito Y, Kanai T, Totsuka T, Okamoto R, Tsuchiya K, Nemoto Y, et al. Blockade of NKG2D signaling prevents the development of murine CD4+ T cell-mediated colitis. Am J Physiol Gastrointest Liver Physiol (2008) 294(1):G199–207. doi:10.1152/ajpgi.00286.2007
13. Ogasawara K, Hamerman JA, Ehrlich LR, Bour-Jordan H, Santamaria P, Bluestone JA, et al. NKG2D blockade prevents autoimmune diabetes in NOD mice. Immunity (2004) 20(6):757–67. doi:10.1016/j.immuni.2004.05.008
14. Groh V, Bruhl A, El-Gabalawy H, Nelson JL, Spies T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc Natl Acad Sci U S A (2003) 100(16):9452–7. doi:10.1073/pnas.1632807100
15. Xia M, Guerra N, Sukhova GK, Yang K, Miller CK, Shi GP, et al. Immune activation resulting from NKG2D/ligand interaction promotes atherosclerosis. Circulation (2011) 124(25):2933–43. doi:10.1161/CIRCULATIONAHA.111.034850
16. Van Belle TL, Ling E, Haase C, Bresson D, Urso B, von Herrath MG. NKG2D blockade facilitates diabetes prevention by antigen-specific Tregs in a virus-induced model of diabetes. J Autoimmun (2013) 40:66–73. doi:10.1016/j.jaut.2012.08.001
17. La Scaleia R, Stoppacciaro A, Oliva S, Morrone S, Di Nardo G, Santoni A, et al. NKG2D/ligand dysregulation and functional alteration of innate immunity cell populations in pediatric IBD. Inflamm Bowel Dis (2012) 18(10):1910–22. doi:10.1002/ibd.22899
18. Zhang J, Basher F, Wu JD. NKG2D ligands in tumor immunity: two sides of a coin. Front Immunol (2015) 6:97. doi:10.3389/fimmu.2015.00097
19. Horng T, Bezbradica JS, Medzhitov R. NKG2D signaling is coupled to the interleukin 15 receptor signaling pathway. Nat Immunol (2007) 8(12):1345–52. doi:10.1038/ni1524
20. Jensen H, Potempa M, Gotthardt D, Lanier LL. Cutting edge: IL-2-induced expression of the amino acid transporters SLC1A5 and CD98 is a prerequisite for NKG2D-mediated activation of human NK cells. J Immunol (2017) 199(6):1967–72. doi:10.4049/jimmunol.1700497
21. Tang F, Chen Z, Ciszewski C, Setty M, Solus J, Tretiakova M, et al. Cytosolic PLA2 is required for CTL-mediated immunopathology of celiac disease via NKG2D and IL-15. J Exp Med (2009) 206(3):707–19. doi:10.1084/jem.20071887
22. Diefenbach A, Tomasello E, Lucas M, Jamieson AM, Hsia JK, Vivier E, et al. Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat Immunol (2002) 3(12):1142–9. doi:10.1038/ni858
23. Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity (2008) 28(4):571–80. doi:10.1016/j.immuni.2008.02.016
24. Carapito R, Bahram S. Genetics, genomics, and evolutionary biology of NKG2D ligands. Immunol Rev (2015) 267(1):88–116. doi:10.1111/imr.12328
25. Eagle RA, Trowsdale J. Promiscuity and the single receptor: NKG2D. Nat Rev Immunol (2007) 7(9):737–44. doi:10.1038/nri2144
26. Baranwal AK, Mehra NK. Major histocompatibility complex class I chain-related A (MICA) molecules: relevance in solid organ transplantation. Front Immunol (2017) 8:182. doi:10.3389/fimmu.2017.00182
27. Carapito R, Aouadi I, Ilias W, Bahram S. Natural killer group 2, member D/NKG2D ligands in hematopoietic cell transplantation. Front Immunol (2017) 8:368. doi:10.3389/fimmu.2017.00368
28. Kim J, Chang CK, Hayden T, Liu FC, Benjamin J, Hamerman JA, et al. The activating immunoreceptor NKG2D and its ligands are involved in allograft transplant rejection. J Immunol (2007) 179(10):6416–20. doi:10.4049/jimmunol.179.10.6416
29. Ogasawara K, Benjamin J, Takaki R, Phillips JH, Lanier LL. Function of NKG2D in natural killer cell-mediated rejection of mouse bone marrow grafts. Nat Immunol (2005) 6(9):938–45. doi:10.1038/ni1236
30. Cerboni C, Zingoni A, Cippitelli M, Piccoli M, Frati L, Santoni A. Antigen-activated human T lymphocytes express cell-surface NKG2D ligands via an ATM/ATR-dependent mechanism and become susceptible to autologous NK-cell lysis. Blood (2007) 110(2):606–15. doi:10.1182/blood-2006-10-052720
31. Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature (2005) 436(7054):1186–90. doi:10.1038/nature03884
32. Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc Natl Acad Sci U S A (1996) 93(22):12445–50. doi:10.1073/pnas.93.22.12445
33. Venkataraman GM, Suciu D, Groh V, Boss JM, Spies T. Promoter region architecture and transcriptional regulation of the genes for the MHC class I-related chain A and B ligands of NKG2D. J Immunol (2007) 178(2):961–9. doi:10.4049/jimmunol.178.2.961
34. Hosomi S, Grootjans J, Tschurtschenthaler M, Krupka N, Matute JD, Flak MB, et al. Intestinal epithelial cell endoplasmic reticulum stress promotes 1 up-regulation and NKG2D-mediated inflammation. J Exp Med (2017) 214(10):2985–97. doi:10.1084/jem.20162041
35. Raulet DH, Marcus A, Coscoy L. Dysregulated cellular functions and cell stress pathways provide critical cues for activating and targeting natural killer cells to transformed and infected cells. Immunol Rev (2017) 280(1):93–101. doi:10.1111/imr.12600
36. Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol (2013) 31:413–41. doi:10.1146/annurev-immunol-032712-095951
37. Gowen BG, Chim B, Marceau CD, Greene TT, Burr P, Gonzalez JR, et al. A forward genetic screen reveals novel independent regulators of ULBP1, an activating ligand for natural killer cells. Elife (2015) 4. doi:10.7554/eLife.08474
38. Nausch N, Florin L, Hartenstein B, Angel P, Schorpp-Kistner M, Cerwenka A. Cutting edge: the AP-1 subunit JunB determines NK cell-mediated target cell killing by regulation of the NKG2D-ligand RAE-1epsilon. J Immunol (2006) 176(1):7–11. doi:10.4049/jimmunol.176.1.7
39. Sauer M, Schuldner M, Hoffmann N, Cetintas A, Reiners KS, Shatnyeva O, et al. CBP/p300 acetyltransferases regulate the expression of NKG2D ligands on tumor cells. Oncogene (2017) 36(7):933–41. doi:10.1038/onc.2016.259
40. Eagle RA, Jafferji I, Barrow AD. Beyond stressed self: evidence for NKG2D ligand expression on healthy cells. Curr Immunol Rev (2009) 5(1):22–34. doi:10.2174/157339509787314369
41. Ebihara T, Masuda H, Akazawa T, Shingai M, Kikuta H, Ariga T, et al. Induction of NKG2D ligands on human dendritic cells by TLR ligand stimulation and RNA virus infection. Int Immunol (2007) 19(10):1145–55. doi:10.1093/intimm/dxm073
42. Hamerman JA, Ogasawara K, Lanier LL. Cutting edge: toll-like receptor signaling in macrophages induces ligands for the NKG2D receptor. J Immunol (2004) 172(4):2001–5. doi:10.4049/jimmunol.172.4.2001
43. Stern-Ginossar N, Gur C, Biton M, Horwitz E, Elboim M, Stanietsky N, et al. Human microRNAs regulate stress-induced immune responses mediated by the receptor NKG2D. Nat Immunol (2008) 9(9):1065–73. doi:10.1038/ni.1642
44. Yadav D, Ngolab J, Lim RS, Krishnamurthy S, Bui JD. Cutting edge: down-regulation of MHC class I-related chain A on tumor cells by IFN-gamma-induced microRNA. J Immunol (2009) 182(1):39–43. doi:10.4049/jimmunol.182.1.39
45. Berhani O, Nachmani D, Yamin R, Schmiedel D, Bar-On Y, Mandelboim O. Vigilin regulates the expression of the stress-induced ligand MICB by interacting with its 5′ untranslated region. J Immunol (2017) 198(9):3662–70. doi:10.4049/jimmunol.1601589
46. Nice TJ, Coscoy L, Raulet DH. Posttranslational regulation of the NKG2D ligand 1 in response to cell stress. J Exp Med (2009) 206(2):287–98. doi:10.1084/jem.20081335
47. Nice TJ, Deng W, Coscoy L, Raulet DH. Stress-regulated targeting of the NKG2D ligand 1 by a membrane-associated RING-CH family E3 ligase. J Immunol (2010) 185(9):5369–76. doi:10.4049/jimmunol.1000247
48. Mincheva-Nilsson L, Baranov V. Cancer exosomes and NKG2D receptor-ligand interactions: impairing NKG2D-mediated cytotoxicity and anti-tumour immune surveillance. Semin Cancer Biol (2014) 28:24–30. doi:10.1016/j.semcancer.2014.02.010
49. Chitadze G, Bhat J, Lettau M, Janssen O, Kabelitz D. Generation of soluble NKG2D ligands: proteolytic cleavage, exosome secretion and functional implications. Scand J Immunol (2013) 78(2):120–9. doi:10.1111/sji.12072
50. Molfetta R, Quatrini L, Santoni A, Paolini R. Regulation of NKG2D-dependent NK cell functions: the yin and the yang of receptor endocytosis. Int J Mol Sci (2017) 18(8):1677. doi:10.3390/ijms18081677
51. Deng W, Gowen BG, Zhang L, Wang L, Lau S, Iannello A, et al. Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science (2015) 348(6230):136–9. doi:10.1126/science.1258867
52. Antonangeli F, Soriani A, Cerboni C, Sciume G, Santoni A. How mucosal epithelia deal with stress: role of NKG2D/NKG2D ligands during inflammation. Front Immunol (2017) 8:1583. doi:10.3389/fimmu.2017.01583
53. Thompson TW, Kim AB, Li PJ, Wang J, Jackson BT, Huang KTH, et al. Endothelial cells express NKG2D ligands and desensitize anti-tumor NK responses. Elife (2017) 6. doi:10.7554/eLife.30881
54. Sharma N, Trinidad CV, Trembath AP, Markiewicz MA. NKG2D signaling between human NK cells enhances TACE-mediated TNF-alpha release. J Immunol (2017) 199(8):2865–72. doi:10.4049/jimmunol.1700647
55. Ogasawara K, Hamerman JA, Hsin H, Chikuma S, Bour-Jordan H, Chen T, et al. Impairment of NK cell function by NKG2D modulation in NOD mice. Immunity (2003) 18(1):41–51. doi:10.1016/S1074-7613(02)00505-8
56. Nakamura K, Nakayama M, Kawano M, Amagai R, Ishii T, Harigae H, et al. Fratricide of natural killer cells dressed with tumor-derived NKG2D ligand. Proc Natl Acad Sci U S A (2013) 110(23):9421–6. doi:10.1073/pnas.1300140110
57. Siren J, Sareneva T, Pirhonen J, Strengell M, Veckman V, Julkunen I, et al. Cytokine and contact-dependent activation of natural killer cells by influenza A or Sendai virus-infected macrophages. J Gen Virol (2004) 85(Pt 8):2357–64. doi:10.1099/vir.0.80105-0
58. Eissmann P, Evans JH, Mehrabi M, Rose EL, Nedvetzki S, Davis DM. Multiple mechanisms downstream of TLR-4 stimulation allow expression of NKG2D ligands to facilitate macrophage/NK cell crosstalk. J Immunol (2010) 184(12):6901–9. doi:10.4049/jimmunol.0903985
59. Kloss M, Decker P, Baltz KM, Baessler T, Jung G, Rammensee HG, et al. Interaction of monocytes with NK cells upon toll-like receptor-induced expression of the NKG2D ligand MICA. J Immunol (2008) 181(10):6711–9. doi:10.4049/jimmunol.181.10.6711
60. Vankayalapati R, Garg A, Porgador A, Griffith DE, Klucar P, Safi H, et al. Role of NK cell-activating receptors and their ligands in the lysis of mononuclear phagocytes infected with an intracellular bacterium. J Immunol (2005) 175(7):4611–7. doi:10.4049/jimmunol.175.7.4611
61. Nowbakht P, Ionescu MC, Rohner A, Kalberer CP, Rossy E, Mori L, et al. Ligands for natural killer cell-activating receptors are expressed upon the maturation of normal myelomonocytic cells but at low levels in acute myeloid leukemias. Blood (2005) 105(9):3615–22. doi:10.1182/blood-2004-07-2585
62. Rausch A, Hessmann M, Holscher A, Schreiber T, Bulfone-Paus S, Ehlers S, et al. Interleukin-15 mediates protection against experimental tuberculosis: a role for NKG2D-dependent effector mechanisms of CD8+ T cells. Eur J Immunol (2006) 36(5):1156–67. doi:10.1002/eji.200535290
63. Borchers MT, Harris NL, Wesselkamper SC, Zhang S, Chen Y, Young L, et al. The NKG2D-activating receptor mediates pulmonary clearance of Pseudomonas aeruginosa. Infect Immun (2006) 74(5):2578–86. doi:10.1128/IAI.74.5.2578-2586.2006
64. Lodoen M, Ogasawara K, Hamerman JA, Arase H, Houchins JP, Mocarski ES, et al. NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules. J Exp Med (2003) 197(10):1245–53. doi:10.1084/jem.20021973
65. Jung H, Hsiung B, Pestal K, Procyk E, Raulet DH. RAE-1 ligands for the NKG2D receptor are regulated by E2F transcription factors, which control cell cycle entry. J Exp Med (2012) 209(13):2409–22. doi:10.1084/jem.20120565
66. Guilloton F, de Thonel A, Jean C, Demur C, Mansat-De Mas V, Laurent G, et al. TNFalpha stimulates NKG2D-mediated lytic activity of acute myeloid leukemic cells. Leukemia (2005) 19(12):2206–14. doi:10.1038/sj.leu.2403952
67. Nausch N, Galani IE, Schlecker E, Cerwenka A. Mononuclear myeloid-derived “suppressor” cells express RAE-1 and activate natural killer cells. Blood (2008) 112(10):4080–9. doi:10.1182/blood-2008-03-143776
68. Diefenbach A, Hsia JK, Hsiung MY, Raulet DH. A novel ligand for the NKG2D receptor activates NK cells and macrophages and induces tumor immunity. Eur J Immunol (2003) 33(2):381–91. doi:10.1002/immu.200310012
69. Jamieson AM, Diefenbach A, McMahon CW, Xiong N, Carlyle JR, Raulet DH. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity (2002) 17(1):19–29. doi:10.1016/S1074-7613(02)00333-3
70. Buhtoiarov IN, Rakhmilevich AL, Lanier LL, Ranheim EA, Sondel PM. Naive mouse macrophages become activated following recognition of L5178Y lymphoma cells via concurrent ligation of CD40, NKG2D, and CD18 molecules. J Immunol (2009) 182(4):1940–53. doi:10.4049/jimmunol.0800443
71. Qian L, Liu Y, Wang S, Gong W, Jia X, Liu L, et al. NKG2D ligand RAE-1epsilon induces generation and enhances the inhibitor function of myeloid-derived suppressor cells in mice. J Cell Mol Med (2017) 21(9):2046–54. doi:10.1111/jcmm.13124
72. Brandstadter JD, Huang X, Yang Y. NK cell-extrinsic IL-18 signaling is required for efficient NK-cell activation by vaccinia virus. Eur J Immunol (2014) 44(9):2659–66. doi:10.1002/eji.201344134
73. Guan H, Moretto M, Bzik DJ, Gigley J, Khan IA. NK cells enhance dendritic cell response against parasite antigens via NKG2D pathway. J Immunol (2007) 179(1):590–6. doi:10.4049/jimmunol.179.1.590
74. Draghi M, Pashine A, Sanjanwala B, Gendzekhadze K, Cantoni C, Cosman D, et al. NKp46 and NKG2D recognition of infected dendritic cells is necessary for NK cell activation in the human response to influenza infection. J Immunol (2007) 178(5):2688–98. doi:10.4049/jimmunol.178.5.2688
75. Jinushi M, Takehara T, Kanto T, Tatsumi T, Groh V, Spies T, et al. Critical role of MHC class I-related chain A and B expression on IFN-alpha-stimulated dendritic cells in NK cell activation: impairment in chronic hepatitis C virus infection. J Immunol (2003) 170(3):1249–56. doi:10.4049/jimmunol.170.3.1249
76. Jinushi M, Takehara T, Tatsumi T, Kanto T, Groh V, Spies T, et al. Autocrine/paracrine IL-15 that is required for type I IFN-mediated dendritic cell expression of MHC class I-related chain A and B is impaired in hepatitis C virus infection. J Immunol (2003) 171(10):5423–9. doi:10.4049/jimmunol.171.10.5423
77. Qiao Y, Liu B, Li Z. Activation of NK cells by extracellular heat shock protein 70 through induction of NKG2D ligands on dendritic cells. Cancer Immun (2008) 8:12.
78. Tsan MF, Gao B. Heat shock proteins and immune system. J Leukoc Biol (2009) 85(6):905–10. doi:10.1189/jlb.0109005
79. Schrama D, Terheyden P, Otto K, Kammerer U, Brocker EB, Luhder F, et al. Expression of the NKG2D ligand UL16 binding protein-1 (ULBP-1) on dendritic cells. Eur J Immunol (2006) 36(1):65–72. doi:10.1002/eji.200535115
80. Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol (2016) 17(7):765–74. doi:10.1038/ni.3489
81. Andoniou CE, van Dommelen SL, Voigt V, Andrews DM, Brizard G, Asselin-Paturel C, et al. Interaction between conventional dendritic cells and natural killer cells is integral to the activation of effective antiviral immunity. Nat Immunol (2005) 6(10):1011–9. doi:10.1038/ni1244
82. Viaud S, Terme M, Flament C, Taieb J, Andre F, Novault S, et al. Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: a role for NKG2D ligands and IL-15Ralpha. PLoS One (2009) 4(3):e4942. doi:10.1371/journal.pone.0004942
83. Gerosa F, Baldani-Guerra B, Nisii C, Marchesini V, Carra G, Trinchieri G. Reciprocal activating interaction between natural killer cells and dendritic cells. J Exp Med (2002) 195(3):327–33. doi:10.1084/jem.20010938
84. Piccioli D, Sbrana S, Melandri E, Valiante NM. Contact-dependent stimulation and inhibition of dendritic cells by natural killer cells. J Exp Med (2002) 195(3):335–41. doi:10.1084/jem.20010934
85. Moretta L, Ferlazzo G, Bottino C, Vitale M, Pende D, Mingari MC, et al. Effector and regulatory events during natural killer-dendritic cell interactions. Immunol Rev (2006) 214:219–28. doi:10.1111/j.1600-065X.2006.00450.x
86. Ferlazzo G, Tsang ML, Moretta L, Melioli G, Steinman RM, Munz C. Human dendritic cells activate resting natural killer (NK) cells and are recognized via the NKp30 receptor by activated NK cells. J Exp Med (2002) 195(3):343–51. doi:10.1084/jem.20011149
87. Alter G, Kavanagh D, Rihn S, Luteijn R, Brooks D, Oldstone M, et al. IL-10 induces aberrant deletion of dendritic cells by natural killer cells in the context of HIV infection. J Clin Invest (2010) 120(6):1905–13. doi:10.1172/JCI40913
88. Schulz U, Kreutz M, Multhoff G, Stoelcker B, Kohler M, Andreesen R, et al. Interleukin-10 promotes NK cell killing of autologous macrophages by stimulating expression of NKG2D ligands. Scand J Immunol (2010) 72(4):319–31. doi:10.1111/j.1365-3083.2010.02435.x
89. Nedvetzki S, Sowinski S, Eagle RA, Harris J, Vely F, Pende D, et al. Reciprocal regulation of human natural killer cells and macrophages associated with distinct immune synapses. Blood (2007) 109(9):3776–85. doi:10.1182/blood-2006-10-052977
90. Campbell AR, Duggan MC, Suarez-Kelly LP, Bhave N, Opheim KS, McMichael EL, et al. MICA-expressing monocytes enhance natural killer cell Fc receptor-mediated antitumor functions. Cancer Immunol Res (2017) 5(9):778–89. doi:10.1158/2326-6066.CIR-16-0005
91. Zhou Z, Zhang C, Zhang J, Tian Z. Macrophages help NK cells to attack tumor cells by stimulatory NKG2D ligand but protect themselves from NK killing by inhibitory ligand Qa-1. PLoS One (2012) 7(5):e36928. doi:10.1371/journal.pone.0036928
92. Coudert JD, Zimmer J, Tomasello E, Cebecauer M, Colonna M, Vivier E, et al. Altered NKG2D function in NK cells induced by chronic exposure to NKG2D ligand-expressing tumor cells. Blood (2005) 106(5):1711–7. doi:10.1182/blood-2005-03-0918
93. Oppenheim DE, Roberts SJ, Clarke SL, Filler R, Lewis JM, Tigelaar RE, et al. Sustained localized expression of ligand for the activating NKG2D receptor impairs natural cytotoxicity in vivo and reduces tumor immunosurveillance. Nat Immunol (2005) 6(9):928–37. doi:10.1038/ni1239
94. Morvan MG, Champsaur M, Reizis B, Lanier LL. Chronic in vivo interaction of dendritic cells expressing the ligand Rae-1epsilon with NK cells impacts NKG2D expression and function. Immunohorizons (2017) 1(3):10–9. doi:10.4049/immunohorizons.1700004
95. Coudert JD, Scarpellino L, Gros F, Vivier E, Held W. Sustained NKG2D engagement induces cross-tolerance of iple distinct NK cell activation pathways. Blood (2008) 111(7):3571–8. doi:10.1182/blood-2007-07-100057
96. Crane CA, Austgen K, Haberthur K, Hofmann C, Moyes KW, Avanesyan L, et al. Immune evasion mediated by tumor-derived lactate dehydrogenase induction of NKG2D ligands on myeloid cells in glioblastoma patients. Proc Natl Acad Sci U S A (2014) 111(35):12823–8. doi:10.1073/pnas.1413933111
97. Ehrlich LI, Ogasawara K, Hamerman JA, Takaki R, Zingoni A, Allison JP, et al. Engagement of NKG2D by cognate ligand or antibody alone is insufficient to mediate costimulation of human and mouse CD8+ T cells. J Immunol (2005) 174(4):1922–31. doi:10.4049/jimmunol.174.4.1922
98. Karimi M, Cao TM, Baker JA, Verneris MR, Soares L, Negrin RS. Silencing human NKG2D, DAP10, and DAP12 reduces cytotoxicity of activated CD8+ T cells and NK cells. J Immunol (2005) 175(12):7819–28. doi:10.4049/jimmunol.175.12.7819
99. Groh V, Rhinehart R, Randolph-Habecker J, Topp MS, Riddell SR, Spies T. Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat Immunol (2001) 2(3):255–60. doi:10.1038/85321
100. Roberts AI, Lee L, Schwarz E, Groh V, Spies T, Ebert EC, et al. NKG2D receptors induced by IL-15 costimulate CD28-negative effector CTL in the tissue microenvironment. J Immunol (2001) 167(10):5527–30. doi:10.4049/jimmunol.167.10.5527
101. Maasho K, Opoku-Anane J, Marusina AI, Coligan JE, Borrego F. NKG2D is a costimulatory receptor for human naive CD8+ T cells. J Immunol (2005) 174(8):4480–4. doi:10.4049/jimmunol.174.8.4480
102. Markiewicz MA, Carayannopoulos LN, Naidenko OV, Matsui K, Burack WR, Wise EL, et al. Costimulation through NKG2D enhances murine CD8+ CTL function: similarities and differences between NKG2D and CD28 costimulation. J Immunol (2005) 175(5):2825–33. doi:10.4049/jimmunol.175.5.2825
103. Rajasekaran K, Xiong V, Fong L, Gorski J, Malarkannan S. Functional dichotomy between NKG2D and CD28-mediated co-stimulation in human CD8+ T cells. PLoS One (2010) 5(9):e12635. doi:10.1371/journal.pone.0012635
104. Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz TN, et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity (2004) 21(3):357–66. doi:10.1016/j.immuni.2004.06.020
105. Correia MP, Cardoso EM, Pereira CF, Neves R, Uhrberg M, Arosa FA. Hepatocytes and IL-15: a favorable microenvironment for T cell survival and CD8+ T cell differentiation. J Immunol (2009) 182(10):6149–59. doi:10.4049/jimmunol.0802470
106. Correia MP, Costa AV, Uhrberg M, Cardoso EM, Arosa FA. IL-15 induces CD8+ T cells to acquire functional NK receptors capable of modulating cytotoxicity and cytokine secretion. Immunobiology (2011) 216(5):604–12. doi:10.1016/j.imbio.2010.09.012
107. Meresse B, Curran SA, Ciszewski C, Orbelyan G, Setty M, Bhagat G, et al. Reprogramming of CTLs into natural killer-like cells in celiac disease. J Exp Med (2006) 203(5):1343–55. doi:10.1084/jem.20060028
108. Allez M, Tieng V, Nakazawa A, Treton X, Pacault V, Dulphy N, et al. CD4+NKG2D+ T cells in Crohn’s disease mediate inflammatory and cytotoxic responses through MICA interactions. Gastroenterology (2007) 132(7):2346–58. doi:10.1053/j.gastro.2007.03.025
109. Upshaw JL, Arneson LN, Schoon RA, Dick CJ, Billadeau DD, Leibson PJ. NKG2D-mediated signaling requires a DAP10-bound Grb2-Vav1 intermediate and phosphatidylinositol-3-kinase in human natural killer cells. Nat Immunol (2006) 7(5):524–32. doi:10.1038/ni1325
110. Jabri B, Abadie V. IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nat Rev Immunol (2015) 15(12):771–83. doi:10.1038/nri3919
111. Wensveen FM, Lenartic M, Jelencic V, Lemmermann NA, ten Brinke A, Jonjic S, et al. NKG2D induces Mcl-1 expression and mediates survival of CD8 memory T cell precursors via phosphatidylinositol 3-kinase. J Immunol (2013) 191(3):1307–15. doi:10.4049/jimmunol.1300670
112. Zloza A, Kohlhapp FJ, Lyons GE, Schenkel JM, Moore TV, Lacek AT, et al. NKG2D signaling on CD8(+) T cells represses T-bet and rescues CD4-unhelped CD8(+) T cell memory recall but not effector responses. Nat Med (2012) 18(3):422–8. doi:10.1038/nm.2683
113. Pariente B, Mocan I, Camus M, Dutertre CA, Ettersperger J, Cattan P, et al. Activation of the receptor NKG2D leads to production of Th17 cytokines in CD4+ T cells of patients with Crohn’s disease. Gastroenterology (2011) 141(1):217–26, 226.e1–2. doi:10.1053/j.gastro.2011.03.061
114. Camus M, Esses S, Pariente B, Le Bourhis L, Douay C, Chardiny V, et al. Oligoclonal expansions of mucosal T cells in Crohn’s disease predominate in NKG2D-expressing CD4 T cells. Mucosal Immunol (2014) 7(2):325–34. doi:10.1038/mi.2013.51
115. Capraru D, Muller A, Csernok E, Gross WL, Holl-Ulrich K, Northfield J, et al. Expansion of circulating NKG2D+ effector memory T-cells and expression of NKG2D-ligand MIC in granulomaous lesions in Wegener’s granulomatosis. Clin Immunol (2008) 127(2):144–50. doi:10.1016/j.clim.2007.12.004
116. de Menthon M, Lambert M, Guiard E, Tognarelli S, Bienvenu B, Karras A, et al. Excessive interleukin-15 transpresentation endows NKG2D+CD4+ T cells with innate-like capacity to lyse vascular endothelium in granulomatosis with polyangiitis (Wegener’s). Arthritis Rheum (2011) 63(7):2116–26. doi:10.1002/art.30355
117. Phoksawat W, Jumnainsong A, Leelayuwat N, Leelayuwat C. Aberrant NKG2D expression with IL-17 production of CD4+ T subsets in patients with type 2 diabetes. Immunobiology (2017) 222(10):944–51. doi:10.1016/j.imbio.2016.05.001
118. Ruck T, Bittner S, Gross CC, Breuer J, Albrecht S, Korr S, et al. CD4+NKG2D+ T cells exhibit enhanced migratory and encephalitogenic properties in neuroinflammation. PLoS One (2013) 8(11):e81455. doi:10.1371/journal.pone.0081455
119. Yang D, Wang H, Ni B, He Y, Li J, Tang Y, et al. Mutual activation of CD4+ T cells and monocytes mediated by NKG2D-MIC interaction requires IFN-gamma production in systemic lupus erythematosus. Mol Immunol (2009) 46(7):1432–42. doi:10.1016/j.molimm.2008.12.010
120. Yang D, Tian Z, Zhang M, Yang W, Tang J, Wu Y, et al. NKG2D(+)CD4(+) T cells kill regulatory T cells in a NKG2D-NKG2D ligand-dependent manner in systemic lupus erythematosus. Sci Rep (2017) 7(1):1288. doi:10.1038/s41598-017-01379-y
121. Groh V, Smythe K, Dai Z, Spies T. Fas-ligand-mediated paracrine T cell regulation by the receptor NKG2D in tumor immunity. Nat Immunol (2006) 7(7):755–62. doi:10.1038/ni1350
122. Garcia-Chagollan M, Jave-Suarez LF, Haramati J, Sanchez-Hernandez PE, Aguilar-Lemarroy A, Bueno-Topete MR, et al. Substantial increase in the frequency of circulating CD4+NKG2D+ T cells in patients with cervical intraepithelial neoplasia grade 1. J Biomed Sci (2013) 20:60. doi:10.1186/1423-0127-20-60
123. Garcia-Chagollan M, Jave-Suarez LF, Haramati J, Bueno-Topete MR, Aguilar-Lemarroy A, Estrada-Chavez C, et al. An approach to the immunophenotypic features of circulating CD4(+)NKG2D(+) T cells in invasive cervical carcinoma. J Biomed Sci (2015) 22:91. doi:10.1186/s12929-015-0190-7
124. Escarra-Senmarti M, Bueno-Topete MR, Jave-Suarez LF, Gomez-Banuelos E, Gutierrez-Franco J, Vega-Magana N, et al. Loss of CD28 within CD4(+) T cell subsets from cervical cancer patients is accompanied by the acquisition of intracellular perforin, and is further enhanced by NKG2D expression. Immunol Lett (2017) 182:30–8. doi:10.1016/j.imlet.2017.01.006
125. Azimi N, Jacobson S, Tanaka Y, Corey L, Groh V, Spies T. Immunostimulation by induced expression of NKG2D and its MIC ligands in HTLV-1-associated neurologic disease. Immunogenetics (2006) 58(4):252–8. doi:10.1007/s00251-006-0082-9
126. Saez-Borderias A, Guma M, Angulo A, Bellosillo B, Pende D, Lopez-Botet M. Expression and function of NKG2D in CD4+ T cells specific for human cytomegalovirus. Eur J Immunol (2006) 36(12):3198–206. doi:10.1002/eji.200636682
127. Schirmer M, Vallejo AN, Weyand CM, Goronzy JJ. Resistance to apoptosis and elevated expression of Bcl-2 in clonally expanded CD4+CD28- T cells from rheumatoid arthritis patients. J Immunol (1998) 161(2):1018–25.
128. Vallejo AN, Schirmer M, Weyand CM, Goronzy JJ. Clonality and longevity of CD4+CD28null T cells are associated with defects in apoptotic pathways. J Immunol (2000) 165(11):6301–7. doi:10.4049/jimmunol.165.11.6301
129. Namekawa T, Snyder MR, Yen JH, Goehring BE, Leibson PJ, Weyand CM, et al. Killer cell activating receptors function as costimulatory molecules on CD4+CD28null T cells clonally expanded in rheumatoid arthritis. J Immunol (2000) 165(2):1138–45. doi:10.4049/jimmunol.165.2.1138
130. Lamprecht P, Moosig F, Csernok E, Seitzer U, Schnabel A, Mueller A, et al. CD28 negative T cells are enriched in granulomatous lesions of the respiratory tract in Wegener’s granulomatosis. Thorax (2001) 56(10):751–7. doi:10.1136/thorax.56.10.751
131. Duftner C, Goldberger C, Falkenbach A, Wurzner R, Falkensammer B, Pfeiffer KP, et al. Prevalence, clinical relevance and characterization of circulating cytotoxic CD4+CD28- T cells in ankylosing spondylitis. Arthritis Res Ther (2003) 5(5):R292–300. doi:10.1186/ar986
132. Duftner C, Dejaco C, Kullich W, Klauser A, Goldberger C, Falkenbach A, et al. Preferential type 1 chemokine receptors and cytokine production of CD28- T cells in ankylosing spondylitis. Ann Rheum Dis (2006) 65(5):647–53. doi:10.1136/ard.2005.042085
133. Fasth AE, Bjorkstrom NK, Anthoni M, Malmberg KJ, Malmstrom V. Activating NK-cell receptors co-stimulate CD4(+)CD28(-) T cells in patients with rheumatoid arthritis. Eur J Immunol (2010) 40(2):378–87. doi:10.1002/eji.200939399
134. Dai Z, Turtle CJ, Booth GC, Riddell SR, Gooley TA, Stevens AM, et al. Normally occurring NKG2D+CD4+ T cells are immunosuppressive and inversely correlated with disease activity in juvenile-onset lupus. J Exp Med (2009) 206(4):793–805. doi:10.1084/jem.20081648
135. Hayday AC. Gammadelta T cells and the lymphoid stress-surveillance response. Immunity (2009) 31(2):184–96. doi:10.1016/j.immuni.2009.08.006
136. Cai Y, Shen X, Ding C, Qi C, Li K, Li X, et al. Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity (2011) 35(4):596–610. doi:10.1016/j.immuni.2011.08.001
137. Pang DJ, Neves JF, Sumaria N, Pennington DJ. Understanding the complexity of gammadelta T-cell subsets in mouse and human. Immunology (2012) 136(3):283–90. doi:10.1111/j.1365-2567.2012.03582.x
138. Hinz T, Wesch D, Halary F, Marx S, Choudhary A, Arden B, et al. Identification of the complete expressed human TCR V gamma repertoire by flow cytometry. Int Immunol (1997) 9(8):1065–72. doi:10.1093/intimm/9.8.1065
139. Caccamo N, La Mendola C, Orlando V, Meraviglia S, Todaro M, Stassi G, et al. Differentiation, phenotype, and function of interleukin-17-producing human Vgamma9Vdelta2 T cells. Blood (2011) 118(1):129–38. doi:10.1182/blood-2011-01-331298
140. Groh V, Steinle A, Bauer S, Spies T. Recognition of stress-induced MHC molecules by intestinal epithelial gammadelta T cells. Science (1998) 279(5357):1737–40. doi:10.1126/science.279.5357.1737
141. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science (1999) 285(5428):727–9. doi:10.1126/science.285.5428.727
142. Zhao J, Huang J, Chen H, Cui L, He W. Vdelta1 T cell receptor binds specifically to MHC I chain related A: molecular and biochemical evidences. Biochem Biophys Res Commun (2006) 339(1):232–40. doi:10.1016/j.bbrc.2005.10.198
143. Wu J, Groh V, Spies T. T cell antigen receptor engagement and specificity in the recognition of stress-inducible MHC class I-related chains by human epithelial gamma delta T cells. J Immunol (2002) 169(3):1236–40. doi:10.4049/jimmunol.169.3.1236
144. Poggi A, Venturino C, Catellani S, Clavio M, Miglino M, Gobbi M, et al. Vdelta1 T lymphocytes from B-CLL patients recognize ULBP3 expressed on leukemic B cells and up-regulated by trans-retinoic acid. Cancer Res (2004) 64(24):9172–9. doi:10.1158/0008-5472.CAN-04-2417
145. Das H, Wang L, Kamath A, Bukowski JF. Vgamma2Vdelta2 T-cell receptor-mediated recognition of aminobisphosphonates. Blood (2001) 98(5):1616–8. doi:10.1182/blood.V98.5.1616
146. Rincon-Orozco B, Kunzmann V, Wrobel P, Kabelitz D, Steinle A, Herrmann T. Activation of V gamma 9V delta 2 T cells by NKG2D. J Immunol (2005) 175(4):2144–51. doi:10.4049/jimmunol.175.4.2144
147. Lanca T, Correia DV, Moita CF, Raquel H, Neves-Costa A, Ferreira C, et al. The MHC class Ib protein ULBP1 is a nonredundant determinant of leukemia/lymphoma susceptibility to gammadelta T-cell cytotoxicity. Blood (2010) 115(12):2407–11. doi:10.1182/blood-2009-08-237123
148. Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein KH, Spies T. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc Natl Acad Sci U S A (1999) 96(12):6879–84. doi:10.1073/pnas.96.12.6879
149. Deniger DC, Maiti SN, Mi T, Switzer KC, Ramachandran V, Hurton LV, et al. Activating and propagating polyclonal gamma delta T cells with broad specificity for malignancies. Clin Cancer Res (2014) 20(22):5708–19. doi:10.1158/1078-0432.CCR-13-3451
150. Kong Y, Cao W, Xi X, Ma C, Cui L, He W. The NKG2D ligand ULBP4 binds to TCRgamma9/delta2 and induces cytotoxicity to tumor cells through both TCRgammadelta and NKG2D. Blood (2009) 114(2):310–7. doi:10.1182/blood-2008-12-196287
151. Girardi M, Oppenheim DE, Steele CR, Lewis JM, Glusac E, Filler R, et al. Regulation of cutaneous malignancy by gammadelta T cells. Science (2001) 294(5542):605–9. doi:10.1126/science.1063916
152. Yoshida S, Mohamed RH, Kajikawa M, Koizumi J, Tanaka M, Fugo K, et al. Involvement of an NKG2D ligand H60c in epidermal dendritic T cell-mediated wound repair. J Immunol (2012) 188(8):3972–9. doi:10.4049/jimmunol.1102886
153. Nielsen MM, Dyring-Andersen B, Schmidt JD, Witherden D, Lovato P, Woetmann A, et al. NKG2D-dependent activation of dendritic epidermal T cells in contact hypersensitivity. J Invest Dermatol (2015) 135(5):1311–9. doi:10.1038/jid.2015.23
154. Nitahara A, Shimura H, Ito A, Tomiyama K, Ito M, Kawai K. NKG2D ligation without T cell receptor engagement triggers both cytotoxicity and cytokine production in dendritic epidermal T cells. J Invest Dermatol (2006) 126(5):1052–8. doi:10.1038/sj.jid.5700112
155. Strid J, Roberts SJ, Filler RB, Lewis JM, Kwong BY, Schpero W, et al. Acute upregulation of an NKG2D ligand promotes rapid reorganization of a local immune compartment with pleiotropic effects on carcinogenesis. Nat Immunol (2008) 9(2):146–54. doi:10.1038/ni1556
156. Whang MI, Guerra N, Raulet DH. Costimulation of dendritic epidermal gammadelta T cells by a new NKG2D ligand expressed specifically in the skin. J Immunol (2009) 182(8):4557–64. doi:10.4049/jimmunol.0802439
157. Ibusuki A, Kawai K, Yoshida S, Uchida Y, Nitahara-Takeuchi A, Kuroki K, et al. NKG2D triggers cytotoxicity in murine epidermal gammadelta T cells via PI3K-dependent, Syk/ZAP70-independent signaling pathway. J Invest Dermatol (2014) 134(2):396–404. doi:10.1038/jid.2013.353
158. Gumperz JE, Miyake S, Yamamura T, Brenner MB. Functionally distinct subsets of CD1d-restricted natural killer T cells revealed by CD1d tetramer staining. J Exp Med (2002) 195(5):625–36. doi:10.1084/jem.20011786
159. Lee PT, Benlagha K, Teyton L, Bendelac A. Distinct functional lineages of human V(alpha)24 natural killer T cells. J Exp Med (2002) 195(5):637–41. doi:10.1084/jem.20011908
160. Kuylenstierna C, Bjorkstrom NK, Andersson SK, Sahlstrom P, Bosnjak L, Paquin-Proulx D, et al. NKG2D performs two functions in invariant NKT cells: direct TCR-independent activation of NK-like cytolysis and co-stimulation of activation by CD1d. Eur J Immunol (2011) 41(7):1913–23. doi:10.1002/eji.200940278
161. Wang H, Yang D, Xu W, Wang Y, Ruan Z, Zhao T, et al. Tumor-derived soluble MICs impair CD3(+)CD56(+) NKT-like cell cytotoxicity in cancer patients. Immunol Lett (2008) 120(1–2):65–71. doi:10.1016/j.imlet.2008.07.001
162. Guo H, Xu B, Gao L, Sun X, Qu X, Li X, et al. High frequency of activated natural killer and natural killer T-cells in patients with new onset of type 2 diabetes mellitus. Exp Biol Med (Maywood) (2012) 237(5):556–62. doi:10.1258/ebm.2012.011272
163. Li WX, Pan HF, Hu JL, Wang CZ, Zhang N, Li J, et al. Assay of T- and NK-cell subsets and the expression of NKG2A and NKG2D in patients with new-onset systemic lupus erythematosus. Clin Rheumatol (2010) 29(3):315–23. doi:10.1007/s10067-009-1322-9
164. Miko E, Szereday L, Barakonyi A, Jarkovich A, Varga P, Szekeres-Bartho J. The role of invariant NKT cells in pre-eclampsia. Am J Reprod Immunol (2008) 60(2):118–26. doi:10.1111/j.1600-0897.2008.00603.x
165. Waldmann TA. Targeting the interleukin-15/interleukin-15 receptor system in inflammatory autoimmune diseases. Arthritis Res Ther (2004) 6(4):174–7. doi:10.1186/ar1202
166. Waldmann TA. The biology of IL-15: implications for cancer therapy and the treatment of autoimmune disorders. J Investig Dermatol Symp Proc (2013) 16(1):S28–30. doi:10.1038/jidsymp.2013.8
167. Van Belle TL, von Herrath MG. The role of the activating receptor NKG2D in autoimmunity. Mol Immunol (2009) 47(1):8–11. doi:10.1016/j.molimm.2009.02.023
168. Ikeshita S, Miyatake Y, Otsuka N, Kasahara M. MICA/B expression in macrophage foam cells infiltrating atherosclerotic plaques. Exp Mol Pathol (2014) 97(1):171–5. doi:10.1016/j.yexmp.2014.07.002
169. Allegretti YL, Bondar C, Guzman L, Cueto Rua E, Chopita N, Fuertes M, et al. Broad MICA/B expression in the small bowel mucosa: a link between cellular stress and celiac disease. PLoS One (2013) 8(9):e73658. doi:10.1371/journal.pone.0073658
Keywords: NKG2D, NKG2D ligands, myeloid cells, natural killer cells, NKG2D+ T cells
Citation: Stojanovic A, Correia MP and Cerwenka A (2018) The NKG2D/NKG2DL Axis in the Crosstalk Between Lymphoid and Myeloid Cells in Health and Disease. Front. Immunol. 9:827. doi: 10.3389/fimmu.2018.00827
Received: 08 February 2018; Accepted: 04 April 2018;
Published: 23 April 2018
Edited by:
Nadia Guerra, Imperial College London, United KingdomReviewed by:
Alessandra Zingoni, Sapienza Università di Roma, ItalyBojan Polić, University of Rijeka, Croatia
Copyright: © 2018 Stojanovic, Correia and Cerwenka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana Stojanovic, YW5hLnN0b2phbm92aWNAbWVkbWEudW5pLWhlaWRlbGJlcmcuZGU=
†These authors have contributed equally to this work.