 Anshu Malik
Anshu Malik Manish Gupta
Manish Gupta Rajesh Mani
Rajesh Mani Himanshu Gogoi
Himanshu Gogoi Rakesh Bhatnagar
Rakesh Bhatnagar
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 20 March 2018
Sec. Vaccines and Molecular Therapeutics
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.00562
This article is part of the Research Topic Nanoparticle Vaccines Against Infectious Diseases View all 16 articles
Anthrax is an era old deadly disease against which there are only two currently available licensed vaccines named anthrax vaccine adsorbed and precipitated (AVP). Though they can provide a protective immunity, their multiple side-effects owing to their ill-defined composition and presence of toxic proteins (LF and EF) of Bacillus anthracis, the causative organism of anthrax, in the vaccine formulation makes their widespread use objectionable. Hence, an anthrax vaccine that contains well-defined and controlled components would be highly desirable. In this context, we have evaluated the potential of various vaccine formulations comprising of protective antigen (PA) encapsulated trimethyl-chitosan nanoparticles (TMC-PA) in conjunction with either CpG-C ODN 2395 (CpG) or Poly I:C. Each formulation was administered via three different routes, viz., subcutaneous (SC), intramuscular (IM), and intraperitoneal in female BALB/c mice. Irrespective of the route of immunization, CpG or Poly I:C adjuvanted TMC-PA nanoparticles induced a significantly higher humoral response (total serum IgG and its isotypes viz., IgG1, IgG2a, and IgG2b), compared to their CpG or Poly I:C PA counterparts. This clearly demonstrates the synergistic behavior of CpG and Poly I:C with TMC nanoparticles. The adjuvant potential of TMC nanoparticles could be observed in all the three routes as the TMC-PA nanoparticles by themselves induced IgG titers (1–1.5 × 105) significantly higher than both CpG PA and Poly I:C PA groups (2–8 × 104). The effect of formulations on T-helper (Th) cell development was assessed by quantifying the Th1-dependant (TNF-α, IFN-γ, and IL-2), Th2-dependant (IL-4, IL-6, and IL-10), and Th17-type (IL-17A) cytokines. Adjuvanation with CpG and Poly I:C, the TMC-PA nanoparticles triggered a Th1 skewed immune response, as suggested by an increase in the levels of total IgG2a along with IFN-γ cytokine production. Interestingly, the TMC-PA group showed a Th2-biased immune response. Upon challenge with the B. anthracis Ames strain, CpG and Poly I:C adjuvanted TMC-PA nanoparticles immunized via the SC and IM routes showed the highest protective efficacy of ~83%. Altogether, the results suggest that CpG or Poly I:C adjuvanted, PA-loaded TMC nanoparticles could be used as an effective, non-toxic, second generation subunit-vaccine candidate against anthrax.
Bacillus anthracis, a Gram-positive microorganism, is the cause of an acute disease Anthrax. It can form spores, which could remain dormant for years and accidental exposure by inhalation may result in death (1). Although inhalational anthrax has been reported to have a high fatality rate near to 100%, few deaths have also been documented due to cutaneous and gastrointestinal anthrax (2). It is also recognized for many bioterror attacks in history, including the famous 2001 bioterror attack of anthrax spores in the US (3). A prophylactic approach would be the best way to cease the disease and protect people against any bioterror attack. The currently existing vaccines BioThrax (also known as anthrax vaccine adsorbed) and anthrax vaccine precipitated (AVP) are the only licensed vaccines present globally (4). The major caveats of these vaccines are the presence of toxic proteins [lethal factor (LF) and edema factor (EF)] and the undefined composition of anthrax proteins (5). At present, only the high-risk population such as defense personnel are vaccinated with BioThrax. However, there have been several reports of reactogenicity in such individuals (6). Another drawback associated with this vaccine is the cumbersome dosing schedule, which comprises of multiple initial doses and subsequent annual boosters for sustained immunity (7).
In recent times, the focus has shifted to second generation subunit vaccines where the composition is pharmaceutically defined, and highly immunogenic proteins are taken into account making it safer for the mass use. In case of anthrax, B. anthracis, the causative organism of anthrax, secrete two toxins comprised of three proteins: the protective antigen (PA), the LF, and the EF. The edema toxin (PA + EF) causes edema and lethal toxin (PA + LF) causes death in animals (8). PA has always been the choice of antigen as it binds to the host receptor and binds to LF and EF to make lethal toxin and edema toxin, which eventually cause the toxicity in the host (9). Hence, the antibodies against PA have been shown to protect against aerosolized B. anthracis Ames spore challenge (10).
A soluble antigen when injected alone has less residence time and often lack essential danger signals necessary to provoke dendritic cells and consequently, T-cells. To surpass these limitations, antigen encapsulation into particulate system helps by increasing the retention time of the antigen in the host system and creating a depot, which releases the antigen into the system for a longer duration in a controlled manner (11, 12). The susceptibility of particles to be phagocytized by the macrophages is 1,000–10,000 times more efficient, and it also allows multimeric antigen presentation and delivery to antigen-presenting cells (APCs), in comparison to the soluble antigens (13).
The intervention of natural, biodegradable polymers-based encapsulation carriers makes them reasonably safe to employ for vaccine delivery (14, 15). Poly (lactic-co-glycolic acid) and chitosan are the most studied among them. Chitosan is derived from chitin, which is a ubiquitous natural polymer and is soluble in water under acidic conditions. Chitosan particles are usually positively charged and irregular in shape. However, they are susceptible to dissociation with ionic fluctuations and unstable at physiological pH. Several chemically modified (at functional groups -OH and -NH2) derivatives of chitosan have been studied. For instance, trimethyl-chitosan (TMC) is preferred over chitosan due to its stability at several pH ranges and its positive surface charge and has been utilized for vaccine delivery against influenza and Hepatitis B (14, 16–21).
Trimethyl-chitosan as well as chitosan have also been claimed to possess mucoadhesive and intrinsic adjuvant properties, and evidently, its nanoparticles can stimulate in vitro T-cell maturation and proliferation (22). Furthermore, it also possesses the ability to open tight junctions and cross epithelial barriers by redistributing the cytoskeletal F-actin and ZO-1 tight junction protein (23–26). The precursor of TMC and chitosan, chitin, has also been demonstrated to be a size-dependent pathogen-associated molecular pattern (PAMP) and shown to interact and activate the macrophages through different TLRs and dectin-1 (27, 28).
In addition to TMC nanoparticles, we sought to investigate further whether the immune response of nanoparticles could be synergistically improved upon addition of an adjuvant. Alum or aluminum salts-based adjuvants are currently licensed by FDA for human use and are most commonly employed in combination with several antigens. Despite this, they too suffer from various shortcomings such as variable antigen adsorption, induction of weak T-cell-mediated immune response, poor maturation of APCs, and intermittent occurrence of hypersensitivity reactions (29–31). Hence, PAMP-based adjuvants CpG-C ODN 2395 and Poly I:C were used in the study, which has been documented to provide protective immunity against anthrax infection (32, 33). CpG interacts with the TLR9 receptor and initiates a cascade of immuno-stimulatory signaling events culminating in secreting various cytokines and chemokines. This, in turn, causes maturation, differentiation, and proliferation of several immune cell types and eventually perpetuates a pro-inflammatory and TH1-biased immune response (34, 35). Similarly, the interaction of Poly I:C with TLR-3 induces dendritic cells maturation and is also reported to promote the in vitro survival of activated CD4+ cells and in vivo survival of antigen-activated CD8+ cells (36–39). Cytokines also perform a crucial part in the generation of humoral and cell-mediated immunity, and hence were examined in this study post immunization. The inflammatory Th1 cytokines, comprising of IFN-γ, TNF, and IL-2, stimulate predominantly cell-mediated immunity and T-cell proliferation, along with T-helper cell differentiation (40, 41). Immune modulating Th2 cytokines, such as IL-4, IL-6, and IL-10, support humoral immunity and adaptive immunity, mediate allergic diseases, and regulate chronic infections (42, 43).
The following parameters are known to affect the immune response elicited by the particulate-antigen delivery system: (i) administration route, (ii) booster administration, and the interval between boosters (iii) delivery system and amount of antigen, (iv) inclusion of immune modulating molecules like toll-like receptors (TLRs) ligands. Hence, in this study, we evaluated the vaccine potential of various TMC-PA nano-formulations with adjuvants through subcutaneous (SC), intramuscular (IM), and intraperitoneal (IP) delivery platforms. The protective efficacy of the various TMC-PA vaccine formulations was also assessed.
Chitosan (75–85% deacetylated, mol wt 50–190 kDa), Sodium tripolyphosphate (TPP), Poly I:C and Tween 80 were purchased from Sigma (Sigma, India). CpG-C ODN 2395 was purchased from Hycult Biotech Pvt. Ltd. All other materials used were of the analytical or pharmaceutical grade.
Trimethyl-chitosan was derived from chitosan as described previously by Muzarelli and Tanfani and Verheul et al. (17, 44), with some modifications. To begin with, for formic acid–formaldehyde methylation (Eschweiler-Clarke), 3 g of chitosan was dissolved in 10 ml of formic acid, 10 ml of formaldehyde, and 60 ml distilled water added one by one in a round bottom flask. Under reflux condensing conditions, it was heated in an oil bath at 70°C for 118 h, under constant magnetic stirring. The resultant viscous liquid was evaporated and treated with 1N NaOH to increase its pH to 12. The so-formed dimethyl-chitosan (DMC) was washed with distilled water and then dissolved in 25 ml of 1-methyl-2-pyrrolidinone in the presence of 0.5 ml of iodomethane. It was heated up to 40°C on an oil bath under constant magnetic stirring for overnight. The solution was then dissolved in ethanol/diethyl ether mixture (50:50) resulting in the precipitation of TMC. The precipitated TMC was separated by centrifugation and washed thrice with diethyl ether. After complete evaporation of diethyl-ether, TMC was dissolved in 50 ml of 10% NaCl and dialyzed against deionized water for 3 days. After dialysis, TMC was lyophilized to obtain a dried powder form. 1H-nuclear magnetic resonance (1H-NMR) spectroscopy was done to assess the presence of trimethyl groups in chitosan. The degree of quaternization (DQ) of the end product was estimated by:
The nanoparticles were prepared by using the ionic gelation method as described previously (22, 45). A schematic representation of the preparation process is illustrated by Figure S1 in Supplementary Material. Briefly, 10 mg of TMC was dissolved in 5 ml 10 mM HEPES (pH 7.4), and PA was also prepared in the same buffer (8 mg/ml). The protein was dissolved in the TMC solution achieving a final concentration of 0.1 mg/ml under constant stirring. While continuous stirring (350 rpm), 1 ml of the crosslinking agent, TPP solution (1.7 mg/ml) was added to the TMC-PA solution drop by drop to induce ionic complexation. Weight ratio TMC:PA:TPP of 10:0.5:1.7 was taken to get the desired size of particles. An opalescent dispersion forms after TPP addition indicating the formation of nanoparticles. The nanoparticles were collected by centrifugation at 12,000 rpm for 15 min on a 10 µl glycerol bed. The particles were stored at −20°C until further use. The supernatant obtained was used for Micro BCA protein estimation. The protein encapsulation efficiency of the particles was further calculated by using Eq. 2:
For size analysis, a separate batch of nanoparticles was prepared with a minor modification. 0.05% of Tween 80 was added after dissolving TMC in the HEPES buffer.
The particles were dissolved in HEPES buffer pH 7.4 to measure the size, polydispersity index and zeta potential of the particles with the help of a Nanosizer ZS apparatus (Malvern Instruments, Malvern, UK). For scanning electron microscopic (SEM) imaging, particles were coated on a carbon tape on an aluminum stub and then coated with gold particles at 2 kV for 200 s under inert Argon condition. SEM images were captured using an electron microscope [Zeiss EVO40 (Carl Zeiss, Thornwood, NY, USA)]. For transmission electron microscopy [JEM 2100 F (Jeol Ltd., Tokyo, Japan), Electron Microscopy Sciences, Hatfield, PA] the nanoparticles dissolved in HEPES buffer were dropped (20 µl) on a copper mesh grid (46). After evaporating the buffer, the grid was inserted in the instrument and imaging was done in a high vacuum, at 200 kV and direct magnification of 3,000X.
For the in vitro protein release profile of the nanoparticles, a definite amount of particles were dissolved in 1× PBS and were stirred at 100 rpm at 37°C in several aliquots. Each aliquot was taken out from stirring at a specific time point and was centrifuged to spin down the nanoparticles, and the supernatant was taken to estimate the protein released by the nanoparticles in that specific time interval. Consequently, at different time points (2, 4, 6, 8, 10, 12, 24, 48, and 96 h), the release of protein was measured to assess the release of nanoparticles. The protein release was monitored for 96 h.
Mice were obtained from National Centre for Laboratory Animal Sciences, NIN, Hyderabad, India, and were maintained in animal holding room of BSL3 laboratory. All the animal experiments including challenge studies were performed in compliance with Institutional Animal Ethics Committee (Jawaharlal Nehru University) and Council for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA, Ministry of Social Justice and Empowerment, Government of India). Female Balb/c mice (6–8 weeks of age) were immunized with the placebo nanoparticles (Blank NP), TMC-PA nanoparticles (TMC-PA), TMC-PA nanoparticles in combination with CpG (CpG TMC-PA), and Poly I:C (Poly I:C TMC-PA) (n = 21 per group). All these formulations were administered in mice via SC, IM and IP routes. The amount of PA administered with every formulation was 20 μg/mice. CpG (20 μg/mice) as well as Poly I:C (10 μg/mice) were mixed with the nanoparticles right before the administration into mice. After an initial prime dose, two successive booster doses were given at an interval of 7 and 15 days, respectively (as depicted in Figure S2 in Supplementary Material). Sera were collected on 28th and 42nd day and stored at −80°C until further use. The sera samples were then used for the estimation of total IgG, IgG1, and IgG2a endpoint antibody titer against PA (anti-PA) by ELISA. Administration routes used in the study and the corresponding volume of dose injected into each animal are detailed in Table S1 in Supplementary Material.
After 42 days of primary immunization, three mice from each group were sacrificed, and their spleens were crushed to make a single cell suspension. 106 cells were seeded in each well of a 96-well round bottom (Nunc) plate. The cells were stimulated with PA (10 µg/ml) to induce the production of cytokines in the supernatant. Unstimulated and Con A (5 μg/well) treated cells were taken as negative and positive control, respectively. After 48 h of stimulation, the supernatant was collected from each well and was used for cytokine measurement using CBA (Cytometric Bead Array) Mouse Th1/Th2/Th17 Cytokine Kit (BD Biosciences), according to manufacturer instructions. The data were acquired on a BD LSR™ II flow cytometer (Becton Dickinson) and analyzed using the FCAP Array software V3.0 (Becton 519 Dickinson).
At 43rd day, remaining mice in each group were challenged with 0.5 × 103 spores of B. anthracis Ames strain. As a control, 20 µg of PA adsorbed on alum was subcutaneously injected into a separate mice group (n = 12) and then challenged simultaneously. Mice were monitored for 15 days for death events in each group. Survival curve was plotted to compare the efficiency of protection in vaccinated mice groups over control blank nanoparticle immunized mice group.
The results are reported as mean ± SE post data preparation and statistical analysis using GraphPad Prism v6.05 software. The statistical significance of antibody titer and cytokine level data was calculated by using one-way ANOVA, followed by Tukey’s multiple comparisons test. The survival curve for anthrax spore challenge experiment was evaluated using Kaplan–Meier survival estimates (GraphPad Prism, La Jolla, CA, USA). Statistically significant differences between the groups are highlighted by following denotations: * for P-value <0.05, ** for P-values <0.01, *** for P-values <0.001, and **** for P-values <0.0001.
First, a dimethyl derivative of chitosan (DMC) was obtained with formic acid and formaldehyde treatment. Followed by derivation of TMC from DMC, 1H-NMR spectrum was analyzed for the presence of trimethyl group in chitosan. The peak present at 3.33 ppm represents the trimethyl group incorporated into the polymer at the -NH2 group of chitosan, imparting a positive charge to the polymer (Figure S3 in Supplementary Material). The final product was completely soluble in water, and the NMR was carried out by dissolving the final product in D2O. The DQ calculated by the Eq. 1 above was 80.4 ± 5.2%. Negligible O-methylation was observed in the product as seen in the NMR profile.
TMC-PA nanoparticles were prepared by ionic gelation method with the help of TPP anion. The particles were in the nanometer range with an average particle size of 254 ± 24 nm as depicted in Figure 1D. The particles shape and morphology was observed by SEM and TEM as shown in Figures 1A,B, and the nanoparticles were found to be smooth in texture but irregular in shape. The encapsulation efficiency of the particles was found to be 78.33 ± 4.75%. The polydispersity index of the particles was found to be 0.2 indicating fine homogeneity of the particles. Figure 1E reveals the zeta potential of the protein-loaded nanoparticles was found to be 8.84 ± 0.25 mV, which confirmed that the particles carried a positive charge.
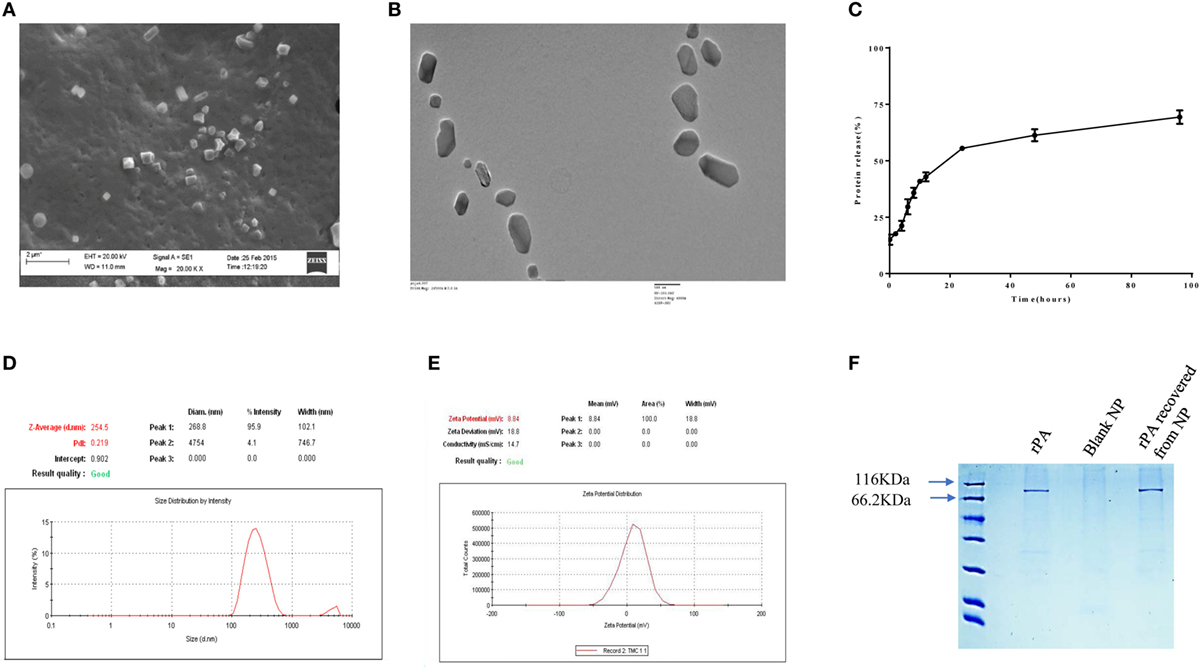
Figure 1. Physical characterization of nanoparticles. (A) scanning electron microscopic and (B) TEM micrographs, (C) in vitro release profile of protective antigen (PA) protein from Trimethyl-chitosan (TMC)-PA nanoparticles after suspension and incubation at 37°C in PBS (50 mg/ml, pH7.4) for indicated time periods and the released protein estimation by micro-BCA assay. (D) Particle size distribution (DLS) and (E) zeta potential of TMC nanoparticles encapsulating PA. (F) SDS PAGE analysis of the purified recombinant PA and the PA recovered from TMC-PA nanoparticles.
The antigen release profile of the TMC-PA nanoparticles was taken into account to estimate the amount of protein released from the particles over a specified period. The release of protein was studied until 96 h, illustrated in Figure 1C. Around 30% of the protein was released in the first 6 h only, and approximately 55% of the protein was released within first 24 h. Afterward, protein release was quite slow, and in next 24 h, only 7% protein was released. For the next 24 h also, only 8% release was observed. Therefore, in the later period, the release was slow and consistent inferring that remaining 30% of the protein would also be released at the same pace at these conditions. This suggests that at physiological pH, PA would be completely released within a week from the particles. Since 50% of the protein was burst released in the first 24 h; therefore, for immunization studies, 20 µg of protein encapsulating nanoparticles were used per mice. The encapsulated protein was analyzed on SDS polyacrylamide upon dissolution of the nanoparticles with 1% NaCl. The protein was observed at its molecular weight of 83KDa, and no protein degradation was observed, as seen in Figure 1F.
The immune response was elucidated in terms of the total IgG titer raised against the antigen. Since the mice were immunized via three routes: subcutaneous, IM, and IP; therefore, the comparison between different groups was also carried out within one frame of the route at a time.
Figure 2 illustrates the highly elevated total IgG titer in almost all the vaccinated mice groups when compared to only PA vaccinated control mice. In all three routes, the descending order for the titers was CpG TMC-PA > Poly I:C TMC-PA > TMC-PA > Poly I:C PA > CpG PA. It could be seen that CpG and Poly I:C adjuvanation increased the total IgG titer from TMC-PA group significantly in almost all the three routes. In the subcutaneous route, the total IgG titer of the CpG TMC-PA group was significantly (P < 0.0001) higher than Poly I:C TMC-PA group on the 28th day (1.45-fold higher), whereas on 42nd day, the two titers were not significantly different. However in IM route, on both 28th and the 42nd day, the total IgG titer of CpG TMC-PA group is significantly (P < 0.01 and P < 0.0001, respectively) higher than Poly I:C TMC-PA group (1.44-fold on the 28th day and 1.5-fold on the 42nd day). Also, the TMC-PA, CpG TMC-PA, and Poly I:C TMC-PA groups showed significantly higher titer than the only PA group. These results elucidate the potential of TMC in mediating a very good humoral response in comparison to the only protein group and upon adjuvanation with CpG or Poly I:C, it increased in a synergistic manner. In all the three routes, the TMC-PA group showed significantly higher titer than CpG PA group and Poly I:C PA group (P < 0.0001). This shows that the TMC acts as a strong and better adjuvant in the form of nanoparticles with the PA protein and CpG as well Poly I:C adjuvanation improves the immune response mounted by TMC-PA alone.
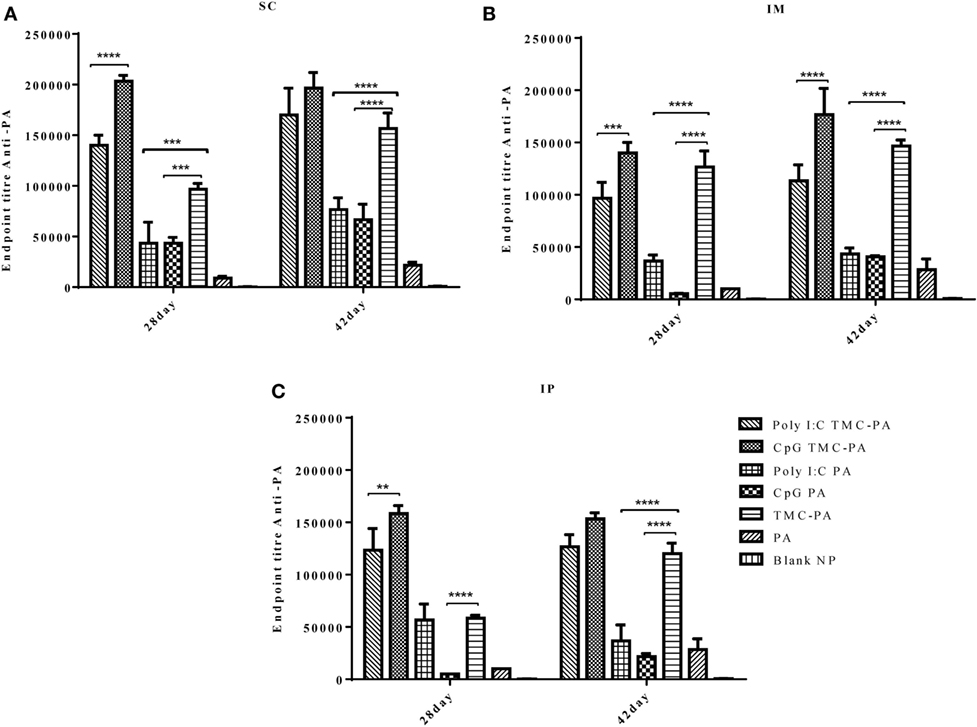
Figure 2. Serum antiprotective antigen (PA) total IgG endpoint titer. Female Balb/c mice (6–8 weeks of age; n = 21) were immunized with 1× PBS, PA, trimethyl-chitosan (TMC) NP-PA, Poly IC PA, Poly I:C TMC NP-PA, CpG PA, and CpG TMC NP-PA via three routes: subcutaneous (SC), intramuscular (IM), and intraperitoneal (IP) with a prime dose and two booster doses with an interval of 7 and 15 days, respectively. The mice were bled on 28th and 42nd day and sera samples were collected from all mice. Serial dilutions of sera from each group were analyzed for anti-PA total IgG titer for (A) SC route, (B) IM route, and (C) IP route. Statistically significant differences between the groups are highlighted (* for P-value <0.05, ** for P-values <0.01, *** for P-values <0.001 and **** for P-values <0.0001).
The levels of IgG1 (Figure 3) were higher in all TMC-PA containing groups in comparison to other groups, and this pattern was similar for all the three routes. On 28th and 42nd day, the TMC-PA group showed the highest titer in SC and IM route. However, for IP route, Poly I:C TMC-PA group showed the highest titer on both 28th and 42nd day followed by the TMC-PA group. The Poly I:C PA and CpG PA groups showed substantially lower levels of IgG1 in comparison to the other nanoformulation vaccinated groups at both the time points and all the routes (P < 0.0001).
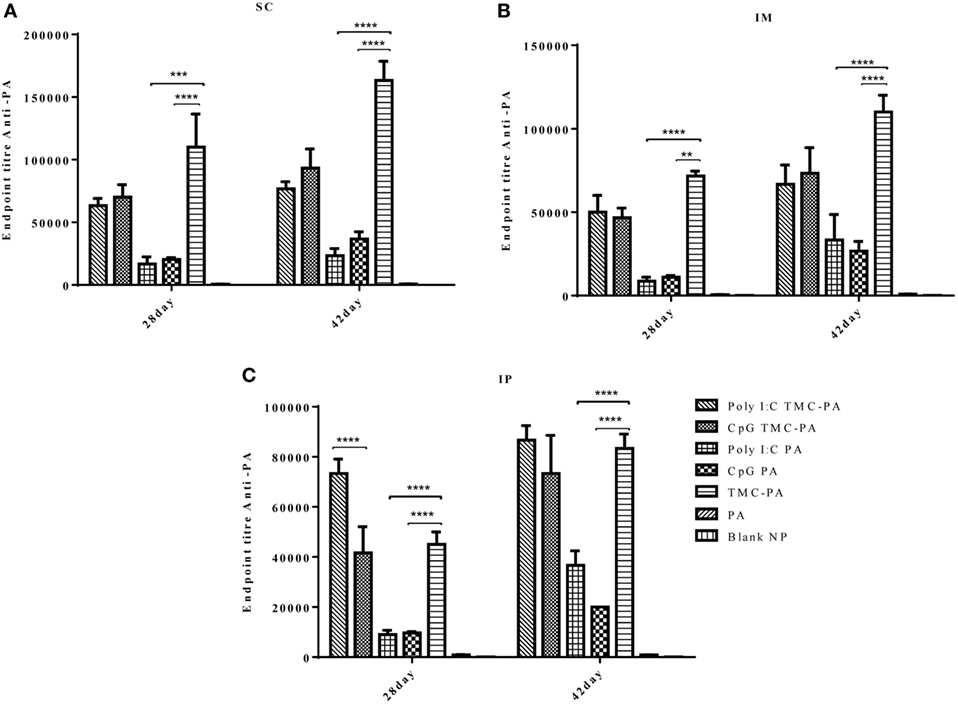
Figure 3. Serum antiprotective antigen (PA) IgG1 endpoint titer. Female Balb/c mice (6–8 weeks of age; n = 21) were immunized with 1× PBS, PA, TMC NP-PA, Poly IC PA, Poly I:C TMC NP-PA, CpG PA, and CpG TMC NP-PA via three routes: subcutaneous (SC), intramuscular (IM), and intraperitoneal (IP) with a prime dose and two booster doses with an interval of 7 and 15 days, respectively. The mice were bled on 28th and 42nd day and sera samples were collected from all mice. Serial dilutions of sera from each group were analyzed for anti-PA IgG1 titer for (A) SC route, (B) IM route, and (C) IP route. Statistically significant differences between the groups are highlighted (* for P-value <0.05, ** for P-values <0.01, *** for P-values <0.001, and **** for P-values <0.0001).
In case of IgG2a subtype (Figure 4), among all routes, on the 28th and 42nd day, the Poly I:C TMC-PA and CpG TMC-PA groups showed the highest titers. The TMC-PA, CpG PA, and Poly I:C PA groups showed significantly lower titers when compared to the CpG and Poly I:C adjuvated TMC-PA groups, through all the routes, and at 42nd day, there was no significant difference in their titers as well, except in SC route. IgG isotypes have been of importance as to determine the Th biasness of the immune response. TMC has been reported to have a Th2-directing effect on the immune response, whereas CpG and Poly I:C have been reported to have a Th1 biasing effect on the immune response.
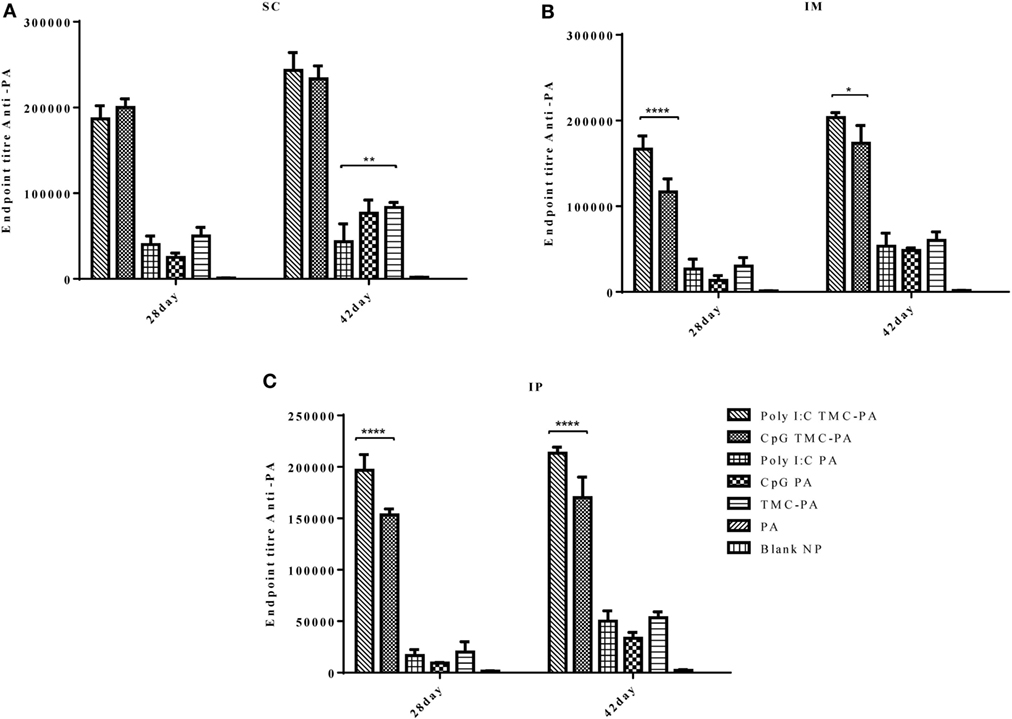
Figure 4. Serum antiprotective antigen (PA) IgG2a endpoint titer. Female Balb/c mice (6–8 weeks of age; n = 21) were immunized with 1× PBS, PA, trimethyl-chitosan (TMC) NP-PA, Poly IC PA, Poly I:C TMC NP-PA, CpG PA, and CpG TMC NP-PA via three routes: subcutaneous (SC), intramuscular (IM), and intraperitoneal (IP) with a prime dose and two booster doses with an interval of 7 and 15 days, respectively. The mice were bled on 28th and 42nd day, and sera samples were collected from all mice. Serial dilutions of sera from each group were analyzed for anti-PA IgG2a titer for (A) SC route, (B) IM route, and (C) IP route. Statistically significant differences between the groups are highlighted (* for P-value <0.05, ** for P-values <0.01, *** for P-values <0.001, and **** for P-values <0.0001).
Based on the cytokine levels and the levels of IgG isotypes (IgG1 and IgG2a), it was seen that CpG TMC-PA SC/IM/IP groups of mice developed a dominant Th1-biased immune response. By 42nd day, the ratio of IgG2a vs IgG1 was found to be 2.4 for SC route, 2.3 for IM route, and 2.3 for IP route (Figures 3 and 4). The IFN-γ levels for these groups for SC, IM, and IP routes were highest among all the groups with no significant difference between them (Figure 5A). IL-4 levels for this group were <10 pg/ml for all three routes with no significant difference among them (Figure 5D). Similarly, IL-10 levels were <150 pg/ml and IL-6 levels were ~200 pg/ml, with no significant difference among all three routes (Figures 5E,F). TNF-α level for the SC, IM, and IP routes were ~500 pg/ml, with no significant difference between different routes (Figure 5B). However, IL-2 levels for IM route were significantly higher (P < 0.0001) than SC and IP routes (Figure 5C).
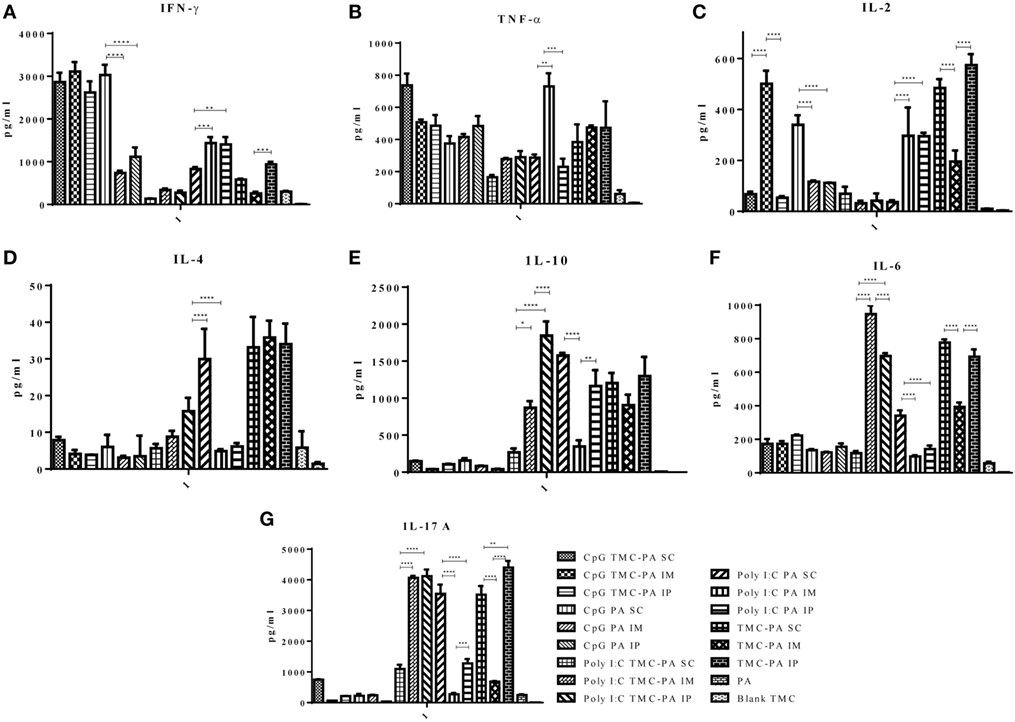
Figure 5. Cytokine levels. Female Balb/c mice (6–8 weeks of age; n = 21) were immunized with 1× PBS, PA, Trimethyl-chitosan (TMC) NP-protective antigen (PA), Poly IC PA, Poly I:C TMC NP-PA, CpG PA, and CpG TMC NP-PA via three routes: subcutaneous (SC), intramuscular (IM), and intraperitoneal (IP) with a prime dose and two booster doses with an interval of 7 and 15 days, respectively. 42 days after the primary immunization, mice (n = 3) from each group were sacrificed, and their spleen single cell suspension was used for (A) IFN-γ, (B) TNF-α, (C) IL-2, (D) IL-4, (E) IL-10, (F) IL-6, and (G) IL-17A cytokines analysis. Statistically significant differences between the groups are highlighted (* for P-value <0.05, ** for P-values <0.01, *** for P-values <0.001, and **** for P-values <0.0001).
This group also showed a Th1-skewed response as the ratio of IgG2a:IgG1 for SC, IM, and IP routes were 2.1, 1.8, and 1.7, respectively at 42nd day (Figures 3 and 4). The IFN-γ levels were significantly different between SC vs IM route (P < 0.0001) and SC vs IP route (P < 0.0001); however, no significant difference was observed between IM vs IP routes (Figure 5A). The Th2 cytokine IL-4 was observed to have very low levels (<10 pg/ml) supporting the Th1 biasness (Figure 5D). TNF-α levels for SC, IM, and IP routes were ~400 pg/ml (Figure 5B). The IL-2 levels of the SC route were significantly higher in comparison to IM (P < 0.0001) and IP (P < 0.0001) routes (Figure 5C). IL-10 and IL-6 levels for all three routes were <160 pg/ml, with no significant difference amongst the three routes (Figures 5E,F).
At 42nd day, the ratios of IgG2a:IgG1 for SC, IM, and IP routes were 3.2, 3.0, and 2.5, respectively, suggesting a dominant Th1 response (Figures 3 and 4). In contrast, the IFN-γ and IL-4 levels do not support a strong Th-1 response (Figures 5A,D). Similarly, the levels of TNF-α and IL-2 were relatively low and exhibited no significant difference between the routes (Figure 5B). Both IL-10 and IL-6 levels were relatively higher with the IL-10 levels of IP route significantly higher than SC (P < 0.0001) and IM route (P < 0.001) (Figures 5E,F). The IL-6 levels for SC, IM and IP routes also revealed a highly significant inter-route difference (P < 0.0001) (Figure 5F).
At 42nd day, the ratios of IgG2a:IgG1 for SC, IM, and IP routes were 1.9, 1.6, and 1.4, respectively, suggesting a moderate Th1 response (Figures 3 and 4). Similarly, the IFN-γ levels were relatively high showing a significant difference between SC vs IM route (P < 0.001) and SC vs IP (P < 0.01) routes whereas the IL-4 levels for all routes were >20 pg/ml, also suggesting a moderate Th-1 response (Figures 5A,D). The levels of TNF-α for the IM route were significantly higher than SC (P < 0.001) and IP (P < 0.0001) routes (Figure 5B). The IL-2 levels of IM and IP routes were found to be significantly higher (P < 0.0001) than SC route (Figure 5C). In contrast to the moderate Th-1 response demonstrated by above parameters, IL-10 values of SC and IP routes were higher, whereas IM route response was moderate (Figure 5E). The IL-6 levels for all three routes were observed to be <350 pg/ml (Figure 5F).
At 42nd day, the ratios of IgG1:IgG2a for SC, IM, and IP routes were 2.9, 3.5, and 1.6, respectively, suggesting a dominant Th2 response (Figures 3 and 4). However, IFN-γ levels and IL-4 levels suggest a Th1 biased immune response (Figures 5A,D). As the values of IL-10 and IL-6 fall in the range of 500–1,200 pg/ml, it suggests a Th2 response, which is also supported by low levels (<500 pg/ml) of TNF-α and IL-2 (Figures 5B–F).
Figure 5G illustrates that the highest levels of IL-17A were observed in the TMC-PA IP group, followed by Poly:IC TMC PA IP group and Poly:IC TMC PA IM being ~4,000 pg/ml. The Poly:IC PA SC and TMC-PA SC groups produced similar levels of this cytokine. Poly:IC PA IP and Poly:IC TMC PA SC groups secreted ~1,000 pg/ml of IL-17. All the remaining groups secreted relatively lower amount of cytokine.
After 6 weeks of immunization, all the mice groups were challenged with 0.5 × 103 spores of B. anthracis Ames strain. The mice were monitored for 2 weeks to observe the protective efficacy of different formulations as shown in Figure 6. The mice vaccinated with Alum and PA were taken as a positive control and exhibited a survival of 83.3%, which was the maximum of all the groups examined. The CpG TMC-PA SC, Poly I:C TMC-PA SC, and Poly I:C TMC-PA IM groups also showed a survival rate of 83.3%, equivalent to the positive control used. Except for the blank nanoparticles and only PA-treated groups, all other mice groups showed a survival rate of more than 50%. The TMC-PA SC group showed a survival of 75% and the TMC-PA IM and TMC-PA IP groups showed a survival of 66.6%. Our study demonstrates that the SC route of immunization showed a better survival rate in comparison to the other two routes.
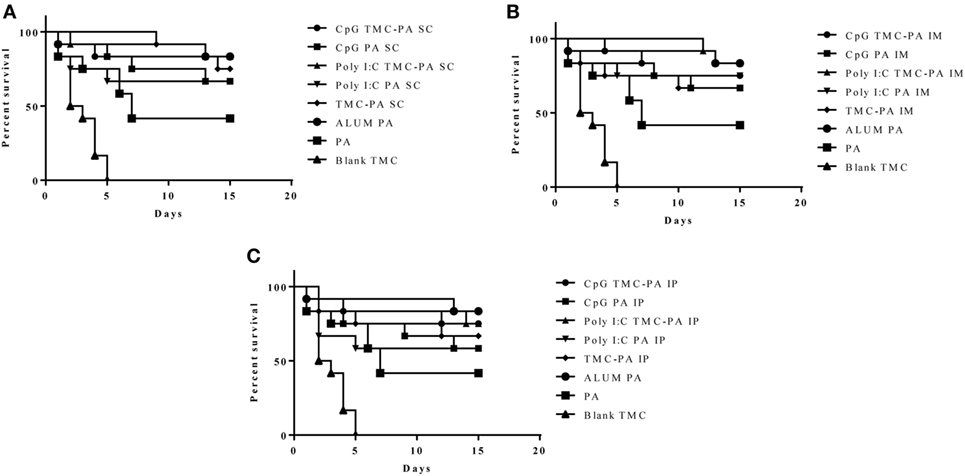
Figure 6. Survival curve. Female Balb/c mice (6–8 weeks) were immunized with 1X PBS, Protective Antigen (PA), Trimethyl-chitosan (TMC) NP-PA, Poly IC PA, Poly I:C TMC NP-PA, CpG PA and CpG TMC NP-PA via three routes: (A) subcutaneous (SC), (B) intramuscular (IM), and (C) intraperitoneal (IP) with a prime dose and two booster doses with an interval of 7 and 15 days, respectively. At 43rd day, mice in each group (n = 12) were challenged with 0.5 × 103 spores of Ames strain of Bacillus anthracis. Mice were monitored for 15 days for death events in each group. Survival curve was plotted to compare the efficiency of protection in vaccinated mice groups over control placebo nanoparticle immunized mice group. As a control, 20 µg PA adsorbed on Alum (group named as ALUMN PA) was subcutaneously injected into a separate mice group (n = 12). The survival curve for anthrax spore challenge experiments was evaluated using Kaplan–Meier survival estimates (GraphPad Prism, La Jolla, CA, USA).
The need to expand the arsenal of adjuvants and development of new subunit vaccine against anthrax with fewer side effects has set out an exploration of the natural, biodegradable polymers with enhanced bioavailability. This study investigated a novel combination of TMC nanoparticles with PA as a potential vaccine candidate against anthrax. To the best of author’s knowledge, this novel approach has not been reported to-date.
The preparation of TMC and its nanoparticles, its subsequent characterization of physicochemical properties, and in vitro release kinetics was studied before performing the murine studies. The synthesis of TMC polymer followed an intermediate step of formation of dimethyl chitosan. This method was adapted to avoid the O-methylation of the chain and chain scission which occurs commonly due to prolonged harsh conditions during the preparation of TMC. A DQ of >30% and lower extent of O-methylation enhances the water solubility of TMC. This property of TMC was leveraged for the preparation of PA-loaded TMC nanoparticles at pH 7 in HEPES buffer (17, 44). The TPP-based ionic gelation method follows the principle of complexation of oppositely charged molecules (protonated amine of TMC and TPP anion), resulting in the formation of nanoparticles (19, 47, 48). Unlike the widely used methods utilizing organic solvents and high temperature, the ionic gelation method is very mild, and thus we did not observe any degradation products in the SDS PAGE analysis (49–51).
The average size of PA-loaded TMC nanoparticles (254 nm) was found to be suitable for an efficient uptake by APCs. As the TMC nanoparticles were positively charged at physiological pH, an electrostatic interaction with the negatively charged cell membrane is highly likely. This is in alignment with a previously reported study where insulin, ovalbumin (OVA), and tetanus toxoid-based chitosan nanoparticles were shown to have a similar particle size, charge distribution (47, 52–54). In terms of the release kinetics of protein, the observed pattern of an initial burst release followed by a controlled release is also supported by other studies (52, 53). Thus, the in vitro antigen release study confirms the long-term antigen-releasing capacity of PA-TMCs without any protein degradation post-encapsulation.
After successful preparation and characterization of TMC nanoparticles, mice studies were conducted. Although the nasal route has been extensively explored for TMC nanoparticle-based immunization (19, 23, 47, 52, 55), we did not achieve a satisfactory immune response modulation. This could potentially be attributed to the large size of TMC-PA nanoparticles and subsequently its poor diffusion across the epithelial layer covering nasal-associated lymphoid tissue. In a similar instance, Boonyo et al. and Hagenaars et al. reported a subdued intranasal immune response for an OVA and HI-loaded TMC nanoparticle system (52, 55). Thus, the SC, IM, and IP routes were subsequently pursued for immunization.
The results of our study, as evident from Figures 2–6, demonstrated an effective immune response for TMC nanoparticles by all three routes (SC, IM, and IP). The striking results were that the TMC-PA groups via all routes were able to produce a sufficient amount of anti-PA titer as well as provided nearly 60–70% protection in anthrax challenged mice. More interestingly, the adjuvants CpG and Poly I:C behaved synergistically with TMC-PA in terms of generating an elevated anti-PA antibody titer and survival efficacy of nearly 70–80%. However, SC route was found be more effective in generating the highest IgG titer. Such response could be attributed to longer persistence, slower clearance, and prolonged antigen presentation after SC administration (56). Consequently, we could relate to the large number of vaccines, which are currently being administered via SC route like mumps, rubella, yellow fever, Japanese encephalitis, measles, Haemophilus influenza type b, inactivated polio vaccine, and S. pneumonia (25, 56).
In line with the previously reported studies, the TMC-PA group generated a Th2-biased immune response (25, 34). On the other hand, Poly I:C and CpG are reportedly Th1 biasing adjuvants (57–60). CpG alone has been reported to establish a Th1-biased immune response with several antigens (61–63). In this study, Poly I:C administration modulated the immune response toward Th1 with PA and TMC-PA nanoparticles. Supporting the earlier evidence, the CpG PA groups generated a Th1 response via all the three routes. Despite the fact that the PA is a Th2 biased antigen and TMC is also a Th2 biased adjuvant, the CpG adjuvated TMC-PA-treated groups also generated a dominant Th1 response via all the three routes. This observation is supported by Slütter and Jiskoot (34), which reported that the TMC-CpG nanoparticles encapsulating OVA as an antigen generated a 10-fold higher Ig2a titer than the TMC-TPP nanoparticles encapsulating OVA, thus provoked a strong Th1 response. The same report also revealed that the antigen-stimulated splenocytes secreted elevated levels of IFN-γ. This was also observed in our study that the CpG PA and CpG adjuvated TMC-PA groups showed highest levels of IFN-γ. In another study by Wang et al., virus-like particles encapsulating CpG-gold nanoparticle conjugates elicited a strong IFN-γ secretion (64). Therefore, we conclude that CpG enhances the production of IFN-γ, upon adjuvanation with an antigen. However, the levels of IL-4 produced by the splenocytes were low for all the groups, but the TMC-PA group relatively secreted the highest IL-4 irrespective of the route of immunization. Although, the CD4+ and CD8+ T-cell count gives a clear indication about the Th1 or Th2 response, but in this study, we did not analyze these parameters. However, the cytokines levels do indicate the direction of biasness of the immune response.
IL-17 T-helper cells are well known to serve as a bridge between innate and acquired immunity. Upon sensing the cytokines in the mucosal sites, the memory effector subset of Th17 cells can transform into Th1 or Th2 phenotype. IL-17A is the hallmark cytokine of the proinflammatory Th cell subset of Th17. As evident from Figure 5, Poly I:C PA and Poly I:C TMC-PA groups generated high levels of IL-17A. The TMC-PA group also secreted high levels of IL-17A, except the IM route. In a study by Holm et al., Poly I:C was reported to stimulate the production of IL-17A and IL-21 directly and consequently, drive the human naïve CD4+ T-cells differentiation, whereas CpG did not affect IL-17A on either mRNA or protein level (65). Similarly, Vultaggio et al. reported a positive correlation between Poly I:C and 1L-17A production (66). The chitin-based adjuvants have also been reported to be an immune response modulator (67) with a size-dependent response on macrophage IL-17A production (28). Thus, the growing scientific evidence indicates that TMC plays a role in stimulating IL-17A production. However, the mechanism of action remains unclear and need further exploration.
CpG and Poly I:C are PAMP-based adjuvants and act by initiating a cascade of innate immune system stimulation by interacting with TLR-3 and TLR-9, respectively. However, the immune modulation mechanism behind TMC nanoparticles remains elusive. We hypothesize that this immune-modulated response is due to the positively charged TMC nanoparticles, which might act as “danger signal” for dendritic cells.
In conclusion, the PA-loaded TMC nanoparticles as well as CpG and Poly I:C adjuvanted TMC-PA formulations elicited strong IgG antibody response via SC, IM, and IP routes in mice. However, the SC route showed the strongest response for IgG titer and survival efficacy. The TMC nanoparticle formulations were able to protect the mice against anthrax challenge and were comparable in protective efficacy to alum. In our opinion, TMC-based formulations possess a great potential as a vaccine candidate against anthrax, and further studies are needed to explore its adjuvant mechanisms.
All mice experiments were performed in compliance with Institutional Animal Ethics Committee (Jawaharlal Nehru University) and Council for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA, Ministry of Social Justice and Empowerment, Government of India). Mice were housed in the individually ventilated animal caging system.
Experiments conceptualized and designed by AM. Performed by AM, MG, and RM. Data analysis by AM. Wrote the article: AM, HG, and RB. Supervised by RB.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank all the members of the laboratory for thoughtful and lively discussions during this work. The author AM would like to thank Council of Scientific and Industrial Research (CSIR), India, Department of Biotechnology (DBT), India, and JC Bose fellowship for providing financial support during this research term.
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.00562/full#supplementary-material.
1. Turnbull PC; World Health Organization. Guidelines for the Surveillance and Control of Anthrax in Humans and Animals. Geneva, Switzerland: World Health Organization, Department of Communicable Diseases Surveillance and Response (1998).
2. Kamal S, Rashid AM, Bakar M, Ahad M. Anthrax: an update. Asian Pac J Trop Biomed (2011) 1(6):496–501. doi:10.1016/S2221-1691(11)60109-3
3. Jernigan DB, Raghunathan PL, Bell BP, Brechner R, Bresnitz EA, Butler JC, et al. Investigation of bioterrorism-related anthrax, United States, 2001: epidemiologic findings. Emerg Infect Dis (2002) 8(10):1019. doi:10.3201/eid0810.020353
4. Plotkin S, Grabenstein JD. Countering anthrax: vaccines and immunoglobulins. Clin Infect Dis (2008) 46(1):129–36. doi:10.1086/523578
5. Taft SC, Weiss AA. Neutralizing activity of vaccine-induced antibodies to two Bacillus anthracis toxin components, lethal factor and edema factor. Clin Vaccine Immunol (2008) 15(1):71–5. doi:10.1128/CVI.00321-07
6. Strom BL, Durch JS, Zwanziger LL, Joellenbeck LM. The Anthrax Vaccine: Is It Safe? Does It Work? Washington, DC: National Academies Press (2002).
7. Friedlander AM, Welkos S, Ivins B. Anthrax vaccines. In: Koehler T, editor. Anthrax. Frederick, MD: Springer (2002). p. 33–60.
8. Brossier F, Mock M. Toxins of Bacillus anthracis. Toxicon (2001) 39(11):1747–55. doi:10.1016/S0041-0101(01)00161-1
9. Liu S, Moayeri M, Leppla SH. Anthrax lethal and edema toxins in anthrax pathogenesis. Trends Microbiol (2014) 22(6):317–25. doi:10.1016/j.tim.2014.02.012
10. Mikszta JA, Sullivan VJ, Dean C, Waterston AM, Alarcon JB, Dekker JP III, et al. Protective immunization against inhalational anthrax: a comparison of minimally invasive delivery platforms. J Infect Dis (2005) 191(2):278–88. doi:10.1086/426865
11. Irvine DJ, Swartz MA, Szeto GL. Engineering synthetic vaccines using cues from natural immunity. Nat Mater (2013) 12(11):978–90. doi:10.1038/nmat3775
12. Gregory AE, Titball R, Williamson D. Vaccine delivery using nanoparticles. Front Cell Infect Microbiol (2013) 3:13. doi:10.3389/fcimb.2013.00013
13. Peek LJ, Middaugh CR, Berkland C. Nanotechnology in vaccine delivery. Adv Drug Deliv Rev (2008) 60(8):915–28. doi:10.1016/j.addr.2007.05.017
14. Amidi M, Mastrobattista E, Jiskoot W, Hennink WE. Chitosan-based delivery systems for protein therapeutics and antigens. Adv Drug Deliv Rev (2010) 62(1):59–82. doi:10.1016/j.addr.2009.11.009
15. Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release (2012) 161(2):505–22. doi:10.1016/j.jconrel.2012.01.043
16. Verheul RJ, Amidi M, van Steenbergen MJ, van Riet E, Jiskoot W, Hennink WE. Influence of the degree of acetylation on the enzymatic degradation and in vitro biological properties of trimethylated chitosans. Biomaterials (2009) 30(18):3129–35. doi:10.1016/j.biomaterials.2009.03.013
17. Verheul RJ, Amidi M, van der Wal S, van Riet E, Jiskoot W, Hennink WE. Synthesis, characterization and in vitro biological properties of O-methyl free N, N, N-trimethylated chitosan. Biomaterials (2008) 29(27):3642–9. doi:10.1016/j.biomaterials.2008.05.026
18. Hagenaars N, Mania M, de Jong P, Que I, Nieuwland R, Slütter B, et al. Role of trimethylated chitosan (TMC) in nasal residence time, local distribution and toxicity of an intranasal influenza vaccine. J Control Release (2010) 144(1):17–24. doi:10.1016/j.jconrel.2010.01.027
19. Amidi M, Romeijn SG, Verhoef JC, Junginger HE, Bungener L, Huckriede A, et al. N-trimethyl chitosan (TMC) nanoparticles loaded with influenza subunit antigen for intranasal vaccination: biological properties and immunogenicity in a mouse model. Vaccine (2007) 25(1):144–53. doi:10.1016/j.vaccine.2006.06.086
20. Verheul RJ, Hagenaars N, van Es T, van Gaal EV, de Jong PH, Bruijns S, et al. A step-by-step approach to study the influence of N-acetylation on the adjuvanticity of N, N, N-trimethyl chitosan (TMC) in an intranasal nanoparticulate influenza virus vaccine. Eur J Pharm Sci (2012) 45(4):467–74. doi:10.1016/j.ejps.2011.10.001
21. Tafaghodi M, Saluja V, Kersten GF, Kraan H, Slütter B, Amorij J-P, et al. Hepatitis B surface antigen nanoparticles coated with chitosan and trimethyl chitosan: impact of formulation on physicochemical and immunological characteristics. Vaccine (2012) 30(36):5341–8. doi:10.1016/j.vaccine.2012.06.035
22. Slütter B, Plapied L, Fievez V, Sande MA, des Rieux A, Schneider Y-J, et al. Mechanistic study of the adjuvant effect of biodegradable nanoparticles in mucosal vaccination. J Control Release (2009) 138(2):113–21. doi:10.1016/j.jconrel.2009.05.011
23. Bal SM, Slütter B, Verheul R, Bouwstra JA, Jiskoot W. Adjuvanted, antigen loaded N-trimethyl chitosan nanoparticles for nasal and intradermal vaccination: adjuvant-and site-dependent immunogenicity in mice. Eur J Pharm Sci (2012) 45(4):475–81. doi:10.1016/j.ejps.2011.10.003
24. Hagenaars N, Verheul RJ, Mooren I, de Jong PH, Mastrobattista E, Glansbeek HL, et al. Relationship between structure and adjuvanticity of N, N, N-trimethyl chitosan (TMC) structural variants in a nasal influenza vaccine. J Control Release (2009) 140(2):126–33. doi:10.1016/j.jconrel.2009.08.018
25. Bal SM, Slütter B, van Riet E, Kruithof AC, Ding Z, Kersten GF, et al. Efficient induction of immune responses through intradermal vaccination with N-trimethyl chitosan containing antigen formulations. J Control Release (2010) 142(3):374–83. doi:10.1016/j.jconrel.2009.11.018
26. Slütter B, Soema PC, Ding Z, Verheul R, Hennink W, Jiskoot W. Conjugation of ovalbumin to trimethyl chitosan improves immunogenicity of the antigen. J Control Release (2010) 143(2):207–14. doi:10.1016/j.jconrel.2010.01.007
27. Da Silva CA, Chalouni C, Williams A, Hartl D, Lee CG, Elias JA. Chitin is a size-dependent regulator of macrophage TNF and IL-10 production. The J Immunol (2009) 182(6):3573–82. doi:10.4049/jimmunol.0802113
28. Da Silva CA, Hartl D, Liu W, Lee CG, Elias JA. TLR-2 and IL-17A in chitin-induced macrophage activation and acute inflammation. J Immunol (2008) 181(6):4279–86. doi:10.4049/jimmunol.181.6.4279
29. Tomljenovic L, Shaw CA. Aluminum vaccine adjuvants: are they safe? Curr Med Chem (2011) 18(17):2630–7. doi:10.2174/092986711795933740
30. Petrovsky N, Aguilar JC. Vaccine adjuvants: current state and future trends. Immunol Cell Biol (2004) 82(5):488–96. doi:10.1111/j.0818-9641.2004.01272.x
31. Lindblad EB. Aluminium compounds for use in vaccines. Immunol Cell Biol (2004) 82(5):497–505. doi:10.1111/j.0818-9641.2004.01286.x
32. Kachura MA, Hickle C, Kell SA, Sathe A, Calacsan C, Kiwan R, et al. A CpG-Ficoll nanoparticle adjuvant for anthrax protective antigen enhances immunogenicity and provides single-immunization protection against inhaled anthrax in monkeys. J Immunol (2016) 196(1):284–97. doi:10.4049/jimmunol.1501903
33. Sloat BR, Cui Z. Nasal immunization with anthrax protective antigen protein adjuvanted with polyriboinosinic–polyribocytidylic acid induced strong mucosal and systemic immunities. Pharm Res (2006) 23(6):1217–26. doi:10.1007/s11095-005-9078-7
34. Slütter B, Jiskoot W. Dual role of CpG as immune modulator and physical crosslinker in ovalbumin loaded N-trimethyl chitosan (TMC) nanoparticles for nasal vaccination. J Control Release (2010) 148(1):117–21. doi:10.1016/j.jconrel.2010.06.009
35. Friede M, Aguado M. Need for new vaccine formulations and potential of particulate antigen and DNA delivery systems. Adv Drug Deliv Rev (2005) 57(3):325–31. doi:10.1016/j.addr.2004.10.001
36. Bardel E, Doucet-Ladeveze R, Mathieu C, Harandi AM, Dubois B, Kaiserlian D. Intradermal immunisation using the TLR3-ligand poly (I: C) as adjuvant induces mucosal antibody responses and protects against genital HSV-2 infection. npj Vaccines (2016) 1:16010.
37. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature (2001) 413(6857):732–8. doi:10.1038/35099560
38. Gelman AE, Zhang J, Choi Y, Turka LA. Toll-like receptor ligands directly promote activated CD4+ T cell survival. J Immunol (2004) 172(10):6065–73. doi:10.4049/jimmunol.172.10.6065
39. Salem ML, Kadima AN, Cole DJ, Gillanders WE. Defining the antigen-specific T-cell response to vaccination and poly (I: C)/TLR3 signaling: evidence of enhanced primary and memory CD8 T-cell responses and antitumor immunity. J Immunother (2005) 28(3):220–8. doi:10.1097/01.cji.0000156828.75196.0d
40. Wei H, Tan K, Sun R, Yin L, Zhang J, Pu Y. Aberrant production of Th1/Th2/Th17-related cytokines in serum of C57BL/6 mice after short-term formaldehyde exposure. Int J Environ Res Public Health (2014) 11(10):10036–50. doi:10.3390/ijerph111010036
41. Hoyer KK, Dooms H, Barron L, Abbas AK. Interleukin-2 in the development and control of inflammatory disease. Immunol Rev (2008) 226(1):19–28. doi:10.1111/j.1600-065X.2008.00697.x
42. Hofmann SR, Ettinger R, Zhou Y-J, Gadina M, Lipsky P, Siegel R, et al. Cytokines and their role in lymphoid development, differentiation and homeostasis. Curr Opin Allergy Clin Immunol (2002) 2(6):495–506. doi:10.1097/00130832-200212000-00004
43. Jones SA. Directing transition from innate to acquired immunity: defining a role for IL-6. J Immunol (2005) 175(6):3463–8. doi:10.4049/jimmunol.175.6.3463
44. Muzzarelli RA, Tanfani F. The N-permethylation of chitosan and the preparation of N-trimethyl chitosan iodide. Carbohydr Polym (1985) 5(4):297–307. doi:10.1016/0144-8617(85)90037-2
45. Slütter B, Bal S, Keijzer C, Mallants R, Hagenaars N, Que I, et al. Nasal vaccination with N-trimethyl chitosan and PLGA based nanoparticles: nanoparticle characteristics determine quality and strength of the antibody response in mice against the encapsulated antigen. Vaccine (2010) 28(38):6282–91. doi:10.1016/j.vaccine.2010.06.121
46. Manish M, Rahi A, Kaur M, Bhatnagar R, Singh S. A single-dose PLGA encapsulated protective antigen domain 4 nanoformulation protects mice against Bacillus anthracis spore challenge. PLoS One (2013) 8(4):e61885. doi:10.1371/journal.pone.0061885
47. Amidi M, Romeijn SG, Borchard G, Junginger HE, Hennink WE, Jiskoot W. Preparation and characterization of protein-loaded N-trimethyl chitosan nanoparticles as nasal delivery system. J Control Release (2006) 111(1):107–16. doi:10.1016/j.jconrel.2005.11.014
48. Vila A, Sánchez A, Janes K, Behrens I, Kissel T, Jato JLV, et al. Low molecular weight chitosan nanoparticles as new carriers for nasal vaccine delivery in mice. Eur J Pharm Biopharm (2004) 57(1):123–31. doi:10.1016/j.ejpb.2003.09.006
49. Xu Y, Du Y, Huang R, Gao L. Preparation and modification of N-(2-hydroxyl) propyl-3-trimethyl ammonium chitosan chloride nanoparticle as a protein carrier. Biomaterials (2003) 24(27):5015–22. doi:10.1016/S0142-9612(03)00408-3
50. Calvo P, Remuñan-López C, Vila-Jato JL, Alonso MJ. Chitosan and chitosan/ethylene oxide-propylene oxide block copolymer nanoparticles as novel carriers for proteins and vaccines. Pharm Res (1997) 14(10):1431–6. doi:10.1023/A:1012128907225
51. Mao S, Bakowsky U, Jintapattanakit A, Kissel T. Self-assembled polyelectrolyte nanocomplexes between chitosan derivatives and insulin. J Pharm Sci (2006) 95(5):1035–48. doi:10.1002/jps.20520
52. Boonyo W, Junginger HE, Waranuch N, Polnok A, Pitaksuteepong T. Chitosan and trimethyl chitosan chloride (TMC) as adjuvants for inducing immune responses to ovalbumin in mice following nasal administration. J Control Release (2007) 121(3):168–75. doi:10.1016/j.jconrel.2007.05.025
53. Siddhapura K, Harde H, Jain S. Immunostimulatory effect of tetanus toxoid loaded chitosan nanoparticles following microneedles assisted immunization. Nanomedicine (2016) 12(1):213–22. doi:10.1016/j.nano.2015.10.009
54. Mukhopadhyay P, Mishra R, Rana D, Kundu PP. Strategies for effective oral insulin delivery with modified chitosan nanoparticles: a review. Prog Polym Sci (2012) 37(11):1457–75. doi:10.1016/j.progpolymsci.2012.04.004
55. Hagenaars N, Mastrobattista E, Verheul RJ, Mooren I, Glansbeek HL, Heldens JG, et al. Physicochemical and immunological characterization of N, N, N-trimethyl chitosan-coated whole inactivated influenza virus vaccine for intranasal administration. Pharm Res (2009) 26(6):1353–64. doi:10.1007/s11095-009-9845-y
56. Hickling J, Jones K, Friede M, Zehrung D, Chen D, Kristensen D. Intradermal delivery of vaccines: potential benefits and current challenges. Bull World Health Organ (2011) 89(3):221–6. doi:10.2471/BLT.10.079426
57. Jin B, Wang RY, Qiu Q, Sugauchi F, Grandinetti T, Alter HJ, et al. Induction of potent cellular immune response in mice by hepatitis C virus NS3 protein with double-stranded RNA. Immunology (2007) 122(1):15–27. doi:10.1111/j.1365-2567.2007.02607.x
58. Salaun B, Greutert M, Romero P. Toll-like receptor 3 is necessary for dsRNA adjuvant effects. Vaccine (2009) 27(12):1841–7. doi:10.1016/j.vaccine.2009.01.044
59. Trumpfheller C, Caskey M, Nchinda G, Longhi MP, Mizenina O, Huang Y, et al. The microbial mimic poly IC induces durable and protective CD4+ T cell immunity together with a dendritic cell targeted vaccine. Proc Natl Acad Sci U S A (2008) 105(7):2574–9. doi:10.1073/pnas.0711976105
60. Navabi H, Jasani B, Reece A, Clayton A, Tabi Z, Donninger C, et al. A clinical grade poly I: C-analogue (Ampligen®) promotes optimal DC maturation and Th1-type T cell responses of healthy donors and cancer patients in vitro. Vaccine (2009) 27(1):107–15. doi:10.1016/j.vaccine.2008.10.024
61. Weeratna RD, Brazolot Millan CL, McCluskie MJ, Davis HL. CpG ODN can re-direct the Th bias of established Th2 immune responses in adult and young mice. FEMS Immunol Med Microbiol (2001) 32(1):65–71. doi:10.1111/j.1574-695X.2001.tb00535.x
62. Millan CLB, Weeratna R, Krieg AM, Siegrist C-A, Davis HL. CpG DNA can induce strong Th1 humoral and cell-mediated immune responses against hepatitis B surface antigen in young mice. Proc Natl Acad Sci U S A (1998) 95(26):15553–8. doi:10.1073/pnas.95.26.15553
63. Chu RS, Targoni OS, Krieg AM, Lehmann PV, Harding CV. CpG oligodeoxynucleotides act as adjuvants that switch on T helper 1 (Th1) immunity. J Exp Med (1997) 186(10):1623–31. doi:10.1084/jem.186.10.1623
64. Wang Y, Wang Y, Kang N, Liu Y, Shan W, Bi S, et al. Construction and immunological evaluation of CpG-Au@ HBc virus-like nanoparticles as a potential vaccine. Nanoscale Res Lett (2016) 11(1):1–9. doi:10.1186/s11671-016-1554-y
65. Holm CK, Petersen CC, Hvid M, Petersen L, Paludan SR, Deleuran B, et al. TLR3 ligand polyinosinic: polycytidylic acid induces IL-17A and IL-21 synthesis in human Th cells. J Immunol (2009) 183(7):4422–31. doi:10.4049/jimmunol.0804318
66. Vultaggio A, Nencini F, Pratesi S, Petroni G, Romagnani S, Maggi E. Poly (I: C) promotes the production of IL-17A by murine CD1d-driven invariant NKT cells in airway inflammation. Allergy (2012) 67(10):1223–32. doi:10.1111/j.1398-9995.2012.02876.x
Keywords: anthrax, vaccine, protective antigen, trimethylchitosan nanoparticles, CpG, PolyI:C
Citation: Malik A, Gupta M, Mani R, Gogoi H and Bhatnagar R (2018) Trimethyl Chitosan Nanoparticles Encapsulated Protective Antigen Protects the Mice Against Anthrax. Front. Immunol. 9:562. doi: 10.3389/fimmu.2018.00562
Received: 18 December 2017; Accepted: 06 March 2018;
Published: 20 March 2018
Edited by:
África González-Fernández, Centro de Investigaciones Biomédicas (CINBIO), SpainReviewed by:
Noemi Csaba, Universidade de Santiago de Compostela, SpainCopyright: © 2018 Malik, Gupta, Mani, Gogoi and Bhatnagar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rakesh Bhatnagar, cmFrZXNoYmhhdG5hZ2FyQGpudS5hYy5pbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.