 Alexandre Vallée
Alexandre Vallée Yves Lecarpentier
Yves Lecarpentier- 1DRCI, Hôpital Foch, Suresnes, France
- 2Centre de Recherche Clinique, Grand Hôpital de l’Est Francilien (GHEF), Meaux, France
Inflammation and oxidative stress are common and co-substantial pathological processes accompanying, promoting, and even initiating numerous cancers. The canonical WNT/β-catenin pathway and peroxisome proliferator-activated receptor gamma (PPARγ) generally work in opposition. If one of them is upregulated, the other one is downregulated and vice versa. WNT/β-catenin signaling is upregulated in inflammatory processes and oxidative stress and in many cancers, although there are some exceptions for cancers. The opposite is observed with PPARγ, which is generally downregulated during inflammation and oxidative stress and in many cancers. This helps to explain in part the opposite and unidirectional profile of the canonical WNT/β-catenin signaling and PPARγ in these three frequent and morbid processes that potentiate each other and create a vicious circle. Many intracellular pathways commonly involved downstream will help maintain and amplify inflammation, oxidative stress, and cancer. Thus, many WNT/β-catenin target genes such as c-Myc, cyclin D1, and HIF-1α are involved in the development of cancers. Nuclear factor-kappaB (NFκB) can activate many inflammatory factors such as TNF-α, TGF-β, interleukin-6 (IL-6), IL-8, MMP, vascular endothelial growth factor, COX2, Bcl2, and inducible nitric oxide synthase. These factors are often associated with cancerous processes and may even promote them. Reactive oxygen species (ROS), generated by cellular alterations, stimulate the production of inflammatory factors such as NFκB, signal transducer and activator transcription, activator protein-1, and HIF-α. NFκB inhibits glycogen synthase kinase-3β (GSK-3β) and therefore activates the canonical WNT pathway. ROS activates the phosphatidylinositol 3 kinase/protein kinase B (PI3K/Akt) signaling in many cancers. PI3K/Akt also inhibits GSK-3β. Many gene mutations of the canonical WNT/β-catenin pathway giving rise to cancers have been reported (CTNNB1, AXIN, APC). Conversely, a significant reduction in the expression of PPARγ has been observed in many cancers. Moreover, PPARγ agonists promote cell cycle arrest, cell differentiation, and apoptosis and reduce inflammation, angiogenesis, oxidative stress, cell proliferation, invasion, and cell migration. All these complex and opposing interactions between the canonical WNT/β-catenin pathway and PPARγ appear to be fairly common in inflammation, oxidative stress, and cancers.
Introduction
Cancer is a complex process that can be defined in term of three steps: initiation, promotion, and progression (1). Several chemical, physical, and biological factors may induce chronic inflammation, thereby increasing the risk of cancers (2). This link between cancer and inflammation has been reported in experimental and epidemiological studies (3, 4) and demonstrated through the efficacy of anti-inflammatory therapies in cancer (5). Chronic inflammation is responsible for various steps involved in carcinogenesis, such as promotion, survival, cellular transformation, invasion, proliferation, angiogenesis, and metastasis (6, 7).
Oxidative stress also operates at these stages by promoting DNA damages and genes mutations (8). In recent years, several studies have shown that the link between inflammation and cancer can involve oxidative stress through reactive oxygen species (ROS) production. Tumor promoters have the capacity to recruit inflammatory factors and then stimulate ROS production (9, 10). Oncogenic transformation is promoted by oxidative stress that acts as a DNA-damaging effector (11). ROS generation, together with oxidative stress, stimulates several signaling pathways that contribute to cancer development by regulating proliferation, invasion, angiogenesis, and metastasis (12).
The canonical WNT/β-catenin pathway regulates several signaling pathways involved in development and tissue homeostasis. This pathway is modulated from transcription level regulations to post-transcriptional modifications. An aberrant WNT/β-catenin pathway is observed in cancers (13, 14). This results in stimulating the expression of numerous WNT target genes involved in tumor development, such as c-Myc, cyclin D1, and HIF-1α (15), the production of ROS (16), and the activation of chronic inflammation (17).
In contrast, peroxisome proliferator-activated receptor gamma (PPARγ) is downregulated in numerous cancers (13). By regulating lipid and glucose homeostasis, differentiation, ROS and inflammation, PPARγ agonists appear to offer interesting therapeutic solution in cancers (18, 19).
In numerous tissues, canonical WNT/β-catenin pathway activation induces inactivation of PPARγ, while PPARγ activation induces inhibition of canonical WNT/β-catenin signaling (20). In most cancers, the canonical WNT/β-catenin pathway is increased while PPARγ is downregulated (13). PPARγ agonists induce repression of the canonical WNT/β-catenin signaling in several pathophysiological states. In this review, we focus on the crosstalk between canonical WNT/β-catenin pathway and PPARγ in chronic inflammation and oxidative stress during carcinogenesis processes.
Peroxisome Proliferator-Activated Receptor Gamma
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors. Four subtypes of PPARγ have been identified: PPARγ1, PPARγ2, PPARγ3, and PPARγ4 (21, 22). After activation by natural or synthetic ligands, PPARγ heterodimerizes with the retinoid X receptor (RXR). Then, the complex PPARγ–RXR translocates to the nucleus to bind PPAR response elements (PPREs) to modulate the expression of several genes involved in immunity, inflammation, metabolism, cell proliferation, and cell differentiation (23–25). Fatty acids derivatives, such as 15-deoxy-delta-12,14-prostaglandin J (15d-PGJ2) hydroxyoctadecadienoic acid (9-HODE, 13-HODE), are endogenous ligands that activate PPARγ (26). PPARγ expression is involved in the development of heart and placenta (27) and during adipogenesis (28, 29). Thiazolidinediones (TZDs, antidiabetic drugs) are synthetic PPARγ ligands, which have been used in the diabetes treatment because of their ability to enhance insulin sensitivity. They also favor adipocyte differentiation and upregulation of adiponectin (30).
Canonical WNT/β-Catenin Pathway
The WNT pathway is involved in numerous pathways that control tissue homeostasis and embryogenesis development. WNT ligands belong to the family of genes observed in humans, Xenopus, mice, drosophilia, and Zebrafish (31). Dysregulation of the canonical WNT pathway activity has been reported in several disorders and cancers (32–34).
Canonical WNT signaling is characterized by the interaction between WNT ligand and specific targets resulting in cytosolic β-catenin accumulation and then its nuclear translocation (Figure 1). The nuclear activation of β-catenin results in the stimulation of downstream factors (35). During the “off state” of the WNT/β-catenin pathway, WNT ligands do not bind specific receptors. Cytosolic β-catenin is maintained at a minimal level through the activation of the β-catenin destruction complex, formed by the combination of AXIN (a cytoplasmic protein regulating G-protein signaling), glycogen synthase kinase-3β (GSK-3β, a serine-theronine kinase), adenomatous polyposis coli (APC, a tumor suppressor gene), and casein kinase 1 (CK-1, a serine/threonine-selective enzyme) (36). CK-1 and GSK-3β target β-catenin by phosphorylating the serine and threonine residues located in the amino acid terminus (37–39). CK-1 phosphorylates an N-terminus of β-catenin and GSK-3β phosphorylates a threonine 41 (Th41), Ser33, and Ser37 sites of β-catenin (35, 40). These phosphorylations result in recruiting APC in the destruction complex. APC modulates the degradation of the cytosolic β-catenin into the proteasome through its tumor suppressor properties (36, 41).
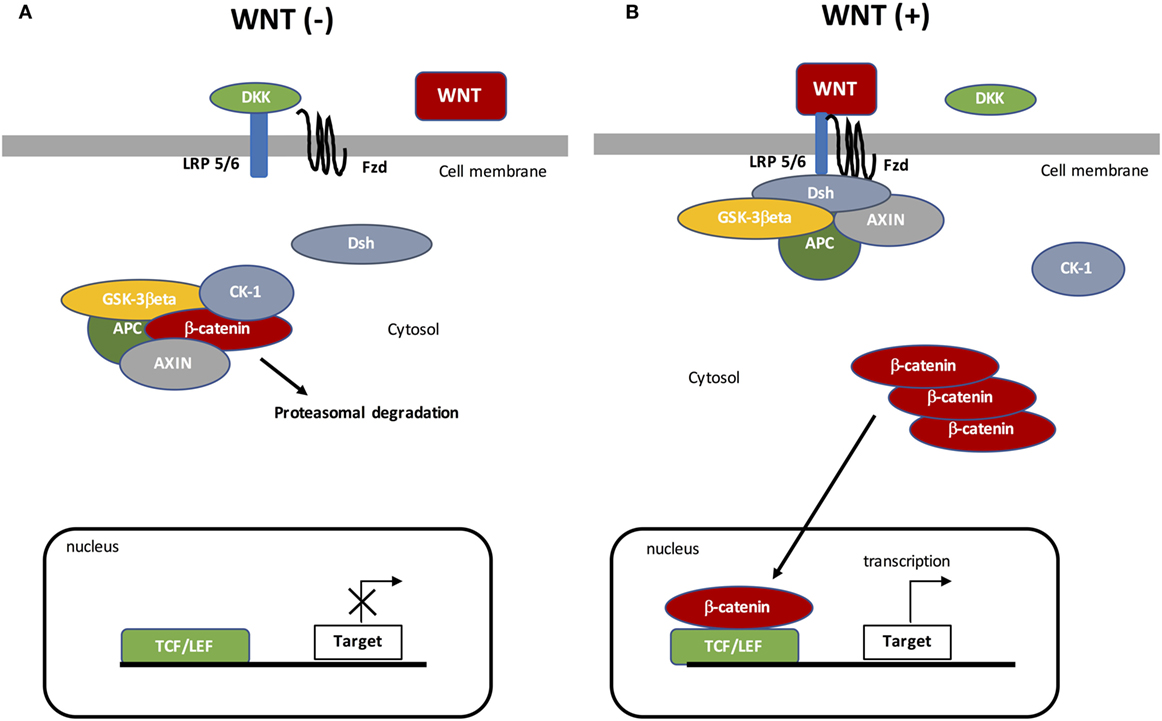
Figure 1. The canonical WNT/β-catenin pathway. (A) Under resting condition, the cytoplasmic β-catenin is bound to its destruction complex, consisting of adenomatous polyposis coli (APC), AXIN, and glycogen synthase kinase-3β (GSK-3β). After CK-1 phosphorylates on Ser45 residue, β-catenin is further phosphorylated on Thr41, Ser37, and Ser33 residues by GSK-3β. Then, phosphorylated β-catenin is degraded into the proteasome. Therefore, the cytosolic level of β-catenin is kept low in the absence of WNT ligands. If β-catenin is not present in the nucleus, the TCF/LEF complex cannot activate the target genes. DKK can inhibit the WNT/β-catenin pathway by binding to WNT ligands or LRP5/6. (B) When WNT ligands bind to both frizzled (FZD) and low-density lipoprotein receptor-related protein 5 (LRP5/6), Disheveled (DSH) is recruited and phosphorylated by FZD. Phosphorylated DSH in turn recruits AXIN, which dissociates the β-catenin destruction complex. Therefore, β-catenin escapes from phosphorylation and subsequently accumulates in the cytosol. The accumulated cytosolic β-catenin goes into the nucleus, where it binds to T-cell factor/lymphoid enhancer factor (TCF/LEF) and activates the transcription of target genes.
The “on state” is characterized by modified WNT/β-catenin signaling. WNT ligands bind Frizzled (FZD, a family of G protein-coupled receptors) and low-density lipoprotein receptor-related protein 5 (LRP 5/6) co-receptors (42). Then, Disheveled (DSH, a phosphoprotein) forms a complex with FZD, which results in the phosphorylation of LRP5/6 by GSK-3β and then the recruitment of the β-catenin destruction complex. DSH phosphorylates LRP6 (43), which inhibits GSK-3β activity leading to the stabilization and then the cytosolic accumulation of β-catenin. Accumulation of β-catenin leads to its nuclear translocation and then β-catenin binds the T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors (44, 45). The nuclear complex formed by β-catenin and TCF/LEF activates several WNT target genes, such as c-Myc (a transcription factor) and cyclin D1 (a protein belonging to the highly conserved cyclin family encoded by the CCND1 gene) (37, 38). The WNT target genes are involved in several processes, such as cell division, proliferation, invasion, and stem cell maintenance (46). Furthermore, β-catenin accumulation is involved in cancer phenotype maintenance (47–49).
Crosstalk between PPARγ and the WNT/β-Catenin Pathway
In several diseases, the WNT/β-catenin pathway and PPARγ act in an opposite manner as in cancers, such as gliomas (15, 50), neurodegenerative diseases, such as Alzheimer’s disease (51, 52), amyotrophic lateral sclerosis (53, 54), multiple sclerosis (55), age-related macular degeneration (56, 57), and fibrosis processes (58–60).
The WNT/β-catenin pathway and PPARγ interact through a TCF/LEF β-catenin domain and a catenin-binding domain within PPARγ (61–64). Downregulation of the WNT/β-catenin pathway leads to the stimulation of PPARγ expression (65), whereas PPARγ upregulation downregulates β-catenin levels in several cellular systems (66–68). PPARγ agonists stimulate synaptic plasticity by interacting with the WNT/β-catenin/phosphatidylinositol 3 kinase/protein kinase B (PI3K/Akt) pathway (69). Moreover, mesenchymal stem cell differentiation also presents this interaction between these two pathways (70).
Indeed, in numerous diseases, β-catenin signaling decreases PPARγ expression (71–80). In many studies, PPARγ operates as a negative β-catenin target gene (81, 82).
Peroxisome proliferator-activated receptor gamma agonists are considered as a promising treatment through the action of this crosstalk (83). Troglitazones (anti-inflammatory drugs) can decrease c-Myc levels (84). Intestinal fibrosis presents an activation of the WNT/β-catenin pathway, and the use of PPARγ agonists can decrease it and diminish fibrosis formation (85). PPARγ agonists activate Dickkopf-1 (DKK1, a WNT inhibitor) activity to decrease the canonical WNT/β-catenin pathway and then inhibit the fibroblasts differentiation (86). In 3T3-L1 cells, the inhibition of the signal transducer protein kinase B (Akt) pathway leads to activation of PPARγ (87). The phosphatidylinositol 3 kinase/protein kinase B (PI3K/Akt) pathway acts by phosphorylating GSK-3β to negatively regulate PPARγ expression (88, 89). Furthermore, PPARγ agonists activate GSK-3β to decrease β-catenin expression (90). Conversely, the β-catenin signaling activates the Akt pathway and this leads to a decrease in PPARγ expression in adipocytes and 2T2-L1 preadipocytes (68, 91). PPARγ agonists downregulate the PI3K/Akt signaling pathway (92, 93) by stimulating PTEN activity in fibrotic process (59).
Numerous inflammatory cytokines, chemokines, or intracellular pathways, such as the canonical WNT/β-catenin signaling, TNF-α, interleukin (IL)-1, and IL-13, downregulate PPARγ expression (94–96). The transcription factor COUP II is a canonical WNT target and downregulates PPARγ expression (97). In adipocytes, adiponectin increases PPARγ expression and then downregulates the LPS-induced NFκB expression and IL-6 production (98). Mesenchymal stem cell differentiation also shows a crosstalk between the WNT pathway and PPARγ (70). Hepatic fatty acid metabolism, fatty acid oxidation, hepatic mitochondrial function, and energy balance are regulated by the interaction between the WNT/β-catenin pathway and PPARγ (62, 99, 100).
Crosstalk Between PPARγ and WNT/β-Catenin Signaling in Cancers
Even if, the molecular mechanisms by which TZDs regulate differentiation and stemness programs have been well studied in adipocytes and normal cells, in cancer cells, they still remain unclear (32). In normal cells, PPARγ suppresses tumorigenesis and WNT signaling by targeting phosphorylated β-catenin at the proteasome by a process involving its catenin-binding domain within PPARγ. In contrast, oncogenic β-catenin resists proteasomal degradation by inhibiting PPARγ activity, which requires its TCF/LEF-binding domain (62). In adipocytes, PPARγ increases differentiation and inhibits proliferation by affecting the WNT/β-catenin pathway. PPARγ interacts with GSK3-β to induce the differentiation factor C/EBPα and this leads to the production of adiponectin (101, 102). PPARγ activation reduces β-catenin at both the mRNA and protein levels to promote differentiation (103). In human metastatic prostate cancer LnCaP cells, PPARγ inhibits the WNT pathway by targeting phosphorylated β-catenin at the proteasome (62, 104). In gastric and colon cancer cells, PPARγ decreases β-catenin expression, subcellular localization, and downstream effectors, resulting in the modulation of several genes, such as telomerase reverse transcriptase, and Sox9, which are involved in cell development, differentiation, and survival processes (105–107). PPARγ agonists, by inhibiting activation of the WNT/β-catenin pathway, could be used in combination with other drugs such as inhibitors of tyrosine kinases (108), PI3K/AKT (109), and mitogen-activated protein kinase (MAPK) cascades to maximize the antitumor and pro-differentiating effect.
Carcinogenesis: Role of Chronic Inflammation and Oxidative Stress
Cancer progression is promoted by an environment rich in inflammatory factors, DNA damages, and genetic or epigenetic mutations (110).
Chronic Inflammation
Several studies have shown that prolonged inflammation leads to DNA damages and tissue injury (111). Chronic inflammation can affect cell homeostasis, metabolism, and genomic regulation, leading to the initiation of tumorigenesis (112). Furthermore, damages induced by chronic inflammation are responsible for the development of malignancy sites (113, 114).
The link between inflammation and cancer initiation has been examined in a recent study (115). Inflammation stimulates the activation of cytotoxic mediators, such as reactive oxygen species (ROS) and reactive nitrogen species (RNS), which have a major role in DNA damages (116). DNA damage accumulation is responsible for the initiation of carcinogenesis through the enhancement of genomic instabilities (117).
Pathogenic stimuli can stimulate inflammation and then eradicate the normal host defense response (118). Pathogens promote carcinogenesis through the recruitment of infections and the inhibition of immune response leading to chronic inflammation (3). Stomach, intestine, liver, colon, and skin are the main sites of common pathogenic infections that are believed to be related to cancer progression (119, 120).
The inflammatory response is regulated by the canonical WNT/β-catenin pathway (111). Moreover, infection pathogens can overexpress the WNT/β-catenin pathway leading to uncontrolled inflammation and then to an increased risk of carcinogenesis (121).
Several inflammatory factors can facilitate the migration and invasion of neoplastic cells (122). Tumor necrosis factor α (TNF-α), interleukin-6 (IL-6), vascular endothelial growth factor (VEGF), and tumor growth factor-β (TGF-β) are inflammatory factors involved in the regulation of the immune system (123). TGF-β and VEGF can suppress the immune response during cancer development (124). TNF-α overexpression induces DNA damage leading to tumor growth (122), angiogenesis, and invasion (125). TNF-α can stimulate other cytokines such as IL-17 to directly promote tumor growth (126). Then, IL-17 activates IL-6 and the signal transducer and activator transcription (STAT) signaling for invasion (127).
In parallel, chronic inflammation stimulates the expression of cyclooxygenase 2 (COX2, a prostaglandin-endoperoxidase synthase) (128). Numerous cytokines (TNF-α, IL-1) induce the activation of COX2 (122). COX2 is involved in the stimulation of ROS, and RNS intermediates found to be overexpressed during carcinogenesis processes (128, 129) and the production of prostaglandins leading to angiogenesis, anti-apoptosis, and metastasis (128, 130). Nuclear factor-kappaB (NFκB) activates numerous pro-inflammatory factors that induce COX2 and inducible nitric oxide synthase (iNOS) (112). NFκB is one of the main factors involved in inflammation in association with carcinogenesis (112, 131). NFκB activates the expression of TNF-α, IL-6, IL-8, COX2, BCL-2 (B-cell lymphoma 2), metalloproteinases (MMPs), and angiogenic factors such as VEGF (112), and ROS production (132).
The STAT3 pathway involved in metastasis, proliferation, and angiogenesis (133) is activated by VEGF and cytokines (IL-6). The STAT3 signaling appears over-activated in numerous cancers, such as colon, breast, stomach, prostate, skin, and head cancers (134). Moreover, iNOS, an enzyme which catalyzes nitric oxide (NO), is overexpressed during inflammation (135) and enhances p53 gene mutations (122).
Oxidative Stress
Oxidative stress is characterized by an imbalance between production and elimination of reactive metabolites and free radicals (ROS and RNS) (8, 136). ROS generation is caused by cell damages through nitration and oxidation of macromolecules, such as proteins, lipids, DNA, and RNA. The NADPH oxidase (NOX) enzyme enhances ROS through the oxidation of intracellular NADPH to NADP+. Then, the transfer of electrons through the mitochondrial membrane reduces molecular oxygen and produces the superoxide anion as a primary product. ROS production has a major role in several pathways and in changes of intracellular and extracellular environmental conditions (137).
Reactive oxygen species are produced by dysregulation of the mitochondrial respiratory chain (138). During carcinogenesis, in a positive feedback, DNA damage and genomic instability can favor ROS production (139). ROS production has been observed in several cancer cells, such as in case of brain (140), breast (141), rachis (142), stomach (143), liver (144), lung (145), skin (146), pancreas (147), and prostate (148) cancers.
Leukocytes during inflammation are recruited from the damage sites and this leads to an increased uptake of oxygen, which induces the release of ROS and subsequently its accumulation (6, 149).
Several redox-regulated transcription factors have a key role in the stimulation of pro-inflammatory mediators, such as NFκB, a signal transducer and activator of transcription (STAT), activator protein-1 (AP-1), and the hypoxia-inducible factors (HIF) (112). The oxidative stress-induced inflammation induces the production of COX2, iNOS, TNF-α, IL-6, and miRNAs (150). A vicious circle operates between inflammation and oxidative stress leading to carcinogenesis (129).
NADPH-oxidase (NOX) is stimulated by inflammation and leads to oxidative stress and alteration of nuclear signaling (151). ROS, activated by NOX, stimulate the canonical WNT/β-catenin pathway through oxidization and inactivation of the nucleoredoxin (a redox-sensitive regulator), resulting in tumor cell proliferation (112). ROS production leads to the activation of c-Myc (152), STAT (153), and PI3K/Akt (154) and the inactivation of PPARγ (155). ROS production activates Akt signaling through the inhibition of the phosphatase and tensin homolog deleted from chromosome (PTEN) (156, 157). The Akt pathway is involved in cellular metabolism and the promotion of cell survival (156, 157).
Interactions between the Canonical WNT/β-Catenin Pathway and Carcinogenesis
Interactions Between the Canonical WNT/β-Catenin Pathway and Inflammation
A positive crosstalk between WNT/β-catenin and NFκB has been reported recently (17). The overexpression of WNT/β-catenin results in the enhancement of IκB-α degradation and then NFκB transactivation (158) (Figure 2). Upregulation of the target gene, CRDBP, by stimulated β-catenin signaling leads to a stabilization of βTrCP mRNA (159). In colon cancer, overexpression of both βTrCP and CRD-BP is associated with the activation of the β-catenin signaling and NFκB, contributing to cell proliferation and metastasis (159, 160). In breast cancer, TLR3 stimulation activates β-catenin signaling simultaneously with activation of the NFκB pathway, in a synergistic manner (161). β-catenin and NFκB pathways act in together diffuse large B-cell lymphomas (162). The WNT/β-catenin pathway leads to an increase in COX expression, which then influences the inflammatory response (163). E-cadherin and GSK-3β are decreased in melanoma cells by the stimulated β-catenin signaling (164). Concomitant GSK-3β and E-cadherin inactivation with cytosolic β-catenin accumulation induces NFκB-dependent iNOS expression in hepatic cells (165). The WNT/β-catenin pathway modulates in a positive manner its downstream target TNFRSF19 in colon cancer, which activates the NFκB signaling (166). However, the synergistic effect between β-catenin and NFκB depends on both the TCF/LEF link and the context of the genes or cell types (167).
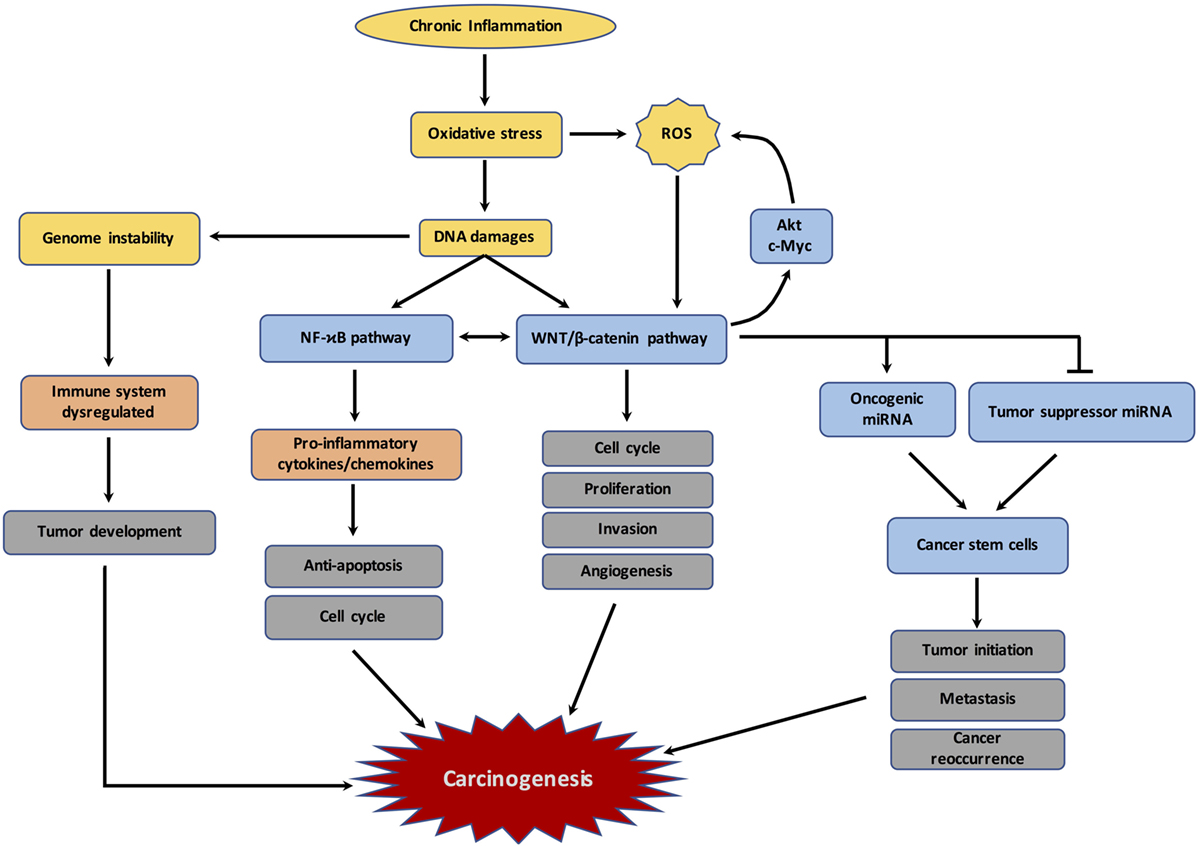
Figure 2. Schematic representation of the links between chronic inflammation, oxidative stress, DNA damages and carcinogenesis. The figure represents the different mechanisms involved by chronic inflammation and resulting in dysregulation of immune response, reactive oxygen species (ROS) production, DNA damages, and subsequently the initiation of carcinogenesis.
Nuclear factor-kappaB inhibits GSK-3β and positively regulates the β-catenin signaling (168, 169). Indeed, the activation of GSK-3β results in the inhibition of TNF-α-induced NFκB stimulation in carcinoma cells (168). IκB is stabilized by GSK-3β overexpression, which results in the inhibition of the NFκB pathway (169).
Nuclear factor-kappaB signaling can regulate WNT/β-catenin signaling through the use of IKKα (170) and RelA (171). IKKα can increase β-catenin signaling, whereas IKKβ downregulates β-catenin signaling (172). IKKα upregulates β-catenin/TCF/LEF activation and then the target gene cyclin D1 (173). GSK3-β and APC are degraded through the activation of IKKα leading to the cytosolic β-catenin accumulation (170). In glioma cells, overexpression of RelA coupled with the knockout of SN50, a NFκB inhibitor, increases the β-catenin nuclear translocation and then enhances β-catenin/TCF/LEF activity (174). The positive crosstalk between the WNT/β-catenin pathway and NFκB pathway participates in the regulation of several pathways involved in cancer development based on inflammation-induced carcinogenesis. This could be explained in part by the synergistic effect observed between β-catenin/TCF4 and NFκB on the overexpression of WNT target genes in colon cancer (171). This positive crosstalk induces several stem cell signature genes, such as Sox9, Ascl2, and Lgr5, leading to tumor growth.
Interactions Between the Canonical WNT/β-Catenin Pathway and ROS
Reactive oxygen species production activates the PI3K/Akt pathway, which is overactivated in numerous cancers (175). PTEN is a phosphoinositide-3-phosphatase, which downregulates the PI3K/Akt pathway (157). NADPH oxidase and superoxide dismutase oxidize PTEN and inactivate it. Then, the inhibition of PTEN activity by oxidative stress increases the activity of Akt and thus enhances the phosphorylation of GSK-3β by Akt. GSK-3β inactivated by Akt does not inhibit the nuclear β-catenin signaling, resulting in cell proliferation in several cancers. Alkylation of PTEN activates Akt and β-catenin (176). In addition, ROS causes the stabilization of HIF-1α and then the activation of the glycolytic enzymes participating in cell proliferation and angiogenesis (50, 175). The WNT/β-catenin pathway can activate HIF-1α by stimulating the PI3K/Akt pathway (15).
A recent study by Zhang et al. (177) has shown that ROS production activates the WNT/β-catenin pathway, but the mechanism involved remains unclear (177) (Figure 2). Furthermore, carcinogenesis of cells can increase the endogenous level of ROS production (175). Indeed, several oncogenes enhance ROS production, such as Akt (16) and c-Myc (178).
Genetic and Epigenetic Regulation of the Canonical WNT/β-Catenin Pathway in Cancers
Several genetic mutations lead to the aberrant activation of the canonical WNT/β-catenin pathway (14). In numerous malignant processes, the regulator genes of CTNNB1, AXIN, and APC have been observed to be mutated (179).
Mutations of CTNNB1, a β-catenin target, have been shown to be involved in the initiation of colon, gastric, ovarian, pancreatic, and prostate cancers, but also in melanoma and medulloblastoma (180, 181). APC mutations have been observed in colon cancer and AXIN mutations in hepatocellular carcinoma and medulloblastoma (182, 183).
Several studies have shown an interaction between miRNAs and the canonical WNT/β-catenin pathway, such as in osteoblast differentiation and cardiac and bone formation (184–186) (Figure 2). An aberrant expression of miR-374a is coupled with cytosolic β-catenin accumulation in breast cancers (187), and degradation of APC leading to the inactivation of the β-catenin destruction complex and enhancing the transcriptional activity of TCF/LEF (188). Moreover, the canonical WNT/β-catenin pathway controls the activity of several cancer-stem cell (CSC)-specific miRNAs. These specific miRNAs play a major role in tumor initiation. Overexpression of the WNT/β-catenin pathway leads to activation of the oncogenic miRNA expression to enhance the self-renewal potential of CSCs, which is involved in the resistance to drug therapy and initiation of new tumor growth (14).
Cancer-stem cells theory is characterized by the fact that cancer cells are derived from certain populations of cells, which possess stem cell properties (189–191). Several studies have shown that the WNT/β-catenin pathway can regulate stem cells and stem progenitors, plethora system maintenance, and cell self-renewal (37). Moreover, in a recent study, it was observed that the canonical WNT/β-catenin pathway plays a major role in the regulation of the activity of stem self-renewal in numerous cells (192).
The role of miRNAs in the regulation of CSCs is currently being investigated. Nevertheless, it has been observed that several miRNAs, such as miR410, can promote tumor growth, invasion, and migration of NSCLC cells through the activation of the canonical WNT/β-catenin pathway (193). miR-451 expression stimulates the upregulation of the macrophage migration inhibitory factor (MIF) and COX-2 expression to activate WNT/β-catenin pathway in CSCs (194, 195).
The interactions observed between WNT/β-catenin and miRNAs are involved in the regulation of tumorigenesis in numerous cancers, such as liver cancer (196), colon cancer (197, 198), brain cancer (199), and several other cancers (200–204).
On the other hand, other miRNAs appear to be tumor suppressors, such as miR-34a. miR-34a directly targets the tumor suppressor p53 and suppresses the expression of several target genes, such as SOX, Nanog, and N-Myc (205, 206). Let-7 is considered as a β-catenin negative regulator (207). Future studies will help us to better understand the role of miRNAs in the inhibition or activation of cancer initiation and its development through interaction with the WNT/β-catenin pathway by regulating the epithelial mesenchymal transition in cancer (208, 209).
Action of PPARγ Agonists in Cancers
Several studies have shown a significant reduction of PPARγ expression in cancers such as colon cancer (210, 211), gastric cancers (212), follicular thyroid cancer (213), cervical carcinoma (214), and esophageal cancer (215) (Figure 3). Numerous studies have shown that PPARγ has antineoplastic actions on lung, breast, prostate, and colon cancers (216, 217). NCOR, a repressor of PPARγ, has been found to increase in prostate cancer and to inhibit the expression of PPARγ (218). Several mutations of PPARγ are correlated with cancer initiation (219, 220). PPARγ agonists, such as rosiglitazone and troglitazone, are involved in cell cycle arrest, differentiation, proliferation, invasion, migration, apoptosis, inflammation, angiogenesis, and oxidative stress (19) (Table 1).
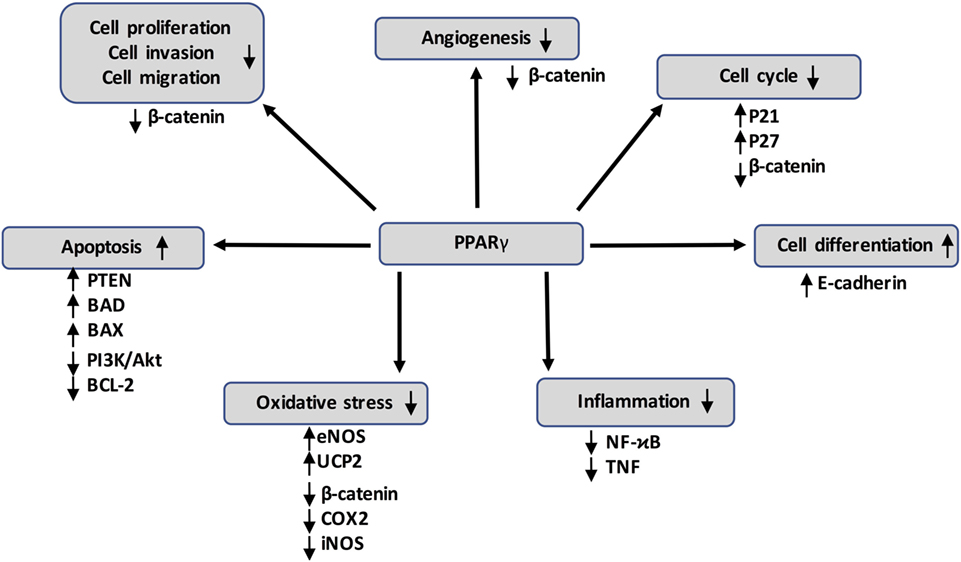
Figure 3. Peroxisome proliferator-activated receptor gamma (PPARγ) activation can inhibit cancer development through several mechanisms by acting on numerous target genes and pathways, such as the canonical WNT/β-catenin pathway. It also interferes with pro-inflammatory signaling by repressing nuclear factor-kappaB (NFκB) and tumor necrosis factor α (TNF-α).
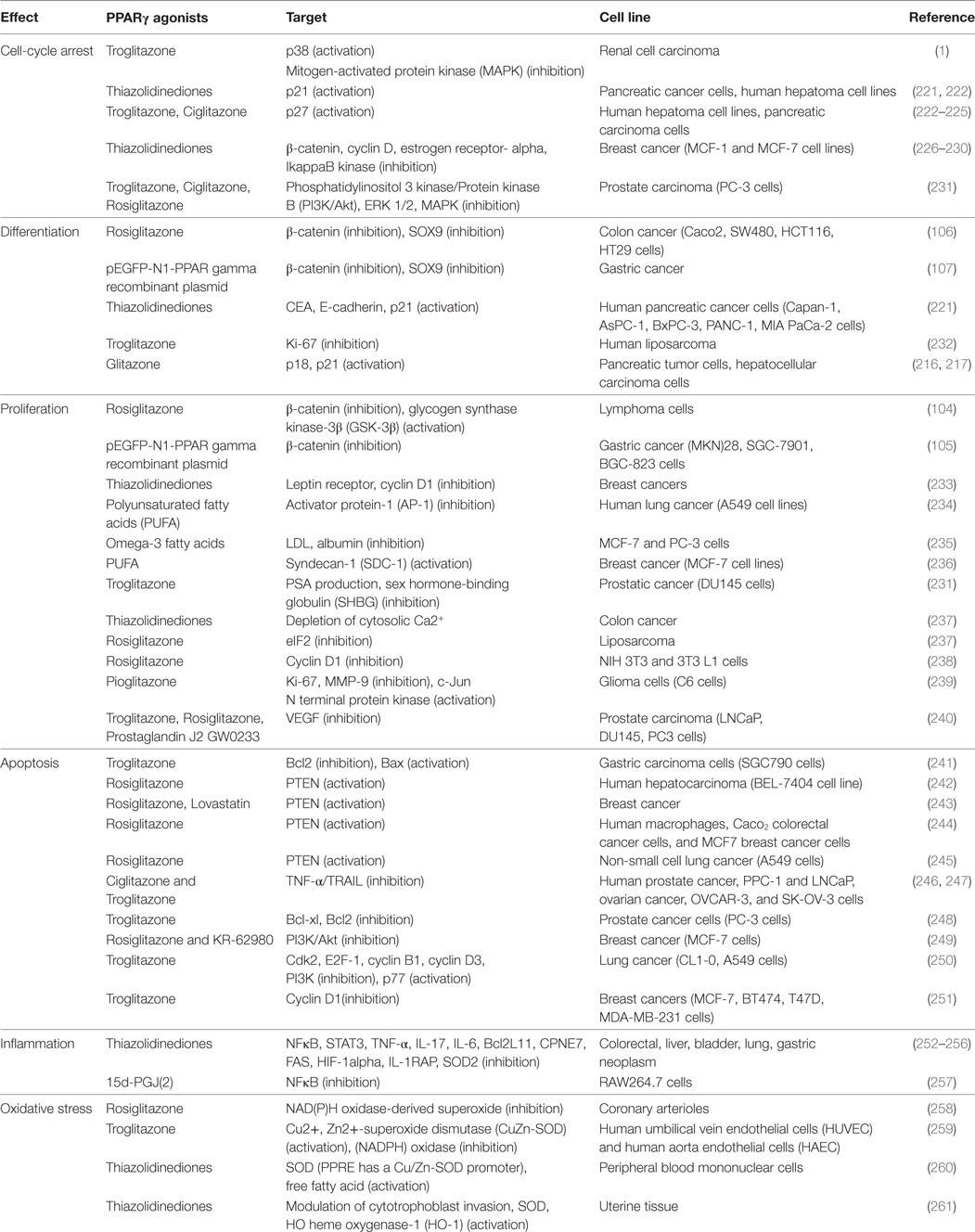
Table 1. Anti-tumoral effects of peroxisome proliferator-activated receptor gamma (PPARγ) agonists.
Cell Cycle Arrest
Peroxisome proliferator-activated receptor gamma agonists can induce G2/M cell cycle arrest through the stimulation of p38 MAPK in carcinoma (1) and in pancreatic cancer cells (221). PPARγ overexpression helps to stimulate the expression of cyclin-dependent kinase inhibitors p27 (222–224) and p21 (221, 222). PPARγ activation stops the cytosolic β-catenin accumulation and then decreases the expression of cyclin D1 (226–230).
Differentiation
Peroxisome proliferator-activated receptor gamma agonists can stimulate molecules involved in well-differentiated cells, such as E-cadherin, alkaline phosphatase, keratin, and carcinoembryonic antigen (CEA). This stimulation works in opposition to the non-differentiation of cells observed in cancers (219, 221, 262–264). PPARγ agonists are involved in the stimulation of terminal differentiation in cells (216–218).
Proliferation
Several studies have observed an anti-proliferative role played by TZDs (233, 265). Moreover, the PPARγ ligand docosahexaenoic acid (DHA) has been shown to have an anti-proliferative role in lung tumor cell cultures (234). In parallel, DHA also downregulates the cell proliferation and angiogenesis processes in breast cancer (231, 236). Rosiglitazone reduces the proliferation time of liposarcoma, but troglitazone has a limited effect in prostate, colon, and breast cancers (266–268).
Invasion and Migration
Thiazolidinediones downregulate tumor growth and the migration of tumor cells in colon cancer cells by inducing cell differentiation (264). In the same way, TZDs arrest cell cycle G1 with a decrease in E-cadherin expression (264). To date, few studies have revealed PPARγ agonists to play a positive role in the inhibition of invasion and the migration of cancer cells.
Apoptosis
The apoptotic process is stimulated by using a TZD in gastric cancer (241). PPARγ agonists can stimulate the expression of PTEN, a PI3K/Akt pathway inhibitor (242–245, 269), BAD, and BAX (239, 270). In the same way, PPARγ agonists can downregulate Bcl-2 expression (248) and PI3K/Akt pathway activity (249, 250).
Inflammation
Peroxisome proliferator-activated receptor gamma agonists, such as DHA and omega-3 fatty acids EPA, are known to induce anti-inflammatory activity (252, 253). Some natural and synthetic PPARγ agonists can have a chemoprotective role by targeting inflammatory agents (257, 271). PPARγ overexpression inhibits the activity of TNF-α and NFκB (257, 272) resulting in a reduction of tumor development (273). PPARγ activation seems to act on the tumor environment, especially on inflammation (19).
Angiogenesis
Peroxisome proliferator-activated receptor gamma agonists can modulate angiogenesis in vitro and in vivo models (274). However, some paradoxical effects of PPARγ agonists have also been observed. PPARγ agonists may enhance VEGF in tumor cells (275, 276) and may have pro- or anti-angiogenic roles depending on the cell environment (277–280).
Oxidative Stress
Superoxide dismutase (SOD) expression is regulated by PPARγ agonists, through a Cu/Zn-SOD promoter on the PPRE (260). Numerous studies have shown PPARγ to act as an antioxidant (258, 259). PPARγ acts on macrophages by reversing and uptaking the transport of cholesterol and then decreasing the oxidative stress initiation (281–283). PPARγ ligands promote antioxidant response through the stimulation of GPx3 (273), manganese SOD (MnSOD) (284), CD36 (a scavenger receptor) (285), endothelial oxide synthase (eNOS) (286), and UCP2 (mitochondrial uncoupling protein 2) (287). In parallel, PPARγ ligands can downregulate the prooxidant response by inhibiting COX2 and iNOS (288–290).
Conclusion
Cancers are readily associated with complex inflammatory phenomena and oxidative stress that may complicate or even initiate them. In cancers, apart from certain exceptions, the canonical WNT/β-catenin signaling is generally upregulated while PPARγ is downregulated. These two major cell pathways work in an opposite manner and this partly explains their unidirectional profile observed in cancers, chronic inflammation, and oxidative stress. This results in an activation of several upstream or downstream pathways involved in carcinogenesis, such as TGF-β, NFκB, TNF-α, TGF-β, IL-6, IL-8, VEGF, iNOS, PI3K/Akt, HIF-1α, and certain target genes such as c-Myc, cyclin D1, COX2, and Bcl2. The use of PPARγ agonists in cancers could reduce both ROS production and chronic inflammation leading to a decrease in the WNT/β-catenin pathway and then an inhibition of carcinogenesis processes (Figure 4). Because of the considerable impact of cancers and inflammatory processes on mortality and morbidity rates worldwide, it is imperative to continue to find new therapeutic pathways by seeking, directly or indirectly, to inhibit the canonical system WN/β-catenin and to activate PPARγ by new agonists free of deleterious effects.
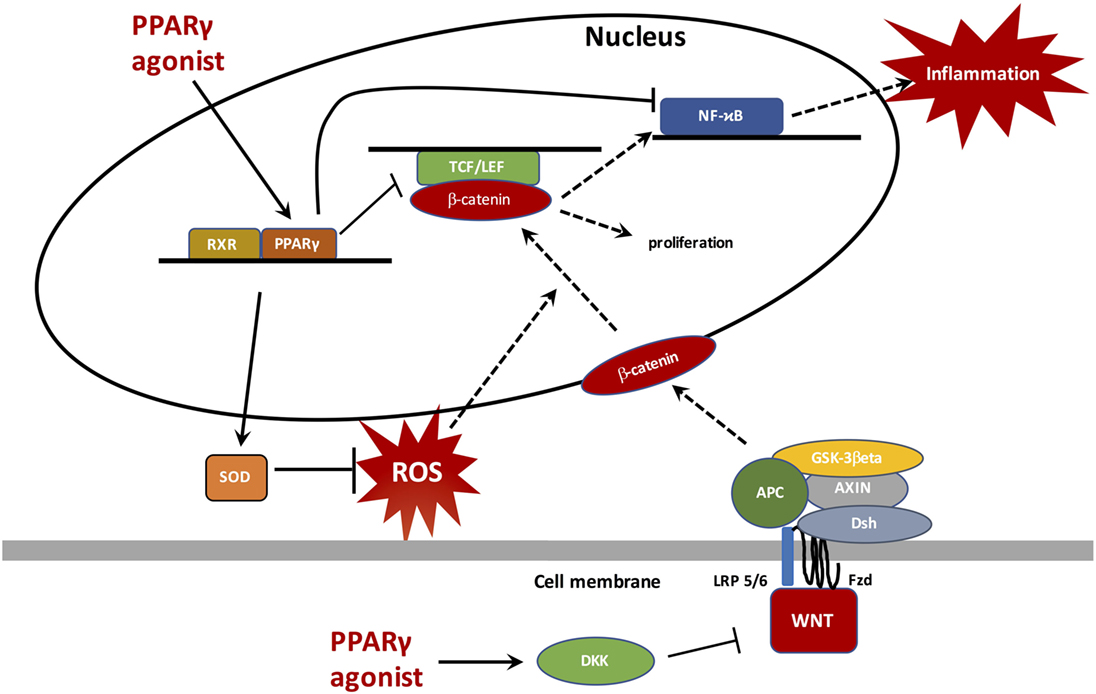
Figure 4. Peroxisome proliferator-activated receptor gamma (PPARγ) translocates to the nucleus to bind with retinoid X receptor (RXR) and then activates PPAR response elements (PPREs), such as superoxide dismutase (SOD) to inhibit reactive oxygen species (ROS) generation. ROS inhibition does not stimulate the β-catenin nuclear transcription and therefore does not activate proliferation processes and the NFκB pathway. PPARγ, through its anti-inflammatory role, inhibits NFκB and decreases inflammation. Through a catenin domain, PPARγ directly inhibits the TCF/LEF/β-catenin nuclear activity. In parallel, PPARγ can activate DKK, a WNT inhibitor.
Author Contributions
All listed authors have made substantial, direct, intellectual contributions to the study and have given their approval for its submission for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Mr. Brian Keogh, PhD, for the proofreading of the manuscript.
Abbreviations
APC, adenomatous polyposis coli; CK1, casein kinase 1; COX-2, cyclooxygenase-2; EMT, epithelial-mesenchymal transition; FZD, frizzled; GSK-3β, glycogen synthase kinase-3β; LRP 5/6, low-density lipoprotein receptor-related protein 5/6; NFκB, nuclear factor-kappaB; NOX, NADPH oxidase; PPARγ, peroxisome proliferator-activated receptor gamma; PI3K-Akt, phosphatidylinositol 3-kinase-protein kinase B; ROS, reactive oxygen species; SOD, superoxide dismutase; TCF/LEF, T-cell factor/lymphoid enhancer factor; TNF- α, tumor necrosis factor alpha; TZD, thiazolidinedione.
References
1. Fujita M, Yagami T, Fujio M, Tohji C, Takase K, Yamamoto Y, et al. Cytotoxicity of troglitazone through PPARγ-independent pathway and p38 MAPK pathway in renal cell carcinoma. Cancer Lett (2011) 312:219–27. doi:10.1016/j.canlet.2011.08.010
2. Bartsch H, Nair J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: role of lipid peroxidation, DNA damage, and repair. Langenbecks Arch Surg (2006) 391:499–510. doi:10.1007/s00423-006-0073-1
3. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140:883–99. doi:10.1016/j.cell.2010.01.025
4. Grivennikov SI, Karin M. Inflammation and oncogenesis: a vicious connection. Curr Opin Genet Dev (2010) 20:65–71. doi:10.1016/j.gde.2009.11.004
5. Gonda TA, Tu S, Wang TC. Chronic inflammation, the tumor microenvironment and carcinogenesis. Cell Cycle (2009) 8:2005–13. doi:10.4161/cc.8.13.8985
7. Mantovani A. Cancer: inflammation by remote control. Nature (2005) 435:752–3. doi:10.1038/435752a
8. Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med (2010) 49:1603–16. doi:10.1016/j.freeradbiomed.2010.09.006
9. Frenkel K. Carcinogen-mediated oxidant formation and oxidative DNA damage. Pharmacol Ther (1992) 53:127–66. doi:10.1016/0163-7258(92)90047-4
10. Shacter E, Beecham EJ, Covey JM, Kohn KW, Potter M. Activated neutrophils induce prolonged DNA damage in neighboring cells. Carcinogenesis (1988) 9:2297–304. doi:10.1093/carcin/9.12.2297
11. Jackson AL, Loeb LA. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat Res (2001) 477:7–21. doi:10.1016/S0027-5107(01)00091-4
12. Storz P. Reactive oxygen species in tumor progression. Front Biosci (2005) 10:1881–96. doi:10.2741/1667
13. Lecarpentier Y, Claes V, Vallée A, Hébert J-L. Thermodynamics in cancers: opposing interactions between PPAR gamma and the canonical WNT/beta-catenin pathway. Clin Transl Med (2017) 6:14. doi:10.1186/s40169-017-0144-7
14. Onyido EK, Sweeney E, Nateri AS. Wnt-signalling pathways and microRNAs network in carcinogenesis: experimental and bioinformatics approaches. Mol Cancer (2016) 15:56. doi:10.1186/s12943-016-0541-3
15. Vallée A, Guillevin R, Vallée J-N. Vasculogenesis and angiogenesis initiation under normoxic conditions through Wnt/β-catenin pathway in gliomas. Rev Neurosci (2017) 29(1):71–91. doi:10.1515/revneuro-2017-0032
16. Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A (2010) 107:8788–93. doi:10.1073/pnas.1003428107
17. Ma B, Hottiger MO. Crosstalk between Wnt/β-Catenin and NF-κB signaling pathway during inflammation. Front Immunol (2016) 7:378. doi:10.3389/fimmu.2016.00378
18. Peters JM, Shah YM, Gonzalez FJ. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat Rev Cancer (2012) 12:181–95. doi:10.1038/nrc3214
19. Bandera Merchan B, Tinahones FJ, Macías-González M. Commonalities in the association between PPARG and vitamin D related with obesity and carcinogenesis. PPAR Res (2016) 2016:2308249. doi:10.1155/2016/2308249
20. Lecarpentier Y, Claes V, Duthoit G, Hébert J-L. Circadian rhythms, Wnt/beta-catenin pathway and PPAR alpha/gamma profiles in diseases with primary or secondary cardiac dysfunction. Front Physiol (2014) 5:429. doi:10.3389/fphys.2014.00429
21. Zhu Y, Qi C, Korenberg JR, Chen XN, Noya D, Rao MS, et al. Structural organization of mouse peroxisome proliferator-activated receptor gamma (mPPAR gamma) gene: alternative promoter use and different splicing yield two mPPAR gamma isoforms. Proc Natl Acad Sci U S A (1995) 92:7921–5. doi:10.1073/pnas.92.17.7921
22. Fajas L, Fruchart JC, Auwerx J. PPARgamma3 mRNA: a distinct PPARgamma mRNA subtype transcribed from an independent promoter. FEBS Lett (1998) 438:55–60. doi:10.1016/S0014-5793(98)01273-3
23. Rogue A, Spire C, Brun M, Claude N, Guillouzo A. Gene expression changes induced by PPAR gamma agonists in animal and human liver. PPAR Res (2010) 2010:325183. doi:10.1155/2010/325183
24. Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs: from orphan receptors to drug discovery. J Med Chem (2000) 43:527–50. doi:10.1021/jm990554g
25. Schupp M, Cristancho AG, Lefterova MI, Hanniman EA, Briggs ER, Steger DJ, et al. Re-expression of GATA2 cooperates with peroxisome proliferator-activated receptor-gamma depletion to revert the adipocyte phenotype. J Biol Chem (2009) 284:9458–64. doi:10.1074/jbc.M809498200
26. Schupp M, Lazar MA. Endogenous ligands for nuclear receptors: digging deeper. J Biol Chem (2010) 285:40409–15. doi:10.1074/jbc.R110.182451
27. Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, et al. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell (1999) 4:585–95. doi:10.1016/S1097-2765(00)80209-9
28. Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell (1994) 79:1147–56. doi:10.1016/0092-8674(94)90006-X
29. Semple RK, Chatterjee VKK, O’Rahilly S. PPAR gamma and human metabolic disease. J Clin Invest (2006) 116:581–9. doi:10.1172/JCI28003
30. Ament Z, Masoodi M, Griffin JL. Applications of metabolomics for understanding the action of peroxisome proliferator-activated receptors (PPARs) in diabetes, obesity and cancer. Genome Med (2012) 4:32. doi:10.1186/gm331
31. Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell (2012) 149:1192–205. doi:10.1016/j.cell.2012.05.012
32. Novellasdemunt L, Antas P, Li VSW. Targeting Wnt signaling in colorectal cancer. A review in the theme: cell signaling: proteins, pathways and mechanisms. Am J Physiol Cell Physiol (2015) 309:C511–21. doi:10.1152/ajpcell.00117.2015
33. De P, Carlson JH, Wu H, Marcus A, Leyland-Jones B, Dey N. Wnt-beta-catenin pathway signals metastasis-associated tumor cell phenotypes in triple negative breast cancers. Oncotarget (2016) 7:43124–49. doi:10.18632/oncotarget.8988
34. Sheikh A, Niazi AK, Ahmed MZ, Iqbal B, Anwer SMS, Khan HH. The role of Wnt signaling pathway in carcinogenesis and implications for anticancer therapeutics. Hered Cancer Clin Pract (2014) 12:13. doi:10.1186/1897-4287-12-13
35. MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell (2009) 17:9–26. doi:10.1016/j.devcel.2009.06.016
36. Miller JR, Hocking AM, Brown JD, Moon RT. Mechanism and function of signal transduction by the Wnt/beta-catenin and Wnt/Ca2+ pathways. Oncogene (1999) 18:7860–72. doi:10.1038/sj.onc.1203245
37. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature (2005) 434:843–50. doi:10.1038/nature03319
38. Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer (2013) 13:11–26. doi:10.1038/nrc3419
39. Kim W, Kim M, Jho E. Wnt/β-catenin signalling: from plasma membrane to nucleus. Biochem J (2013) 450:9–21. doi:10.1042/BJ20121284
40. Rao TP, Kühl M. An updated overview on Wnt signaling pathways: a prelude for more. Circ Res (2010) 106:1798–806. doi:10.1161/CIRCRESAHA.110.219840
41. Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. J Cell Sci (2007) 120:3327–35. doi:10.1242/jcs.03485
42. Clevers H. Wnt/β-catenin signaling in development and disease. Cell (2006) 127:469–80. doi:10.1016/j.cell.2006.10.018
43. Zeng X, Huang H, Tamai K, Zhang X, Harada Y, Yokota C, et al. Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Devolepment (2008) 135:367–75. doi:10.1242/dev.013540
44. Nusse R. Wnt signaling. Cold Spring Harb Perspect Biol (2012) 4:a011163. doi:10.1101/cshperspect.a011163
45. Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol (2009) 10(7):468–77. doi:10.1038/nrm2717
46. ten Berge D, Kurek D, Blauwkamp T, Koole W, Maas A, Eroglu E, et al. Embryonic stem cells require Wnt proteins to prevent differentiation to epiblast stem cells. Nat Cell Biol (2011) 13:1070–5. doi:10.1038/ncb2314
47. Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol (2004) 20:781–810. doi:10.1146/annurev.cellbio.20.010403.113126
48. Pinto D, Gregorieff A, Begthel H, Clevers H. Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev (2003) 17:1709–13. doi:10.1101/gad.267103
49. Nateri AS, Spencer-Dene B, Behrens A. Interaction of phosphorylated c-Jun with TCF4 regulates intestinal cancer development. Nature (2005) 437:281–5. doi:10.1038/nature03914
50. Vallée A, Lecarpentier Y, Guillevin R, Vallée J-N. Thermodynamics in gliomas: interactions between the canonical WNT/beta-catenin pathway and PPAR gamma. Front Physiol (2017) 8:352. doi:10.3389/fphys.2017.00352
51. Vallée A, Lecarpentier Y, Guillevin R, Vallée J-N. Effects of Cannabidiol interactions with Wnt/β-catenin pathway and PPARγ on oxidative stress and neuroinflammation in Alzheimer’s disease. Acta Biochim Biophys Sin (2017) 49(10):853–66. doi:10.1093/abbs/gmx073
52. Vallée A, Lecarpentier Y. Alzheimer disease: crosstalk between the canonical Wnt/beta-catenin pathway and PPARs alpha and gamma. Front Neurosci (2016) 10:459. doi:10.3389/fnins.2016.00459
53. Lecarpentier Y, Vallée A. Opposite interplay between PPAR gamma and canonical Wnt/beta-catenin pathway in amyotrophic lateral sclerosis. Front Neurol (2016) 7:100. doi:10.3389/fneur.2016.00100
54. Vallée A, Lecarpentier Y, Guillevin R, Vallée J-N. Aerobic glycolysis in amyotrophic lateral sclerosis and Huntington’s disease. Rev Neurosci (2018). doi:10.1515/revneuro-2017-0075
55. Vallée A, Vallée J-N, Guillevin R, Lecarpentier Y. Interactions between the canonical WNT/beta-catenin pathway and PPAR gamma on neuroinflammation, demyelination, and remyelination in multiple sclerosis. Cell Mol Neurobiol (2018) 38:783–95. doi:10.1007/s10571-017-0550-9
56. Vallée A, Lecarpentier Y, Guillevin R, Vallée J-N. Aerobic glycolysis hypothesis through WNT/beta-catenin pathway in exudative age-related macular degeneration. J Mol Neurosci (2017) 62:368–79. doi:10.1007/s12031-017-0947-4
57. Vallée A, Lecarpentier Y, Guillevin R, Vallée J-N. PPARγ agonists: potential treatments for exudative age-related macular degeneration. Life Sci (2017) 188:123–30. doi:10.1016/j.lfs.2017.09.008
58. Vallée A, Lecarpentier Y, Vallée J-N. Thermodynamic aspects and reprogramming cellular energy metabolism during the fibrosis process. Int J Mol Sci (2017) 18:E2537. doi:10.3390/ijms18122537
59. Vallée A, Lecarpentier Y, Guillevin R, Vallée J-N. Interactions between TGF-β1, canonical WNT/β-catenin pathway and PPAR γ in radiation-induced fibrosis. Oncotarget (2017) 8:90579–604. doi:10.18632/oncotarget.21234
60. Lecarpentier Y, Schussler O, Claes V, Vallée A. The myofibroblast: TGFβ-1, a conductor which plays a key role in fibrosis by regulating the balance between PPARγ and the canonical WNT pathway. Nucl Recept Res (2017) 4:23. doi:10.11131/2017/101299
61. Sharma C, Pradeep A, Wong L, Rana A, Rana B. Peroxisome proliferator-activated receptor gamma activation can regulate beta-catenin levels via a proteasome-mediated and adenomatous polyposis coli-independent pathway. J Biol Chem (2004) 279:35583–94. doi:10.1074/jbc.M403143200
62. Liu J, Wang H, Zuo Y, Farmer SR. Functional interaction between peroxisome proliferator-activated receptor gamma and beta-catenin. Mol Cell Biol (2006) 26:5827–37. doi:10.1128/MCB.00441-06
63. Takada I, Kouzmenko AP, Kato S. Wnt and PPARgamma signaling in osteoblastogenesis and adipogenesis. Nat Rev Rheumatol (2009) 5:442–7. doi:10.1038/nrrheum.2009.137
64. Lu D, Carson DA. Repression of beta-catenin signaling by PPAR gamma ligands. Eur J Pharmacol (2010) 636:198–202. doi:10.1016/j.ejphar.2010.03.010
65. Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, et al. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest (2006) 116:2012–21. doi:10.1172/JCI27751
66. Elbrecht A, Chen Y, Cullinan CA, Hayes N, Leibowitz MD, Moller DE, et al. Molecular cloning, expression and characterization of human peroxisome proliferator activated receptors gamma 1 and gamma 2. Biochem Biophys Res Commun (1996) 224:431–7. doi:10.1006/bbrc.1996.1044
67. Fajas L, Auboeuf D, Raspé E, Schoonjans K, Lefebvre AM, Saladin R, et al. The organization, promoter analysis, and expression of the human PPARgamma gene. J Biol Chem (1997) 272:18779–89. doi:10.1074/jbc.272.30.18779
68. Moldes M, Zuo Y, Morrison RF, Silva D, Park B-H, Liu J, et al. Peroxisome-proliferator-activated receptor gamma suppresses Wnt/beta-catenin signalling during adipogenesis. Biochem J (2003) 376:607–13. doi:10.1042/BJ20030426
69. Farshbaf MJ, Ghaedi K, Shirani M, Nasr-Esfahani MH. Peroxisome proliferator activated receptor gamma (PPARγ) as a therapeutic target for improvement of cognitive performance in Fragile-X. Med Hypotheses (2014) 82:291–4. doi:10.1016/j.mehy.2013.12.012
70. Xu C, Wang J, Zhu T, Shen Y, Tang X, Fang L, et al. Cross-talking between PPAR and WNT signaling and its regulation in mesenchymal stem cell differentiation. Curr Stem Cell Res Ther (2016) 11:247–54. doi:10.2174/1574888X10666150723145707
71. Drygiannakis I, Valatas V, Sfakianaki O, Bourikas L, Manousou P, Kambas K, et al. Proinflammatory cytokines induce crosstalk between colonic epithelial cells and subepithelial myofibroblasts: implication in intestinal fibrosis. J Crohns Colitis (2013) 7:286–300. doi:10.1016/j.crohns.2012.04.008
72. Farmer SR. Regulation of PPARgamma activity during adipogenesis. Int J Obes (Lond) (2005) 29(Suppl 1):S13–6. doi:10.1038/sj.ijo.0802907
73. Jeon K-I, Kulkarni A, Woeller CF, Phipps RP, Sime PJ, Hindman HB, et al. Inhibitory effects of PPARγ ligands on TGF-β1-induced corneal myofibroblast transformation. Am J Pathol (2014) 184:1429–45. doi:10.1016/j.ajpath.2014.01.026
74. Kumar V, Mundra V, Mahato RI. Nanomedicines of hedgehog inhibitor and PPAR-γ agonist for treating liver fibrosis. Pharm Res (2014) 31:1158–69. doi:10.1007/s11095-013-1239-5
75. Lee Y, Kim SH, Lee YJ, Kang ES, Lee B-W, Cha BS, et al. Transcription factor Snail is a novel regulator of adipocyte differentiation via inhibiting the expression of peroxisome proliferator-activated receptor γ. Cell Mol Life Sci (2013) 70:3959–71. doi:10.1007/s00018-013-1363-8
76. Li Q, Yan Z, Li F, Lu W, Wang J, Guo C. The improving effects on hepatic fibrosis of interferon-γ liposomes targeted to hepatic stellate cells. Nanotechnology (2012) 23:265101. doi:10.1088/0957-4484/23/26/265101
77. Liu J, Farmer SR. Regulating the balance between peroxisome proliferator-activated receptor gamma and beta-catenin signaling during adipogenesis. A glycogen synthase kinase 3beta phosphorylation-defective mutant of beta-catenin inhibits expression of a subset of adipogenic genes. J Biol Chem (2004) 279:45020–7. doi:10.1074/jbc.M407050200
78. Qian J, Niu M, Zhai X, Zhou Q, Zhou Y. β-Catenin pathway is required for TGF-β1 inhibition of PPARγ expression in cultured hepatic stellate cells. Pharmacol Res (2012) 66:219–25. doi:10.1016/j.phrs.2012.06.003
79. Segel MJ, Izbicki G, Cohen PY, Or R, Christensen TG, Wallach-Dayan SB, et al. Role of interferon-gamma in the evolution of murine bleomycin lung fibrosis. Am J Physiol Lung Cell Mol Physiol (2003) 285:L1255–62. doi:10.1152/ajplung.00303.2002
80. Shim CY, Song B-W, Cha M-J, Hwang K-C, Park S, Hong G-R, et al. Combination of a peroxisome proliferator-activated receptor-gamma agonist and an angiotensin II receptor blocker attenuates myocardial fibrosis and dysfunction in type 2 diabetic rats. J Diabetes Investig (2014) 5:362–71. doi:10.1111/jdi.12153
81. Ajmone-Cat MA, D’Urso MC, di Blasio G, Brignone MS, De Simone R, Minghetti L. Glycogen synthase kinase 3 is part of the molecular machinery regulating the adaptive response to LPS stimulation in microglial cells. Brain Behav Immun (2016) 55:225–35. doi:10.1016/j.bbi.2015.11.012
82. Jansson EA, Are A, Greicius G, Kuo I-C, Kelly D, Arulampalam V, et al. The Wnt/beta-catenin signaling pathway targets PPARgamma activity in colon cancer cells. Proc Natl Acad Sci U S A (2005) 102:1460–5. doi:10.1073/pnas.0405928102
83. Sabatino L, Pancione M, Votino C, Colangelo T, Lupo A, Novellino E, et al. Emerging role of the β-catenin-PPARγ axis in the pathogenesis of colorectal cancer. World J Gastroenterol (2014) 20:7137–51. doi:10.3748/wjg.v20.i23.7137
84. Akinyeke TO, Stewart LV. Troglitazone suppresses c-Myc levels in human prostate cancer cells via a PPARγ-independent mechanism. Cancer Biol Ther (2011) 11:1046–58. doi:10.4161/cbt.11.12.15709
85. Di Gregorio J, Sferra R, Speca S, Vetuschi A, Dubuquoy C, Desreumaux P, et al. Role of glycogen synthase kinase-3β and PPAR-γ on epithelial-to-mesenchymal transition in DSS-induced colorectal fibrosis. PLoS One (2017) 12:e0171093. doi:10.1371/journal.pone.0171093
86. Gustafson B, Eliasson B, Smith U. Thiazolidinediones increase the wingless-type MMTV integration site family (WNT) inhibitor Dickkopf-1 in adipocytes: a link with osteogenesis. Diabetologia (2010) 53:536–40. doi:10.1007/s00125-009-1615-1
87. Park HJ, Yun J, Jang S-H, Kang SN, Jeon B-S, Ko Y-G, et al. Coprinus comatus cap inhibits adipocyte differentiation via regulation of PPARγ and Akt signaling pathway. PLoS One (2014) 9:e105809. doi:10.1371/journal.pone.0105809
88. Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol (2001) 65:391–426. doi:10.1016/S0301-0082(01)00011-9
89. Ross SE, Erickson RL, Hemati N, MacDougald OA. Glycogen synthase kinase 3 is an insulin-regulated C/EBPalpha kinase. Mol Cell Biol (1999) 19:8433–41. doi:10.1128/MCB.19.12.8433
90. Jeon M, Rahman N, Kim Y-S. Wnt/β-catenin signaling plays a distinct role in methyl gallate-mediated inhibition of adipogenesis. Biochem Biophys Res Commun (2016) 479:22–7. doi:10.1016/j.bbrc.2016.08.178
91. Huelsken J, Behrens J. The Wnt signalling pathway. J Cell Sci (2002) 115:3977–8. doi:10.1242/jcs.00089
92. Aljada A, O’Connor L, Fu Y-Y, Mousa SA. PPAR gamma ligands, rosiglitazone and pioglitazone, inhibit bFGF- and VEGF-mediated angiogenesis. Angiogenesis (2008) 11:361–7. doi:10.1007/s10456-008-9118-0
93. Goetze S, Eilers F, Bungenstock A, Kintscher U, Stawowy P, Blaschke F, et al. PPAR activators inhibit endothelial cell migration by targeting Akt. Biochem Biophys Res Commun (2002) 293:1431–7. doi:10.1016/S0006-291X(02)00385-6
94. Simon MF, Daviaud D, Pradère JP, Grès S, Guigné C, Wabitsch M, et al. Lysophosphatidic acid inhibits adipocyte differentiation via lysophosphatidic acid 1 receptor-dependent down-regulation of peroxisome proliferator-activated receptor gamma2. J Biol Chem (2005) 280:14656–62. doi:10.1074/jbc.M412585200
95. Tan JTM, McLennan SV, Song WW, Lo LW-Y, Bonner JG, Williams PF, et al. Connective tissue growth factor inhibits adipocyte differentiation. Am J Physiol Cell Physiol (2008) 295:C740–51. doi:10.1152/ajpcell.00333.2007
96. Yamasaki S, Nakashima T, Kawakami A, Miyashita T, Tanaka F, Ida H, et al. Cytokines regulate fibroblast-like synovial cell differentiation to adipocyte-like cells. Rheumatology (Oxford) (2004) 43:448–52. doi:10.1093/rheumatology/keh092
97. Okamura M, Kudo H, Wakabayashi K, Tanaka T, Nonaka A, Uchida A, et al. COUP-TFII acts downstream of Wnt/beta-catenin signal to silence PPARgamma gene expression and repress adipogenesis. Proc Natl Acad Sci U S A (2009) 106:5819–24. doi:10.1073/pnas.0901676106
98. Ajuwon KM, Spurlock ME. Adiponectin inhibits LPS-induced NF-kappaB activation and IL-6 production and increases PPARgamma2 expression in adipocytes. Am J Physiol Regul Integr Comp Physiol (2005) 288:R1220–5. doi:10.1152/ajpregu.00397.2004
99. Gebhardt R, Hovhannisyan A. Organ patterning in the adult stage: the role of Wnt/beta-catenin signaling in liver zonation and beyond. Dev Dyn (2010) 239:45–55. doi:10.1002/dvdy.22041
100. Lehwald N, Tao G-Z, Jang KY, Papandreou I, Liu B, Liu B, et al. β-Catenin regulates hepatic mitochondrial function and energy balance in mice. Gastroenterology (2012) 143:754–64. doi:10.1053/j.gastro.2012.05.048
101. Ohta T, Elnemr A, Yamamoto M, Ninomiya I, Fushida S, Nishimura G-I, et al. Thiazolidinedione, a peroxisome proliferator-activated receptor-gamma ligand, modulates the E-cadherin/beta-catenin system in a human pancreatic cancer cell line, BxPC-3. Int J Oncol (2002) 21:37–42.
102. Piwien-Pilipuk G, Van Mater D, Ross SE, MacDougald OA, Schwartz J. Growth hormone regulates phosphorylation and function of CCAAT/enhancer-binding protein beta by modulating Akt and glycogen synthase kinase-3. J Biol Chem (2001) 276:19664–71. doi:10.1074/jbc.M010193200
103. Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL, et al. Inhibition of adipogenesis by Wnt signaling. Science (2000) 289:950–3. doi:10.1126/science.289.5481.950
104. Liu J-J, Dai X-J, Xu Y, Liu P-Q, Zhang Y, Liu X-D, et al. Inhibition of lymphoma cell proliferation by peroxisomal proliferator-activated receptor-γ ligands via Wnt signaling pathway. Cell Biochem Biophys (2012) 62:19–27. doi:10.1007/s12013-011-9253-x
105. Guo F, Ren X, Dong Y, Hu X, Xu D, Zhou H, et al. Constitutive expression of PPARγ inhibits proliferation and migration of gastric cancer cells and down-regulates Wnt/β-catenin signaling pathway downstream target genes TERT and ENAH. Gene (2016) 584:31–7. doi:10.1016/j.gene.2016.03.003
106. Panza A, Pazienza V, Ripoli M, Benegiamo G, Gentile A, Valvano MR, et al. Interplay between SOX9, β-catenin and PPARγ activation in colorectal cancer. Biochim Biophys Acta (2013) 1833:1853–65. doi:10.1016/j.bbamcr.2013.04.004
107. Ren X, Zheng D, Guo F, Liu J, Zhang B, Li H, et al. PPARγ suppressed Wnt/β-catenin signaling pathway and its downstream effector SOX9 expression in gastric cancer cells. Med Oncol (2015) 32:91. doi:10.1007/s12032-015-0536-8
108. Prost S, Relouzat F, Spentchian M, Ouzegdouh Y, Saliba J, Massonnet G, et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARγ agonists. Nature (2015) 525:380–3. doi:10.1038/nature15248
109. Liu L, Yang Z, Xu Y, Li J, Xu D, Zhang L, et al. Inhibition of oxidative stress-elicited AKT activation facilitates PPARγ agonist-mediated inhibition of stem cell character and tumor growth of liver cancer cells. PLoS One (2013) 8:e73038. doi:10.1371/journal.pone.0073038
110. Meira LB, Bugni JM, Green SL, Lee C-W, Pang B, Borenshtein D, et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest (2008) 118:2516–25. doi:10.1172/JCI35073
111. Anuja K, Roy S, Ghosh C, Gupta P, Bhattacharjee S, Banerjee B. Prolonged inflammatory microenvironment is crucial for pro-neoplastic growth and genome instability: a detailed review. Inflamm Res (2017) 66:119–28. doi:10.1007/s00011-016-0985-3
112. Wu Y, Antony S, Meitzler JL, Doroshow JH. Molecular mechanisms underlying chronic inflammation-associated cancers. Cancer Lett (2014) 345:164–73. doi:10.1016/j.canlet.2013.08.014
113. Kundu JK, Surh Y-J. Inflammation: gearing the journey to cancer. Mutat Res (2008) 659:15–30. doi:10.1016/j.mrrev.2008.03.002
114. Clevers H. At the crossroads of inflammation and cancer. Cell (2004) 118:671–4. doi:10.1016/j.cell.2004.09.005
116. Roessner A, Kuester D, Malfertheiner P, Schneider-Stock R. Oxidative stress in ulcerative colitis-associated carcinogenesis. Pathol Res Pract (2008) 204:511–24. doi:10.1016/j.prp.2008.04.011
117. Rouse J, Jackson SP. Interfaces between the detection, signaling, and repair of DNA damage. Science (2002) 297:547–51. doi:10.1126/science.1074740
118. Medzhitov R, Janeway C. Innate immunity. N Engl J Med (2000) 343:338–44. doi:10.1056/NEJM200008033430506
119. Itzkowitz SH, Yio X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol (2004) 287:G7–17. doi:10.1152/ajpgi.00079.2004
120. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin (2005) 55:74–108. doi:10.3322/canjclin.55.2.74
121. Padhi S, Saha A, Kar M, Ghosh C, Adhya A, Baisakh M, et al. Clinico-pathological correlation of β-catenin and telomere dysfunction in head and neck squamous cell carcinoma patients. J Cancer (2015) 6:192–202. doi:10.7150/jca.9558
122. Lu H, Ouyang W, Huang C. Inflammation, a key event in cancer development. Mol Cancer Res (2006) 4:221–33. doi:10.1158/1541-7786.MCR-05-0261
123. Lin W-W, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest (2007) 117:1175–83. doi:10.1172/JCI31537
124. Smyth MJ, Cretney E, Kershaw MH, Hayakawa Y. Cytokines in cancer immunity and immunotherapy. Immunol Rev (2004) 202:275–93. doi:10.1111/j.0105-2896.2004.00199.x
125. Szlosarek P, Charles KA, Balkwill FR. Tumour necrosis factor-alpha as a tumour promoter. Eur J Cancer (2006) 42:745–50. doi:10.1016/j.ejca.2006.01.012
126. Charles KA, Kulbe H, Soper R, Escorcio-Correia M, Lawrence T, Schultheis A, et al. The tumor-promoting actions of TNF-alpha involve TNFR1 and IL-17 in ovarian cancer in mice and humans. J Clin Invest (2009) 119:3011–23. doi:10.1172/JCI39065
127. Tang Q, Li J, Zhu H, Li P, Zou Z, Xiao Y. Hmgb1-IL-23-IL-17-IL-6-Stat3 axis promotes tumor growth in murine models of melanoma. Mediators Inflamm (2013) 2013:713859. doi:10.1155/2013/713859
128. Sobolewski C, Cerella C, Dicato M, Ghibelli L, Diederich M. The role of cyclooxygenase-2 in cell proliferation and cell death in human malignancies. Int J Cell Biol (2010) 2010:215158. doi:10.1155/2010/215158
129. Federico A, Morgillo F, Tuccillo C, Ciardiello F, Loguercio C. Chronic inflammation and oxidative stress in human carcinogenesis. Int J Cancer (2007) 121:2381–6. doi:10.1002/ijc.23192
130. Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol (2011) 31:986–1000. doi:10.1161/ATVBAHA.110.207449
131. Dvorak HF. Rous-Whipple Award Lecture. How tumors make bad blood vessels and stroma. Am J Pathol (2003) 162:1747–57.
132. Bubici C, Papa S, Pham CG, Zazzeroni F, Franzoso G. The NF-kappaB-mediated control of ROS and JNK signaling. Histol Histopathol (2006) 21:69–80. doi:10.14670/HH-21.69
133. Herrero R, Castellsagué X, Pawlita M, Lissowska J, Kee F, Balaram P, et al. Human papillomavirus and oral cancer: the international agency for research on cancer multicenter study. J Natl Cancer Inst (2003) 95:1772–83. doi:10.1093/jnci/djg107
134. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer (2014) 14:736–46. doi:10.1038/nrc3818
135. Kim Y-H, Woo KJ, Lim JH, Kim S, Lee TJ, Jung EM, et al. 8-Hydroxyquinoline inhibits iNOS expression and nitric oxide production by down-regulating LPS-induced activity of NF-kappaB and C/EBPbeta in Raw 264.7 cells. Biochem Biophys Res Commun (2005) 329:591–7. doi:10.1016/j.bbrc.2005.01.159
137. Jabs T. Reactive oxygen intermediates as mediators of programmed cell death in plants and animals. Biochem Pharmacol (1999) 57:231–45. doi:10.1016/S0006-2952(98)00227-5
138. Poyton RO, Ball KA, Castello PR. Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinol Metab (2009) 20:332–40. doi:10.1016/j.tem.2009.04.001
139. Visconti R, Grieco D. New insights on oxidative stress in cancer. Curr Opin Drug Discov Devel (2009) 12:240–5.
140. Salganik RI, Albright CD, Rodgers J, Kim J, Zeisel SH, Sivashinskiy MS, et al. Dietary antioxidant depletion: enhancement of tumor apoptosis and inhibition of brain tumor growth in transgenic mice. Carcinogenesis (2000) 21:909–14. doi:10.1093/carcin/21.5.909
141. Brown NS, Bicknell R. Hypoxia and oxidative stress in breast cancer. Oxidative stress: its effects on the growth, metastatic potential and response to therapy of breast cancer. Breast Cancer Res (2001) 3:323–7. doi:10.1186/bcr315
142. Sharma A, Rajappa M, Satyam A, Sharma M. Oxidant/anti-oxidant dynamics in patients with advanced cervical cancer: correlation with treatment response. Mol Cell Biochem (2010) 341:65–72. doi:10.1007/s11010-010-0437-2
143. Oliveira CPMS, Kassab P, Lopasso FP, Souza HP, Janiszewski M, Laurindo FRM, et al. Protective effect of ascorbic acid in experimental gastric cancer: reduction of oxidative stress. World J Gastroenterol (2003) 9:446–8. doi:10.3748/wjg.v9.i3.446
144. Calvisi DF, Ladu S, Hironaka K, Factor VM, Thorgeirsson SS. Vitamin E down-modulates iNOS and NADPH oxidase in c-Myc/TGF-alpha transgenic mouse model of liver cancer. J Hepatol (2004) 41:815–22. doi:10.1016/j.jhep.2004.07.030
145. Azad N, Rojanasakul Y, Vallyathan V. Inflammation and lung cancer: roles of reactive oxygen/nitrogen species. J Toxicol Environ Health B Crit Rev (2008) 11:1–15. doi:10.1080/10937400701436460
146. Fruehauf JP, Trapp V. Reactive oxygen species: an Achilles’ heel of melanoma? Expert Rev Anticancer Ther (2008) 8:1751–7. doi:10.1586/14737140.8.11.1751
147. Edderkaoui M, Hong P, Vaquero EC, Lee JK, Fischer L, Friess H, et al. Extracellular matrix stimulates reactive oxygen species production and increases pancreatic cancer cell survival through 5-lipoxygenase and NADPH oxidase. Am J Physiol Gastrointest Liver Physiol (2005) 289:G1137–47. doi:10.1152/ajpgi.00197.2005
148. Khandrika L, Kumar B, Koul S, Maroni P, Koul HK. Oxidative stress in prostate cancer. Cancer Lett (2009) 282:125–36. doi:10.1016/j.canlet.2008.12.011
149. Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer (2003) 3:276–85. doi:10.1038/nrc1046
150. Hussain SP, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer (2007) 121:2373–80. doi:10.1002/ijc.23173
151. Weyemi U, Lagente-Chevallier O, Boufraqech M, Prenois F, Courtin F, Caillou B, et al. ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene (2012) 31:1117–29. doi:10.1038/onc.2011.327
152. Park J-H, Mangal D, Frey AJ, Harvey RG, Blair IA, Penning TM. Aryl hydrocarbon receptor facilitates DNA strand breaks and 8-oxo-2’-deoxyguanosine formation by the aldo-keto reductase product benzo[a]pyrene-7,8-dione. J Biol Chem (2009) 284:29725–34. doi:10.1074/jbc.M109.042143
153. Simon AR, Rai U, Fanburg BL, Cochran BH. Activation of the JAK-STAT pathway by reactive oxygen species. Am J Physiol (1998) 275:C1640–52. doi:10.1152/ajpcell.1998.275.6.C1640
154. Clerkin JS, Naughton R, Quiney C, Cotter TG. Mechanisms of ROS modulated cell survival during carcinogenesis. Cancer Lett (2008) 266:30–6. doi:10.1016/j.canlet.2008.02.029
155. Rusyn I, Rose ML, Bojes HK, Thurman RG. Novel role of oxidants in the molecular mechanism of action of peroxisome proliferators. Antioxid Redox Signal (2000) 2:607–21. doi:10.1089/15230860050192350
156. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell (2007) 129:1261–74. doi:10.1016/j.cell.2007.06.009
157. Plas DR, Thompson CB. Akt-dependent transformation: there is more to growth than just surviving. Oncogene (2005) 24:7435–42. doi:10.1038/sj.onc.1209097
158. Spiegelman VS, Slaga TJ, Pagano M, Minamoto T, Ronai Z, Fuchs SY. Wnt/beta-catenin signaling induces the expression and activity of betaTrCP ubiquitin ligase receptor. Mol Cell (2000) 5:877–82. doi:10.1016/S1097-2765(00)80327-5
159. Noubissi FK, Elcheva I, Bhatia N, Shakoori A, Ougolkov A, Liu J, et al. CRD-BP mediates stabilization of betaTrCP1 and c-myc mRNA in response to beta-catenin signalling. Nature (2006) 441:898–901. doi:10.1038/nature04839
160. Ougolkov A, Zhang B, Yamashita K, Bilim V, Mai M, Fuchs SY, et al. Associations among beta-TrCP, an E3 ubiquitin ligase receptor, beta-catenin, and NF-kappaB in colorectal cancer. J Natl Cancer Inst (2004) 96:1161–70. doi:10.1093/jnci/djh219
161. Jia D, Yang W, Li L, Liu H, Tan Y, Ooi S, et al. β-Catenin and NF-κB co-activation triggered by TLR3 stimulation facilitates stem cell-like phenotypes in breast cancer. Cell Death Differ (2015) 22:298–310. doi:10.1038/cdd.2014.145
162. Bognar MK, Vincendeau M, Erdmann T, Seeholzer T, Grau M, Linnemann JR, et al. Oncogenic CARMA1 couples NF-κB and β-catenin signaling in diffuse large B-cell lymphomas. Oncogene (2016) 35:4269–81. doi:10.1038/onc.2015.493
163. Phelps RA, Broadbent TJ, Stafforini DM, Jones DA. New perspectives on APC control of cell fate and proliferation in colorectal cancer. Cell Cycle (2009) 8:2549–56. doi:10.4161/cc.8.16.9278
164. Kuphal S, Poser I, Jobin C, Hellerbrand C, Bosserhoff AK. Loss of E-cadherin leads to upregulation of NFkappaB activity in malignant melanoma. Oncogene (2004) 23:8509–19. doi:10.1038/sj.onc.1207831
165. Bandino A, Compagnone A, Bravoco V, Cravanzola C, Lomartire A, Rossetto C, et al. Beta-catenin triggers nuclear factor kappaB-dependent up-regulation of hepatocyte inducible nitric oxide synthase. Int J Biochem Cell Biol (2008) 40:1861–71. doi:10.1016/j.biocel.2008.01.029
166. Schön S, Flierman I, Ofner A, Stahringer A, Holdt LM, Kolligs FT, et al. β-catenin regulates NF-κB activity via TNFRSF19 in colorectal cancer cells. Int J Cancer (2014) 135:1800–11. doi:10.1002/ijc.28839
167. Ma B, Zhong L, van Blitterswijk CA, Post JN, Karperien M. T cell factor 4 is a pro-catabolic and apoptotic factor in human articular chondrocytes by potentiating nuclear factor κB signaling. J Biol Chem (2013) 288:17552–8. doi:10.1074/jbc.M113.453985
168. Saegusa M, Hashimura M, Kuwata T, Hamano M, Okayasu I. Crosstalk between NF-kappaB/p65 and beta-catenin/TCF4/p300 signalling pathways through alterations in GSK-3beta expression during trans-differentiation of endometrial carcinoma cells. J Pathol (2007) 213:35–45. doi:10.1002/path.2198
169. Buss H, Dörrie A, Schmitz ML, Frank R, Livingstone M, Resch K, et al. Phosphorylation of serine 468 by GSK-3beta negatively regulates basal p65 NF-kappaB activity. J Biol Chem (2004) 279:49571–4. doi:10.1074/jbc.C400442200
170. Carayol N, Wang C-Y. IKKalpha stabilizes cytosolic beta-catenin by inhibiting both canonical and non-canonical degradation pathways. Cell Signal (2006) 18:1941–6. doi:10.1016/j.cellsig.2006.02.014
171. Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Göktuna SI, Ziegler PK, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell (2013) 152:25–38. doi:10.1016/j.cell.2012.12.012
172. Lamberti C, Lin KM, Yamamoto Y, Verma U, Verma IM, Byers S, et al. Regulation of beta-catenin function by the IkappaB kinases. J Biol Chem (2001) 276:42276–86. doi:10.1074/jbc.M104227200
173. Albanese C, Wu K, D’Amico M, Jarrett C, Joyce D, Hughes J, et al. IKKalpha regulates mitogenic signaling through transcriptional induction of cyclin D1 via Tcf. Mol Biol Cell (2003) 14:585–99. doi:10.1091/mbc.02-06-0101
174. Cho HH, Song JS, Yu JM, Yu SS, Choi SJ, Kim DH, et al. Differential effect of NF-kappaB activity on beta-catenin/Tcf pathway in various cancer cells. FEBS Lett (2008) 582:616–22. doi:10.1016/j.febslet.2008.01.029
175. Jiao R, Liu Y, Gao H, Xiao J, So KF. The anti-oxidant and antitumor properties of plant polysaccharides. Am J Chin Med (2016) 44:463–88. doi:10.1142/S0192415X16500269
176. Covey TM, Edes K, Coombs GS, Virshup DM, Fitzpatrick FA. Alkylation of the tumor suppressor PTEN activates Akt and β-catenin signaling: a mechanism linking inflammation and oxidative stress with cancer. PLoS One (2010) 5:e13545. doi:10.1371/journal.pone.0013545
177. Zhang D-Y, Pan Y, Zhang C, Yan B-X, Yu S-S, Wu D-L, et al. Wnt/β-catenin signaling induces the aging of mesenchymal stem cells through promoting the ROS production. Mol Cell Biochem (2013) 374:13–20. doi:10.1007/s11010-012-1498-1
178. Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, et al. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell (2002) 9:1031–44. doi:10.1016/S1097-2765(02)00520-8
180. Lee S-Y, Saito T, Mitomi H, Hidaka Y, Murakami T, Nomura R, et al. Mutation spectrum in the Wnt/β-catenin signaling pathway in gastric fundic gland-associated neoplasms/polyps. Virchows Arch (2015) 467:27–38. doi:10.1007/s00428-015-1753-4
181. Abdelmaksoud-Damak R, Miladi-Abdennadher I, Triki M, Khabir A, Charfi S, Ayadi L, et al. Expression and mutation pattern of β-catenin and adenomatous polyposis coli in colorectal cancer patients. Arch Med Res (2015) 46:54–62. doi:10.1016/j.arcmed.2015.01.001
182. Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet (2000) 24:245–50. doi:10.1038/73448
183. da Silva R, Marie SKN, Uno M, Matushita H, Wakamatsu A, Rosemberg S, et al. CTNNB1, AXIN1 and APC expression analysis of different medulloblastoma variants. Clinics (Sao Paulo) (2013) 68:167–72. doi:10.6061/clinics/2013(02)OA08
184. Lin GL, Hankenson KD. Integration of BMP, Wnt, and notch signaling pathways in osteoblast differentiation. J Cell Biochem (2011) 112:3491–501. doi:10.1002/jcb.23287
185. Vimalraj S, Selvamurugan N. MicroRNAs: synthesis, gene regulation and osteoblast differentiation. Curr Issues Mol Biol (2013) 15:7–18.
186. Olson EN. Gene regulatory networks in the evolution and development of the heart. Science (2006) 313:1922–7. doi:10.1126/science.1132292
187. Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res (1992) 52:1399–405.
188. He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, et al. A microRNA polycistron as a potential human oncogene. Nature (2005) 435:828–33. doi:10.1038/nature03552
189. Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer (2013) 13:727–38. doi:10.1038/nrc3597
190. Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer (2012) 12:133–43. doi:10.1038/nrc3184
191. Ibrahim EE, Babaei-Jadidi R, Saadeddin A, Spencer-Dene B, Hossaini S, Abuzinadah M, et al. Embryonic NANOG activity defines colorectal cancer stem cells and modulates through AP1- and TCF-dependent mechanisms. Stem Cells (2012) 30:2076–87. doi:10.1002/stem.1182
192. Yang K, Wang X, Zhang H, Wang Z, Nan G, Li Y, et al. The evolving roles of canonical WNT signaling in stem cells and tumorigenesis: implications in targeted cancer therapies. Lab Invest (2016) 96:116–36. doi:10.1038/labinvest.2015.144
193. Zhang X, Ke X, Pu Q, Yuan Y, Yang W, Luo X, et al. MicroRNA-410 acts as oncogene in NSCLC through downregulating SLC34A2 via activating Wnt/β-catenin pathway. Oncotarget (2016) 7:14569–85. doi:10.18632/oncotarget.7538
194. Bitarte N, Bandres E, Boni V, Zarate R, Rodriguez J, Gonzalez-Huarriz M, et al. MicroRNA-451 is involved in the self-renewal, tumorigenicity, and chemoresistance of colorectal cancer stem cells. Stem Cells (2011) 29:1661–71. doi:10.1002/stem.741
195. de Sousa EMF, Vermeulen L, Richel D, Medema JP. Targeting Wnt signaling in colon cancer stem cells. Clin Cancer Res (2011) 17:647–53. doi:10.1158/1078-0432.CCR-10-1204
196. Wang X, Chen J, Li F, Lin Y, Zhang X, Lv Z, et al. MiR-214 inhibits cell growth in hepatocellular carcinoma through suppression of β-catenin. Biochem Biophys Res Commun (2012) 428:525–31. doi:10.1016/j.bbrc.2012.10.039
197. Kim NH, Kim HS, Kim N-G, Lee I, Choi H-S, Li X-Y, et al. p53 and microRNA-34 are suppressors of canonical Wnt signaling. Sci Signal (2011) 4:ra71. doi:10.1126/scisignal.2001744
198. Kim NH, Cha YH, Kang SE, Lee Y, Lee I, Cha SY, et al. p53 regulates nuclear GSK-3 levels through miR-34-mediated Axin2 suppression in colorectal cancer cells. Cell Cycle (2013) 12:1578–87. doi:10.4161/cc.24739
199. Saydam O, Shen Y, Würdinger T, Senol O, Boke E, James MF, et al. Downregulated microRNA-200a in meningiomas promotes tumor growth by reducing E-cadherin and activating the Wnt/beta-catenin signaling pathway. Mol Cell Biol (2009) 29:5923–40. doi:10.1128/MCB.00332-09
200. Williams Z, Ben-Dov IZ, Elias R, Mihailovic A, Brown M, Rosenwaks Z, et al. Comprehensive profiling of circulating microRNA via small RNA sequencing of cDNA libraries reveals biomarker potential and limitations. Proc Natl Acad Sci U S A (2013) 110:4255–60. doi:10.1073/pnas.1214046110
201. Du WW, Fang L, Li M, Yang X, Liang Y, Peng C, et al. MicroRNA miR-24 enhances tumor invasion and metastasis by targeting PTPN9 and PTPRF to promote EGF signaling. J Cell Sci (2013) 126:1440–53. doi:10.1242/jcs.118299
202. Huang K, Zhang J-X, Han L, You Y-P, Jiang T, Pu P-Y, et al. MicroRNA roles in beta-catenin pathway. Mol Cancer (2010) 9:252. doi:10.1186/1476-4598-9-252
203. Su W-L, Kleinhanz RR, Schadt EE. Characterizing the role of miRNAs within gene regulatory networks using integrative genomics techniques. Mol Syst Biol (2011) 7:490. doi:10.1038/msb.2011.23
204. Chen T-H, Chang S-W, Huang C-C, Wang K-L, Yeh K-T, Liu C-N, et al. The prognostic significance of APC gene mutation and miR-21 expression in advanced-stage colorectal cancer. Colorectal Dis (2013) 15:1367–74. doi:10.1111/codi.12318
205. Choi YJ, Lin C-P, Ho JJ, He X, Okada N, Bu P, et al. miR-34 miRNAs provide a barrier for somatic cell reprogramming. Nat Cell Biol (2011) 13:1353–60. doi:10.1038/ncb2366
206. Takahashi R-U, Miyazaki H, Ochiya T. The role of microRNAs in the regulation of cancer stem cells. Front Genet (2014) 4:295. doi:10.3389/fgene.2013.00295
207. Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of microRNA processing by Lin28. Science (2008) 320:97–100. doi:10.1126/science.1154040
208. Song JL, Nigam P, Tektas SS, Selva E. MicroRNA regulation of Wnt signaling pathways in development and disease. Cell Signal (2015) 27:1380–91. doi:10.1016/j.cellsig.2015.03.018
209. Ghahhari NM, Babashah S. Interplay between microRNAs and WNT/β-catenin signalling pathway regulates epithelial-mesenchymal transition in cancer. Eur J Cancer (2015) 51:1638–49. doi:10.1016/j.ejca.2015.04.021
210. Ogino S, Shima K, Baba Y, Nosho K, Irahara N, Kure S, et al. Colorectal cancer expression of peroxisome proliferator-activated receptor gamma (PPARG, PPARgamma) is associated with good prognosis. Gastroenterology (2009) 136:1242–50. doi:10.1053/j.gastro.2008.12.048
211. Lecarpentier Y, Claes V, Vallée A, Hébert J-L. Interactions between PPAR gamma and the canonical Wnt/beta-catenin pathway in type 2 diabetes and colon cancer. PPAR Res (2017) 2017:1–9. doi:10.1155/2017/5879090
212. He Q, Chen J, Lin H, Hu P, Chen M. Expression of peroxisome proliferator-activated receptor gamma, E-cadherin and matrix metalloproteinases-2 in gastric carcinoma and lymph node metastases. Chin Med J (Engl) (2007) 120:1498–504.
213. Aldred MA, Morrison C, Gimm O, Hoang-Vu C, Krause U, Dralle H, et al. Peroxisome proliferator-activated receptor gamma is frequently downregulated in a diversity of sporadic nonmedullary thyroid carcinomas. Oncogene (2003) 22:3412–6. doi:10.1038/sj.onc.1206400
214. Jung T-I, Baek W-K, Suh S-I, Jang B-C, Song D-K, Bae J-H, et al. Down-regulation of peroxisome proliferator-activated receptor gamma in human cervical carcinoma. Gynecol Oncol (2005) 97:365–73. doi:10.1016/j.ygyno.2005.01.019
215. Terashita Y, Sasaki H, Haruki N, Nishiwaki T, Ishiguro H, Shibata Y, et al. Decreased peroxisome proliferator-activated receptor gamma gene expression is correlated with poor prognosis in patients with esophageal cancer. Jpn J Clin Oncol (2002) 32:238–43. doi:10.1093/jjco/hyf056
216. Grommes C, Landreth GE, Heneka MT. Antineoplastic effects of peroxisome proliferator-activated receptor gamma agonists. Lancet Oncol (2004) 5:419–29. doi:10.1016/S1470-2045(04)01509-8
217. Koeffler HP. Peroxisome proliferator-activated receptor gamma and cancers. Clin Cancer Res (2003) 9:1–9.
218. Battaglia S, Maguire O, Thorne JL, Hornung LB, Doig CL, Liu S, et al. Elevated NCOR1 disrupts PPARalpha/gamma signaling in prostate cancer and forms a targetable epigenetic lesion. Carcinogenesis (2010) 31:1650–60. doi:10.1093/carcin/bgq086
219. Sarraf P, Mueller E, Smith WM, Wright HM, Kum JB, Aaltonen LA, et al. Loss-of-function mutations in PPAR gamma associated with human colon cancer. Mol Cell (1999) 3:799–804. doi:10.1016/S1097-2765(01)80012-5
220. Capaccio D, Ciccodicola A, Sabatino L, Casamassimi A, Pancione M, Fucci A, et al. A novel germline mutation in peroxisome proliferator-activated receptor gamma gene associated with large intestine polyp formation and dyslipidemia. Biochim Biophys Acta (2010) 1802:572–81. doi:10.1016/j.bbadis.2010.01.012
221. Elnemr A, Ohta T, Iwata K, Ninomia I, Fushida S, Nishimura G, et al. PPARgamma ligand (thiazolidinedione) induces growth arrest and differentiation markers of human pancreatic cancer cells. Int J Oncol (2000) 17:1157–64.
222. Koga H, Sakisaka S, Harada M, Takagi T, Hanada S, Taniguchi E, et al. Involvement of p21(WAF1/Cip1), p27(Kip1), and p18(INK4c) in troglitazone-induced cell-cycle arrest in human hepatoma cell lines. Hepatology (2001) 33:1087–97. doi:10.1053/jhep.2001.24024
223. Chen F, Kim E, Wang C-C, Harrison LE. Ciglitazone-induced p27 gene transcriptional activity is mediated through Sp1 and is negatively regulated by the MAPK signaling pathway. Cell Signal (2005) 17:1572–7. doi:10.1016/j.cellsig.2005.03.012
224. Chen F, Harrison LE. Ciglitazone-induced cellular anti-proliferation increases p27kip1 protein levels through both increased transcriptional activity and inhibition of proteasome degradation. Cell Signal (2005) 17:809–16. doi:10.1016/j.cellsig.2004.11.002
225. Motomura W, Okumura T, Takahashi N, Obara T, Kohgo Y. Activation of peroxisome proliferator-activated receptor gamma by troglitazone inhibits cell growth through the increase of p27KiP1 in human. Pancreatic carcinoma cells. Cancer Res (2000) 60:5558–64.
226. Wang C, Fu M, D’Amico M, Albanese C, Zhou JN, Brownlee M, et al. Inhibition of cellular proliferation through IkappaB kinase-independent and peroxisome proliferator-activated receptor gamma-dependent repression of cyclin D1. Mol Cell Biol (2001) 21:3057–70. doi:10.1128/MCB.21.9.3057-3070.2001
227. Qin C, Burghardt R, Smith R, Wormke M, Stewart J, Safe S. Peroxisome proliferator-activated receptor gamma agonists induce proteasome-dependent degradation of cyclin D1 and estrogen receptor alpha in MCF-7 breast cancer cells. Cancer Res (2003) 63:958–64.
228. Huang J-W, Shiau C-W, Yang Y-T, Kulp SK, Chen K-F, Brueggemeier RW, et al. Peroxisome proliferator-activated receptor gamma-independent ablation of cyclin D1 by thiazolidinediones and their derivatives in breast cancer cells. Mol Pharmacol (2005) 67:1342–8. doi:10.1124/mol.104.007732
229. Lapillonne H, Konopleva M, Tsao T, Gold D, McQueen T, Sutherland RL, et al. Activation of peroxisome proliferator-activated receptor gamma by a novel synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid induces growth arrest and apoptosis in breast cancer cells. Cancer Res (2003) 63:5926–39.
230. Yin F, Wakino S, Liu Z, Kim S, Hsueh WA, Collins AR, et al. Troglitazone inhibits growth of MCF-7 breast carcinoma cells by targeting G1 cell cycle regulators. Biochem Biophys Res Commun (2001) 286:916–22. doi:10.1006/bbrc.2001.5491
231. Mueller E, Smith M, Sarraf P, Kroll T, Aiyer A, Kaufman DS, et al. Effects of ligand activation of peroxisome proliferator-activated receptor gamma in human prostate cancer. Proc Natl Acad Sci U S A (2000) 97:10990–5. doi:10.1073/pnas.180329197
232. Demetri GD, Fletcher CD, Mueller E, Sarraf P, Naujoks R, Campbell N, et al. Induction of solid tumor differentiation by the peroxisome proliferator-activated receptor-gamma ligand troglitazone in patients with liposarcoma. Proc Natl Acad Sci U S A (1999) 96:3951–6. doi:10.1073/pnas.96.7.3951
233. Bambury RM, Iyer G, Rosenberg JE. Specific PPAR gamma agonists may have different effects on cancer incidence. Annu Oncol Off (2013) 24:854. doi:10.1093/annonc/mdt003
234. Trombetta A, Maggiora M, Martinasso G, Cotogni P, Canuto RA, Muzio G. Arachidonic and docosahexaenoic acids reduce the growth of A549 human lung-tumor cells increasing lipid peroxidation and PPARs. Chem Biol Interact (2007) 165:239–50. doi:10.1016/j.cbi.2006.12.014
235. Edwards IJ, Berquin IM, Sun H, O’flaherty JT, Daniel LW, Thomas MJ, et al. Differential effects of delivery of omega-3 fatty acids to human cancer cells by low-density lipoproteins versus albumin. Clin Cancer Res (2004) 10:8275–83. doi:10.1158/1078-0432.CCR-04-1357
236. Sun H, Berquin IM, Owens RT, O’Flaherty JT, Edwards IJ. Peroxisome proliferator-activated receptor gamma-mediated up-regulation of syndecan-1 by n-3 fatty acids promotes apoptosis of human breast cancer cells. Cancer Res (2008) 68:2912–9. doi:10.1158/0008-5472.CAN-07-2305
237. Palakurthi SS, Aktas H, Grubissich LM, Mortensen RM, Halperin JA. Anticancer effects of thiazolidinediones are independent of peroxisome proliferator-activated receptor gamma and mediated by inhibition of translation initiation. Cancer Res (2001) 61:6213–8.
238. Debrock G, Vanhentenrijk V, Sciot R, Debiec-Rychter M, Oyen R, Van Oosterom A. A phase II trial with rosiglitazone in liposarcoma patients. Br J Cancer (2003) 89:1409–12. doi:10.1038/sj.bjc.6601306
239. Bae M-A, Song BJ. Critical role of c-Jun N-terminal protein kinase activation in troglitazone-induced apoptosis of human HepG2 hepatoma cells. Mol Pharmacol (2003) 63:401–8. doi:10.1124/mol.63.2.401
240. Jozkowicz A, Dulak J, Piatkowska E, Placha W, Dembinska-Kiec A. Ligands of peroxisome proliferator-activated receptor-gamma increase the generation of vascular endothelial growth factor in vascular smooth muscle cells and in macrophages. Acta Biochim Pol (2000) 47:1147–57.
241. Liu Y, Zhu Z, Zhang S-N, Mou J, Liu L, Cui T, et al. Combinational effect of PPARγ agonist and RXR agonist on the growth of SGC7901 gastric carcinoma cells in vitro. Tumour Biol (2013) 34:2409–18. doi:10.1007/s13277-013-0791-2
242. Zhang W, Wu N, Li Z, Wang L, Jin J, Zha X-L. PPARgamma activator rosiglitazone inhibits cell migration via upregulation of PTEN in human hepatocarcinoma cell line BEL-7404. Cancer Biol Ther (2006) 5:1008–14. doi:10.4161/cbt.5.8.2887
243. Teresi RE, Shaiu C-W, Chen C-S, Chatterjee VK, Waite KA, Eng C. Increased PTEN expression due to transcriptional activation of PPARgamma by lovastatin and rosiglitazone. Int J Cancer (2006) 118:2390–8. doi:10.1002/ijc.21799
244. Patel L, Pass I, Coxon P, Downes CP, Smith SA, Macphee CH. Tumor suppressor and anti-inflammatory actions of PPARgamma agonists are mediated via upregulation of PTEN. Curr Biol (2001) 11:764–8. doi:10.1016/S0960-9822(01)00225-1
245. Lee SY, Hur GY, Jung KH, Jung HC, Lee SY, Kim JH, et al. PPAR-gamma agonist increase gefitinib’s antitumor activity through PTEN expression. Lung Cancer (2006) 51:297–301. doi:10.1016/j.lungcan.2005.10.010
246. Kim Y, Suh N, Sporn M, Reed JC. An inducible pathway for degradation of FLIP protein sensitizes tumor cells to TRAIL-induced apoptosis. J Biol Chem (2002) 277:22320–9. doi:10.1074/jbc.M202458200
247. Schultze K, Böck B, Eckert A, Oevermann L, Ramacher D, Wiestler O, et al. Troglitazone sensitizes tumor cells to TRAIL-induced apoptosis via down-regulation of FLIP and urvivin. Apoptosis (2006) 11:1503–12. doi:10.1007/s10495-006-8896-3
248. Shiau C-W, Yang C-C, Kulp SK, Chen K-F, Chen C-S, Huang J-W, et al. Thiazolidenediones mediate apoptosis in prostate cancer cells in part through inhibition of Bcl-xL/Bcl-2 functions independently of PPARgamma. Cancer Res (2005) 65:1561–9. doi:10.1158/0008-5472.CAN-04-1677
249. Kim KY, Kim SS, Cheon HG. Differential anti-proliferative actions of peroxisome proliferator-activated receptor-gamma agonists in MCF-7 breast cancer cells. Biochem Pharmacol (2006) 72:530–40. doi:10.1016/j.bcp.2006.05.009
250. Yan K-H, Yao C-J, Chang H-Y, Lai G-M, Cheng A-L, Chuang S-E. The synergistic anticancer effect of troglitazone combined with aspirin causes cell cycle arrest and apoptosis in human lung cancer cells. Mol Carcinog (2010) 49:235–46. doi:10.1002/mc.20593
251. Toyota M, Miyazaki Y, Kitamura S, Nagasawa Y, Kiyohara T, Shinomura Y, et al. Peroxisome proliferator-activated receptor gamma reduces the growth rate of pancreatic cancer cells through the reduction of cyclin D1. Life Sci (2002) 70:1565–75. doi:10.1016/S0024-3205(01)01524-7
252. Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol (2010) 10:365–76. doi:10.1038/nri2748
253. Straus DS, Glass CK. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol (2007) 28:551–8. doi:10.1016/j.it.2007.09.003
254. Atsumi T, Singh R, Sabharwal L, Bando H, Meng J, Arima Y, et al. Inflammation amplifier, a new paradigm in cancer biology. Cancer Res (2014) 74:8–14. doi:10.1158/0008-5472.CAN-13-2322
255. Gagliani N, Hu B, Huber S, Elinav E, Flavell RA. The fire within: microbes inflame tumors. Cell (2014) 157:776–83. doi:10.1016/j.cell.2014.03.006
256. Shah YM, Morimura K, Gonzalez FJ. Expression of peroxisome proliferator-activated receptor-gamma in macrophage suppresses experimentally induced colitis. Am J Physiol Gastrointest Liver Physiol (2007) 292:G657–66. doi:10.1152/ajpgi.00381.2006
257. Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH, et al. 15-deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in the NF-kappa B signaling pathway. Proc Natl Acad Sci U S A (2000) 97:4844–9. doi:10.1073/pnas.97.9.4844
258. Bagi Z, Koller A, Kaley G. PPARgamma activation, by reducing oxidative stress, increases NO bioavailability in coronary arterioles of mice with Type 2 diabetes. Am J Physiol Heart Circ Physiol (2004) 286:H742–8. doi:10.1152/ajpheart.00718.2003
259. Inoue I, Goto S, Matsunaga T, Nakajima T, Awata T, Hokari S, et al. The ligands/activators for peroxisome proliferator-activated receptor alpha (PPARalpha) and PPARgamma increase Cu2+,Zn2+-superoxide dismutase and decrease p22phox message expressions in primary endothelial cells. Metabolism (2001) 50:3–11. doi:10.1053/meta.2001.19415
260. Garcia-Fuentes E, Murri M, Garrido-Sanchez L, Garcia-Serrano S, García-Almeida JM, Moreno-Santos I, et al. PPARgamma expression after a high-fat meal is associated with plasma superoxide dismutase activity in morbidly obese persons. Obesity (Silver Spring) (2010) 18:952–8. doi:10.1038/oby.2009.314
261. Bilban M, Haslinger P, Prast J, Klinglmüller F, Woelfel T, Haider S, et al. Identification of novel trophoblast invasion-related genes: heme oxygenase-1 controls motility via peroxisome proliferator-activated receptor gamma. Endocrinology (2009) 150:1000–13. doi:10.1210/en.2008-0456
262. Tontonoz P, Singer S, Forman BM, Sarraf P, Fletcher JA, Fletcher CD, et al. Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator-activated receptor gamma and the retinoid X receptor. Proc Natl Acad Sci U S A (1997) 94:237–41. doi:10.1073/pnas.94.1.237
263. Gupta RA, Brockman JA, Sarraf P, Willson TM, DuBois RN. Target genes of peroxisome proliferator-activated receptor gamma in colorectal cancer cells. J Biol Chem (2001) 276:29681–7. doi:10.1074/jbc.M103779200
264. Yoshizumi T, Ohta T, Ninomiya I, Terada I, Fushida S, Fujimura T, et al. Thiazolidinedione, a peroxisome proliferator-activated receptor-gamma ligand, inhibits growth and metastasis of HT-29 human colon cancer cells through differentiation-promoting effects. Int J Oncol (2004) 25:631–9.
265. Terrasi M, Bazan V, Caruso S, Insalaco L, Amodeo V, Fanale D, et al. Effects of PPARγ agonists on the expression of leptin and vascular endothelial growth factor in breast cancer cells. J Cell Physiol (2013) 228:1368–74. doi:10.1002/jcp.24295
266. Smith MR, Manola J, Kaufman DS, George D, Oh WK, Mueller E, et al. Rosiglitazone versus placebo for men with prostate carcinoma and a rising serum prostate-specific antigen level after radical prostatectomy and/or radiation therapy. Cancer (2004) 101:1569–74. doi:10.1002/cncr.20493
267. Burstein HJ, Demetri GD, Mueller E, Sarraf P, Spiegelman BM, Winer EP. Use of the peroxisome proliferator-activated receptor (PPAR) gamma ligand troglitazone as treatment for refractory breast cancer: a phase II study. Breast Cancer Res Treat (2003) 79:391–7. doi:10.1023/A:1024038127156
268. Kulke MH, Demetri GD, Sharpless NE, Ryan DP, Shivdasani R, Clark JS, et al. A phase II study of troglitazone, an activator of the PPARgamma receptor, in patients with chemotherapy-resistant metastatic colorectal cancer. Cancer J (2002) 8:395–9. doi:10.1097/00130404-200209000-00010
269. Farrow B, Evers BM. Activation of PPARgamma increases PTEN expression in pancreatic cancer cells. Biochem Biophys Res Commun (2003) 301:50–3. doi:10.1016/S0006-291X(02)02983-2
270. Zander T, Kraus JA, Grommes C, Schlegel U, Feinstein D, Klockgether T, et al. Induction of apoptosis in human and rat glioma by agonists of the nuclear receptor PPARgamma. J Neurochem (2002) 81:1052–60. doi:10.1046/j.1471-4159.2002.00899.x
271. Kapadia R, Yi J-H, Vemuganti R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front Biosci (2008) 13:1813–26. doi:10.2741/2802
272. Sharma AM, Staels B. Review: peroxisome proliferator-activated receptor gamma and adipose tissue – understanding obesity-related changes in regulation of lipid and glucose metabolism. J Clin Endocrinol Metab (2007) 92:386–95. doi:10.1210/jc.2006-1268
273. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454:436–44. doi:10.1038/nature07205
274. Margeli A, Kouraklis G, Theocharis S. Peroxisome proliferator activated receptor-gamma (PPAR-gamma) ligands and angiogenesis. Angiogenesis (2003) 6:165–9. doi:10.1023/B:AGEN.0000021377.13669.c0
275. Fauconnet S, Lascombe I, Chabannes E, Adessi G-L, Desvergne B, Wahli W, et al. Differential regulation of vascular endothelial growth factor expression by peroxisome proliferator-activated receptors in bladder cancer cells. J Biol Chem (2002) 277:23534–43. doi:10.1074/jbc.M200172200
276. Chintalgattu V, Harris GS, Akula SM, Katwa LC. PPAR-gamma agonists induce the expression of VEGF and its receptors in cultured cardiac myofibroblasts. Cardiovasc Res (2007) 74:140–50. doi:10.1016/j.cardiores.2007.01.010
277. Biscetti F, Gaetani E, Flex A, Aprahamian T, Hopkins T, Straface G, et al. Selective activation of peroxisome proliferator-activated receptor (PPAR)alpha and PPAR gamma induces neoangiogenesis through a vascular endothelial growth factor-dependent mechanism. Diabetes (2008) 57:1394–404. doi:10.2337/db07-0765
278. Chu K, Lee S-T, Koo J-S, Jung K-H, Kim E-H, Sinn D-I, et al. Peroxisome proliferator-activated receptor-gamma-agonist, rosiglitazone, promotes angiogenesis after focal cerebral ischemia. Brain Res (2006) 1093:208–18. doi:10.1016/j.brainres.2006.03.114
279. Huang P-H, Sata M, Nishimatsu H, Sumi M, Hirata Y, Nagai R. Pioglitazone ameliorates endothelial dysfunction and restores ischemia-induced angiogenesis in diabetic mice. Biomed Pharmacother (2008) 62:46–52. doi:10.1016/j.biopha.2007.06.014
280. Bishop-Bailey D. PPARs and angiogenesis. Biochem Soc Trans (2011) 39:1601–5. doi:10.1042/BST20110643
281. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature (2007) 447:1116–20. doi:10.1038/nature05894
282. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol (2010) 72:219–46. doi:10.1146/annurev-physiol-021909-135846
283. Moore KJ, Fitzgerald ML, Freeman MW. Peroxisome proliferator-activated receptors in macrophage biology: friend or foe? Curr Opin Lipidol (2001) 12:519–27. doi:10.1097/00041433-200110000-00007
284. Pancione M, Forte N, Sabatino L, Tomaselli E, Parente D, Febbraro A, et al. Reduced beta-catenin and peroxisome proliferator-activated receptor-gamma expression levels are associated with colorectal cancer metastatic progression: correlation with tumor-associated macrophages, cyclooxygenase 2, and patient outcome. Hum Pathol (2009) 40:714–25. doi:10.1016/j.humpath.2008.08.019
285. Tsugane S, Inoue M. Insulin resistance and cancer: epidemiological evidence. Cancer Sci (2010) 101:1073–9. doi:10.1111/j.1349-7006.2010.01521.x
286. Wang F, Mullican SE, DiSpirito JR, Peed LC, Lazar MA. Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of PPARγ. Proc Natl Acad Sci U S A (2013) 110:18656–61. doi:10.1073/pnas.1314863110
287. Hevener AL, Olefsky JM, Reichart D, Nguyen MTA, Bandyopadyhay G, Leung H-Y, et al. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J Clin Invest (2007) 117:1658–69. doi:10.1172/JCI31561
288. Cristancho AG, Lazar MA. Forming functional fat: a growing understanding of adipocyte differentiation. Nat Rev Mol Cell Biol (2011) 12:722–34. doi:10.1038/nrm3198
289. Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer (2004) 4:579–91. doi:10.1038/nrc1408
Keywords: canonical WNT/β-catenin pathway, PPARγ, carcinogenesis, chronic inflammation, oxidative stress, reactive oxygen species, cancer
Citation: Vallée A and Lecarpentier Y (2018) Crosstalk Between Peroxisome Proliferator-Activated Receptor Gamma and the Canonical WNT/β-Catenin Pathway in Chronic Inflammation and Oxidative Stress During Carcinogenesis. Front. Immunol. 9:745. doi: 10.3389/fimmu.2018.00745
Received: 06 January 2018; Accepted: 26 March 2018;
Published: 13 April 2018
Edited by:
Judit Pongracz, University of Pécs, HungaryReviewed by:
Masahide Tone, Cedars-Sinai Medical Center, United StatesMaria Cecilia G. Marcondes, San Diego Biomedical Research Institute, United States
Copyright: © 2018 Vallée and Lecarpentier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandre Vallée, YWxleGFuZHJlLmcudmFsbGVlQGdtYWlsLmNvbQ==