 Didem Ağaç
Didem Ağaç Michelle A. Gill
Michelle A. Gill J. David Farrar
J. David Farrar- 1Department of Immunology, The University of Texas Southwestern Medical Center, Dallas, TX, United States
- 2Department of Pediatrics, The University of Texas Southwestern Medical Center, Dallas, TX, United States
Upper respiratory viral infections are a major etiologic instigator of allergic asthma, and they drive severe exacerbations of allergic inflammation in the lower airways of asthma sufferers. Rhinovirus (RV), in particular, is the main viral instigator of these pathologies. Asthma exacerbations due to RV infections are the most frequent reasons for hospitalization and account for the majority of morbidity and mortality in asthma patients. In both critical care and disease control, long- and short-acting β2-agonists are the first line of therapeutic intervention, which are used to restore airway function by promoting smooth muscle cell relaxation in bronchioles. While prophylactic use of β2-agonists reduces the frequency and pathology of exacerbations, their role in modulating the inflammatory response is only now being appreciated. Adrenergic signaling is a component of the sympathetic nervous system, and the natural ligands, epinephrine and norepinephrine (NE), regulate a multitude of autonomic functions including regulation of both the innate and adaptive immune response. NE is the primary neurotransmitter released by post-ganglionic sympathetic neurons that innervate most all peripheral tissues including lung and secondary lymphoid organs. Thus, the adrenergic signaling pathways are in direct contact with both the central and peripheral immune compartments. We present a perspective on how the adrenergic signaling pathway controls immune function and how β2-agonists may influence inflammation in the context of virus-induced asthma exacerbations.
Rhinovirus (RV)-Induced Asthma Exacerbations
Asthma is a debilitating chronic disease that has a significant impact on society, including decreased quality of life, work productivity, and increased utilization of health-care resources. With total annual costs reported at $81.9 billion in the U.S. alone (1), asthma represents an enormous economic burden. Approximately 2 million annual emergency room visits and 500,000 hospitalizations have been attributed to acute asthma management in the U.S. (2), highlighting the substantial contribution of asthma exacerbations to the morbidity associated with this disease. Respiratory viral infections are commonly associated with asthma exacerbation episodes (3–6), and RVs have long been recognized as the most frequent viral contributors. The seasonality of RV-associated asthma exacerbations has also been well described, with predictable peaks of hospitalizations for asthma occurring during September epidemics of RV infection (7).
The mechanisms underlying this association between RV and exacerbations of asthma represent an area of intense investigation. The impact of the infection itself on the lung represents one potential mechanism. Although most commonly detected in upper airway samples, RVs have also been demonstrated in lower airway fluids and cells following experimental infection of the upper airway (8–10). Paired with clinical evidence linking RV to lower respiratory tract infections in children (11–13), it is possible that RV infection directly injures airway tissues in the lower airway (14), potentially contributing to exacerbations of asthma. RV infection of airway epithelial cells (ECs) induces the expression of a range of chemokines and cytokines that promote ensuing inflammatory responses. These include such pro-inflammatory molecules as IL-8/CXCL8 (15–17), IL-6 (17, 18), CCL11/eotaxin-1, RANTES/CCL5 (19), IP-10/CXCL10 (20), and ICAM-1 (21). In turn, inflammatory cells recruited by these chemokines secrete IFN-γ and TNF-α, which in some cases can provide a direct antiviral activity in target cells mimicking type I interferon (22). Increased concentrations of inflammatory cytokines have also been demonstrated in airway samples (nasal samples and sputum) obtained from RV-infected individuals (21, 23, 24). RV-induced secretion of such chemokines may also promote asthma exacerbations by promoting an influx of immune cells such as eosinophils, neutrophils, lymphocytes, and macrophages (Mɸs) into the airway (25). Immune cells themselves have also been shown to contribute to the epithelial RV response; human monocytic cells amplify bronchial epithelial cell (BEC) chemokine production during RV infection (26) and could thus also influence asthma pathogenesis in the setting of RV infection.
In addition to the chemokines listed above, RV also induces type I IFN (IFN) expression in airway ECs. The demonstration of decreased IFN-β responses in RV-infected BECs from asthmatics led to the hypothesis that defective IFN antiviral responses could contribute to the pathogenesis of asthma exacerbations (27). While virtually all somatic cells have the capacity to produce IFN-α/β in response to infection, specialized plasmacytoid dendritic cells (pDCs) are the primary cell type to secrete IFN at high levels in response to viral infection. Furthermore, human pDCs also express the high affinity IgE receptor, enabling them to respond to both viral and allergic signals. Deficient viral-induced IFN responses have been demonstrated in virus-simulated whole-blood cultures (28, 29) and purified pDCs (30) from individuals with allergic asthma, providing further evidence for a potential role of IFN in asthma exacerbations. In addition, the link between IgE and pDC antiviral IFN responses could explain the increased risk of asthma exacerbations seen in the presence of atopy and respiratory viral infections. Allergic sensitization and elevated IgE levels are known risk factors for asthma exacerbations with RV infection (3). The magnitude of pDC IFN responses to in vitro viral challenge is inversely correlated with serum IgE levels. In addition, IgE cross-linking abrogates viral-induced pDC IFN production (30, 31). In a recent NIAID-sponsored trial of omalizumab in children with allergic asthma, RV-induced pDC IFN responses were significantly increased in the group who received this IgE-reducing treatment, and this improved antiviral response was associated with lower exacerbations (31, 32).
Since pDCs represent the major source of this antiviral cytokine (33), a defect in IFN production, this cell type could explain how viral infection promotes severe disease in patients with asthma. Another potentially significant effect of reduced pDC antiviral IFN production includes the effect on T helper type 2 responses. IFN has recently been shown to reverse the Th2 phenotype of CD4 lymphocytes via suppression of the Th2 transcription factor GATA-3 (34, 35) and to acutely inhibit IL-5 and IL-13 secretion from memory Th2 cells (36). Thus, a deficient IFN response during respiratory RV infection could contribute to the increased Th2 inflammation observed in individuals with allergic asthma.
Control of Immune Function by Adrenergic Signaling
While the use of corticosteroids and long-term β2-agonists are used for maintenance therapy for asthma sufferers, the front-line intervention for acute exacerbations driven by RV infections is the short-acting β2-agonist, ventolin (nebulized albuterol). The β2-adrenergic receptor (ADRB2) is expressed on smooth muscle cells surrounding the bronchioles, and activation of this receptor by both the natural ligand, epinephrine and norepinephrine (NE), as well as β2-agonists promotes smooth muscle cell relaxation and restored breathing capacity. Signaling through adrenergic receptors controls a myriad of physiological responses, including heart rate, respiratory capacity, and lung turgor. As such, both natural and synthetic ligands for adrenergic receptors have been chiefly used to control sepsis, heart disease, COPD, and asthma.
Innervating throughout most tissues and organs, post-ganglionic sympathetic neurons release the major neurotransmitter NE in response to various intrinsic and external stimuli. Diurnal fluctuations in the release of NE link the sympathetic nervous system to circadian rhythms. Sympathetic neurons also control the “fight or flight” response during periods of stress or fear. Upon ligand binding, adrenergic receptors can activate various G-proteins, depending upon the class of receptor and the specific cell types that express them. For example, the binding of adrenaline and noradrenaline to β2AR results in activation of Gαs (the stimulatory subunit of heterotrimeric G protein) and subsequently activation of adenylyl cyclase, increase in cyclic AMP (cAMP) concentration, and activation of cAMP-dependent protein kinase A (PKA). Depending on the cell that the receptor is engaged, PKA activation can lead to several physiological changes, including muscle contraction, cytokine secretion, and so on. Moreover, the same receptor can couple to the inhibitory Gαi and or signal through MAP kinase pathways (37–39). This complex behavior of the adrenergic receptors enables these receptors to induce cell- and context-specific physiological changes.
The ADRB2 is expressed widely on many types of immune cells, albeit at different levels of cell surface ligand binding sites (40). For example, Maisel et al. identified expression of beta-adrenergic receptor density on lymphocytes ranging from 1,000 to 2,000 receptors/cell (41). In general, ADRB2 signaling acts to suppress the level of inflammation and cytokine secretion in both innate and adaptive T cells (diagrammed in Figure 1). For example, recent studies demonstrated that CD8+ T cell effector function was impaired in response to adrenergic receptor signaling (42–44). Presence of β2-agonists such as albuterol reduced TCR-induced IFNγ and TNFα production, as well as cytolytic activity of both human and murine T cells (43). Similarly, use of beta-blockers increased the frequency of intratumoral CD8+ T cells and increased the efficacy of anti-PD-1 treatment (45). In CD4+ T cells, the presence of NE increases IFN-γ production from Th1 cells (46). Although Th1 cells have been reported to be affected by NE, Th2 cells are less responsive to NE due to the reduction of ADRB2 expression during differentiation and lack of the receptor expression on mature Th2 cells (47, 48). In addition to suppressing T cell effector function, previous studies have demonstrated that ADRB2 signaling can also inhibit TNF-α and IL-12 secretion from innate cells including dendritic cells and Mɸs (49–52) perhaps through direct inhibition of TLR-mediated NF-κB activation (53, 54). Finally, ADRB2 signaling has been shown to enhance the suppressive function of Treg cells (55), which may have significance for clinical effectiveness in asthma.
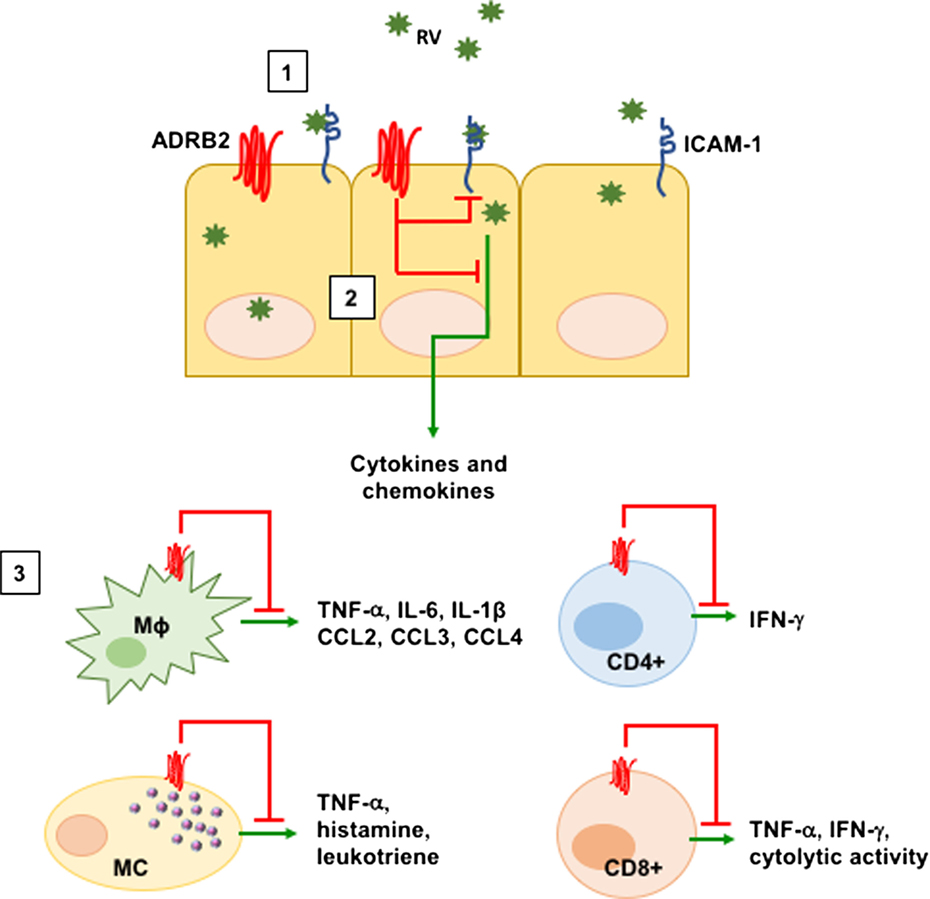
Figure 1. ADRB2-mediated suppression of inflammatory processes. Adrenergic signaling through the ADRB2 inhibits various virus-induced immune mediators. [1] Rhinovirus (RV) infects the upper airways by binding to ICAM-1 on the surface of lung ECs. RV infection of ECs upregulates ICAM-1 as well as IL-8, IL-6, CCL5, CCL11, and CXCL10 to recruit inflammatory cells. [2] Activation of the ADRB2 by either the natural ligands epinephrine and norepinephrine or by β2-agonists downregulates ICAM-1 as well as IL-8, CCL5, and GM-CSF from ECs. [3] ADRB2 signaling additionally inhibits pro-inflammatory mediators in innate and adaptive immune cells. Abbreviations: EC, epithelial cell; MC, mast cell; Mɸ, macrophage.
Role of β2-Agonists in the Context of RV-Mediated Inflammation
Rhinovirus infects human airway ECs by binding to ICAM-1 (Figure 1). As discussed earlier, the natural course of inflammation and cytokine expression increases ICAM-1 expression, allowing additional migration of inflammatory cells into sites of infection. This process likely contributes to RV-induced exacerbations in allergic subjects. Interestingly, both natural ligands of adrenergic receptors (epinephrine and NE), as well as synthetic agonists of the ADRB2 (salbutamol and terbutaline) downregulate ICAM-1 expression on monocytes (56, 57). Furthermore, human BECs reduced the expression of ICAM-1 in response to fenoterol, a β2-agonist (58), suggesting the use of β2-agonists might help patients with RV-induced exacerbations by downregulating the entry receptor on various cell types. Moreover, human airway parasympathetic neurons also downregulate expression of ICAM-1 (59), raising the possibility that the use of β2-agonists has a broader effect than previously appreciated.
Smooth muscle cell responsiveness to β2-agonists is critical for emergency intervention during exacerbations. However, RV infection has been shown to reduce expression of the ADRB2 on airway smooth muscle cells via indirect actions on infected ECs. RV drives secretion of prostaglandins from ECs, which act in a paracrine fashion on smooth muscle cells to suppress ADRB2 expression (60). In this context, COX2 inhibitors tended to restore adrenergic responsiveness. Airway ECs express functional adrenergic receptors (61, 62). Stimulation of the ADRB2 increased the beat frequency of cilia (63), and fenoterol, a β2-agonist downregulates ICAM-1 (58). Sabatini and colleagues reported that salmeterol downregulated VCAM-1 in addition to ICAM-1. In addition, RANTES, IL-8, and GM-CSF were inhibited in response to adrenergic stimulation (64). In murine models, airway EC-specific expression of ADRB2 can recapitulate IL-13-induced airway hyperresponsiveness, mucus production, and cellular infiltration (65), which contrasts to the suppressive effects of β2-agonists seen in human cells.
Victoni et al. demonstrated that β2-agonists can downregulate TNFα, IL-6, and IL-1β from human monocyte-derived Mɸs; however, lung Mɸs are resistant to suppressive effects of β2-agonists (66). Similarly, chemokines CCL2, CCL3, and CCL4 were downregulated in human monocyte-derived Mɸs, yet lung Mɸs were not affected. One possible mechanism of cytokine suppression may involve targeting cytokine mRNA transcripts. For example, β2-agonist salbutamol increases the expression of tristetraproline (TTP) in murine and human Mɸ cell lines. TTP can bind to AU-rich elements in 3′UTR of several pro-inflammatory cytokine transcripts, including TNF and GM-CSF. This interaction might account for the reduction in pro-inflammatory cytokines in response to adrenergic signaling (67). Although lung Mɸs had similar levels of ADRB2 transcript, the ADRB2 protein was not expressed, which can explain why lung Mɸs may not respond to β2-agonists as efficiently as their splenic and circulating counterparts (66). β2-Agonists inhibit release of histamine and leukotriene from mast cells (MCs) in vitro and in vivo (68–70). Similarly, β2-agonists reduce histamine release from human lung MCs when cocultured with airway smooth muscle cells (71). IgE-mediated release of TNFα is also reduced in response to β2-agonists (72). These findings suggest that MC mediators that are involved in acute inflammatory responses can be controlled by adrenergic receptor agonists.
In IFN-γ-primed human dendritic cells, salbutamol inhibited IL-12, IL-1α, IL-1β, IL-6, and TNFα; however, IL-10 was unaffected. When naive T cells were primed with dendritic cells exposed to salbutamol, commitment to Th1 lineage significantly reduced (possibly due to the reduction in IL-12) (49). This is accompanied by an increase in IL-4+ Th2 cells in the coculture. This suggests that use of β2-agonists may skew lung T cells to the pathogenic Th2 lineage. Similarly, in murine bone marrow-derived dendritic cells, epinephrine enhanced differentiation of IL-4- and IL-17A-producing T cells (73). In addition to T cell priming, β2-agonists also alter phagosomal degradation of antigens and cross-presentation of dendritic cells (74). Finally, Yewdell and colleagues recently demonstrated that chemical sympathectomy increased CD8+ T cell responses to influenza infection in mice (42). Furthermore, ADRB2 antagonists enhanced CD8+ responses, and while a direct role for the ADRB2 on CD8+ T cells was not examined, this study suggests that adrenergic signaling acts to limit the response to viral infections.
Final Comments and Future Areas of Interest
Although β2-agonists are widely used in the management of asthma and COPD, many questions remain regarding their ability to suppress inflammation in the context of exacerbations. As mentioned earlier, stimulation of ECs in vitro with β2-agonists downregulates ICAM-1 expression. This indicates that the use of β2-agonists can potentially reduce RV entry and spread within the lungs. Yamaya and colleagues reported that pretreatment of human tracheal ECs with tulabuterol, a long-acting β2-agonist, for 3 days before RV-14 exposure reduced the expression of ICAM-1 and viral replication in ECs (75). By contrast, Bochkov and colleagues reported that pretreatment of BECs with budesonide (a corticosteroid), formoterol (a β2-agonist), or in combination for 24 h did not alter replication of RV-16 in asthmatics and healthy subjects (76). However, the authors did not present data on the level of ICAM-1 protein. It would be beneficial to assess the role of β2-agonists ex vivo during RV infections to eliminate the variation from in vitro settings. Also, no studies to date have investigated the role of adrenergic receptor signaling on expression of RV viral proteins. Moreover, β2-agonists promote an anti-inflammatory phenotype in innate and adaptive immune cells by suppressing production of antiviral cytokines (43) and downregulate a plethora of chemokines (64) that can contribute to recruitment of inflammatory cells to the lungs. This raises the issue of the benefits versus costs of the use of long-term β2-agonists to control asthma symptoms. If β2-agonists generally suppress innate immune function, does their use allow for a more receptive environment for infection? By contrast, in the contest of overt RV-driven inflammation, β2-agonists can certainly dampen the magnitude of inflammation, which is also thought to be the main benefit of corticosteroids. Additional studies are warranted to determine the long-range effects of β2-agonists in the context of both RV susceptibility and the acute effects these drugs have on suppressing inflammation during exacerbations.
Author Contributions
DA wrote drafts of Sections “Control of Immune Function by Adrenergic Signaling” and “Role of β2-Agonists in the Context of RV-Mediated Inflammation.” MG wrote Section “Rhinovirus (RV)-Induced Asthma Exacerbations.” JF conceived of the subject, wrote the abstract, and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was supported by the NIH (NIAID) and by the Beecherl Endowment (UT Southwestern Medical Center).
References
1. Nurmagambetov T, Kuwahara R, Garbe P. The economic burden of asthma in the United States, 2008 – 2013. Ann Am Thorac Soc (2018) 15(3):348–56. doi:10.1513/AnnalsATS.201703-259OC
2. Dougherty RH, Fahy JV. Acute exacerbations of asthma: epidemiology, biology and the exacerbation-prone phenotype. Clin Exp Allergy (2009) 39(2):193–202. doi:10.1111/j.1365-2222.2008.03157.x
3. Heymann PW, Carper HT, Murphy DD, Platts-Mills TA, Patrie J, McLaughlin AP, et al. Viral infections in relation to age, atopy, and season of admission among children hospitalized for wheezing. J Allergy Clin Immunol (2004) 114(2):239–47. doi:10.1016/j.jaci.2004.04.006
4. Johnston SL, Pattemore PK, Sanderson G, Smith S, Lampe F, Josephs L, et al. Community study of role of viral infections in exacerbations of asthma in 9-11 year old children. BMJ (1995) 310(6989):1225–9. doi:10.1136/bmj.310.6989.1225
5. Atmar RL, Guy E, Guntupalli KK, Zimmerman JL, Bandi VD, Baxter BD, et al. Respiratory tract viral infections in inner-city asthmatic adults. Arch Intern Med (1998) 158(22):2453–9. doi:10.1001/archinte.158.22.2453
6. Dawood FS, Kamimoto L, D’Mello TA, Reingold A, Gershman K, Meek J, et al. Children with asthma hospitalized with seasonal or pandemic influenza, 2003–2009. Pediatrics (2011) 128(1):e27–32. doi:10.1542/peds.2010-3343
7. Johnston NW, Johnston SL, Duncan JM, Greene JM, Kebadze T, Keith PK, et al. The September epidemic of asthma exacerbations in children: a search for etiology. J Allergy Clin Immunol (2005) 115(1):132–8. doi:10.1016/j.jaci.2004.09.025
8. Mosser AG, Vrtis R, Burchell L, Lee WM, Dick CR, Weisshaar E, et al. Quantitative and qualitative analysis of rhinovirus infection in bronchial tissues. Am J Respir Crit Care Med (2005) 171(6):645–51. doi:10.1164/rccm.200407-970OC
9. Gern JE, Galagan DM, Jarjour NN, Dick EC, Busse WW. Detection of rhinovirus RNA in lower airway cells during experimentally induced infection. Am J Respir Crit Care Med (1997) 155(3):1159–61. doi:10.1164/ajrccm.155.3.9117003
10. Papadopoulos NG, Bates PJ, Bardin PG, Papi A, Leir SH, Fraenkel DJ, et al. Rhinoviruses infect the lower airways. J Infect Dis (2000) 181(6):1875–84. doi:10.1086/315513
11. Miller EK, Lu X, Erdman DD, Poehling KA, Zhu Y, Griffin MR, et al. Rhinovirus-associated hospitalizations in young children. J Infect Dis (2007) 195(6):773–81. doi:10.1086/511821
12. Lee WM, Kiesner C, Pappas T, Lee I, Grindle K, Jartti T, et al. A diverse group of previously unrecognized human rhinoviruses are common causes of respiratory illnesses in infants. PLoS One (2007) 2(10):e966. doi:10.1371/journal.pone.0000966
13. Regamey N, Kaiser L. Rhinovirus infections in infants: is respiratory syncytial virus ready for the challenge? Eur Respir J (2008) 32(2):249–51. doi:10.1183/09031936.00076508
14. Gern JE. Rhinovirus and the initiation of asthma. Curr Opin Allergy Clin Immunol (2009) 9(1):73–8. doi:10.1097/ACI.0b013e32831f8f1b
15. Johnston SL, Papi A, Bates PJ, Mastronarde JG, Monick MM, Hunninghake GW. Low grade rhinovirus infection induces a prolonged release of IL-8 in pulmonary epithelium. J Immunol (1998) 160(12):6172–81.
16. Zhu Z, Tang W, Gwaltney JM Jr, Wu Y, Elias JA. Rhinovirus stimulation of interleukin-8 in vivo and in vitro: role of NF-kappaB. Am J Physiol (1997) 273(4 Pt 1):L814–24.
17. Subauste MC, Jacoby DB, Richards SM, Proud D. Infection of a human respiratory epithelial cell line with rhinovirus. Induction of cytokine release and modulation of susceptibility to infection by cytokine exposure. J Clin Invest (1995) 96(1):549–57. doi:10.1172/JCI118067
18. Zhu Z, Tang W, Ray A, Wu Y, Einarsson O, Landry ML, et al. Rhinovirus stimulation of interleukin-6 in vivo and in vitro. Evidence for nuclear factor kappa B-dependent transcriptional activation. J Clin Invest (1996) 97(2):421–30. doi:10.1172/JCI118431
19. Schroth MK, Grimm E, Frindt P, Galagan DM, Konno SI, Love R, et al. Rhinovirus replication causes RANTES production in primary bronchial epithelial cells. Am J Respir Cell Mol Biol (1999) 20(6):1220–8. doi:10.1165/ajrcmb.20.6.3261
20. Spurrell JC, Wiehler S, Zaheer RS, Sanders SP, Proud D. Human airway epithelial cells produce IP-10 (CXCL10) in vitro and in vivo upon rhinovirus infection. Am J Physiol Lung Cell Mol Physiol (2005) 289(1):L85–95. doi:10.1152/ajplung.00397.2004
21. Papi A, Johnston SL. Rhinovirus infection induces expression of its own receptor intercellular adhesion molecule 1 (ICAM-1) via increased NF-kappaB-mediated transcription. J Biol Chem (1999) 274(14):9707–20. doi:10.1074/jbc.274.14.9707
22. Davis AM, Hagan KA, Matthews LA, Bajwa G, Gill MA, Gale M Jr, et al. Blockade of virus infection by human CD4+ T cells via a cytokine relay network. J Immunol (2008) 180(10):6923–32. doi:10.4049/jimmunol.180.10.6923
23. Grunberg K, Smits HH, Timmers MC, de Klerk EP, Dolhain RJ, Dick EC, et al. Experimental rhinovirus 16 infection. Effects on cell differentials and soluble markers in sputum in asthmatic subjects. Am J Respir Crit Care Med (1997) 156(2 Pt 1):609–16. doi:10.1164/ajrccm.156.2.9610079
24. Pizzichini MM, Pizzichini E, Efthimiadis A, Chauhan AJ, Johnston SL, Hussack P, et al. Asthma and natural colds. Inflammatory indices in induced sputum: a feasibility study. Am J Respir Crit Care Med (1998) 158(4):1178–84. doi:10.1164/ajrccm.158.4.9712082
25. Jackson DJ, Johnston SL. The role of viruses in acute exacerbations of asthma. J Allergy Clin Immunol (2010) 125(6):1178–87;quiz1188–9. doi:10.1016/j.jaci.2010.04.021
26. Korpi-Steiner NL, Bates ME, Lee WM, Hall DJ, Bertics PJ. Human rhinovirus induces robust IP-10 release by monocytic cells, which is independent of viral replication but linked to type I interferon receptor ligation and STAT1 activation. J Leukoc Biol (2006) 80(6):1364–74. doi:10.1189/jlb.0606412
27. Wark PA, Johnston SL, Bucchieri F, Powell R, Puddicombe S, Laza-Stanca V, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med (2005) 201(6):937–47. doi:10.1084/jem.20041901
28. Bufe A, Gehlhar K, Grage-Griebenow E, Ernst M. Atopic phenotype in children is associated with decreased virus-induced interferon-alpha release. Int Arch Allergy Immunol (2002) 127(1):82–8. doi:10.1159/000048173
29. Gehlhar K, Bilitewski C, Reinitz-Rademacher K, Rohde G, Bufe A. Impaired virus-induced interferon-alpha2 release in adult asthmatic patients. Clin Exp Allergy (2006) 36(3):331–7. doi:10.1111/j.1365-2222.2006.02450.x
30. Gill MA, Bajwa G, George TA, Dong CC, Dougherty II, Jiang N, et al. Counterregulation between the FcepsilonRI pathway and antiviral responses in human plasmacytoid dendritic cells. J Immunol (2010) 184(11):5999–6006. doi:10.4049/jimmunol.0901194
31. Teach SJ, Gill MA, Togias A, Sorkness CA, Arbes SJ Jr, Calatroni A, et al. Preseasonal treatment with either omalizumab or an inhaled corticosteroid boost to prevent fall asthma exacerbations. J Allergy Clin Immunol (2015) 136(6):1476–85. doi:10.1016/j.jaci.2015.09.008
32. Gill MA, Liu AH, Calatroni A, Krouse RZ, Shao B, Schiltz A, et al. Enhanced plasmacytoid dendritic cell antiviral responses after omalizumab. J Allergy Clin Immunol (2017). doi:10.1016/j.jaci.2017.07.035
33. Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol (2005) 23:275–306. doi:10.1146/annurev.immunol.23.021704.115633
34. Huber JP, Ramos HJ, Gill MA, Farrar JD. Cutting edge: type I IFN reverses human Th2 commitment and stability by suppressing GATA3. J Immunol (2010) 185(2):813–7. doi:10.4049/jimmunol.1000469
35. Huber JP, Gonzales-van Horn SR, Roybal KT, Gill MA, Farrar JD. IFN-alpha suppresses GATA3 transcription from a distal exon and promotes H3K27 trimethylation of the CNS-1 enhancer in human Th2 cells. J Immunol (2014) 192(12):5687–94. doi:10.4049/jimmunol.1301908
36. Gonzales-van Horn SR, Estrada LD, van Oers NS, Farrar JD. STAT4-mediated transcriptional repression of the IL5 gene in human memory Th2 cells. Eur J Immunol (2016) 46(6):1504–10. doi:10.1002/eji.201546050
37. Rosenbaum DM, Rasmussen SGF, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature (2009) 459(7245):356–63. doi:10.1038/nature08144
38. El-Zohairy SN, Oriowo MA, Ezeamuzie CI. Cyclic adenosine monophosphate-mediated enhancement of vascular endothelial growth factor released by differentiated human monocytic cells: the role of protein kinase A. Med Princ Pract (2015) 24(6):548–54. doi:10.1159/000433540
39. Tan KS, Nackley AG, Satterfield K, Maixner W, Diatchenko L, Flood PM. Beta2 adrenergic receptor activation stimulates pro-inflammatory cytokine production in macrophages via PKA- and NF-kappaB-independent mechanisms. Cell Signal (2007) 19(2):251–60. doi:10.1016/j.cellsig.2006.06.007
40. Nance DM, Sanders VM. Autonomic innervation and regulation of the immune system (1987–2007). Brain Behav Immun (2007) 21(6):736–45. doi:10.1016/j.bbi.2007.03.008
41. Maisel AS, Fowler P, Rearden A, Motulsky HJ, Michel MC. A new method for isolation of human lymphocyte subsets reveals differential regulation of beta-adrenergic receptors by terbutaline treatment. Clin Pharmacol Ther (1989) 46(4):429–39. doi:10.1038/clpt.1989.161
42. Grebe KM, Hickman HD, Irvine KR, Takeda K, Bennink JR, Yewdell JW. Sympathetic nervous system control of anti-influenza CD8+ T cell responses. Proc Natl Acad Sci U S A (2009) 106(13):5300–5. doi:10.1073/pnas.0808851106
43. Estrada LD, Ağaç D, Farrar JD. Sympathetic neural signaling via the β2-adrenergic receptor suppresses T-cell receptor-mediated human and mouse CD8(+) T-cell effector function. Eur J Immunol (2016) 46(8):1948–58. doi:10.1002/eji.201646395
44. Kalinichenko VV, Mokyr MB, Graf LH, Cohen RL, Chambers DA. Norepinephrine-mediated inhibition of antitumor cytotoxic T lymphocyte generation involves a beta-adrenergic receptor mechanism and decreased TNF-alpha gene expression. J Immunol (1999) 163(5):2492–9.
45. Bucsek MJ, Qiao G, MacDonald CR, Giridharan T, Evans L, Niedzwecki B, et al. β-Adrenergic signaling in mice housed at standard temperatures suppresses an effector phenotype in CD8+ T cells and undermines checkpoint inhibitor therapy. Cancer Res (2017) 77(20):5639–51. doi:10.1158/0008-5472.CAN-17-0546
46. Swanson MA, Lee WT, Sanders VM. IFN-gamma production by Th1 cells generated from naive CD4+ T cells exposed to norepinephrine. J Immunol (2001) 166(1):232–40. doi:10.4049/jimmunol.166.1.232
47. McAlees JW, Smith LT, Erbe RS, Jarjoura D, Ponzio NM, Sanders VM. Epigenetic regulation of beta2-adrenergic receptor expression in T(H)1 and T(H)2 cells. Brain Behav Immun (2011) 25(3):408–15. doi:10.1016/j.bbi.2010.10.019
48. Sanders VM, Baker RA, Ramer-Quinn DS, Kasprowicz DJ, Fuchs BA, Street NE. Differential expression of the beta2-adrenergic receptor by Th1 and Th2 clones: implications for cytokine production and B cell help. J Immunol (1997) 158(9):4200–10.
49. Panina-Bordignon P, Mazzeo D, Lucia PD, D’Ambrosio D, Lang R, Fabbri L, et al. Beta2-agonists prevent Th1 development by selective inhibition of interleukin 12. J Clin Invest (1997) 100(6):1513–9. doi:10.1172/JCI119674
50. Donnelly LE, Tudhope SJ, Fenwick PS, Barnes PJ. Effects of formoterol and salmeterol on cytokine release from monocyte-derived macrophages. Eur Respir J (2010) 36(1):178–86. doi:10.1183/09031936.00158008
51. Tanaka S, Tsutsui M, Kishida T, Souma S, Kuroda J, Yoshida T. Salbutamol inhibits lipopolysaccharide-induced inflammatory responses in rat peritoneal macrophages. J Toxicol Sci (2010) 35(3):327–34. doi:10.2131/jts.35.327
52. Hu Z, Chen R, Cai Z, Yu L, Fei Y, Weng L, et al. Salmeterol attenuates the inflammatory response in asthma and decreases the pro-inflammatory cytokine secretion of dendritic cells. Cell Mol Immunol (2012) 9(3):267–75. doi:10.1038/cmi.2011.56
53. Gao H, Sun Y, Wu Y, Luan B, Wang Y, Qu B, et al. Identification of beta-arrestin2 as a G protein-coupled receptor-stimulated regulator of NF-kappaB pathways. Mol Cell (2004) 14(3):303–17. doi:10.1016/S1097-2765(04)00216-3
54. Fan H, Luttrell LM, Tempel GE, Senn JJ, Halushka PV, Cook JA. Beta-arrestins 1 and 2 differentially regulate LPS-induced signaling and pro-inflammatory gene expression. Mol Immunol (2007) 44(12):3092–9. doi:10.1016/j.molimm.2007.02.009
55. Guereschi MG, Araujo LP, Maricato JT, Takenaka MC, Nascimento VM, Vivanco BC, et al. Beta2-adrenergic receptor signaling in CD4+ Foxp3+ regulatory T cells enhances their suppressive function in a PKA-dependent manner. Eur J Immunol (2013) 43(4):1001–12. doi:10.1002/eji.201243005
56. Tamura R, Takahashi HK, Iwagaki H, Yagi T, Mori S, Yoshino T, et al. Effect of beta2-adrenergic receptor agonists on intercellular adhesion molecule (ICAM)-1, B7, and CD40 expression in mixed lymphocyte reaction. Transplantation (2004) 77(2):293–301. doi:10.1097/01.TP.0000101517.48541.7B
57. Kuroki K, Takahashi HK, Iwagaki H, Murakami T, Kuinose M, Hamanaka S, et al. Beta2-adrenergic receptor stimulation-induced immunosuppressive effects possibly through down-regulation of co-stimulatory molecules, ICAM-1, CD40 and CD14 on monocytes. J Int Med Res (2004) 32(5):465–83. doi:10.1177/147323000403200503
58. Oddera S, Silvestri M, Lantero S, Sacco O, Rossi GA. Downregulation of the expression of intercellular adhesion molecule (ICAM)-1 on bronchial epithelial cells by fenoterol, a β2-adrenoceptor agonist. J Asthma Allergy (1998) 35(5):401–8. doi:10.3109/02770909809048948
59. Nie Z, Fryer AD, Jacoby DB. Beta2-agonists inhibit TNF-alpha-induced ICAM-1 expression in human airway parasympathetic neurons. PLoS One (2012) 7(9):e44780. doi:10.1371/journal.pone.0044780
60. Van Ly D, Faiz A, Jenkins C, Crossett B, Black JL, McParland B, et al. Characterising the mechanism of airway smooth muscle beta2 adrenoceptor desensitization by rhinovirus infected bronchial epithelial cells. PLoS One (2013) 8(2):e56058. doi:10.1371/journal.pone.0056058
61. Kelsen SG, Higgins NC, Zhou S, Mardini IA, Benovic JL. Expression and function of the beta-adrenergic receptor coupled-adenylyl cyclase system on human airway epithelial cells. Am J Res Crit Care Med (1995) 152(6 Pt 1):1774–83. doi:10.1164/ajrccm.152.6.8520736
62. Davis PB, Silski CL, Kercsmar CM, Infeld M. Beta-adrenergic receptors on human tracheal epithelial cells in primary culture. Am J Physiol (1990) 258(1 Pt 1):C71–6. doi:10.1152/ajpcell.1990.258.1.C71
63. Sanderson MJ, Dirksen ER. Mechanosensitive and beta-adrenergic control of the ciliary beat frequency of mammalian respiratory tract cells in culture. Am Rev Respir Dis (1989) 139(2):432–40. doi:10.1164/ajrccm/139.2.432
64. Sabatini F, Silvestri M, Sale R, Serpero L, Di Blasi P, Rossi GA. Cytokine release and adhesion molecule expression by stimulated human bronchial epithelial cells are downregulated by salmeterol. Respir Med (2003) 97(9):1052–60. doi:10.1016/S0954-6111(03)00137-9
65. Nguyen LP, Al-Sawalha NA, Parra S, Pokkunuri I, Omoluabi O, Okulate AA, et al. β2-Adrenoceptor signaling in airway epithelial cells promotes eosinophilic inflammation, mucous metaplasia, and airway contractility. Proc Natl Acad Sci U S A (2017) 114(43):E9163–71. doi:10.1073/pnas.1710196114
66. Victoni T, Salvator H, Abrial C, Brollo M, Porto LCS, Lagente V, et al. Human lung and monocyte-derived macrophages differ with regard to the effects of β2-adrenoceptor agonists on cytokine release. Respir Res (2017) 18(1):126. doi:10.1186/s12931-017-0613-y
67. Jalonen U, Leppänen T, Kankaanranta H, Moilanen E. Salbutamol increases tristetraprolin expression in macrophages. Life Sci (2007) 81(25–26):1651–8. doi:10.1016/j.lfs.2007.09.022
68. Church MK, Hiroi J. Inhibition of IgE-dependent histamine release from human dispersed lung mast cells by anti-allergic drugs and salbutamol. Br J Pharmacol (1987) 90(2):421–9. doi:10.1111/j.1476-5381.1987.tb08972.x
69. Chong LK, Cooper E, Vardey CJ, Peachell PT. Salmeterol inhibition of mediator release from human lung mast cells by β-adrenoceptor-dependent and independent mechanisms. Br J Pharmacol (1998) 123(5):1009–15. doi:10.1038/sj.bjp.0701703
70. Nials AT, Ball DI, Butchers PR, Coleman RA, Humbles AA, Johnson M, et al. Formoterol on airway smooth muscle and human lung mast cells: a comparison with salbutamol and salmeterol. Eur J Pharmacol (1994) 251(2–3):127–35. doi:10.1016/0014-2999(94)90392-1
71. Lewis RJ, Chachi L, Newby C, Amrani Y, Bradding P. Bidirectional counterregulation of human lung mast cell and airway smooth muscle β2 adrenoceptors. J Immunol (2016) 196(1):55–63. doi:10.4049/jimmunol.1402232
72. Bissonnette EY, Befus AD. Anti-inflammatory effect of b-agonists: inhibition of TNF- release from human mast cells. J Allergy Clin Immunol (1997) 100(6 Pt 1):825–31. doi:10.1016/S0091-6749(97)70280-X
73. Kim B-J, Jones HP. Epinephrine-primed murine bone marrow-derived dendritic cells facilitate production of IL-17A and IL-4 but not IFN-γ by CD4+ T cells. Brain Behav Immun (2010) 24(7):1126–36. doi:10.1016/j.bbi.2010.05.003
74. Hervé J, Dubreil L, Tardif V, Terme M, Pogu S, Anegon I, et al. β2-Adrenoreceptor agonist inhibits antigen cross-presentation by dendritic cells. J Immunol (2013) 190(7):3163–71. doi:10.4049/jimmunol.1201391
75. Yamaya M, Nishimura H, Nadine L, Kubo H, Ryoichi N. Tulobuterol inhibits rhinovirus infection in primary cultures of human tracheal epithelial cells. Physiol Rep (2013) 1(3):e00041. doi:10.1002/phy2.41
Keywords: adrenergic receptor, asthma, rhinovirus, inflammation, cytokine
Citation: Ağaç D, Gill MA and Farrar JD (2018) Adrenergic Signaling at the Interface of Allergic Asthma and Viral Infections. Front. Immunol. 9:736. doi: 10.3389/fimmu.2018.00736
Received: 01 February 2018; Accepted: 26 March 2018;
Published: 11 April 2018
Edited by:
Andrea Sant, David H. Smith Center for Vaccine Biology and Immunology, United StatesReviewed by:
Shahram Salek-Ardakani, Pfizer, United StatesKarl Kai McKinstry, University of Central Florida, United States
Copyright: © 2018 Ağaç, Gill and Farrar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: J. David Farrar, ZGF2aWQuZmFycmFyQHV0c291dGh3ZXN0ZXJuLmVkdQ==