 Wen Chean Lim
Wen Chean Lim Michael Olding
Michael Olding Eugene Healy1,2
Eugene Healy1,2 Timothy M. Millar
Timothy M. Millar
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 23 March 2018
Sec. Immunological Tolerance and Regulation
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.00565
Endothelial cells (ECs) line the luminal surface of blood vessels and have an active role in the recruitment of leukocytes, including immune cell activation. Regulatory T cells (Tregs) are immune suppressor cells that maintain peripheral tolerance and must interact with the endothelium as they traffic into tissue. We hypothesized that human ECs could modulate Tregs and their suppressor function. Cocultures of CD4+ T cells with human umbilical vein ECs (HUVECs) or dermal microvascular ECs (HDMECs) were conducted and analyzed for activation and proliferation after 72 and 120 h using flow cytometry. In monocyte-depleted cultures, human ECs were found to support CD4+ T cell proliferation in the presence of external mitogens phytohemagglutinin or anti-CD3/28 antibodies (aCD3/28). Activation was shown by CD25 expression in these cells that also transiently expressed the Treg transcription factor FOXP3. HUVECs supported the specific concurrent proliferation of both effector T cells and Tregs when cocultured with aCD3/28. Purified Tregs were also functionally activated by prior coculture with EC to suppress effector T (Teff) cell proliferation. Both direct coculture and indirect coculture of EC and Treg showed activation of the Treg suppressive phenotype. However, whereas HUVEC showed enhancement of suppression by both mechanisms, HDMEC only supported Treg suppressive activity via the contact-independent mechanism. In the contact-independent cultures, the soluble mediators IL-6, GM-CSF, or G-CSF released from ECs following interferon-γ activation were not responsible for the enhanced Treg suppressor function. Following direct coculture, Treg expression of inhibitory receptors PD-1 and OX40 was elevated while activated EC expressed the counter ligands programmed death ligand (PD-L)1 and PD-L2. Therefore, human ECs have a role in supporting T cell proliferation and increasing Treg suppressor function. This ability of EC to enhance Treg function could offer novel targets to boost Treg activity during inflammatory disorders.
The endothelium is a biologically active cellular site lining the luminal surface of blood vessels. Endothelial cells (ECs) contribute to the many physiological roles of the vasculature including coagulation, forming a selective barrier to regulate vessel permeability, and controlling vascular tone. In addition to these functions, ECs also have important immunological roles, in particular the control of leukocyte recruitment and activation (1). ECs express a variety of adhesion molecules, including selectins and integrins, which allow them to regulate recruitment and trafficking of leukocytes into an inflammatory site (2). ECs can also express surface molecules that mediate antigen presentation and provide costimulatory signals, including class I and class II major histocompatibility complexes and costimulatory molecules such as inducible T cell costimulator (ICOS)-ligand, programmed death ligand (PD-L) -1 and -2, OX40-ligand and 4-1BB-ligand (1). Evidence for the involvement of human ECs in regulating T cell activation include cytokine production and proliferation of T cells mediated via ICOS-L expression on human ECs and costimulation of resting memory CD4+ T cells to produce T helper (Th) -1 and Th2 cytokines (3). Conversely, inhibitory signals mediated through PD-L1 expression on human umbilical vein endothelial cells (HUVECs) have been shown to negatively regulate IL-2 and interferon (IFN)-γ production of phytohemagglutinin (PHA)-stimulated T cells (4).
Endothelial cells also have a role in the recruitment of regulatory T cells (Tregs). These cells were initially characterized by Sakaguchi et al. (5) as IL-2 receptor α-chain (CD25) expressing CD4+ T cells and subsequently the transcription factor Foxhead Box P3 (Foxp3) was shown to be necessary for Treg development and function (6, 7). Treg function is regulated by multiple mechanisms, including direct interaction with cells via costimulatory signals through PD-1 and OX40 and their corresponding ligands (8, 9) and indirectly by cytokine signaling via IL-6 and IL-10 (10, 11). Treg recruitment and migration into lymph nodes and peripheral tissue is pivotal in regulating their role in peripheral tolerance (12). In mice, Krupnick et al. (13) demonstrated that ECs derived from the thoracic aorta could selectively expand CD4+CD25+Foxp3+ Tregs in cocultures with CD4+ T cells. Later, Bedke et al. (14) showed that activated murine lung ECs increased the capacity of CD4+CD25+ Tregs to suppress effector T cell proliferation. More recently, human dermal ECs have been shown to induce expansion of Tregs and proinflammatory Th17 populations in cocultures with CD4+ T cells (15) but did not investigate the suppressive function of Tregs following endothelial interaction. While a further study showed that in rapamycin-treated HUVECs Treg suppressive activity was increased potentially through increased PD-L1 and PD-L2 expression (16).
Endothelial cells have, therefore, been proposed to induce Treg expansion and enhance Treg suppressive capacities but the evidence in humans and chronic inflammatory models is limited. We hypothesized that under chronic inflammatory cytokine activation the endothelium could potentially modulate T cell function in a manner that relates to chronic diseases of the skin. This aim of this present study was to demonstrate the capabilities of cytokine stimulated human ECs to modulate T cell differentiation and Treg function. This paper uses in vitro EC-T cell cocultures and shows that EC–Treg interactions are important for Treg activation and that differences exist between ECs of different lineages. We also show that ECs are capable of the induction and expansion of Tregs and that the potential mechanism(s) by which this occurs involves both direct contact and indirect signals to enhance the suppressive activity of Tregs. In the light of these and previous findings, the endothelium has a potential role to play in controlling chronic inflammation via both Teff and Treg activation and presents itself as a potential target for immune modulation in inflammation, cancer and autoimmune disease.
PerCP-Cy5.5 conjugated anti-CD4 (RPA-T4) mAb, eFluor 450 conjugated anti-CD127 (eBioRDR5) mAb, and APC conjugated anti-FOXP3 (PCH101) mAb (eBioscience, UK). PE conjugated anti-CD25 (CD25-3G10) mAb (Life Technologies, UK). PE-Cy7 conjugated PD-1 (EH12.2H7) mAb and Brilliant Violet 421 conjugated OX40 (Ber-ACT35) mAb (Biolegend, UK). IFN-γ (human, leukocyte-derived) and tumor necrosis factor (TNF)-α (human, rDNA derived) (The National Institute for Biological Standards and Control, NIBSC, UK).
M199 media (Life Technologies, UK) with 20% pooled off the clot human serum (TCS Bioscience, UK), 100 U/mL penicillin, 100 µg/mL streptomycin, and 292 μg/mL l-glutamine (Life Technologies, UK) was used for HUVEC culture, whereas EC Growth Medium MV2 (Promocell; Heidelberg, Germany) was used for HDMEC culture. Porcine gelatin (0.2%, Sigma Aldrich, UK) was used as the coating matrix for EC culture.
Methods for isolating HUVECs were adapted from protocols published by Jaffe et al. (17) and Gimbrone et al. (18). Briefly, umbilical cords were collected from the Princess Anne Hospital, Southampton, UK from non-complicated natural vaginal births following agreed ethical collection protocols [Local Research Ethical Committee (LREC); Ref: 07/H0502/83]. Veins were cannulated and incubated with collagenase (1 mg/mL of type 1 collagenase ~250 U/mg, Worthington Biochemical; Lakewood, US) for 10 min in a 37°C water bath. HUVECs were collected by centrifugation and plated on to gelatin coated plastics and grown to confluence. HUVECs were used at passage 1 in assays. Primary juvenile foreskin HDMECs were purchased from Promocell and cell numbers expanded in culture until passage 6 before being used in assays (passage 6–8). Both cell types showed coexpression of both CD31 and CD105 (Figure A in Supplementary Material)
Whole blood was collected from healthy donors with signed consent following ethically approved protocols (LREC Ref: 07/H0504/93) into BD Vacutainer® K2E tubes (BD Biosciences, UK). PBMCs were isolated from whole blood using density gradient sedimentation techniques (Lymphoprep™, Axis-Shield, UK) after dilution in sterile PBS. Buffy coats were harvested, washed, and cell counts were conducted. Cells were resuspended in PBS + 1% bovine serum albumin for cell surface staining.
PBMCs were stained with PerCP-Cy5.5 conjugated anti-CD4 (RPA-T4) mAb, PE conjugated anti-CD25 (CD25-3G10) mAb, and eFluor 450 conjugated anti-CD127 (eBioRDR5) mAb for 30 min at 4°C. Cell sorting was conducted using BD FACSAria Cell Sorter (BD Bioscience, UK). CD4+ T cells were gated as live singlet CD4+ T cells, Tregs were gated as live singlet CD4+CD25hiCD127low T cells and Teff were gated as live singlet CD4+CD25− T cells. Cells were sorted in to RPMI culture media, washed, and used immediately in functional assays. For FOXP3 analysis, cells were fixed and permeabilized using the FOXP3/Transcription factor staining buffer set (eBioscience, UK) and stained with APC conjugated anti-FOXP3 (PCH101) mAb for 30 min at 4°C.
Confluent ECs were plated onto gelatinized 24-well culture plates at 1 × 105 cells/well and cultured overnight to achieve confluence. ECs were used unstimulated or stimulated with 10 U/mL IFN-γ or 1 ng/mL TNF-α for 24 h. Isolated CD4+ T cells were stained with 5 µM CFSE. ECs were washed twice with warm RPMI media to remove stimulating cytokine media and CFSE-stained CD4+ T cells were plated at 1 × 105 cells/well an EC-CD4 ratio of 1:1. Cocultures were stimulated with 3 µg/mL PHA or 5 µg/mL aCD3 and 10 µg/mL aCD28 incubated together for 72 or 120 h. Appropriate controls of unstimulated EC or CD4+ T cell alone cultures and cocultures were also included.
At 72 or 120 h, cells and supernatants were collected and adherent cells were trypsinised and collected into FACS tubes. LIVE/DEAD fixable violet dead cell stain (405 nm; Life Technologies, UK) was added to allow for dead cell exclusion and cells were stained with antibodies for CD4, CD25, and FOXP3 expression. Flow cytometric analysis of the cocultures was completed on the BD FACSAria and data were analyzed using FlowJo software v7.6.5.
Confluent ECs were seeded at 1 × 105 cells/well 24 h before use in 24-well plates and left either unstimulated or stimulated separately with 10 U/mL IFN-γ and 1 ng/mL TNF-α or in combination for 24 h prior to coculture with T cells. CD4+CD25hiCD127low cells (Tregs), CD4+CD25− cells (Teffs) and CD4+ T cells were isolated from PBMCs using cell sorting as described. ECs were washed to remove exogenous cytokines and Tregs (30,000 cells/well) were added and cocultured with ECs in complete RPMI media for a further 24 h. Transwell assays were also conducted where Tregs were plated on to the upper chamber of a Transwell (Corning 3470 Transwell; pore size 0.4 µm; Corning Life Sciences) suspended above a monolayer of ECs. On the same day, Teffs and CD4+ cells were stained with 5 µM CFSE and rested for 24 h whereas accessory cells (47 Gy gamma-irradiated autologous PBMCs) were plated at 1 × 105 cells/well with plate-bound aCD3 antibody (2.5 µg/mL) in 96-well round-bottom plates (kept in 100 µL complete RPMI media) for the same 24 h.
After 24 h coculture, Tregs were recovered by gentle washing of EC monolayers with complete RPMI media and supernatants with detached Tregs were collected. CFSE-stained Teffs and CD4+ cells were also collected from culture plastic where they had been allowed to incubate for an equivalent time as Tregs with ECs. CFSE-stained Teffs or CD4+ T cells were then plated with the autologous aCD3-stimulated accessory cells at 25,000 cells/well and Tregs were added to Teffs at a ratio of 1:1 in the wells, with appropriate controls. After a further 72 h incubation, cells were harvested and stained with LIVE/DEAD and for CD4 and CD25 expression, then fixed and permeabilized for FOXP3 intracellular staining. Expression of PD-1 and OX40 on Tregs was also interrogated via staining cells with PE-Cy7 conjugated PD-1 (EH12.2H7) mAb and Brilliant Violet 421 conjugated OX40 (Ber-ACT35) mAb for 30 min at 4°C. Cells were analyzed by flow cytometry for Teff proliferation as indicated by dilution of CFSE fluorescence and antigen expression on the BD FACSAria. Data was analyzed using FlowJo software v7.6.5.
To detect possible soluble mediators, HUVECs or HDMECs were plated on to gelatinized 24-well plates at 1 × 105 cells/well and cultured overnight to reach confluence. Cells were stimulated for 24 h with either 10 U/mL IFN-γ, 1 ng/mL TNF-α or a combination of both cytokines with appropriate controls. Cell monolayers were then washed twice and media was replaced with 1 mL warm complete RPMI media. Cells were cultured for a further 24 h before the conditioned media were collected. Conditioned media were analyzed for cytokine, chemokine, and growth factor using ELISA or multiplex-based assays [Luminex® performance assay human cytokine panel base kit A (LUH000) and individual bead sets, Human granulocyte colony-stimulating factor (G-CSF) DuoSet® ELISA development systems (R&D systems), Human IL-6 ELISA Ready-SET-Go! ®, and Human granulocyte macrophage colony-stimulating factor (GM-CSF) ELISA Ready-SET-Go! ® (2nd Generation) (eBioscience, UK)].
Statistical significance was determined by carrying out statistical analysis using the GraphPad Prism 6 software (GraphPad Software, Inc.; La Jolla, CA, USA). One-way analysis of variance (ANOVA) was used when comparing three or more groups, whereas two-way ANOVA analysis was used for data with two affecting factors. Appropriate post hoc tests were completed, including Dunnett’s, Tukey, and Sidak methods, where mentioned.
Cocultures of CSFE-labeled CD4+ T cells with human ECs were analyzed at 72 and 120 h as shown in Figure 1A, where cells with low CFSE fluorescence were gated as proliferated cells and expression of markers including CD25 and FOXP3 were measured. Initial experiments showed that in the absence of any accessory cells, CD4+ T cells did not proliferate in response to PHA or soluble aCD3/28 (Figure 1B). When CD4+ T cells were cocultured with human ECs for 72 h in the absence of mitogens, low but significant levels of proliferation were observed in cocultures with IFN-γ-stimulated ECs. Similarly, when CD4+ T cells were allowed to interact with human ECs for 120 h, significant CD4+ T cell proliferation was seen in the absence of mitogens only in cocultures where the EC had been previously stimulated by IFN-γ or TNF-α (Figure 1C). In the presence of PHA or soluble aCD3/28 and EC acting as accessory cells, high levels of CD4+ T cell proliferation were observed (P < 0.0001, n = 10).
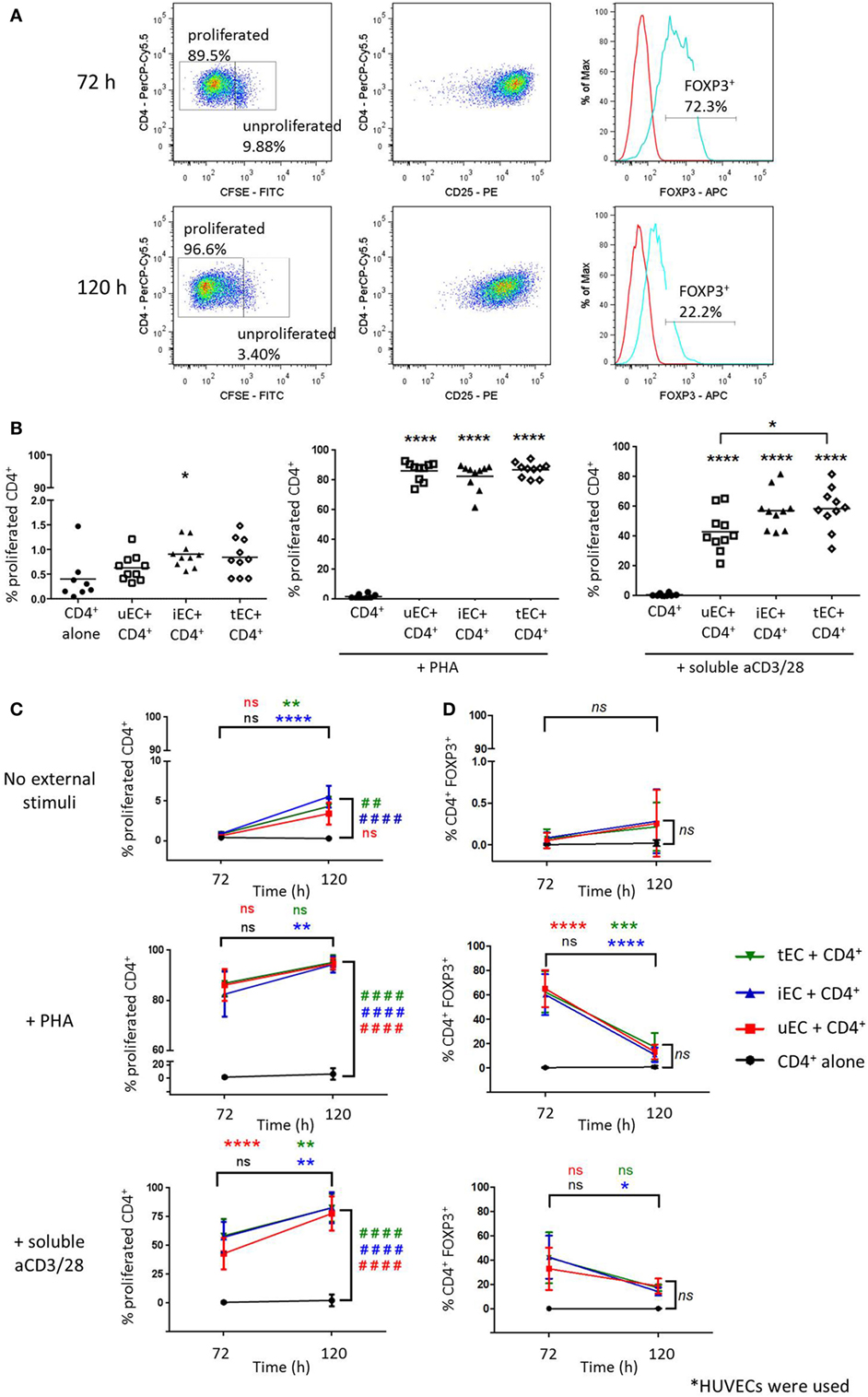
Figure 1. Proliferation of CD4+ T cells in endothelial cell (EC) cocultures and expression of FOXP3 of proliferated CD4+ T cells. Cells were harvested from cocultures of CD4+ T cells with HUVECs at 72 or 120 h and analyzed by flow cytometry, where T cells were discriminated based on CD4 expression. (A) CD4+ T cells with the highest CFSE fluorescence were gated as unproliferated whereas those with diluted/low CFSE fluorescence were gated as the proliferated population. The CD25 and FOXP3 expression of proliferated cells (blue line) were analyzed with the aid of the isotype control (red line). (B) Proliferation of CD4+ T cells cultured with unstimulated (uEC) or IFN-γ (iEC) or tumor necrosis factor (TNF)-α-stimulated (tEC) HUVECs were analyzed by CFSE dilution at 72 h, in which coculture had no external stimuli or were stimulated with phytohemagglutinin (PHA) or aCD3/28 antibodies. One-way analysis of variance (ANOVA) with Tukey test was conducted; ****P < 0.0001, *P < 0.05 cf. control; n = 10. (C) Comparison of CD4+ T cell proliferation in cocultures with uEC, iEC, or tEC at 72 and 120 h, in which cocultures had no external stimuli or were stimulated with PHA or aCD3/28 antibodies. Two-way ANOVA analysis with Tukey test was conducted; ****P < 0.0001; **P < 0.01 for 120 h cf. 72 h data; ####P < 0.0001, ##P < 0.01 cf. control for 120 h data; n = 10/6. (D) FOXP3 expression of the proliferated CD4+ T cell populations at 72 and 120 h from the different HUVEC-CD4+ T cell cocultures were compared, where data are expressed as percentage of CD4+FOXP3+ cells from the total number of CD4+ T cell analyzed, with mean ± SD shown. Two-way ANOVA with Tukey test was conducted, where ****P < 0.0001, ***P < 0.001, *P < 0.05 for data at 120 h cf. data at 72 h; n = 4.
These cells were interrogated for their intracellular FOXP3 expression. In control CD4+ T cell HUVEC cocultures, with no added mitogen, there was no significant FOXP3 expression in CD4+ T cells observed at either 72 or 120 h of culture nor was this seen when cytokine stimulated HUVEC were used as accessory cells (Figure 1D). However, high expression levels of FOXP3 were seen with proliferated T cells in mitogen-stimulated CD4+ T cell HUVEC cocultures at 72 h with both unstimulated and cytokine stimulated HUVECs. This FOXP3 expression however, was transient because at 120 h the expression levels in cocultured, mitogen activated proliferated CD4+ T cells was significantly reduced compared to 72 h and was similar to the levels seen in control mitogen-free cultures.
Cocultures of CFSE-stained CD4+ T cells with HDMECs with or without aCD3/28 were conducted (Figure 2A). In the absence of external stimulation, there was no significant T cell proliferation in these cocultures by 72 h (Figure 2B). In cocultures of CD4+ T cells and IFN-γ-stimulated HDMECs with soluble aCD3/28, there was a significant proliferation of CD4+ T cells. A significant increase in FOXP3 expression was also found when CD4+ T cells were cocultured with IFN-γ-stimulated HDMECs and soluble aCD3/28 but not in control non-cytokine stimulated cocultures (Figure 2C). Additionally, IFN-γ-activated HUVECs were capable of supporting proliferation of purified Teff and Treg populations separately in the presence of soluble aCD3/28 (Figure B in Supplementary Material).
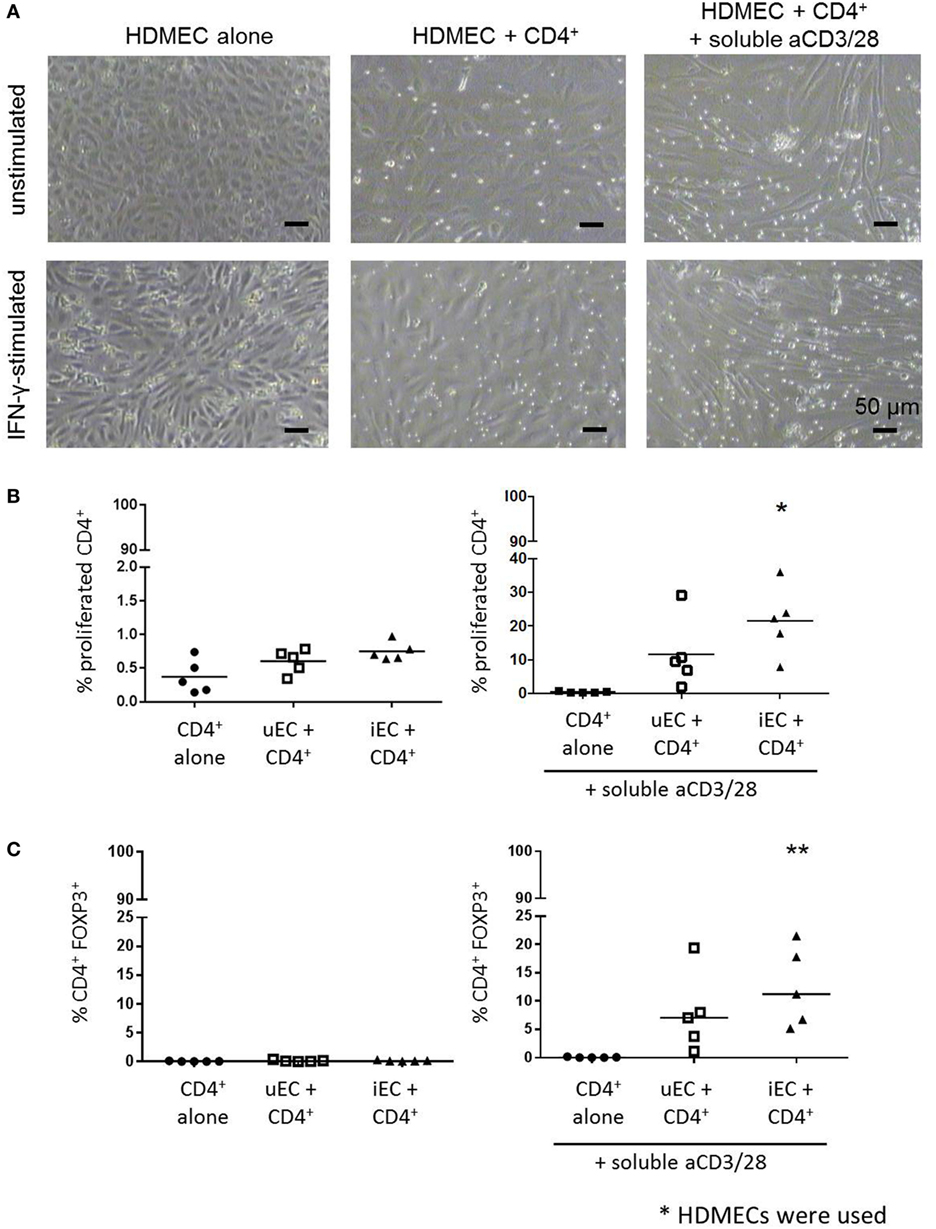
Figure 2. Proliferation of CD4+ T cells in HDMEC cocultures and their expression of FOXP3. (A) Phase contrast micrographs of HDMEC-CD4+ T cell cocultures using unstimulated or interferon (IFN)-γ-stimulated HDMECs in the absence or presence of stimulatory aCD3/28 antibodies were taken at 72 h; micrographs were taken at 100× original magnification; scale bar of 50 µm included. (B) CFSE-labeled CD4+ T cells were cocultured with HDMECs used either unstimulated (uEC) or stimulated with 10 U/mL IFN-γ (iEC) for 24 h; proliferation of CD4+ T cells was analyzed by CFSE dilution at 72 h. Proliferation of CD4+ T cells in HDMEC-CD4+ T cells cocultures stimulated with aCD3/28 antibodies were also measured. One-way analysis of variance (ANOVA) with Dunnett’s test was conducted; *P < 0.05 cf. control, n = 5. (C) FOXP3 expression of proliferated CD4+ T cells from the same HDMEC-CD4+ T cell cocultures was also analyzed at 72 h, where data are expressed as percentage of CD4+FOXP3+ cells from the total CD4+ population with means shown. One-way ANOVA with Dunnett’s test was used; **P < 0.01 cf. control; n = 5.
Cocultures of HUVECs with Tregs were conducted, where CD4+CD25hiCD127low cells were plated in direct cocultures with unstimulated (uEC, 1:1 ratio), IFN-γ-stimulated (iEC, 1:1 ratio), TNF-α-stimulated (tEC, 1:1 ratio), or both IFN-γ + TNF-α-stimulated (itEC, 1:1 ratio) HUVECs, for 24 h, then Tregs recovered and plated in a suppression assay. Purity of each population is shown in supplementary Figure C in Supplementary Material. In an assay of Treg function, despite some variation in the suppression by Tregs from different donors, control Tregs which had not been cultured with ECs suppressed Teff proliferation in the expected manner (Figure 3A, P < 0.0001; n = 16).
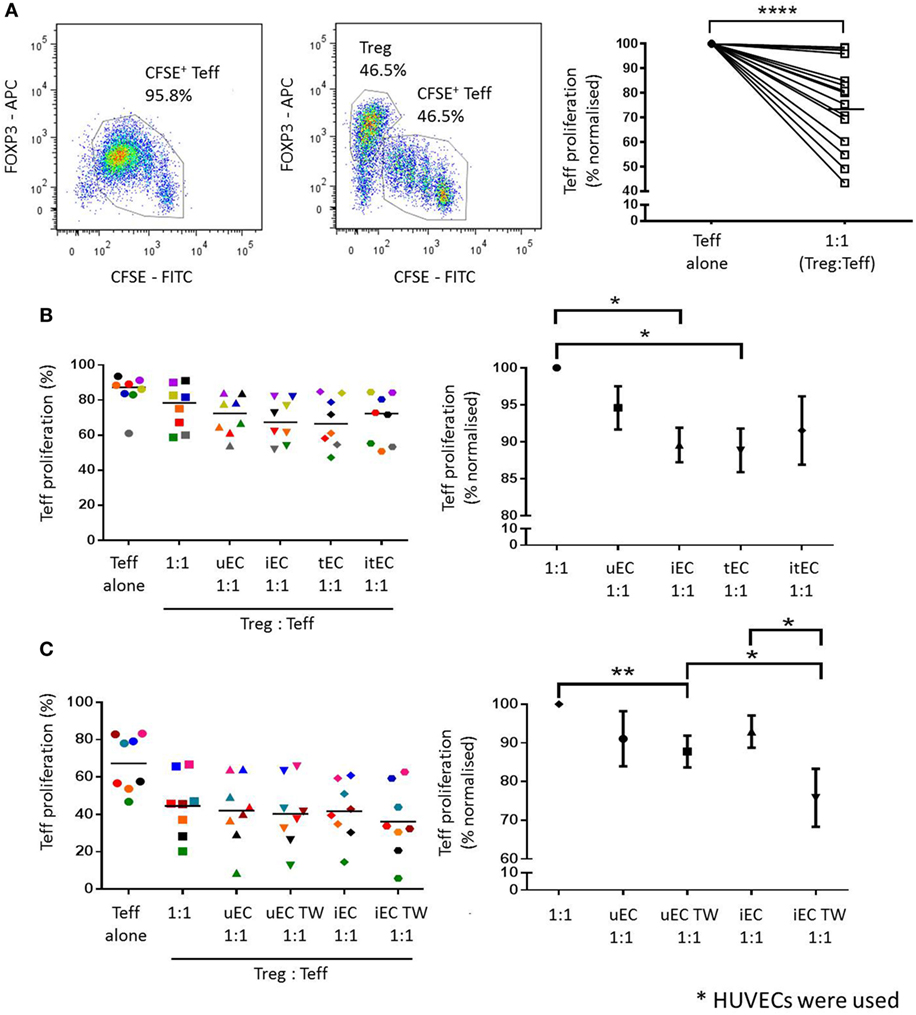
Figure 3. Enhancement of Treg suppressor function after coculture with activated human umbilical vein ECs (HUVECs). (A) Treg suppression assays were set up where CFSE-labeled Teffs were either stimulated with 5 µg/mL plate-bound aCD3 antibody (in the presence of gamma-irradiated autologous PBMCs) or with CD4+CD25hiCD127low Tregs at a 1:1 ratio. Teff proliferation was analyzed by CFSE dilution and the suppressive capacity of Tregs was measured by comparing the reduction of Teff proliferation in the presence of Tregs. Tregs from 16 different donors were found to suppress Teff proliferation in varying capacity. Two-tailed paired T test was conducted; ****P < 0.0001; n = 16. (B) CD4+CD25hiCD127low cells (Tregs) were cocultured with unstimulated (uEC), interferon (IFN)-γ- (iEC), tumor necrosis factor (TNF)-α- (tEC), or both IFN-γ and TNF-α (itEC) HUVECs for 24 h, before they were recovered and plated in a Treg suppression assay to measure suppressive capacity. Raw data is expressed as % proliferated Teff, with each experiment represented by a different color. Data are also expressed where Teff proliferation is normalized to proliferation in 1:1 cultures as 100%, showing mean ± SEM. One-way analysis of variance (ANOVA) with Dunnett’s test was used; *P < 0.05, n = 8. (C) The contact-dependent nature of the modulation of Treg function by HUVEC was investigated where Tregs were plated in direct contact with uEC or iEC, or suspended above unstimulated (uEC TW) or IFN-γ-stimulated (iEC TW) HUVECs in Transwells for 24 h, before plating in a Treg suppression assay. Raw data are expressed as % proliferated Teffs, with each experiment represented by a different color. Data are also expressed where Teff proliferation is normalized to proliferation in 1:1 cultures as 100%, showing mean ± SEM. One-way ANOVA with Dunnett’s test was used; **P < 0.01, *P < 0.05, n = 8.
After direct coculture with HUVECs, Tregs were shown to have an enhanced suppressor function as indicated by a decrease in Teff proliferation compared to controls where Tregs were not previously cocultured with HUVEC (Figure 3B). Tregs cocultured with unstimulated HUVECs (uEC 1:1) produced an additional mean reduction of 4.4% (normalized to individual control 5.4%) in Teff proliferation compared to control Tregs (1:1) in the absence of ECs. Tregs cocultured with IFN-γ- (iEC 1:1) or TNF-α-stimulated (tEC 1:1) HUVECs showed a further enhancement in their suppressive capacity causing an 8.0% (normalized 10.4%) and 8.2% (normalized 11.2%) further reduction in Teff proliferation compared to control Tregs (1:1) (P < 0.05; n = 8). Tregs cocultured with IFN-γ plus TNF-α-stimulated HUVECs also showed increased suppressive function compared to unstimulated Treg but was not statistically significant compared to the suppressor function of Tregs cocultured with HUVEC stimulated with IFN-γ or TNF-α alone.
The requirement for direct contact on the modulation of Treg function by HUVEC was next investigated using purified and isolated Tregs cultured in Transwells suspended above control or cytokine-activated ECs. Following coculture of Tregs either in contact with or suspended above control ECs, the suspended Tregs showed a small but non-significant (1.8%, normalized 3.3%) reduction in Teff proliferation (Figure 3C). However, in Tregs previously suspended over IFN-γ stimulated HUVEC, a significantly greater reduction in Teff proliferation of 5.6% (normalized 17.1%) was demonstrated compared to direct contact coculture of Tregs with IFN-γ-stimulated EC (P < 0.05; n = 8). In the contact-independent conditions, Tregs previously suspended above IFN-γ stimulated HUVECs showed a greater suppressive effect on Teff proliferation than those suspended above unstimulated HUVECs [increase of a further 4.2% (normalized 12.0%) (P < 0.05; n = 8)].
After direct coculture of Tregs with unstimulated HDMECs (uEC 1:1; Figure 4A), the suppressive capacity of Tregs on Teff proliferation was not significantly increased when compared to control Tregs (1:1). This was also the case for Tregs cultured on Transwells above unstimulated HDMECs (uEC TW 1:1). When Tregs were cocultured directly with IFN-γ-stimulated HDMECs, their suppressive capacity was not enhanced compared to control Treg suppressor function. However, Tregs cultured in Transwells above previously IFN-γ-stimulated HDMECs showed an increase in their suppressor function compared to those Tregs in direct contact with previously IFN-γ-stimulated HDMEC. Teff proliferation was suppressed by these suspended Tregs by 8.2% (17.9% normalized to control 1:1) (P < 0.05; n = 8).
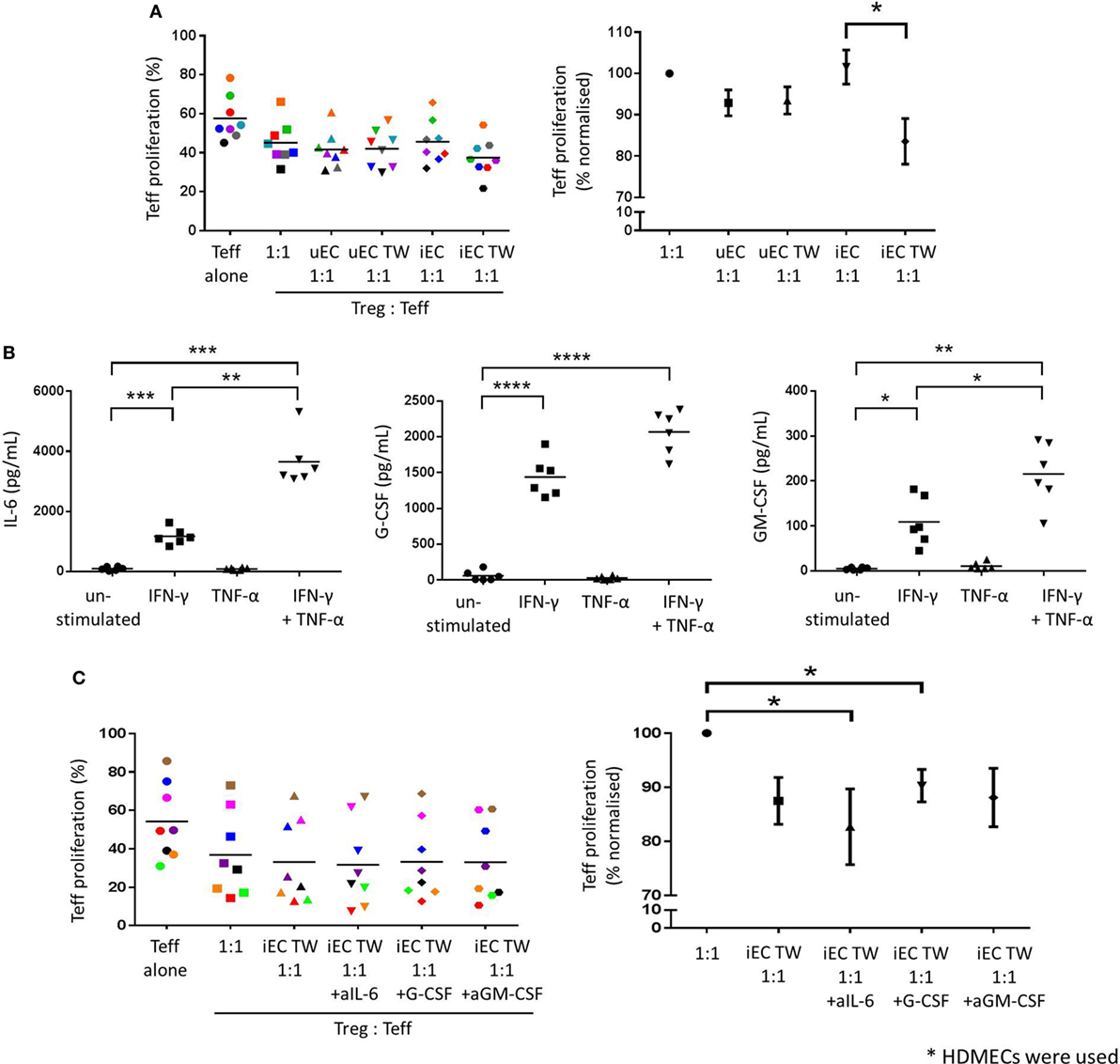
Figure 4. Increased Treg suppressive capacity through contact-independent interaction with activated HDMECs. (A) The effect of HDMEC interaction on Treg function was investigated whereby CD4+CD25hiCD127low were plated in direct contact with unstimulated (uEC) or IFN-γ-stimulated (iEC) HDMECs, or suspended above unstimulated (uEC TW) or IFN-γ-stimulated (iEC TW) HDMECs in Transwells for 24 h, then recovered and plated in Treg suppression assays. Data are expressed as % proliferated Teffs, with each experiment represented by a different color. Data are also expressed where Teff proliferation is normalized to proliferation in 1:1 cultures as 100%, showing mean ± SEM. One-way analysis of variance (ANOVA) with Dunnett’s test was used; *P < 0.05; n = 8. (B) HDMECs were left unstimulated or stimulated with 10 U/mL IFN-γ, 1 ng/mL TNF-α, or a combination of both cytokines for 24 h. Cells were washed and media replaced with 1 mL complete RPMI. Conditioned media was collected after 24 h and secreted cytokines detected using bead-based multiplex assays or ELISAs. Production of IL-6, G-CSF, and GM-CSF by HDMECs are shown with means drawn. One-way ANOVA with Dunnett’s test was conducted; ****P < 0.0001, ***P < 0.001, **P < 0.01, and *P < 0.05 cf. control; n = 6. (C) Tregs were cocultured in the upper chambers of Transwells with IFN-γ-stimulated (iEC TW) HDMECs in the lower chambers for 24 h, where 5 µg/mL of anti-IL-6, anti-G-CSF, or anti-GM-CSF blocking antibody was added in the lower chambers. Tregs were recovered and plated in a Treg suppression assay where Teff proliferation was measured by CFSE dilution. Data are expressed as % proliferated Teffs, with each experiment represented by a different color. Data are also expressed where Teff proliferation is normalized to proliferation in 1:1 cultures as 100%, showing mean ± SEM. One-way ANOVA with Dunnett’s test was used; *P < 0.05; n = 8.
To study potential factors responsible for the indirect nature of the enhanced Treg suppressor function, conditioned media from HDMECs was analyzed using cytokine multiplex assay and ELISAs. Of all the cytokines produced by ECs, (Figure D in Supplementary Material) only IL-6, G-CSF, and GM-CSF were modulated by IFN-γ; therefore, these factors were studied in greater detail (Figure 4B). Unstimulated HDMEC cultures produced low levels of IL-6, G-CSF, and GM-CSF, whereas IFN-γ stimulation significantly increased IL-6 (1,166 ± 274.1 pg/mL, P < 0.001; n = 6), G-CSF (1,439 ± 278.1 pg/mL, P < 0.0001; n = 6), and GM-CSF (108.9 ± 54.2 pg/mL, P < 0.05; n = 6). Production of these cytokines was not significantly stimulated by TNF-α alone; however, stimulation with both IFN-γ and TNF-α in combination significantly increased levels of IL-6, G-CSF, and GM-CSF, compared to unstimulated controls. Furthermore, IL-6 and GM-CSF secretion was significantly elevated above the levels stimulated by IFN-γ alone. Combined cytokine treatment did not, however, significantly increase G-CSF secretion above that of IFN-γ alone.
The function of the soluble mediators IL-6, G-CSF, and GM-CSF on the interaction between HDMEC and Tregs, in terms of the modulation of Treg suppressive capacity, was investigated by the use of blocking antibodies specific for each of the three cytokines. Blocking antibodies against IL-6, G-CSF, or GM-CSF were added at 5 µg/mL in the lower chamber with the IFN-γ-stimulated HDMEC in the Treg-EC cocultures followed by assays of Treg suppressor function with appropriate isotype controls. Inclusion of blocking antibodies against IL-6 appeared to significantly increase Treg suppressor function as shown with the greater decrease in Teff proliferation in Figure 4C, when compared to control Tregs (1:1) (P < 0.05; n = 8). However, this was not significant when compared with Tregs cocultured above IFN-γ-stimulated HDMECs alone. Blockade of G-CSF or GM-CSF in indirect coculture of Tregs with IFN-γ-stimulated HDMECs had no significant effect on Treg suppressive capacity against Teffs proliferation.
PD-1 and its ligands have been implicated in the interaction between Tregs and ECs or other cell types, which can lead to activation of Treg function or induction of Treg populations (14, 16). In our system, unstimulated HUVECs showed low levels of PD-L1 and PD-L2 expression, whereas IFN-γ stimulation caused significant increases in expression of both ligands on HUVECs, indicated by both their mean fluorescence index (MFI) values and percentages of positive cells (P < 0.001 or P < 0.05 cf. unstimulated control; n = 3) (Figures 5A,B). In contrast, TNF-α treatment of HUVECs did not affect either PD-L1 or PD-L2 expression. However, PD-L1 and PD-L2 expression was increased significantly on HUVECs with combined IFN-γ and TNF-α cytokine stimulation but not significantly above IFN-γ alone cultures suggesting only IFN-γ regulated PD-L1 and -2 expression.
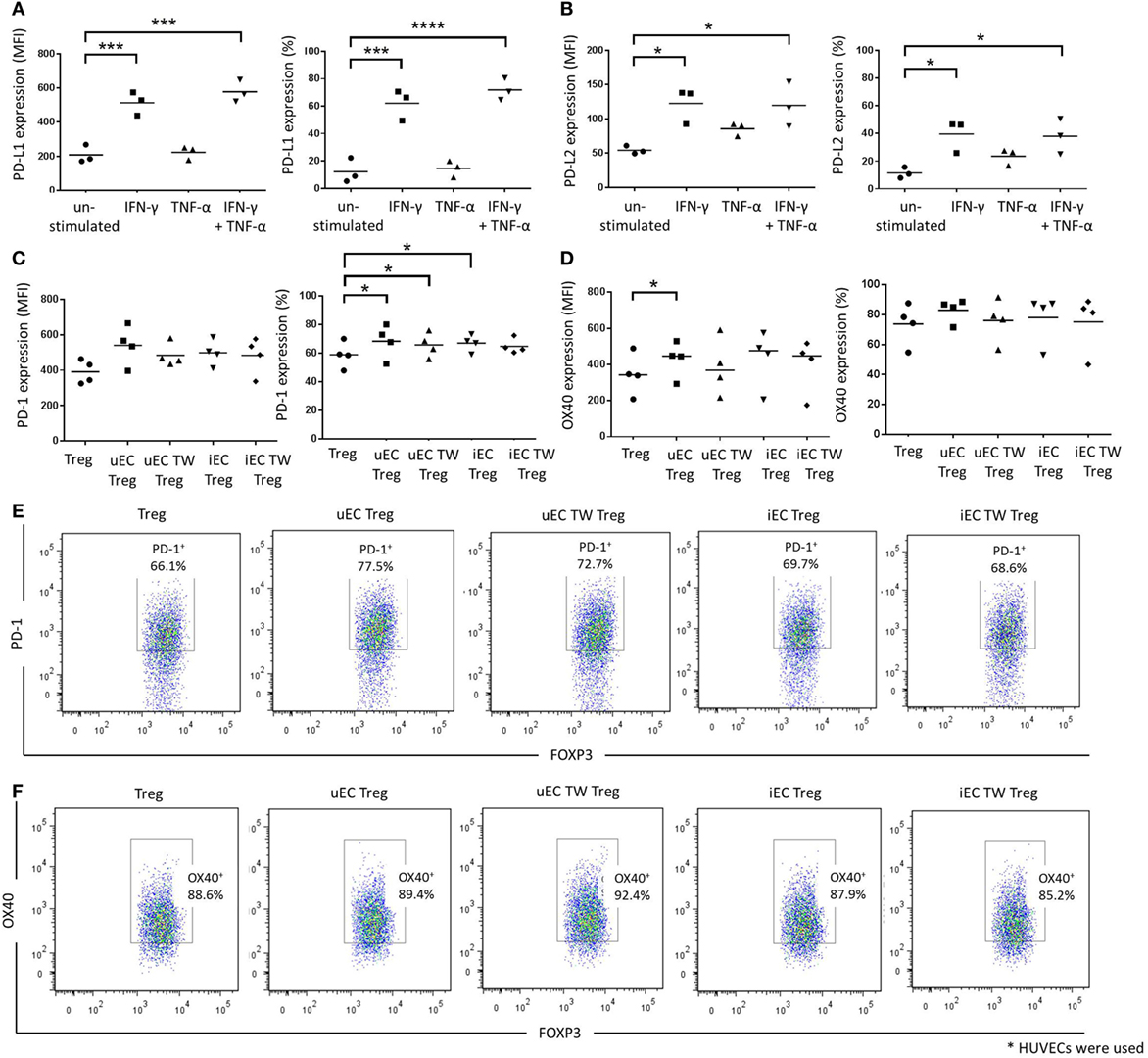
Figure 5. Involvement of PD-1/programmed death ligand (PD-L)1/2 in the contact-dependent interactions between HUVEC and Tregs. Human umbilical vein ECs (HUVECs) were stimulated with 10 U/mL IFN-γ, 1 ng/mL tumor necrosis factor (TNF)-α, or combination of both cytokines for 24 h. Cells were then stained and analyzed for (A) PD-L1 or (B) PD-L2 expression using flow cytometry. Data are expressed as mean fluorescence index (MFI) and percentage of positive cells, with means shown. One-way analysis of variance (ANOVA) with Dunnett’s test was conducted; ****P < 0.0001, ***P < 0.001, *P < 0.05; n = 3. Modulation of Treg phenotype after HUVEC interactions were also investigated whereby Tregs were plated in direct contact with unstimulated (uEC Treg) or IFN-γ-stimulated (iEC Treg) HUVECs, or in the upper chambers of Transwells with unstimulated (uEC TW Treg) or IFN-γ-stimulated (iEC TW Treg) HUVECs. Tregs were then recovered and plated in a Treg suppression assay and analyzed for (C) PD-1 and (D) OX40 expression after 72 h using flow cytometry. One-way ANOVA with Dunnett’s test was conducted; *P < 0.05; n = 3. Plots of a representative experiment are shown in Figures (E,F).
The expression of costimulatory molecules PD-1 and OX40 on Tregs after HUVEC coculture was examined as part of the possible mechanism of action in the Treg suppression assay. There were no significant changes in PD-1 MFI values for Tregs regardless of coculture conditions with HUVECs (Figures 5C,E). However, the percentage of PD-1+ Tregs was increased in Tregs that were in direct contact with unstimulated HUVECs (uEC Treg) or IFN-γ-stimulated HUVECs (iEC Treg) or in Transwells above unstimulated HUVEC (uEC TW Treg) (P < 0.05; n = 5). This was not observed, however, in Tregs that were cocultured above IFN-γ-stimulated HUVECs (iEC TW Treg). In contrast, there was a slight increase in OX40 MFI values in Tregs that were in direct contact with unstimulated HUVECs (uEC Treg) (P < 0.05; n = 4) but there was no difference in OX40 expression profiles of Tregs (MFI values and percentage of positive cells) for other HUVEC coculture conditions (Figures 5D,F).
Endothelial cells can promote CD4+ T cell activation and proliferation in the absence of professional antigen presenting cells. In the presence of an external mitogen including PHA or stimulatory aCD3/28 antibodies, strong proliferation was shown with CD4+ T cells in cocultures with the endothelium regardless of prior EC activation with inflammatory cytokines. This immunological function, seen with ECs from different lineages suggests a further functional role for the endothelium in immune cell activation. In the absence of any external mitogen, T cell proliferation occurred only in cocultures of CD4+ T cells with IFN-γ activated ECs which supports previous literature that demonstrates the ability of ECs in the activation, cytokine production, and proliferation of CD4+ T cells (19, 20). Although ECs increased FOXP3 expression within the previously CD25 −ve FOXP3 −ve population, the temporal nature of the FOXP3 expressing population suggests that these cells are likely to represent activated effector T cells which can express FOXP3 without acquiring a regulatory phenotype (21). Taken in conjunction with previous work on ECs, this suggests that the endothelium can provide partial activating signals that may be further augmented by direct contact or cytokine activation from stromal cells (including immune cells) following transmigration into the tissue.
Previous research has shown that rapamycin activated EC can enhance EC dependent Treg suppressor function (16) and that TGF-β and retinoic acid along with rapamycin can induce human iTregs with superior suppressive activity (22). Our data using purified Tregs further demonstrate a role for human ECs in enhancing Treg suppressor function. Both direct cocultures and indirect cocultures of Tregs with cytokine-activated HUVECs enhanced the suppressive capacity of Tregs on Teff proliferation. This augmented Treg activity was greatest in the indirect coculture of IFN-γ-stimulated HUVECs and Tregs and, in the case of HDMECs, the higher Treg suppressive function was only observed in the indirect cultures. This suggests a potentially complex interplay between ECs and Tregs when both cells are in direct contact and that some of the direct interactions may regulate or indeed negate the stimulatory effects of ECs on Tregs. The differences in Treg activation between HUVECs and HDMECs may reflect the nature of the endothelial response to cytokine and the vascular bed from which the cells are derived. Protecting the fetus from insult including the mothers own immunity may explain why HUVEC possess both contact-dependent and independent mechanisms to regulate Tregs and may have evolved to regulate maternal–fetal tolerance (23). In contrast, Tregs in the skin often reside in the tissue (24) and have a greater diversity of roles in relation to resolution of inflammation after exogenous and endogenous stimuli. Interestingly, a previous study also showed that HMEC-1 cells and HDMECs were able to expand Treg populations (15), providing additional support for a role of the skin endothelium in Treg activity. Overall, the combination of murine and human EC studies, including our data, suggests an adaptive immune function for ECs above the simple recruitment of T cells from the circulation.
Roles for the involvement of ECs (HUVECs) in upregulating GITR, CTLA-4, and PD-1 on Tregs (25), antigen presentation, and co-stimulation of T cells by the endothelium (1–4), the suppression of endothelial function by Tregs (26) and Treg inhibition of Teff recruitment that defines the kinetics of an immune response (27) have been reported. PD-1 (and its ligands PD-L1 and PD-L2) and OX40 have also been studied as targets for cancer immunotherapy to boost antitumor immunity (28). The PD-1 signaling pathway has also been implicated in EC–Treg interactions (14, 16) and increased PD-1 expression on Tregs after EC modulation was observed in the current study which concurs with the work of Chen et al. (25). The exact role for PD-1 on Tregs has been debated, with evidence to support both PD-1 promoted Treg function and inhibition of Treg function in other scenarios (8, 25). Our data showing that ECs increased the percentage of PD-1+ Tregs and also augmented Treg suppressive activity suggests that EC-induced PD-1 expression on Tregs is likely involved in the increased suppressor function of the Tregs. Furthermore, the upregulation of PD-1 ligands expressed on activated HUVECs suggests that ECs possess the relevant mechanisms to activate Tregs via the PD-1/PD-L1 signaling pathway during direct interactions and supports the findings of Wang et al. (16) who were able to show increased Treg differentiation in a rapamycin activated endothelial PD-L1-dependent manner.
A limitation to this present study is that the PD-1/PD-L1 interaction was not studied further with blocking antibodies to better define the role of these ligands in the Treg/EC interaction. These studies will need to be performed to show the definitive role of PD-1/PD-L1 in Treg/EC activity. However, in the light of other research, the nature of the endothelial regulation of PD-1 expression on Tregs suggests that the response may depend partly on the Treg subset in question as well as on other mediators. For instance, in a chronic HCV infection model (25) the PD-1/PD-L1 interaction was shown to lower the expansion of Treg phenotype compared to Teff whereas the same ligand interaction was shown to regulate induced Tregs and helped to maintain their Treg phenotype and suggests a role in Treg plasticity (8). Indeed, Lion et al. (29) have recently shown that ECs can amplify Tregs in a model of alloactivation via HLA-DR, the expression of which can be modified by immunosuppressants. Despite this group showing expression of endothelial PD-L1 in their model, immunosuppressants were not shown to affect PD-L1 levels. In addition, a previous study by the same group did not observe a role for PD-L1 in Treg expansion under inflammatory conditions (15). This suggests that complex interactions and potentially multiple pathways exist that are dependent on the microenvironment, vascular bed, and T cell subsets available and requires further study. It is not entirely clear, therefore, that the contact-dependent mechanism is solely due to the PD-1/PD-L1 axis and it is unlikely to describe the full nature of endothelial specific Treg activation especially in the context of the non-contact mediated Treg activation.
In vivo, the length of time that T cells interact with the endothelium would be an important factor in considering the mechanisms as detailed here. In in vitro flow based assays, T cell residence on the luminal side of the endothelium can last for some considerable time before full transmigration occurs. In the presence of antigen pulsed endothelium, this interaction is significantly slowed in memory/effector T cells (30) suggesting a range of time dependent signaling events prior to transmigration. Following attachment, complete transmigration took up to 2 h and T cells were shown to use invadosome-like processes in their interaction with the endothelium. Migrating T cells may then either remain in the subendothelial environment in close contact with the endothelium or travel further in to tissue and interact with additional stromal cells. Indeed, subendothelial pericytes and smooth muscle have been suggested to present antigen and provide signals that may regulate T cell function and localize them in close proximity to the endothelium (31). Using, confocal intravital microscopy, Deane et al. showed that in a contact sensitivity model, using mice, Tregs were found to accumulate in the inflamed skin by 24 h, peaking at 48 h (32). This suggests a prolonged response where, due to the high vascularity of the skin, the proximity of ECs and Treg is such that the endothelium is likely to influence Treg function over a prolonged period similar to that used in the present study.
On interrogating the contact-dependent and contact-independent nature of Treg activation the initial screening of HUVEC and HDMEC-conditioned media showed IL-6, G-CSF and GM-CSF as potential soluble mediator candidates. These mediators were produced only after IFN-γ stimulation of ECs, indicating their potential specific involvement in the indirect interactions of IFN-γ-stimulated ECs and Tregs. However, individual blockade of these mediators did not abolish the boosted Treg function in indirect coculture with activated HDMECs and suggests that the interaction is multifaceted or via further soluble mediators. A role for IL-6 was suggested, however, in contact-independent effects by ECs on Tregs as the IL-6 blocking antibody increased Treg suppressor function during EC–Treg interactions. This is in line with studies which reported that IL-6 can dampen Treg suppressive activity and skew Treg induction toward Th17 differentiation (33, 34). This suggests that IL-6, in combination with other co-stimulator mechanisms, is partly suppressive on Treg function as this effect is lost when both contact signals and IL-6 signaling were removed, leading to the enhancement of Treg function.
Our initial studies (Figure F in Supplementary Material) have ruled out nitric oxide signaling from the endothelium as a soluble mediator despite NO being generated by EC and a known regulator of T cell function (35). Additionally, TGF-β can regulate Tregs and can be secreted by the endothelium either onto the endothelial surface or out into the media (36, 37). In our hands (Figure F in Supplementary Material) the endothelium constitutively secretes TGF-β but this was not enhanced by either of the stimulating cytokines used thus suggesting that TGF-β is unlikely to mediate the EC/Treg suppressive activity at a distance in this model. However, there may be a role for endothelial surface bound TGF-β when EC and Treg are in close proximity and this could link both contact and non-contact-dependent activities. A further possibility for signaling at a distance lies in the release of endothelial microvesicles. These lipid and protein rich vesicles can be induced by inflammatory cytokines (38) and can carry molecules across distances and appear to mediate responses via a soluble mediator(s) when in fact, a contact-like effect is in use (39, 40). The discovery of vascular derived microvesicles in disease and their ability to control T cell activity via a range of mechanisms including carriage of microRNAs, particularly miRs 146a and 155 and adhesion molecules suggests this hybrid contact/non-contact mechanism as a further possibility (41–45). Indeed, PD-L1 within microvesicles has been observed released from mesenchymal stem cells (46) and microvesicles have been shown to transfer materials between T cells and APCs (47) suggesting a common mechanism of control form a range of cell types.
Human ECs exhibit important functions over and above leukocyte recruitment that contributes to immune homeostasis, with the distinct mechanisms responsible for these immune functions dependent on the vascular bed from which the endothelium is derived. Based on the current study, it is likely that human ECs activated during inflammation modulate both Teff and Treg populations by enhancing their proliferation and suppressive capacity, respectively. It is also possible that dysregulation of EC–Treg interaction could potentially be a source of unregulated inflammation as not only the control of Treg function by EC interaction is important but the reverse situation where EC function and/or phenotype can be controlled by Tregs. Tregs can exert protective effects during inflammation by downregulating production of proinflammatory mediators by ECs, as suggested by He et al., who showed downregulation of LPS-induced VCAM-1, monocyte chemoattractant protein-1 and IL-6 on HUVECs by Tregs (48). Furthermore, Tregs can also affect EC activation and leukocyte recruitment by dampening EC E- and P-selectin expression that subsequently decreased effector T cell adhesion (26, 49). Finally, blockade of E-selectin in a contact sensitivity model in mice showed increased swelling and neutrophil infiltration due to a lack of Treg recruitment (50). Disruption of EC–Treg interaction can, therefore, have wide ranging consequences.
These studies and ours suggest a complex, tripartite interaction of Teff, Treg, and EC with various activating and inhibitory signals passing between them. The competing signals are likely to evolve with the microenvironment to balance tolerance and inflammation. Where perturbation of any of these mediators is sought, an understanding of the consequences of immune modulation strategies is required. While the current study has provided the groundwork for understanding the nature of the interactions (i.e., direct and indirect) between human ECs and Tregs, future studies identifying the exact mechanisms of indirect EC–Treg interaction may have the potential to identify novel targets for modulation of T cell function controlled by the endothelium.
| aCD28 | Anti-CD28 |
| aCD3 | Anti-CD3 |
| aCD3/28 | Anti-CD3 and anti-CD28 |
| ANOVA | Analysis of variance |
| APC | Allophycocyanin |
| CFSE | Carboxyfluorescein succinimidyl ester |
| Cy | Cyanine |
| EC | Endothelial cell |
| FITC | Fluorescein isothiocyanate |
| FOXP3 | Foxhead Box P3 |
| G-CSF | Granulocyte colony-stimulating factor |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| HDMEC | Human dermal microvascular endothelial cell |
| HMEC-1 | Human dermal microvascular endothelial cell-1 |
| HUVEC | Human umbilical vein endothelial cell |
| ICOS | Inducible T cell costimulator |
| iEC | IFN-γ-stimulated endothelial cell |
| IFN | Interferon |
| MFI | Mean fluorescence intensity |
| PBMC | Peripheral blood mononuclear cell |
| PBS | Phosphate buffered saline |
| PD-1 | Programmed death-1 |
| PD-L1/L2 | Programmed death ligand-1/2 |
| PE | R-phycoerythrin |
| PerCP | Peridinin chlorphyll protein |
| PHA | Phytohemagglutinin |
| tEC | TNF-stimulated endothelial cell |
| Teff | Effector T |
| Th | T helper |
| TNF | Tumor necrosis factor |
| Treg | Regulatory T cell |
| uEC | Unstimulated endothelial cell |
This study was carried out in accordance with the recommendations of Southampton & South West Hampshire Research Ethics Committee B with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the Southampton & South West Hampshire Research Ethics Committee B. Umbilical cords were collected from the Princess Anne Hospital, Southampton, UK from non-complicated natural vaginal births following agreed ethical collection protocols (Local Research Ethical Committee (LREC); Ref: 07/H0502/83). Whole blood was collected from healthy donors with signed consent following ethically approved protocols (LREC Ref: 07/H0504/93).
WL, EH, and TM conceived and designed experiments. WL performed the experiments and carried out the acquisition and analysis. MO provided isolated primary cells. WL, EH, and TM interpreted data and wrote the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to acknowledge the help and support of Dr. Carolann McGuire and Mr. Richard Jewell in the flow cytometry facility for their excellent technical help and Dr. Laurie Lau for his assistance in measuring cytokine levels. The authors would also like to thank Prof. Martin Feelisch and his laboratory staff for their expertise in the measurement of nitric oxide. The authors also extend their thanks to the blood and umbilical cord donors of the Faculty of Medicine, University of Southampton and Princess Anne Hospital, Southampton, respectively, for the primary materials to conduct the study.
This work was supported by a British Skin Foundation Ph.D. studentship, BSF Large Grant 3035s (WCL). MO was funded by a Psoriasis Association Ph.D. studentship and TM is funded by a Higher Education Funding Council for England (HEFCE) Lectureship and a Wellcome Investment in People award. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.00565/full#supplementary-material.
1. Pober JS, Tellides G. Participation of blood vessel cells in human adaptive immune responses. Trends Immunol (2012) 33:49–57. doi:10.1016/j.it.2011.09.006
2. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol (2007) 7:678–89. doi:10.1038/nri2156
3. Khayyamian S, Hutloff A, Büchner K, Gräfe M, Henn V, Kroczek RA, et al. ICOS-ligand, expressed on human endothelial cells, costimulates Th1 and Th2 cytokine secretion by memory CD4+ T cells. Proc Natl Acad Sci U S A (2002) 99:6198–203. doi:10.1073/pnas.092576699
4. Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, et al. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol (2003) 33:3117–26. doi:10.1002/eji.200324270
5. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor. J Immunol (1995) 155:1151–64.
6. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4:330–6. doi:10.1038/ni904
7. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299:1057–61. doi:10.1126/science.1079490
8. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med (2009) 206:3015–29. doi:10.1084/jem.20090847
9. Kitamura N, Murata S, Ueki T, Mekata E, Reilly R, Jaffee E, et al. OX40 costimulation can abrogate Foxp3+ mediated suppression of antitumor immunity regulatory T cell-mediated suppression of antitumor immunity. Int J Cancer (2009) 125:630–8. doi:10.1002/ijc.24435.OX40
10. Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+ CD25+ T cell-mediated suppression by dendritic cells. Science (2003) 299:1033–6. doi:10.1126/science.1078231
11. Chaudhry A, Samstein RM, Treuting P, Liang Y, Marina C, Heinrich J, et al. Interleukin-10 signalling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity (2011) 34:566–78. doi:10.1016/j.immuni.2011.03.018.Interleukin-10
12. Sather BD, Treuting P, Perdue N, Miazgowicz M, Fontenot JD, Rudensky AY, et al. Altering the distribution of Foxp3(+) regulatory T cells results in tissue-specific inflammatory disease. J Exp Med (2007) 204:1335–47. doi:10.1084/jem.20070081
13. Krupnick AS, Gelman AE, Barchet W, Richardson S, Kreisel FH, Turka L, et al. Cutting edge: murine vascular endothelium activates and induces the generation of allogeneic CD4+25+Foxp3+ regulatory T cells. J Immunol (2005) 175:6265–70. doi:10.4049/jimmunol.175.10.6265
14. Bedke T, Pretsch L, Karakhanova S, Enk AH, Mahnke K. Endothelial cells augment the suppressive function of CD4+ CD25+ Foxp3+ regulatory T cells: involvement of programmed death-1 and IL-10. J Immunol (2010) 184:5562–70. doi:10.4049/jimmunol.0902458
15. Taflin C, Favier B, Baudhuin J, Savenay A, Hemon P, Bensussan A, et al. Human endothelial cells generate Th17 and regulatory T cells under inflammatory conditions. Proc Natl Acad Sci U S A (2011) 108:2891–6. doi:10.1073/pnas.1011811108
16. Wang C, Yi T, Qin L, Maldonado R, von Andrian U, Kulkarni S, et al. Rapamycin-treated human endothelial cells preferentially activate allogeneic regulatory T cells. J Clin Invest (2013) 123:1677–93. doi:10.1172/JCI66204
17. Jaffe E, Nachman R, Becker C, Miinick C. Culture of human endothelial cells derived from umbilical veins: identification by morphologic and immunologic criteria. J Clin Invest (1973) 52:2745–56. doi:10.1172/JCI107470
18. Gimbrone M, Cotran R, Folkman J, March P. Human vascular endothelial cells in culture: growth and DNA synthesis. J Cell Biol (1974) 60:673–84. doi:10.1083/jcb.60.3.673
19. Ma W, Pober JS. Human endothelial cells effectively costimulate cytokine production by, but not differentiation of, naive CD4+ T cells. J Immunol (1998) 161:2158–67.
20. Kunitomi A, Hori T, Imura A, Uchiyama T. Vascular endothelial cells provide T cells with costimulatory signals via the OX40/gp34 system. J Leukoc Biol (2000) 68:111–8. doi:10.1189/jlb.68.1.111
21. Wang J, Ioan-Facsinay A, van der Voort EIH, Huizinga TWJ, Toes REM. Transient expression of FOXP3 in human activated nonregulatory CD4+ T cells. Eur J Immunol (2007) 37:129–38. doi:10.1002/eji.200636435
22. Schmidt A, Eriksson M, Shang MM, Weyd H, Tegnér J. Comparative analysis of protocols to induce human CD4+Foxp3+ regulatory T cells by combinations of IL-2, TGF-beta, retinoic acid, rapamycin and butyrate. PLoS One (2016) 11:e0148474. doi:10.1371/journal.pone.0148474
23. La Rocca C, Carbone F, Longobardi S, Matarese G. The immunology of pregnancy: regulatory T cells control maternal immune tolerance toward the fetus. Immunol Lett (2014) 162:41–8. doi:10.1016/j.imlet.2014.06.013
24. Lai C, August S, Albibas A, Behar R, Cho SY, Polak ME, et al. Ox40+ regulatory T cells in cutaneous squamous cell carcinoma suppress effector T-cell responses and associate with metastatic potential. Clin Cancer Res (2016) 22:4236–48. doi:10.1158/1078-0432.CCR-15-2614
25. Chen WJ, Hu XF, Yan M, Zhang WY, Mao XB, Shu YW. Human umbilical vein endothelial cells promote the inhibitory activation of CD4+CD25+Foxp3+regulatory T cells via PD-L1. Atherosclerosis (2016) 244:108–12. doi:10.1016/j.atherosclerosis.2015.11.002
26. Maganto-García E, Bu D-X, Tarrio ML, Alcaide P, Newton G, Griffin GK, et al. Foxp3+-inducible regulatory T cells suppress endothelial activation and leukocyte recruitment. J Immunol (2011) 187:3521–9. doi:10.4049/jimmunol.1003947
27. Fu H, Kishore M, Gittens B, Wang G, Coe D, Komarowska I, et al. Self-recognition of the endothelium enables regulatory T-cell trafficking and defines the kinetics of immune regulation. Nat Commun (2014) 5:1–14. doi:10.1038/ncomms4436
28. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi:10.1038/nrc3239
29. Lion J, Burbach M, Cross A, Poussin K, Taflin C, Kaveri S, et al. Endothelial cell amplification of regulatory t cells is differentially modified by immunosuppressors and intravenous immunoglobulin. Front Immunol (2017) 8:1761. doi:10.3389/fimmu.2017.01761
30. Sage PT, Varghese LM, Martinelli R, Sciuto TE, Kamei M, Dvorak AM, et al. Antigen recognition is facilitated by invadosome-like protrusions formed by memory/effector T cells. J Immunol (2012) 188:3686–99. doi:10.4049/jimmunol.1102594
31. Pober JS, Merola J, Liu R, Manes TD. Antigen presentation by vascular cells. Front Immunol (2017) 8:1907. doi:10.3389/fimmu.2017.01907
32. Deane JA, Abeynaike LD, Norman MU, Wee JL, Kitching AR, Kubes P, et al. Endogenous regulatory T cells adhere in inflamed dermal vessels via ICAM-1: association with regulation of effector leukocyte adhesion. J Immunol (2012) 188:2179–88. doi:10.4049/jimmunol.1102752
33. Goodman WA, Levine AD, Massari JV, Sugiyama H, McCormick TS, Cooper KD. IL-6 signaling in psoriasis prevents immune suppression by regulatory T cells. J Immunol (2009) 183:3170–6. doi:10.4049/jimmunol.0803721
34. Zhou L, Lopes JE, Chong MMW, Ivanov II, Min R, Victora GD, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature (2008) 453:236–40. doi:10.1038/nature06878
35. Niedbala W, Cai B, Liew FY. Role of nitric oxide in the regulation of T cell functions. Ann Rheum Dis (2006) 65(Suppl 3):iii37–40. doi:10.1136/ard.2006.058446
36. Iwashima M, Love R. Potential of targeting TGF-beta for organ transplant patients. Future Med Chem (2013) 5:281–9. doi:10.4155/fmc.12.215
37. Hynes RO. Extracellular matrix: not just prety fibrils. Science (2009) 326:1216–9. doi:10.1126/science.1176009.Extracellular
38. Pelletier F, Garnache-Ottou F, Biichlé S, Vivot A, Humbert P, Saas P, et al. Effects of anti-TNF-α agents on circulating endothelial-derived and platelet-derived microparticles in psoriasis. Exp Dermatol (2014) 23:924–5. doi:10.1111/exd.12551
39. Wheway J, Latham SL, Combes V, Grau GER. Endothelial microparticles interact with and support the proliferation of T cells. J Immunol (2014) 193:3378–87. doi:10.4049/jimmunol.1303431
40. Walker JD, Maier CL, Pober JS. Cytomegalovirus-infected human endothelial cells can stimulate allogeneic CD4+ memory T cells by releasing antigenic exosomes. J Immunol (2009) 182:1548–59. doi:10.4049/jimmunol.182.3.1548
41. Chironi GN, Boulanger CM, Simon A, Dignat-George F, Freyssinet J-M, Tedgui A. Endothelial microparticles in diseases. Cell Tissue Res (2009) 335:143–51. doi:10.1007/s00441-008-0710-9
42. Stahl HF, Fauti T, Ullrich N, Bopp T, Kubach J, Rust W, et al. miR-155 inhibition sensitizes CD4+ Th cells for TREG mediated suppression. PLoS One (2009) 4:e7158. doi:10.1371/journal.pone.0007158
43. Rodríguez-Muñoz A, Martínez-Hernández R, Ramos-Leví AM, Serrano-Somavilla A, González-Amaro R, Sánchez-Madrid F, et al. Circulating microvesicles regulate treg and Th17 differentiation in human autoimmune thyroid disorders. J Clin Endocrinol Metab (2015) 100:E1531–9. doi:10.1210/jc.2015-3146
44. Muturi HT, Dreesen JD, Nilewski E, Jastrow H, Giebel B, Ergun S, et al. Tumor and endothelial cell-derived microvesicles carry distinct CEACAMs and influence T-cell behavior. PLoS One (2013) 8:e74654. doi:10.1371/journal.pone.0074654
45. Yee D, Shah KM, Coles MC, Sharp TV, Lagos D. MicroRNA-155 induction via TNF-α and IFN-γ suppresses expression of programmed death ligand-1 (PD-L1) in human primary cells. J Biol Chem (2017) 292:20683–93. doi:10.1074/jbc.M117.809053
46. Mokarizadeh A, Delirezh N, Morshedi A, Mosayebi G, Farshid A-A, Mardani K. Microvesicles derived from mesenchymal stem cells: potent organelles for induction of tolerogenic signaling. Immunol Lett (2012) 147:47–54. doi:10.1016/j.imlet.2012.06.001
47. Gutiérrez-Vázquez C, Villarroya-Beltri C, Mittelbrunn M, Sánchez-Madrid F. Transfer of extracellular vesicles during immune cell-cell interactions. Immunol Rev (2013) 251:125–42. doi:10.1111/imr.12013
48. He S, Li M, Ma X, Lin J, Li D. CD4+CD25+Foxp3+ regulatory T cells protect the proinflammatory activation of human umbilical vein endothelial cells. Arterioscler Thromb Vasc Biol (2010) 30:2621–30. doi:10.1161/ATVBAHA.110.210492
49. Ring S, Oliver SJ, Cronstein BN, Enk AH, Mahnke K. CD4+CD25+ regulatory T cells suppress contact hypersensitivity reactions through a CD39, adenosine-dependent mechanism. J Allergy Clin Immunol (2009) 123:1287–96.e2. doi:10.1016/j.jaci.2009.03.022
Keywords: regulatory T cells, endothelial cells, immune regulation, cell–cell interaction, CD4+ T cells
Citation: Lim WC, Olding M, Healy E and Millar TM (2018) Human Endothelial Cells Modulate CD4+ T Cell Populations and Enhance Regulatory T Cell Suppressive Capacity. Front. Immunol. 9:565. doi: 10.3389/fimmu.2018.00565
Received: 27 October 2017; Accepted: 06 March 2018;
Published: 23 March 2018
Edited by:
Shohei Hori, The University of Tokyo, JapanReviewed by:
Ana Claudia Zenclussen, Universitätsklinikum Magdeburg, GermanyCopyright: © 2018 Lim, Olding, Healy and Millar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Timothy M. Millar, VC5NLk1pbGxhckBzb3Rvbi5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.