 Valéria Bumiller-Bini1
Valéria Bumiller-Bini1 Gabriel Adelman Cipolla1
Gabriel Adelman Cipolla1 Rodrigo Coutinho de Almeida1†
Rodrigo Coutinho de Almeida1† Maria Luiza Petzl-Erler1
Maria Luiza Petzl-Erler1 Danillo Gardenal Augusto1,2
Danillo Gardenal Augusto1,2 Angelica Beate Winter Boldt1,3*
Angelica Beate Winter Boldt1,3*
- 1Laboratory of Human Molecular Genetics, Department of Genetics, Universidade Federal do Paraná, Curitiba, Brazil
- 2Departamento de Ciências Biológicas, Universidade Estadual de Santa Cruz, Ilhéus, Brazil
- 3Laboratory of Molecular Immunopathology, Department of Clinical Pathology, Hospital de Clínicas, Universidade Federal do Paraná, Curitiba, Brazil
Skin blisters of pemphigus foliaceus (PF) present concomitant deposition of autoantibodies and components of the complement system (CS), whose gene polymorphisms are associated with susceptibility to different autoimmune diseases. To investigate these in PF, we evaluated 992 single-nucleotide polymorphisms (SNPs) of 44 CS genes, genotyped through microarray hybridization in 229 PF patients and 194 controls. After excluding SNPs with minor allele frequency <1%, out of Hardy–Weinberg equilibrium in controls or in strong linkage disequilibrium (r2 ≥ 0.8), 201 SNPs remained for logistic regression. Polymorphisms of 11 genes were associated with PF. MASP1 encodes a crucial serine protease of the lectin pathway (rs13094773: OR = 0.5, p = 0.0316; rs850309: OR = 0.23, p = 0.03; rs3864098: OR = 1.53, p = 0.0383; rs698104: OR = 1.52, p = 0.0424; rs72549154: OR = 0.55, p = 0.0453). C9 (rs187875: OR = 1.46, p = 0.0189; rs700218: OR = 0.12, p = 0.0471) and C8A (rs11206934: OR = 4.02, p = 0.0323) encode proteins of the membrane attack complex (MAC) and C5AR1 (rs10404456: OR = 1.43, p = 0.0155), a potent anaphylatoxin-receptor. Two encode complement regulators: MAC-blocking CD59 (rs1047581: OR = 0.62, p = 0.0152) and alternative pathway-blocking CFH (rs34388368: OR = 2.57, p = 0.0195). One encodes opsonin: C3 (rs4807895: OR = 2.52, p = 0.0239), whereas four encode receptors for C3 fragments: CR1 (haplotype with rs6656401: OR = 1.37, p = 0.0382), CR2 (rs2182911: OR = 0.23, p = 0.0263), ITGAM (CR3, rs12928810: OR = 0.66, p = 0.0435), and ITGAX (CR4, rs11574637: OR = 0.63, p = 0.0056). Associations reinforced former findings, regarding differential gene expression, serum levels, C3, and MAC deposition on lesions. Deregulation of previously barely noticed processes, e.g., the lectin and alternative pathways and opsonization-mediated phagocytosis, also modulate PF susceptibility. The results open new crucial avenues for understanding disease etiology and may improve PF treatment through additional therapeutic targets.
Introduction
Pemphigus are blistering autoimmune diseases causing painful bullous lesions, resulting from keratinocyte detachment (acantholysis), through the loss of desmosomes (1). In pemphigus foliaceus (PF), they occur in the superficial granular layer, affecting the skin. Yet in pemphigus vulgaris (PV), they locate in suprabasal stratum, also damaging mucosa. Lesions’ localization correlate with tissue distribution of the main antigens: desmoglein 1 (DSG1) in PF and DSG3 in PV (2). Non-lesional skin may present blisters in the subgranular spinous layer, when submitted to mechanical friction (Nikolsky’s sign) (3). Epithelial PF lesions may be restricted to sun-exposed seborrheic trunk and head areas (localized form) or be ubiquitously distributed (generalized form) (4, 5).
PF—An Epidemiological and Etiopathological Puzzle
Pemphigus occurs sporadically around the world, with incidence of 0.75–5 cases/million per year (6, 7). Despite this, PF is the only autoimmune disease known to be endemic in certain regions, as South America and Tunisia (5, 8), but epidemiology is puzzling, exhibiting wide differences even in neighboring countries. Midwestern Brazilian Amerindian populations actually present prevalences as high as 3.04% (9, 10). There is no sexual disproportion for PF in Brazil; most patients are young (10–40 years old) and have affected relatives. In Colombia, PF affects male mine workers and post-menopausal women (11), whereas young women are predominantly affected in Tunisia (9 female:1 male) (8). Endemic and non-endemic PF are indistinguishable (12), with the exception of higher anti-DSG1 IgM and IgE serum levels in endemic PF (13–15).
The epidemiological puzzle adds to the lack of understanding regarding PF etiology, since postulated major causes differ among countries (11). Brazilian PF patients are usually rural low-wage workers (4), exposed to acantholysis-fostering factors such as UVB (16), thiol and other calcium-sequestering components (11). Most present frequent bites of black (Simuliidae) and sand flies (Phlebotominae), vectors of onchocerciasis and leishmaniasis, respectively. Bites were suggested to increase up to almost five times the susceptibility to PF (17, 18), and components of the fly saliva may trigger a cross-reaction against keratinocyte surface epitopes (19). Viral or bacterial etiology was also suggested (5, 11). Genetic susceptibility involves differential gene expression (20, 21), variants in genes encoding antigen-presenting molecules HLA-DR and HLA-DQ (22, 23) and their corresponding regulatory transcription factor CIITA (24). Several other associations with genes of the immune response have been reported (25–31).
Pemphigus foliaceus patients have higher serum immunoglobulin G (IgG) levels against desmocollins 1 and 2 and all four desmogleins (32). Most pathogenic antibodies, able to induce acantholysis in vitro and in vivo (33–35), are of the IgG4 subclass (36–38), directed against the DSG1 N-terminal ectodomains (39, 40). Anti-DSG1 IgG1 are common in asymptomatic individuals of endemic regions, but can be the only pathogenic antibodies in a subset of PF patients (19). In contrast to IgG4, they initiate the classical pathway of the complement system (CS). This agrees with the frequent concomitant deposition of antibodies and CS components in PF lesions (41–45). Administration of corticosteroids is crucial to achieve disease control in the acute stage. Due to numerous and severe side effects, pemphigus patients are in desperate need of new, specifically targeted therapeutic strategies to substitute common therapy [reviewed in Ref. (46)].
PF and Complement: A Controversial Issue
Complement includes more than 50 plasma and membrane-bound proteins working in the forefront of host defense, killing pathogens and altered cells, and connecting innate to adaptive immune responses. Classical activation begins with the recognition of IgG or IgM, molecules on microbial and apoptotic cells, and C-reactive protein (CRP) by the C1 complex (C1q complexed with serine proteases C1r and C1s). The alternative activation pathway unleashes by spontaneous proteolysis of component C3. The lectin pathway follows recognition of sugar moieties or acetylated residues by colectins (as mannose-binding lectin—MBL) or ficolins (FCNs), respectively, complexed with another set of serine proteases (MASP-1 and MASP-2) (47, 48). All pathways converge in the formation of C3 convertase, which produces opsonic fragments that enhance antigen clearance by phagocytosis. C3b opsonin may be incorporated in the C5 convertase, which leads to the release of C5a anaphylatoxins and to pores opening on target cells, by insertion of the membrane attack complex (C5b-9 complex or MAC). Recognition of CS fragments leads to phagocytosis or blockage of the cascade, which is constantly activated at low levels, being continuously controlled to avoid tissue damage. Far beyond these well-known roles, CS also accomplishes critical functions in regulating inflammation, nervous system development and maturation, coagulation and hemostasis (49, 50).
In a series of five 1980s articles, entitled “Complement fixation by pemphigus antibody,” the Jordon’s group chased the hypothesis that complement has an important role in PV blister formation (51–55). They were closely followed by others who argued the same for PF. Strong granular C3 deposition was repeatedly reported along the basement membrane zone and in intercellular spaces of the epidermal strata (41–45). C3 was also reported to colocalize with IgG1 deposits in the upper epidermis intercellular spaces, in intact as well as injured skin, with a trend for higher deposits in perilesional tissue (42, 43, 45, 56). In fact, C1q and C4 fragments (reported in one patient), and MAC deposits distinguish injured skin, since IgG4 also occurs abundantly in non-acantholytic tissue (42, 43). In cell culture, complement does not seem necessary for acantholysis, but enhances keratinocyte detachment (55, 57).
Serum levels of C3 and CRP (opsonins), Ba and C4d factors (indicative of activated alternative and classical/lectin pathways, respectively), are increased in PF patients with active disease (58–60). CD4+ T cells of PF patients present upregulated C1QA gene expression, compared to controls (20). Protein levels and C1QA expression fall with therapeutic intervention (20, 59, 60). By contrast, anti-DSG1 IgG levels remain high during disease remission (61, 62). MASP-2 levels tend to decline in PF patients, but MBL serum concentrations seem unaffected (63). In PV biopsies, MBL and FCN2, but not C1q nor FCN3, recognize antigens in the basal membrane zone and intercellular spaces of the epidermis (64).
Nevertheless, C5-deficient mice or complement-depleted mice (after inoculation with cobra venom factor) develop the disease when injected with non-endemic PF IgG4 or its F(ab′)2 fragments (65). Both models did not affect C3 upstream components of the classical and lectin pathways, meaning that any roles played by these initiator molecules in the acantholytic process were not appreciated. In addition, the abundant but non-pathogenic anti-DSG1 human IgG1 does not cross-react with murine epidermis (66). By contrast, anti-DSG3 autoantibodies of PF patients, with cutaneous disease only, induce PV-like lesions in mice (67). Conversely, anti-DSG1 autoantibodies of PV patients without superficial epithelial lesions induce PF-like lesions (68). Adding to this picture, DSG expression pattern greatly differs between human and mouse (69) and differences in the genetic background of mouse models, which may deliver completely different outcomes for cutaneous inflammation, were not accounted for (70). Thus, although the mouse model reproduces acantholysis, it cannot reproduce the natural history of the disease itself, and pathological mechanisms may be quite different. For example, murine lesions present apoptotic cells (71), an uncommon finding in human biopsies (3, 72–74), with one reported exception (75). These results undeniably places complement in the disease, but its possible roles are still an issue to be solved.
Genetic Association Between Complement Genes and PF
In the late 90s, the observations from experimental models seemed to have settled the interest on the role of complement in pemphigus. Nevertheless, tissue damage and inflammation, through over-activation and/or deficiency of complement components, play a key role in many dermatological diseases (76). These host-offensive actions may be exacerbated by genetic variation (77), but the extensive polymorphism of complement components impairs the comprehension of their overall impact in any given disease. In addition to the great genetic variation, the pleiotropic effects observed for complement genes add another layer of complexity.
Knowing that genetic associations may reveal new elements that play pivotal roles in disease susceptibility, we intend to reignite this discussion with new results of a PF case–control study that encompass tag polymorphisms within CS genes (Table S1 in Supplementary Material). We analyzed 992 single-nucleotide polymorphisms (SNPs) distributed within 44 genes, out of a subset of 551,839 SNPs genotyped in 229 endemic PF patients and 194 controls, through microarray hybridization (CoreExome-24 v1.1 Illumina). Included patients presented confirmed clinical PF diagnosis, according to physical examination and immunohistochemistry results. Controls were individuals of the endemic region, with no diagnosis or familial history of autoimmune diseases and unrelated to the patients. This study was carried out in accordance with the recommendations of the guidelines of the Conselho Nacional de Ética em Pesquisa (CONEP) with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by CONEP (number 505.988). The statistical analyses were done with PLINK v1.1.9 (78). After excluding those SNPs with minor allele frequencies >1%, genotypic distributions deviating from those expected by Hardy–Weinberg equilibrium in controls (p < 0.05) and high linkage disequilibrium (r2 ≥ 0.8), 201 SNPs remained for subsequent analyses. For haplotypic analysis, 35 additional SNPs with r2 > 0.8 were included. Association analysis was carried out by binary logistic regression, using two principal components (PCA) as covariables, which efficiently eliminates spurious associations due to ethnical differences. Thus, significance level was set to p = 0.05. Rather than exhausting the debate, our purpose is to launch new hypotheses that could be further validated through functional studies, which will link the pathogenic role of the PF autoantibodies to CS underexplored arms.
The Long Known Versus the Unexpected: Complement in PF
We found evidence of association with gene variants of almost all complement elements previously detected in the epidermis or with altered serum levels in PF patients (Table 1; Figure 1). Among them, homozygotes for the intronic rs4807895*T allele within the C3 gene were more susceptible to the disease (OR = 2.52; p = 0.0239). C3 fragments have been consistently reported in PF lesions (41–45, 56, 79, 80) and necessarily result from the activation of proteolytic cascades that converge in its enzymatic cleavage. Given the lack of functional evidence for this association, we speculate that it could be partly explained by increased C3 gene regulation. This would not only increase phagocytosis and MAC deposition, but also T cell-mediated skin inflammation, as reported in other autoimmune diseases (81, 82).
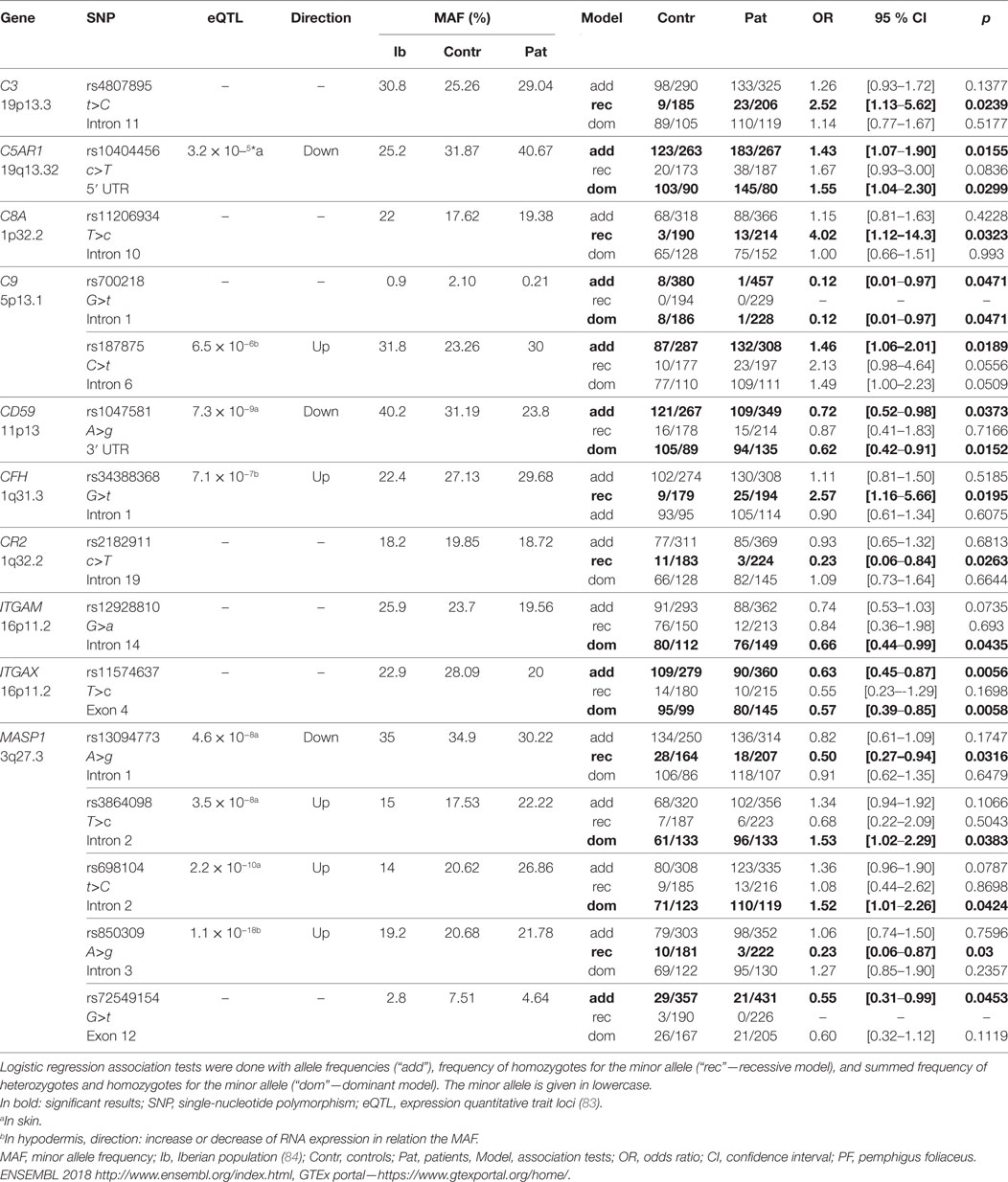
Table 1. Complement gene variants associated with PF.
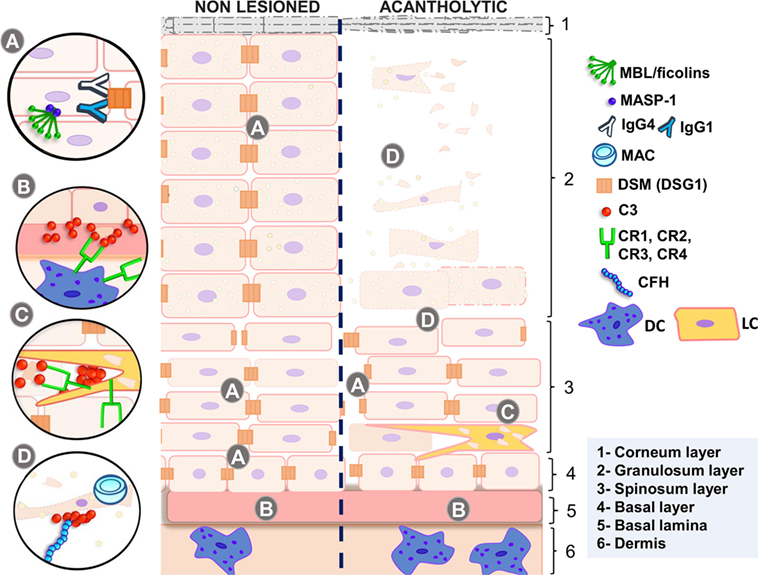
Figure 1. Complement in non-lesioned and acantholytic PF lesions. (A) IgG1 and IgG4 autoantibodies binding to desmosomes causes shrinkage of keratinocytes, increasing intercellular spaces. This process is fostered by activation of the p38 MAPK signaling cascade, which may be unleashed by MASP-1, the serine protease associated with initiating molecules of the lectin pathway. (B) This is further accompanied by the release of antigens, most probably recognized by MBL or ficolins, leading to granular deposition of C3 fragments in the basal lamina. These deposits, as well as deposits of C3 fragments in the intercellular spaces, may also be caused by activation of the alternative pathway, inhibited by CFH. (C) Complement receptors recognize C3 fragments, leading to phagocytosis of autoantigens and increasing antigen presentation to T lymphocytes, thus feedbacking and diversifying autoantibody production. (D) Acantholytic lesions present formation of the membrane attack complex (blocked by CD59 expression), which may protect cells against apoptosis, if present in sublytic amounts. Active disease is also followed by increased C5a release, the latter recognized by C5A receptors in dendritic cells. MBL, mannose-binding lectin; MASP-1, mannose-binding lectin serine protease 1; MASP-2, mannose-binding lectin serine protease 2; MAC, membrane attack complex; IgG4, immunoglobulin 4; IgG1, immunoglobulin 1; DSM (DSG1), desmosome (desmoglein 1); C3, complement component 3; CR1, complement receptor type 1; CR2, complement receptor type 2; CR3, complement receptor type 3; CR4, complement receptor type 4; CFH, complement factor H; DC, dendritic cell; LC, Langerhans cell; PF, pemphigus foliaceus. Source: the author (2018).
Among the pathways held responsible for generating C3 fragments, we found association with genetic variants within genes of the alternative and lectin pathways, but not with the classical pathway. This agrees with the almost complete absence of C1q in human biopsies (42, 64). It also argues against the traditional hypothesis that activation of the classical pathway by anti-DSG1 IgG1 would play an important role in PF (42, 52, 85). In fact, the most abundant pathogenic IgG subclass in pemphigus is IgG4, which is unable to activate complement (33, 34).
Regarding the alternative pathway, we found a surprising genetic association with factor H, its most important regulator. Homozygotes for CFH rs34388368*T, an intronic allele associated with higher CFH mRNA levels in the hypodermis (83), were more susceptible to PF (OR = 2.57; p = 0.0195). These results contradict the conception that uncontrolled complement activation would be one of the underlying causes of PF.
We also found association between PF and five MASP1 polymorphisms, four of them associated with differential mRNA levels in sun-exposed skin and/or in the hypodermis (83). They can potentially interfere with alternative pre-mRNA splicing, which generates three MASP1 products—the collectin/ficolin-associated serine proteases 1 and 3 (MASP-1 and MASP-3) and the truncated non-catalytic MAp44 (also called MAp1, only expressed in cardiac tissue). These products play important roles in competitive activation and blockage of the lectin and alternative pathways, intracellular signaling, coagulation, and bradykinin/kinin systems (86). In our setting, homozygotes for rs13094773*G and rs850309*G (within an intronic region recognized by multiple regulatory proteins) (87) were more protected against PF (OR = 0.5; p = 0.0316 and OR = 0.23; p = 0.03, respectively). Yet individuals with intronic rs3864098*C (OR = 1.53; p = 0.0383) or rs698104*T (OR = 1.52; p = 0.0424) presented increased susceptibility to the disease. Of note, the rs3864098*C allele occurs in linkage disequilibrium with rs710469*C, an allele associated with lower MASP-3 levels in pre-admission critically ill children (88) (Table S2 in Supplementary Material). Finally, we found a protective association (OR = 0.55; p = 0.0453) with a missense variant affecting exclusively the serine protease domain of MASP-3 (rs72549154*T in exon 12, encoding p.Arg576Met). Heterozygotes for rs72549154*T present proportionally increased MASP-3 and decreased MASP-1 serum levels (89). Co-occurring MASP1 alleles increase susceptibility to PF (not necessarily within the same haplotype): rs13094773*A combined with rs3864098*C (OR = 2.51 [95% CI = 1.26–4.97], p = 0.0063), rs13094773*A, and rs698104*T (OR = 2.37 [95% CI = 1.22–4.59], p = 0.0074) and between rs3864098*C and rs698104*T (OR = 1.67 [95% CI = 1.09–2.55], p = 0.0141). All the three variants are associated with higher MASP1 levels (83). Thus, it is conceivable that higher MASP-1 levels contribute to PF, while higher MASP-3 levels are protective. From the physiological point of view, altered MASP-1 levels would affect activation of the lectin pathway, which relies entirely on MASP-1 autoactivation (90). Additionally, MASP-1 activates MASP-3, which cleaves pro-factor D and launches the alternative complement cascade under non-inflammatory conditions (91). It further activates the p38 MAPK pathway in endothelial cells, which leads to IL-8 secretion and neutrophil recruitment (92, 93), both reported to occur in different forms of pemphigus (94–96). Most importantly, activation of the p38 MAPK signaling cascade causes acantholysis in keratinocytes and may be initiated by MASP-1 as well (97, 98).
Genetic variants of MAC components were also associated with PF. Homozygotes for the less common C8A allele rs11206934*C (OR = 4.02; p = 0.0323) in intron 10 and individuals with a C9 haplotype harboring intronic variants consistently associated with increased gene expression in hypodermis and mucosa—rs187875*T (which disrupts a methylated CpG) (87) presented higher susceptibility to PF (Table S2 in Supplementary Material). By contrast, individuals with rs700218*A (intron 1 of C9) were more protected (OR = 0.12; p = 0.0471). We found no association with C5 polymorphisms, as reported by others (who investigated only one SNP) (99). Nevertheless, individuals with the rs10404456*C allele (located in the 5′ UTR of the C5AR1 gene and associated with decreased mRNA levels in sun-exposed skin) (83) presented increased susceptibility to PF (OR = 1.43 p = 0.0155). This gene encodes the major receptor for C5a anaphylatoxin (49) and its deficiency has been rather associated with protection against several immune complex-mediated diseases, including epidermolysis bullosa acquisita (70, 100).
Keratinocytes may keep MAC formation at sublytic levels, eliciting pro-survival signal transduction, hence inhibiting apoptosis—instead of promoting cell destruction (101). This may be achieved by expressing low CD59 levels, MAC’s most important inhibitor. In fact, rs1047581*G in the 3′UTR region of the CD59 gene, associated with reduced mRNA levels in sun-exposed skin (83), protected against PF (OR = 0.62; p = 0.0152). This result agrees with a recent study of our group, where the alternative allele of this same polymorphism occurs within a haplotype increasing CD59 mRNA expression and PF susceptibility (32).
Among complement main roles, the removal of immune complexes and cellular debris is of critical importance for autoimmunity prevention (70). Within the context of the other associations, we suggest that protection may be explained by higher scavenging efficiency of acantholytic cell debris. Furthermore, we found associations with four opsonin-binding complement receptors (CR1-4, encoded by CR1, CR2, ITGAM, and ITGAX). Interestingly, we found a susceptibility association with a CR1 haplotype that includes the major rs6656401*G allele (Table S2 in Supplementary Material), also associated with protection against Alzheimer’s disease (102). The binding of CR2 to iC3b, C3dg, and C3d lowers the threshold for B cell activation (103) and homozygotes for rs2182911*C of the CR2 gene were more protected against the disease (OR = 0.23; p = 0.0263). The products of ITGAM (CR3) and ITGAX (CR4) genes recognize iC3b (48). Individuals with the rs12928810*A (disrupts a CpG in intron 14 of ITGAM) or rs11574637*C (a missense variant—p.Phe180Leu—in exon 4 of ITGAX) were more resistant against PF (OR = 0.66; p = 0.0435 and OR = 0.63; p = 0.0056, respectively). Remarkably, the same ITGAX allele was associated with higher susceptibility to IgA nephropathy and systemic lupus erythematosus (104, 105). The rs11574637*C (ITGAX) and rs4807895*T (C3) combined are protective against PF (OR = 0.55 [95% CI = 0.32–0.95], p = 0.0276). The same occurs with the rs11574637*C (ITGAX) and the rs12928810*A (ITGAM) (OR = 0.59 [95% CI = 0.38–0.90], p = 0.0115). By contrast, individuals presenting both the rs10404456*C (C5AR1) and rs12928810*G/G (ITGAM) are more susceptible to PF (OR = 2.33 [95% CI = 1.27–4.28], p = 0.0035), as were those with rs10404456*C (C5AR1) and rs11574637*T/T (ITGAX) (OR = 2.64 [95% CI = 1.49–4.66], p = 0.0006). In a previous study of our group, the mRNA expression levels of ITGAM were increased in CD4+ T cells of PF patients with generalized lesions, whereas ITGAX mRNA expression decreased after treatment (20).
Perspectives
Complement gene associations reinforced the findings of former studies, regarding the alternative pathway, C3 and MAC deposition on epidermal cells. Our results shed light on previously barely noticed processes, notably CS-mediated signaling, especially by MASP-1, and removal of opsonized elements, through complement receptors. The role of antigen-presenting phagocytes bearing CR1, CR2, CR3, and CR4, as dendritic and Langerhans cells, should be deeper investigated, since they probably exert crucial roles in the events preceding B cell activation and autoantibody production. Furthermore, lectin and alternative pathways, activated at low levels, are probably important to prevent the disease. Taken together, the results on these pathways lead us to suggest caution on the possible use of the two available complement-inhibiting drugs, able to prevent classical/lectin pathway initiation (C1INH) and MAC generation (Eculizumab), since complement activation appears desirable to PF prevention. Strong evidence for MASP1 association, but not for MASP2 or other genes of the lectin pathway, favor a pathogenic role carried out by MASP-1 in eliciting p38MAPK signaling and consequent Dsg1 clustering on the keratinocyte cell membrane. Functional validation of the pathogenic roles exerted by this wide-reaching network of complement components will open new windows to understand PF etiology and development, hopefully improving therapeutic interventions.
Ethics Statement
This study was carried out in accordance with the recommendations of the guidelines of the Conselho Nacional de Ética em Pesquisa (CONEP) with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by CONEP (number 505.988).
Author Contributions
MP-E, AB, GC, RA, and DA contributed to conception of the work. AB and VB-B designed the study. MP-E provided the samples. DA performed microarray hybridization. VB-B and RA did the statistical analysis. VB-B, AB, and GC drafted the manuscript. All authors revised the work critically for intellectual content and approved the final version of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer HB and handling Editor declared their shared affiliation.
Acknowledgments
We gratefully acknowledge the patients for their participation in this study. We also thank the staff of the Laboratório de Genética Molecular Humana/UFPR for their assistance and helpful discussions. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from https://www.gtexportal.org on 16/01/2018.
Funding
This work was supported by grants of the following funding agencies: Fundação Araucária (FA protocol 39894.413.43926.1904/2013), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq protocol 470483/2014-8), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (a post-doc scholarship with bench rate for DA: CAPES protocol 400648/2014-8).
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.00695/full#supplementary-material.
References
1. Amagai M, Stanley JR. Desmoglein as a target in skin disease and beyond. J Invest Dermatol (2012) 132:776–84. doi:10.1038/jid.2011.390
2. Spindler V, Drenckhahn D, Zillikens D, Waschke J. Pemphigus IgG causes skin splitting in the presence of both desmoglein 1 and desmoglein 3. Am J Pathol (2007) 171(3):906–16. doi:10.2353/ajpath.2007.070028
3. Sokol E, Kramer D, Diercks GFH, Kuipers J, Jonkman MF, Pas HH, et al. Large-scale electron microscopy maps of patient skin and mucosa provide insight into pathogenesis of blistering diseases. J Invest Dermatol (2015) 135(7):1763–70. doi:10.1038/jid.2015.109
4. Diaz LA, Sampaio SA, Rivitti EA, Martins CR, Cunha PR, Lombardi C, et al. Endemic pemphigus foliaceus (fogo selvagem): I. Clinical features and immunopathology. J Am Acad Dermatol (1989) 20(4):657–69. doi:10.1016/S0190-9622(89)70079-7
5. Castro RM, Roscoe JT, Sampaio SAP. Brazilian pemphigus foliaceus. Clin Dermatol (1983) 1(2):22–41. doi:10.1016/0738-081X(83)90021-4
6. Meyer N, Misery L. Geoepidemiologic considerations of auto-immune pemphigus. Autoimmun Rev (2010) 9(5):379–82. doi:10.1016/j.autrev.2009.10.009
7. Alpsoy E, Akman-Karakas A, Uzun S. Geographic variations in epidemiology of two autoimmune bullous diseases: pemphigus and bullous pemphigoid. Arch Dermatol Res (2015) 307(4):291–8. doi:10.1007/s00403-014-1531-1
8. Bastuji-Garin S, Souissi R, Blum L, Turki H, Nouira R, Jomaa B, et al. Comparative epidemiology of pemphigus in Tunisia and France: unusual incidence of pemphigus foliaceus in young Tunisian women. J Invest Dermatol (1995) 104(2):302–5. doi:10.1111/1523-1747.ep12612836
9. Friedman H, Campbell I, Rocha-Alvarez R, Ferrari I, Coimbra CE, Moraes JR, et al. Endemic pemphigus foliaceus (fogo selvagem) in native Americans from Brazil. J Am Acad Dermatol (1995) 32:949–56. doi:10.1016/0190-9622(95)91330-0
10. Hans-Filho G, dos Santos V, Katayama JH, Aoki V, Rivitti EA, Sampaio SAP, et al. An active focus of high prevalence of fogo selvagem on an Amerindian reservation in Brazil. J Invest Dermatol (1996) 107:68–75. doi:10.1111/1523-1747.ep12298213
11. Abréu-vélez AM, Messias-Reason IJ, Howard MS, Roselino A. Endemic pemphigus foliaceus over a century: part I. N Am J Med Sci (2010) 2(3):51–9.
12. Stanley JR, Klaus-Kovtun V, Sampaio SAP. Antigenic specificity of fogo selvagem autoantibodies is similar to North American pemphigus foliaceus and distinct from pemphigus vulgaris autoantibodies. J Invest Dermatol (1986) 87(2):197–201. doi:10.1111/1523-1747.ep12695334
13. Diaz LA, Prisayanh PS, Dasher DA, Li N, Evangelista F, Aoki V, et al. The IgM anti-desmoglein 1 response distinguishes Brazilian pemphigus foliaceus (fogo selvagem) from other forms of pemphigus. J Invest Dermatol (2008) 128(3):667–75. doi:10.1038/sj.jid.5701121
14. Qian Y, Prisayanh P, Andraca E, Qaqish BF, Aoki V, Hans-Filhio G, et al. IgE, IgM, and IgG4 anti-desmoglein 1 autoantibody profile in endemic pemphigus foliaceus (fogo selvagem). J Invest Dermatol (2011) 131(4):985–7. doi:10.1038/jid.2010.403
15. Qian Y, Jeong JS, Abdeladhim M, Valenzuela JG, Aoki V, Hans-Filhio G, et al. IgE anti-LJM11 sand fly salivary antigen may herald the onset of fogo selvagem in endemic Brazilian regions. J Invest Dermatol (2015) 135(3):913–5. doi:10.1038/jid.2014.430
16. Reis VM, Toledo RP, Lopez A, Diaz LA, Martins JE. UVB-induced acantholysis in endemic pemphigus foliaceus (fogo selvagem) and pemphigus vulgaris. J Am Acad Dermatol (2000) 42:571–6. doi:10.1016/S0190-9622(00)90167-1
17. Lombardi C, Borges PC, Chaul A, Sampaio SA, Rivitti EA, Friedman H, et al. Environmental risk factors in the endemic pemphigus foliaceus (fogo selvagem). J Invest Dermatol (1992) 98(6):847–50. doi:10.1111/1523-1747.ep12456932
18. Vernal S, Pepinelli M, Casanova C, Goulart TM, Kim O, De Paula NA, et al. Insights into the epidemiological link between biting flies and pemphigus foliaceus in Southeastern Brazil. Acta Trop (2017) 176:455–62. doi:10.1016/j.actatropica.2017.09.015
19. Qian Y, Jeong JS, Maldonado M, Valenzuela JG, Gomes R, Evangelista F, et al. Brazilian pemphigus foliaceus anti-desmoglein 1 autoantibodies cross-react with sand fly salivary LJM11 antigen. J Immunol (2012) 189(4):1535–9. doi:10.4049/jimmunol.1200842
20. Malheiros D, Panepucci RA, Roselino AM, Araújo AG, Zago MA, Petzl-Erler ML. Genome-wide gene expression profiling reveals unsuspected molecular alterations in pemphigus foliaceus. Immunology (2014) 143(3):381–95. doi:10.1111/imm.12315
21. Camargo CM, Augusto DG, Petzl-Erler M. Differential gene expression levels might explain association of LAIR2 polymorphisms with pemphigus. Hum Genet (2016) 135(2):233–44. doi:10.1007/s00439-015-1626-6
22. Petzl-Erler ML, Santamaria J. Are HLA class II genes controlling susceptibility and resistance to Brazilian pemphigus foliaceus (fogo selvagem)? Tissue Antigens (1989) 33(3):408–14. doi:10.1111/j.1399-0039.1989.tb01684.x
23. Pavoni DP, Roxo VMMS, Marquart Filho A, Petzl-Erler ML. Dissecting the associations of endemic pemphigus foliaceus (fogo selvagem) with HLA-DRB1 alleles and genotypes. Genes Immun (2003) 4(2):110–6. doi:10.1038/sj.gene.6363939
24. Piovezan BZ, Petzl-Erler ML. Both qualitative and quantitative genetic variation of MHC class II molecules may influence susceptibility to autoimmune diseases: the case of endemic pemphigus foliaceus. Hum Immunol (2013) 74(9):1134–40. doi:10.1016/j.humimm.2013.06.008
25. Pereira NF, Hansen JA, Lin MT, Roxo VMMS, Braun K, Petzl-Erler ML. Cytokine gene polymorphisms in endemic pemphigus foliaceus: a possible role for IL6 variants. Cytokine (2004) 28(6):233–41. doi:10.1016/j.cyto.2004.08.006
26. Malheiros D, Petzl-Erler ML. Individual and epistatic effects of genetic polymorphisms of B-cell co-stimulatory molecules on susceptibility to pemphigus foliaceus. Genes Immun (2009) 10(6):547–58. doi:10.1038/gene.2009.36
27. Dalla-Costa R, Pincerati MR, Beltrame MH, Malheiros D, Petzl-Erler ML. Polymorphisms in the 2q33 and 3q21 chromosome regions including T-cell coreceptor and ligand genes may influence susceptibility to pemphigus foliaceus. Hum Immunol (2010) 71(8):809–17. doi:10.1016/j.humimm.2010.04.001
28. Augusto DG, Lobo-Alves SC, Melo MF, Pereira NF, Petzl-Erler ML. Activating KIR and HLA Bw4 ligands are associated to decreased susceptibility to pemphigus foliaceus, an autoimmune blistering skin disease. PLoS One (2012) 7(7):e39991. doi:10.1371/journal.pone.0039991
29. Augusto DG, O’Connor GM, Lobo-Alves SC, Bass S, Martin MP, Carrington M, et al. Pemphigus is associated with KIR3DL2 expression levels and provides evidence that KIR3DL2 may bind HLA-A3 and A11 in vivo. Eur J Immunol (2015) 45(7):2052–60. doi:10.1002/eji.201445324
30. Cipolla GA, Park JK, de Oliveira LA, Lobo-Alves SC, de Almeida RC, Farias TDJ, et al. A 3′UTR polymorphism marks differential KLRG1 mRNA levels through disruption of a miR-584-5p binding site and associates with pemphigus foliaceus susceptibility. Biochim Biophys Acta (2016) 1859(10):1306–13. doi:10.1016/j.bbagrm.2016.07.006
31. Salviano-Silva A, Petzl-Erler ML, Boldt ABW. CD59 polymorphisms are associated with gene expression and different sexual susceptibility to pemphigus foliaceus. Autoimmunity (2017) 50(6):377–85. doi:10.1080/08916934.2017.1329830
32. Flores G, Culton DA, Prisayanh P, Qaqish BF, James K, Maldonado M, et al. IgG autoantibody response against keratinocyte cadherins in endemic pemphigus foliaceus (fogo selvagem). J Invest Dermatol (2012) 132(11):2573–80. doi:10.1038/jid.2012.232
33. Roscoe JT, Diaz L, Sampaio SAP, Castro RM, Labib RS, Takahashi Y, et al. Brazilian pemphigus foliaceus autoantibodies are pathogenic to BALB/c mice by passive transfer. J Invest Dermatol (1985) 85(6):538–41. doi:10.1111/1523-1747.ep12277362
34. Rock B, Martins CR, Theofilopoulos AN, Balderas RS, Anhalt GJ, Labib RS, et al. The pathogenic effect of IgG4 autoantibodies in endemic pemphigus foliaceus (fogo selvagem). N Engl J Med (1989) 320(22):1463–9. doi:10.1056/NEJM198906013202206
35. Rock B, Labib RS, Diaz LA. Monovalent Fab’ immunoglobulin fragments from endemic pemphigus foliaceus autoantibodies reproduce the human disease in neonatal Balb/c mice. J Clin Invest (1990) 85:296–9. doi:10.1172/JCI114426
36. Warren SJ, Arteaga LA, Rivitti EA, Aoki V, Hans-Filho G, Qaqish BF, et al. The role of subclass switching in the pathogenesis of endemic pemphigus foliaceus. J Invest Dermatol (2003) 120(1):104–8. doi:10.1046/j.1523-1747.2003.12017.x
37. Qaqish BF, Prisayanh P, Qian Y, Andraca E, Li N, Aoki V, et al. Development of an IgG4-based classifier/predictor of endemic pemphigus foliaceus (fogo selvagem). J Invest Dermatol (2009) 129(1):110–8. doi:10.1038/jid.2008.189
38. Maldonado M, Diaz LA, Prisayanh P, Yang J, Qaqish BF, Aoki V, et al. Divergent specificity development of IgG1 and IgG4 autoantibodies in endemic pemphigus foliaceus (fogo selvagem). Immunohorizons (2017) 1(6):71–80. doi:10.4049/immunohorizons.1700029
39. Li N, Aoki V, Hans-Filho G, Rivitti EA, Diaz LA. The role of intramolecular epitope spreading in the pathogenesis of endemic pemphigus foliaceus (fogo selvagem). J Exp Med (2003) 197(11):1501–10. doi:10.1084/jem.20022031
40. Aoki V, Millikan RC, Rivitti EA, Hans-Filho G, Eaton DP, Warren SJP, et al. Environmental risk factors in endemic pemphigus foliaceus (fogo selvagem). J Investig Dermatol Symp Proc (2004) 9(1):34–40. doi:10.1111/1346-8138.12675
41. Messias-Reason IJ, von Kuster LC, Santamaria J, Kajdacsy-Balla A. Complement and antibody deposition in Brazilian pemphigus foliaceus and correlation of disease activity with circulating antibodies. Arch Dermatol (1988) 124(11):1664–8. doi:10.1001/archderm.1988.01670110024005
42. Kawana S, Geoghegan WD, Jordon RE, Nishiyanu S. Deposition of membrane attack complex of complement in pemphigus vulgaris and in pemphigus foliaceus skin. J Invest Dermatol (1989) 92(4):588–92. doi:10.1111/1523-1747.ep12709624
43. Pegas JR, dos Reis VMS. Direct immunofluorescence on uninvolved, lesional and perilesional skin in patients with endemic pemphigus foliaceus (fogo selvagem). Med Sci Monit (2004) 10(12):657–62.
44. Odo MEY, Rodrigues RML, Miyauchi L. Direct immunofluorescence in skin biopsy of Brazilian pemphigus foliaceus. An Bras Dermatol (1981) 56(2):1–5.
45. Hernandez C, Amagai M, Chan L. Pemphigus foliaceus: preferential binding of IgGl and C3 at the upper epidermis. Br J Dermatol (1997) 136:249–52. doi:10.1111/j.1365-2133.1997.tb14907.x
46. Grando SA. Pemphigus autoimmunity: hypotheses and realities. Autoimmunity (2012) 45(1):1–35. doi:10.3109/08916934.2011.606444
47. Beltrame MH, Catarino SJ, Goeldner I, Boldt ABW, Messias-Reason IJ. The lectin pathway of complement and rheumatic heart disease. Front Pediatr (2015) 2:148. doi:10.3389/fped.2014.00148
48. Holmskov U, Thiel S, Jensenius JC. Collectins and ficolins: humoral lectins of the innate immune defense. Annu Rev Immunol (2003) 21(1):547–78. doi:10.1146/annurev.immunol.21.120601.140954
49. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol (2010) 11(9):785–97. doi:10.1038/ni.1923
50. Gros P, Milder FJ, Janssen BJC. Complement driven by conformational changes. Nat Rev Immunol (2008) 8(1):48–58. doi:10.1038/nri2231
51. Kawana S, Janson M, Jordon RE. Complement fixation by pemphigus antibody. I. In vitro fixation to organ and tissue culture skin. J Invest Dermatol (1984) 48(5):506–10. doi:10.1111/1523-1747.ep12261058
52. Kawana S, Geoghegan WD, Jordon RE. Complement fixation by pemphigus antibody. II. Complement enhanced detachment of epidermal cells. Clin Exp Immunol (1985) 61(3):517–25.
53. Kawana S, Geoghegan WD, Jordon R. Complement fixation by pemphigus antibody. III. Altered epidermal cell membrane integrity mediated by pemphigus antibody and complement. J Invest Dermatol (1986) 86(1):29–33. doi:10.1111/1523-1747.ep12283762
54. Doubleday CW, Geoghegan WD, Jordon R. Complement fixation by pemphigus antibody. IV. Enhanced epidermal cell detachment in the absence of human plasminogen. J Lab Clin Med (1988) 111(1):28–34.
55. Xia P, Jordon RE, Geoghegan W. Complement fixation by pemphigus antibody. V. Assembly of the membrane attack complex on cultured human keratinocytes. J Clin Invest (1988) 82(6):1939–47. doi:10.1172/JCI113813
56. Hahn K, Kippes W, Amagai M, Rzany B, Bröcker EB, Zillikens D. Clinical aspects and immunopathology in 48 patients with pemphigus. Hautarzt (2000) 51(9):670–7. doi:10.1007/s001050051193
57. Hashimoto K, Shafran KM, Webber PS, Lazarus GS, Singer K. Anti-cell surface pemphigus autoantibody stimulates plasminogen activator activity of human epidermal cells – a mechanism for the loss of epidermal cohesion and blister formation. J Exp Med (1983) 157(1):259–72. doi:10.1084/jem.157.1.259
58. Ablin RJ. Levels of C’3 in the serum of patients with pemphigus. J Invest Dermatol (1971) 56(6):450–3. doi:10.1111/1523-1747.ep12261375
59. Messias-Reason IJ, Santamaria J, Ragiotto R, Doi EM, Kajdacsy-Balla A. Complement activation in Brazilian pemphigus foliaceus. Clin Exp Dermatol (1989) 14(1):51–5. doi:10.1111/j.1365-2230.1989.tb00883.x
60. Franquini J, Adad SJ, Murta AH, de Morais CA, Teixeira Vde P, Júnior VR. Tests of inflammatory activity in endemic pemphigus foliaceus. Rev Soc Bras Med Trop (1994) 27(1):25–9.
61. Harman KE, Seed PT, Gratian MJ, Bhogal BS, Challacombe SJ, Black MM. The severity of cutaneous and oral pemphigus is related to desmoglein 1 and 3 antibody levels. Br J Dermatol (2001) 144(4):775–80. doi:10.1046/j.1365-2133.2001.04132.x
62. Kwon EJ, Yamagami J, Nishikawa T, Amagai M. Anti-desmoglein IgG autoantibodies in patients with pemphigus in remission. J Eur Acad Dermatol Venereol (2008) 22(9):1070–5. doi:10.1111/j.1468-3083.2008.02715.x
63. Messias-Reason IJ, Bosco DG, Nisihara RM, Jakobsen LH, Petzl-Erler ML, Jensenius JC. Circulating levels of mannan-binding lectin (MBL) and MBL-associated serine protease 2 in endemic pemphigus foliaceus. Clin Exp Dermatol (2008) 33(4):495–7. doi:10.1111/j.1365-2230.2008.02743.x
64. Messias-Reason IJ, Nisihara RM, Mocelin V. Mannan-binding lectin and ficolin deposition in skin lesions of pemphigus. Arch Dermatol Res (2011) 303:521–5. doi:10.1007/s00403-011-1132-1
65. España A, Diaz LA, Mascaró JM, Giudice GJ, Fairley JA, Till GO, et al. Mechanisms of acantholysis in pemphigus foliaceus. Clin Immunol Immunopathol (1997) 85(1):83–9. doi:10.1006/clin.1997.4407
66. Hacker MK, Janson M, Fairley JA, Lin MS. Isotypes and antigenic profiles of pemphigus foliaceus and pemphigus vulgaris autoantibodies. Clin Immunol (2002) 105(1):64–74. doi:10.1006/clim.2002.5259
67. Arteaga LA, Prisayanh PS, Warren SJ, Liu Z, Diaz LA, Lin M-S. A subset of pemphigus foliaceus patients exhibits pathogenic autoantibodies against both desmoglein-1 and desmoglein-3. J Invest Dermatol (2002) 118(5):806–11. doi:10.1046/j.1523-1747.2002.01743.x
68. Ding X, Diaz LA, Fairley JA, Giudice GJ, Liu Z. The anti-desmoglein 1 autoantibodies in pemphigus vulgaris sera are pathogenic. J Invest Dermatol (1999) 112(5):739–43. doi:10.1046/j.1523-1747.1999.00585.x
69. Mahoney MG, Hu Y, Brennan D, Bazzi H, Christiano AM, Wahl JK. Delineation of diversified desmoglein distribution in stratified squamous epithelia: implications in diseases. Exp Dermatol (2006) 15(2):101–9. doi:10.1111/j.1600-0625.2006.00391.x
70. Karsten CM, Köhl J. The immunoglobulin, IgG Fc receptor and complement triangle in autoimmune diseases. Immunobiology (2012) 217:1067–79. doi:10.1016/j.imbio.2012.07.015
71. Li N, Zhao M, Wang J, Liu Z, Diaz LA. Involvement of the apoptotic mechanism in pemphigus foliaceus autoimmune injury of the skin. J Immunol (2009) 182(1):711–7. doi:10.4049/jimmunol.182.1.711
72. Janse IC, Wier VDG, Jonkman MF, Pas HH, Diercks GFH. No evidence of apoptotic cells in pemphigus acantholysis. J Invest Dermatol (2014) 134(7):2039–41. doi:10.1038/jid.2014.60
73. van der Wier G, Jonkman MF, Pas HH, Diercks GF. Ultrastructure of acantholysis in pemphigus foliaceus re-examined from the current perspective. Br J Dermatol. (2012) 167(6):1265–71. doi:10.1111/j.1365-2133.2012.11173.x
74. Zuccolotto I, Roselino AM, Ramalho LNZ, Zucoloto S. Apoptosis and p63 expression in the pathogenesis of bullous lesions of endemic pemphigus foliaceus. Arch Dermatol Res (2003) 295(7):284–6. doi:10.1007/s00403-003-0434-3
75. Rodrigues DBR, Pereira SAL, dos Reis MA, Adad SJ, Caixeta JE, Chiba AM, et al. In situ detection of inflammatory cytokines and apoptosis in pemphigus foliaceus patients. Arch Pathol Lab Med (2009) 133(1):97–100. doi:10.1043/1543-2165-133.1.97
76. Palianus J, Meri S. Complement system in dermatological diseases – fire under the skin. Front Med (2015) 2:3. doi:10.3389/fmed.2015.00003
77. Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turning offensive. Nat Rev Nephrol (2016) 12(7):383–401. doi:10.1038/nrneph.2016.70
78. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet (2007) 81(3):559–75. doi:10.1086/519795
79. Amin MN, Islam AZMM. Clinical, histologic and immunologic features of pemphigus in Bangladesh. Int J Dermatol (2006) 45(11):1317–8. doi:10.1111/j.1365-4632.2006.02942.x
80. Júnior JVO, Maruta CW, Sousa JX, Santi CG, Valente NYS, Ichimura LMF, et al. Clinical and immunological profile of umbilical involvement in pemphigus vulgaris and pemphigus foliaceus. Clin Exp Dermatol (2013) 38(1):20–4. doi:10.1111/j.1365-2230.2012.04468.x
81. Schonthaler HB, Guinea-Viniegra J, Wculek SK, Ruppen I, Ximénez-Embún P, Guío-Carrión A, et al. S100A8-S100A9 protein complex mediates psoriasis by regulating the expression of complement factor C3. Immunity (2013) 39(6):1171–81. doi:10.1016/j.immuni.2013.11.011
82. Ma Q, Li D, Carreño R, Patenia R, Tsai KY, Xydes-Smith M, et al. Complement component C3 mediates Th1/Th17 polarization in human T cell activation and cutaneous graft-versus-host disease. Bone Marrow Transplant (2014) 49(7):972–6. doi:10.1038/bmt.2014.75
83. Consortium TG. The genotype-tissue expression (GTEx) project. Nat Genet (2013) 45(6):580–5. doi:10.1038/ng.2653
84. Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, et al. Ensembl 2018. Nucleic Acids Res (2018) 46:754–61. doi:10.1371/journal.pone.0051647
85. Nishikawa T, Kurihara S, Harada T, Sugawara M, Hatano H. Capability of complement fixation of pemphigus antibodies in vitro. Arch Dermatol Res (1977) 260:1–6. doi:10.1007/BF00558008
86. Boldt ABW, Boschmann SE, Catarino SJ, Andrade FA, Messias-Reason IJ. MASP1 and MASP2. In: Choi S, editor. Encyclopedia of Signaling Molecules. 2nd ed. Springer (2018). p. 2972–89. doi:10.1007/978-3-319-67199-4
87. Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res (2002) 12(6):996–1006. doi:10.1101/gr.229102
88. Ingels C, Vanhorebeek I, Steffensen R, Derese I, Jensen L, Wouters PJ, et al. Lectin pathway of complement activation and relation with clinical complications in critically ill children. Pediatr Res (2014) 75(1):99–108. doi:10.1038/pr.2013.180
89. Ammitzboll CG, Steffensen R, Nielsen HJ, Thiel S, Stengaard-Pedersen K, Bogsted M, et al. Polymorphisms in the MASP1 gene are associated with serum levels of MASP-1, MASP-3, and MAp44. PLoS Genet (2013) 8(9):e73317. doi:10.1371/journal.pone.0073317
90. Degn SE, Jensen L, Hansen AG, Duman D, Tekin M, Jensenius JC, et al. Mannan-binding lectin-associated serine protease (MASP)-1 is crucial for lectin pathway activation in human serum, whereas neither MASP-1 nor MASP-3 is required for alternative pathway function. J Immunol (2012) 189(8):3957–69. doi:10.4049/jimmunol.1201736
91. Dobó J, Pál G, Cervenak L, Gál P. The emerging roles of mannose-binding lectin-associated serine proteases (MASPs) in the lectin pathway of complement and beyond. Immunol Rev (2016) 274(1):98–111. doi:10.1111/imr.12460
92. Jani PK, Kajdácsi E, Megyeri M, Dobó J, Doleschall Z, Futosi K, et al. MASP-1 induces a unique cytokine pattern in endothelial cells: a novel link between complement system and neutrophil granulocytes. PLoS One (2014) 9(1):e87104. doi:10.1371/journal.pone.0087104
93. Megyeri M, Mako V, Beinrohr L, Doleschall Z, Prohaszka Z, Cervenak L, et al. Complement protease MASP-1 activates human endothelial cells: PAR4 activation is a link between complement and endothelial function. J Immunol (2009) 183(5):3409–16. doi:10.4049/jimmunol.0900879
94. Lee SH, Hong WJ, Kim S. Analysis of serum cytokine profile in pemphigus. Ann Dermatol (2017) 29(4):438–45. doi:10.5021/ad.2017.29.4.438
95. Timóteo RP, Silva MV, Alves-Silva DA, Catarino JS, Alves FHC, Junior VR, et al. Cytokine and chemokines alterations in the endemic form of pemphigus foliaceus (fogo selvagem). Front Immunol (2017) 8:978. doi:10.3389/fimmu.2017.00978
96. Hoss DM, Shea CR, Grant-Kels J. Neutrophilic spongiosis in pemphigus. Arch Dermatol (1996) 132(3):315–8. doi:10.1001/archderm.132.3.315
97. Berkowitz P, Chua M, Liu Z, Diaz LA, Rubenstein DS. Autoantibodies in the autoimmune disease pemphigus foliaceus induce blistering via p38 mitogen-activated protein kinase-dependent signaling in the skin. Am J Pathol (2008) 173(6):1628–36. doi:10.2353/ajpath.2008.080391
98. Yoshida K, Ishii K, Shimizu A, Yokouchi M, Amagai M, Shiraishi K, et al. Non-pathogenic pemphigus foliaceus (PF) IgG acts synergistically with a directly pathogenic PF IgG to increase blistering by p38MAPK-dependent desmoglein 1 clustering. J Dermatol Sci (2017) 85(3):197–207. doi:10.1016/j.jdermsci.2016.12.010
99. Mejri K, Mbarek H, Kallel-Sellami M, Petit-Teixeira E, Zerzeri Y, Abida O, et al. TRAF1/C5 polymorphism is not associated with pemphigus. Br J Dermatol (2009) 160(6):1348–50. doi:10.1111/j.1365-2133.2009.09136.x
100. Karsten CM, Laumonnier Y, Eurich B, Ender F, Bröker K, Roy S, et al. Monitoring and cell-specific deletion of C5aR1 using a novel floxed GFP-C5aR1 reporter knock-in mouse. J Immunol (2015) 194:1841–55. doi:10.4049/jimmunol.1401401
101. Tegla CA, Cudrici C, Patel S, Trippe R, Rus V, Niculescu F, et al. Membrane attack by complement: the assembly and biology of terminal complement complexes. Immunol Res (2011) 51(1):45–60. doi:10.1007/s12026-011-8239-5
102. Shen N, Chen B, Jiang Y, Feng R, Liao M, Zhang L, et al. An updated analysis with 85,939 samples confirms the association between CR1 rs6656401 polymorphism and Alzheimer’s disease. Mol Neurobiol (2015) 51(3):1017–23. doi:10.1007/s12035-014-8761-2
103. Hannan J. The structure-function relationships of complement receptor type 2 (CR2; CD21). Curr Protein Pept Sci (2016) 17(5):463–87. doi:10.2174/1389203717666151201192124
104. Kiryluk K, Li Y, Sanna-Cherchi S, Rohanizadegan M, Suzuki H, Eitner F, et al. Geographic differences in genetic susceptibility to IgA nephropathy: GWAS replication study and geospatial risk analysis. PLoS Genet (2012) 8(6):e1002765. doi:10.1371/journal.pgen.1002765
Keywords: pemphigus foliaceus, complement, lectin pathway, acantholysis, membrane attack complex, alternative pathway, opsonin, complement receptors
Citation: Bumiller-Bini V, Cipolla GA, de Almeida RC, Petzl-Erler ML, Augusto DG and Boldt ABW (2018) Sparking Fire Under the Skin? Answers From the Association of Complement Genes With Pemphigus Foliaceus. Front. Immunol. 9:695. doi: 10.3389/fimmu.2018.00695
Received: 29 January 2018; Accepted: 21 March 2018;
Published: 09 April 2018
Edited by:
Ralf J. Ludwig, Universität zu Lübeck, GermanyReviewed by:
Abhigyan Satyam, Harvard Medical School, United StatesHauke Busch, Universität zu Lübeck, Germany
Copyright: © 2018 Bumiller-Bini, Cipolla, de Almeida, Petzl-Erler, Augusto and Boldt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angelica Beate Winter Boldt, YW5nZWxpY2Fib2xkdEBnbWFpbC5jb20=
†Present address: Rodrigo Coutinho de Almeida, Molecular Epidemiology, Department of Biomedical Data Sciences, Leiden University Medical Center, Leiden, Netherlands